

Abstract
Decarboxylative functionalization via hydrogen-atom transfer offers an attractive alternative to standard redox approaches to this important class of transformations. Herein, we report a direct decarboxylative functionalization of aliphatic carboxylic acids using N-xanthylamides. The unique reactivity of amidyl radicals in hydrogen-atom transfer enables decarboxylative xanthylation under redox-neutral conditions. This platform provides expedient access to a range of derivatives through subsequent elaboration of the xanthate group.
Graphical Abstract
Decarboxylative functionalizations constitute a diverse class of synthetic transformations that leverage the wide availability of carboxylic acids as substrates to deliver a variety of useful products.1 For example, the classic Barton decarboxylation via thiohydroxamate esters is a powerful approach to the modification of carboxylic acids.2 Oxidative processes are also featured in many reactions, from the Kolbe electrolysis to an array of Ag(II)-mediated transformations. Recently, photoredox catalysis has significantly expanded the breadth of decarboxylative transformations. These typically involve either oxidations of carboxylate salts,3 or reductions of activated carboxylate derivatives (Figure 1).4 N-hydroxyphthalimide esters have found much use in this reductive platform.5
Figure 1.

Decarboxylative functionalizations of carboxylic acids.
A complementary approach to decarboxylative functionalization would involve O─H hydrogen-atom transfer (HAT) of a carboxylic acid to generate a carboxyl radical under neutral conditions. Such a process presents a significant challenge, however: the O─H bond dissociation energy is approximately 112 kcal/mol, providing a large barrier to reaction.6 Unsurprisingly, a ground state O─H HAT of a carboxylic acid is unknown and would require a highly reactive species to occur.7
We have demonstrated previously that N-functionalized amides can serve as precursors of amidyl radicals for achieving a range of intermolecular, site-selective aliphatic C─H bond functionalizations.8 The key thermodynamic driving force for these reactions is the formation of a strong amide N─H bond (BDE ~ 111 kcal/mol) from an unactivated aliphatic C─H bond (BDE = 96–101 kcal/mol).9 Considering that the range of BDEs for carboxylic acid O─H bonds (BDE ~ 112 kcal/mol) is similar to that of an amide N─H bond, we hypothesized that amidyl radicals could engage carboxylic acids directly via O─H HAT to facilitate decarboxylative transformations. Herein, we report a direct, decarboxylative xanthylation of carboxylic acids as a representative reaction of this type, demonstrating the unique ability of amidyl radicals to perform O─H HAT.10
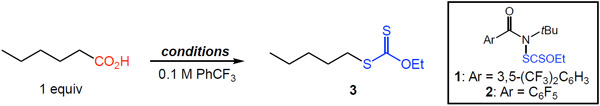
Our studies commenced with hexanoic acid as substrate using 440 nm blue LED photoinitiation and our previously reported N-xanthylamide 1.8c We found that under these conditions, alkyl xanthate 3 was formed in moderate yield (59%, Table 1, entry 1). The use of dilauroyl peroxide (DLP) as initiator provided 3 with similar efficiency (entry 2). Switching to the use of a pentafluorophenyl-substituted N-xanthylamide (2) significantly increased the reaction yield (entries 3 and 4).
Table 1.
Decarboxylative xanthylation of hexanoic acid.
 | |||
|---|---|---|---|
| entry | xanthylamide reagent | initiation | % yielda |
| 1 | 1 (1 equiv) | 440 nm LED | 59 |
| 2 | 1 (1 equiv) | 10 mol % DLP | 49 |
| 3 | 2 (1 equiv) | 440 nm LED | 81 |
| 4 | 2 (1.2 equiv) | 440 nm LED | 86 |
| 5 | 2 (1 equiv) | 10 mol % DLP | 55 |
| 6 | 2 (2 equiv) | 10 mol % DLP | 70 |
| 7b | 2 (1 equiv) | 10 mol % DLP | <2 |
| 8 | 2 (1 equiv) | - | <2 |
Determined by 1H NMR spectroscopy of the crude reaction mixtures using hexamethyldisiloxane as an internal standard.
Reaction was performed in the presence of 1 equiv Cs2CO3.
Chemical initiation of the xanthylation using reagent 2 was also successful, albeit with slightly decreased efficiency (entries 5 and 6). The reaction of a carboxylate salt using Cs2CO3 as base (entry 7) or performing a reaction in the absence of initiator (entry 8) led to no conversion of the carboxylic acid. We continued our studies using reagent 2 owing to its superior performance and ease of large-scale reagent preparation.
Figure 2 details our studies demonstrating the considerable scope of the decarboxylative xanthylation. Both chemical (10 mol % DLP) and photochemical (440 nm LEDs) modes of initiation were successful with virtually all of the substrates shown. The highest reaction yield of the two methods is provided in Figure 2; all yields and the corresponding conditions are provided in the Supporting Information. Primary carboxylic acids were converted to alkyl xanthates 3-8 in good yields. Alkyl bromides are tolerated (4), which is notable considering that substitution of alkyl halides is the most common method for the preparation of alkyl xanthates and thiols.11 The presence of aryl, ester, ketone, or alkene functionalities was also permitted (5-8). The functionalization of an N-Boc indole-substituted primary carboxylic acid delivered xanthate 9 in 62% yield.
Figure 2.

Decarboxylative xanthylation of diverse substrates. a10 mol % DLP used as initiator. b440 nm LEDs used as initiator. cYield refers to NMR yield with hexamethyldisiloxane as an internal standard. d50 mol % DLP used as initiator. eGC yield using dodecane as an internal standard. fReaction performed using reagent 1.
Secondary carboxylic acids were also excellent substrates (Figure 2). The xanthylation of endo-norbornane-2-carboxylic acid provided solely the exo diastereomer of xanthate 12 in 92% yield. The reaction of 1,4-cyclohexanedicarboxylic acid with 2 equiv of 2 provided dixanthate 13 in excellent yield (94%) as a mixture of diastereomers. Decarboxylative xanthylation of 3-oxocyclobutanecarboxylic acid delivered bifunctional cyclobutane 14 (85% yield), which is an attractive derivative for medicinal chemistry applications.12 Transformations of 4-substituted tetrahydropyran and piperidine carboxylic acids were also efficient, providing 15 and 16 in 73 and 69% yield, respectively. Tertiary carboxylic acids were likewise excellent substrates, providing xanthates 17-20 in nearly quantitative yield. Ketopinic acid yielded xanthate 21 in 67% yield, which is notable considering that decarboxylation of this substrate is known to be challenging.13
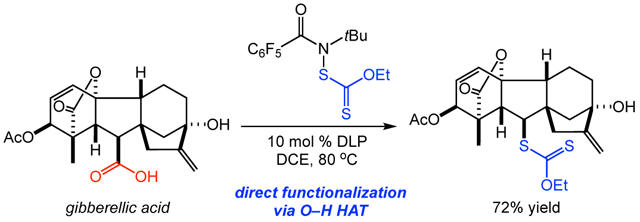
We next applied the decarboxylative xanthylation as a tool for late-stage modification of a range of natural products and drug derivatives (Figure 2). Xanthylation of clotting agent tranexamic acid provided 22 in 90% yield as a mixture of diastereomers. Reactions of the nonsteroidal anti-inflammatory drugs (NSAIDs) ibuprofen and indomethacin delivered xanthates 23 and 24, respectively. We hypothesize that the decreased efficiency of these reactions is due to the increased stability of the intermediate conjugated radicals, which may lead to reversible xanthate transfer.14 Interestingly, ibuprofen was the sole substrate which we examined that gave a higher yield using xanthylamide 1. A number of complex terpenoids and steroids were also good substrates for the xanthylation (25-29), indicating compatibility with unprotected alcohol, ketone, and enone functionalities.15 The plant hormone gibberellic acid efficiently provided xanthate 30 in 72% yield as a single diastereomer. In addition, protected amino acids provided decarboxylative functionalization products 31 and 32 in good yields. Notably, glutamic acid provides opportunities for both unnatural amino acid synthesis and late-stage functionalization of peptides via the carboxylic acid side chain. As a representative example, we performed the decarboxylative xanthylation on a tripeptide to deliver an analogue of the antioxidant glutathione xanthate 33 in moderate yield, albeit with good recovery of the remaining starting material. In light of the importance of cysteine-containing peptides in maintaining regulatory and metabolic functions in plants, animals, fungi and bacteria, we view the decarboxylative xanthylation as an attractive method for peptide modification in future biological studies.16
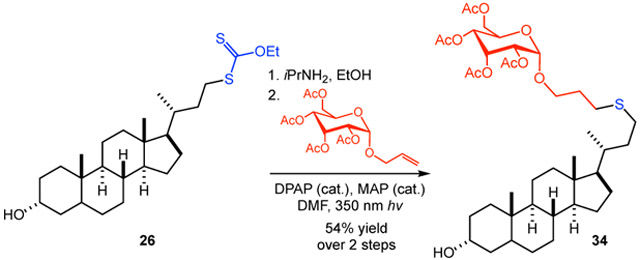
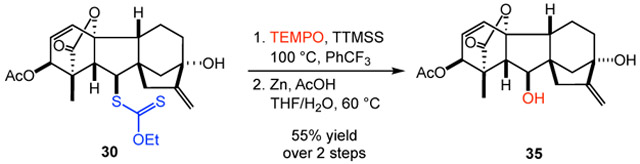
The xanthate functional group is readily elaborated to a wide range of derivatives.8c,17 For example, aminolysis of lithocholic acid-derived xanthate 26 with isopropylamine provided the corresponding thiol in excellent yield.18 Subsequent photochemical thiol-ene coupling with a glucose-derived allyl glycoside produced the conjugation product 34 in 54% yield over two steps. In addition, hydroxylation of gibberellic acid-derived xanthate 30 proceeded in 55% yield using conditions we previously developed for accessing hydroxyl or ketone functionality from alkyl xanthates via an alkoxyamine intermediate.8c Alternative approaches to accessing the products of a formal decarboxylative oxidation include the Barton decarboxylation via thiohydroxamate esters and decarboxylative borylation using N-hydroxyphthalimide esters.19,20
 |
(1) |
 |
(2) |
We envision the decarboxylative xanthylation herein to significantly expand the synthetic capabilities in decarboxylative functionalization via the versatile alkyl xanthate products.
In order to assess the unique reactivity of the amidyl radical in the O─H HAT process, we analyzed the reactivity of related xanthylsulfonamide 36 (Figure 3). The parent sulfonamide has a calculated N─H bond strength of 104 kcal/mol,9 which is somewhat lower than that of alkyl carboxylic acid O─H bonds. Indeed, in an attempted xanthylation of hexanoic acid using 36, no decarboxylative products were observed–a mixture of C─H functionalization products was formed instead.21 Furthermore, in an intermolecular competition between hexanoic acid and cyclohexane, xanthylsulfonamide 36 provided only cyclohexyl xanthate 10 (Figure 3A). In contrast, the same experiments using xanthylamides were selective for decarboxylative xanthylation with a selectivity (kO─H/kC─H) of 10 and 24 for reagents 1 and 2, respectively, correcting for the number of hydrogen atoms. This marked difference in reactivity profile between the amide and sulfonamide reagents offers opportunities for multi-site xanthylations of carboxylic acids. As an example, adamantane carboxylic acid was transformed to either the decarboxylative xanthylation product 18 or the C─H xanthylation product 37 simply by selecting the appropriate reagent (Figure 3B).
Figure 3.

Chemoselective O─H versus C─H HAT studies.
While our results are consistent with a reaction mechanism involving direct O─H HAT, other potential mechanisms were considered, such as oxidation of a carboxylate salt by the xanthylamide reagent. No reaction was observed in the presence of a non-coordinating base (Table 1, entry 7) or with a cesium carboxylate salt as substrate, which is inconsistent with an oxidative pathway. Furthermore, no reaction was observed upon heating hexanoic acid and xanthylamide 2 in the dark with no initiator. As further evidence for the intermediacy of carbon-centered radicals, an enantioenriched carboxylic acid provided the corresponding xanthate as a racemate (see Supporting Information for details).
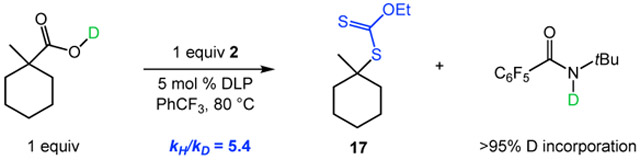
With respect to the O─H HAT step, a primary kinetic isotope effect (KIE) of 5.4 was determined from a comparison of the initial rates of decarboxylative xanthylation between O─H and O─D forms of 1-methyl-1-cyclohexanecarboxylic acid, consistent with an irreversible O─H HAT (eq 3). The present study clearly indicates the possibility for HAT of a stronger O─H bond in the presence of weaker C─H bonds, such as activated benzylic, allylic, and α-heteroatom positions. We hypothesize that this chemoselectivity stems from the kinetic facility of HAT between heteroatoms.22
 |
(3) |
We additionally undertook a computational analysis of a model O─H HAT process involving the relevant nitrogen-centered radicals to support the proposed pathway. As depicted in Figure 4A, we evaluated the O─H HAT of n-propanoic acid by the pentafluorophenyl-substituted amidyl radical 2’ (from reagent 2) and the 3,5-bis(trifluoromethyl)phenyl-substituted sulfonamidyl radical 36’ (from reagent 36). We optimized the transition state structures 38 (see Figure 4B) and 39, and tracked the evolution of the systems along the corresponding intrinsic reaction coordinates (IRC; see Figures S7 and S8). We also determined the associated Gibbs free energy barriers (ΔG‡) and changes (ΔG).23
Figure 4.

A) O─H HAT processes modeled by computational means; the reported ΔG and ΔG‡ values were obtained at the SMD(CH2Cl2)-UωB97XD/def2TZVP//UωB97XD/def2TZVP level of theory (see SI for further details). B) Optimized structure of transition state 38 at the UωB97XD/def2TZVP level of theory (gas phase optimization).
Our calculations suggested that the O─H HAT involving 2’ is slightly exergonic (ΔG = −1.68 kcal•mol−1), while that of 36’ is endergonic (ΔG = +5.90 kcal•mol−1). Similarly, the reaction of 2’ proceeds with a lower energy barrier (ΔG‡ = +20.04 kcal•mol−1) than that promoted by 36’ (ΔG‡ = +25.39 kcal•mol−1), with a ΔΔG‡ value of 5.35 kcal•mol−1. Overall, these computational data support a facile O─H HAT by amidyl radical 2’, with a somewhat larger barrier for the reaction of 36’. We also speculate that hydrogen bonding to the carboxylic acid may play a role in facilitating the O─H HAT step. Investigations targeting these mechanistic details are underway.24
In conclusion, we have developed a direct decarboxylative xanthylation of aliphatic carboxylic acids using N-xanthylamides. This transformation exhibits a broad substrate scope, excellent functional group tolerance, and serves as a general platform for decarboxylative functionalization via the synthetic versatility of alkyl xanthates. Mechanistic data support a pathway involving direct O─H HAT, which complements standard redox-based approaches to decarboxylative chemistry. We anticipate that this unique mode of direct hydrogen-atom transfer will prove valuable in a variety of synthetic contexts.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by Award No. R35 GM131708 from the National Institute of General Medical Sciences. C.G.N. thanks the NSF for a Graduate Research Fellowship and the Graduate School for a Royster Fellowship. We additionally would like to thank the University of North Carolina at Chapel Hill and the Research Computing group for providing computational resources.
Footnotes
Supporting Information. Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Schwarz J; König B Decarboxylative Reactions with and without Light – a Comparison. Green Chem. 2018, 20, 323–361. [Google Scholar]
- (2).(a) For reviews, see: Motherwell WB; Imboden C Radicals in Organic Synthesis, Volume 1: Basic Principles; Renaud P, Sibi MP, Eds.; Wiley-VCH: Weinheim, 2001; p 107; [Google Scholar]; (b) Barton DHR; Motherwell WB Some Recent Progress in Natural Product Chemistry. Heterocycles 1984, 21, 1–19. [Google Scholar]; (c) Barton DHR; Zard SZ nventions of New Reactions Useful in the Chemistry of Natural Products. Pure Appl. Chem 1986, 58, 675–684. [Google Scholar]; (d) Crich D; Quintero L Radical Chemistry Associated with the Thiocarbonyl Group. Chem. Rev 1989, 89, 1413–1432. [Google Scholar]; (e) Barton DHR The Invention of Chemical Reactions: the Last Five Years. Tetrahedron 1992, 48, 2529–2544. [Google Scholar]; (f) Barton DHR The Invention of Chemical Reactions of Relevance to the Chemistry of Natural Products. Pure Appl. Chem 1994, 66, 1943–1954. [Google Scholar]
- (3).(a) For recent examples of oxidative decarboxylations, see Liang Y; Zhang X; MacMillan DWC Decarboxylative sp3 C─N Coupling via Dual Copper and Photoredox Catalysis. Nature 2018, 559, 83–88; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Garza-Sanchez RA; Tlahuext-Aca A; Tavakoli G; Glorius F Visible Light-Mediated Direct Decarboxylative C─H Functionalization of Heteroarenes. ACS Catal. 2017, 7, 4057–4061; [Google Scholar]; (c) Johnston CP; Smith RT; Allmendinger S; MacMillan DWC Metallaphotoredox-Catalysed sp3–sp3 Cross-Coupling of Carboxylic Acids with Alkyl Halides. Nature 2016, 536, 322–325; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Griffin JD; Zeller MA; Nicewicz DA Hydrodecarboxylation of Carboxylic and Malonic Acid Derivatives via Organic Photoredox Catalysis: Substrate Scope and Mechanistic Insight. J. Am. Chem. Soc 2015, 137, 11340–11348; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Merging Photoredox with Nickel Catalysis: Coupling of α-Carboxyl sp3-Carbons with Aryl Halides. Science 2014, 345, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Liu X; Wang Z; Cheng X; Li C Silver-Catalyzed Decarboxylative Alkynylation of Aliphatic Carboxylic Acids in Aqueous Solution. J. Am. Chem. Soc 2012, 134, 14330–14333; [DOI] [PubMed] [Google Scholar]; (g) Cui L; Chen H; Liu C; Li C Silver-Catalyzed Decarboxylative Allylation of Aliphatic Carboxylic Acids in Aqueous Solution. Org. Lett 2016, 18, 2188–2191; [DOI] [PubMed] [Google Scholar]; (h) Tan X; Song T; Wang Z; Chen H; Cui L; Li C Silver-Catalyzed Decarboxylative Bromination of Aliphatic Carboxylic Acids. Org. Lett 2017, 19, 1634–1637; [DOI] [PubMed] [Google Scholar]; (i) Wang Z; Zhu L; Yin F; Su Z; Li Z; Li C Silver-Catalyzed Decarboxylative Chlorination of Aliphatic Carboxylic Acids. J. Am. Chem. Soc 2012, 134, 4258–4263; [DOI] [PubMed] [Google Scholar]; (j) Yin F; Wang Z; Li Z; Li C Silver-Catalyzed Decarboxylative Fluorination of Aliphatic Carboxylic Acids in Aqueous Solution. J. Am. Chem. Soc 2012, 134, 10401–10404; [DOI] [PubMed] [Google Scholar]; (k) Liu C; Wang X; Li Z; Cui L; Li C Silver-Catalyzed Decarboxylative Radical Azidation of Aliphatic Carboxylic Acids in Aqueous Solution. J. Am. Chem. Soc 2015, 137, 9820–9823; [DOI] [PubMed] [Google Scholar]; (l) Tan X; Liu Z; Shen H; Zhang P; Zhang Z; Li C Silver-Catalyzed Decarboxylative Trifluoromethylation of Aliphatic Carboxylic Acids. J. Am. Chem. Soc 2017, 139, 12430–12433. [DOI] [PubMed] [Google Scholar]
- (4).(a) For recent examples of reductive decarboxylations, see Zeng X; Yan W; Zacate SB; Chao T-H; Sun X; Cao Z; Bradford KGE; Paeth M; Tyndall SB; Yang K; Kuo T-C; Cheng M-J; Liu W Copper-Catalyzed Decarboxylative Difluoromethylation. J. Am. Chem. Soc. 2019, 141, 11398–11403; [DOI] [PubMed] [Google Scholar]; (b) Fu M-C; Shang R; Zhao B; Wang B; Fu Y Photocatalytic Decarboxylative Alkylations Mediated by Triphenylphosphine and Sodium Iodide. Science 2019, 363, 1429–1434; [DOI] [PubMed] [Google Scholar]; (c) Patra T; Mukherjee S; Ma J; Strieth-Kalthoff F; Glorius F Visible-Light-Photosensitized Aryl and Alkyl Decarboxylative Functionalization Reactions. Angew. Chem. Int. Ed 2019, 58, 10514–10520; [DOI] [PubMed] [Google Scholar]; (d) Fawcett A; Pradeilles J; Wang Y; Mutsuga T; Myers EL; Aggarwal VK Photoinduced Decarboxylative Borylation of Carboxylic Acids. Science 2017, 357, 283–286; [DOI] [PubMed] [Google Scholar]; (e) Li C; Wang J; Barton LM; Yu S; Tian M; Peters DS; Kumar M; Yu AW; Johnson KA; Chatterjee AK; Yan M; Baran PS Decarboxylative Borylation. Science 2017, 356, eaam7355; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Edwards JT; Merchant RR; McClymont KS; Knouse KW; Qin T; Malins LR; Vokits B; Shaw SA; Bao D-H; Wei F-L; Zhou T; Eastgate MD; Baran PS Decarboxylative Alkenylation. Nature 2017, 545, 213–218; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Huihui KMM; Caputo JA; Melchor Z; Olivares AM; Spiewak AM; Johnson KA; DiBenedetto TA; Kim S; Ackerman LKG; Weix DJ Decarboxylative Cross-Electrophile Coupling of N-Hydroxyphthalimide Esters with Aryl Iodides. J. Am. Chem. Soc. 2016, 138, 5016–5019; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Pratsch G; Lackner GL; Overman LE Constructing Quaternary Carbons from N-(Acyloxy)Phthalimide Precursors of Tertiary Radicals Using Visible-Light Photocatalysis. J. Org. Chem 2015, 80, 6025–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Murarka S N-(Acyloxy)Phthalimides as Redox-Active Esters in Cross-Coupling Reactions. Adv. Synth. Catal 2018, 360, 1735–1753. [Google Scholar]
- (6).Blanksby SJ; Ellison GB Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- (7).An article proposing an excited state O─H HAT was recently published: Nguyen VT; Nguyen VD; Haug GC; Dang HT; Jin S; Li Z; Flores-Hansen C; Benavides BS; Arman HD; Larionov OV Alkene Synthesis by Photocatalytic Chemoenzymatically Compatible Dehydrodecarboxylation of Carboxylic Acids and Biomass. ACS Catal. 2019, 9, 9485–9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8) (a).Schmidt VA; Quinn RK; Brusoe AT; Alexanian EJ Site-Selective Aliphatic C─H Bromination Using N–Bromoamides and Visible Light. J. Am. Chem. Soc 2014, 136, 14389–14392; [DOI] [PubMed] [Google Scholar]; (b) Quinn RK; Könst ZA; Michalak SE; Schmidt Y; Szklarski AR; Flores AR; Nam S; Horne DA; Vanderwal CD; Alexanian EJ Site-Selective Aliphatic C─H Chlorination Using N–Chloroamides Enables a Synthesis of Chlorolissoclimide. J. Am. Chem. Soc 2016, 138, 696–702; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Czaplyski WL; Na CG; Alexanian EJ C─H Xanthylation: A Synthetic Platform for Alkane Functionalization. J. Am. Chem. Soc 2016, 138, 13854–13857; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Carestia AM; Ravelli D; Alexanian EJ Reagent-Dictated Site Selectivity in Intermolecular Aliphatic C─H Functionalizations Using Nitrogen-Centered Radicals. Chem. Sci 2018, 9, 5360–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tierney MM; Crespi S; Ravelli D; Alexanian EJ Identifying Amidyl Radicals for Intermolecular C─H Functionalizations. J. Org. Chem 2019, 84, 12983–12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) For examples of decarboxylative xanthylations using the Barton method, see: Barton DHR; George MV; Tomoeda M Photochemical Transformations. Part XIII. A New Method for the Production of Acyl Radicals. J. Chem. Soc 1962, 1967–1974; [Google Scholar]; (b) Delduc P; Tailhan C; Zard SZ A Convenient Source of Alkyl and Acyl Radicals. J. Chem. Soc., Chem. Commun 1988, 4, 308; [Google Scholar]; (c) Jenkins EN; Czaplyski WL; Alexanian EJ A General Approach to Quaternary Center Construction from Couplings of Unactivated Alkenes and Acyl Xanthates. Org. Lett 2017, 19, 2350–2353; [DOI] [PubMed] [Google Scholar]; (d) Zard SZ The Radical Chemistry of Thiocarbonylthio Compounds: An Overview In Handbook of RAFT Polymerization; Barner-Kowollik C, Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp 151–187. [Google Scholar]
- (11).Rayner CM Synthesis of Thiols, Sulfides, Sulfoxides and Sulfones. Contemp. Org. Synth 1995, 2, 409–440. [Google Scholar]
- (12).Marson CM New and Unusual Scaffolds in Medicinal Chemistry. Chem. Soc. Rev 2011, 40, 5514–5533. [DOI] [PubMed] [Google Scholar]
- (13).Fawcett FS Bredt’s Rule of Double Bonds in Atomic-Bridged-Ring Structures. Chem. Rev 1950, 47, 219–274. [DOI] [PubMed] [Google Scholar]
- (14).Chiefari J; Mayadunne RT; Moad CL; Moad G; Rizzardo E; Postma A; Skidmore MA; Thang SH Thiocarbonylthio Compounds (SC(Z)S-R) in Free Radical Polymerization with Reversible Addition-Fragmentation Chain Transfer (RAFT Polymerization). Effect of the Activating Group Z. Macromolecules 2003, 36, 2273–2283. [Google Scholar]
- (15).Comparative studies indicated that decarboxylative xanthylations via Barton thiohydroxamate esters were less efficient with simple aliphatic substrates and ineffective with substrates containing increased functionality. See the Supporting Information for details.
- (16).Poole LB The Basics of Thiols and Cysteines in Redox Biology and Chemistry. Free Radic. Biol. Med 2015, 80, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17) (a).Quiclet-Sire B; Zard SZ Some Aspects of Radical Chemistry in the Assembly of Complex Molecular Architectures. Beilstein J. Org. Chem 2013, 9, 557–576; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Quiclet-Sire B; Zard SZ Powerful Carbon–Carbon Bond Forming Reactions Based on a Novel Radical Exchange Process. Chem. - Eur. J 2006, 12, 6002–6016; [DOI] [PubMed] [Google Scholar]; (c) Quiclet-Sire B; Zard SZ Fun with radicals: Some new perspectives for organic synthesis. Pure Appl. Chem 2011, 83, 519–551. [Google Scholar]
- (18).(a) For examples of C—S bond forming decarboxylations, see ref. (17c) and Wei L; Wu C; Tung C-H; Wang W; Xu Z Decarboxylative Sulfenylation of Amino Acids via Metallaphotoredox Catalysis. Org. Chem. Front 2019, 6, 3224–3227; [Google Scholar]; (b) Nyfeler E; Renaud P Decarboxylative Radical Azidation Using MPDOC and MMDOC Esters. Org. Lett 2008, 10, 985–988; [DOI] [PubMed] [Google Scholar]; (c) Jin Y; Yang H; Fu H An N-(Acetoxy)Phthalimide Motif as a Visible-Light pro-Photosensitizer in Photoredox Decarboxylative Arylthiation. Chem. Commun 2016, 52, 12909–12912; [DOI] [PubMed] [Google Scholar]; (d) Wang P-F; Wang X-Q; Dai J-J; Feng Y-S; Xu H-J Silver-Mediated Decarboxylative C–S Cross-Coupling of Aliphatic Carboxylic Acids under Mild Conditions. Org. Lett 2014, 16, 4586–4589. [DOI] [PubMed] [Google Scholar]
- (19) (a).Barton DHR; Crich D; Motherwell WB Conversion of Aliphatic and Alicyclic Carboxylic Acids into nor-Hydroperoxides, nor-Alcohols, and nor-Oxo Derivatives using Radical Chemistry. J. Chem. Soc., Chem. Commun 1984, 242–242; [Google Scholar]; (b) Barton DHR; Crich D; Motherwell WB The Invention of New Radical Chain Reactions. Part VIII. Radical Chemistry of Thiohydroxamic Esters; A New Method for the Generation of Carbon Radicals from Carboxylic Acids. Tetrahedron 1985, 41, 3901–3924; [Google Scholar]; (c) Barton DHR; Géro SD; Holliday P; Quiclet-Sire B; Zard SZ A Practical Decarboxylative Hydroxylation of Carboxylic Acids. Tetrahedron 1998, 54, 6751–6756. [Google Scholar]
- (20).(a) For examples of decarboxylative oxidations with limited scope, see Song H-T; Ding W; Zhou Q-Q; Liu J; Lu L-Q; Xiao W-J Photocatalytic Decarboxylative Hydroxylation of Carboxylic Acids Driven by Visible Light and Using Molecular Oxygen. J. Org. Chem 2016, 81, 7250–7255; [DOI] [PubMed] [Google Scholar]; (b) Sakakibara Y; Cooper P; Murakami K; Itami K Photoredox-Catalyzed Decarboxylative Oxidation of Arylacetic Acids. Chem.-Asian J 2018, 13, 2410–2413; [DOI] [PubMed] [Google Scholar]; (c) Kiyokawa K; Yahata S; Kojima T; Minakata S Hypervalent Iodine(III)-Mediated Oxidative Decarboxylation of β,γ-Unsaturated Carboxylic Acids. Org. Lett 2014, 16, 4646–4649; [DOI] [PubMed] [Google Scholar]; (d) Xu K; Wang Z; Zhang J; Yu L; Tan J Cobalt-Catalyzed Decarboxylative Acetoxylation of Amino Acids and Arylacetic Acids. Org. Lett 2015, 17, 4476–4478. [DOI] [PubMed] [Google Scholar]
- (21).As attempts to synthesize the pentafluorobenzenesulfonyl xanthylamide were unsuccessful, comparisons were made with the 3,5-bis(trifluoromethyl)phenyl derivatives.
- (22).Mayer JM Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res 2011, 44, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).For the spin density plots of the transition states studied, see Figure S9.
- (24).O─H HAT of a carboxylic acid by the tBuO radical formed from tBuOCl has been previously speculated as a mechanistic possibility, along with polar pathways: Shigemitsu Y; Odaira Y; Tsutsumi S Studies of the Chlorination of the Carboxylic Acids with t-Butyl Hypochlorite. Bull. Chem. Soc. Jpn. 1965, 38, 1450–1455. We thank a reviewer for bringing this paper to our attention. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.