

Introduction
Atopic dermatitis (AD) is a common inflammatory skin disease associated with significant medical burden, affecting 15% to 20% of children and 1% to 3% of adults worldwide.1 The prevalence of AD has increased by 2-fold to 3-fold during the past decade in Western countries. Although the cause of this increase remains unknown, meta-analyses have indicated that the risk of AD is lower when associated with potentially protective environmental factors during childhood, such as 3 or more siblings, daycare, pet ownership, and farm residence.2,3 In contrast, the risk of AD increases with at least 1 clinically apparent infectious disease in the first 6 months of life.2 Antibiotic use has also been associated with an increase in AD risk.4 These environmental associations strongly support the role of microbes in the cause of AD. This concept is not new, because the hygiene hypothesis posed over 30 years ago suggested that exposure to a diverse range of nonpathogenic microbes in early life is necessary for development of functional immune systems to appropriately respond to environmental stimuli. Today we are in an exciting time in which the molecular understanding of host–microbe interactions is providing a rational explanation for such clinical associations with the environment, and the opportunity to develop new approaches to therapy.
Atopic dermatitis is a complex and heterogeneous disease affected by a variety of factors such as host genetics, altered skin barrier function/structure, immunological abnormalities, and environmental factors, including exposure to specific pathogens such as Staphylococcus aureus. Evidence accumulated in the past decade has also shown that specific strains of other microbes within the community of resident skin bacteria suppress inflammation, stimulate the adaptive and innate immune system, and produce diverse molecules with antimicrobial activity. These actions of the microbial community are essential to maintain cutaneous homeostasis and defend against pathogenic microorganisms. Recent evidence has also shown that a loss of commensal microbes with capacity to produce antimicrobial activity increases the risk of skin colonization by S aureus. This review provides an overview of the current evidence that the microbial ecosystem of the skin is a critical part of the pathophysiology of AD.
The Microbiome and Human Health
An abundant and diverse population of microbial organisms colonizes epithelial surfaces of mammals.5 This community has been called the “microbiome,” and it includes bacteria, fungi, protozoa, and viruses. Most current research has focused on the function of bacteria residing on and within the host, but evidence also exists for important roles of viruses as well as other microbial kingdoms. Microbe–host interactions occur on all of the epithelial surfaces of the body, including airway, intestine, and skin, but most microbiome research has focused on the intestinal bacterial community. This emphasis may have arisen because of the accessibility of fecal samples as a surrogate for understanding bacteria in the gut, the relative ease of DNA extraction and sequencing from feces, and the assumption that the large and absorptive surface area in the intestine would be the most likely site for microbes to influence human health. Indeed, the probiotic industry has been highly successful, with many strong believers in these interventions despite minimal supporting evidence from randomized controlled trials. In contrast, skin has been less well studied. This may be in part based on the assumption that the total surface area of adult human skin was only 2 m2, which is much smaller than that of other epithelial tissues such as gut and lung. However, this estimation ignored the inner follicular surfaces. Given the complex structure of skin with appendages such as hair follicles, sebaceous glands, and sweat glands, the human skin provides an organ with an enormous epithelial surface area and a unique anatomy that is optimally designed to harbor and protect microbes.6 Furthermore, as is discussed later, the epidermis normally enables penetration of a fraction of surface bacteria into the dermis.7 This indicates that skin is not a barrier that separates us from the microbiome, but it is indeed a large interface where the microbiome interacts with the host and can influence systemic health.
Given the abundant evidence showing that the community of symbiotic bacteria at multiple epithelial surfaces is necessary for human health, hologenome theory is a useful concept for better understanding its role in homeostasis and diseases.8,9 In this theory, a holobiont (a host along with all of its associated symbiotic microbes) should be considered as a single unit of life and a joint process of evolution. Many clear examples exist of the function of the holobiont in ecology (eg, clownfish and sea anemone, plants and nitrogen-fixing bacteria), but the understanding of mutualistic interactions between the genomes of humans and our microbiome is vastly incomplete.
The Microbiome and the Skin Immune System
Our group first demonstrated a unique beneficial interaction between the skin and Staphylococcus epidermidis, a predominant bacterial species found on human skin. In 2009, it was quite unexpected when a bacterium was found to be anti-inflammatory, and that a lipoteichoic acid with unique structure produced by this bacterial species could block proinflammatory signaling triggered by small double-stranded noncoding RNA released from cells after epithelial damage.10,11 In this case, the bacterial product prevents excessive inflammation after tissue injury. Other examples of beneficial functions from S epidermidis have been subsequently discovered. For example, a lipopeptide from S epidermidis defends the host from pathogen infection by enhancing production of antimicrobial peptides (AMPs) in human keratinocytes and mouse skin.12,13 Other groups also have shown that colonization with S epidermidis enhances CD8+ skin-resident T cell functions via activation of interleukin (IL)-1 signaling.14 This event promotes up-regulation of AMP production in keratinocytes, limiting pathogen invasion.15 Indeed, mono-colonization of germ-free mouse skin by S epidermidis was more protective against pathogen infection in comparison with germ-free skin.14 In addition, S epidermidis–specific CD8+ T-cells express genes with immunoregulatory and tissue repair signatures, facilitating wound repair.16 Microbes residing within the hair follicle promote differentiation of regulatory T-cells and appear to be necessary for differentiation of skin stem cells and establishment of immune tolerance to commensal microbes.17 Most recently, our group has demonstrated that selected strains of S epidermidis protect the skin from neoplasia induced by ultraviolet through the production of a nucleobase analog with potent antimetabolite activity.18
Dysbiosis in Atopic Dermatitis
Environmental factors, including pH, temperature, dryness, host genetics, antibiotic use, and hygiene practices, play a critical role in the maintenance and stability of our microbiome. Dysregulation of these systems can disrupt the structure of the microbial community, a condition known as “dysbiosis,” which often reflects dominance by 1 microbe and a decrease in the richness and diversity of microbes. In many cases, dysbiosis can trigger the disruption of skin homeostasis and the development of disease not only by detrimental effects from a dominant pathogen, but also from loss of the symbiotic interactions from other beneficial microbes.
Dysbiosis has been well characterized in AD (Table 1). In particular, subjects with AD are highly colonized by S aureus19 and show a loss in bacterial diversity on the skin.20 Depending on methods used for detecting S aureus, 30% to 100% of the subjects with AD are colonized by S aureus, which is 1 of the most influential environmental factors in the pathogenesis of this disease.19,21 Patients with AD who had a history of infection by herpes simplex virus especially have greater frequency and abundance of skin colonization by S aureus than those without such a history.22 S aureus exacerbates skin inflammation and allergic reactions by diverting both adaptive and innate immune responses through multiple mechanisms. S aureus isolated from the skin of subjects with AD release staphylococcal enterotoxin B, which acts as a superantigen and causes inflammation by inducing uncontrolled activation of lymphocytes and macrophages.23 Phenol-soluble modulin (PSM)-α from S aureus stimulate production of IL-36α and IL-1α in keratinocytes, which in turn induces IL-17 production in γδT cells, innate lymphoid cell type 3, and CD4+ T cells, and enhances neutrophil recruitment.24,25 S aureus also triggers TH2 skewing by initiating the production of thymic stromal lymphopoietin and stimulating mast cell degranulation through TLR2-dependent mechanism.26–28 In addition, S aureus disrupts the proteolytic balance in the skin by producing various extracellular proteases and enhancing production of serine proteases by keratinocytes and metalloproteases in dermal fibroblasts, which can further disrupt the skin barrier.29–31 Colonization by S aureus strains isolated from patients with severe AD elicits more inflammation in mouse skin than that by strains isolated from less severe AD.32 Strains isolated from severe AD patients also have been found to produce higher extracellular proteolytic activity than isolates from less severe patients or healthy subjects.33 These findings indicate that the capacity of S aureus to trigger the development of AD can be strain-dependent.
Table 1.
Compared Skin Characterization between Normal and AD Skin
| Normal skin | AD skin | |
|---|---|---|
| Skin microbiome | Colonized by CoNS-AM+ | Less colonized by CoNS-AM+ |
| High diversity | Low diversity | |
| High prevalence of antimicrobials and anti-inflammatory substances from CoNS | Increased colonization by S aureus High prevalence of virulence factors from S aureus, such as δ-toxin, PSMα |
|
| Protection by AMPs | Inducible on injury or bacterial stimuli | Inhibited induction by Th2 cytokines Insufficient to suppress S aureus Degraded by aureolysin from S aureus |
| Immune system | No inflammation | Proinflammatory |
| Optimal innate immune potential | Th2 predominant Suppressed innate immune potential |
|
| pH | Acidic | Neutral-Alkaline |
| Barrier function | High | Host genetic defects in barrier |
| Regulated entry of commensal microbes | Enhanced entry of microbes and allergens | |
| Low water evaporation | High water evaporation S aureus further disrupt barrier Mechanical disruption by scratch |
Abbreviations: CoNS-AM+, Coagulase-negative staphylococcal (CoNS) strains that show a capacity to produce antimicrobials (AM); PSM, phenol soluble modulin.
Skin Barrier and Microbiome in AD
Primary functions of the skin barrier are to maintain fluid and electrolyte homeostasis, thermoregulate, and protect the host from environmental dangers such as microbial pathogens and chemical contaminants. Previously, microbial organisms residing on the skin surface had been assumed to be totally separated from the host by the epidermal barrier. It was unexpected when our research provided evidence that microbial communities exist in an equilibrium across the basement membrane of epidermis and populate the dermal stroma of normal skin.7 These observations showed that the surface microbiome can penetrate into a position for direct priming with live cells below the epithelial surface, therefore influencing cutaneous immunity without the need for sampling by dendrites from antigen-presenting cells. This observation that microbes enter the dermis was not evidence of infection. In contrast, this finding demonstrated that subepidermal microbes can be well controlled by host defenses and that products made by these microbes are communicating with various types of live cells in the dermis. Most of the microbes in the dermis of human skin are not present within CD11c+ phagocytic immune cells, suggesting other routes of bacterial penetration into the dermis. The bacterial penetration through the epidermis was dependent on bacterial viability and protease activity, because killed bacteria or a protease-null mutant was unable to penetrate.34 Evidence suggests the holobiont enabled bacteria to evolve genes for epidermal penetration and the host to evolve genes to suppress proliferation but not completely exclude their entry. Such an observation suggests a mutualistic advantage for host and microbe.
The observation that the microbiome exists across the epidermal barrier also may provide a better understanding of how the skin microbial ecology can trigger disease. Indeed, S aureus is abundantly detected in the dermis of lesional skin of patients with AD, but less abundant in nonlesional skin.34 Filaggrin is a structural protein that is fundamental in the development and maintenance of the physical skin barrier.35 Loss-of-function mutations in the filaggrin gene (FLG) represent a significant genetic factor predisposing some populations to developing AD in some populations.36–39 Two of the most studied mutations, R501X and 2282del4, are common in European populations and result in loss-of-function.40 Therefore, mice with loss-of-function mutation (5303delA) in FLG (FLGft/ft), of which the Tmem79/Matt mutation are removed from the original flaky-tail mouse, have been used for an experimental murine model associated with human AD.41 Using this mouse model, we have demonstrated that ovalbuminsensitized skin of FLGft/ft mice permitted more S aureus to penetrate into the dermis than that of wild-type mice. Subsequently, the enhanced entry by S aureus directly correlated with a Th2 immune phenotype, such as enhanced expression of IL-4, IL-13, IL-17, and thymic stromal lymphopoietin, and decreased expression of AMPs.34 These results are consistent with previous reports showing that Th2 cytokines directly down-regulate the induction of AMPs in the skin.42 FLG mutation is not the only pathogenic mechanism of AD, because only 27% of human populations with a FLG-null mutation develop AD.43 Other epidermal barrier proteins, such as envoplakin, periplakin, and involucrin, also may control bacterial penetration across the epidermal barrier and provide a similar pathway for inflammatory activation.44 Such changes are characteristic of AD and illustrate how skin barrier defects confer a risk of AD by enabling the abnormal entry of microbes into the dermis. Indeed, optimized lipid mixture to restore skin barrier function (cholesterol, ceramide, and free fatty acids) improves clinical symptoms in patients with AD.45,46 In FLGft/ft mice, barrier restoration with this lipid mixture decreased S aureus entry into the skin and resulted in decreased proinflammatory cytokines and increased expression of cathelicidin and β-defensins.34
AD Has Abnormal Antimicrobial Defense from Both Host and Microbiome
The human skin actively regulates colonization by microbial organisms by producing diverse molecules, such as cathelicidin and b-defensin AMPs, fatty acids, and reactive oxygen species that directly inhibit growth of bacteria.47–49 Therefore, dysregulation in these innate antimicrobial defense systems increases the risk of dysbiosis and infection. In the case of AD, skin colonization by S aureus is in part attributed to an inadequate induction of cathelicidin and β-defensins. Relative expression of these AMPs is much less in AD skin than in other skin inflammatory conditions such as psoriasis, rosacea, and wounds, and the amount of AMPs is not enough to suppress the growth of S aureus.42,50–53 In vitro experiments have demonstrated that Th2 cytokines, including IL-4 and IL-13, suppress induction of cathelicidin in keratinocytes or grafted human skin.42 In addition, aureolysin, an extracellular metal-loprotease produced by S aureus, degrades cathelicidin to inactive shorter fragments, further facilitating S aureus survival.54
Recent studies by multiple groups also have demonstrated that antimicrobials from some strains of coagulase-negative staphylococcus (CoNS) within the human microbiome could be beneficial to the host and thus serve as an additional antimicrobial barrier on normal skin surface (Fig 1). For example, our group has proposed that the unique peptides PSMγ and PSMδ produced by S epidermidis limit survival of pathogenic bacteria on the skin surface.55,56 These PSMs caused membrane leakage and membrane perturbation in bacteria, suggesting that these peptides function by a mechanism similar to that of innate human AMPs. Adding to the function of the human AMPs, we have observed that lantibiotics, a class of cyclic AMP that contain lanthionine and methyllanthionine, are produced by Staphylococcus hominis and other CoNS strains isolated from normal human skin.57 Importantly, these prokaryotic AMPs are abundantly detectable on the surface of normal skin and can synergize with host AMPs, including cathelicidin and β-defensin-2 against pathogenic bacteria, further enhancing antimicrobial defense.55–57 These peptides selectively exhibited bactericidal activity against skin pathogens, such as S aureus, group A Streptococcus, and Escherichia coli, whereas they are not active against S epidermidis. This selectivity may be important in establishing normal microbial ecosystem, because they do not bother microbes that coexist within the same ecosystem.
Figure 1.
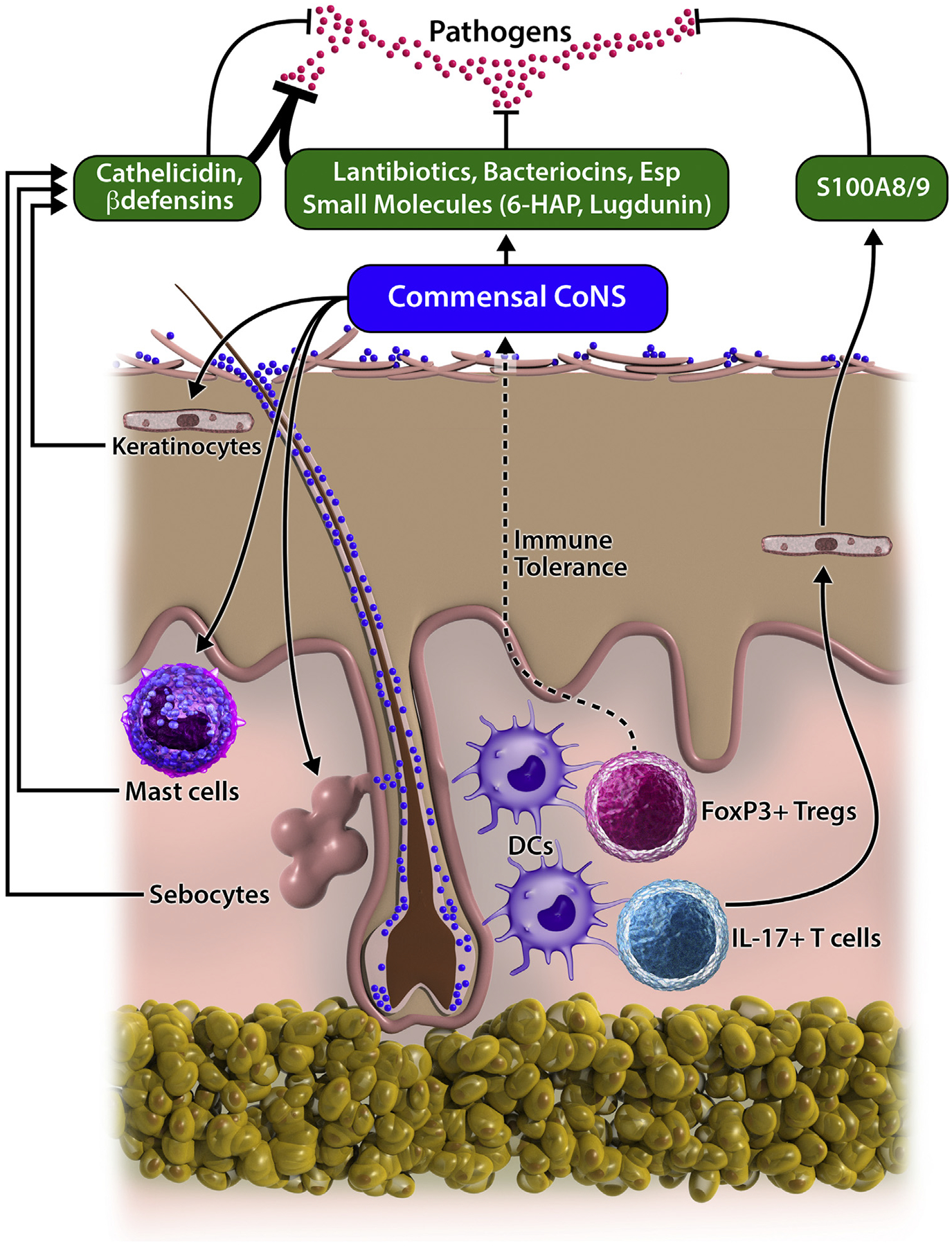
Mutualistic interaction between commensal microbes and cutaneous immune system to defend against pathogenic bacteria. Selected strains of coagulase-negative staphylococci (CoNS) that reside on the human skin produce diverse molecules with antimicrobial activity that protect skin from pathogens. Bacterial products or metabolites from commensal microbes defend the host against pathogens by enhancing production of antimicrobial peptides (AMPs) in keratinocytes, mast cells, and sebocytes. Induced host AMPs synergize with bacterial antimicrobials, further enhancing the antimicrobial barrier. Staphylococcus epidermidis primed by antigen-presenting cells within hair follicles stimulate differentiation of IL-17+ T cells and FoxP3+ Tregs, which contribute to antimicrobial defense and establishment of immune tolerance against commensal microbes, respectively.
Other strains of CoNS that reside in the human nasal cavity exert similar actions. Some strains of S epidermidis are capable of producing a specific serine protease that destroys biofilm formed by S aureus.58 A Staphylococcus lugdunensis strain produces a non-ribosomally synthesized peptide with antimicrobial activity against S aureus.59 Clinical observations have shown that the presence of these inhibitory S epidermidis or S lugdunensis strains significantly correlates to a reduction in S aureus number in the nasal bacterial community. Although gram-negative bacteria are a very small fraction and are rarely detected on normal human skin, application of gram-negative bacteria that were selectively expanded from skin swabs could suppress the growth of S aureus on AD subjects.60 Because gram-negative bacteria are typically killed by a healthy skin innate immune system, whether the presence of such gram-negative bacteria influences normal host defense or whether their presence on skin swabs represented transient contamination from the environment are unclear. In addition to compounds with antimicrobial activity, a thiolactone-containing peptide (also called autoinducing peptide), and its derivatives produced by S epidermidis, blocks the S aureus agr quorum sensing system that controls production of various virulence factors.61 Thus, current evidence strongly supports a conclusion that some strains within the human microbiome could protect the host from detrimental actions of microbial pathogens.
We have investigated the frequency of CoNS strains with capacity to inhibit growth of S aureusin the skin of normal subjects and patients with AD.57 Most CoNS strains that colonize normal human skin were capable of suppressing or decreasing survival of S aureus. In contrast, skin of patients with AD lacked these protective strains of CoNS. In particular, subjects who were colonized by S aureus were the least colonized by CoNS with anti–S aureus activity. These observations suggest that a combined deficiency to induce human cathelicidins and β-defensins, and low colonization by CoNS strains with capacity to produce antimicrobials, enable survival and dominance by S aureus in the skin of patients with AD. Other supporting evidence for this conclusion has come with findings of less skin colonization by CoNS during infancy being significantly associated with the development of AD at a later age.62 It has been also demonstrated in mice that skin colonization by S epidermidis during neonatal life enhanced differentiation of FoxP3-positive regulatory T-cells after the second exposure to the same bacterial species during adult life.63 These data from human and mice suggest that exposure to beneficial CoNS strains at an early age may protect against the later development of AD.
New Generation Therapeutics from the Microbiome
Because a decreased abundance of S aureus in the skin directly correlates to improvement of skin inflammation in mice and patients with AD,20,64 therapies using anti–S aureus agents have been attempted to manage AD. For example, bleach baths have been proposed as a treatment for decreasing the severity of AD. Although these may be effective in decreasing severity of AD, whether this is more effective than water bath alone is unclear, and it does not appear to be an effective antimicrobial.65 Diluted bleach has been suggested to be directly anti-inflammatory by blocking nuclear factor-κB signaling, and this may explain some therapeutic effect.66 Topical therapy using systemic pharmaceutical antibiotics also has not been very successful in reducing severity of AD and typically fail to reduce skin colonization by S aureus.67 The failure of direct antimicrobial therapy may be attributed to the habitat of skin bacteria. Skin bacteria abundantly reside within the appendages, such as hair follicles and eccrine glands, and many bacteria exist beneath the epidermal barrier,7,34 locations in which antimicrobial agents are difficult to reach. Therefore, alternative strategies to inhibit S aureus survival are required to manage dysbiosis in AD.
Some nonpathogenic or commensal bacteria directly suppress skin inflammation, which may be beneficial in future use for treatment for AD. Topical application of lysate of a nonpathogenic gram-negative bacterium Vitreoscilla filiformis significantly improved local inflammation in the skin with AD.68,69 It was later demonstrated that V filiformis extracts modulated the cutaneous inflammatory response through induction of IL-10–producing dendritic cells and priming of regulatory T cells in mouse skin.70 In addition, epicutaneous application of live S epidermidis to neonatal skin triggers an abrupt influx of highly activated regulatory T cells after the second exposure to the same bacterial species in adult life of mice.17,63 Lipoteichoic acid produced by S epidermidis suppresses skin inflammation from tissue damage.10 Intradermal inoculation or epicutaneous application of Staphylococcus caprae inhibited skin injury induced by S aureus or skin colonization by S aureus, respectively.71 These findings advance observations of links between bacteria and maintenance of host immune homeostasis and provide further evidence for novel strategies to control skin inflammation.
The beneficial functions of skin commensal bacteria are now leading to early clinical trials to test the capacity to treat S aureus colonization and potentially inflammation in AD. We have suggested biotherapy to adapt beneficial actions from screened bacterial strains to restore normal functions of the skin microbial ecosystem. Indeed, clinical trials of biotherapy to reconstitute the healthy intestinal microbial ecosystems have shown promise. Transplantation of purified bacterial cultures from stool of healthy donors is sometimes effective to treat colitis induced by antibiotic-resistant Clostridium difficile infection.72,73 In mice, introduction of bacteriocin-producing bacterial strains can eliminate pathogenic bacteria in the intestinal tract.74 These successful observations support the applicability of a similar approach for skin disorders. The unique opportunity that biotherapy may offer in the skin over conventional antimicrobial therapy is also the capacity for selective killing of pathogenic bacteria over the normal microflora. This could both kill the pathogen and help shape the normal bacterial community to enable additional factors to become active from the microbiome as a whole. To test this, our group conducted the first human clinical trial to attempt to compensate for the decreased numbers of CoNS strains with capacity to inhibit S aureus on the skin surface of patients with AD.57 We screened CoNS strains with anti–S aureus activity by a nonbiased high-throughput screening from each patient with AD who was colonized by S aureus. Reintroduction of these clones with anti–S aureus activity successfully decreased survival of S aureus on the lesional skin of patients with AD within 24 hours. In another study, application of a strain of Roseomonas mucosa decreased S aureus colonization and improved severity of AD, although the effect of vehicle was not tested.75 These findings suggest that biotherapy using skin commensal bacteria may be superior to the existing use of existing pharmaceutical antibiotics or antiseptics that are often ineffective or nonspecifically disrupt homeostasis provided by the normal microflora.76
Conclusion
Recent evidence strongly supports the concept that the skin immune system works together with specific microbes within the skin microbiome to protect against overgrowth of opportunistic pathogens. Dysbiosis contributes to the pathogenesis of AD by both detrimental effects from S aureus and by loss of beneficial effects from some members of the microbiome. Pharmaceutical antibiotics have been used to inhibit S aureus in the management of AD, but their efficacyon the skin is limited and has disadvantages in that they may kill beneficial strains and break mutualistic interactions between the skin and microbial communities. Highly potent antimicrobials may result in a short-term improvement, but they can subsequently increase a long-term risk by causing dysbiosis and promoting antibiotic resistance. Because ongoing studies have brought better understanding of the communication between the skin immune system and our essential microbial symbionts, we should expect that breakthroughs in therapeutics from this field will be developed in the near future.
Key Messages.
Patients with atopic dermatitis have dysbiosis of the skin microbiome with Staphylococcus aureus as the dominant species.
Colonization by Staphylococcus aureus is associated with increased disease severity.
Th2 cytokines present in atopic dermatitis suppress production of antimicrobial peptides (AMPs) by the skin that inhibit Staphylococcus aureus.
Many coagulase-negative staphylococcal (CoNS) strains found on healthy skin produce antimicrobials that kill Staphylococcus aureus.
Atopic dermatitis patients have a combined deficiency in AMPs made by the skin and antimicrobials from commensal CoNS strains.
Bacteria that produce antimicrobials can be used to manage skin colonization by Staphylococcus aureus and improve disease symptoms of atopic dermatitis.
Funding Sources:
RLG and TN are supported by NIH R01AR06781, R01AI052453, U19 AI117673.
Footnotes
Disclosures: RLG is a consultant and has equity interest in MatriSys Biosciences and Sente Inc.
References
- 1.Nutten S. Atopic dermatitis: global epidemiology and risk factors. Ann Nutr Metab. 2015;66(Suppl 1):8–16. [DOI] [PubMed] [Google Scholar]
- 2.Benn CS, Melbye M, Wohlfahrt J, Bjorksten B, Aaby P. Cohort study of sibling effect, infectious diseases, and risk of atopic dermatitis during first 18 months of life. BMJ. 2004;328:1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flohr C, Yeo L. Atopic dermatitis and the hygiene hypothesis revisited. Curr Probl Dermatol. 2011;41:1–34. [DOI] [PubMed] [Google Scholar]
- 4.Flohr C, Pascoe D, Williams HC. Atopic dermatitis and the ‘hygiene hypothesis‘: too clean to be true? Br J Dermatol. 2005;152:202–216. [DOI] [PubMed] [Google Scholar]
- 5.Grice EA, Kong HH, Conlan S, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallo RL. Human skin is the largest epithelial surface for interaction with microbes. J Invest Dermatol. 2017;137:1213–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, Gallo RL. The microbiome extends tosubepidermalcompartments ofnormalskin. Nat Commun. 2013;4:1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg E, Zilber-Rosenberg I. The hologenome concept of evolution after 10 years. Microbiome. 2018;6:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bordenstein SR, Theis KR. Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PLoS Biol. 2015;13:e1002226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lai Y, Di Nardo A, Nakatsuji T, et al. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med. 2009;15:1377–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernard JJ, Cowing-Zitron C, Nakatsuji T, et al. Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nat Med. 2012;18:1286–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai Y, Cogen AL, Radek KA, et al. Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J Invest Dermatol. 2010;130:2211–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li D, Lei H, Li Z, Li H, Wang Y, Lai Y. A novel lipopeptide from skin commensal activates TLR2/CD36-p38 MAPK signaling to increase antibacterial defense against bacterial infection. PLoS One. 2013;8:e58288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naik S, Bouladoux N, Wilhelm C, et al. Compartmentalized control of skin immunity by resident commensals. Science. 2012;337:1115–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naik S, Bouladoux N, Linehan JL, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature. 2015;520: 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Linehan JL, Harrison OJ, Han SJ, et al. Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell. 2018;172:784–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scharschmidt TC, Vasquez KS, Pauli ML, et al. Commensal microbes and hair follicle morphogenesis coordinately drive treg migration into neonatal skin. Cell Host Microbe. 2017;21:467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakatsuji T, Chen TH, Butcher AM, et al. A commensal strain of Staphylococcus epidermidis protects against skin neoplasia. Sci Adv. 2018;4:eaao4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leyden JJ, Marples RR, Kligman AM. Staphylococcus aureus in the lesions of atopic dermatitis. Br J Dermatol. 1974;90:525–530. [DOI] [PubMed] [Google Scholar]
- 20.Kong HH, Oh J, Deming C, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22:850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leung DY. Infection in atopic dermatitis. Curr Opin Pediatr. 2003;15:399–404. [DOI] [PubMed] [Google Scholar]
- 22.Beck LA, Boguniewicz M, Hata T, et al. Phenotype of atopic dermatitis subjects with a history of eczema herpeticum. J Allergy Clin Immunol. 2009;124:260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spaulding AR, Salgado-Pabon W, Kohler PL, Horswill AR, Leung DY, Schlievert PM. Staphylococcal and streptococcal superantigen exotoxins. Clin Microbiol Rev. 2013;26:422–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakagawa S, Matsumoto M, Katayama Y, et al. Staphylococcus aureus virulent PSMalpha peptides induce keratinocyte alarmin release to orchestrate IL-17-dependent skin inflammation. Cell Host Microbe. 2017;22:667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu H, Archer NK, Dillen CA, et al. Staphylococcus aureus epicutaneous exposure drives skin inflammation via IL-36-mediated T cell responses. Cell Host Microbe. 2017;22:653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bekeredjian-Ding I, Inamura S, Giese T, et al. Staphylococcus aureus protein A triggers T cell-independent B cell proliferation by sensitizing B cells for TLR2 ligands. J Immunol. 2007;178:2803–2812. [DOI] [PubMed] [Google Scholar]
- 27.Vu AT, Baba T, Chen X, et al. Staphylococcus aureus membrane and diacylated lipopeptide induce thymic stromal lymphopoietin in keratinocytes through the Toll-like receptor 2-Toll-like receptor 6 pathway. J Allergy Clin Immunol. 2010;126:985–993. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura Y, Oscherwitz J, Cease KB, et al. Staphylococcus delta-toxin induces allergic skin disease by activating mast cells. Nature. 2013;503:397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanangat S, Postlethwaite A, Hasty K, et al. Induction of multiple matrix metalloproteinases in human dermal and synovial fibroblasts by Staphylococcus aureus: implications in the pathogenesis of septic arthritis and other soft tissue infections. Arthritis Res Ther. 2006;8:R176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williams MR, Nakatsuji T, Sanford JA, Vrbanac AF, Gallo RL. Staphylococcus aureus induces increased serine protease activity in keratinocytes. J Invest Dermatol. 2017;137:377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw L, Golonka E, Potempa J, Foster SJ. The role and regulation of the extracellular proteases of Staphylococcus aureus. Microbiology. 2004;150:217–228. [DOI] [PubMed] [Google Scholar]
- 32.Byrd AL, Deming C, Cassidy SKB, et al. Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miedzobrodzki J, Kaszycki P, Bialecka A, Kasprowicz A. Proteolytic activity of Staphylococcus aureus strains isolated from the colonized skin of patients with acute-phase atopic dermatitis. Eur J Clin Microbiol Infect Dis. 2002;21: 269–276. [DOI] [PubMed] [Google Scholar]
- 34.Nakatsuji T, Chen TH, Two AM, et al. Staphylococcus aureus exploits epidermal barrier defects in atopic dermatitis to trigger cytokine expression. J Invest Dermatol. 2016;136:2192–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandilands A, Sutherland C, Irvine AD, McLean WH. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci. 2009;122:1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith FJ, Irvine AD, Terron-Kwiatkowski A, et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet. 2006;38:337–342. [DOI] [PubMed] [Google Scholar]
- 37.Sandilands A, Terron-Kwiatkowski A, Hull PR, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet. 2007;39:650–654. [DOI] [PubMed] [Google Scholar]
- 38.Palmer CN, Irvine AD, Terron-Kwiatkowski A, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–446. [DOI] [PubMed] [Google Scholar]
- 39.Bisgaard H, Simpson A, Palmer CN, et al. Gene-environment interaction in the onset of eczema in infancy: filaggrin loss-of-function mutations enhanced by neonatal cat exposure. PLoS Med. 2008;5:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weidinger S, Illig T, Baurecht H, et al. Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. J Allergy Clin Immunol. 2006;118:214–219. [DOI] [PubMed] [Google Scholar]
- 41.Leisten S, Oyoshi MK, Galand C, Hornick JL, Gurish MF, Geha RS. Development of skin lesions in filaggrin-deficient mice is dependent on adaptive immunity. J Allergy Clin Immunol. 2013;131:1247–1250. [DOI] [PubMed] [Google Scholar]
- 42.Howell MD, Gallo RL, Boguniewicz M, et al. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity. 2006;24:341–348. [DOI] [PubMed] [Google Scholar]
- 43.Morar N, Cookson WO, Harper JI, Moffatt MF. Filaggrin mutations in children with severe atopic dermatitis. J Invest Dermatol. 2007;127:1667–1672. [DOI] [PubMed] [Google Scholar]
- 44.Natsuga K, Cipolat S, Watt FM. Increased bacterial load and expression of antimicrobial peptides in skin of barrier-deficient mice with reduced cancer susceptibility. J Invest Dermatol. 2016;136:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chamlin SL, Frieden IJ, Fowler A, et al. Ceramide-dominant, barrier-repair lipids improve childhood atopic dermatitis. Arch Dermatol. 2001;137:1110–1112. [PubMed] [Google Scholar]
- 46.Lowe AJ, Tang ML, Dharmage SC, et al. A phase I study of daily treatment with a ceramide-dominant triple lipid mixture commencing in neonates. BMC Dermatol. 2012;12:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol. 2012;12:503–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drake DR, Brogden KA, Dawson DV, Wertz PW. Thematic review series: skin lipids. Antimicrobial lipids at the skin surface. J Lipid Res. 2008;49:4–11. [DOI] [PubMed] [Google Scholar]
- 49.Bogdan C. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol. 2015;36:161–178. [DOI] [PubMed] [Google Scholar]
- 50.Mallbris L, Carlen L, Wei T, et al. Injury downregulates the expression of the human cathelicidin protein hCAP18/LL-37 in atopic dermatitis. Exp Dermatol. 2010;19:442–449. [DOI] [PubMed] [Google Scholar]
- 51.Ong PY, Ohtake T, Brandt C, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347:1151–1160. [DOI] [PubMed] [Google Scholar]
- 52.Hata TR, Kotol P, Boguniewicz M, et al. History of eczema herpeticum is associated with the inability to induce humanbeta-defensin (HBD)-2, HBD-3 and cathelicidin in the skin of patients with atopic dermatitis. Br J Dermatol. 2010;163:659–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Howell MD, Boguniewicz M, Pastore S, et al. Mechanism of HBD-3 deficiency in atopic dermatitis. Clin Immunol. 2006;121:332–338. [DOI] [PubMed] [Google Scholar]
- 54.Sieprawska-Lupa M, Mydel P, Krawczyk K, et al. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Anti-microb Agents Chemother. 2004;48:4673–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cogen AL, Yamasaki K, Sanchez KM, et al. Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J Invest Dermatol. 2010;130:192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cogen AL, Yamasaki K, Muto J, et al. Staphylococcus epidermidis antimicrobial delta-toxin (phenol-soluble modulin-gamma) cooperates with host antimicrobial peptides to kill group A Streptococcus. PLoS One. 2010;5:e8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakatsuji T, Chen TH, Narala S, et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iwase T, Uehara Y, Shinji H, et al. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature. 2010; 465:346–349. [DOI] [PubMed] [Google Scholar]
- 59.Zipperer A, Konnerth MC, Laux C, et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature. 2016;535:511–516. [DOI] [PubMed] [Google Scholar]
- 60.Myles IA, Williams KW, Reckhow JD, et al. Transplantation of human skin microbiota in models of atopic dermatitis. JCI Insight. 2016;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Otto M, Sussmuth R, Vuong C, Jung G, Gotz F. Inhibition of virulence factor expression in Staphylococcus aureus by the Staphylococcus epidermidis agr pheromone and derivatives. FEBS Lett. 1999;450:257–262. [DOI] [PubMed] [Google Scholar]
- 62.Kennedy EA, Connolly J, Hourihane JO, et al. Skin microbiome before development of atopic dermatitis: early colonization with commensal staphylococci at 2 months is associated with a lower risk of atopic dermatitis at 1 year. J Allergy Clin Immunol. 2017;139:166–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scharschmidt TC, Vasquez KS, Truong HA, et al. A wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity. 2015;43:1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kobayashi T, Glatz M, Horiuchi K, et al. Dysbiosis and Staphylococcus aureus colonization drives inflammation in atopic dermatitis. Immunity. 2015;42:756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chopra R, Vakharia PP, Sacotte R, Silverberg JI. Efficacy of bleach baths in reducing severity of atopic dermatitis: a systematic review and meta-analysis. Ann Allergy Asthma Immunol. 2017;119:435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leung TH, Zhang LF, Wang J, Ning S, Knox SJ, Kim SK. Topical hypochlorite ameliorates NF-kappaB-mediated skin diseases in mice. J Clin Invest. 2013;123: 5361–5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bath-Hextall FJ, Birnie AJ, Ravenscroft JC, Williams HC. Interventions to reduce Staphylococcus aureus in the management of atopic eczema: an updated Cochrane review. Br J Dermatol. 2010;163:12–26. [DOI] [PubMed] [Google Scholar]
- 68.Gueniche A, Hennino A, Goujon C, et al. Improvement of atopic dermatitis skin symptoms by Vitreoscilla filiformis bacterial extract. Eur J Dermatol. 2006;16: 380–384. [PubMed] [Google Scholar]
- 69.Gueniche A, Knaudt B, Schuck E, et al. Effects of nonpathogenic gram-negative bacterium Vitreoscilla filiformis lysate on atopic dermatitis: a prospective, randomized, double-blind, placebo-controlled clinical study. Br J Dermatol. 2008;159:1357–1363. [DOI] [PubMed] [Google Scholar]
- 70.Volz T, Skabytska Y, Guenova E, et al. Nonpathogenic bacteria alleviating atopic dermatitis inflammation induce IL-10-producing dendritic cells and regulatory Tr1 cells. J Invest Dermatol. 2014;134:96–104. [DOI] [PubMed] [Google Scholar]
- 71.Paharik AE, Parlet CP, Chung N, et al. Coagulase-negative Staphylococcal strain prevents Staphylococcus aureus colonization and skin infection by blocking quorum sensing. Cell Host Microbe. 2017;22:746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Petrof EO, Gloor GB, Vanner SJ, et al. Stool substitute transplant therapy for the eradication of Clostridium difficileinfection: “RePOOPulating” the gut. Microbiome. 2013;1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J Clin Gastroenterol 2010;44:354–360. [DOI] [PubMed] [Google Scholar]
- 74.Corr SC, Li Y, Riedel CU, O‘Toole PW, Hill C, Gahan CG. Bacteriocin production as a mechanism for the antiinfective activity of Lactobacillus salivarius UCC118. Proc Natl Acad Sci U S A. 2007;104:7617–7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Myles IA, Earland NJ, Anderson ED, et al. First-in-human topical microbiome transplantation with Roseomonas mucosa for atopic dermatitis. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim YG, Udayanga KG, Totsuka N, Weinberg JB, Nunez G, Shibuya A. Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi-induced PGE2. Cell Host Microbe. 2014;15:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]