

Abstract
Background:
Exposure to estrogen is strongly associated with increased breast cancer risk. While all women are exposed to estrogen, only 12% are expected to develop breast cancer during their lifetime. These women may be more sensitive to estrogen, as rodent models have demonstrated variability in estrogen sensitivity. Our objective was to determine individual variation in expression of estrogen receptor (ER) and estrogen-induced responses in the normal human breast.
Methods:
Human breast tissue from female donors undergoing reduction mammoplasty surgery were collected for microarray analysis of ER expression. To examine estrogen-induced responses, breast tissue from 23 female donors were cultured ex vivo in basal or 10nM 17β-estradiol (E2) media for 4 days.
Results:
Expression of ER genes (ESR1 and ESR2) increased significantly with age. E2 induced consistent increases in global gene transcription, but expression of target genes AREG, PGR, and TGFβ2 increased significantly only in explants from nulliparous women. E2-treatment did not induce consistent changes in proliferation or radiation induced apoptosis.
Conclusion:
Responses to estrogen are highly variable among women and not associated with levels of ER expression, suggesting differences in intracellular signaling among individuals. The differences in sensitivity to E2-stimulated responses may contribute to variation in risk of breast cancer.
Keywords: Human breast explants, Estrogen Receptor, estrogen signaling
Introduction:
Epidemiological studies have identified lifestyle, environmental, and reproductive risk factors for breast cancer. Reproductive risk factors are linked to lifetime exposure to endogenous estrogens, including early menarche (RR = 1.3), late menopause (RR = 1.2–2.0), or having the highest quartile of serum estrogen levels (RR = 1.8 – 5.0) [1–4]. Exogenous estrogens and use of hormone replacement therapy containing both estrogen and progesterone also increases breast cancer risk (RR = 1.2) [5]. These risks factors would suggest that longer lifetime exposure to estrogen or periods of increased estrogen exposure increase breast cancer risk. Paradoxically, estrogen may also protect against breast cancer. While a full term pregnancy after age 35 increases breast cancer risk, a full term pregnancy before age 20 decreases breast cancer risk by up to 50% [6–9]. This protective effect of early pregnancy has been mimicked in rodent models with estrogen and progesterone treatment [10–13]. For postmenopausal women with breast cancer, high dose estrogen treatment can be an effective antitumor therapy [14, 15]. These contradictory outcomes to estrogen exposure suggest that high levels of estrogen can have antagonistic effects on breast cancer risk.
Estrogen acts in target tissues by binding to estrogen receptor dimers to initiate gene transcription [16–18]. There are two estrogen receptors: Estrogen Receptor alpha (ERα) and Estrogen Receptor beta (ERβ). ERα protein is encoded by ESR1, located on chromosome 6q24–27 and ERβ is encoded by ESR2, located on chromosome 14 q22–24. Gene transcription is modulated by the estrogen receptor dimer combination (ERα homodimers, ERβ homodimers or heterodimers of ERα with ERβ) and the recruitment of transcriptional co-activators or repressors [19]. In the breast, ERα expression is restricted to the luminal mammary epithelia, whereas ERβ is expressed in both the luminal and basal epithelial cells and in the stroma [20]. The expression of ERα in the normal breast epithelium is positively correlated with risk of breast cancer [21, 22] and with age [23, 24]. The variation in ERα positivity among individuals, suggest that women vary, not only in their exposure to estrogen, but also their sensitivity.
All women are exposed to estrogens, but only a subset (~12%) are expected to develop breast cancer if their natural life expectancy is 80 years of age. Why are a subset of women more sensitive to their estrogens? Intracellular signals elicited by E2 may differ among women and determine the impact on breast cancer risk. Hormone studies using rodent models demonstrate variability to estrogen sensitivity in the mammary gland. Different strains of mice exhibit differences in proliferation, apoptosis and tumor development in response to estrogen [25–27]. Likewise, in rat models, both the Copenhagen and Brown Norway rat are resistant to estrogen-mediated mammary tumors, while the ACI rat is susceptible [28].
Human models for estrogen response have relied primarily on population studies that have obtained human breast tissue at various life stages or different stages of the menstrual cycle. Several studies revealed wide variation in ER levels in breast tissues among women, but did not measure levels of estrogen or responses [29, 30]. 2D primary breast epithelial cell culture systems also have limitations because primary human breast epithelia lose estrogen receptor expression and consequently, their estrogen response [31, 32]. Estrogen receptor positive breast cancer cell lines, such as MCF7 and T47D cells, express high levels of estrogen receptor, and although exquisitely sensitive to estrogens, do not reflect responses of the normal breast. These estrogen receptor positive breast cancer cell lines have homogenous expression of ERα and they proliferate in response to estrogen-treatment [33, 34]. However, expression of ERα in normal breast epithelium is limited to a subset of cells and receptor expression does not overlap with cells that are actively proliferating [35, 36]. 3D human breast organoids are comprised of both basal and luminal epithelial cells and retain expression of estrogen receptors in a subset of cells [37], but are devoid of the endogenous stromal cells and connective tissues that may impact tissue responses. In contrast, human breast explants can be maintained in culture for a limited time (2–4 weeks), retain complex cell interactions and are responsive to steroid hormones ex-vivo [38–41]. The ex-vivo culture system eliminates confounding effects of other systemic hormones and factors.
Our goal was to determine variation of estrogen responses in women. Our first objective was to determine the range of endogenous estrogen receptor expression in a sample of women spanning 14 to 70 years of age. Our second objective was to utilize the ex-vivo human breast tissue explant model to examine the effects of a 4-day treatment with 10nM 17β-estradiol (E2) in breast explants from individual women. Using this ex-vivo breast culture system, we can compare responses among women, as well as comparing specific responses within individual donor tissue cultured in basal or E2 media.
Transcriptional activation of genes containing estrogen response elements and formation of R-loops provide measures of estrogen-stimulated intracellular signaling. R-loops are DNA: RNA hybrids that form as a consequence of gene transcription [42, 43]. ERα has been shown to stimulate increases in R-loops providing a global indicator of E2-stimulated transcription [44, 45]. As a more specific indication of E2-mediated transcription, we also compared the expression of known estrogen receptor target genes (AREG, PGR, and TGFβ2) [46–48]. Changes in the proportions of cells with detectable levels of progesterone receptor (PR) and proliferating cell nuclear antigen (PCNA) provided additional measures of functional responses to E2-treatment. Previous work with human breast xenografts in mice [49, 50] and human breast explants cultured ex-vivo [39] demonstrated increases in both PR and proliferation. As a consequence of E2-mediated proliferation, apoptotic rates in the mammary epithelium may be altered. Eigeleine et. al. [39] determined that E2-treatment decreased spontaneous apoptosis the human mammary epithelium. However, in mice, radiation-induced apoptosis is increased in response treatment with estrogen and progesterone [51]. A prior pregnancy or treatment with exogenous estrogen and progesterone at levels to mimic pregnancy also resulted in persistent increases in radiation induced apoptosis (spontaneous and radiation-induced) in the mammary epithelium of mice [13]. The human explant model system provides an opportunity to compare both spontaneous and radiation-induced apoptosis in human breast epithelium in response to E2 from both nulliparous and parous donors.
In the present study, significant age-related changes in estrogen receptors were observed in breast tissues. Although levels of ESR1 and ESR2 varied by greater than 80-fold in breast explants from donors, levels of estrogen receptor expression did not correlate with functional responses in the breast explant cultures. Tissue from nulliparous donors exhibited a greater sensitivity to E2 induced expression of target genes (AREG, PGR, TGFβ2), but expression was inconsistent among parous women. Levels of estrogen receptors do not appear to be rate-limiting for either induction of transcription-associated R-loops or expression of target genes. These data suggest that components of intracellular signal transduction differ among individuals resulting in variable patterns of gene expression and proliferation among individuals.
Materials and Methods
Sample Collection
Human breast tissue samples used for microarray analysis were collected from female donors, age 14–70 years, undergoing reduction mammoplasty surgery at Baystate Medical Center in accordance with the Institutional Review Boards at Baystate Medical Center and the University of Massachusetts, Amherst [52–54]. Donated tissue was snap frozen in liquid nitrogen and stored at −80°C at the Pioneer Valley Life Sciences Institute (PVLSI). Donor demographics and reproductive history were recorded after tissue collection. Human breast tissue for explants was gathered through the Rays of Hope Center for Breast Cancer Research at the PVLSI according to IRB approval #286173 and #132204. Tissue was collected from female donors (n=23) ages 18–62 years undergoing reduction mammoplasty surgery. Donor information including age and parity status were recorded.
Microarray Analysis
Microarrays were performed at the University of North Carolina at Chapel Hill as described previously [52–54]. Briefly, 100mg of frozen tissues were homogenized for RNA isolation using RNeasy kits. RNA quality and concentration were determined using an Agilent 2100 Bioanalyzer and a ND-1000 Nanodrop spectrophotometer. Two-color 4×44K Agilent whole genome arrays were performed. Data are publicly available through the Gene Expression Omnibus (GSE:16113 and GSE:33526). When multiple probe sets for a gene were present, we used the probe with expression values for the most donor samples. Trends were analyzed using linear regression analysis in Graphpad Prism (GraphPad Prism version 7, La Jolla, CA).
Explant Culture
Fresh breast tissue was cut into approximately 1×1×4mm sections using a Stadie-Riggs microtome. For each donor, tissue sections were frozen or fixed immediately for time zero (T=0) samples. The remaining sections were placed on top of surgical foam (Ethicon) in 60mm plastic dishes and cultured for a total of 7 days. The explants were kept in a humidified incubator with 5% CO2 at 37°C. Explants were cultured in basal media for 3 days to clear endogenous hormones and then cultured in basal or E2 media for an additional 4 days. Basal culture media consisted of phenol red free DMEM:F12 (Sigma-Aldrich), 10% charcoal stripped FBS (FB-04, Omega Scientific), 10ng/mL human EGF (21–8356-U100, Tonobo Biosciences), and antibiotic-antimycotic (15240062, Gibco). E2 treatment media included basal media with the addition of 10nM 17β-estradiol (E2758–250MG, Sigma-Aldrich). 4,4’,4”-(4-Propyl-[1H]-pyrazole-1,3,4-triyl)trisphenol-treatment media (PPT; ERα specific agonist) or 7-Ethenyl-2-(3-fluoro-4-hydroxyphenyl)-5-benzoxazolol (ERB041; ERβ specific agonist) included basal media with the addition of 200nM PPT or 200nM ERB041; respectively (Tocris, Minneapolis, MN). Half of the tissue from each donor was irradiated with 5Gy 6 hours prior to tissue collection on day 7 of culture. Half of the explant sections intended for histological endpoints were fixed in 10% NBF overnight and transferred to 70% ethanol before processing and paraffin embedding. The other half of the explant samples were snap frozen and stored at −80°C for later RNA isolation. Donor information for the explants used in this study are included in Table 1 and an overview of the breast explant model is detailed in Figure 2a.
Table 1:
Donor information. Donor number, age, parity status, and age at first birth for cultured breast explants. Mean age for nulliparous donor tissue is 33.4 years (sem +/−5.4); mean age for parous is 42 years (sem +/−3.5). There is no statistical difference in age between the nulliparous and parous donors (p=0.19).
| Nulliparous | Parous | |||
|---|---|---|---|---|
| Donor Number | Age | Donor Number | Age | Age at first birth |
| 237 | 18 | 186 | 26 | 20 |
| 238 | 18 | 189 | 27 | 25 |
| 240 | 18 | 225 | 35 | 27 |
| 231 | 26 | 234 | 35 | 23 |
| 184 | 27 | 181 | 39 | 19 |
| 249 | 28 | 178 | 43 | 39 |
| 187 | 29 | 243 | 45 | 21 |
| 180 | 47 | 185 | 49 | 17 |
| 179 | 61 | 241 | 50 | 18 |
| 250 | 62 | 244 | 56 | 26 |
| 177 | 58 | 21 | ||
| 239 | 35 | unknown | ||
| 176 | 37 | unknown | ||
Fig. 2: Tissue architecture of human breast explants are intact after 7 days in culture.
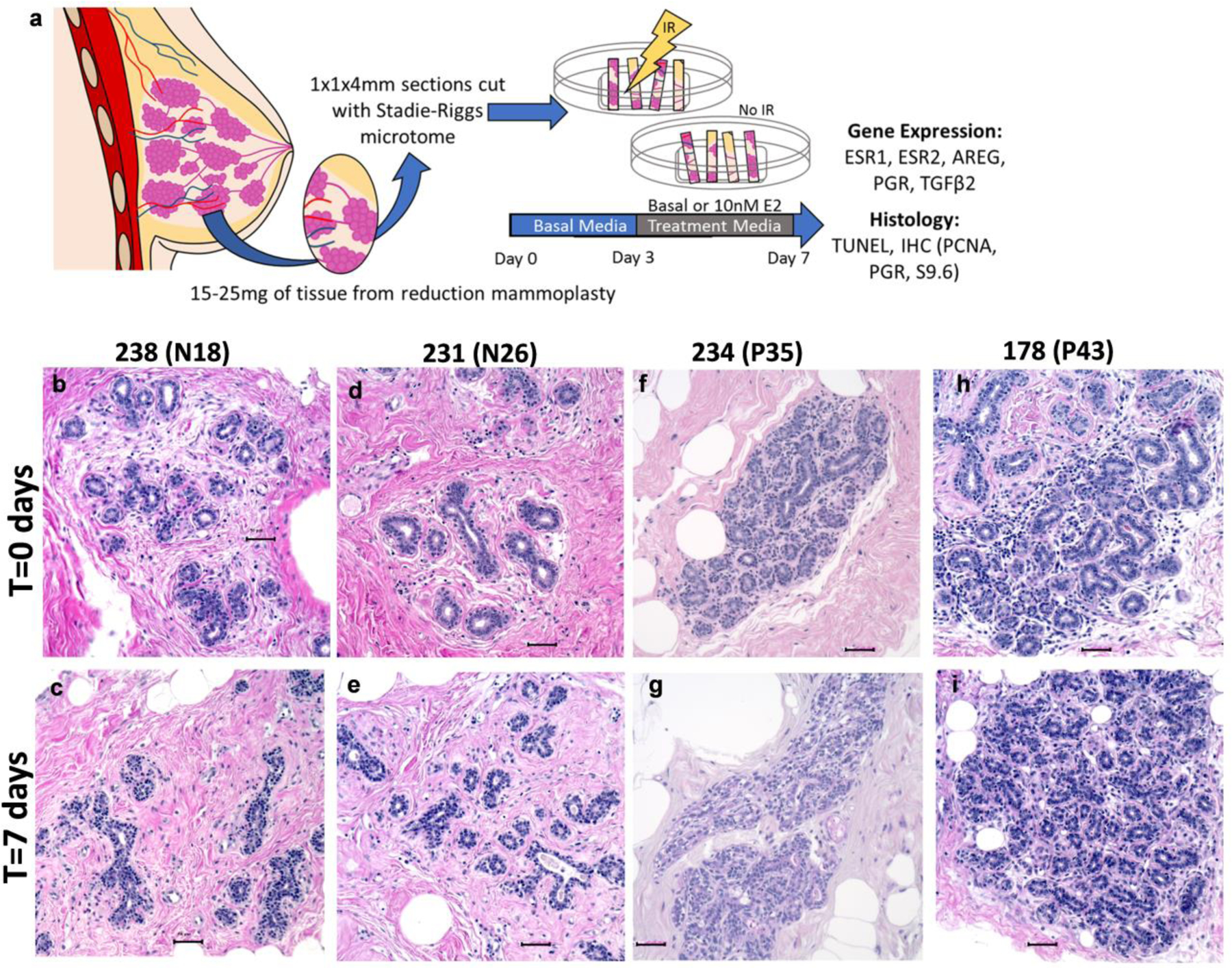
(a) Overview of breast explant model. Tissue obtained from women undergoing reduction mammoplasties was sectioned using a Stadie-Riggs microtome. After clearing in Basal media for 3 days, explants were treated with Basal or E2 containing media for an additional 4 days. On day 7, half of the samples were subjected to 5Gy of irradiation 6 hours prior to tissue collection, and FFPE or freezing in liquid nitrogen for RNA analysis. (b-i) Representative hematoxylin and eosin staining of nulliparous (b-e) and parous (f-i) explant tissue. Comparison of fresh tissue T=0 (b,d,f,h) today 7 tissue (c,e,g,i) demonstrate that tissue architecture is maintained in culture conditions. Scale bars 50μm.
RT-qPCR
RNA isolation was performed using TRIzol according to manufacturer’s recommendation (15596018, ThermoFisher Scientific). cDNA was synthesized using 2 μg of RNA with the Transcriptor First Strand cDNA synthesis kit (04–379-012–001, Roche). cDNA was diluted 1:10 before use in RT-qPCR. RT-qPCR was performed in a thermocycler (Mastercyler Epgradient S model 5345, Eppendorf). See Supplemental Table 1 for primers used. Statistical analysis was performed using 2-way ANOVA in Graphpad Prism (GraphPad Prism version 7, La Jolla, CA). Expression is relative to mean control or to an inter run calibrator (IRC) consisting of pooled cDNA from a subset of T=0 (not cultured) samples.
Hematoxylin and Eosin Staining
Hematoxylin and eosin (H&E) staining were performed on paraffin-embedded 4-micron sections. Briefly, sections were deparaffinized in xylenes and rehydrated in graded ethanols. Sections were stained with Mayer’s modified hematoxylin (S216–32OZ, Poly Scientific) and eosin phloxine alcohol working solution (S176–32OZ, Poly Scientific). After dehydration in graded ethanols and clearing with xylenes, slides were cover-slipped. Images were obtained under 200x magnification (BZ-X700 microscope, Keyence, Itasca, IL).
Promoter Methylation for ESR1 and ESR2
Donor tissues obtained from reduction mammoplasty conducted at Baystate Medical Center in Springfield, MA (IRB 132304) were minced and digested in digestion media with collagen immediately after collection as described [55]. Isolated mammary epithelia were enriched in primary culture in mammocult media. DNA extraction, bisulfite modification, PCR amplification and pyrosequencing were conducted as previously described [56]. Briefly, 2μl treated with DNA bisulfide modification reagents (ZymoResearch, Irving, CA). PCR products for each gene were generated using 1μl of bisulfite-modified DNA and sequenced by Pyrosequencing PyroMark Q24 System (Qiagen Valencia, CA). Genomic target sequences for ERα and ERβ are listed in supplementary table 2 [57, 58].
Immunohistochemistry
Immunohistochemistry (IHC) was performed on paraffin-embedded 4-micron sections using a DakoCytomation autostainer (Dako, Carpinteria, CA) and the antibodies listed in Supplemental Table 3. Sections were deparaffinized in xylenes, rehydrated in graded ethanols, and rinsed in phosphate buffered saline (PBS) before antigen retrieval. For ERβ detection, antigen retrieval was performed in heated 0.01M citrate buffer for 10 minutes. Using the Acuity polymer detection system, primary antibody (1:200, Dako M7292 PPG5/10) incubation was performed at room temperature for 30 minutes, followed by incubation with horse-radish peroxidase (HRP) polymer and chromogen detection. For ERα, PR and PCNA detection the Envision HRP Detection system was used (Dako). Antigen retrieval was performed by heating in 0.01M citrate buffer (pH 6) for 10 minutes for PR and 20 minutes for ERα and PCNA. Primary antibody incubation was performed for 30 minutes using anti-ERα (1:20 MA5–13191, ThermoFisher) anti-PR (1:500 D8Q2J Cell Signaling), or anti-PCNA (1:10000 ab29 Abcam). Incubation with secondary antibody (K4001 or K4003, Dako) was performed according to manufacturer’s instructions (Dako, Carpinteria, CA). Immunoreactivity was visualized with chromogen diaminobenzidine (DAB) incubation for 10 minutes. Sections were counterstained with hematoxylin. Images were obtained under 200x magnification (model BZ-X700 microscope). Positive cells were quantified (ImageJ software, https://imagej.nih.gov/ij/) as the percentage of positive luminal cell nuclei in a total of 600–1200 cells) [59].
Immunofluorescence
Freshly cut 4μM paraffin-embedded sections were deparaffinized/rehydrated (3X xylenes 5 min, 2X 100% ethanol for 5 min, 95% ethanol for 3 min, 70% ethanol for 3 min). Samples were rinsed with PBS. Antigen retrieval was performed by boiling slides in 0.1mM EDTA for 30 minutes. Samples were cooled to RT and treated with SSC 0.2X with gentle shaking at RT for 20 minutes. Samples were blocked in 5% BSA/PBS with 0.5% Tween-20 for 1 hr. Primary antibody incubation was done with monoclonal S9.6 antibody (1:100 ENH0001, Kerafast) or anti-γH2AX (1:100, Cell Signaling #9718S) overnight at 4°C. After primary incubation, samples were washed 3 times with PBS containing 0.5% Tween-20 and then incubated with secondary antibody for 1 hr. Samples were washed 2–3 times with PBS containing 0.5% Tween-20 and then mounted with Vectashield mounting medium containing DAPI (H-1200, Vector Laboratories). Slides were imaged at 60X with Nikon A1 Spectral Confocal microscope in the Light Microscopy Facility and Nikon Center of Excellence at Umass Amherst. Analysis of S9.6 or γH2AX foci per nucleus were calculated using Nikon analysis software.
TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was performed on 4-micron sections. Sections were deparaffinized in xylenes and rehydrated in graded ethanols. The Apoptag Plus Peroxidase in Situ Apoptosis Detection kit was used according to manufacturer’s instructions to label apoptotic cells (S7101, MilliporeSigma). Sections were rehydrated in ethanols and cleared in xylenes before adding cover slips. Images were taken at 200x (model BX40 microscope, Olympus, Tokyo, Japan) using a MicroPublisher 3.3RTV camera (QImaging, Surrey, British Columbia, Canada). Positive cells were quantified using ImageJ as the percentage of positive luminal epithelial cells.
RESULTS
Variation in estrogen receptor expression
Previous studies have demonstrated that ERα protein levels in normal human breast tissue increase with age. Because quantification of ERα expression by IHC is subjective due to heterogeneous distribution and intensity in the mammary gland, we used microarray expression data from normal breast tissue to examine the relationship between age of donor and mRNA levels of ESR1 and ESR2. To account for variation in epithelial content between samples, cytokeratin 18 (KRT18) was used as a normalizer. Linear regression analysis of ESR1 expression normalized to KRT18 demonstrate that ESR1 levels, although highly variable among women, significantly increase with age (p<0.0003; Figure 1a), corresponding with previously reported ERα protein expression data. Variation in ESR1 mRNA level was high, as indicated by a low R2-value for the line of best fit (R2=0.115). ESR2 expression normalized to KRT18 (Figure 1b) in these same donors also revealed a significant increase in expression with age (p<0.0081). There is a weak, but positive correlation between ESR1 and ESR2 (Pearson Correlation Coefficient 0.378, p<0.001) in this data set. The data was also stratified by parity status to assess whether the effect of age was limited to one subgroup. There is no difference in ESR1 expression between parous and nulliparous donors (Figure 1c) and age-related changes in expression were similar for both nulliparous and parous donors (Figure 1e p=0.064 and 0.0016, respectively). For ESR2, the overall expression was significantly higher among parous women (Figure 1 d), but the effect of age did not reach statistical significance in either nulliparous or parous donors (Figure 1f; p=0.43 and 0.12, respectively). However, age is confounded with parity because there are significant differences in age between the nulliparous and parous donors (Supplementary Figure 1a and 1b; mean nulliparous = 26.46+/−1.92 and mean parous = 44.45+/−1.46; p<0.001). Therefore, age is a major factor affecting levels of ESR1 mRNA, but the apparent increase in ESR2 expression (Figure 1b) is likely due to parity, rather than age.
Fig. 1: Estrogen receptor expression diversity among women.
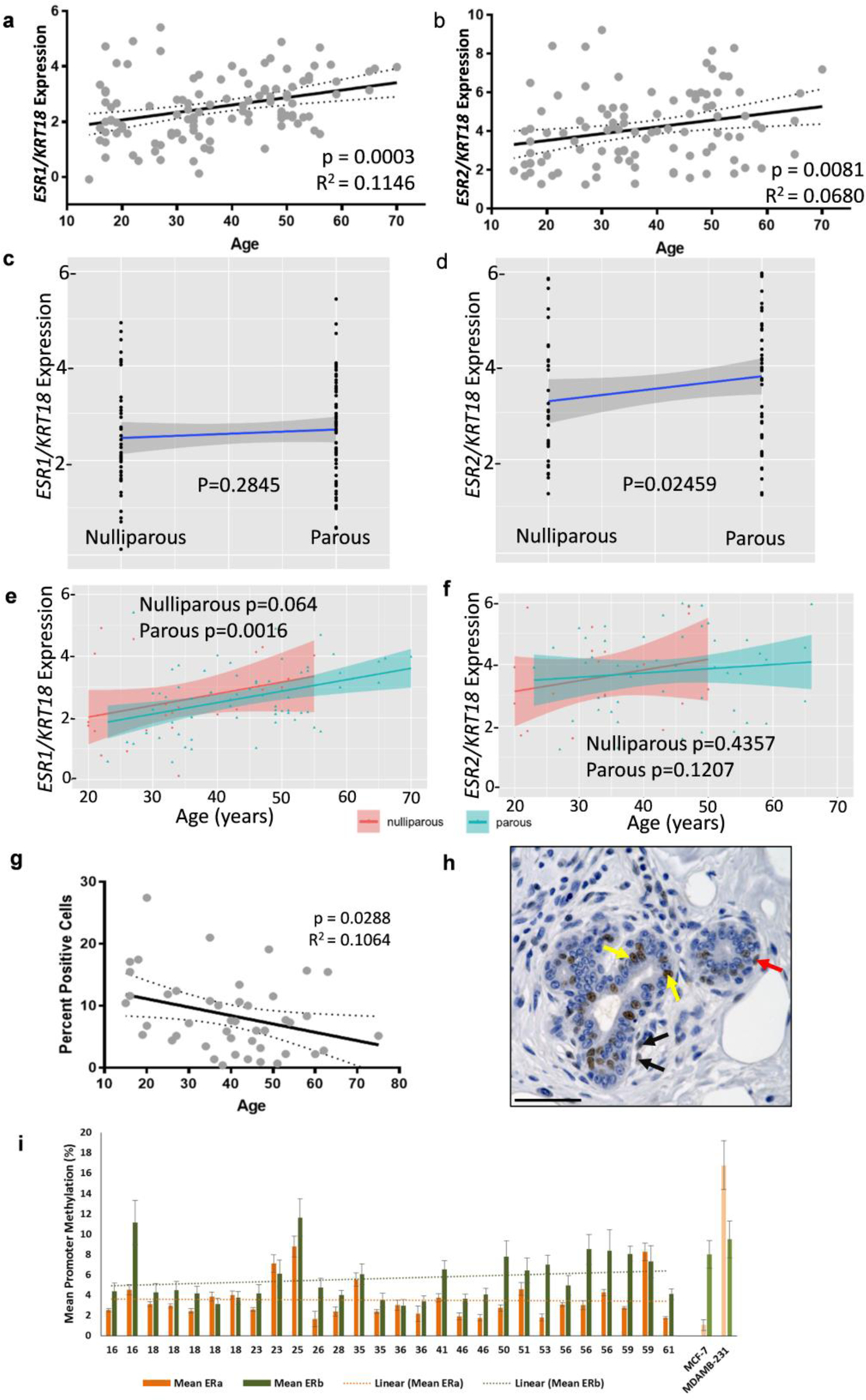
(a-b) Agilent microarray data showing relative (a) ESR1 mRNA levels from 111 donor samples and (b) ESR2 mRNA levels from 102 samples, normalized to KRT18 expression. Linear regression analysis showed a significant increase in both ESR1 (P < 0.001) and ESR2 mRNA levels (P < 0.01) with age. Dotted lines indicate 95% confidence intervals and solid line indicates the best fit line. R2 values for best fit lines are indicated. (c-d) Relative ESR1 and ESR2 expression in nulliparous and parous women. (c) There is no difference in relative normalized ESR1 expression between nulliparous and parous donors. (d) ESR2 expression is increased by parity. (e) Normalized ESR1 expression increases with age in parous donors and is approaching significance in nulliparous donors. (f) Normalized ESR2 expression does not increase with age for either nulliparous or parous donors. (g-h) IHC using ERβ antibody (PPG5/10) was performed on paraffin-embedded sections from 45 patient samples. (g) Quantification of ERβ positive ductal cells. Linear regression analysis of ERβ protein levels shows a significant decrease in ERβ with age (P < 0.05). 95% confidence intervals indicated by dotted lines and solid line indicates the best fit line. (h) Representative image of IHC. Arrows indicate representative positive cells: yellow arrows indicate positive luminal cells, red indicate positive basal cells, and black indicate positive stromal cells. Scale bar 50μM. (i) Methylation in the promoters for ESR1 and ESR2 show there is no association with promoter methylation and age for either ESR1 or ESR2.
While ERα expression is restricted to the luminal epithelial cells of the breast, ERβ expression has been observed in luminal epithelial cells, myoepithelial cells, stromal cells, endothelial cells and lymphocytes. To examine ERβ expression in luminal epithelial cells, we performed IHC using the PGG5/10 mouse anti-human ERβ1 monoclonal antibody in a subset of 45 samples (Figure 1g&h). Linear regression analysis of ERβ positive luminal epithelial cells revealed a significant decrease in ERβ protein expression with age (p<0.0288). Both ESR2 mRNA and ERβ protein levels had a high level of variance (R2-values of 0.068 and 0.106, respectively). These data demonstrate age-related changes in expression of estrogen receptors, but that the effect is highly variable among individuals.
Promoter methylation at CpG sites silences gene expression and differences in promoter methylation for ESR1 and ESR2 may account for the differences in receptor expression between individuals. For instance, percent methylation will be lower in cells with high ESR1 expression and methylation will be higher in cells devoid of ER. This is demonstrated by comparing the high promoter methylation at 7 CpG sites at the ESR1 promoter in MDA-MB-231 cells, which lack ERα, relative to the low ESR1 promoter methylation in ERα-positive MCF7 (Figure 1i; last two samples) and ZR751 cells (supplementary Figure 2a). Likewise, comparison of 9 CpG sites in the promoter of ESR2 demonstrates almost complete promoter methylation for ZR751 and a similar degree of promoter methylation for MCF7 and MDA-MB-231 breast cancer cells (Supplementary Figure 2b). Primary cultures of breast epithelial cells were used to interrogate methylation of these promoters. Comparing the 7 CpG promoter methylation sites for ESR1 and the 9 CpG promoter methylation sites for ESR2 in cells from 28 individuals spanning 16 to 61 years of age shows individual variation in the promoter methylation for both genes (Figure 1i & Supplementary Figure 2 c & d). Linear regression analysis shows no correlation with promoter methylation for either gene with age (Figure 1i) suggesting DNA methylation is not likely to be responsible for age-related changes in ESR1 or ESR2 mRNA. ESR2 methylation was generally increased among women greater than 50 years of age, however two younger individuals also had elevated methylation of ESR2.
Human breast explant model for studying E2 responses in vitro
The analysis of ESR1 and ESR2 expression for figure 1 were performed from fresh tissue obtained from reduction mammoplasties. Some of these women were postmenopausal, but the majority were premenopausal and not controlled for stages of the menstrual cycle. Estrogen receptor expression has been reported to fluctuate with the menstrual cycle such that ER expression is higher at the follicular phase of the estrus cycle. There is also a general consensus that increasing E2 decreases the expression of ERα. The human breast explant system allows us to compare estrogenic responses in the breast tissues from each donor with their own basal control and without confounding systemic factors (Figure 2a). An additional benefit is that stromal, luminal, epithelial, myoepithelial, adipose and even immune cells are present in the culture, so cell interactions can still occur between these cell types. In attempting to restrict effects in explants due to growth factors, we have determined a requirement for EGF in the media; culturing in suboptimal explant conditions resulted in small rounded apoptotic nuclei in the ducts of epithelium that are indicative of tissue degradation (data not shown). In order to confirm that our culture conditions were not leading to degradation of the breast tissue, we compared tissue architecture from tissue that was fixed within two hours of surgery (T=0) to explant tissue after 7 days in culture (Figure 2 b–i). In the minimal media used for the explant samples in this study, we did not observe tissue degradation; nor did we detect gross apoptotic nuclei in the epithelial ducts. The explants retained normal breast architecture. Luminal epithelial cells maintained cuboidal shape and the surrounding stromal cells and adipocytes were intact. As reported by Russo J et al [60], the lobules of nulliparous individuals most often consisted of smaller lobular structures (Figure 2 b–e) whereas tissue from parous individuals had more complex Terminal Duct Lobular Unit (TDLU) structure (Figure 2 g–i) TDLU complexity (or lack of complexity) is retained by the tissue after 7 days in explant culture.
Optimization for gene expression in explant tissues
The expression of ERα protein in the human breast is heterogeneous, in both expression and intensity, and therefore, quantification is complicated. Patterns of distribution within individuals can be scattered or contiguous in the mammary epithelium of one lobule (Figure 3a), while neighboring lobules have absolutely no expression at all (Figure 3b). In addition, the explant fragments are small and tissue is limiting, specifically collagenous portions with good epithelial content. The combined heterogeneity and tissue limitations makes quantification of ERα immunohistochemistry unreliable in explant cultures. Therefore, we quantified ESR1 and ESR2 mRNA levels in response to estrogen treatment in the explant cultures. The breast tissue composition collected from the donors also varies greatly between individuals, which affects the amount of epithelium collected in each tissue fragment and the overall RNA yield. The breast tissue from some donors is enriched with collagenous stroma and epithelial cells, while the stroma of other donors has a greater adipose to collagenous tissue ratio and fewer epithelial cells. To account for differing amounts of epithelium between donors and between tissue fragments, we used KRT18 as a normalizer for epithelial content. We prepared multiple fragments of flash-frozen un-treated breast tissue from three donors and used each piece in separate RNA isolations. We then used RT-qPCR to determine ESR1 and ESR1 normalized to KRT18 (Figure 3 c & d). Donor 178 demonstrates how variable epithelial content of the tissue fragments can be. KRT18 expression (light bars) in fragment 178b is ~6-fold greater and fragment 178d is ~2.7-fold greater than in fragment 178a. ESR1 expression (striped bars) in the four fragments from donor 178 is also variable. However, when ESR1 is normalized to KRT18 (black bars), the ESR1 expression is similar between the four fragments from the same donor. The tissue composition is less variable in donors 185 and 177, which have consistent KRT18 and ESR1 expression between fragments. However, comparison of ESR1 normalized to KRT18 shows that donor 177 had 3.8-fold higher expression of ESR1 than donor 178 (p<0.01) and 1.8-fold higher expression than donor 185 (p<0.05; Figure 3d). Donor 185 had 2-fold higher expression than donor 178 (p<0.05).
Fig. 3: Optimization for gene expression in explant tissues.
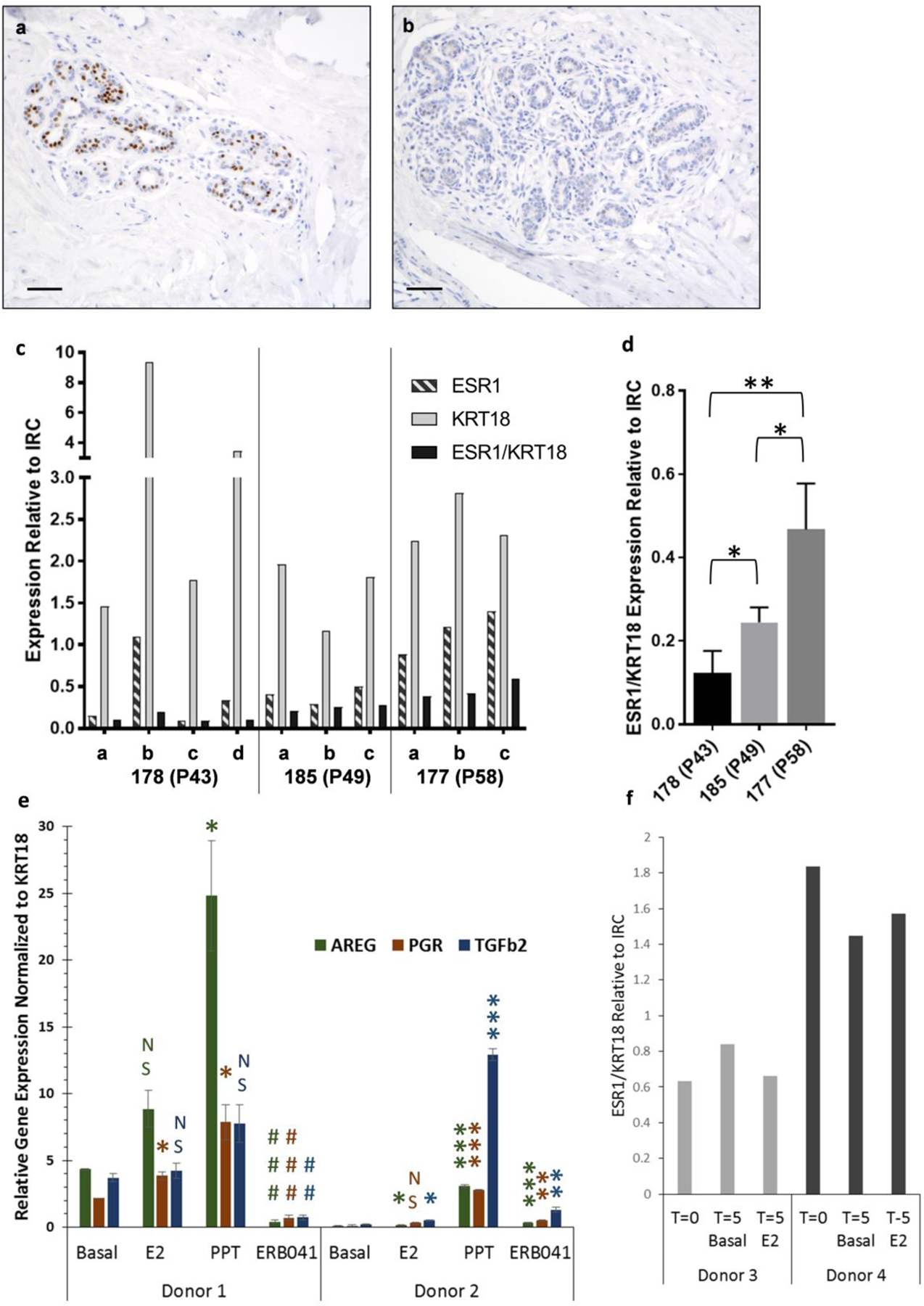
Representative ERα IHC for donor 234 T=0 of two different lobules from same section demonstrating (a) high ERα expression versus (b) no ERα expression. Scale bar is 50μM. (c) 3–4 separate 100ug pieces of flash-frozen tissue from three donors were treated as separate samples and used for RT-qPCR analysis of ESR1, KRT18 and ESR1 expression normalized to KRT18. (d) Normalized ESR1/KRT18 expression demonstrates variation between donors. (e) Comparison of estrogen response genes (AREG, PGR, TGFβ2) in tissue from two donors (3 replicates per donor) collected after treatment with basal media, E2, PPT (ERα specific agonist) or ERB041 (ERβ specific agonist). Genes are normalized to KRT18 and relative to inter run calibrator (IRC). (f) Tissue from two donors was collected at day 0 (T=0) and day 5 (T5) for RT-qPCR analysis. ESR1 levels normalized to KRT18 and relative to a pool of fresh breast tissue which was used as an inter-run calibrator (IRC) are shown.
Both ERαand ERβ contribute to the regulation of estrogen-stimulated responses in breast tissue. The promoters of amphiregulin (AREG) and progesterone receptor (PGR) have estrogen responsive elements that result in increased transcription, while transforming growth factor beta 2 (TGFβ2) is a direct ER target that may be transcriptionally repressed by E2 [61]. Expression of these genes may be modulated by ERβ. Therefore, we wanted to compare the expression of each gene, each normalized to KRT18, in response to basal media, E2-treated media, or media treated with selective agonists for ERα (PPT) or ERβ (ERB041) in tissue obtained from two different donors (Figure 3e). Basal expression of all 3 genes was significantly greater in donor 1 compared to donor 2, but the relative levels varied for each gene. For donor 1, only PGR expression was significantly increased by E2 whereas AREG and PGR expression were significantly increased by PPT (p<0.05). Treatment with the ERβ agonist ERB041 significantly decreased expression of all three genes in donor 1 (p<0.01). Therefore, both ERα and ERβ appear to participate in the regulation of these target genes. Although the basal expression of all three genes was very low in donor 2, E2 stimulated significant increases in AREG and TGFβ2 (p<0.05). PPT-treatment significantly increased all three genes: AREG by almost 40-fold, PGR by 24-fold and TGFβ2 by 60-fold (p<0.001). Whereas ERB041-treatment decreased gene expression in donor 1, expression of all 3 genes were increased by the ERβ agonist in donor 2 (p<0.01). These two donors demonstrate that basal gene expression differs between individuals as well as transcriptional responses mediated by ERα or ERβ.
To confirm that ESR1 expression is retained in the explant cultures, we compared normalized ESR1 expression from two separate donors that had been cultured for 5 days in basal media (T5 Basal) or 10nM E2 (T5 E2) to the normalized ESR1 expression in the fresh donor tissue (T=0; Figure 3f). ESR1 levels were similar between the T=0 fresh tissue and tissue that had been cultured for five days, although there was a consistent 2-fold difference in normalized ESR1 expression between tissue from the two different donors.
Transcriptional responses to estrogen
We used immunofluorescence for the S9.6 antibody against DNA:RNA hybrids as a measure of global gene transcription (Figure 4). There was no difference between the number of R-loop foci/nucleus between T=0 and the explants maintained in basal media (Figure 4c), indicating that basal transcription between the two conditions were similar. However, the E2-treated explants had significantly more R-loop foci/nucleus than either the T=0 tissues or the tissues cultured in basal media (p<0.01). For each donor sampled, E2-treatment increased R-loop formation similarly relative to each basal control (compare representative Figure 4a with 4b), albeit donor 237 had significantly lower R-loop foci/nuclei due to E2-treatment than each of the other four E2-treated donor samples (p<0.01; Figure 4d). These results indicate that 10nM E2-treatment activated significant increases in R-loop formation relative to basal levels, indicating greater transcription compared to the fresh tissue or the explants maintained in basal media.
Fig. 4: E2-induces R-loop formation in human explants.
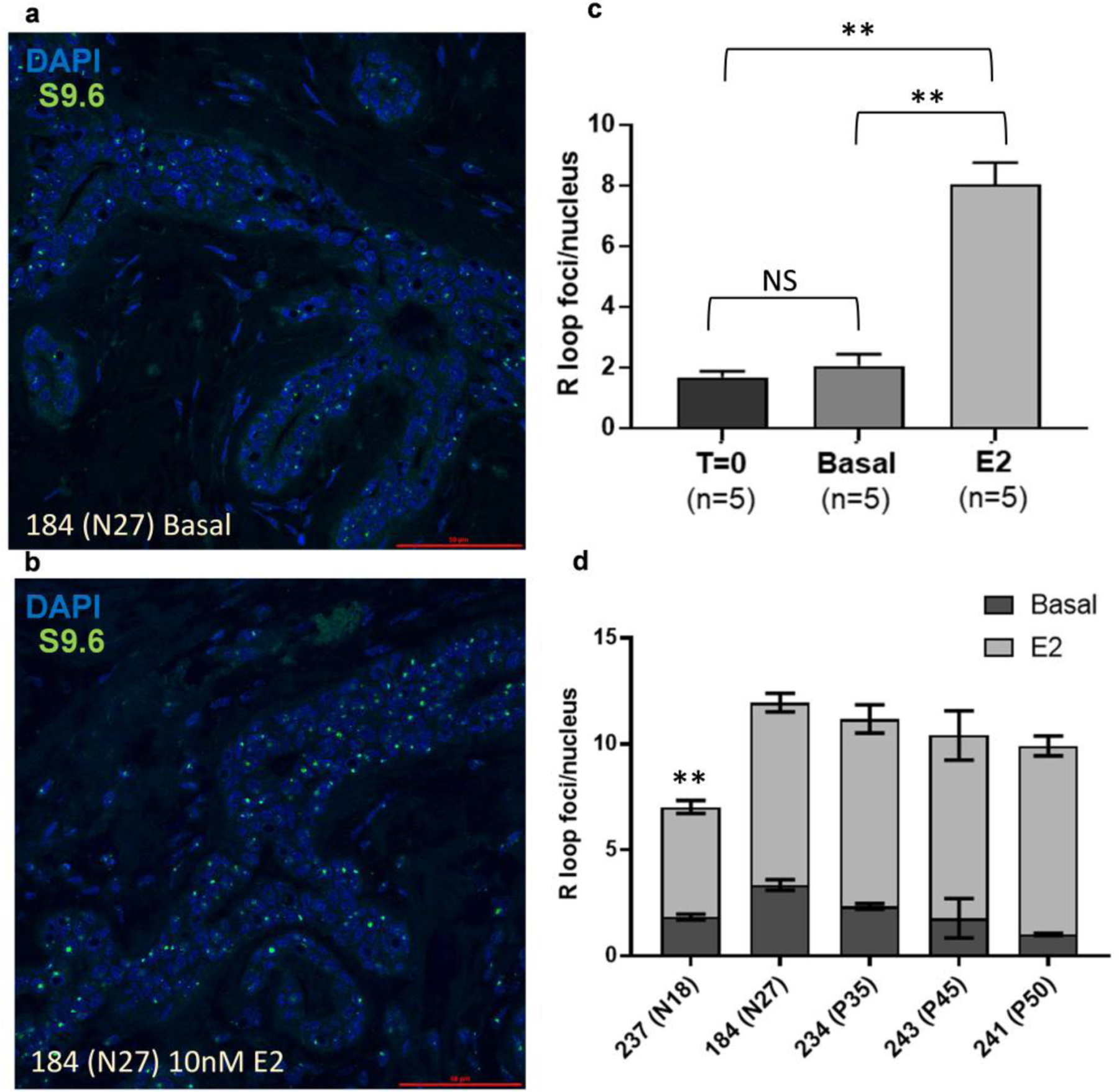
Representative images of S9.6 immunofluorescence in (a) basal and (b) E2-treated explants. (c) Quantification of R-loop foci per nucleus in basal and luminal epithelial cells demonstrates increased R-loop formation with E2-treatment relative to T=0 or basal explants (n =5 for T=0 and each treatment group). (d) R-loop foci per nucleus in Basal and 10nM E2 explants from 5 donors. Error bars indicate SEM. Significance was determined using ANOVA (** p<0.01).
E2-induced transcriptional activation of AREG, PGR, and TGFβ2 were examined using RT-qPCR as more specific endpoints of E2-ER mediated transcriptional responsiveness (Figure 5a–c). Using 2-way ANOVA to account for donor variation, we compared expression levels between the basal and E2-treated samples. In our combined sample set of nulliparous and parous women, there were no significant changes in AREG, PGR, or TGFβ2 expression induced by E2-treatment. However, after comparing responses from nulliparous and parous groups separately, we noted significant increases in E2-mediated expression in tissue from nulliparous donors. In nulliparous samples, mean AREG expression increased from 0.39+/−0.23 to 1.14+/−0.34 (p<0.05; Figure 5a). PGR expression increased from 0.30+/−0.15 to 1.53+/−0.37 (p<0.06; Figure 5b). TGFβ2 expression increased from 0.46+/−0.25 to 1.68+/−0.34 (p<0.05; Figure 5c). In parous samples, AREG, PGR and TGFβ2 expression were not significantly increased and, in some individuals, expression decreased.
Fig. 5: AREG, PGR, and TGFβ2 expression in nulliparous and parous explants.
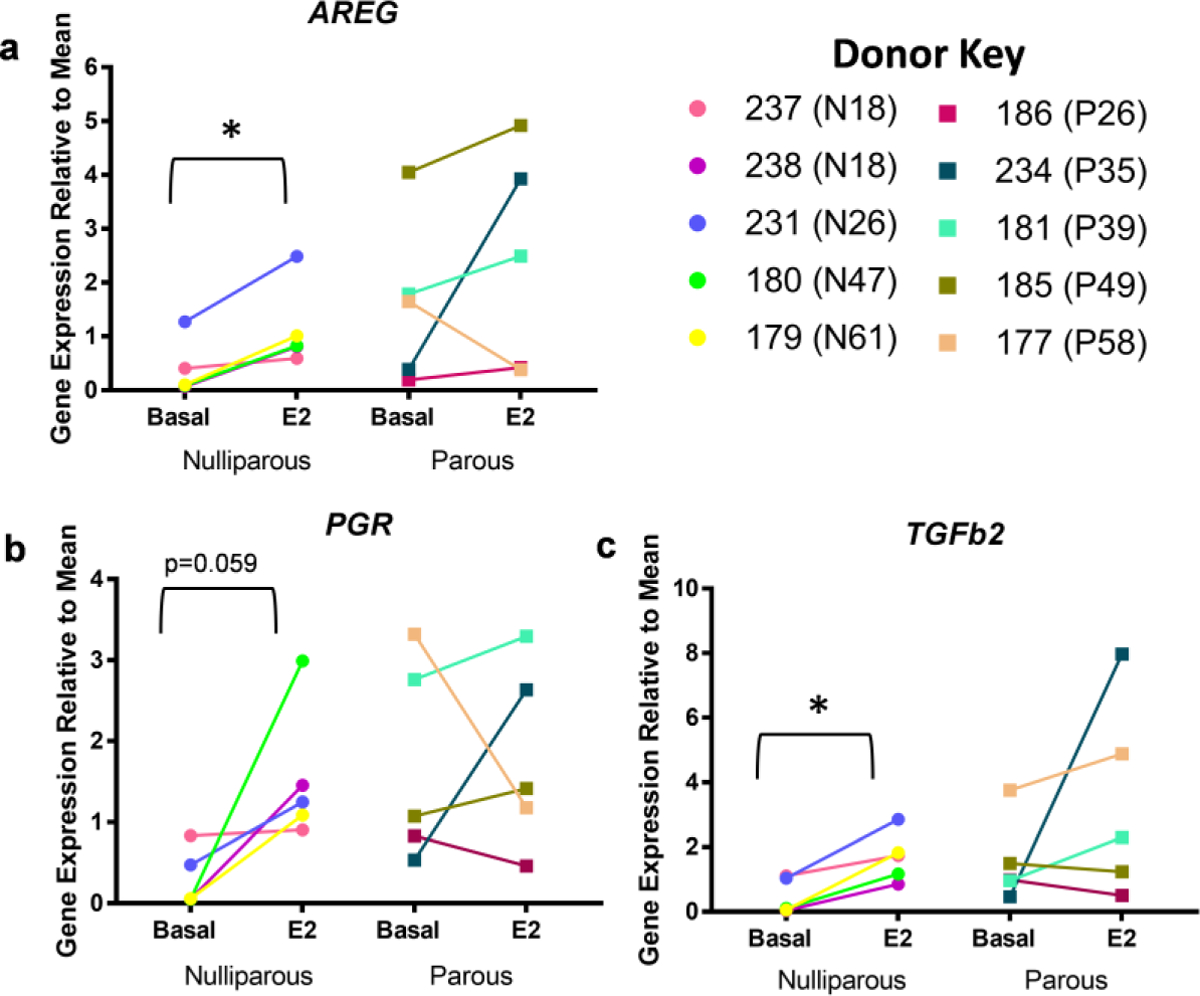
RT-qPCR analysis of basal and 10nM E2 treated explant samples for AREG, PGR, and TGFβ2 using KRT18 to normalize. Gene expression shown relative to basal mean. Circles represent nulliparous explants and squares parous explants. Parity status and age shown in brackets next to donor ID. 4 day E2 treatment significantly increased (a) AREG (p < 0.05), (b) increased PGR (p = 0.0596), and significantly increased (c) TGFβ2 (p < 0.05) in nulliparous explants, but not in parous explants using 2-way ANOVA analysis.
To determine if 4-day E2-treatment exerts a repressive effect on the estrogen receptors, we compared normalized ESR1 and ESR2 expression between explants that were either maintained in basal media or media with 10nM E2 from nulliparous and parous donors. RT-qPCR for normalized ESR1 shows a trend of decreased expression in explants after 4-day E2-treatment relative to basal explants (Figure 6), but was not significant (p=0.1805). Expression of ESR1 was decreased or not changed in response to E2-treatment for 66% (8 of 12) donors, but increased for four donors. There was also no significant change in ESR2 expression between the basal or E2-treated explants, although two donors with high ESR2 expression did demonstrate a reduction of ESR2 expression after culturing in E2-media. Parity also did not affect ESR1 and ESR2 expression. Although E2-treatment did not repress expression of these receptors, we observed over 80-fold variation in ESR1 expression and over 100-fold variation in ESR2 expression between the explants maintained in basal media from this donor population. Pearson correlation analysis found no correlation between either ESR1 or ESR2 mRNA levels and fold-increase in expression of AREG, PGR, or TGFβ2 with E2-treatment. However, ESR1 is positively correlated with ESR2 expression (0.702; p=0.02; Supplementary Figure 3). Therefore, levels of estrogen receptors do not appear to be rate-limiting in determining estrogen-induced transcriptional responses in breast tissues.
Fig. 6: ESR1 and ESR2 expression normalized to KRT18 in nulliparous and parous explants.
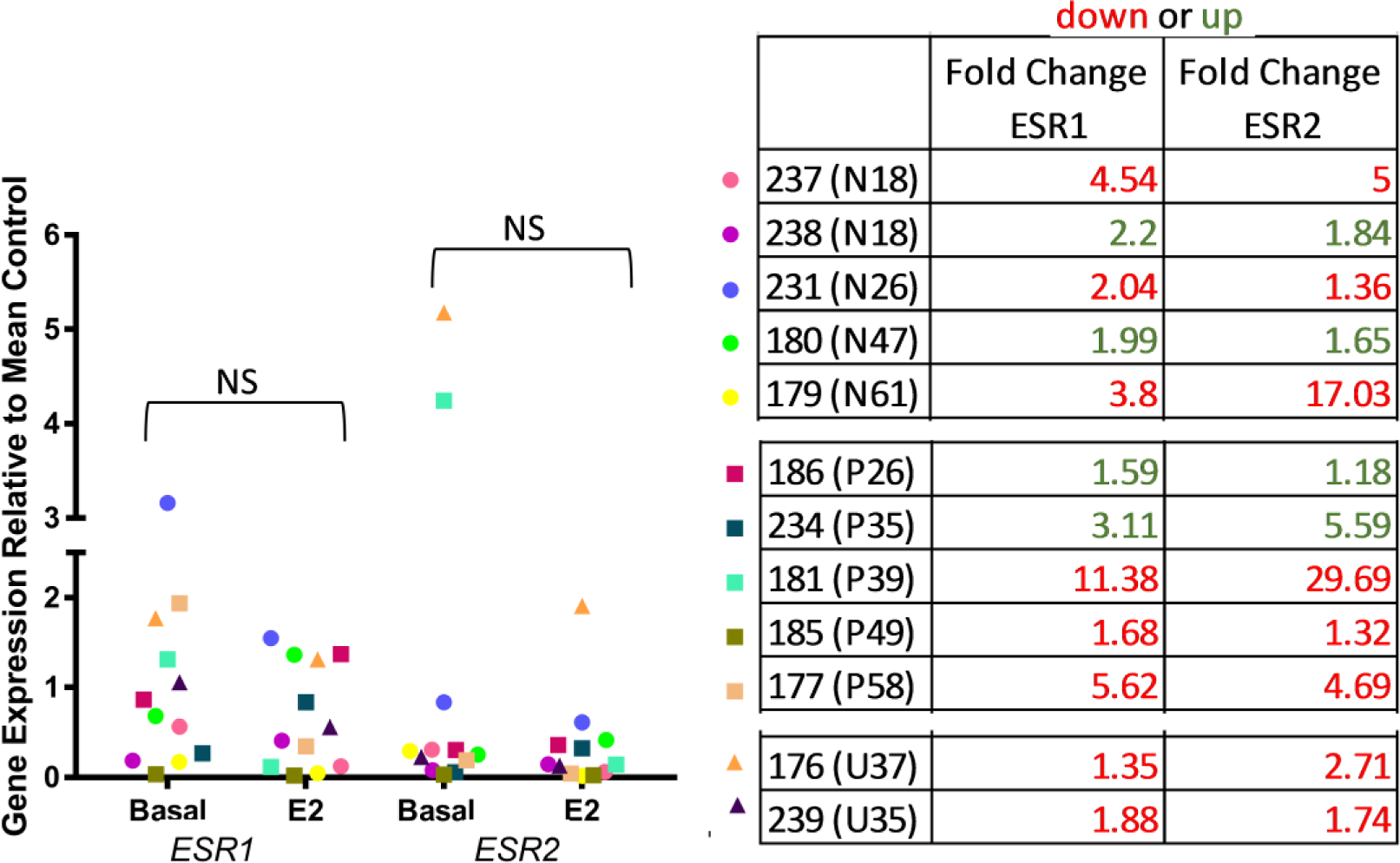
RT-qPCR analysis of basal and 10nM E2 treated explant samples for ESR1 and ESR2 using KRT18 to normalize for differing amounts of epithelium. Gene expression shown relative to control mean. Circles represent nulliparous explants, squares parous explants, and triangles explants with unknown parity status. Parity status and age shown in brackets next to patient ID and individual fold change in gene expression is indicated (red is decreased expression in E2-treated; green is increased expression in E2-treated). No significant difference in ESR1 or ESR2 expression between basal and E2 treated explants (P > 0.05).
Functional responses to estrogen
We compared PR protein expression and proliferation in the human explants to assess functional responses of the E2-treatment ex-vivo. PR was detected in T=0 and cultured explants samples (Figure 7a). Quantification of PR positive luminal epithelial cells revealed 13-fold variation in PR expression level between donors in T=0, with a mean expression of 11.52%+/−2.55 (Figure 7b). PR expression was maintained in the cultured explants. Basal control treated explants expressed mean PR levels of 7.9%+/−1.38, which is not significantly decreased from T=0 samples. PR expression in explants treated with 10nM E2 were not different from basal-treated, but showed a significant decrease (3.44%+/−0.94) compared to T=0 samples (p<0.05). Likewise, we did not detect significant differences in proliferation as measured by PCNA expression between T=0 (24.64%+/−5.78), basal (17.21%+/−3.6) or E2-treated (26.74+/−3.47) explants, although the E2-treated almost reached significance relative to basal (Figure 7c & d; p = 0.067). Linear regression analysis shows no significant change in proliferation with either age or parity for either the basal or E2-treated explants (Figure 7e). However, it is interesting to note, that some donors were insensitive to E2-mediated proliferation (black arrows, same color circle and square are close together) regardless of whether they had a high or low basal proliferation index. In particular, the oldest parous donor (58 yrs.) had the highest percent proliferation (basal and E2-treated), while the nulliparous donor of similar age (61 yrs.) had low basal and E2-treated proliferation. Both of these donor tissues were insensitive to the E2-mediated proliferation in culture. In contrast, explants from other donors were sensitive to E2-mediated proliferation (white arrows) with high percent PCNA positive cells (squares) relative to the basal-treatment (same color circles).
Fig. 7: Immunohistochemical analysis of E2-ER function in human breast explants.
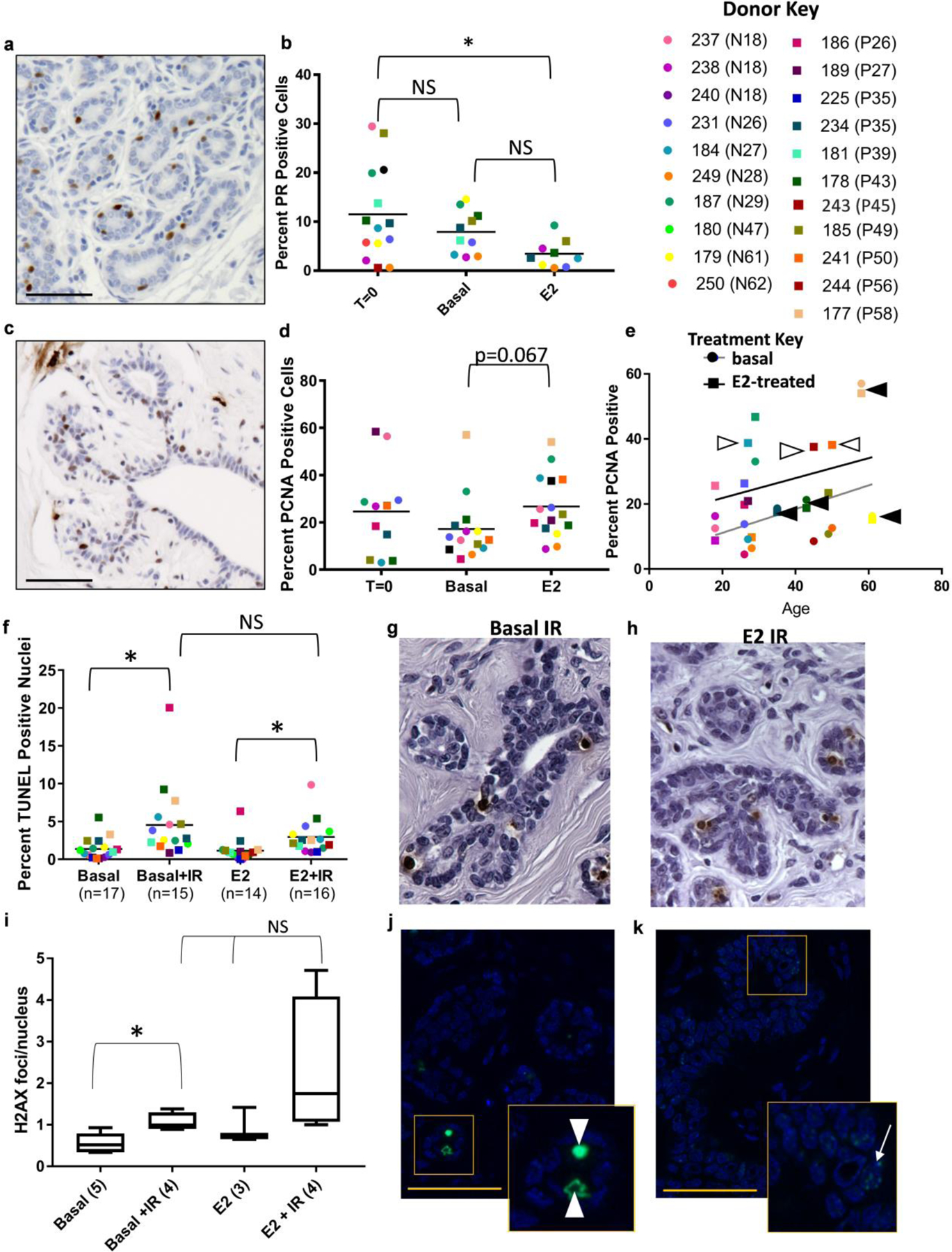
(a) Representative progesterone receptor and (b) quantification. (c) Representative PCNA with (d) quantification. (e) Linear Regression Analysis shows no significant changes in E2-mediated proliferation (PCNA) due to age. Each individual donor is indicated by a color (see key): square indicates E2-treated, circle indicates basal-treated. Black arrowhead identifies individuals that are insensitive to E2-mediated proliferation. White arrow identifies individuals that are highly sensitive to E2-mediated proliferation (compare to same color circle below square). (f) Quantification of radiation induced apoptosis (TUNEL) and representative images for basal IR (g) and E2 IR (h). (i) Quantification of γH2AX foci/nucleus and representative images for basal IR (j) and E2 IR (k) with inset indicated. Scale bars are 50μM. Mean indicated in each column with a line. Donor key: Circles represent nulliparous explants and squares parous explants. Parity status and age shown in brackets next to patient ID.
We irradiated half of the tissue from each explant sample with 5Gy ionizing irradiation (IR) 6 hours prior to collection in order to compare both spontaneous and radiation-induced apoptotic responses in human breast explants between basal and E2-treated. Irradiation significantly increased the percent apoptotic cells in basal-treatment from 1.36%+/−0.34 to 4.53%+/−1.2 and increased the number of apoptotic cells in the E2-treatment group from 1.14%+/−0.33 to 2.94%+/−0.55 (Figure 7f–h). Although radiation-induced apoptosis was greater than spontaneous apoptosis, E2-treatment did not enhance or reduce the apoptotic response in these explant samples. There was no significant difference in irradiation response between nulliparous and parous explants. In order to determine if there was also an E2-mediated accumulation of DNA damage due to IR, we quantified γH2AX foci/nucleus in each of the four explant treatments (Figure 7i–k). For this analysis, we quantified foci in epithelial cells (white arrow, E2 IR inset; Figure 7k) because they represent cells surviving with DNA damage, but not apoptotic nuclei (white arrowhead, basal IR inset; Figure 7j) because these cells are being eliminated. The apoptotic cells were often rounded with intense nuclear signal and usually located within the ductal lumen. Approximately 0.56 γH2AX foci/nucleus were observed in the basal treated explants and γH2AX was significantly increased to 1.06 foci/nucleus with IR (p=0.02). There was no significant increase in γH2AX foci/nucleus for the E2-treatment (0.93 foci/nucleus) relative to the basal treatment. Although the E2+IR-treatment (2.3 foci/nucleus) was significantly greater than the basal no IR (p=0.05), it was not significantly different from the E2 no IR. Pearson correlation analysis of the functional responses to E2-treatment in the explants found that spontaneous apoptosis was correlated with proliferation (Correlation index 0.817; p = 0.013; data not shown), although none of the E2-mediated functional assessments were correlated with ESR1 or ESR2 expression.
Discussion:
Exposure to estrogen is a risk factor for breast cancer [1]. Extended lifetime exposures to estrogens, including early menarche, late menopause, or hormone replacement therapy post menopause are factors associated with breast cancer risk. Similarly, women with the highest quartile of serum E2 levels have increased risk relative to those with the lowest quartile of serum E2. Women vary in their estrogen exposure, but also in their sensitivity to estrogens. Increased breast density is also associated with increased risk of breast cancer and because density is positively correlated with higher serum estrogens [62], breast density may signify estrogen sensitivity. Likewise, specific genetic polymorphisms in the gene ESR1, including 908A/G (K303R) and 1608T/A (Y537N), code for an ERα protein that is hypersensitive to E2 [63] or constitutively active [64], respectively. These SNPs (and others) are associated with increased risk of breast cancer, resistance to anti-estrogens and poor prognosis [65–68].
Because E2 exposure and E2 sensitivity are associated with breast cancer risk, our goal was to utilize the breast explant model to normalize a panel of breast tissue specimens, from both pre-and post-menopausal women, to a specific dose of E2: either none (basal) or pregnancy levels (10nM). Our objective with these treatments was to establish ranges of E2 sensitivity in a population and determine if it was correlated to ER expression. To ascertain estrogen sensitivity, we compared E2-mediated transcription and proliferation. The effects of estrogen on radiation-induced apoptosis was also examined as an indicator of the potential to accumulate DNA damage and the bioactivity of the p53 tumor suppressor pathway.
Expression of ERα protein has been extensively studied in human breast tissue and the protein expression is highly variable, such that detection of any receptor positivity may be limited to between 35–60% of individual samples [29, 69, 70]. ERα detection and intensity are also highly variable within each individual specimen and one lobule may exhibit high ER expression while several neighboring lobules express none. Likewise, ER expression patterns throughout the tissue may be singular in a lobule, scattered, or a duct may express a contiguous pattern in several adjacent cells [71, 29]. Because explant tissue is limiting and immunohistochemistry for ERα is semi-quantitative, we sought to quantify ESR1 and ESR2 expression in the treated and un-treated breast tissue explants to compare individual responses. Our data demonstrates considerable diversity in the estrogen receptor expression in the explant donor population. ESR1 varies by greater than 80-fold and ESR2 by greater than 100-fold among individual donors.
Previous studies quantifying immunohistochemical detection of ERα in the epithelial cells of normal human breast tissue demonstrate that ERα expression increases with age [22, 72, 73, 23], particularly among postmenopausal women. Through microarray analysis of normal human breast tissue from reduction mammoplasties, we observed that ESR1 mRNA also increases with age. We observe a similar age associated increase for ESR2 expression (Figure 1b), but the proportion of breast epithelial cells expressing ERβ protein was decreased with age (Figure 1g). ERβ is known to be expressed in stromal and basal epithelial cells in addition to luminal epithelial cells [20]. It has been reported that the epithelium: stroma ratio decreases with age [71]. Therefore, the increased proportion of stroma with age could account for the overall increase in ESR2 mRNA when expressed relative to the luminal marker KRT18 mRNA. There are some inconsistencies with regards to ER expression in parous versus nulliparous individuals. Some report that parity is associated with increased ER protein [74], but others report decreases in ERα and ESR1 in samples from parous women [75]. We, however, did not find any differences in ESR1 expression in the breast between nulliparous and parous individuals.
To modulate cellular sensitivity to hormone, negative-feedback of ligand-activated receptors represses the expression of their own receptors. The human explant system offers a unique opportunity to examine negative-feedback and repression of estrogen receptors because each E2-treated donor sample can be compared to its own basal control. E2-treatment did not significantly reduce ESR1 or ESR2 expression as a whole, but there was a trend towards decreased ER expression in 8 of the 12 donor tissues (Figure 6). Similar differences were observed among human breast cancer cell lines [76–80]. Other investigators demonstrate that MCF7 cells that have been maintained in media with serum will have E2-mediated repression of ER transcript. However, when MCF7 cells are maintained in media with serum depleted of estrogens for an extended time, they no longer repress ER transcript during estrogen-treatment. Additionally, the T47D breast cancer cell line does not repress ER transcript in the presence of estrogen. The literature demonstrates that the regulation of estrogen receptor transcript by E2 depends on the cell line and prior estrogen exposures and not all epithelial cells will repress ER transcripts in the presence of estrogen.
The estrogen receptors initiate ligand-dependent transcription in target cells [16–18]. As a consequence of estrogen-mediated transcription, DNA-RNA hybrids, referred to as R-loops, are formed [44, 42, 43]. In MCF7 cells, E2 stimulates R-loop formation at E2-responsive genes [44]. We have also demonstrated that E2-stimulates R-loop formation in T47D cells and in the mammary epithelium in mice [45]. MCF10A cells [44] and 76NTert cells [45] lack estrogen receptors and fail to increase R-loop formation in response to E2. But R-loops are induced by E2 in 76NTert cells that have been transduced with a doxycycline inducible ERα. Because E2-mediated increases in R-loop formation are dependent on ER-mediated transcription, we can use the S9.6 antibody to demonstrate global E2-ER-mediated gene transcription in the breast tissue. We detected a significant increase in estrogen-mediated R-loop formation, signifying increased transcription in the E2-treated explants that was consistent among donors (Figure 4).
Although E2-treatment consistently increased global ER-mediated gene transcription in donors, the expression of specific estrogen response genes (AREG, PGR, and TGFβ2) was highly variable (Figure 3e and 5). AREG and PGR are induced in response to E2 in mice [46] and human breast cancer cell lines [47]. TGFβ2 is also a direct target of ER and but E2 represses its expression in MCF7 cells [48]. Multiple components of the TGFβ pathway are also repressed by E2 and may be modulated by ERβ [61]. Therefore, we compared the expression of these estrogen response genes using ER specific agonists: PPT for ERα and ERB041 for ERβ. The ERα specific agonist induced expression of AREG and PGR in both sets of donor tissue and strongly induced TGFβ2 expression for donor 2. The responses to the ERβ specific agonist were more surprising. For the donor with modest basal expression of the three genes, the ERβ specific agonist repressed expression of AREG, PGR and TGFβ2, agreeing with multiple reports that ERβ opposes the activity of ERα [81–83]. However, gene expression was increased for all three of the genes by the ERβ specific agonist in donor 2. In both of these donors, gene activation by E2, the agonist for both receptors, was more modest relative to either PPT or ERB041 alone and may be due to ratios between the two receptors in the breast tissue [19, 81].
Significant increases in AREG and TGFβ2 mRNA were observed in the nulliparous subset, but not among the parous donor tissues (Figure 5). That we did not find significant increases in PGR was surprising because E2 has been shown to up-regulate PGR in estrogen responsive human organoids, whereas AREG was not [84]. Likewise, expression of PGR transcript is significantly greater in ER+ breast tumors taken from premenopausal women at phases of the menstrual cycle where serum E2 levels are highest [85]. Meng et. al. proposed that in organoids, only a subset (7–10%) of cells are ER+, whereas ER expression is more homogenous in ER+ breast cancers. Therefore, there is likely a dilution effect that obscures detection of increases in estrogen responsive genes in the heterogeneous organoids, and likely in our more cellularly heterogeneous explants. More to the point, these genes were identified as ER targets in ER-positive breast cancer cell lines; cells with homogeneous expression of ERα Human tissue has heterogeneous ERα expression. Therefore, the expression of these particular genes may be modest and more variable in relation to breast cancer cell lines. Although it is hypothesized that individuals with greater ER expression are more sensitive to estrogen, we found no correlation between ESR1 and ESR2 expression and E2-stimulated expression of AREG, PGR, and TGFβ2. Rather, genes that are consistently regulated by ER in breast cancer cell lines are more diversely regulated in the normal human breast, indicating the presence of “personal patterns” of ER-mediated transcription that differ among individuals.
The ESR1 and ESR2 expression also was not correlated to functional estrogen responses: PR protein expression, proliferation or apoptosis. Unlike ER which is detected in less than 50% of specimens, PR is readily detected in greater than 80% of human breast specimens by IHC [29, 30]. Human explants in nude mice had initial mean increases in PR expression in response to E2-treatment [49]. In vitro human explant experiments by Eigeliene et. al. demonstrated E2-mediated increases in proliferation [39], although PR expression was not changed relative to control. The PR expression in our E2-treated explants was also not different from basal-treated (Figure 7b). This agrees with human PR comparisons that demonstrate equivalent expression during both follicular and luteal phases of the menstrual cycle [29, 30].
Ki67 is used widely to quantify proliferation in human breast cancer. However, it is subject to inter-laboratory variability [86–88] and the range of Ki67 positive cells is very low (0.3–2.6%) in the normal breast [21]. Likewise, phospho-histone H3, an S/M-phase specific marker for proliferative index, is also very low (<0.05%) and in normal epithelium does not differentiate proliferation between E2-treated and control [41]. In the explant cultures, we found Ki67 expression was low (<2% of cells). Given the limited numbers of epithelial cells in the small fragments of explant tissues, it was not possible to accurately quantify differences between proliferation in basal and E2-treated. PCNA is a proliferation marker expressed more broadly across the S and G2 phases of the cell cycle [89, 90]. We used PCNA to mark proliferation in mammary epithelium from ovariectomized mice treated with estrogen and progesterone for 4 days. The percent PCNA positive in the E+P-treated mice (60%) is significantly increased relative to basal (<5%) [51]. In the human explants, PCNA positivity had a greater range amongst individuals (range 3.0–58.4%). In the explant cultures, there was a trend for an increase in PCNA-positivity in the E2-treated relative to basal control. Although several of the donors were insensitive to E2-mediated PCNA expression, most of the donor explants increased PCNA in response to E2 to varying degrees. Similar variability in proliferative responses to hormone-treatment of human breast tissue determined that ~35% responded to progesterone only, ~20% responded to estrogen only, ~34% responded to either hormone and ~10% were non-responsive [41]. It has also been reported that proliferation in the human mammary epithelium is reduced in parous individuals, declines with age and is correlated with increased serum progesterone during the luteal phase of the menstrual cycle [91–93]. However, E2, not progesterone, induced proliferation in human explants implanted in nude mice [49]. In the explant model, we, did not find differences in percent PCNA-positive epithelial cells due to age (Figure 7e) or parity.
Parous BALB/c mice are more sensitive to radiation-induced apoptosis than nulliparous [13] and 4-day E2+P4-treatment enhances radiation-induced apoptosis [51]. In the human breast tissues, spontaneous apoptosis did not vary during the menstrual cycle [93, 91], although E2-treatment repressed spontaneous apoptosis in explants [39]. However, there was no difference in either spontaneous (no IR) or radiation-induced (+ IR) apoptosis in in the explants in response to E2 (Figure 7f). Although radiation enhanced apoptosis regardless of hormone treatment, there were no differences in apoptosis between nulliparous or parous donors. DNA damage induced by irradiation may accumulate in cells if E2 were to repress apoptosis. Additionally, E2 also induces DNA damage by producing free-radicals, forming DNA adducts, and causing DNA strand breaks [94–96]. ER-mediated formation of R-loops are also susceptible to DNA damage [44, 45]. Our results do show an increase in both apoptosis and DNA damage (γH2AX foci/nucleus) due to IR in the breast explants. But neither apoptosis, nor DNA damage are increased by E2 alone. Likewise, there is no additional accumulation of IR-induced DNA damage in the human breast explants treated with E2. Therefore, from these two experiments we could conclude that E2-treatment for four days neither induces significant DNA damage, nor does it repress apoptosis to allow the accumulation of epithelial cells with DNA damage.
These results demonstrate the diversity in expression of estrogen receptors in breast tissue among women. ESR1 and ESR2 expression increases significantly with age, following the previously reported trend for increased protein expression. E2-induces significant increases in global transcription-associated R-loops in the explants, but the pattern of gene transcription appears unique for each individual. ER-target genes were consistently induced in nulliparous explants, but expression was heterogeneous among tissues from parous women. Estrogen receptor mRNA expression is highly variable in the human breast, as is the estrogen response, and ESR1 and ESR2 expression were not correlated with either transcriptional responses or proliferation in the human breast. The data suggest that the complement of transcriptional co-activators and co-repressors may differ among individuals and modify the sensitivity and patterns of responses to estrogen in human breast epithelium. Differences in estrogen-induced transcriptional responses among women may be important for defining for whom exposure to endogenous estrogens or environmental xenoestrogens may pose a substantial risk for breast cancer.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements:
Research reported in this publication was supported, in part, by the National Institute of Environmental Health Sciences of the National Institutes of Health under Award Number U01ES026140 (DJJ, SSS) and R01ES015739 (DJJ). Funding was also provided by the Department of Defense under contract #W81XWH-15-1-0217 (DJJ) and the Rays of Hope Center for Breast Cancer Research (KAD, GMJ, DJJ).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References Cited:
- 1.Clemons M, Goss P. Estrogen and the risk of breast cancer. N Engl J Med. 2001;344(4):276–85. doi: 10.1056/nejm200101253440407. [DOI] [PubMed] [Google Scholar]
- 2.Brinton LA, Hoover R, Fraumeni JF Jr. Reproductive factors in the aetiology of breast cancer. Br J Cancer. 1983;47(6):757–62. doi: 10.1038/bjc.1983.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brinton LA, Schairer C, Hoover RN, Fraumeni JF Jr. Menstrual factors and risk of breast cancer. Cancer Invest. 1988;6(3):245–54. [DOI] [PubMed] [Google Scholar]
- 4.Dall GV, Britt KL. Estrogen Effects on the Mammary Gland in Early and Late Life and Breast Cancer Risk. Front Oncol. 2017;7:110. doi: 10.3389/fonc.2017.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. Jama. 2002;288(3):321–33. [DOI] [PubMed] [Google Scholar]
- 6.MacMahon B, Cole P, Lin TM, Lowe CR, Mirra AP, Ravnihar B et al. Age at first birth and breast cancer risk. Bull World Health Organ. 1970;43(2):209–21. [PMC free article] [PubMed] [Google Scholar]
- 7.Trichopoulos D, Hsieh CC, MacMahon B, Lin TM, Lowe CR, Mirra AP et al. Age at any birth and breast cancer risk. Int J Cancer. 1983;31(6):701–4. [DOI] [PubMed] [Google Scholar]
- 8.Albrektsen G, Heuch I, Hansen S, Kvale G. Breast cancer risk by age at birth, time since birth and time intervals between births: exploring interaction effects. Br J Cancer. 2005;92(1):167–75.doi: 10.1038/sj.bjc.6602302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lambe M, Hsieh CC, Chan HW, Ekbom A, Trichopoulos D, Adami HO. Parity, age at first and last birth, and risk of breast cancer: a population-based study in Sweden. Breast Cancer Res Treat. 1996;38(3):305–11. [DOI] [PubMed] [Google Scholar]
- 10.Guzman RC, Yang J, Rajkumar L, Thordarson G, Chen X, Nandi S. Hormonal prevention of breast cancer: mimicking the protective effect of pregnancy. Proc Natl Acad Sci U S A. 1999;96(5):2520–5. doi: 10.1073/pnas.96.5.2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thordarson G, Jin E, Guzman RC, Swanson SM, Nandi S, Talamantes F. Refractoriness to mammary tumorigenesis in parous rats: is it caused by persistent changes in the hormonal environment or permanent biochemical alterations in the mammary epithelia? Carcinogenesis. 1995;16(11):2847–53. doi: 10.1093/carcin/16.11.2847. [DOI] [PubMed] [Google Scholar]
- 12.Sivaraman L, Stephens LC, Markaverich BM, Clark JA, Krnacik S, Conneely OM et al. Hormone-induced refractoriness to mammary carcinogenesis in Wistar-Furth rats. Carcinogenesis. 1998;19(9):1573–81. doi: 10.1093/carcin/19.9.1573. [DOI] [PubMed] [Google Scholar]
- 13.Dunphy KA, Blackburn AC, Yan H, O’Connell LR, Jerry DJ. Estrogen and progesterone induce persistent increases in p53-dependent apoptosis and suppress mammary tumors in BALB/c-Trp53+/−mice. Breast Cancer Res. 2008;10(3): R43. doi: 10.1186/bcr2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lonning PE, Taylor PD, Anker G, Iddon J, Wie L, Jorgensen LM et al. High-dose estrogen treatment in postmenopausal breast cancer patients heavily exposed to endocrine therapy. Breast Cancer Res Treat. 2001;67(2):111–6. [DOI] [PubMed] [Google Scholar]
- 15.Coelingh Bennink HJ, Verhoeven C, Dutman AE, Thijssen J. The use of high-dose estrogens for the treatment of breast cancer. Maturitas. 2017;95:11–23.doi: 10.1016/j.maturitas.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 16.Gruber CJ, Tschugguel W, Schneeberger C, Huber JC. Production and actions of estrogens. N Engl J Med. 2002;346(5):340–52. doi: 10.1056/NEJMra000471. [DOI] [PubMed] [Google Scholar]
- 17.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87(3):905–31. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 18.Yasar P, Ayaz G, User SD, Gupur G, Muyan M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod Med Biol. 2017;16(1):4–20. doi: 10.1002/rmb2.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang EC, Charn TH, Park SH, Helferich WG, Komm B, Katzenellenbogen JA et al. Estrogen Receptors alpha and beta as determinants of gene expression: influence of ligand, dose, and chromatin binding. Mol Endocrinol. 2008;22(5):1032–43. doi: 10.1210/me.2007-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Speirs V, Skliris GP, Burdall SE, Carder PJ. Distinct expression patterns of ER alpha and ER beta in normal human mammary gland. J Clin Pathol. 2002;55(5):371–4. doi: 10.1136/jcp.55.5.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shoker BS, Jarvis C, Clarke RB, Anderson E, Hewlett J, Davies MP et al. Estrogen receptor-positive proliferating cells in the normal and precancerous breast. Am J Pathol. 1999;155(6):1811–5. doi: 10.1016/s0002-9440(10)65498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gulbahce HE, Blair CK, Sweeney C, Salama ME. Quantification of Estrogen Receptor Expression in Normal Breast Tissue in Postmenopausal Women With Breast Cancer and Association With Tumor Subtypes. Appl Immunohistochem Mol Morphol. 2017;25(8):548–52. doi: 10.1097/pai.0000000000000337. [DOI] [PubMed] [Google Scholar]
- 23.Shoker BS, Jarvis C, Sibson DR, Walker C, Sloane JP. Oestrogen receptor expression in the normal and pre-cancerous breast. J Pathol. 1999;188(3):237–44. doi:. [DOI] [PubMed] [Google Scholar]
- 24.Umekita Y, Souda M, Ohi Y, Rai Y, Sagara Y, Sagara Y et al. Expression of estrogen receptor alpha and progesterone receptor in normal human breast epithelium. In Vivo. 2007;21(3):535–9. [PubMed] [Google Scholar]
- 25.Jerry DJ, Shull JD, Hadsell DL, Rijnkels M, Dunphy KA, Schneider SS et al. Genetic variation in sensitivity to estrogens and breast cancer risk. Mamm Genome. 2018;29(1–2):24–37. doi: 10.1007/s00335-018-9741-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montero Girard G, Vanzulli SI, Cerliani JP, Bottino MC, Bolado J, Vela J et al. Association of estrogen receptor-alpha and progesterone receptor A expression with hormonal mammary carcinogenesis: role of the host microenvironment. Breast Cancer Res. 2007;9(2): R22. doi: 10.1186/bcr1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aupperlee MD, Drolet AA, Durairaj S, Wang W, Schwartz RC, Haslam SZ. Strain-specific differences in the mechanisms of progesterone regulation of murine mammary gland development. Endocrinology. 2009;150(3):1485–94. doi: 10.1210/en.2008-1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shull JD, Dennison KL, Chack AC, Trentham-Dietz A. Rat models of 17beta-estradiol-induced mammary cancer reveal novel insights into breast cancer etiology and prevention. Physiol Genomics. 2018;50(3):215–34. doi: 10.1152/physiolgenomics.00105.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Battersby S, Robertson BJ, Anderson TJ, King RJ, McPherson K. Influence of menstrual cycle, parity and oral contraceptive use on steroid hormone receptors in normal breast. Br J Cancer. 1992;65(4):601–7. doi: 10.1038/bjc.1992.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soderqvist G, von Schoultz B, Tani E, Skoog L. Estrogen and progesterone receptor content in breast epithelial cells from healthy women during the menstrual cycle. Am J Obstet Gynecol. 1993;168(3 Pt 1):874–9. doi: 10.1016/s0002-9378(12)90837-6. [DOI] [PubMed] [Google Scholar]
- 31.Roskelley CD, Desprez PY, Bissell MJ. Extracellular matrix-dependent tissue-specific gene expression in mammary epithelial cells requires both physical and biochemical signal transduction. Proc Natl Acad Sci U S A. 1994;91(26):12378–82. doi: 10.1073/pnas.91.26.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kass L, Erler JT, Dembo M, Weaver VM. Mammary epithelial cell: influence of extracellular matrix composition and organization during development and tumorigenesis. Int J Biochem Cell Biol. 2007;39(11):1987–94. doi: 10.1016/j.biocel.2007.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenblatt AE, Garcia MI, Lyons L, Xie Y, Maiorino C, Desire L et al. Inhibition of the Rho GTPase, Rac1, decreases estrogen receptor levels and is a novel therapeutic strategy in breast cancer. Endocr Relat Cancer. 2011;18(2):207–19. doi: 10.1677/erc-10-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Padro M, Louie RJ, Lananna BV, Krieg AJ, Timmerman LA, Chan DA. Genome-independent hypoxic repression of estrogen receptor alpha in breast cancer cells. BMC Cancer. 2017;17(1):203. doi: 10.1186/s12885-017-3140-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clarke RB. Steroid receptors and proliferation in the human breast. Steroids. 2003;68(10–13):789–94. doi: 10.1016/s0039-128x(03)00122-3. [DOI] [PubMed] [Google Scholar]
- 36.Russo J, Ao X, Grill C, Russo IH. Pattern of distribution of cells positive for estrogen receptor alpha and progesterone receptor in relation to proliferating cells in the mammary gland. Breast Cancer Res Treat. 1999;53(3):217–27. [DOI] [PubMed] [Google Scholar]
- 37.Sokol ES, Miller DH, Breggia A, Spencer KC, Arendt LM, Gupta PB. Growth of human breast tissues from patient cells in 3D hydrogel scaffolds. Breast Cancer Res. 2016;18(1):19. doi: 10.1186/s13058-016-0677-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eigeliene N, Harkonen P, Erkkola R. Effects of estradiol and medroxyprogesterone acetate on expression of the cell cycle proteins cyclin D1, p21 and p27 in cultured human breast tissues. Cell Cycle. 2008;7(1):71–80. doi: 10.4161/cc.7.1.5102. [DOI] [PubMed] [Google Scholar]
- 39.Eigeliene N, Harkonen P, Erkkola R. Effects of estradiol and medroxyprogesterone acetate on morphology, proliferation and apoptosis of human breast tissue in organ cultures. BMC Cancer. 2006;6:246. doi: 10.1186/1471-2407-6-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhuang YH, Saaristo R, Ylikomi T. An in vitro long-term culture model for normal human mammary gland: expression and regulation of steroid receptors. Cell Tissue Res. 2003;311(2):217–26. doi: 10.1007/s00441-002-0683-z. [DOI] [PubMed] [Google Scholar]
- 41.Tanos T, Sflomos G, Echeverria PC, Ayyanan A, Gutierrez M, Delaloye JF et al. Progesterone/RANKL is a major regulatory axis in the human breast. Sci Transl Med. 2013;5(182):182ra55. doi: 10.1126/scitranslmed.3005654. [DOI] [PubMed] [Google Scholar]
- 42.Vanoosthuyse V Strengths and Weaknesses of the Current Strategies to Map and Characterize R-Loops. Noncoding RNA. 2018;4(2). doi: 10.3390/ncrna4020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belotserkovskii BP, Tornaletti S, D’Souza AD, Hanawalt PC. R-loop generation during transcription: Formation, processing and cellular outcomes. DNA Repair (Amst). 2018;71:69–81. doi: 10.1016/j.dnarep.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stork CT, Bocek M, Crossley MP, Sollier J, Sanz LA, Chedin F et al. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife. 2016;5. doi: 10.7554/eLife.17548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Majhi PD, Sharma A, Roberts AL, Daniele E, Majewski AR, Chuong LM, Black AL, Vandenberg LN, Schneider SS, Dunphy KA, Jerry DJ. Effects of benzophenone-3 and propylparaben on estrorogen-receptor-dependent R-loops and DNA damage in breast epithelial cells and mice. Environmental Health Perspectives. 2020; January 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ciarloni L, Mallepell S, Brisken C. Amphiregulin is an essential mediator of estrogen receptor alpha function in mammary gland development. Proc Natl Acad Sci U S A. 2007;104(13):5455–60. doi: 10.1073/pnas.0611647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peterson EA, Jenkins EC, Lofgren KA, Chandiramani N, Liu H, Aranda E et al. Amphiregulin Is a Critical Downstream Effector of Estrogen Signaling in ERalpha-Positive Breast Cancer. Cancer Res. 2015;75(22):4830–8. doi: 10.1158/0008-5472.Can-15-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Putnik M, Zhao C, Gustafsson JA, Dahlman-Wright K. Global identification of genes regulated by estrogen signaling and demethylation in MCF-7 breast cancer cells. Biochem Biophys Res Commun. 2012;426(1):26–32. doi: 10.1016/j.bbrc.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 49.Laidlaw IJ, Clarke RB, Howell A, Owen AW, Potten CS, Anderson E. The proliferation of normal human breast tissue implanted into athymic nude mice is stimulated by estrogen but not progesterone. Endocrinology. 1995;136(1):164–71. doi: 10.1210/endo.136.1.7828527. [DOI] [PubMed] [Google Scholar]
- 50.McManus MJ, Welsch CW. The effect of estrogen, progesterone, thyroxine, and human placental lactogen on DNA synthesis of human breast ductal epithelium maintained in athymic nude mice. Cancer. 1984;54(9):1920–7. doi:. [DOI] [PubMed] [Google Scholar]
- 51.Becker KA, Lu S, Dickinson ES, Dunphy KA, Mathews L, Schneider SS et al. Estrogen and progesterone regulate radiation-induced p53 activity in mammary epithelium through TGF-beta-dependent pathways. Oncogene. 2005;24(42):6345–53. doi: 10.1038/sj.onc.1208787. [DOI] [PubMed] [Google Scholar]
- 52.Rotunno M, Sun X, Figueroa J, Sherman ME, Garcia-Closas M, Meltzer P et al. Parity-related molecular signatures and breast cancer subtypes by estrogen receptor status. Breast Cancer Res. 2014;16(4): R74. doi: 10.1186/bcr3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun X, Casbas-Hernandez P, Bigelow C, Makowski L, Joseph Jerry D, Smith Schneider S et al. Normal breast tissue of obese women is enriched for macrophage markers and macrophage-associated gene expression. Breast Cancer Res Treat. 2012;131(3):1003–12. doi: 10.1007/s10549-011-1789-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Troester MA, Lee MH, Carter M, Fan C, Cowan DW, Perez ER et al. Activation of host wound responses in breast cancer microenvironment. Clin Cancer Res. 2009;15(22):7020–8. doi: 10.1158/1078-0432.Ccr-09-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sturgeon SR, Arcaro KF, Johnson MA, Balasubramanian R, Zorn M, Jerry DJ et al. DNA methylation in paired breast epithelial and white blood cells from women undergoing reduction mammoplasty. Anticancer Res. 2014;34(6):2985–90. [PubMed] [Google Scholar]
- 56.Browne EP, Punska EC, Lenington S, Otis CN, Anderton DL, Arcaro KF. Increased promoter methylation in exfoliated breast epithelial cells in women with a previous breast biopsy. Epigenetics. 2011;6(12):1425–35. doi: 10.4161/epi.6.12.18280. [DOI] [PubMed] [Google Scholar]
- 57.Zhao C, Lam EW, Sunters A, Enmark E, De Bella MT, Coombes RC et al. Expression of estrogen receptor beta isoforms in normal breast epithelial cells and breast cancer: regulation by methylation. Oncogene. 2003;22(48):7600–6. doi: 10.1038/sj.onc.1207100. [DOI] [PubMed] [Google Scholar]
- 58.Al-Ghnaniem R, Peters J, Foresti R, Heaton N, Pufulete M. Methylation of estrogen receptor alpha and mutL homolog 1 in normal colonic mucosa: association with folate and vitamin B-12 status in subjects with and without colorectal neoplasia. Am J Clin Nutr. 2007;86(4):1064–72. doi: 10.1093/ajcn/86.4.1064. [DOI] [PubMed] [Google Scholar]
- 59.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Russo J, Rivera R, Russo IH. Influence of age and parity on the development of the human breast. Breast Cancer Res Treat. 1992;23(3):211–8. [DOI] [PubMed] [Google Scholar]
- 61.Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 2006;147(10):4831–42. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- 62.Brooks JD, Sung JS, Pike MC, Orlow I, Stanczyk FZ, Bernstein JL et al. MRI background parenchymal enhancement, breast density and serum hormones in postmenopausal women. Int J Cancer. 2018;143(4):823–30. doi: 10.1002/ijc.31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Conway K, Parrish E, Edmiston SN, Tolbert D, Tse CK, Moorman P et al. Risk factors for breast cancer characterized by the estrogen receptor alpha A908G (K303R) mutation. Breast Cancer Res. 2007;9(3):R36. doi: 10.1186/bcr1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20(7):1757–67. doi: 10.1158/1078-0432.Ccr-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Conway K, Parrish E, Edmiston SN, Tolbert D, Tse CK, Geradts J et al. The estrogen receptor-alpha A908G (K303R) mutation occurs at a low frequency in invasive breast tumors: results from a population-based study. Breast Cancer Res. 2005;7(6): R871–80. doi: 10.1186/bcr1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abbasi S, Rasouli M, Nouri M, Kalbasi S. Association of estrogen receptor-alpha A908G (K303R) mutation with breast cancer risk. Int J Clin Exp Med. 2013;6(1):39–49. [PMC free article] [PubMed] [Google Scholar]
- 67.Herynk MH, Parra I, Cui Y, Beyer A, Wu MF, Hilsenbeck SG et al. Association between the estrogen receptor alpha A908G mutation and outcomes in invasive breast cancer. Clin Cancer Res. 2007;13(11):3235–43. doi: 10.1158/1078-0432.Ccr-06-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schubert EL, Lee MK, Newman B, King MC. Single nucleotide polymorphisms (SNPs) in the estrogen receptor gene and breast cancer susceptibility. J Steroid Biochem Mol Biol. 1999;71(1–2):21–7. doi: 10.1016/s0960-0760(99)00126-0. [DOI] [PubMed] [Google Scholar]
- 69.Gabrielson M, Chiesa F, Paulsson J, Strell C, Behmer C, Ronnow K et al. Amount of stroma is associated with mammographic density and stromal expression of oestrogen receptor in normal breast tissues. Breast Cancer Res Treat. 2016;158(2):253–61. doi: 10.1007/s10549-016-3877-x. [DOI] [PubMed] [Google Scholar]
- 70.Chamberlin T, D’Amato JV, Arendt LM. Obesity reversibly depletes the basal cell population and enhances mammary epithelial cell estrogen receptor alpha expression and progenitor activity. Breast Cancer Res. 2017;19(1):128. doi: 10.1186/s13058-017-0921-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goyal R, Gupta T, Gupta R, Aggarwal A, Sahni D, Singh G. Histological and immunohistochemical study of estrogen and progesterone receptors in normal human breast tissue in adult age groups vulnerable to malignancy. Clin Anat. 2016;29(6):729–37. doi: 10.1002/ca.22723. [DOI] [PubMed] [Google Scholar]
- 72.Quong J, Eppenberger-Castori S, Moore D 3rd, Scott GK, Birrer MJ, Kueng W et al. Age-dependent changes in breast cancer hormone receptors and oxidant stress markers. Breast Cancer Res Treat. 2002;76(3):221–36. [DOI] [PubMed] [Google Scholar]
- 73.Oh H, Eliassen AH, Wang M, Smith-Warner SA, Beck AH, Schnitt SJ et al. Expression of estrogen receptor, progesterone receptor, and Ki67 in normal breast tissue in relation to subsequent risk of breast cancer. NPJ Breast Cancer. 2016;2. doi: 10.1038/npjbcancer.2016.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gabrielson M, Chiesa F, Behmer C, Ronnow K, Czene K, Hall P. Association of reproductive history with breast tissue characteristics and receptor status in the normal breast. Breast Cancer Res Treat. 2018;170(3):487–97. doi: 10.1007/s10549-018-4768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Asztalos S, Gann PH, Hayes MK, Nonn L, Beam CA, Dai Y et al. Gene expression patterns in the human breast after pregnancy. Cancer Prev Res (Phila). 2010;3(3):301–11. doi: 10.1158/1940-6207.Capr-09-0069. [DOI] [PubMed] [Google Scholar]
- 76.Pink JJ, Jordan VC. Models of estrogen receptor regulation by estrogens and antiestrogens in breast cancer cell lines. Cancer Res. 1996;56(10):2321–30. [PubMed] [Google Scholar]
- 77.Read LD, Greene GL, Katzenellenbogen BS. Regulation of estrogen receptor messenger ribonucleic acid and protein levels in human breast cancer cell lines by sex steroid hormones, their antagonists, and growth factors. Mol Endocrinol. 1989;3(2):295–304. doi: 10.1210/mend-3-2-295. [DOI] [PubMed] [Google Scholar]
- 78.Saceda M, Lippman ME, Chambon P, Lindsey RL, Ponglikitmongkol M, Puente M et al. Regulation of the estrogen receptor in MCF-7 cells by estradiol. Mol Endocrinol. 1988;2(12):1157–62. doi: 10.1210/mend-2-12-1157. [DOI] [PubMed] [Google Scholar]
- 79.Eckert RL, Mullick A, Rorke EA, Katzenellenbogen BS. Estrogen receptor synthesis and turnover in MCF-7 breast cancer cells measured by a density shift technique. Endocrinology. 1984;114(2):629–37. doi: 10.1210/endo-114-2-629. [DOI] [PubMed] [Google Scholar]
- 80.Berkenstam A, Glaumann H, Martin M, Gustafsson JA, Norstedt G. Hormonal regulation of estrogen receptor messenger ribonucleic acid in T47Dco and MCF-7 breast cancer cells. Mol Endocrinol. 1989;3(1):22–8. doi: 10.1210/mend-3-1-22. [DOI] [PubMed] [Google Scholar]
- 81.Sotoca AM, van den Berg H, Vervoort J, van der Saag P, Strom A, Gustafsson JA et al. Influence of cellular ERalpha/ERbeta ratio on the ERalpha-agonist induced proliferation of human T47D breast cancer cells. Toxicol Sci. 2008;105(2):303–11. doi: 10.1093/toxsci/kfn141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hodges-Gallagher L, Valentine CD, El Bader S, Kushner PJ. Estrogen receptor beta increases the efficacy of antiestrogens by effects on apoptosis and cell cycling in breast cancer cells. Breast Cancer Res Treat. 2008;109(2):241–50. doi: 10.1007/s10549-007-9640-6. [DOI] [PubMed] [Google Scholar]
- 83.Paruthiyil S, Cvoro A, Tagliaferri M, Cohen I, Shtivelman E, Leitman DC. Estrogen receptor beta causes a G2 cell cycle arrest by inhibiting CDK1 activity through the regulation of cyclin B1, GADD45A, and BTG2. Breast Cancer Res Treat. 2011;129(3):777–84. doi: 10.1007/s10549-010-1273-5. [DOI] [PubMed] [Google Scholar]
- 84.Meng P, Vaapil M, Tagmount A, Loguinov A, Vulpe C, Yaswen P. Propagation of functional estrogen receptor positive normal human breast cells in 3D cultures. Breast Cancer Res Treat. 2019;176(1):131–40. doi: 10.1007/s10549-019-05229-5. [DOI] [PubMed] [Google Scholar]
- 85.Haynes BP, Viale G, Galimberti V, Rotmensz N, Gibelli B, Smith IE et al. Differences in expression of proliferation-associated genes and RANKL across the menstrual cycle in estrogen receptor-positive primary breast cancer. Breast Cancer Res Treat. 2014;148(2):327–35. doi: 10.1007/s10549-014-3181-6. [DOI] [PubMed] [Google Scholar]
- 86.Lanng MB, Moller CB, Andersen AH, Palsdottir AA, Roge R, Ostergaard LR et al. Quality assessment of Ki67 staining using cell line proliferation index and stain intensity features. Cytometry A. 2019;95(4):381–8. doi: 10.1002/cyto.a.23683. [DOI] [PubMed] [Google Scholar]
- 87.Polley MY, Leung SC, Gao D, Mastropasqua MG, Zabaglo LA, Bartlett JM et al. An international study to increase concordance in Ki67 scoring. Mod Pathol. 2015;28(6):778–86. doi: 10.1038/modpathol.2015.38. [DOI] [PubMed] [Google Scholar]
- 88.Mengel M, von Wasielewski R, Wiese B, Rudiger T, Muller-Hermelink HK, Kreipe H. Inter-laboratory and inter-observer reproducibility of immunohistochemical assessment of the Ki-67 labelling index in a large multi-centre trial. J Pathol. 2002;198(3):292–9. doi: 10.1002/path.1218. [DOI] [PubMed] [Google Scholar]
- 89.Jurikova M, Danihel L, Polak S, Varga I. Ki67, PCNA, and MCM proteins: Markers of proliferation in the diagnosis of breast cancer. Acta Histochem. 2016;118(5):544–52. doi: 10.1016/j.acthis.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 90.Qiu X, Wang H, Wang Z, Fu Y, Yin J. Expression of PCNA, Ki-67 and COX-2 in breast cancer based on DCE-MRI image information. J Infect Public Health. 2019. doi: 10.1016/j.jiph.2019.06.024. [DOI] [PubMed] [Google Scholar]
- 91.Potten CS, Watson RJ, Williams GT, Tickle S, Roberts SA, Harris M et al. The effect of age and menstrual cycle upon proliferative activity of the normal human breast. Br J Cancer. 1988;58(2):163–70. doi: 10.1038/bjc.1988.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ramakrishnan R, Khan SA, Badve S. Morphological changes in breast tissue with menstrual cycle. Mod Pathol. 2002;15(12):1348–56. doi: 10.1097/01.Mp.0000039566.20817.46. [DOI] [PubMed] [Google Scholar]
- 93.Navarrete MA, Maier CM, Falzoni R, Quadros LG, Lima GR, Baracat EC et al. Assessment of the proliferative, apoptotic and cellular renovation indices of the human mammary epithelium during the follicular and luteal phases of the menstrual cycle. Breast Cancer Res. 2005;7(3): R306–13. doi: 10.1186/bcr994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354(3):270–82. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 95.Roy D, Liehr JG. Estrogen, DNA damage and mutations. Mutat Res. 1999;424(1–2):107–15. doi: 10.1016/s0027-5107(99)00012-3. [DOI] [PubMed] [Google Scholar]
- 96.Santen R, Cavalieri E, Rogan E, Russo J, Guttenplan J, Ingle J et al. Estrogen mediation of breast tumor formation involves estrogen receptor-dependent, as well as independent, genotoxic effects. Ann N Y Acad Sci. 2009;1155:132–40. doi: 10.1111/j.1749-6632.2008.03685.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.