

Publisher Summary
The achievement of robust and regulated protein production in mammalian cells is a complex process that requires careful consideration of many factors, including transcriptional and translational control elements, RNA processing, gene copy number, mRNA stability, the chromosomal site of gene integration, potential toxicity of recombinant proteins to the host cell, and the genetic properties of the host. Gene transfer into mammalian cells may be effected either by infection with virus that carries the recombinant gene of interest, or by direct transfer of plasmid DNA. This chapter discusses the molecular architecture of non-viral vectors for high-level protein production. Virus-based vectors for gene therapy, protein production, vaccine development and other applications are summarized in a table and described.
Abbreviations: BHK, baby hamster kidney; CHO, Chinese hamster ovary; EBNA, Epstein-Barr virus nuclear antigen; ER, endoplasmic reticulum; HEK, human embryonic kidney; IRES, internal ribosome entry site; LTR, long terminal repeat; UTR, untranslated region
1. Introduction
Achievement of robust and regulated protein production in mammalian cells is a complex process that requires careful consideration of many factors, including transcriptional and translational control elements, RNA processing, gene copy number, mRNA stability, the chromosomal site of gene integration, potential toxicity of recombinant proteins to the host cell, and the genetic properties of the host. Some of these topics are covered in detail elsewhere [1] and in other chapters in this book, therefore, only brief discussion will be provided here. Gene transfer into mammalian cells may be effected either by infection with virus that carries the recombinant gene of interest, or by direct transfer of plasmid DNA (Chapters 4 and 5). This chapter provides an overview of the molecular architecture of non-viral vectors for high-level protein production. Virus-based vectors for gene therapy, protein production, vaccine development and other applications are summarized in Table 1 and discussed in Chapters 3.1–3.13. In addition, inducible vector systems are examined in Chapter 22. Due to space limitations, many original publications regrettably could not be included, and the reader is referred to cited reviews and other chapters in this book.
Table 1.
Virus-based vectors for gene delivery and expression in mammalian cells
| Virus | Family | Vector features |
|---|---|---|
| DNA viruses Herpes simplex virus (HSV) | Herpesviridae. A heterogeneous family of viruses that contain linear dsDNA (130–230 kb) and infect man and many other vertebrates. Virions are enveloped, 180–200 nm in diameter. An icosadeltahedral capsid 100–110 nm in diameter contains 162 capsomers | The HSV-1 genome is 152 kb long and accomodates ∼30 kb exogenous DNA. Broad mammalian host and cell type range. Potential for gene therapy. Difficulties with vector targeting and long-term transgene expression in certain tissues |
| Epstein-Barr virus (EBV) | Herpesviridae. See above | EBV has a large (∼172 kb) dsDNA genome. Maintenance as a plasmid requires the viral origin of replication (oriP) and the viral gene encoding the trans-acting factor EBNA-1. OriP-based vectors can be maintained extrachromosomally in human, monkey, bovine, canine, and feline cells, but not in murine and rat cells in culture. Used as recombinant DNA shuttle vectors, screening of cDNA libraries, and production of recombinant proteins. EBV can accommodate up to 180 kb DNA. Potential for gene therapy |
| Simian virus 40 (SV40) | Polyomaviridae. This family was previously considered to be a subfamily of Papovaviridae. Small, antigenically distinct viruses that replicate in nuclei of infected cells; most have oncogenic properties. Virions are nonenveloped, 45–55 nm in diameter. The icosahedral capsids contain three virus-encoded proteins, VP1-3, with 72 pentameric capsomers, surrounding a molecule of circular dsDNA (5.2 kb) | Integrates in host genome, and provides stable transgene expression. In the presence of SV40 ori and large T antigen it replicates episomally at high copy number. Transduces both dividing and nondividing cells. Broad mammalian host range. Used in gene therapy. Nonimmunogenic, high yield and transduction efficiency. Principal limitation is the size of packageable insert (5 kb) |
| Adenovirus (Ad) | Adenoviridae. Viruses that replicate in the cell nuclei of mammals and birds. Virions are nonenveloped, 70–100 nm in diameter; the icosahedral capsids are composed of 252 capsomers, of which 240 are hexons and 12 are pentons. Contain linear dsDNA (30–38 kb). No integration into host genome. The family includes two genera | Broad mammalian host range. Used in gene therapy. Infects both dividing and non-dividing cells. Immunogenic and toxic. Vector is maintained as a nuclear episome, which may lead to loss of DNA during cell division. New Ad vectors deleted in most viral genes are less immunogenic and accomodate ∼35 kb insert |
| Adeno-associated virus (AAV) | Parvoviridae. Small viruses containing linear ssDNA (∼5.0 kb), which converts to dsDNA after infection. Virions are nonenveloped, 18–26 nm in diameter, composed of three capsid proteins, VP1-3. The particle is icosahedral, and the capsid consists of 60 protein subunits. The inverted terminal repeats (ITRs) can pair to form hairpins, which are required for replication and packaging. Replication and assembly occur in the nucleus of infected cells. The family includes two subfamilies, each containing three genera. AAV (a member of the genus Dependovirus) normally requires a helper virus (Ad or herpes virus) to proceed through replication and lytic infection | AAV replication requires extra genes from a helper virus to mediate infection, but vectors can also be constructed that do not require the input of helper virus. Broad mammalian host range. Used in gene therapy. Vectors transduce cells through both episomal transgene expression and by random chromosomal integration. Infects both dividing and non-dividing cells with minimal cell-mediated immune response or toxicity. Prevalence of neutralizing antibodies against wild-type AAV may limit vector re-administration. Major limitation is the packaging capacity (∼5 kb) that precludes the use of large genes, but which may be increased through viral DNA heterodimerization, concatemerization, or AAV/Ad hybrid vector constructs |
| Vaccinia virus (VV) | Poxviridae. Virions are enveloped, 200–400 nm long. Replication occurs in the cytoplasm of infected cells. Capsids are of complex symmetry and contain linear, dsDNA (130–300 kb) with a hairpin loop at each end. The family includes two subfamilies containing eight and three genera, respectively | Used for expression of heterologous genes and for vaccination. Broad mammalian host range.Vector can accomodate 25 kb exogenous DNA |
| Baculovirus | Baculoviridae. Insect, arachnid and crustacean viruses with a large circular dsDNA genome (90–160 kb), which is packaged in a rod-shaped capsid. Baculoviruses are divided into two genera: the nucleopolyhedroviruses (NPVs) and granuloviruses (GVs) | Mammalian promoters in baculovirus vectors enable heterologous gene expression in mammalian cells. Broad host range, no overt cytotoxicity, may be used for transient and stable gene expression. Its rapid inactivation by human complement is disadvantageous for in vivo gene delivery. Protein fusions to the amino terminus of the membrane glycoprotein gp64 may facilitate surface display applications, complement inactivation, and virus targeting to specific cell types. Vector can accommodate 40 kb exogenous DNA |
| RNA viruses Coronavirus | Coronaviridae. Viruses contain positive-sense, capped and polyadenylated ssRNA (27–32 kb). Virions are enveloped, 60–220 nm in diameter. The family includes two genera, Coronavirus and Torovirus | Virus replicates in cytoplasm without DNA intermediate, making its integration into host genome unlikely. Potential for vaccine development and gene therapy |
| Poliovirus | Picornaviridae. Nonenveloped viruses, 27–30 nm in diameter, with one molecule of positive-sense polyadenylated ssRNA (7.2–8.5 kb) enclosed in a capsid of icosahedral symmetry with 60 protomers. Each protomer consists of four polypeptides, VP1-4. Replication occurs in the cytoplasm. The family includes six genera. | Primarily used for vaccination. |
| Sindbis virus (SIN) | Togaviridae. Virions are enveloped, spherical, 60–70 nm in diameter. The capsid is of icosahedral symmetry. The family consists of two genera, Alphavirus and Rubivirus. Alphaviruses (SIN and SFV) contain one molecule of linear, positive-sense, capped with 7-methylguanosine, polyadenylated ssRNA (11–12 kb) | Mosquito-borne, with broad host range including mammals, birds, reptiles and amphibia. Used for expression of heterologous genes, production of retrovirus vectors, detection and identification of other human viruses, construction of libraries of sequences inserted into SIN replicons to identify specific protease-cleavage sites, and development of high-throughput cloning systems. Potential applications include the control of mosquito-transmitted diseases, and vaccination for infectious diseases and cancer |
| Semliki Forest virus (SFV) | Togaviridae. Genus Alphavirus. See above | Used for expression of heterologous genes, production of retrovirus vectors, vaccination and potentially in gene therapy. Broad host range. Cloning capacity is ∼8 kb. In DNA-based SFV vectors expression is RNA polymerase II-dependent |
| Retrovirus (RV) | Retroviridae. Virions are about 100 nm in diameter, enveloped, and contain two identical molecules of linear, positive-sense ssRNA (each monomer 7–13 kb), which have a 5′ cap and 3′ poly(A). RVs possess RNA-dependent DNA polymerases (reverse transcriptases). Upon entry into the host cell, the virion genomic RNA is reverse-transcribed into DNA, which is integrated into the host chromosomal DNA. The preintegration complex requires disruption of the nuclear membrane during mitosis to access the chromatin, thus they transduce only dividing cells. The family includes seven genera, according to recent taxonomic criteria [65] | Used in gene therapy. Accomodates ∼9 kb insert. Host range: ecotropic virus replicates in cells derived from the host species; amphotropic virus replicates in a range of mammalian host cells. Minor immune response. Safety concerns. RV long terminal repeat (LTR) (used as the promoter) attenuates transgene expression in transduced cells. In general, RV-mediated high-level and tissue-specific transgene expression using non-LTR promoters is difficult to achieve |
| Lentivirus (LV) | Retroviridae. LVs rely on active transport of the preintegration complex through the nuclear pores for translocation into the nucleus of the target cell. They transduce dividing and non-dividing cells | Replication-deficient vectors derived from human immunodeficiency virus (HIV) and from non-human lentiviruses that may not be infectious to humans. Cloning capacity is ∼9 kb. Potential for gene therapy. Minor immune response. Vector improvements include minimizing HIV sequences and eliminating viral accessory proteins for enhanced transduction efficiency and safety. In self-inactivating LVs, a deletion in the U3 region of the 3′ LTR results in transcriptional inactivation of the 5′ LTR after integration, enabling transgene expression to be regulated solely by an internal promoter, without reducing viral titers. This diminishes the risk of vector mobilization and recombination, and facilitates high-level targeted transgene expression |
Virus vector systems are reviewed in Chapters 3.1–3.13. Other RNA virus vectors are examined by Palese [66]. ds, double-stranded; ss, single-stranded.
2. Transient gene expression
Transient gene expression is typically used for rapid production of small quantities of protein for initial characterization, testing of vector functionality, and optimization of different combinations of promoters and other elements in expression vectors. Newly developed transient expression systems facilitate high-level production of recombinant proteins on a larger scale [2]. There are several cell types used for transient expression, including COS, baby hamster kidney (BHK), and human embryonic kidney (HEK)-293 cells, as well as genetically modified HEK-293 cells (see Table 1 in Chapter 1). COS cells were generated by transfection of African green monkey kidney CV1 cells with an origin-defective SV40 [3]. COS cells express the SV40 T antigen, which allows replication of plasmids containing the SV40 origin of replication. This host/vector system facilitates high-level plasmid amplification and protein production, followed by lysis of the cells several days from the time of transfection. Transient gene expression, therefore, permits rapid production of recombinant proteins, but does not enable preparation of “permanent” cell lines. Thus, transfection of the gene of interest must be repeated, as necessary. In contrast, stable transformants may be prepared by a more labor-intensive procedure, as discussed below. Virus-based vectors that are useful for transient gene expression include adenovirus, adeno-associated virus, Epstein-Barr virus, Semliki Forest virus, baculovirus, Sindbis virus, lentivirus, Herpes simplex virus, and vaccinia virus (Table 1).
3. Stable gene expression
In contrast to transient gene expression, preparation of stable cell lines usually depends on integration of plasmid into the host chromosome. Transformants must be cloned in order to ensure that all cells in the culture are genetically identical. Typically, DNA-transfected cells are maintained in non-selective medium for about two days, followed by transfer to selective medium. Marker-containing cells that survive the selection are allowed to proliferate, and single transformants are then isolated and characterized using a variety of techniques, including cloning cylinders, soft agar, limiting dilution, or flow cytometry. It is also possible, however, to generate stable cell lines that harbor vectors extrachromosomally. For example, vectors that carry the Epstein-Barr virus nuclear antigen-1 (EBNA-1) and the origin of replication (oriP) can be maintained episomally in primate and canine cell lines but not in rodent cell lines [4]. An episomal replicating vector has been described that does not express any viral proteins, thus avoiding cell transformation [5]. The vector contains the SV40 origin of replication and the scaffold/matrix attachment region (S/MAR) (Chapters 10 and 20) from the human interferon-β gene. The vector was shown to replicate at very low copy numbers (below 20) in CHO cells and was stably maintained without selection for more than 100 generations [5].
The host cell (see Table 1 in Chapter 1) may have a significant impact on gene expression levels. For example, myeloma cells, such as NS0 and Sp2/0, have been used mainly for high-level production of monoclonal antibodies. An epithelial cell line, Madin-Darby canine kidney, was shown to be capable of producing large amounts of protein, comparable to those obtained from CHO amplification systems [6]. The human cell line PER.C6 [7] has recently generated considerable interest for commercial production of therapeutic proteins. Amplifiable gene expression using CHO cells has been widely used for protein production (Chapter 7). The two most widely used amplification systems rely on the dihydrofolate reductase and glutamine synthetase genes. Typically, the selectable marker and the cDNA are under the control of separate transcription units. By growing cells in increasing concentrations of selection drugs it is possible to amplify the copy number of the cotransfected (and cointegrated) gene of interest and concomitantly elevate the amount of protein produced. An alternative method for high-level production of recombinant proteins in CHO cells utilizes an expression vector that produces both selectable marker and cDNA from a single primary transcript via differential splicing [8].
Generation of stable cell lines, particularly the selection of amplified and high-expressing clonal cells, involves screening of large numbers of transfected cells, both during the initial transfection as well as at each subsequent amplification step. This arduous exercise is necessitated by the wide variation in the level of expression and amplification of the transfected gene in different cells, an outcome that reflects the chromosomal site of plasmid integration (reviewed in [1]). An alternative strategy for efficient preparation of stable cell lines is site-specific gene integration using recombination systems (Chapter 20) such as Cre/loxP and FLP/FRT. Cre (cyclization recombination) recombinase of bacteriophage P1 recombines DNA at 34-bp sites called loxP (locus of crossover of P1). The FLP recombinase from the 2-μm circle of Saccharomyces cerevisiae recognizes FRT (the FLP recombination target). It should be possible to engineer a cell line using a reporter gene to select a transcriptionally active chromosomal locus. Such a cell line could then be used for the routine excision and replacement of the reporter construct with the gene of interest. A commercially available vector–host system makes use of the FLP/FRT elements (Flp-In™ expression vectors; Invitrogen, Carlsbad, CA). In this case, different mammalian cell lines were engineered to contain a single FRT site integrated at a transcriptionally active locus. These cells can be used with targeting vectors to prepare recombinant cell lines containing the gene of interest.
Other integrases that hold promise for the engineering of mammalian stable cell lines include those derived from phages R4 and φC31 of Streptomyces spp. [9]. These enzymes function in mammalian cells with no added cofactors. Unlike Cre and FLP, which catalyze reversible recombination between two identical sites, R4 and φC31 integrases mediate unidirectional site-specific recombination between two attachment sites with dissimilar sequences, at higher net integration frequencies than is possible with Cre and FLP [9]. Olivares et al. [10] used the integrase from φC31 to achieve site-specific integration of the gene encoding the human blood clotting Factor IX into the chromosomes of mice, resulting in the stable production of normal levels of the protein. Recent work using DNA shuffling and screening aims at the generation of phage integrases that exhibit improved integration frequency and sequence specificity in human cells [11].
An alternative vector system for gene expression involves receptor-mediated endocytosis of recombinant protein vehicles that target cell-surface receptors ([12] and references therein). The construct in this case comprises a modified β-galactosidase gene containing an insertion of a viral peptide that binds the integrin αvβ3, and an amino-terminal DNA-condensing poly-L-lysine domain. The construct is expressed in Escherichia coli, and when the purified protein is mixed with plasmid DNA, it facilitates transfection of cells expressing αvβ3 receptors [12]. This approach exploits the cell-targeting specificity of viruses without the disadvantages of virus-based vectors.
4. Genetic elements of mammalian expression vectors
Vectors for protein production in mammalian cells comprise a variety of genetic elements with distinct functionalities (Fig. 1 ): (1) a constitutive or inducible promoter that is capable of robust transcriptional activity; (2) a transcription terminator that stabilizes the transcript and prevents transcription interference; (3) optimized mRNA processing and translational signals that include the Kozak sequence, translation termination codon, mRNA cleavage and polyadenylation signals, as well as mRNA splicing signals for higher levels of expression; (4) prokaryotic origin of replication and selection marker for vector propagation in bacteria; and (5) selection markers for the preparation of stable cell lines and for gene amplification. The inclusion of the SV40 origin of replication facilitates transient gene expression in COS cells. Other genetic elements for specific applications include sequences for gene or protein targeting, signal peptides for protein secretion, fusion moieties and protease cleavage sites (see Section 6), and ribosome- or protease-recognition sites that facilitate the expression of multiple genes from polycistronic (Fig. 1C) or monocistronic (Fig. 1D) constructs, respectively (see Section 8). An extensive list of mammalian expression vectors has been published [1].
Fig. 1.
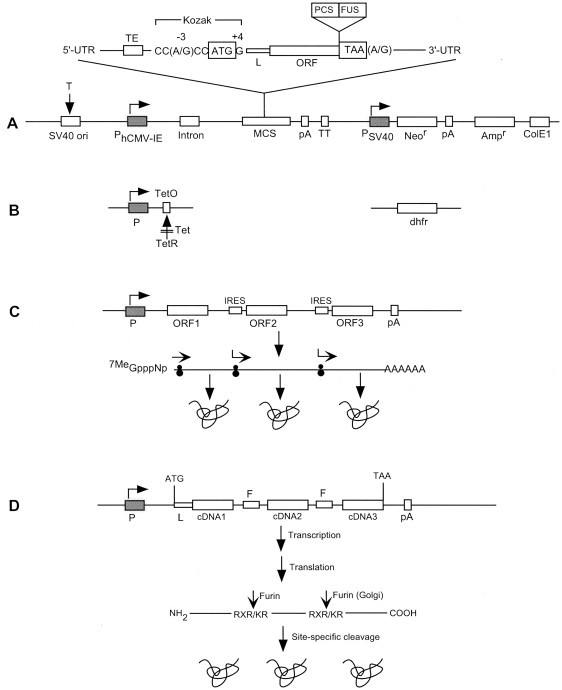
Configuration of model genetic elements in mammalian expression vectors. The combination of different elements (not drawn to scale) may vary in order to meet specific objectives. SV40 ori facilitates transient gene expression in COS cells. Promoters (P) facilitate constitutive (A) or inducible (B) expression. The optimal translational initiation sequence (Kozak) and termination tetranucleotide are shown. The ColE1 origin and the Ampr gene allow plasmid replication and selection, respectively, in bacteria. The Neor gene facilitates selection, and the dhfr gene allows both selection and gene amplification in cells. Multiple gene expression utilizes polycistronic constructs (C) where IRES elements enable ORFs to be translated from a single transcript (see Section 8). Alternatively, a monocistronic construct (D) contains in-frame cDNAs joined by linkers encoding recognition sites (Arg-X-Arg/Lys-Arg) for the endoprotease furin, thus facilitating the post-synthetic cleavage of different proteins (see Section 8). Abbreviations: Ampr, ampicillin-resistance gene (β-lactamase); ColE1, prokaryotic origin of replication; dhfr, dihydrofolate reductase (methotrexate resistance); F, furin-recognition sequence; FUS, fusion moiety; hCMV-IE, human cytomegalovirus immediate early enhancer/promoter; IRES, internal ribosome entry site; L, leader (targeting sequence); MCS, multiple cloning site; Neor, neomycin-resistance gene (aminoglycoside phosphotransferase, aph); ORF, open reading frame; ori, origin of replication; P, promoter; pA, polyadenylation signal; PCS, protease cleavage site; T, SV40 large tumor (T) antigen; TE, translational enhancer; Tet, tetracycline; TetO, tetracycline operator; TetR, tetracycline repressor protein; TT, transcription terminator; UTR, untranslated region.
4.1. Transcriptional control elements
Regulation of transcription in eukaryotic genomes involves the coordinated interaction of multiple genetic elements, a remarkably complex process that is understood in great detail [13]. The promoter is defined as the region proximal to the transcription start site. Transcription initiation is mediated through interactions of transcription factors with their cognate promoter and enhancer elements. Enhancers are sequences which may be located thousands of bases upstream or downstream of the promoter that enhance transcriptional activity when bound by transcription factors. In addition, upstream activation sequences, located within a few hundred bases of the promoter, influence transcription activity. The variability in expression levels observed in different clones during the preparation of stable cell lines is caused by several factors, collectively referred to as position effects. These include the proximity of the target gene to heterochromatin, orientation/location relative to other endogenous genes, and proximity to chromosomal structural elements. Chromatin elements that may abrogate position effects include S/MARs (Chapter 10), chromatin insulators (Chapter 11), and Locus Control Regions (Chapter 12).
Promoters: Promoters used for gene expression in mammalian cells are listed in Table 2 . Some promoters are transcriptionally active in a wide range of cell types and tissues. Most, however, exhibit tissue selectivity, a property that must be carefully considered prior to the construction of expression vectors for high-level production of proteins. Strong constitutive promoters, which drive expression in many cell types, include the adenovirus major late promoter, the human cytomegalovirus immediate early promoter (hCMV-IE), the SV40 and Rous Sarcoma virus promoters, the murine 3-phosphoglycerate kinase promoter, the translation elongation factor 1α (EF-1α) promoter, and the human ubiquitin C promoter. Tissue-selective promoters [14] may facilitate gene targeting and expression in specific organs and tissues.
Table 2.
Selected promoter elements for gene expression in mammalian cells
| Promoter | Source | Properties |
|---|---|---|
| SV40 | Simian virus 40 | Constitutive expression; in some cell lines inducible with phorbol ester. Broad host and cell type range. In COS cell lines expressing the T antigen, high vector copy number is achieved |
| hCMV-IE mCMV-IE | Human and mouse cytomegalovirus immediate-early promoter genes | High-level constitutive expression. Broad host and cell type range |
| RSV-LTR | Rous sarcoma virus long terminal repeat | High-level constitutive expression in murine and avian cell lines |
| MMTV-LTR | Mouse mammary tumor virus | Inducible with glucocorticoids. Moderate level of transcriptional induction |
| MoMLV-LTR | Moloney murine leukemia virus | Moderate to strong transcriptional induction |
| Ad2MLP-TPL | Adenovirus major late promoter and tripartite leader | High-level constitutive expression. Broad host range |
| hUbC | Human ubiquitin C gene | High-level constitutive expression in a broad range of tissues and cell types |
| hEF-1α | Human translation elongation factor 1α subunit gene | High-level constitutive expression. Broad host and cell type range |
| mPGK | Mouse phosphoglycerate kinase gene | High-level constitutive expression. Broad host and cell type range |
| mMT-I | Mouse metallothionein I gene | Inducible with Cd++, Zn++, phorbol esters. “Leaky” promoter |
| hMT-II | Human metallothionein II gene | Inducible with Cd++, Zn++, phorbol esters. “Leaky” promoter |
| hMT-IIA (mutant) | Human metallothionein II gene | Inducible with Cd++, Zn++, phorbol esters. High inducibility, low basal activity |
| hIFN-α | Human interferon-α gene | Inducible with virus |
| β-actin | Chicken β-actin gene | High-level constitutive expression in a broad range of tissues and cell types |
| β-globin | β-globin gene | Tissue-specific for adult β erythroid cells. Potential gene therapy applications |
| ET-1 | Endothelin-1 gene | Endothelium-specific |
| vWf | von Willebrand factor gene | Endothelium-specific |
| Endoglin | Gene encoding human endoglin (CD105), a component of the TGF-β complex | Endothelium-specific |
| GFAP | Gene encoding human glial fibrillary acidic protein | Brain-specific |
| Synapsin I | Rat synapsin I gene | Brain-specific |
A list of promoters that are tissue-specific, tumor-selective, treatment-responsive or cell cycle-regulated has been published [14].
Promoters can be divided into two classes, those that function constitutively, and those that are regulated by induction or derepression (Chapter 22). Promoters used for high-level production of proteins in mammalian cells should be strong and, preferably, active in a wide range of cell types to permit qualitative and quantitative evaluation of the recombinant protein. Inducible promoters should exhibit a minimal level of basal transcriptional activity, and be capable of substantial induction with a non-toxic inducer in a simple and cost-effective manner. Weaker promoters may be desirable in specific applications. For example, in studies of intracellular targeting of antibodies, the powerful EF-1α promoter led to aggregation of expressed antibody, an effect that was avoided by using a weaker promoter [15]. Inducible promoters are desirable for the production of proteins that may be toxic to the host cell, for the study of gene regulation during development in transgenic animals, and for experimental and therapeutic applications of gene transfer.
Introns: Most genes from higher eukaryotes contain introns, which are removed during RNA processing. Genomic constructs have been shown to be expressed more efficiently in transgenic animals than identical constructs lacking introns [16], an effect thought to be due to an enhanced rate of RNA polyadenylation and nuclear transport coupled to RNA splicing [17]. Although many cDNA constructs lacking introns can be expressed efficiently in mammalian cells, the inclusion of introns can enhance expression 10- to 20-fold, and some sequences, such as the β-globin cDNA, show a virtual requirement for the presence of an intron [18]. The placement of introns at the 3′ end of the transcription unit may lead to aberrant splicing [[19], [20]], therefore, it is preferable to place introns at the 5′ end of the open reading frame. A synthetic intron, SIS, generated by the fusion of an adenovirus splice donor site and an immunoglobulin G splice acceptor site was very active in a variety of cell types [21]. The insertion of the human EF-1α first intron downstream of the human or murine CMV-IE promoters strongly enhanced the level of reporter gene expression in several cell lines [22]. The use of introns, however, demands careful vector design, as it is possible that cryptic splicing signals may cause aberrant processing of the mRNA transcript, resulting in reduced expression levels and defective protein products (e.g., [[23], [24]]).
Polyadenylation signals: Most eukaryotic nascent mRNAs possess a poly(A) tail (n≈200) at their 3′ ends, which is added during cleavage of the primary transcript and a coupled polyadenylation reaction [25]. The poly(A) tract is important for mRNA stability and translatability [26]. The signals for polyadenylation of mammalian mRNAs are well defined: One component consists of a highly conserved AAUAAA sequence, which is located about 20–30 nucleotides upstream of the 3′ end of the mRNA, and the other element consists of an unconserved GU-rich sequence immediately downstream of the polyadenylation site [27]. Among the more efficient poly(A) signal sequences to insert in mammalian expression vectors are those derived from bovine growth hormone, mouse β-globin, the SV40 early transcription unit, and the herpes simplex virus thymidine kinase gene.
Transcription terminators: Continued transcription from a promoter through a second transcription unit reduces expression of the second gene, a phenomenon known as transcriptional interference [28]. This has been documented in bacteria, yeast, mammalian and plant cells, but the mechanism is poorly understood. The placement of transcription termination signals between two transcription units, along with the designing of gene orientation, can minimize transcriptional interference. Prokaryotic transcription terminators are well characterized, and their incorporation in expression vectors has multiple beneficial effects on gene expression [29]. In eukaryotes, a consensus sequence consisting of ATCAAA(A/T)TAGGAAGA has been identified in the termination region of nine genes [30].
4.2. Translational control elements
Optimal expression of eukaryotic cDNAs requires careful consideration of several structural features, including the nucleotide context around the translation initiation codon, and the 5′- and 3′-untranslated regions (UTRs), which are involved in many posttranscriptional processes that control mRNA localization, stability and translation efficiency [31]. In addition, codon usage can have a significant impact on the translation efficiency of some heterologous genes in mammalian cells.
5′-Untranslated region: Based on comparison of eukaryotic mRNA sequences and systematic mutagenesis of specific genes, Kozak proposed the “scanning model” of translation initiation in higher eukaryotes (Chapter 16). The initiation complex, consisting of the 40S ribosomal subunit and cap-binding proteins, forms at the mRNA 5′ terminal cap (m7GpppN) followed by movement of the ribosome to the “correct” initiating AUG codon, which is surrounded by an optimal consensus sequence, GCC(A/G)CCaugG (the Kozak sequence, [32]). The purines A or G in position -3 (i.e. three nucleotides upstream from the AUG codon) and G immediately following the AUG codon are the most influential in facilitating optimal translation initiation. The presence of AUG codons in the 5′-UTR of the transcript can severely depress translation initiation at the “authentic” start codon, although the extent of inhibition depends on sequences surrounding the upstream AUG. The design of plasmids for the expression of heterologous genes should therefore consider the 5′ sequence context of the gene of interest and, preferably, avoid the presence of upstream AUGs in the 5′ UTR.
Another concern in the design of expression vectors involves the potential ability of the 5′-UTR to form secondary structure. GC-rich regions have the potential to form stable hairpin structures, which can inhibit translation initiation, a phenomenon that has been documented in eukaryotic [32] and prokaryotic [29] expression systems. One remedy to this potential problem is the removal of the 5′-UTR prior to the insertion of cDNAs into expression vectors, with the caveat that the 5′-UTR may contain translational enhancer elements, such as the SP163 element of the vascular endothelial growth factor mRNA [33]. The SP163 sequence has been shown to enhance translation of different mRNAs 25- to 40-fold in several mammalian cell types [33].
3 ′ -Untranslated region: mRNA destabilization can be influenced by specific sequences present in the 3′-UTR (see Section 7 and Chapter 17). In addition, translational regulation of certain mRNAs is mediated by protein-binding AU-rich elements located in the 3′-UTR (Chapter 17).
Termination codon: Translational termination in mammalian genes may be modulated by nucleotides additional to those of the trinucleotide stop codons. Statistical analysis of the context of termination codons in 5208 mammalian genes showed a highly significant bias in the position immediately following the stop codon [34]. Experimental evidence determined that the base following the stop codon influences the efficiency of translation termination both in vitro and in vivo. Thus, tetranucleotides with a purine in the fourth position are more effective as termination signals than those with a pyrimidine [34].
Codon usage: Genes from both prokaryotic and eukaryotic organisms exhibit a nonrandom usage of synonymous codons. In general, highly expressed genes exhibit a greater degree of codon bias than do poorly expressed ones, and the frequency of use of synonymous codons is strongly correlated with the abundance of their cognate tRNAs within each pool of isoaccepting tRNAs. These observations led to the hypothesis that the main reason for codon bias is translational efficiency. An alternative view holds that the abundance of tRNAs is probably a consequence of and not a reason for codon bias and, furthermore, the primary reason for codon bias is selection for the accuracy of protein synthesis on the ribosome [35]. In E. coli, transcripts of heterologous genes enriched with codons that are rarely used by E. coli may not be translated efficiently, or may result in polypeptides containing misincorporated amino acid residues (reviewed in [[29], [36]]). Similarly, mammalian codon usage can adversely affect translation efficiency of heterologous genes. In some cases, codon optimization has been demonstrated to enhance expression levels of the target genes by 10- to 50-fold (reviewed in [1]).
5. Selectable markers
In addition to the presence of a selectable marker for vector propagation in bacteria, mammalian expression vectors contain markers for the selection of transfected cells, preparation of stable cell lines and for gene amplification. There are several amplifiable genes, the most commonly used ones being dihydrofolate reductase and glutamine synthetase. A compilation of markers, including their mechanism of action, has been published [1].
6. Signal peptides and fusion moieties
Mammalian proteins that are targeted for secretion are translocated from the endoplasmic reticulum (ER) to Golgi to the extracellular medium [[37], [38]] (Chapter 13). Secreted proteins are synthesized as precursor proteins possessing a signal or leader peptide composed of 15–30 amino acid residues, usually located at the amino-terminus, which is subsequently cleaved by a signal peptidase in the ER lumen. Signal peptides typically consist of three regions: a positively charged amino-terminal region (N-region), a central hydrophobic region (H-region), and a polar carboxy-terminal region (C-region) followed by the signal peptidase cleavage site [[39], [40]]. Signal peptides are usually interchangeable and have been widely used to effect protein secretion in mammalian cells. Moreover, signal sequences from bacteria (e.g., [41]) and yeast (e.g., [42]) are recognized by mammalian cells. However, signal peptides can vary significantly in their ability to promote protein secretion. In fact, the presence of a signal sequence per se does not necessarily ensure protein secretion, as has been documented in both prokaryotic (reviewed in [29]) and eukaryotic cells (e.g., [43]). There are many examples of efficient signal peptides including those derived from erythropoietin [44], tissue plasminogen activator [45], interleukin-2 [46], albumin [47], and immunoglobulin sequences [48].
Fusion partners, have a wide range of applications in both prokaryotic [[29], [49]] and eukaryotic [[1], [49]] expression systems. Fusion moieties are used as affinity handles for the facile isolation and purification of proteins (Chapter 25); as reporters (Chapter 6) in studies of promoter activity or localization of proteins in cellular compartments; as protein dimerization domains; as immunogens for the production of antibodies; to target antibodies (Chapter 21); to increase expression, folding, solubility, and secretion of proteins; or to display polypeptides on the surface of cells for vaccine development, protein–protein interactions, drug screening, and other potential applications. Fusion moieties have also been used to increase the half-life of target proteins for potential therapeutic applications ([50] and references therein). Several factors must be carefully considered in the design of fusion proteins. For example, a linker may be inserted between two protein partners in order to optimize protein folding and stability. The length and sequence composition of the linker can impact protein folding. An affinity tag is often fused to the N-terminus of a protein to facilitate purification. This is less desirable, in theory, than a C-terminal tag, which has the advantage that only fully translated proteins can be purified. The latter strategy, however, requires that the C-terminus be structurally accessible. Interestingly, the widely used (His)6 tag has been shown recently to modify the properties of certain proteins expressed in E. coli [51]. Separation of the fusion moiety from the target protein is facilitated by a protease cleavage site engineered between the two components. Selection of an appropriate protease enables regeneration of the native terminus of the target protein upon proteolytic digestion. Fusion proteins are examined in a recent comprehensive review [49].
7. mRNA and protein stability
Turnover of mRNA is an important posttranscriptional mechanism for the physiological control of gene expression (Chapter 17). The potential ability to extend significantly the half-life of transcripts offers an attractive means to enhance protein production in mammalian cells. One determinant of eukaryotic mRNA lability is an AU-rich sequence in the 3′-UTR of many unstable mammalian mRNAs [52]. Insertion of an AU-rich element into the 3′-UTR of a stable mRNA destabilizes the chimeric transcript [53]. The optimal sequence for this destabilizing determinant is UUAUUUAUU [53] or UUAUUUA(U/A)(U/A) [54]. Removal of these sequences from the 3′-UTR of unstable mRNAs can prolong the half-life of transcripts and enhance protein production.
Synthetic 5′ secondary structures have been shown to increase mRNA half-lives in E. coli. In seeking to maximize transcript stability and protein production in mammalian cells, investigators have substituted the UTRs of stable mRNAs, such as β-globin, for the UTRs of target transcripts. This strategy, effective in specific cases, may not have universal application, as mRNA degradation is effected by multiple pathways in mammalian cells. Thus, in addition to exonucleolytic activity at both the 5′ and 3′ termini, determinants of mRNA half-life have been mapped to the coding regions of several mRNA species. Furthermore, mRNA stability is modulated by a variety of cell-specific proteins that act in trans to destabilize or stabilize transcripts (Chapter 17). The use of a specific UTR for the purpose of stabilizing a heterologous transcript in mammalian cells assumes the presence of the cognate UTR-binding proteins in the same cells. At present our knowledge of the distribution of such proteins in different mammalian cell lines used for protein production is incomplete.
Levels of heterologous proteins are also affected by protein degradation pathways (Chapter 18). Recent work has shown that the Gly-Ala repeat of the Epstein-Barr virus nuclear antigen-1 is a cis-acting transferable element that inhibits ubiquitin/proteasome-dependent proteolysis. It has been suggested that the viral Gly-Ala repeat might be used for the prolongation of protein half-life in gene therapy (Chapter 19).
8. Coordinated expression of multiple genes
Coordinated expression of two or more heterologous genes is an important requirement in many applications, including establishment of stable mammalian cell lines that require coexpression of the gene of interest and a selectable marker; characterization of antibody responses in DNA immunization protocols; coexpression of genes for positive-negative (suicide) selections in gene therapy; gene trapping for the identification of developmentally regulated genes; gene targeting; in vitro and in vivo imaging using reporter genes; and coordinated constitutive or inducible high-level expression of several genes in mammalian cells (for references see [1]). As briefly outlined below, a variety of methods exist for the coordinated expression of two or more genes. The suitability of each strategy will depend on the experimental context:
-
(a)
Different expression vectors may be used, each carrying a different gene of interest. This approach is widely used for the production of equimolar amounts of target proteins or protein chains, e.g., antibodies, and it also allows for the ability to evaluate different protein ratios for optimal results.
-
(b)
A single vector can be constructed containing multiple genes each with its own promoter. This type of construct may be subject to promoter interference, a problem that is usually avoided using transcription terminators.
-
(c)
In a translational fusion, two proteins are genetically joined in-frame (see Section 6). The success of this strategy depends on the accessibility of the termini of the two fusion partners. Potential problems related to steric hindrance, misfolding, instability and loss of activity of one or both of the protein partners are addressed through the insertion of an appropriate peptide linker between the joined proteins. A key advantage of this approach is the production of stoichiometric amounts of both proteins.
-
(d)
Two or more genes may be connected via virus-derived elements, known as internal ribosome entry sites (IRES), which facilitate ribosome binding to the second and subsequent transcription units (e.g., [55]) (Fig. 1C). The use of IRES elements, however, presents its own problems, as often the first gene is favored, and the efficiency of translation initiation from different IRES elements varies substantially [56]. Moreover, tissue tropism determinants of IRES activity are poorly understood, and evidence exists for internal ribosome entry dependence on cellular factors that are differentially expressed in different cell types (reviewed in [57]). Consequently, for specific applications, e.g., in vivo imaging in transgenic animals [58], translational processivity may vary among different tissues. Incidentally, although many studies have reported the presence of IRES elements in cellular mRNAs, the experimental evidence in this body of work has been questioned [59] and continues to be vigorously debated [60] (Chapter 16).
-
(e)
Another approach utilizes monocistronic transcripts [61] (Fig. 1D). In this case, the construct contains several in-frame cDNAs joined by linkers encoding cleavage sites for furin, a Golgi-localized ubiquitous endoprotease. The encoded polypeptides are post-synthetically cleaved and processed into biologically active proteins. Processing of the fusion protein, however, may be suboptimal in cells with low levels of furin. This type of construct could potentially utilize tissue-specific delivery and transcription elements, as well as cleavage sites for tissue-specific endoproteases, rather than furin, to achieve a high level of targeting [61].
-
(f)
The use of the 2A sequence from the foot and mouth disease virus, which functions as a ribosome slippage site [[62], [63]]. This sequence, previously thought to be an autocatalytic proteolytic cleavage site [64], facilitates the stoichiometric production of two joined open reading frames.
Acknowledgements
I thank Holly Prentice for her critical reading of the manuscript. James Barsoum and William Sisk made things happen, and I am much obliged to them.
References
- 1.Makrides S.C. Protein Expr. Purif. 1999;17:183–202. doi: 10.1006/prep.1999.1137. [DOI] [PubMed] [Google Scholar]
- 2.Meissner P., Pick H., Kulangara A., Chatellard P., Friedrich K., Wurm F.M. Biotechnol. Bioeng. 2001;75:197–203. doi: 10.1002/bit.1179. [DOI] [PubMed] [Google Scholar]
- 3.Mellon P., Parker V., Gluzman Y., Maniatis T. Cell. 1981;27:279–288. doi: 10.1016/0092-8674(81)90411-6. [DOI] [PubMed] [Google Scholar]
- 4.Yates J.L., Warren N., Sugden B. Nature. 1985;313:812–815. doi: 10.1038/313812a0. [DOI] [PubMed] [Google Scholar]
- 5.Piechaczek C., Fetzer C., Baiker A., Bode J., Lipps H.J. Nucleic Acids Res. 1999;27:426–428. doi: 10.1093/nar/27.2.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pei D., Yi J. Protein Expr. Purif. 1998;13:277–281. doi: 10.1006/prep.1998.0894. [DOI] [PubMed] [Google Scholar]
- 7.Pau M.G., Ophorst C., Koldijk M.H., Schouten G., Mehtali M., Uytdehaag F. Vaccine. 2001;19:2716–2721. doi: 10.1016/s0264-410x(00)00508-9. [DOI] [PubMed] [Google Scholar]
- 8.Lucas B.K., Giere L.M., DeMarco R.A., Shen A., Chisholm V., Crowley C.W. Nucleic Acids Res. 1996;24:1774–1779. doi: 10.1093/nar/24.9.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olivares E.C., Hollis R.P., Calos M.P. Gene. 2001;278:167–176. doi: 10.1016/s0378-1119(01)00711-9. [DOI] [PubMed] [Google Scholar]
- 10.Olivares E.C., Hollis R.P., Chalberg T.W., Meuse L., Kay M.A., Calos M.P. Nat. Biotechnol. 2002;20:1124–1128. doi: 10.1038/nbt753. [DOI] [PubMed] [Google Scholar]
- 11.Sclimenti C.R., Thyagarajan B., Calos M.P. Nucleic Acids Res. 2001;29:5044–5051. doi: 10.1093/nar/29.24.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alcalá P., Feliu J.X., Arís A., Villaverde A. Biochem. Biophys. Res. Commun. 2001;285:201–206. doi: 10.1006/bbrc.2001.5157. [DOI] [PubMed] [Google Scholar]
- 13.Lemon B., Tjian R. Genes Dev. 2000;14:2551–2569. doi: 10.1101/gad.831000. [DOI] [PubMed] [Google Scholar]
- 14.Nettelbeck D.M., Jérôme V., Müller R. Trends Genet. 2000;16:174–181. doi: 10.1016/s0168-9525(99)01950-2. [DOI] [PubMed] [Google Scholar]
- 15.Persic L., Righi M., Roberts A., Hoogenboom H.R., Cattaneo A., Bradbury A. Gene. 1997;187:1–8. doi: 10.1016/s0378-1119(96)00627-0. [DOI] [PubMed] [Google Scholar]
- 16.Choi T., Huang M., Gorman C., Jaenisch R. Mol. Cell. Biol. 1991;11:3070–3074. doi: 10.1128/mcb.11.6.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang M.T.F., Gorman C.M. Nucleic Acids Res. 1990;18:937–947. doi: 10.1093/nar/18.4.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buchman A.R., Berg P. Mol. Cell. Biol. 1988;8:4395–4405. doi: 10.1128/mcb.8.10.4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wise R.J., Orkin S.H., Collins T. Nucleic Acids Res. 1989;17:6591–6601. doi: 10.1093/nar/17.16.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang M.T.F., Gorman C.M. Mol. Cell. Biol. 1990;10:1805–1810. doi: 10.1128/mcb.10.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petitclerc D., Attal J., Théron M.C., Bearzotti M., Bolifraud P., Kann G., Stinnakre M.-G., Pointu H., Puissant C., Houdebine L.-M. J. Biotechnol. 1995;40:169–178. doi: 10.1016/0168-1656(95)00047-t. [DOI] [PubMed] [Google Scholar]
- 22.Kim S.-Y., Lee J.-H., Shin H.-S., Kang H.-J., Kim Y.-S. J. Biotechnol. 2002;93:183–187. doi: 10.1016/s0168-1656(01)00388-1. [DOI] [PubMed] [Google Scholar]
- 23.Hall J., Hirst B.H., Hazlewood G.P., Gilbert H.J. Biochim. Biophys. Acta. 1992;1130:259–266. doi: 10.1016/0167-4781(92)90438-6. [DOI] [PubMed] [Google Scholar]
- 24.Zaboikin M.M., Schuening F.G. Hum. Gene Ther. 1998;9:2263–2275. doi: 10.1089/hum.1998.9.15-2263. [DOI] [PubMed] [Google Scholar]
- 25.Proudfoot N. Cell. 1996;87:779–781. doi: 10.1016/s0092-8674(00)81982-0. [DOI] [PubMed] [Google Scholar]
- 26.Gray N.K., Wickens M. Annu. Rev. Cell Develop. Biol. 1998;14:399–458. doi: 10.1146/annurev.cellbio.14.1.399. [DOI] [PubMed] [Google Scholar]
- 27.Proudfoot N. Cell. 1991;64:671–674. doi: 10.1016/0092-8674(91)90495-k. [DOI] [PubMed] [Google Scholar]
- 28.Proudfoot N.J. Nature. 1986;322:562–565. doi: 10.1038/322562a0. [DOI] [PubMed] [Google Scholar]
- 29.Makrides S.C. Microbiol. Rev. 1996;60:512–538. doi: 10.1128/mr.60.3.512-538.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maa M.-C., Chinsky J.M., Ramamurthy V., Martin B.D., Kellems R.E. J. Biol. Chem. 1990;265:12513–12519. [PubMed] [Google Scholar]
- 31.Pesole G., Mignone F., Gissi C., Grillo G., Licciulli F., Liuni S. Gene. 2001;276:73–81. doi: 10.1016/s0378-1119(01)00674-6. [DOI] [PubMed] [Google Scholar]
- 32.Kozak M. Gene. 1999;234:187–208. doi: 10.1016/s0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- 33.Stein I., Itin A., Einat P., Skaliter R., Grossman Z., Keshet E. Mol. Cell. Biol. 1998;18:3112–3119. doi: 10.1128/mcb.18.6.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCaughan K.K., Brown C.M., Dalphin M.E., Berry M.J., Tate W.P. Proc. Natl. Acad. Sci. USA. 1995;92:5431–5435. doi: 10.1073/pnas.92.12.5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fedorov A., Saxonov S., Gilbert W. Nucleic Acids Res. 2002;30:1192–1197. doi: 10.1093/nar/30.5.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kane J.F. Curr. Opin. Biotechnol. 1995;6:494–500. doi: 10.1016/0958-1669(95)80082-4. [DOI] [PubMed] [Google Scholar]
- 37.Ellgaard L., Molinari M., Helenius A. Science. 1999;286:1882–1888. doi: 10.1126/science.286.5446.1882. [DOI] [PubMed] [Google Scholar]
- 38.Sakaguchi M. Curr. Opin. Biotechnol. 1997;8:595–601. doi: 10.1016/s0958-1669(97)80035-3. [DOI] [PubMed] [Google Scholar]
- 39.Perlman D., Halvorson H.O. J. Mol. Biol. 1983;167:391–409. doi: 10.1016/s0022-2836(83)80341-6. [DOI] [PubMed] [Google Scholar]
- 40.von Heijne G. J. Membr. Biol. 1990;115:195–201. doi: 10.1007/BF01868635. [DOI] [PubMed] [Google Scholar]
- 41.Clément J.-M., Jehanno M. J. Biotechnol. 1995;43:169–181. doi: 10.1016/0168-1656(95)00127-1. [DOI] [PubMed] [Google Scholar]
- 42.Kamiya T., Sugio S., Yamanouchi K., Kagitani Y. Tohoku J. Exp. Med. 1996;180:297–308. doi: 10.1620/tjem.180.297. [DOI] [PubMed] [Google Scholar]
- 43.Farrell P.J., Behie L.A., Iatrou K. Proteins: Structure, Function and Genetics. 2000;41:144–153. [PubMed] [Google Scholar]
- 44.Herrera A.M., Musacchio A., Fernandez J.R., Duarte C.A. Biochem. Biophys. Res. Commun. 2000;273:557–559. doi: 10.1006/bbrc.2000.2974. [DOI] [PubMed] [Google Scholar]
- 45.Chapman B.S., Thayer R.M., Vincent K.A., Haigwood N.L. Nucleic Acids Res. 1991;19:3979–3986. doi: 10.1093/nar/19.14.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J., O'Kane D.J., Escher A. Gene. 1997;203:141–148. doi: 10.1016/s0378-1119(97)00505-2. [DOI] [PubMed] [Google Scholar]
- 47.Maeda Y., Soda M., Ito K., Sato K. Biochem. Mol. Biol. Int. 1997;42:825–832. doi: 10.1080/15216549700203261. [DOI] [PubMed] [Google Scholar]
- 48.Lo K.-M., Sudo Y., Chen J., Li Y., Lan Y., Kong S.-M., Chen L.L., An Q., Gillies S.D. Protein Eng. 1998;11:495–500. doi: 10.1093/protein/11.6.495. [DOI] [PubMed] [Google Scholar]
- 49.Hearn M.T.W., Acosta D. J. Mol. Recognit. 2001;14:323–369. doi: 10.1002/jmr.555. [DOI] [PubMed] [Google Scholar]
- 50.Makrides S.C., Nygren P.-Å., Andrews B., Ford P.J., Evans K.S., Hayman E.G., Adari H., Levin J., Uhlén M., Toth C.A. J. Pharmacol. Exp. Ther. 1996;277:534–542. [PubMed] [Google Scholar]
- 51.Rumlová M., Benedíková J., Cubínková R., Pichová I., Ruml T. Protein Expr. Purif. 2001;23:75–83. doi: 10.1006/prep.2001.1488. [DOI] [PubMed] [Google Scholar]
- 52.Chen C.-Y.A., Shyu A.-B. Trends Biochem. Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 53.Zubiaga A.M., Belasco J.G., Greenberg M.E. Mol. Cell. Biol. 1995;15:2219–2230. doi: 10.1128/mcb.15.4.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lagnado C.A., Brown C.Y., Goodall G.J. Mol. Cell. Biol. 1994;14:7984–7995. doi: 10.1128/mcb.14.12.7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fussenegger M., Mazur X., Bailey J.E. Biotechnol. Bioeng. 1998;57:1–10. doi: 10.1002/(sici)1097-0290(19980105)57:1<1::aid-bit1>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 56.Hennecke M., Kwissa M., Metzger K., Oumard A., Kröger A., Schirmbeck R., Reimann J., Hauser H. Nucleic Acids Res. 2001;29:3327–3334. doi: 10.1093/nar/29.16.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martinez-Salas E. Curr. Opin. Biotechnol. 1999;10:458–464. doi: 10.1016/s0958-1669(99)00010-5. [DOI] [PubMed] [Google Scholar]
- 58.Contag C.H., Bachmann M.H. Annu. Rev. Biomed. Eng. 2002;4:235–260. doi: 10.1146/annurev.bioeng.4.111901.093336. [DOI] [PubMed] [Google Scholar]
- 59.Kozak M. Mol. Cell. Biol. 2001;21:1899–1907. doi: 10.1128/MCB.21.6.1899-1907.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schneider R., Kozak M. Mol. Cell. Biol. 2001;21:8238–8246. doi: 10.1128/MCB.21.23.8238-8246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gäken J., Jiang J., Daniel K., van Berkel E., Hughes C., Kuiper M., Darling D., Tavassoli M., Galea-Lauri J., Ford K., Kemeny M., Russell S., Farzaneh F. Gene Ther. 2000;7:1979–1985. doi: 10.1038/sj.gt.3301341. [DOI] [PubMed] [Google Scholar]
- 62.Donnelly M.L.L., Hughes L.E., Luke G., Mendoza H., ten Dam E., Gani D., Ryan M.D. J. Gen. Virol. 2001;82:1027–1041. doi: 10.1099/0022-1317-82-5-1027. [DOI] [PubMed] [Google Scholar]
- 63.Donnelly M.L.L., Luke G., Mehrotra A., Li X.J., Hughes L.E., Gani D., Ryan M.D. J. Gen. Virol. 2001;82:1013–1025. doi: 10.1099/0022-1317-82-5-1013. [DOI] [PubMed] [Google Scholar]
- 64.Donnelly M.L.L., Gani D., Flint M., Monaghan S., Ryan M.D. J. Gen. Virol. 1997;78:13–21. doi: 10.1099/0022-1317-78-1-13. [DOI] [PubMed] [Google Scholar]
- 65.Goff S.P. In: Fields Virology Fourth Edition. Knipe D.M., Howley P.M., Griffin D.E., Lamb R.A., Martin M.A., Roizman B., Straus S.E., editors. Lippincott Williams & Wilkins; Philadelphia, PA: 2001. [Google Scholar]
- 66.Palese P. Proc. Natl. Acad. Sci. USA. 1998;95:12750–12752. doi: 10.1073/pnas.95.22.12750. [DOI] [PMC free article] [PubMed] [Google Scholar]