

Summary
Sirtuin 1 (Sirt1) is a NAD+-dependent deacetylase capable of countering age-related neurodegeneration, yet the basis for Sirt1 neuroprotection remains elusive. Spinocerebellar ataxia type 7 (SCA7) is an inherited CAG-polyglutamine repeat disorder. Transcriptome analysis of SCA7 mice revealed down-regulation of calcium flux genes, accompanied by abnormal calcium-dependent cerebellar membrane excitability. Transcription factor binding site analysis of down-regulated genes yielded Sirt1 target sites, and we observed reduced Sirt1 activity in SCA7 mouse cerebellum, with NAD+ depletion. SCA7 patients displayed increased poly(ADP-ribose) in cerebellar neurons, supporting poly(ADP-ribose) polymerase-1 up-regulation. We crossed Sirt1 over-expressing mice with SCA7 mice, and noted rescue of neurodegeneration and calcium flux defects. NAD+ repletion via nicotinamide riboside ameliorated disease phenotypes in SCA7 mice and patient stem cell-derived neurons. Sirt1 thus achieves neuroprotection by promoting calcium regulation, and NAD+ dysregulation underlies Sirt1 dysfunction in SCA7, indicating cerebellar ataxias exhibit altered calcium homeostasis due to metabolic dysregulation, suggesting shared therapy targets.
Graphical Abstract
eTOC summary
How does Sirtuin-1 achieve neuroprotection? By studying SCA7, an inherited cerebellar degenerative disorder, Stoyas, Bushart, et al. discover Sirtuin-1 mediates transcriptional activation of calcium regulatory genes in a NAD-dependent fashion, revealing a number of shared therapy targets for cerebellar ataxia.
Introduction
Spinocerebellar ataxia type 7 (SCA7) is an inherited neurodegenerative disorder caused by the expansion of a CAG/polyglutamine (polyQ) repeat tract (La Spada and Taylor, 2010). Included in the polyQ disease category are: X-linked spinal & bulbar muscular atrophy (SBMA), Huntington’s disease, dentatorubral-pallidoluysian atrophy, and six forms of inherited ataxia (SCA1, 2, 3, 6, 7 & 17). Expansion of the polyQ tract results in misfolding of the disease protein, and it is this misfolding event that initiates a pathogenic cascade, culminating in the degeneration and death of neurons located in different regions of the CNS in the different disorders. The causal protein in SCA7 is ataxin-7, and production of polyQ-expanded ataxin-7 protein results in cerebellar and retinal degeneration in affected patients (Martin et al., 1994; Michalik et al., 2004). Akin to other polyQ repeat diseases, SCA7 is dominantly inherited and displays remarkable genetic instability.
A leading hypothesis in the polyQ disease field, proposed at the discovery of the first repeat expansion mutation in SBMA (La Spada et al., 1991), is that polyQ disease proteins produce toxicity by disrupting transcription. The various polyQ disease proteins share no obvious homologies or functional domains, other than the polyQ repeat tract, but almost all polyQ disease proteins are transcription factors (La Spada and Taylor, 2003). In 2001, we created a representative transgenic mouse model for SCA7, and documented that SCA7 cone-rod dystrophy resulted from interference with the retinal transcription factor CRX (La Spada et al., 2001). Discovery of a physical and functional interaction between normal ataxin-7 and CRX suggested that ataxin-7 is a transcription factor (Chen et al., 2004). When studies of Sgf73, the yeast orthologue of ataxin-7, revealed that Sgf73 is part of the yeast SAGA co-activator complex, we purified a protein in the corresponding human STAGA complex that turned out to be ataxin-7 (Palhan et al., 2005). Independent biochemical studies confirmed that ataxin-7 is a core component of STAGA (Helmlinger et al., 2004).
Cerebellar Purkinje cells are a class of specialized autonomous pacemaker neurons, and are among the most metabolically active of all neurons, with high rates of spiking even in the absence of synaptic input. Purkinje cells receive continuous synaptic input from brainstem and cerebellar neurons, which they integrate to modulate intrinsic pacemaking to provide the only efferent output from cerebellar cortex for precise control of voluntary movement. Autonomous pacemaking is dependent on regulation of calcium entry and intracellar calcium (Carter and Bean, 2009). Furthermore, synaptic activity that modulates Purkinje neuron spiking relies on regulation of calcium flux, via ion channels, mitochondrial calcium channels, endoplasmic reticulum (ER) calcium channels, and calcium responsive enzymes (Egorova et al., 2015). Degeneration of Purkinje cells is a common feature of inherited ataxia in humans and mice, and one predominant theme is mutations in genes encoding calcium regulatory proteins (Mark et al., 2017). Interestingly, many ataxia disease genes converge upon the inositol (1,4,5) triphosphate receptor signaling pathway, which modulates calcium release from ER (Schorge et al., 2010).
The sirtuin family of proteins are nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases first identified in yeast. After the discovery that over-expression of Sir2 promotes lifespan extension in yeast (Houtkooper et al., 2012), attention turned to Sirtuin-1 (Sirt1), its mammalian orthologue, and six closely related proteins (Sirt2 – Sirt7) that comprise this family. Sirtuins can promote lifespan, and as caloric restriction in mouse models of neurodegeneration yields amelioration of disease (Duan et al., 2003; Halagappa et al., 2007; Patel et al., 2005; Wang et al., 2005), Sirt1 can achieve neuroprotection in Huntington’s disease (Jeong et al., 2012; Jiang et al., 2012). Furthermore, a number of Sirt1 targets promote neuroprotective pathways in CNS (Donmez and Outeiro, 2013), but the basis for Sirt1 neuroprotection remains uncertain.
Utilizing unbiased transcriptome analysis of SCA7 mice, we identified an enrichment of gene expression alterations in the phosphatidyl-inositol and calcium homeostasis pathways in SCA7 cerebellum. Through a combination of studies in SCA7 model mice, post-mortem human patient material, and patient stem cell-derived neurons, we implicate altered calcium homeostasis in SCA7 disease pathogenesis, and document that NAD+-dependent Sirt1 neuroprotection involves regulation of calcium homeostasis.
Results
SCA7 transcriptome analysis reveals altered expression of genes that regulate calcium flux
To determine the molecular basis of SCA7 cerebellar degeneration, we performed unbiased transcriptome analysis on cerebellar RNAs isolated from presymptomatic (12 week-old) and symptomatic (29 week-old) fxSCA7 92Q transgenic mice. We selected fxSCA7 92Q mice, because they display widespread modest expression of ataxin-7-92Q protein throughout the CNS, exhibit gradually progressive cerebellar degeneration with symptom onset at 20 weeks, and recapitulate key aspects of SCA7 neurodegeneration (Furrer et al., 2011; Furrer et al., 2012). RNA-Seq analysis of SCA7 cerebellar RNAs yielded a list of 100 genes with significantly altered expression levels in the cerebellum of both presymptomatic and symptomatic fxSCA7 92Q mice (Figure S1A and Table S1). When we completed pathway analysis using DAVID v6.7 (https://david.ncifcrf.gov/), among the most significantly overrepresented gene ontology and KEGG pathways were phosphatidyl-inositol signaling (P = 3.2E-4) and calcium signaling (P = 5.0E-3), both of which were coordinately down-regulated (Figure 1A). Interestingly, many of the identified calcium homeostasis genes are mutated in human and mouse ataxias (Figure 1B), underscoring their importance for cerebellar function. To validate these findings, we performed qRT-PCR analysis on RNAs from the cerebellum of fxSCA7 92Q mice, and confirmed expression reductions in all tested genes (Figure 1C), including the voltage-dependent, T-type, calcium channel alpha-1G subunit gene, Cacna1g (also known as Cav3.1), and large-conductance calcium-activated potassium channel gene, Kcnmal (also known as BK), two ion channel genes crucial for Purkinje cell function. To determine the physiological relevance of these findings, we performed immunoblot analysis on cerebellar protein lysates from symptomatic fxSCA7 92Q mice, and confirmed significant expression reductions (Figure S1B–D). We also performed immunostaining for calbindin, a calcium binding protein that regulates calcium levels, and Cav3.1, and observed marked reductions in calbindin and Cav3.1 immunoreactivity in the posterior cerebellum of fxSCA7 92Q mice (Figure 1D–E and Figure S2A–B).
Figure 1. SCA7 transgenic mice exhibit decreased expression of calcium regulatory genes.
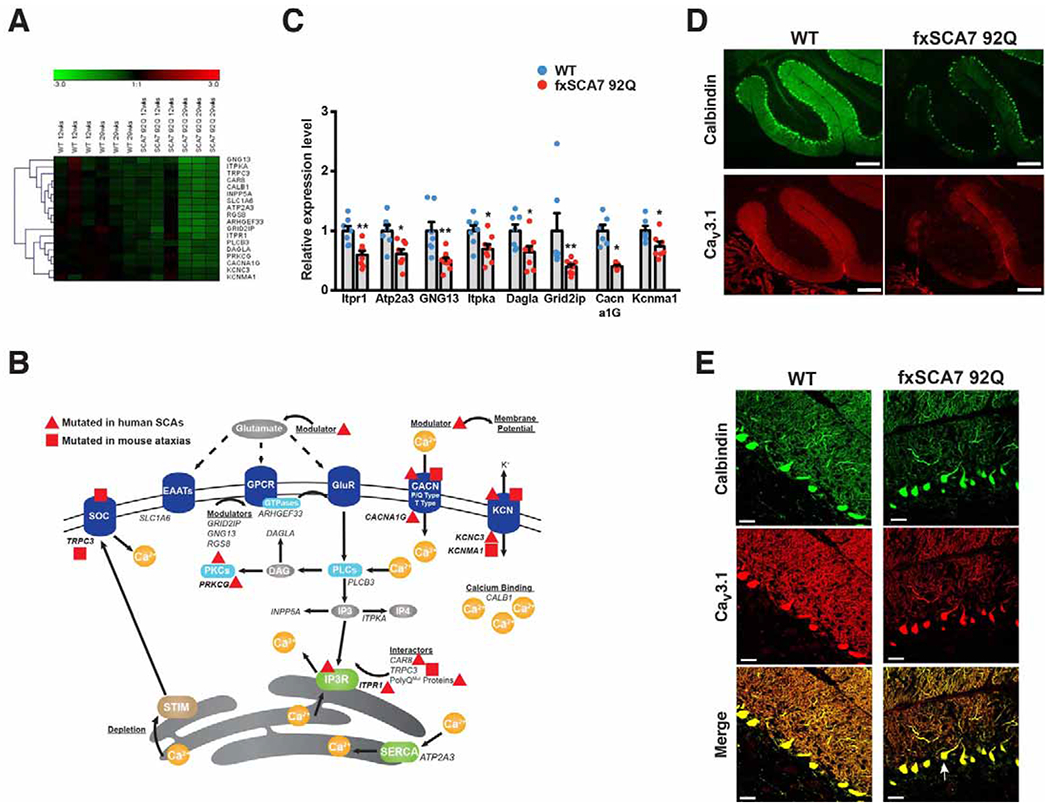
(A) Hierarchical clustering of enriched pathways from DAVID analysis of cerebellar transcriptome data from SCA7 mice and controls (WT) at presymptomatic and symptomatic ages reveals genes involved in calcium signaling and phosphatidyl-inositol signaling. See also Figure S1A and Table S1.
(B) Here we see a diagram of the cell membrane, membrane-based cell signaling pathways, and the ER, indicating proteins involved in regulating calcium homeostasis. Genes that encode these proteins or regulators are shown in italic font. Genes, which when mutated, produce a phenotype of ataxia are shown in bold. A red triangle indicates gene mutations that yield ataxia in humans, while a red square indicates genes that yield ataxia in mice. Note that all listed genes (in italics) were found to be down-regulated in cerebellum of SCA7 transgenic mice.
(C) We performed qRT-PCR analysis on RNA’s isolated from cerebellar tissue from 30 week-old fxSCA7 92Q and WT control mice. WT: n=7, fxSCA7 92Q: n=8; three technical replicates; two-tailed t-test, *P <0.05, ** P <0.01. Error bars = s.e.m.
(D) Representative immunostaining of cerebellar sections from 25 week-old fxSCA7 92Q and WT control mice for calbindin (green) and Cav3.1 (red). Scale bar = 100 μm
(E) Representative higher magnification images of cerebellar sections from 25 week-old fxSCA7 92Q and WT control mice immunostained for calbindin (green) and Cav3.1 (red). Note mislocalization of Purkinje cell soma (arrow). Scale bar = 60 μm. See also Figure S1 and Table S1.
Calcium flux defects result in impaired Purkinje neuron excitability in SCA7 mice
To functionally characterize neurodegeneration in fxSCA7 92Q mice, we sought to quantify changes in Purkinje neuron morphology. As fxSCA7 92Q mice exhibit progressive but only moderate cerebellar degeneration without frank loss of Purkinje neurons (Furrer et al., 2011), we opted to measure capacitance, a highly sensitive metric of Purkinje neuron degeneration (Shuvaev et al., 2017). Moreover, as Purkinje cell degeneration is often non-uniform across the cerebellum (Elrick et al., 2010), we assessed capacitance in both the anterior and posterior cerebellum, and documented a significant reduction in capacitance in only the posterior cerebellum of early symptomatic mice (Figure 2A), consistent with reduced calbindin and Cav3.1 immunostaining at this time (Figure 1D–E). When we measured Purkinje cell capacitance at a later time point, we observed marked reductions in both the anterior and posterior cerebellum of fxSCA7 92Q mice (Figure 2B), consistent with greater spatial involvement with disease progression. To determine if SCA7 cerebellar neurodegeneration results in Purkinje cell loss, we examined Purkinje cell density in the anterior and posterior cerebellum. While we did not detect evidence for Purkinje cell loss in 25 week-old fxSCA7 92Q mice (Figure S2C), we observed significant Purkinje cell loss in the posterior cerebellum of 40 week-old fxSCA7 92Q mice (Figure 2C).
Figure 2. Altered calcium regulatory gene expression is accompanied by cerebellar degeneration and abnormal Purkinje cell electrophysiology.
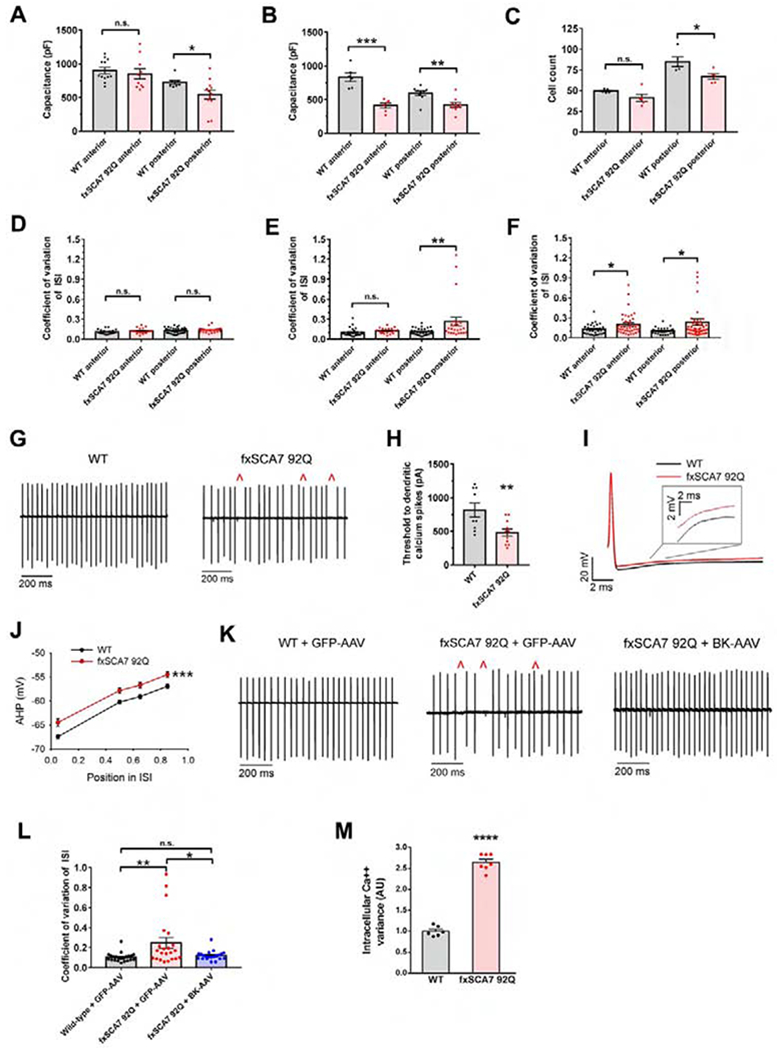
(A) In acute cerebellar slices from 25 week-old fxSCA7 92Q and WT mice (n = 3 /group), we measured total Purkinje cell capacitance, which is a function of membrane surface area, separately in the anterior lobe and posterior nodular zone of the cerebellum. WT cells: n=8-12, fxSCA7 92Q cells: n =10-12; two-tailed t-test, *P <0.05.
(B) Measurement of Purkinje cell capacitance in fxSCA7 92Q and WT mice (n = 3 /group) at 40 weeks of age. WT cells: n=6-9, fxSCA7 92Q cells: n =6-9; two-tailed t-test, ***P < 0.001, **P < 0.01.
(C) To quantify Purkinje cell neuron loss, we measured linear Purkinje cell density in the anterior and posterior cerebellum of 40 week-old fxSCA7 92Q and WT mice (n = 5 / group). Two-tailed t-test, *P <0.05.
(D) Summary of acute cerebellar slice recordings from 14 week-old fxSCA7 92Q and WT control mice (n = 5-6 / group). The coefficient of variation (CV) of the interspike interval (ISI) of Purkinje cell firing is similar in SCA7 and control mice. WT cells: n = 22-38, fxSCA7 92Q cells: n = 19-25.
(E) Summary of acute cerebellar slice recordings from 25 week-old fxSCA7 92Q and WT control mice (n = 4-5 / group). The ISI CV is significantly higher in the posterior cerebellum of SCA7 mice at this age. WT cells: n = 29-32, fxSCA7 92Q cells: n = 19-24; Mann-Whitney U-test, **P <0.01.
(F) Summary of acute cerebellar slice recordings from 40 week-old fxSCA7 92Q and WT control mice (n = 5-7 / group). The ISI CV is significantly higher in both the anterior and the posterior cerebellum of SCA7 mice at this age. WT cells: n = 29-34, fxSCA7 92Q cells: n = 41-49; Mann-Whitney U-test, *P <0.05.
(G) Representative traces of Purkinje cell firing from the posterior nodular zone of the cerebellum of 25 week-old fxSCA7 92Q and WT mice using extracellular recordings from acute cerebellar slices, illustrating irregular spiking in fxSCA7 92Q Purkinje cells (red carats).
(H) In acute cerebellar slices from 25 week-old fxSCA7 92Q mice (n = 3) and WT mice (n = 2), we performed whole-cell patch-clamp recordings from Purkinje neurons from the posterior cerebellum in the presence of tetrodotoxin and determined the threshold to dendritic calcium spikes to increasing depolarizing current steps. WT cells: n=9, fxSCA7 92Q cells: n=11; two-tailed t-test, **P <0.01.
(I) Representative traces of individual spikes from Purkinje cells of fxSCA7 92Q and WT mice at 25 weeks of age, illustrating greater AHP decay in fxSCA7 92Q Purkinje cells from the nodular zone of the cerebellum.
(J) In spikes observed upon whole-cell patch-clamp of Purkinje cells in the nodular zone of midline cerebellar slices from 25 week-old fxSCA7 92Q and WT mice (n = 5 / group), AHP decay was more rapid in fxSCA7 92Q mice. The AHP amplitude was measured at defined points in the ISI: maximal AHP, mean ISI*0.5, mean ISI*0.65, and mean ISI*0.85. WT cells: n=30, fxSCA7 92Q cells: n=23; two-way repeated-measures ANOVA with Holm-Sidak post-test, ***P <0.001.
(K) Representative extracellular recordings from acute cerebellar slices at 26 weeks, showing Purkinje cell spiking from neurons in the nodular zone of the cerebellum from mice following cerebellar delivery of an adeno-associated virus (AAV) encoding either BK or GFP. Irregular spiking is indicated with red carats.
(L) Quantification of data in (K) for 26-week old WT mice treated with GFP-AAV (n = 3), fxSCA7 92Q mice treated with GFP-AAV (n = 3), and fxSCA7 92Q mice treated with BK-AAV (n = 4). WT + GFP-AAV cells: n=22, fxSCA7 92Q + GFP-AAV cells: n=22, fxSCA7 92Q + BK-AAV cells: n=21; one-way ANOVA with Holm-Sidak post-test, **P <0.01.
(M) We cultured primary granule cell neurons from cerebella from P7 fxSCA7 92Q and WT mice, and performed live cell imaging with a calcium-sensitive dye after membrane depolarization with 40 mM KCl. The variance of the resulting calcium amplitude curve was calculated to determine calcium handling. WT: n=6, fxSCA7 92Q: n=7; three technical replicates; two-tailed t-test, ****P <0.0001. Error bars = s.e.m. See also Figure S2 and S3.
When modeling spinocerebellar ataxia in mice, neuronal dysfunction typically precedes overt neuron loss, and changes in neuronal activity often correlate with onset of motor dysfunction (Dell’Orco et al., 2015; Hansen et al., 2013; Shakkottai et al., 2011). Purkinje neurons rely on the precise activity of a multitude of ion channels to maintain spontaneous and regular spiking. Purkinje neuron spiking is sensitive to perturbations in calcium buffering (Edgerton and Reinhart, 2003; Schiffmann et al., 1999), intracellular calcium stores (Ryu et al., 2017), and plasma membrane calcium channels (Gao et al., 2012; Walter et al., 2006), many of whose transcripts are reduced in fxSCA7 92Q cerebellum (Figure 1). Proper intracellular calcium concentration regulates the activity of calcium-activated potassium channels, whose functions are critical for Purkinje neuron spiking (Gao et al., 2012; Walter et al., 2006). We therefore directly evaluated Purkinje cell spiking in acute cerebellar slices, and although we did not observe differences in the coefficient of variation in Purkinje cell firing in presymptomatic fxSCA7 92Q mice (Figure 2D), we documented a significant increase in the coefficient of variation in Purkinje cell firing in early symptomatic fxSCA7 92Q mice in posterior cerebellum (Figure 2E and Figure S2D). With disease progression, an increase in the coefficient of variation in Purkinje cell firing became apparent in both the anterior and posterior cerebellum of late symptomatic mice (Figure 2F). In early symptomatic fxSCA7 92Q Purkinje neurons, we observed a prominent loss of spiking regularity (Figure 2G). The irregular spiking in fxSCA7 92Q Purkinje neurons was not associated with a significant change in firing frequency (Figure S2E–G), consistent with a defect in calcium-activated potassium channel function (Walter et al., 2006). In another SCA mouse model, early changes in dendritic morphology are associated with potassium channel dysfunction resulting in increased intrinsic dendritic excitability (Bushart et al., 2018; Chopra et al., 2018). To determine if increased dendritic excitability is also present in SCA7, we measured the threshold of injected current required to elicit dendritic calcium spikes. fxSCA7 92Q Purkinje neurons demonstrated a significantly lower threshold to dendritic calcium spikes than littermate controls (Figure 2H), with no change in input resistance (Figure S2H), suggesting dendritic hyperexcitability in fxSCA7 92Q Purkinje neurons in addition to altered irregular somatic spiking.
Dysfunction of calcium-activated potassium channels accounts for electrophysiological abnormalities
Calcium-activated potassium channel function depends on intracellular calcium concentrations and mediates a key component of the spike in Purkinje neurons: the after-hyperpolarization (AHP). Consistent with calcium-activated potassium channel dysfunction, the AHP decayed more rapidly in fxSCA7 92Q Purkinje neurons (Figure 2I–J). The altered firing patterns and loss of the AHP in Purkinje cells in SCA7 mice is consistent with calcium-activated potassium channel dysfunction, which in turn reflects aberrant calcium homeostasis secondary to variable intracellular calcium (Dell’Orco et al., 2015; Gao et al., 2012; Walter et al., 2006). These data provide functional validation of the calcium regulatory gene expression alterations revealed by transcriptome analysis (Figure 1A).
Profound spiking irregularity in fxSCA7 92Q Purkinje neurons results from modest reductions in expression of calcium homeostasis genes. Indeed, transcripts of the large-conductance calcium-activated potassium (BK) channel are reduced in fxSCA7 92Q cerebellum (Figure 1C), suggesting that BK channel dysfunction could cause irregular spiking. We thus hypothesized that this irregular spiking is due to a synergistic effect on calcium-activated potassium channel function, and tested this hypothesis by pharmacological blockade of BK channels alone, pharmacological blockade of the T-type calcium channel Cav3 subfamily alone, or combined pharmacological blockade of BK and Cav3 subfamily channels in acute cerebellar slices. Neither pharmacological blockade of BK channels alone nor pharmacological blockade of the Cav3 subfamily alone affected the coefficient of variation (Figure S3A–B); however, combined pharmacological blockade of BK channels and the T-type calcium channel Cav3 subfamily together yielded a significant increase in the coefficient of variation in Purkinje cell firing and produced an obvious loss of spiking regularity (Figure S3C–D).
To determine whether BK channels are central to the irregular spiking in SCA7, we generated an adeno-associated virus (AAV) vector containing the cDNA for the BK channel (Dell’Orco et al., 2015), performed pilot injections into the deep cerebellar nucleus, and confirmed >80% Purkinje neuron transduction (Figure S3E). When we overexpressed BK channel transcripts in fxSCA7 92Q cerebellum, BK-AAV restored fxSCA7 92Q Purkinje neuron spike regularity to wild-type levels, while a control GFP-AAV did not (Figure 2K–L). Importantly, AAV expression alone did not affect firing frequency (Figure S3F). These results indicate that the synergistic effects of impaired calcium sources necessary for BK channel activation are responsible for irregular spiking in fxSCA7 92Q Purkinje neurons, and reduced calcium availability for BK channel function can be overcome by increasing BK channel expression. To determine if aberrant calcium dynamics are a general feature of SCA7 cerebellar degeneration, we cultured primary cerebellar granule neurons. Upon depolarization, we monitored changes in intracellular calcium in individual neurons (Figure S3G–H), and noted dramatically increased variance in calcium concentrations in fxSCA7 92Q cerebellar granule neurons (Figure 2M), which is indicative of imprecisely regulated calcium flux, consistent with cerebellar transcriptome alterations (Figure 1).
Sirt1 dysfunction accounts for altered expression of calcium regulatory genes
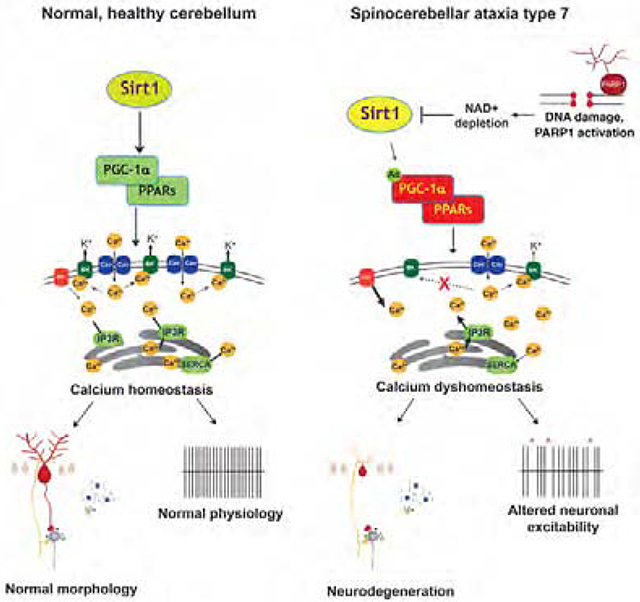
To determine the mechanism of transcription dysregulation, we compiled a list of coordinately down-regulated genes from the transcriptome analysis, and performed bioinformatics analysis to identify enriched transcription factor binding sites (TFBSs) in promoter regions using oPOSSUM v3.0 (http://opossum.cisreg.ca/oPOSSUM3/), applying a ‘z’ score threshold of ≥ 10. Among the top TFBS hits were PPARG::RXRA (Peroxisome Proliferator Response Element [PPRE]) and HIF1::ARNT (Hypoxia Response Element [HRE]) (Table S2), which are respectively positively regulated by PGC-1α and HIF-1α. Both PGC-1α and HIF-1α are Sirt1 substrates, which upon Sirt1 deacetylation are stabilized and activated (Joo et al., 2015; Rodgers et al., 2005). Of the calcium homeostasis and signaling genes found to be altered, 11 contain putative PPREs, including the Itpr1 gene, which encodes the IP3 receptor (Figure 3A). To determine if Sirt1 regulation of PGC-1α is impaired in SCA7, we examined the acetylation status of PGC-1α in the cerebellum of 30 week-old fxSCA7 92Q mice, and observed markedly increased PGC-1α acetylation in SCA7 mice (Figure 3B). To explain the dramatic decrease in Sirt1 enzymatic function in SCA7, we considered a role for PARP1, as PARP1 can consume large amounts of NAD+, the necessary co-factor for Sirt1 (Verdin, 2015). Indeed, previous studies have shown that decreased PARP1 activity, due to genetic reduction or chemical inhibition, yielded a marked increase in NAD+ levels accompanied by enhanced Sirt1 activity (Bai et al., 2011; Pirinen et al., 2014), indicating that PARP1 and Sirt1 compete for NAD+ in the nucleus. To test this hypothesis, we performed immunoblot analysis of PARP1 in the cerebellum of SCA7 266Q knock-in mice (Yoo et al., 2003), and documented a significant increase in PARP1 levels in SCA7 cerebellar protein lysates (Figure 3C). We then measured NAD+ levels in the CNS by mass spectrometry. We documented a significant reduction in NAD+ in the cerebellum, and observed a trend toward NAD+ reduction in cortex (Figure 3D). To identify the basis for enhanced PARP1 utilization of NAD+, we cultured primary cortical neurons from SCA7 r210Q knock-in mice (Niu et al., 2018), quantified the DNA damage indicator γH2Ax at baseline and upon hydrogen peroxide stress, and noted a marked increase in γH2Ax in cortical neurons from SCA7 mice (Figure 3E). These results indicate that DNA damage may contribute to the metabolic abnormalities that promote Sirt1 dysfunction, which likely underlies the transcription abnormalities observed in SCA7 cerebellar degeneration.
Figure 3. SCA7 transcriptome abnormalities stem from Sirt1 dysfunction secondary to NAD+ depletion.
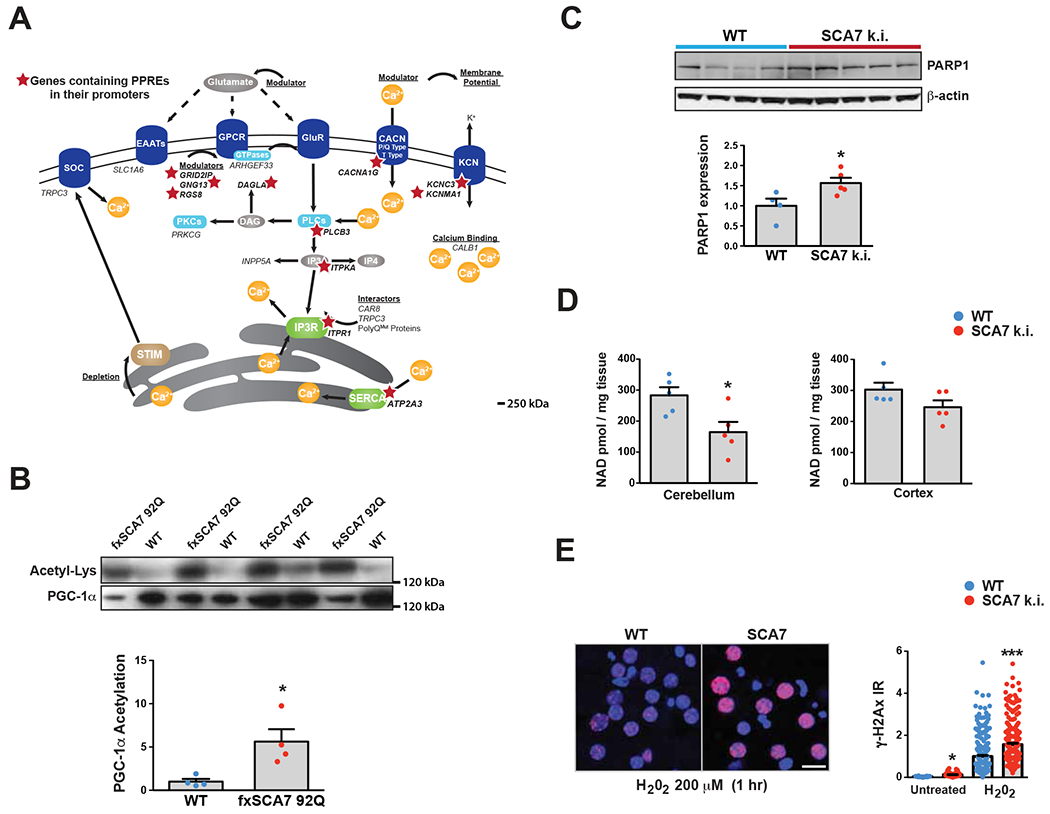
(A) Here we see a diagram of the cell membrane, membrane-based cell signaling pathways, and the ER, indicating proteins involved in regulating calcium homeostasis and flux. Genes that encode these proteins or regulators are shown in italic font. Red stars demarcate the 11 calcium regulatory genes that contain PPAR response elements (PPREs) in their promoters.
(B) We prepared cerebellar protein lysates from 30 week-old fxSCA7-92Q mice and WT controls, and then performed immunoprecipitation of PGC-1α. Immunoblotting of PGC-1α IP’s with antibodies directed against either anti-acetyl-lysine or anti-PGC-1α was performed, revealing a marked increase in acetylated PGC-1α in SCA7 cerebellum. We quantified these results by densitometry analysis of the acetyl-lysine and PGC-1α bands in the graph below. WT: n=4, fxSCA7 92Q: n=4; two-tailed t-test, *P <0.05.
(C) PARP1 immunoblot analysis of cerebellar protein lysates from 8.5 week-old SCA7 266Q mice and WT controls, quantified by densitometry analysis of PARP1 and β-actin (loading control) bands in the graph below. WT: n=4, SCA7 266Q: n=5; two-tailed t-test, *P <0.05.
(D) Mass spectrometry measurement of NAD+ levels in the cerebellum and cortex of 8.5 week-old SCA7-266Q mice and WT controls. WT: n=5, SCA7 266Q: n=5; two-tailed t-test, *P <0.05.
(E) γH2Ax immunostaining of cortical neurons cultured from SCA7 210Q k.i. mice and WT littermates, at baseline and after 1 hr of H202. LEFT: Representative image set. γH2Ax (red), DAPI (blue). RIGHT: Quantification of γH2Ax+ signal / soma area; n = 3 mice/genotype; n = 3 cultures / mouse; n = 50 neurons / culture; ***P <.001, *P <.05, two-tailed t-test. Scale bar = 20 μm. Error bars = s.e.m.
Sirt1 overexpression rescues disease phenotypes in SCA7 mice
To determine if enhanced Sirt1 function can ameliorate SCA7 neurodegeneration, we pursued a genetic rescue in two different SCA7 mouse models, our fxSCA7 92Q mice and in SCA7 266Q knock-in mice (Yoo et al., 2003), which recapitulate a severe, juvenile-onset-like phenotype. To achieve Sirt1 over-expression, we obtained a Sirt1 targeted transgenic model, where the Sirt1 cDNA is regulated by a strong ubiquitous promoter-enhancer with a floxed STOP cassette, at the Collagen A1 locus (Firestein et al., 2008); we crossed these mice with CMV-Cre driver mice to derive mice that ubiquitously overexpress Sirt1 (“Sirt1 uOE” line), and quantified expression at ~3-fold endogenous mouse Sirt1 in CNS (Figure S4A). We then crossed the resultant Sirt1 uOE mice with fxSCA7 92Q mice, and with SCA7 266Q knock-in mice, and examined motor function, neuropathology, and survival in the bigenic progeny. Using a neurological screening battery, we observed marked improvements in motor function in Sirt1 uOE – fxSCA7 92Q mice, as the bigenic mice exhibited delayed disease progression (Figure 4A), and noted that impaired rotarod performance progressed less rapidly in the bigenic mice (Figure S4B). In crosses with SCA7 266Q knock-in mice, we documented marked amelioration of cerebellar degeneration in Sirt1 uOE – SCA7 266Q mice, based upon increased Purkinje cell calbindin immunoreactivity and reduced gliosis (Figure 4B–C). This improvement in cerebellar histology was accompanied by rescue of Purkinje cell body size (Figure 4D). A Kaplan-Meier plot revealed that Sirt1 uOE – SCA7 266Q mice also display improved survival (Fig. 4E), which was corroborated by comparing average lifespans (Figure S5A).
Figure 4. Sirt1 transgenic over-expression ameliorates SCA7 disease phenotypes.
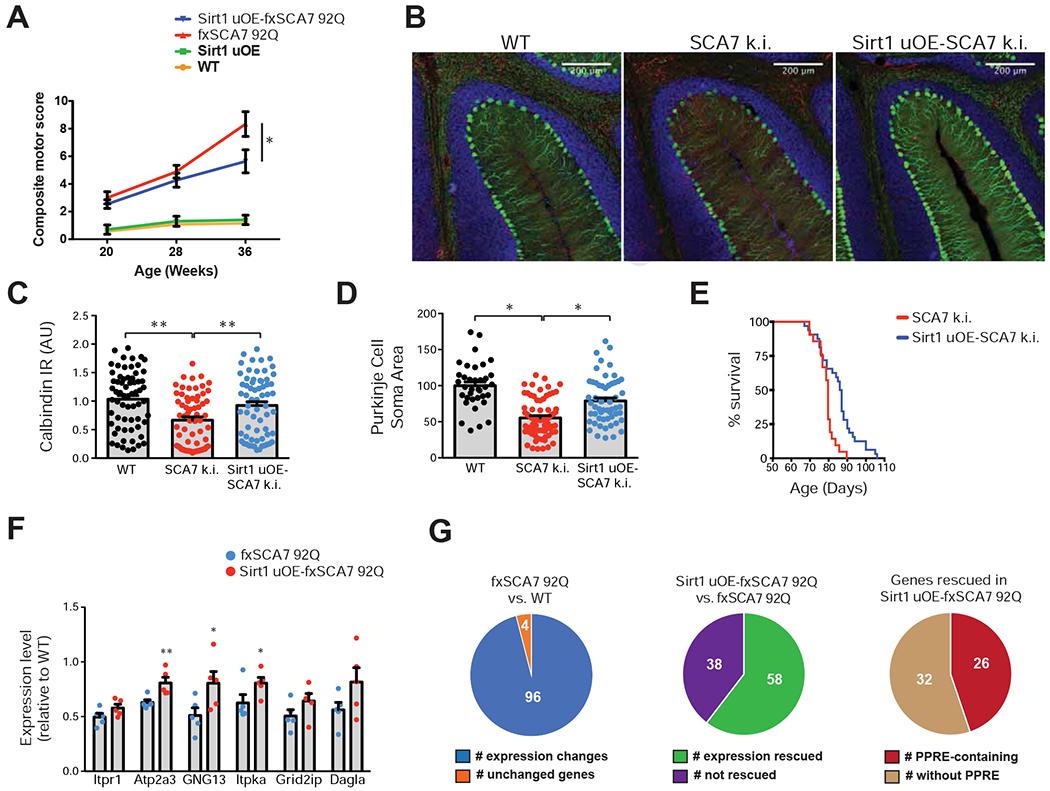
(A) We performed a neurological screening battery on cohorts of mice (n = 8 - 12 / group), of indicated genotypes at indicated ages. One-way ANOVA with Tukey’s post-hoc test, *P<0.05.
(B) Sirt1 uOE – SCA7 266Q mice display increased calbindin immunoreactivity and decreased gliosis. Green = calbindin; Red = Glial Fibrillary Acidic Protein; Blue = DAPI. Scale bar = 200 μm
(C) Quantification of Purkinje cell calbindin immunoreactivity, based upon (B). n = 3 mice / group, 66 neurons / genotype; one-way ANOVA with Tukey post-hoc test, *P <0.05.
(D) Quantification of Purkinje cell soma area, based upon (B). n = 3 mice / group, 36 - 82 neurons / genotype; one-way ANOVA with Tukey post-hoc test, *P <0.05.
(E) Kaplan-Meier plot shows that Sirt1 over-expression significantly extends the survival compared to SCA7-266Q mice. SCA7: n = 21, Sirt1-SCA7: n = 32; log-rank test, P <0.01.
(F) We performed qRT-PCR analysis on RNA’s isolated from cerebellar tissue from 36 week-old fxSCA7 92Q, Sirt1 uOE - fxSCA7 92Q, and WT control mice. n = 5 mice / group; three technical replicates; two-tailed t-test, *P <0.05, ** P <0.01. For comparisons not achieving significance, there was a strong trend (P < 0.08).
(G) Validation of SCA7 transcriptome analysis. LEFT: Nanostring analysis of the 100 genes found to display significant expression alterations comparing cerebellar RNAs from fxSCA7 92Q mice (n = 5) and WT littermate controls (n = 6); two-tailed t-test, P < 0.05. MIDDLE: Nanostring analysis of the 96 expression-altered genes in the Sirt1 uOE-fxSCA7 92Q mice (n = 5) and fxSCA7 92Q mice (n = 5). Fisher’s exact test, P < 0.00001. RIGHT: Inspection of the promoters of the 58 rescued genes for the presence or absence of the PPRE. Fisher’s exact test, P = 0.0003. Error bars = s.e.m.
To assess the effect of Sirt1 over-expression on transcriptional abnormalities, we surveyed key down-regulated calcium homeostasis and signaling genes, and detected significant increases or increase trends in all tested genes in Sirt1 uOE – fxSCA7 92Q mice (Figure 4F). We also interrogated Sirt1 at progressive time points in Sirt1 uOE – fxSCA7 92Q mice, and confirmed that Sirt1 expression remains elevated (Figure S5B). When we compared this gene subset in adult Sirt1 uOE cerebellum and matched WT littermates, we noted modest to moderate increases in expression (Figure S5C), suggesting that Sirt1 over-expression induces calcium regulatory genes. To permit a comprehensive examination of the transcriptome effects of Sirt1 over-expression in fxSCA7 92Q mice, we developed a custom array for digital qPCR quantification of the l00 genes whose expression was significantly altered in fxSCA7 92Q cerebellum (Figure S1), and validated expected gene expression alterations for 96 of the l00 genes (Figure 4G, Table S3). Furthermore, we interrogated expression levels of these 100 genes in cerebellum of littermate age-matched Sirt1 uOE – fxSCA7 92Q mice using the digital qPCR platform, and found that 58 of the 96 gene expression changes were rescued (P < 0.00001, Fisher’s exact test) (Figure 4G, Table S3). We then inspected the promoters of the 58 rescued genes, and noted PPREs in 26 genes (P = 0.0003, Fisher’s exact test) (Figure 4G). These results indicate that enhanced Sirt1 function is sufficient to rescue the molecular genetic abnormalities in SCA7 mice.
Sirt1 overexpression restores Purkinje neuron electrophysiological function
As Sirt1 over-expression rescues calcium regulatory gene expression alterations, we next assessed cerebellar electrophysiological function (Figure S6A). When we recorded Purkinje neuron firing at an early symptomatic time point, we documented a restoration in spiking regularity in cerebellar slices from Sirt1 uOE – fxSCA7 92Q mice (Figure 5A–C), with no change in firing frequency (Figure S6A). When we measured the dynamics of Purkinje neuron AHP, given its dependence on proper calcium mobilization, we noted a significant improvement in Sirt1 uOE – fxSCA7 92Q Purkinje neurons (Figure 5D–E). These read-outs of Purkinje neuron membrane excitability were similar between wild-type and Sirt1 uOE littermate controls (Figure S6B–G). Taken together, these results indicate that Sirt1 overexpression ameliorates the changes in Purkinje neuron excitability in fxSCA7 92Q mice, suggesting that restored expression of calcium homeostasis genes is sufficient to improve Purkinje neuron excitability. Since Sirt1 overexpression produced improvements in spiking in fxSCA7 92Q Purkinje neurons, we additionally explored the relationship between improved calcium homeostasis and neurodegeneration. We used capacitance as a measure for neuron dysfunction and found that Purkinje neuron capacitance is restored in Sirt1 uOE-fxSCA7 92Q mice (Figure 5F). This indicates that the transcriptional rescue elicited by Sirt1 overexpression yields measurable improvements in both Purkinje neuron structure and function.
Figure 5. Purkinje neuron membrane excitability is improved upon Sirt1 over-expression.
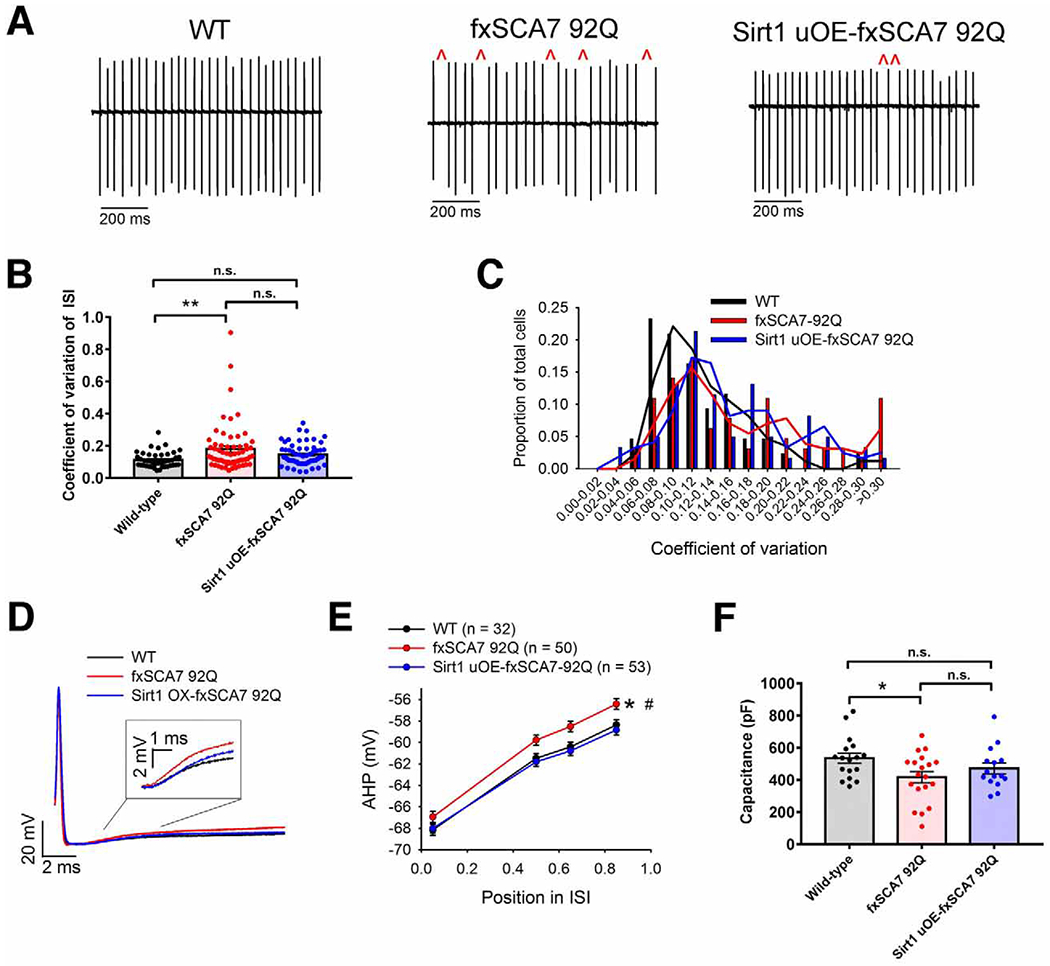
(A) Representative extracellular recordings from acute cerebellar slices of Purkinje cells from ~30 week-old fxSCA7 92Q, Sirt1 uOE-fxSCA7 92Q, and WT mice, illustrating irregular spiking (red carats), which is improved upon Sirt1 over-expression.
(B) Quantification of data from (A) for fxSCA7 92Q mice (n = 11), Sirt1 uOE-fxSCA7 92Q mice (n = 9), and WT mice (n = 8), indicating the CV of the ISI of Purkinje cell firing from the nodular zone of the cerebellum. WT cells: n=43, fxSCA7 92Q cells: n=64, Sirt1 uOE-fxSCA7 92Q cells: n=61; one-way ANOVA with Holm-Sidak post-test, *P <0.05, n.s. = not significant.
(C) Distribution of ISI CV of Purkinje cell firing from fxSCA7 92Q, Sirt1 uOE-fxSCA7 92Q, and WT mice, showing a shift of the distribution towards WT in Sirt1 uOE-fxSCA7 92Q mice.
(D) Overlaid whole-cell patch-clamp traces of individual spikes from Purkinje cells in the nodular zone of the cerebellum of fxSCA7 92Q, Sirt1 uOE-fxSCA7 92Q, and WT mice at ~30 weeks of age, illustrating greater AHP decay in fxSCA7 92Q Purkinje cells, which is normalized in Sirt1 uOE-fxSCA7 92Q Purkinje cells.
(E) Quantification of data from (D) for fxSCA7 92Q mice (n = 11), Sirt1 uOE-fxSCA7 92Q mice (n = 9), and WT mice (n = 8), measuring the decay of Purkinje cell spike-AHP. The AHP amplitude was measured at defined points in the Inter Spike Interval (ISI): maximal AHP, mean ISI*0.5, mean ISI*0.65, and mean ISI*0.85. WT cells: n=32, fxSCA7 92Q cells: n=50, Sirt1 uOE-fxSCA7 92Q cells: n=53; two-way ANOVA with Holm-Sidak post-test, *P <0.05: fxSCA7 92Q compared to WT; #P <0.01: fxSCA7 92Q compared to Sirt1 uOE-fxSCA7 92Q.
(F) In acute cerebellar slices from 35 week-old fxSCA7 92Q mice (n = 5), Sirt1 uOE-fxSCA7 92Q mice (n = 6), and WT mice (n = 7), we measured total Purkinje cell capacitance, which is a function of membrane surface area, in the nodular zone of the cerebellum. WT cells: n=17, fxSCA7 92Q cells: n=19, Sirt1 uOE-fxSCA7 92Q cells: n=15; one-way ANOVA with Hold-Sidak post-test, *P <0.05, n.s. = not significant. Error bars = s.e.m. See also Figure S6.
NAD+ replenishment rescues SCA7 neurodegeneration in mice
Sirt1 is a NAD+-dependent deacetylase, whose enzymatic function is impaired in SCA7 likely due to reduced availability of its co-factor NAD+. As cellular production of NAD+ is achieved primarily through a salvage pathway, wherein nicotinamide is converted to nicotinamide mononucleoside (NMN), which is acted upon by the enzyme nicotinamide mononucleotide adenylyltransferase (NMNAT) to produce NAD+, we considered the role of transcriptional dysregulation, and documented dramatically reduced expression of nuclear NMNAT1 specifically, but not cytosolic NMNAT2 or mitochondrial NMNAT3 (Figure S7A). Knock-down of NMNAT1 in cerebellar granule neurons elicited a marked decrease in the level of NAD+ (Figure S7B). We then measured NAD+ levels in littermate Sirt1 uOE – fxSCA7 92Q mice, fxSCA7 92Q mice, Sirt1 uOE mice, and controls, and while NAD+ was decreased in the cerebellum of fxSCA7 92Q mice, NAD+ was elevated in the cerebellum of Sirt1 uOE – fxSCA7 92Q mice (Figure S7C). As we recently found that NAD+ is reduced in the nuclear compartment of neurons derived from SCA7 patient stem cells (Ward et al., 2019), and as Sirt1 resides in the nucleus, these findings indicate that NAD+ depletion contributes to SCA7 neurodegeneration, and prompted us to pursue a preclinical trial of NAD+ repletion. To achieve NAD+ repletion, we supplemented the diet of SCA7 266Q mice with nicotinamide riboside (NR), which is a direct precursor to NMN in the salvage pathway, and we observed amelioration of motor dysfunction in SCA7 266Q mice receiving NR (Figure 6A). Rescue of neurological abnormalities in SCA7 266Q mice receiving NR was accompanied by significantly extended lifespan (Figure 6B).
Figure 6. Nicotinamide riboside and Sirt1 rescue disease phenotypes in SCA7 mice and patient neurons.
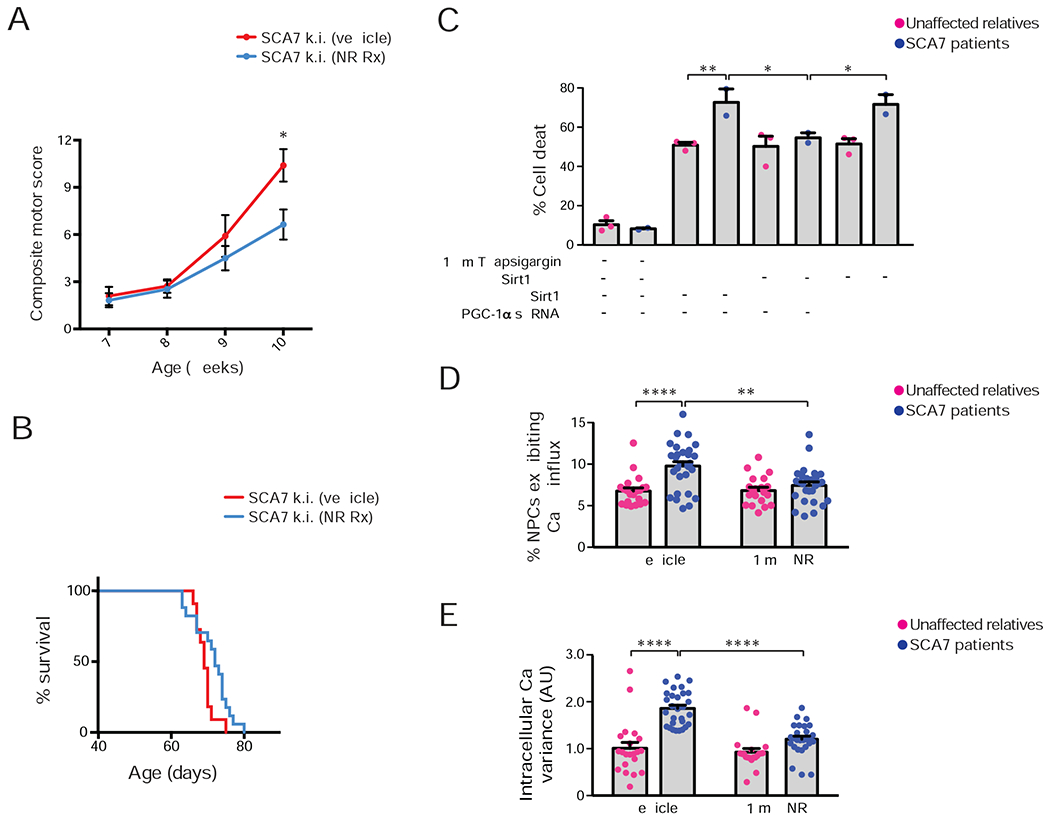
(A) We performed a neurological screening battery on cohorts of SCA7 266Q knock-in mice (n = 11 – 17 / group), maintained on a diet supplemented with nicotinamide riboside (NR) or vehicle, at the indicated ages. Two-tailed t-test, *P<0.05.
(B) Kaplan-Meier plot of SCA7-266Q mice. Vehicle-supplemented diet: n = 11, NR-supplemented diet: n = 17; log-rank test, P <0.05.
(C) We cultured NPCs from SCA7 patients and related, unaffected controls (n = 2 individuals / genotype), treated and/or transfected the cultures as indicated, and quantified cell death. Three technical replicates; two-way ANOVA with post-hoc Tukey test, *P <0.05, **P <0.01.
(D) We cultured NPCs from a SCA7 patient and related, unaffected control in the presence of 1 mM NR or vehicle, performed live cell imaging with a calcium-sensitive dye after 100 mM KCl depolarization, and quantified NPCs that exhibited a sustained increase in calcium concentration. Two unique clones / individual; two technical replicates; two-tailed t-test, ****P <0.0001.
(E) We cultured NPCs from a SCA7 patient and related, unaffected control in the presence of 1 mM NR or vehicle, performed live cell imaging with a calcium-sensitive dye after 100 mM KCl depolarization, and calculated the variance of the resulting calcium amplitude curve. Two unique clones / individual; two technical replicates; two-tailed t-test, ****P <0.0001. Error bars = s.e.m.
Sirt1 and NAD+ prevent calcium handling defects in SCA7 patient-derived neurons
We next sought to address these mechanisms in human SCA7 patients. To do so, we derived induced pluripotent stem cells (iPSCs) from SCA7 patients and related, unaffected controls (Ward et al., 2019), and after confirming retention of repeat expansions in SCA7 iPSCs, we generated neuronal progenitor cells (NPCs). To examine the role of impaired Sirt1 function in calcium homeostasis, we stressed SCA7 patient-derived NPCs and control NPCs with thapsigargin, which inhibits the ER membrane calcium pump, and observed a marked increase in cell death in SCA7 NPCs (Figure 6C). Accentuated neuron cell death elicited by thapsigargin was not remedied by introduction of enzymatically dead Sirt1-HY, but was reduced by normal Sirt1 (Figure 6C and Figure S7D). Moreover, Sirt1 amelioration of SCA7 patient NPC demise required its target PGC-1α, as shRNA knock-down of PGC-1α ablated Sirt1 rescue (Figure 6C and Figure S7E). To determine if metabolic dysregulation underlies Sirt1 dysfunction-driven calcium flux defects in this model system, we treated SCA7 and control NPCs with potassium chloride in the presence or absence of NR. Membrane depolarization yielded a pronounced elevation in calcium, and resulted in a 50% increase in SCA7 NPCs that exhibited sustained elevation of intracellular calcium, but this aberrant calcium handling response with NR (Figure 6D). We recorded the variability in calcium concentration in response to depolarization, and noted marked increases in calcium variability in SCA7 NPCs (Figure 6E), indicative of aberrant calcium handling. NR treatment of SCA7 NPCs blunted this increase in calcium variability (Figure 6E), indicating that metabolic dysregulation lies upstream of calcium flux abnormalities stemming from Sirt1 dysfunction.
PARP1 activation is elevated in the cerebellum of SCA7 patients
To determine if PARP1 activation is contributing to SCA7 cerebellar disease in human SCA7 patients, we obtained post-mortem cerebellum from four SCA7 patients and three control individuals (Table S4), immunostained cerebellar sections with an antibody against poly(ADP-ribose), which is synthesized from NAD+ by PARP1, and detected visibly greater numbers of poly(ADP-ribose) immunoreactive neuronal nuclei in SCA7 patients (Figure 7A). When we counted poly(ADP-ribose) positive nuclei in cerebellar sections, we documented a substantial increase in poly(ADP-ribose) positive nuclei in SCA7 patient cerebellar neurons (Figure 7B). We also quantified poly(ADP-ribose) immunoreactivity in cellular nuclei in cerebellar sections, and observed markedly increased poly(ADP-ribose) staining intensity in SCA7 patient cerebellum (Figure 7C). These results indicate that PARP1 activation may deplete NAD+ levels in the cerebellum of SCA7 patients to promote the demise of cerebellar neurons.
Figure 7. Human SCA7 patients display increased poly(ADP-ribose) in cerebellar neuronal nuclei.
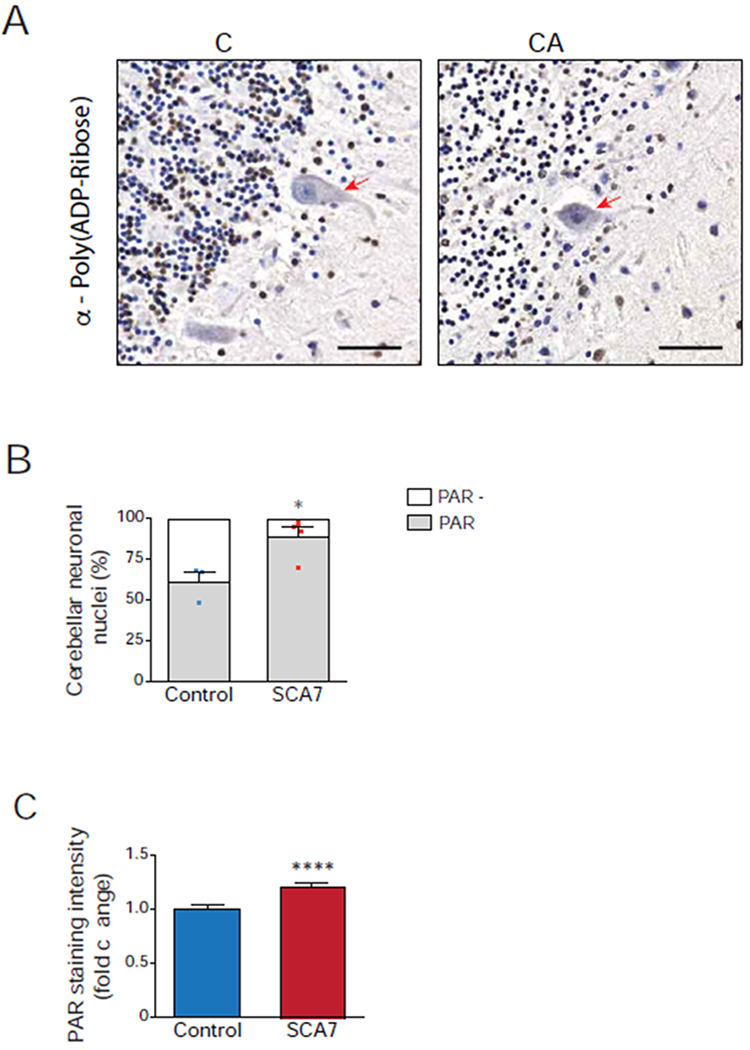
(A) Representative images of control and SCA7 patient post-mortem cerebellar sections immunostained for poly(ADP-ribose), followed by 3,3-diaminobenzidine detection (brown) and counterstained with hematoxylin (blue). Poly(ADP-ribose)-positive nuclei stain brown, while poly(ADP-ribose)-negative nuclei stain blue, and Purkinje cells are indicated with a red arrow. Note greater fraction of cerebellar neuronal nuclei stain brown in SCA7 patient. Scale bar = 50 μM
(B) We determined the percentage of poly(ADP-ribose)-positive neuronal nuclei in cerebellar sections from four SCA7 patients and three unaffected control individuals by counting at least 300 cerebellar neuronal nuclei / individual (n ≥ 100 neuronal nuclei / section, 3 section images / individual) as either poly(ADP-ribose)-positive (brown) or poly(ADP-ribose)-negative (blue). Two-tailed t-test, *P <0.05.
(C) We quantified poly(ADP-ribose) immunoreactivity in cerebellar sections from four SCA7 patients and three unaffected control individuals by measuring 3,3-diaminobenzidine (DAB) pixel intensity in at least 300 cerebellar neuronal nuclei / individual (n ≥ 100 neuronal nuclei / section, 3 section images / individual). Two-tailed t-test, ****P <0.0001. Error bars = s.e.m.
Discussion
Spinocerebellar ataxias (SCAs) share clinical and pathological features and are often caused by mutations in ion channel and transporter genes (Bushart and Shakkottai, 2019). Indeed, unbiased network analysis identified calcium homeostasis as a central pathway in ataxia (Bettencourt et al., 2014). While not previously described in SCA7, alterations in calcium electrophysiology have been reported for eight other SCAs, including SCA1, 2, 3, 5, 6, 14, 15 & 16 (reviewed in (Kasumu and Bezprozvanny, 2012)). Using unbiased transcriptome analysis, we identified decreased expression of calcium regulatory genes in the cerebellum of SCA7 mice. When we interrogated the promoters of differentially expressed calcium regulatory genes for shared TFBSs, we noted significant enrichment of the PPAR response element (PPRE) and the hypoxia-inducible factor response element (HRE). These results led us to consider a role for Sirt1 in SCA7, as PGC-1α-PPARγ and HIF-1α bind these regulatory elements and are substrates of Sirt1. We chose to examine Sirt1 deacetylation of PGC-1α, because PGC-1α is highly expressed in the cerebellum (Cowell et al., 2007), PGC-1α knock-out mice exhibit severe motor incoordination phenotypes (Lucas et al., 2012), and all PPRE-containing genes down-regulated in SCA7 cerebellum, except for one, are significantly decreased in expression in SCA1 mice (Ingram et al., 2016). When we evaluated PGC-1α acetylation status in SCA7, we observed an increase in PGC-1α acetylation, consistent with impaired Sirt1 enzymatic activity in SCA7.
Sirt1, the closest mammalian orthologue of yeast Sir2, is an important nutrient-sensor, and can promote lifespan extension. Furthermore, Sirt1 over-expression in mice confers benefits of caloric restriction, including improved metabolic function, decreased lipids, and enhanced glucose homeostasis. Of particular interest for neurotherapeutics, increased Sirt1 expression can ameliorate phenotypes in HD (Jeong et al., 2012; Jiang et al., 2012). Sirt1 neuroprotection may occur via regulation of key targets, with prior studies implicating RARβ, tau, FOXO3a, and mTORC1 as possible mediators of neuroprotection (Donmez and Outeiro, 2013). Our results indicate that Sirt1 neuroprotection also involves promotion of calcium homeostasis by transactivation of calcium regulatory genes. Sirt1 regulation of calcium homeostasis is especially relevant for brain regions with onerous metabolic demands and rapid, ongoing neurotransmission, where the challenges of calcium flux regulation are particularly acute. The cerebellum and Purkinje cell neurons resident therein certainly fulfill these criteria, as Purkinje cells are among the most metabolically active of all neurons, and selectively express high levels of calcium-binding proteins, such as calbindin. Here we extended our understanding of SCA7 pathobiology by documenting abnormal calcium dynamics and electrophysiology in model mice and in neurons derived from patient stem cells, thereby establishing a role for calcium dysregulation in SCA7. However, Sirt1 neuroprotection in SCA7 and related disorders assuredly involves a myriad of targets, including resistance to cell stress and inflammation (Donmez and Outeiro, 2013). Indeed, as Sirt1 regulates metabolic and mitochondrial function, and we observed impaired oxidative metabolism in association with mitochondrial network abnormalities in SCA7 (Ward et al., 2019), Sirt1’s ability to counter mitochondrial dysfunction likely contributes to neuroprotection in SCA7.
What accounts for impaired Sirt1 function in SCA7? To address this question, we considered its enzymatic co-factor NAD+, and evaluated PARP1, a major consumer of nuclear NAD+. We found that PARP1 levels are increased in cerebellum of SCA7 mice, and using mass spectrometry, we documented a significant reduction of NAD+. We immunostained post-mortem human SCA7 patient cerebellum and detected increased poly(ADP-ribose) staining. Accumulation of poly(ADP-ribose) is consistent with increased PARP1 activation; however, future studies of PARP enzymatic activity will be required, as our findings do not rule out impaired catabolism. We also interrogated DNA integrity in SCA7 mice, and observed evidence for a predisposition to DNA damage, which would favor PARP1 utilization of NAD+. These findings in SCA7 echo another theme in cerebellar ataxia – vulnerability to DNA damage. Indeed, mutations in DNA repair protein genes account for numerous inherited ataxias, including ataxia telangiectasia (AT), AT-like disease, ataxia with oculomotor apraxia types 1 and 2, Cockayne syndrome, and spinocerebellar ataxia with axonal neuropathy (Paulson and Miller, 2005). What is particularly intriguing about these disorders is that post-mitotic cerebellar neurons are exquisitely vulnerable. Recent work has extended this view by identifying loss-of-function mutations in the XRCC1 gene in a patient with ataxia, ocular apraxia, and neuropathy, and implicated PARP1 activation by documenting rescue of cerebellar neuron demise in XRCC1 null mice crossed onto a PARP1 null background (Hoch et al., 2017). As Purkinje cells are very metabolically active, they experience increased oxidative stress, making them highly susceptible to DNA damage. Thus, altered DNA repair and increased DNA damage may be contributing to SCA7 disease pathogenesis and should be the focus of in-depth investigation.
Depletion of NAD+ is emerging as a putative explanation for decreased Sirt1 activity. Here we documented marked reductions in NAD+ in cerebellum of two different SCA7 mouse models, accompanied by decreased expression of nuclear NMNAT1. We have shown that SCA7 patients display altered tryptophan metabolism and thus may have reduced levels of the NAD+ precursor quinolate (Ward et al., 2019); hence, NMNAT1 transcription interference and altered tryptophan metabolism likely combine with PARP1 activation to deplete NAD+ levels in SCA7. To determine if NAD+ depletion is driving SCA7 neurodegeneration, we delivered NR to SCA7 mice and observed rescue of disease phenotypes and shortened lifespan. We also studied SCA7 patient neuron derivatives, and documented a role for Sirt1 dysfunction and NAD+ depletion in calcium-related phenotypes. Our results reinforce a role for NAD+ in promoting cellular homeostasis and link Sirt1 neuroprotection to its ability to utilize NAD+ in the nucleus. As NAD+ can freely redistribute between nucleus and cytosol (Cambronne et al., 2016), Sirt1 in the nucleus likely competes for NAD+ with Sirt2 in the cytosol, explaining why Sirt1 overexpression is neuroprotective in SCA7 despite reduced NAD+, and is consistent with previous work showing inhibition of cytosolic Sirt2 can promote neuroprotection (Outeiro et al., 2007), presumably by enhancing NAD+ availability in the nucleus. However, reduced NAD+ availability could have deleterious effects beyond the nucleus, and could affect neuroprotection by other Sirtuins, including mitochondrial Sirt3 (Houtkooper et al., 2012). Indeed, reduction of NAD+ in both the nucleus and mitochondria of neurons, derived from SCA7 patient iPSCs (Ward et al., 2019), suggests that SCA7 neurodegeneration due to NAD+ depletion involves not only calcium homeostasis, but also other key cellular processes.
In this study, we uncovered a series of defects that represent targets for rational therapy development for SCA7 and related disorders. One appealing target is calcium signaling, as repetitive firing of cerebellar Purkinje cells is mediated by calcium-activated potassium channels (SK and BK) (Sausbier et al., 2004; Walter et al., 2006). Notably, BK channel re-expression improved irregular Purkinje neuron spiking in fxSCA7 92Q mice. Impaired BK channel function has been implicated in SCA2 (Dell’Orco et al., 2017), and increased BK channel expression can improve motor function in SCA1 (Dell’Orco et al., 2015). As pharmacological activation of SK channels may compensate for BK dysfunction (Bushart et al., 2018), and SK channels are more tractable targets, SK channel modulators could counter aberrant calcium homeostasis in numerous SCAs – not just SCA7. Another attractive therapeutic strategy is to boost the function of Sirt1, but promising small molecules are lacking. As supplementation with NR can rescue SCA7, another approach would be to increase NAD+ levels. In addition to NR, other options for NAD+ repletion include vitamin B3 (nicotinamide) and NMN itself. Indeed, NAD+ repletion may have therapeutic efficacy in neuromuscular and neurodegenerative diseases, such as Alzheimer’s and glaucoma (Long et al., 2015; Ryu et al., 2016; Wang et al., 2016; Williams et al., 2017). Furthermore, NAD+ replenishment can promote lifespan extension and counter age-related impairments in stem cell maintenance and function (Ryu et al., 2016; Zhang et al., 2016), as NAD+ levels decline with age. Yet another potential target is PARP1, for which pharmacological inhibitors, many already in clinical trials for cancer, are available (Lin and Kraus, 2017). As recent findings in the neurodegenerative disease field underscore the intriguing possibility that these pathways are dysregulated in multiple disorders (Hoch et al., 2017), our study of SCA7 highlights the potential for therapies targeting shared pathogenic processes with broad applicability to degenerative CNS diseases.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Albert La Spada (al.laspada@duke.edu). This study did not generate any new plasmid constructs or mouse lines, but did produce a new set of human SIRT1 lentivirus expression vectors (pHage-SIRT1 and pHage-SIRT1HY), which are available upon request after completion of a material transfer agreement.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Animals
All animal experimentation adhered to NIH guidelines and was approved by, and performed, in accordance with the University of California San Diego and Duke University Institutional Animal Care and Use Committees, as well as the University of Michigan Committee on the Use and Care of Animals. The Sirt1 overexpression line was derived by mating SIRT1STOP transgenic mice (Firestein et al., 2008) to Cre-hemizygous mice to generate SIRT1ΔSTOP or Sirt1 uOE mice. These mice were mated with two previously characterized models of SCA7-the mPrP-fx SCA7-92Q BAC mouse line (Furrer et al., 2011) and SCA7 266Q (Yoo et al., 2003) to be utilized in rescue studies. Sirt1 OX and fxSCA7 92Q lines were maintained on the C57BL/6J background, and SCA7 266Q animals were maintained on the FVB/NJ background and then backcrossed onto the C57BL/6J strain background, prior to the genetic rescue experiment. After genotyping mice, we performed motor baseline assessment prior to group assignments, excluded any outliers from cohort assignment, and randomly divided littermates to balance genders between experimental groups, in accordance with guidelines intended to avoid spurious results (Landis et al., 2012; Perrin, 2014). Consequently, females and males were used in equal numbers, or as close to equal numbers as possible, and studies were performed by a blinded examiner. In all experiments, we examined the effect of sex within and between groups, and we did not observe a significant difference in outcome measures based upon sex. The age of animals is indicated in each experiment. Mice were housed in groups of 2-5 per cage under 12 hr dark-light cycle.
Cell lines
All work done with fibroblasts and resulting iPSC-derived NPCs was pursued after obtaining informed consent from SCA7 patients and unaffected family members, and was approved by the Institutional Review Board in accordance with the requirements of the Code of Federal Regulations on the Protection of Human Subjects (45 CFR 46 and 21 CFR 50 and 56), including its relevant Subparts, and the UCSD Embryonic Stem Cell Research Oversight (ESCRO) Committee (Project #130337ZF). Demographics and generation of each IPS cell line has been extensively documented by (Ward et al., 2019). NPCs were generated with STEMdiff Neural Induction Medium (NIM) (StemCell Technologies) according to manufacturer’s monolayer culture protocol instructions. Briefly, iPSCs were gently dissociated with Accutase (StemPro, ThermoFisher), pelleted, and resuspended in NIM at 1-2 x 106 cells/mL, then plated on Matrigel coated dishes. Cells were passaged one more time in NIM and plated in dishes coated with poly-L-ornithine (15 ug/mL) and laminin (10 ug/mL) (Sigma-Aldrich). Cells tested negative for mycoplasma, and were thereafter passaged and expanded in STEMdiff Neural Progenitor Medium (NPM) (StemCell Technologies) on poly-L-ornithine/laminin-coated dishes at a density of 0.5 x 105 cells per well for cell death assay (96 well plate), and 0.25-0.5 x 105 cells per well for Fluo-4 NW imaging calcium experiments (96 well plate). For flow cytometry analysis, cells were plated and propagated on Matrigel coated dishes in Advanced DMEM/F12:Neurobasal media (1:1) (ThermoFisher) with Heparin (H3149, Sigma 1:10000 ), recombinant hEGF (E9644, Sigma 1:10000), human basic FGF (571506, Biolegend 1:10000) and B27-A (A3353501, ThermoFisher 1:50) supplements.
METHOD DETAILS
Transcriptome analysis
Total RNA from the cerebellum of fxSCA7 92Q mice and wild-type littermates aged 12 and 29 weeks (n=3 per group) was isolated using TRIzol (ThermoFisher) and purified using an RNeasy kit (Qiagen). Samples were then sent to BGI Americas for deep sequencing on the Illumina HiSeqTM 2000 system (50SE). Analysis of genome-wide expression data was performed by aligning raw reads of biologically independent samples to the reference mouse genome (mm10) using TopHat (Trapnell et al., 2009). Cufflinks software package (Trapnell et al., 2010) was used to assemble individual transcripts from the mapped reads. Cuffdiff, a part of the Cufflinks package, was used to calculate gene expression levels and test for the statistical significance of differences in gene expression. Reads per kilobase per million mapped reads (RPKM) were calculated for each gene and used as an estimate of expression levels. Heatmaps and hierarchical clustering were generated using Genesis software (Sturn et al., 2002).
Real-time quantitative RT-PCR
Mice were euthanized under isoflurane anesthesia, and cerebella were rapidly removed and flash-frozen in liquid nitrogen. Tissue was stored at −80°C until processing. RNA was isolated from the whole cerebellum of animals using TRIzol (ThermoFisher), and treated with DNAse I in the form of TURBO-DNAse (ThermoFisher) to remove traces of genomic DNA, or purified with the RNeasy mini-kit (Qiagen). Reverse transcription was performed with Superscript Reverse Transcriptase (ThermoFisher) or with the iScript cDNA synthesis kit (Bio-Rad). Quantitative PCR was performed using TaqMan probes (ThermoFisher) and TaqMan Universal PCR Mix (ThermoFisher) on a CFX384 Touch system (Bio-Rad), or with the iQ SYBR Green Supermix (Bio-Rad) in a MyiQ Single Color Real-Time PCR Detection System (Bio-Rad). Gene expression was normalized to GAPDH levels. Delta CT values were calculated as Cttarget – CtGAPDH. All experiments were performed with at least three technical replicates. Relative fold changes in gene expression were calculated using the 2−ΔΔCt method (Schmittgen and Livak, 2008). Data are presented as the average of the biological replicates + standard error of the mean (s.e.m.). Specific PCR conditions and primer sequences are available upon request.
Western blot analysis
For Western blot visualization of Cav3.1, cerebellar tissue was homogenized using Igepal lysis buffer, containing: 50 mM Tns-HCl, 150 mM NaCl, 5 mM EDTA, 1 mM EGTA, pH to 8.0, 1% Igepal CA-630 (cat. no. 18896, Sigma-Aldrich) with phosphatase inhibitor (PhosSTOP, cat. no. 4906845001, Sigma-Aldrich) and cOmplete EDTA-free protease inhibitor cocktail (cat. no. 4693132001, Sigma-Aldrich) added immediately before use. Samples were homogenized and sonicated. For all other samples, mouse cerebella were lysed in RIPA buffer (ThermoFisher) and homogenized by trituration.
We used the following antibodies for immunoblot analysis: Sirt1 (07-131, Millipore; 1:1000); PARP1 (9542, Cell Signaling; 1:1000); PCG-1α (SC-13067, Santa Cruz; 1:1000); acetyl-lysine (9441, Cell Signaling; 1:1000); β-actin (ab8226, Abcam; 1:10000), mouse anti-Cav3.1, clone N178A/9 (1μg/mL, ref. no. 75-206, Neuromab), and rabbit anti-α-Tubulin (11H10) (1:500, cat. no. 2125, Cell Signaling). Immunoprecipitation was performed with antibody for PCG-1α (SC-13067, Santa Cruz; 1:20), and densitometry was performed using the Fiji distribution of ImageJ.
Animal behavioral studies
For motor studies, mice were visually inspected by a blinded examiner for obvious neurological signs and examined using a composite neurological evaluation tool (ledge test, clasping, kyphosis and gait were scored on a scale of 0 (normal) to 3 (severely impaired)), as described previously (Guyenet et al., 2010). Females and males were used in equal numbers, or as close to equal numbers as possible, and studies were performed by a blinded examiner. Changes in score data are presented as an average of biological replicates ± standard error of the mean (s.e.m.). For survival studies, animals were monitored daily and pronounced dead when respiration or heartbeat was no longer present. Cohort sizes were designated based upon power analysis for threshold effects of at least 25% difference.
Neuropathology
Deeply anesthetized mice were transcardially perfused with PBS, then 4% paraformaldehyde (PFA) in 0.1M phosphate buffer, pH 7.4. Brains were removed and postfixed with 4.0% PFA in in O.1M PB overnight, then moved to 15% followed by 30% sucrose solutions. Free-floating 30μm sagittal brain sections were cut on a vibratome and incubated with antibodies to calbindin (C9848, Sigma; 1:500) and glial fibrillary acidic protein (GFAP) (G9269, Sigma; 1:1000), and imaged with a Zeiss LSM 780 inverted microscope. For calbindin immunoreactivity quantification, each Purkinje cell soma was manually encircled based on Calbindin staining and the mean pixel value was recorded using ImageJ (Abramoff, 2004; Schneider et al., 2012). Background noise was calculated by randomly selecting 10 regions of interest in cerebellar molecular layer without dendritic staining, and averaging their mean pixel value. Background noise was then subtracted from each PC mean pixel value.
Immunohistochemistry
Mice were anesthetized under isoflurane inhalation and brains were removed, fixed in 1% paraformaldehyde for 1 hour, and then placed in 30% sucrose in PBS for 48 hours. Parasagittal sections of 14 μm were made on a CM1850 cryostat (Leica). For double-labeling experiments, Cav3.1 was labeled with mouse anti-Cav3.1 (1:150, clone N178A/9, Cat. No. 75-206, Neuromab) and goat anti-mouse Alexa594-conjugated secondary antibody (1:200, Ref. no. A11005, ThermoFisher). To label Purkinje neurons, rabbit anti-calbindin (1:200, Cat. No. 13176, Cell Signaling) and goat anti-rabbit Alexa488-conjugated secondary (1:200, Ref. no. A11008, ThermoFisher) were used. Sections were imaged using an Axioskop 2 plus microscope (Zeiss) at 4x and 10x magnification. Sample preparation and imaging was performed with the experimenter blind to genotype.
Fluorescence intensity measurements
To measure relative intensity of calbindin and Cav3.1 staining, images acquired at 10x magnification were used. Fluorescence intensity analysis was performed using ImageJ. A rectangular box was placed in the molecular layer, spanning the dendritic arbors of Purkinje neurons. Mean pixel intensity was measured for each rectangle, and this mean value was used as the relative fluorescence value for each section. The box was an identical size in all cases, and was placed in the same location for calbindin and Cav3.1 analysis in a single section. Two sections were imaged per animal, and the mean of those two fluorescence values were used as the fluorescence intensity value for that animal. All tissue processing and imaging was performed at the same session, and microscope settings were identical for all acquired images. During imaging and analysis, the experimenter was blind to genotype.
Confocal microscopy
Imaging was performed on a Nikon C2+ confocal microscope. Images were acquired at 60x magnification with an oil-immersion lens. Single-plane images were acquired, with microscope settings kept constant between all samples under a specific set of antibodies. Samples were prepared and imaged with the experimenter blind to genotype.
Purkinje celI counts
Sections were taken from 25 and 40 week-old fxSCA7 92Q mice and wild-type controls and stained with rabbit anti-calbindin (1:200, cat. no. 13176, Cell Signaling) and goat anti-rabbit Alexa488-conjugated secondary (1:200, ref. no. A11008, ThermoFisher) to label Purkinje neurons. A blinded reviewer counted the number of Purkinje cell somata in a 400 μM line in lobule 4 (anterior cerebellum) and the entirety of lobule 10 (posterior cerebellum).
PAR DAB staining
Formalin-fixed, paraffin-embedded 10μm tissue sections from control (n=3) and SCA7 (n=4) individuals were deparrafinized in xylene and cleared in 100% and 95% ETOH. Endogenous peroxidase was blocked using 1.875% hydrogen peroxide in methanol for 8 minutes. The slides were hydrated in deionized water before placing in a 5% w/v nonfat milk solution for 20 minutes. The slides were washed in DH2O for 5 min before adding the primary mAb. Poly (ADP-ribose) 10H (ENZO cat#ALX-804-22-R100 was applied at 1:300 10ug/mL. The slides were incubated at 37-40 degrees Celsius for 45 minutes. The secondary and HRP was applied using Agilent technologies Dako EnVision Dual Link System-HRP (Cat# K406189-2). The slides were allowed to incubate at 37-40 degrees Celsius for 30 minutes. Slides were washed and then developed using Agilent technologies Dako DAB solution (Cat# K346811-2) for 5 minutes. Slides were counter-stained with Fisherfinest Hematoxylin+ (Cat#220-100) and blued in ammonical water. Slides were dehydrated through graded and absolute alcohol, cleared in xylene before using permount to coverslip.
NAD+ Quantification
NAD+ was extracted from mouse brain using acidic then alkaline extraction methods and analyzed with mass spectrometry. Frozen tissues taken from the −80°C freezer were immediately extracted in 1 M perchloric acid and neutralized in 3 M K2CO3 on ice. After centrifugation, the supernatant was mixed with buffer A [H20 + 20 mM ammonium acetate (pH 9.4)] and loaded onto a column (150 × 2.1 mm; Kinetex EVO C18, 100 Å). HPLC was run for 2 min at a flow rate of 300 μl/min with 100% buffer A. Then, a linear gradient to 100% buffer B [methanol + 5 mM ammonium acetate (pH 8.5)] was performed (at 2 to 11 min). Buffer B (100%) was maintained for 4 min (at 11 to 15 min), and then a linear gradient back to 100% buffer A (at 15 to 17 min) began. Buffer A was then maintained at 100% until the end (at 17 to 25 min). NAD+ eluted as a sharp peak at 3.3 min and was quantified on the basis of the peak area compared to a standard curve and normalized to tissue weight of frozen tissues (Gariani et al., 2016; Ryu et al., 2016; Yang and Sauve, 2006). For quantification of NAD+ in cultured primary cerebellar granule neurons, we used the EnzyChrom NAD/NADH Assay Kit (E2ND-100, BioAssay Systems), where optical density values obtained with a plate reader at 565 nm wavelength were first standardized using an NAD+ calibration curve and then normalized to the protein concentration in the analyzed lysates.
Metabolite Quantification
Sample Preparation
Mouse cerebellum samples were collected, frozen in liquid nitrogen, transferred to the CK14 tubes, approximately 52 +/− 4.5 mg and stored at −80C. Energy metabolites were extracted using a Bligh-Dyer method as follows. 190 uL 2:1 v/v MeOH:chloroform was added to the tubes, and samples were subjected to bead blasting on a Precellys homogenizer for 3 cycles of 10 seconds at 10,000 rpm, with 60 seconds cooling between. Cooling was provided using the CryoLys unit filled with dry ice. Extraction was continued with 20 minute incubation on a Thermomixer at room temperature and 1000 rpm. Next, 63 uL chloroform was added to each sample, samples were vortexed. Finally, 63 uL water was added to each sample, samples were vortexed, then centrifuged for 10 minutes at 15,000 rcf and 4C to separate the phases. One hundred microliters of the aqueous supernatant from each sample was removed, transferred to glass total recovery vials (Waters Corp.), and dried in the Vacufuge (Eppendorf) at 30C. The dry samples were resuspended in 20 uL of 80:20 v/v MeOH/water resuspension solvent containing the C13 labeled EColi lysate as internal standards. A study pool quality control sample was prepared from 2 uL of each sample. The SPQC was analyzed in triplicate throughout the study. Five microliters from each sample was injected for analysis on LC-MS/MS using a custom method developed in-house and detailed below.
Analytical Conditions
Liquid chromatographic separation was performed using a Waters Acquity UPLC with a 2.1 mm x 30 mm 1.7 um BEH Amide column (Waters P/N 186004839). Mobile phase A was composed of water with 10 mM ammonium hydrogen carbonate (AmBic) (Millipore Sigma, St. Louis, MO) containing 0.2% ammonium hydroxide (NH4OH) generated as follows: 3.34 mL of 30% ACS grade NH4OH was added to 1 L water, followed by the addition of 0.3982 g AmBic. Mobile phase B was neat acetonitrile (Optima LCMS grade Thermo). The weak needle wash was mobile phase B and the strong needle wash was mobile phase A. The outlet of the analytical column was connected directly via electrospray ionization into a Xevo TQ-S mass spectrometer (Waters) with positive/negative mode switching. Retention time scheduleing with 30 second windows was used to minimize concurrent MRM transitions, and automatic dwell calculation was used to maximize dwell time while maintaining at least 8 points across the chromatographic peak. 80msec was set as the polarity-switching delay. Positive and negative ion electrospray were alternated during the entirety of an LC gradient program for one injection. In ESI+ mode, capillary voltage was 3.0kV, source offset was 50V, desolvation temperature was 400C, desolvation gas flow was 650 L/Hr N2, cone gas was 150 L/hr N2, and nebulizer pressure of 7.0 bar was used. Source parameters for ESIionization were the same as ESI+, with the exception of the capillary voltage was set to −2.0 kV. Each sample was analyzed in Multiple Reaction Monitoring (MRM) mode in the mass spectrometer during the LC gradient program as ions eluted from the LC column. Intensity values are reported as the ratio of the analyte to C13 internal standard. The data provided is in the format of relative quantification.
Patch-clamp electrophysiology
Solutions
Artificial cerebrospinal fluid (aCSF) contained the following: 125 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 2 mM CaCl2, 10 mM HEPES, and 10 mM glucose. For all recordings, other than dendritic capacitance measurements, pipettes were filled with internal recording solution containing the following: 119 mM K-Gluconate, 2 mM Na-Gluconate, 6 mM NaCl, 2 mM MgCl2, 0.9 mM EGTA, 10 mM HEPES, 14 mM Tris-phosphocreatine, 4 mM MgATP, 0.2 mM Tris-GTP, at pH 7.3 and osmolarity 290 mOsm. For capacitance measurements, internal recording solution contained: 140 mM CsCl, 2 mM MgCl2, 1 mM CaCl2, 10 mM EGTA, 10 mM HEPES, 4 mM Na2ATP, at pH 7.3 and osmolarity 287 mOsm.
Preparation of brain slices for acute electrophysiological recordings
Mice were anesthetized by isoflurane inhalation and decapitated. The brain was removed and submerged in pre-warmed (33°C) aCSF. Acute parasagittal slices were prepared in aCSF held at 32.5-34°C on a VT1200 vibratome (Leica) to a thickness of 300 μm. Once slices were obtained, they were incubated in carbogen-bubbled (95% O2, 5% CO2) aCSF at 33°C for 45 min. Slices were then stored in carbogen-bubbled aCSF at room temperature until use. During recording, slices were placed in a recording chamber and continuously perfused with carbogen-bubbled aCSF at 33°C at a flow rate of 2.5 mL/min.
Patch-clamp recordings
Purkinje neurons were visually identified for patch-clamp recordings using a 40x water immersion objective and a Nikon Eclipse FN1 upright microscope with infrared differential interference contrast (IR-DIC) optics. Identified cells were visualized using NIS Elements image analysis software. Borosilicate glass patch pipettes were pulled to resistances of 3-4 MΩ for all recordings. Recordings were performed 1-5 hours after slice preparation. Data were acquired using an Axopatch 200B amplifier, Digidata 1440A interface (MDS Analytical Technologies), and pClamp-10 software (Molecular Devices). All data were digitized at 100 kHz. Whole-cell recordings were rejected if the series resistances changed by >20% during the course of recording, or if the whole-cell series resistance rose above 15 MΩ. All voltages are corrected for the liquid gap junction potential, which was calculated to be 10 mV (Dell’Orco et al., 2015). All recordings performed in Sirt1 crosses were performed with the experimenter blind to genotype.
Capacitance measurements
Acute cerebellar slices were obtained as described above. Capacitance measurements were performed in the presence of 50 μM picrotoxin to block spontaneous GABAA synaptic currents, and recording pipettes were filled with a cesium chloride-based internal pipette solution as described above. Recordings were performed at RT. Capacitative transients were obtained in voltage-clamp mode using 1 second steps to −70 mV from a holding potential of −80 mV. Recordings were excluded if the measured input resistance was under 100 MΩ. Dendritic capacitance was determined using a method for the analysis of an equivalent circuit which represents Purkinje neurons (Llano et al., 1991a; Llano et al., 1991b). Input resistance was corrected offline and the decay of the capacitative transient was fit using a two-exponential decay function:
The constants obtained from fitting the decay function of each cell was then used to obtain four parameters: C1 (capacitance of the soma and main proximal dendrites), C2 (capacitance of the distal dendritic arbor), R1 (pipette access resistance), and R2 (composite resistance of dendritic segments separating the main proximal dendritic segments from the distal dendritic arbor). The equations for this analysis are as follows:
In our measurements, total capacitance was indicated by C1 + C2.
Analysis of firing properties
Electrophysiology data were analyzed offline using Clampfit 10.2 software (Molecular Devices). Firing frequency and coefficient of variation (CV) calculations were performed in the cell-attached configuration on spikes in a 150 second time interval obtained ~5 minutes after formation of a stable seal. The CV was calculated as follows:
The firing frequency distribution was obtained by identifying the percentage of cells in each incrementing 10 spike/second bin. The CV distribution was similarly obtained by sorting CV values into incrementing 0.02 bins. A moving average trend-line was added to the CV distribution histogram to outline the shape of the distribution.
AHP decay
Analysis of the after-hyperpolarization (AHP) was performed by analyzing spikes in a 10 second interval ~1 minute after break-in. The AHP value was calculated as the maximum antipeak voltage, and the mean value over the 10 second interval is reported as the AHP for each neuron. To measure decay of the AHP during the Inter Spike Interval (ISI), the mean ISI duration was determined in the same 10 sec interval for each cell. AHP amplitude was then measured at different fractional intervals of the ISI (0.5*ISI, 0.65*ISI, 0.85*ISI) in order to characterize AHP decay.
Dendritic calcium spikes
Threshold to elicit dendritic calcium spikes was performed as described previously (Zagha et al., 2008). Briefly, cells were held at −80 mV in whole-cell current clamp mode and injected with current in +50 pA increments in the presence of tetrodotoxin to block somatic sodium spikes. The amount of injected current to elicit calcium spikes was recorded. Input resistance for each cell was calculated by generating an input-output curve for injected current vs. membrane potential, with only membrane potential values of under −75 mV in an effort to minimize active conductances during measurements (Zagha et al., 2008).
Pharmacology
In some electrophysiology recordings, mibefradil dihydrochloride hydrate (cat. no. M5441, Sigma Aldrich) was used at 4 μM to inhibit Cav3 family T-type calcium channels and iberiotoxin (cat. no. STI-400, Alomone Labs) was used at 100 nM to partially inhibit BK channels.
Stereotaxic cerebellar delivery of adeno-associated virus (AAV)
AAV-mediated expression was performed as described previously (Dell’Orco et al., 2015). Recombinant serotype AAV2/5 vectors, encoding either a BK channel or GFP transcript under the control of the CMV promoter were generated by the University of Iowa Vector Core facility (http://www.medicine.uiowa.edu/vectorcore/). Under isoflurane anesthesia, fxSCA7 92Q and littermate control mice were injected bilaterally with either BK-AAV or GFP-AAV into both medial and lateral deep cerebellar nucleus, which results in efficient Purkinje neuron expression(Dell’Orco et al., 2015). Injection coordinates were as follows, as measured from bregma: Medial DCN = −6.4 mm anterior-posterior, ±1.3 mm medial-lateral, −1.9 mm dorsal-ventral; Lateral DCN = −6.0 mm anterior-posterior, ±2.0 mm medial-lateral, −2.2 mm dorsal-ventral. At each injection coordinate, a total volume of 3.0 μL containing 2.2 x 1012 vg/mL (GFP-AAV) or 2.3 x 1012 vg/mL (BK-AAV) of virus was delivered at a rate of 0.5 μL/min, using a 10 μL Hamilton syringe (BD Biosciences). Mice were given 14 days for the virus to fully express before being sacrificed for patch-clamp electrophysiology studies.
To determine efficiency of AAV expression in the posterior cerebellum, a separate cohort of 3 wild-type mice at 30 weeks of age were injected with AAV-GFP at the same volume and injection coordinates as above. After 14 days of expression, mice were euthanized and parasagittal cerebellar sections were immunostained for with rabbit anti-calbindin (1:200, Cat. No. 13176, Cell Signaling) and goat anti-rabbit Alexa594-conjugated secondary (1:200, Ref. no. A11005, ThermoFisher). In 60x confocal images, regions were identified with calbindin and total Purkinje neuron number was measured. GFP-positive neurons were counted and the percentage of calbindin-positive neurons also expressing GFP was calculated. Assessments were performed by a blinded reviewer.
NPC cell death assay
80-90% confluent NPCs were transfected with PPARGC1A Human shRNA Plasmid Kit (TG310260, Origene) using Lipofectamine 3000 (ThermoFisher). After 4h media was replaced for a media containing lentiviral particles encoding SIRT1 or SIRT1 HY (enzymatically inactive) proteins. 48 hrs after transfection NPCs were transferred into the 96-well or kept on a 24-well palte. After another 24 hrs cells were given fresh media containing 1:1000 DMSO or 1 μM thapsigargin (Sigma-Aldrich), and incubated for another 24 hrs. The media was then replaced with PBS containing a staining solution of 6.75 μM propidum iodide (ThermoFisher), and 6.75 μM Hoechst 33342 (ThermoFisher). Cells were incubated in the dark at room temperature for 15 min and then imaged using the inverted fluorescence scope (Keyence BZ-X700). Images were analyzed to determine the number of dead cells presented as a fraction of all cells in the subset of eGFP positive cells (eGFP was present on a PPARGC1A shRNA plasmid). Images were analyzed using Fiji distribution of ImageJ Analysis workflow included steps such as channel splitting, channel masking, threshold adjusting, particle separation, particle analysis, and particle counting. Adequate threshold and size limits were implemented to exclude background signal fluctuations from being counted as cells. For counting the number of dead cells with flow cytometry, cells were harvested and incubated with PBS containing a staining solution of 6.75 μM propidum iodide (ThermoFisher) for 15 min at room temperature. Samples were collected with FACScanto (BD Biosciences) and analyzed with FlowJo Software (FlowJo, LLC, Ashland, OR). Three independent experiments per treatment per cell line were performed where death indices were obtained and averaged. Gene expression analysis was performed on FACS-sorted (FACSDiva (BD Biosciences), eGFP positive cells. RNA was isolated with Quick-RNA mini-prep kit (Zymo). Reverse transcription was performed with Superscript Reverse Transcriptase (ThermoFisher). Quantitative PCR was performed SYBR Green Supermix (Bio-Rad) in a QuantStudio 3 Real-Time PCR Systems (ThermoFisher).
Primary neuron culture for DNA damage
Cortical neurons were extracted from P0 pups from a FVB wild-type crossed with a male R210 C57BL/6. Cells were seeded to 50k cells / 6mm poly-D-lysine coated coverslip. After culturing in neuronal media for 13 days, cells were treated for 1 hour with 200μM H2O2. Media was removed, washed once in PBS, and then cells were fixed with 4% paraformaldehyde for 15 mins RT. Cells were permeabilized with 0.1% Triton X-100 for 10 mins RT. γH2A.x rabbit primary antibody (BioLegend Clone 2F3 cat #613402) was used at 1:1000 dilution in 5% BSA / PBS, staining for 90 mins RT. Cells were washed 4×5min PBS, then stained with Hoescht 33342 (1:10,000) and AlexaFluor 594 goat anti-rabbit (1:1,000) in 5% BSA / PBS. Cells were washed 3×5mins PBS, then immersed in PBS for 45 mins. Stained cells were preserved in Vectashield and imaged on a fluorescent confocal Leica microscope. Quantitation of fluorescence was performed in NIH ImageJ using automated methods. At least 100 neurons were analyzed for each condition, with at least two mice per condition.
Primary neuron culture for calcium imaging
Primary cerebellar granule neurons (CGNs) were derived from 7-day old fxSCA7 92Q pups and wild-type littermates and authenticated, as previously described (Young et al., 2007). Briefly, cerebella from 7-day old fxSCA7 92Q pups and wild-type littermates were digested with 0.25% Trypsin (ThermoFisher). After neutralization with 10% serum, cells were tritirated and centrifuged for 5 min at 800 x g. The pellet was resuspended in Neurobasal-A Media (ThermoFisher), 10% B27 serum-free supplement (ThermoFisher), 25 mM KCl and added to polystyrene flat-bottomed 96-well plates (Grenier) coated with poly-L-ornithine/laminin at a concentration of 28,000 cells per well, six wells per animal. Neurons were aged seven days prior to performing calcium imaging experiments.
Calcium imaging and neuron / NPC electrophysiology
For real-time recordings of intracellular calcium dynamics, cells were plated in 96-well plates at the densities described above. Prior to measurements, cells were loaded with Fluo-4 NW Calcium Assay Kit (ThermoFisher), then we added 200 ng/ml (650 nM) Hoechst 33342 (ThermoFisher). Plates were incubated with the dye at 37°C for 30 minutes, then at room temperature for an additional 15 min. After extensive wash, cells were incubated in neuronal media for 10 min. To perform optical recordings, we utilized ImageXpress® Micro XLS System (Molecular Devices) equipped with a Spectra X light engine (Lumencor) and standard filter sets. Raw movies were acquired at 50 fps, and pre-processed using MetaExpress Imaging software (Molecular Devices).
Image analysis and physiological parameter calculation
The image analysis and physiological parameter calculation was conducted using ImageJ analysis program. The analysis was performed in a similar manner on fluorescence traces generated from each neuron. Parameter tables were analyzed using Excel 2013 and OriginPro software (OriginLab, CA). Variance analysis of the individual neurons / NPCs was performed for each calcium signal for a minimum of 4 individual neurons per well. Mathematical variance (σ2) is defined as the sum of the squared distances of each term (χ) in the distribution of the mean (μ) divided by the number of samples (N) and calculated as follows:
Resulting variance data were plotted as mean with S.E.M. Single cell traces were gated to remove non-responding and low responding cells by selecting only those cells with a [Ca2+]i value at the peak of the transient that was included in the top 80% range of cells measured in the well. The setting was maintained for the entire analysis. At least five wells were recorded for each test condition.
Nicotinamide Riboside (NR) diet administration
SCA7 266Q knock-in mice were randomly assigned to two groups, one receiving NR supplemented diet (n = 17; 9 males, 8 females) and one receiving control diet (n = 11; 6 males, 5 females). Mice were housed in groups of 2-5 per cage under 12h dark-light cycle. Mice were fed with Open Standard Diet with 2.4g of Nicotinamide Riboside/kg (NR group) or Open Standard Diet with 15 kcal% fat (control group) (Research Diet, New Brunswick, USA) with ad libitum access to the food pellets and water. Dietary supplementation started at 4 weeks of age and continued until the end of the experiment.
QUANTIFICATION AND STATISTICAL ANALYSIS
All sample sizes, details, and statistical tests can be found in accompanying Figure legends. Statistical analysis was done using Microsoft Excel, Prism 6.0 (GraphPad), SigmaPlot (Systat Software), or Origin (Origin Labs). Statistical significance was defined at P<0.05. For one-way and two-way analysis of variance (ANOVA), if statistical significance (P <0.05) was achieved, then we performed post-hoc analysis corresponding to the experiment, as specified, to account for multiple comparisons. All t-tests were two-tailed Student’s t-tests, and level of significance (alpha) was always set to 0.05.
DATA AND CODE AVAILABILITY
The transcriptome data set obtained from RNA-Seq analysis of cerebellum RNAs isolated from 12 week-old and 29 week-old SCA7 mice and littermate control mice is available at the NCBI GEO website, accession #GSE139090.
Supplementary Material
Table S1. Differentially expressed genes from cerebellar transcriptome analysis. [related to Figure 1; comprehensive table of all transcriptome changes, which are highlighted in Figure 1]
Table S2. Transcription Factor Binding Site analysis of differentially expressed genes. [related to Figure 3; comprehensive set of results of oPOSSUM analysis, the key result of which is highlighted in Figure 3]
Table S3. Nanostring analysis of SCA7 cerebellum differentially expressed genes. ]related to Figure 4; raw data for digital qPCR analysis of the 100 differentially expressed genes performed on cerebellar RNAs from fxSCA7 92Q, Sirt1 uOE, Sirt1 uOE – fxSCA7 92Q, and WT control mice, and comparative analysis, the results of which are presented in Figure 4]
Table S4. Post-mortem sample information. [related to Figure 7; provides descriptive information for the seven human cerebellum samples used in the poly(ADP-ribose) immunostaining experiment, the results of which are shown in Figure 7]
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Sirt1(Sir2) Antibody (Rabbit Polyclonal Antibody) | EMD Millipore | Cat# 07-131; RRID:AB_11214517 |
| Anti-PARP1 Antibody (Rabbit Polyclonal Antibody) | Cell Signaling Technology | Cat# 9542; RRID:AB_2160739 |
| Anti-PGC-1α (H-300) Antibody (Rabbit Polyclonal Antibody) | Santa Cruz Biotechnology | Cat# sc-13067; RRID:AB_2166218 |
| Anti-Acetylated-Lysine Antibody (Rabbit Polyclonal Antibody) | Cell Signaling Technology | Cat# 9441; RRID:AB_331805 |
| ITPR1-specific antibody (Rabbit Polyclonal Antibody) | Proteintech Group | Cat# 19962-1-AP; RRID:AB_10638786 |
| ITPKA-specific antibody (Rabbit Polyclonal Antibody) | Proteintech Group | Cat# 14270-1-AP; RRID:AB_2129841 |
| Anti-Poly(ADP-ribose) Monoclonal Antibody, Unconjugated, Clone 10H | Enzo Life Sciences | Cat# ALX-804-220-R100; RRID:AB_2052275 |
| Mouse Anti-Actin, beta Monoclonal Antibody, Unconjugated, Clone mAbcam 8226 | Abcam | Cat# ab8226; RRID:AB_306371 |
| Monoclonal Anti-Calbindin-D-28K antibody produced in mouse | Sigma Aldrich | Cat# C9848; RRID:AB_476894 |
| Rabbit anti-Calbindin (D1I4Q) | Cell Signaling | Cat# 13176; RRID:AB_2687400 |
| Anti-Glial Fibrillary Acidic Protein antibody produced in rabbit | Sigma Aldrich | Cat# G9269; RRID:AB_477035 |
| Mouse anti-Cav3.1 | Neuromab | Cat# 75-206; RRID:AB_2069421 |
| Rabbit anti-α-Tubulin (11H10) | Cell Signaling | Cat# 2125; RRID:AB_2619646 |
| Goat anti-mouse IgG (H+L) Cross-Absorbed Secondary Antibody, Alexa Fluor 594 | ThermoFisher | Ref# A-11005; RRID:AB_2534073 |
| Goat anti-rabbit IgG (H+L) Cross-Absorbed Secondary Antibody, Alexa Fluor 488 | ThermoFisher | Ref# A-11008; RRID:AB_143165 |
| Goat-anti mouse IgG (H+L) Superclonal™ Secondary Antibody, Alexa Fluor 488 | ThermoFisher | Cat# A28175; RRID:AB 2536161 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | ThermoFisher | Cat# A-11012; RRID:AB_2534079 |
| Peroxidase AffiniPure goat anti-rabbit IgG (H+L) | Jackson ImmunoResearch | Cat# 111-035-003; RRID:AB_2313567 |
| Peroxidase AffiniPure goat anti-mouse IgG (H+L) | Jackson ImmunoResearch | Cat# 115-035-003; RRID:AB_10015289 |
| Bacterial and Virus Strains | ||
| AAV2/5 CMVeGFP/BK | University of Iowa Viral Vector Core | UIowa-138 |
| AAV2/5 CMVmSlo1 | University of Iowa Viral Vector Core | Custom |
| pHage-SIRT1 lentivirus | University of California San Diego / Bennett and La Spada labs | N/A |
| pHage-SIRT1HY (enzymatically inactive) lentivirus | University of California San Diego / Bennett and La Spada labs | N/A |
| Biological Samples | ||
| Human SCA7 cerebellar brain sections | NAF Brain Bank | https://ataxia.org/ |
| Human healthy cerebellar brain sections | The University of Michigan Brain Bank | http://www.brainbank.umich.edu/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TRIzol | ThermoFisher | Cat#15596026 |
| Tetrodotoxin citrate | Tocris | Cat# 1069; CAS: 18660-81-6 |
| Iberiotoxin | Alomone Labs | Cat# STI-400; CAS: 129203-60-7 |
| Picrotoxin | Sigma Aldrich | Cat# P1675; CAS: 124-87-8 |
| Mibefradil dihydrochloride hydrate | Sigma Aldrich | Cat# M5441; CAS: 116666-63-8 |
| Igepal CA-630 | Sigma Aldrich | Cat# I8896 |
| PhosSTOP | Sigma Aldrich | Cat# 4906845001 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Sigma Aldrich | Cat# 4693132001 |
| cOmplete ULTRA mini protease inhibitor Cocktail | Roche | Cat# 5892791001 |
| Western Lightning ECL Pro | Perkin Elmer | Cat# NEL121001EA |
| GE Healthcare Amersham ECL Prime Western Blotting Detection Reagent | Denville Scientific | Cat #E3018 |
| Neurobasal-A Media | ThermoFisher | Cat# 10888022 |
| B27 serum-free supplement | ThermoFisher | Cat# 17504044 |
| Matrigel | BD Biosciences | Cat# 356234 |
| Prolong Gold anti-fade reagent | ThermoFisher | Cat# P36930 |
| 0.25% Trypsin- EDTA | ThermoFisher | Cat# 25200056 |
| Accutase | ThermoFisher | Cat# A1110501 |
| STEMdiff Neural Induction Medium | StemCell Technologies | Cat# 05835 |
| STEMdiff Neural Progenitor Medium | StemCell Technologies | Cat# 05833 |
| Hoechst 33342 | Life Technologies | Cat# H3570 |
| Thapsigargin | Sigma Aldrich | Cat# T9033; CAS# 67526-95-8 |
| L-ornithine | Sigma Aldrich | Cat# P4957; CAS# 27378-49-0 |
| Laminin | Sigma Aldrich | Cat# L4544-100 |
| RIPA Buffer | ThermoFisher | Cat# 89900 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| Turbo DNA-free Kit | ThermoFisher | Cat# AM1907 |
| High-Capacity cDNA Reverse Transcriptase Kit | ThermoFisher | Cat# 4368814 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat# 1708890 |
| TaqMAN Universal Master Mix II | ThermoFisher | Cat# 4440040 |
| iQ SYBR Green Supermix | Bio-Rad | Cat# 1708882 |
| Pierce BCA Protein Assay Kit | ThermoFisher | Cat# 23225 |
| Fluo-4AM NW Calcium Assay Kit | ThermoFisher | Cat# F36206 |
| Dako EnVision Dual Link System-HRP | Agilent | Cat# K406189-2 |
| Dako DAB solution | Agilent | Cat# K346811-2 |
| Hematoxylin Stain+ | Fisher Scientific | Cat#220-100 |
| Deposited Data | ||
| Raw and analyzed RNA-Seq data | This paper | GEO: GSE139090 |
| Experimental Models: Cell Lines | ||
| Human: IPSC-derived neuron progenitor cells | Ward et al., 2019 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: mPRP-BAC-fxSCA92Q mice | Furrer et al., 2011 | N/A |
| Mouse: SCA7 266Q mice | Yoo et al., 2003 | N/A |
| Mouse: Sirt1STOP | Firestein et al., 2008 | N/A |
| Oligonucleotides | ||
| Kcnma1 SYBR Probe 5’- AGC CAA CGA TAA GCT GTG GT -3’ | IDT | N/A |
| Kcnma1 SYBR Probe 5’- AAT CTC AAG CCA AGC CAA CT -3’ | IDT | N/A |
| Cacna1g SYBR Probe 5’- GTC GCT TTG GGT ATC TTT GG -3’ | IDT | N/A |
| Cacna1g SYBR Probe 5’- TAC TCC AGC ATC CCA GCA AT -3’ | IDT | N/A |
| Gng13 Taqman Probe | ThermoFisher | Mm00458153_g1 |
| Atp2a3 Taqman Probe | ThermoFisher | Mm00443898_m1 |
| Itpr1 Taqman Probe | ThermoFisher | Mm004399907_m1 |
| Dagla Taqman Probe | ThermoFisher | Mm00813830_m1 |
| Itpka Taqman Probe | ThermoFisher | Mm00525139_m1 |
| Cacna1g Taqman Probe | ThermoFisher | Mm00486572_m1 |
| Grid2ip Taqman Probe | ThermoFisher | Mm00504815_m1 |
| Sirt1 Taqman Probe | ThermoFisher | Mm00490758_m1 |
| Nmnat1 Taqman Probe | ThermoFisher | Mm01257929_m1 |
| Nmnat2 Taqman Probe | ThermoFisher | Mm00615393_m1 |
| Nmnat3 Taqman Probe | ThermoFisher | Mm00513791_m1 |
| SIRT1 SYBR Probe 5’- CCT GGA CAA TTC CAG CCA TCT -3’ | IDT | N/A |
| SIRT1 SYBR Probe 5’- CAA CCT GTT CCA GCG TGT CT -3’ | IDT | N/A |
| PGC-1a SYBR Probe 5’- GTG GAC GCA AGC AAT TTT TC -3’ | IDT | N/A |
| PGC-1a SYBR Probe 5’- TTA CCT GCG CAA GCT TCT CT -3’ | IDT | N/A |
| Recombinant DNA | ||
| PPARGC1A Human shRNA Plasmid Kit (Locus ID 10891) | Origene | TG310260 |
| pBScMXT containing mSlo1 MBr5/3 | Larry Salkoff (through Addgene) | Cat# 16149 |
| pFBAAVCMVmcsBgHpA | University of Iowa Viral Vector Core | Cat# G0347 |
| Software and Algorithms | ||
| TopHat | Trapnell et al, 2009 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks | Trapnell et al, 2010 | http://cole-trapnell-lab.github.io/cufflinks/ |
| Genesis | Sturn et al, 2002 | https://genome.tugraz.at/genesisclient/genesisclient_download.shtml |
| NIS Elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| pClamp-10 | Molecular Devices | https://www.moleculardevices.com/ |
| Clampfit 10.2 | Molecular Devices | https://www.moleculardevices.com/ |
| SigmaPlot | Systat Software | https://systatsoftware.com/products/sigmaplot/ |
| oPPOSSUM 3.0 | Ho-Sui et al, 2005 | http://opossum.cisreg.ca/oPOSSUM3/ |
| OriginPro | OriginLab | https://www.originlab.com/index.aspx?go=Products/Origin |
| GraphPad Prism 6.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Fiji distribution of ImageJ | Schindelin et al, 2012 | https://fiji.sc/#download |
| FlowJo | FlowJo, LLC, Ashland, OR | https://www.flowjo.com/ |
| Other | ||
Highlights.
Transcriptomes of SCA7 mice revealed down-regulation of genes controlling Ca++ flux
Altered membrane excitability in SCA7 involves NAD+ depletion and PARP1 activation
Increased Sirt1 rescued cerebellar degeneration and calcium defects in SCA7 mice
NAD+ repletion prevented disease in SCA7 mice and patient stem cell-derived neurons
Acknowledgments
We are grateful to J. Olefsky (UCSD) for Sirt1 transgenic mice; J. Cleary, F. Fitzgerald, and L. Ranum (Center for NeuroGenetics, University of Florida), and M. Perkins (University of Michigan Brain Bank) for post-mortem materials; and M. Mercola (Stanford) for comments and equipment use. This work was supported by the US National Institutes of Health (R01 EY014061 [A.R.L.S.], R01 AG033082 [A.R.L.S.], R01 NS085054 [V.G.S.], R01AG043930 [J.A.], P01 AG053760 to University of Michigan, and P30 EY022589 to UCSD), École Polytechnique Fédérate de Lausanne [J.A.], and Swiss Initiative for Systems Biology (51RTP0-151019 [J.A.]. P.M.S. was supported by mobility grant from Polish Ministry of Science and Higher Education (1303/MOB/IV/2015/0: Mobilnosc Plus).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Abramoff MD, Magalhaes PJ, Ram SJ (2004), Image Processing with ImageJ. Biophotonics International 11, 36–42. [Google Scholar]
- Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper Riekelt H., et al. (2011), PARP-1 Inhibition Increases Mitochondrial Metabolism through SIRT1 Activation. Cell Metabolism 13, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt C, Ryten M, Forabosco P, Schorge S, Hersheson J, Hardy J, Houlden H, and United Kingdom Brain Expression, C. (2014), Insights from cerebellar transcriptomic analysis into the pathogenesis of ataxia. JAMA Neurol 71, 831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushart DD, Chopra R, Singh V, Murphy GG, Wulff H, and Shakkottai VG (2018). Targeting potassium channels to treat cerebellar ataxia. Ann Clin Transl Neurol 5, 297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushart DD, and Shakkottai VG (2019). Ion channel dysfunction in cerebellar ataxia. Neurosci Lett 688, 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, Cohen MS, and Goodman RH (2016). Biosensor reveals multiple sources for mitochondrial NAD(+], Science 352,1474–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BC, and Bean BP (2009). Sodium entry during action potentials of mammalian neurons: incomplete inactivation and reduced metabolic efficiency in fast-spiking neurons. Neuron 64, 898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Peng GH, Wang X, Smith AC, Grote SK, Sopher BL, and La Spada AR (2004). Interference of Crx-dependent transcription by ataxin-7 involves interaction between the glutamine regions and requires the ataxin-7 carboxy-terminal region for nuclear localization. Hum Mol Genet 13, 53–67. [DOI] [PubMed] [Google Scholar]
- Chopra R, Bushart DD, and Shakkottai VG (2018). Dendritic potassium channel dysfunction may contribute to dendrite degeneration in spinocerebellar ataxia type 1. PLoS One 13, eO198040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell RM, Blake KR, and Russell JW (2007). Localization of the transcriptional coactivator PGC-lalpha to GABAergic neurons during maturation of the rat brain. J Comp Neurol 502,1–18. [DOI] [PubMed] [Google Scholar]
- Dell’Orco JM, Pulst SM, and Shakkottai VG (2017). Potassium channel dysfunction underlies Purkinje neuron spiking abnormalities in spinocerebellar ataxia type 2. Hum Mol Genet 26, 3935–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’Orco JM, Wasserman AH, Chopra R, Ingram MA, Hu YS, Singh V, Wulff H, Opal P, Orr HT, and Shakkottai VG (2015). Neuronal Atrophy Early in Degenerative Ataxia Is a Compensatory Mechanism to Regulate Membrane Excitability. J Neurosci 35,11292–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez G, and Outeiro TF (2013). SIRT1 and SIRT2: emerging targets in neurodegeneration. EMBO Mol Med 5, 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ware M, Li XJ, and Mattson MP (2003). Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci U S A 100, 2911–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgerton JR, and Reinhart PH (2003). Distinct contributions of small and large conductance Ca2+-activated K+ channels to rat Purkinje neuron function. J Physiol 548, 53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorova P, Popugaeva E, and Bezprozvanny I (2015). Disturbed calcium signaling in spinocerebellar ataxias and Alzheimer’s disease. Semin Cell Dev Biol 40,127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrick MJ, Pacheco CD, Yu T, Dadgar N, Shakkottai VG, Ware C, Paulson HL, and Lieberman AP (2010). Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum Mol Genet 19, 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, Bhimavarapu A, Luikenhuis S, de Cabo R, Fuchs C, et al. (2008). The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS One 3, e2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furrer SA, Mohanachandran MS, Waldherr SM, Chang C, Damian VA, Sopher BL, Garden GA, and La Spada AR (2011). Spinocerebellar ataxia type 7 cerebellar disease requires the coordinated action of mutant ataxin-7 in neurons and glia, and displays non-cell-autonomous bergmann glia degeneration. J Neurosci 31,16269–16278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furrer SA, Waldherr SM, Mohanachandran MS, Baughn TD, Nguyen KT, Sopher BL, Damian VA, Garden GA, and La Spada AR (2012). Reduction of mutant ataxin-7 expression restores motor function and prevents cerebellar synaptic reorganization in a conditional mouse model of SCA7. Hum Mol Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Todorov B, Barrett CF, van Dorp S, Ferrari MD, van den Maagdenberg AM, De Zeeuw CI, and Hoebeek FE (2012). Cerebellar ataxia by enhanced Ca(V]2.1 currents is alleviated by Ca2+-dependent K+-channel activators in Cacna1a(S218L] mutant mice. J Neurosci 32,15533–15546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariani K, Menzies KJ, Ryu D, Wegner CJ, Wang X, Ropelle ER, Moullan N, Zhang H, Perino A, Lemos V, et al. (2016). Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 63,1190–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, and Garden GA (2010). A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp, doi: 10.3791/1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halagappa VK, Guo Z, Pearson M, Matsuoka Y, Cutler RG, Laferla FM, and Mattson MP (2007). Intermittent fasting and caloric restriction ameliorate age-related behavioral deficits in the triple-transgenic mouse model of Alzheimer’s disease. Neurobiol Dis 26, 212–220. [DOI] [PubMed] [Google Scholar]
- Hansen ST, Meera P, Otis TS, and Pulst SM (2013). Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet 22, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger D, Hardy S, Sasorith S, Klein F, Robert F, Weber C, Miguet L, Potier N, Van-Dorsselaer A, Wurtz JM, et al. (2004). Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum Mol Genet 13,1257–1265. [DOI] [PubMed] [Google Scholar]
- Hoch NC, Hanzlikova H, Rulten SL, Tetreault M, Komulainen E, Ju L, Hornyak P, Zeng Z, Gittens W, Rey SA, et al. (2017). XRCC1 mutation is associated with PARP1 hyper activation and cerebellar ataxia. Nature 541, 87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, and Auwerx J (2012). Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram M, Wozniak EA, Duvick L, Yang R, Bergmann P, Carson R, O’Callaghan B, Zoghbi HY, Henzler C, and Orr HT (2016). Cerebellar Transcriptome Profiles of ATXN1 Transgenic Mice Reveal SCA1 Disease Progression and Protection Pathways. Neuron 89,1194–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Cohen DE, Cui L, Supinski A, Savas JN, Mazzulli JR, Yates JR 3rd, Bordone L, Guarente L, and Krainc D (2012). Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat Med 18, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Wang J, Fu J, Du L, Jeong H, West T, Xiang L, Peng Q, Hou Z, Cai H, et al. (2012). Neuroprotective role of Sirt1 in mammalian models of Huntington’s disease through activation of multiple Sirt1 targets. Nat Med 18,153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo HY, Yun M, Jeong J, Park ER, Shin HJ, Woo SR, Jung JK, Kim YM, Park JJ, Kim J, et al. (2015). SIRT1 deacetylates and stabilizes hypoxia-inducible factor-lalpha (HIF-lalpha] via direct interactions during hypoxia. Biochem Biophys Res Commun 462, 294–300. [DOI] [PubMed] [Google Scholar]
- Kasumu A, and Bezprozvanny I (2012). Deranged calcium signaling in Purkinje cells and pathogenesis in spinocerebellar ataxia 2 (SCA2] and other ataxias. Cerebellum 11, 630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Spada AR, Fu Y, Sopher BL, Libby RT, Wang X, Li LY, Einum DD, Huang J, Possin DE, Smith AC, et al. (2001). Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron 31, 913–927. [DOI] [PubMed] [Google Scholar]
- La Spada AR, and Taylor JP (2003). Polyglutamines placed into context. Neuron 38, 681–684. [DOI] [PubMed] [Google Scholar]
- La Spada AR, and Taylor JP (2010). Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet 11, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, and Fischbeck KH (1991). Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352, 77–79. [DOI] [PubMed] [Google Scholar]
- Landis SC, Amara SG, Asadullah K, Austin CP, Blumenstein R, Bradley EW, Crystal RG, Darnell RB, Ferrante RJ, Fillit H, et al. (2012). A call for transparent reporting to optimize the predictive value of preclinical research. Nature 490,187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KY, and Kraus WL (2017). PARP Inhibitors for Cancer Therapy. Cell 169, 183. [DOI] [PubMed] [Google Scholar]
- Llano I, Leresche N, and Marty A (1991a). Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron 6, 565–574. [DOI] [PubMed] [Google Scholar]
- Llano I, Marty A, Armstrong CM, and Konnerth A (1991b). Synaptic- and agonist-induced excitatory currents of Purkinje cells in rat cerebellar slices. J Physiol 434,183–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long AN, Owens K, Schlappal AE, Kristian T, Fishman PS, and Schuh RA (2015). Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol 15,19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EK, Dougherty SE, McMeekin LJ, Trinh AT, Reid CS, and Cowell RM (2012). Developmental alterations in motor coordination and medium spiny neuron markers in mice lacking pgc-lalpha. PLoS One 7, e42878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark MD, Schwitalla JC, Groemmke M, and Herlitze S (2017). Keeping Our Calcium in Balance to Maintain Our Balance. Biochem Biophys Res Commun 483, 1040–1050. [DOI] [PubMed] [Google Scholar]
- Martin JJ, Van Regemorter N, Krols L, Brucher JM, de Barsy T, Szliwowski H, Evrard P, Ceuterick C, Tassignon MJ, Smet-Dieleman H, et al. (1994). On an autosomal dominant form of retinal-cerebellar degeneration: an autopsy study of five patients in one family. Acta Neuropathol (Berl) 88, 277–286. [DOI] [PubMed] [Google Scholar]
- Michalik A, Martin JJ, and Van Broeckhoven C (2004). Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet 12, 2–15. [DOI] [PubMed] [Google Scholar]
- Niu C, Prakash TP, Kim A, Quach JL, Huryn LA, Yang Y, Lopez E, Jazayeri A, Hung G, Sopher BL, et al. (2018). Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I, Strathearn KE, Amore AM, Volk CB, Maxwell MM, Rochet JC, McLean PJ, et al. (2007). Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317, 516–519. [DOI] [PubMed] [Google Scholar]
- Palhan VB, Chen S, Peng GH, Tjernberg A, Gamper AM, Fan Y, Chait BT, La Spada AR, and Roeder RG (2005). Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci U SA 102, 8472–8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel NV, Gordon MN, Connor KE, Good RA, Engelman RW, Mason J, Morgan DG, Morgan TE, and Finch CE (2005). Caloric restriction attenuates Abeta-deposition in Alzheimer transgenic models. Neurobiol Aging 26, 995–1000. [DOI] [PubMed] [Google Scholar]
- Paulson HL, and Miller VM (2005). Breaks in coordination: DNA repair in inherited ataxia. Neuron 46, 845–848. [DOI] [PubMed] [Google Scholar]
- Perrin S (2014). Preclinical research: Make mouse studies work. Nature 507, 423–425. [DOI] [PubMed] [Google Scholar]
- Pirinen E, Cantó C, Jo Young S., Morato L, Zhang H, Menzies, Keir J, Williams, Evan G, Mouchiroud L, Moullan N, Hagberg C, et al. (2014). Pharmacological Inhibition of Poly(ADP-Ribose] Polymerases Improves Fitness and Mitochondrial Function in Skeletal Muscle. Cell Metabolism 19,1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, and Puigserver P (2005). Nutrient control of glucose homeostasis through a complex of PGC-lalpha and SIRT1. Nature 434,113–118. [DOI] [PubMed] [Google Scholar]
- Ryu C, Jang DC, Jung D, Kim YG, Shim HG, Ryu HH, Lee YS, Linden DJ, Worley PF, and Kim SJ (2017). STIM1 Regulates Somatic Ca(2+] Signals and Intrinsic Firing Properties of Cerebellar Purkinje Neurons. J Neurosci 37, 8876–8894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D, Zhang H, Ropelle ER, Sorrentino V, Mazala DA, Mouchiroud L, Marshall PL, Campbell MD, Ali AS, Knowels GM, et al. (2016). NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci Transl Med 8, 361ral39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H, Sausbier U, Sailer CA, Feil R, Hofmann F, et al. (2004). Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci U S A 101, 9474–9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann SN, Cheron G, Lohof A, d’Alcantara P, Meyer M, Parmentier M, and Schurmans S (1999). Impaired motor coordination and Purkinje cell excitability in mice lacking calretinin. Proc Natl Acad Sci U S A 96, 5257–5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, and Livak KJ (2008). Analyzing real-time PCR data by the comparative C(T] method. Nat Protoc 3,1101–1108. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012], NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorge S, van de Leemput J, Singleton A, Houlden H, and Hardy J (2010). Human ataxias: a genetic dissection of inositol triphosphate receptor (ITPR1]-dependent signaling. Trends Neurosci 33, 211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakkottai VG, do Carmo Costa M, Dell’Orco JM, Sankaranarayanan A, Wulff H, and Paulson HL (2011). Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci 31, 13002–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuvaev AN, Hosoi N, Sato Y, Yanagihara D, and Hirai H (2017). Progressive impairment of cerebellar mGluR signalling and its therapeutic potential for cerebellar ataxia in spinocerebellar ataxia type 1 model mice. J Physiol 595,141–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturn A, Quackenbush J, and Trajanoski Z (2002). Genesis: cluster analysis of microarray data. Bioinformatics 18, 207–208. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25,1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E (2015). NAD(+] in aging, metabolism, and neurodegeneration. Science 350,1208–1213. [DOI] [PubMed] [Google Scholar]
- Walter JT, Alvina K, Womack MD, Chevez C, and Khodakhah K (2006). Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci 9, 389–397. [DOI] [PubMed] [Google Scholar]
- Wang J, Ho L, Qin W, Rocher AB, Seror I, Humala N, Maniar K, Dolios G, Wang R, Hof PR, et al. (2005). Caloric restriction attenuates beta-amyloid neuropathology in a mouse model of Alzheimer’s disease. Faseb J 19, 659–661. [DOI] [PubMed] [Google Scholar]
- Wang X, Hu X, Yang Y, Takata T, and Sakurai T (2016). Nicotinamide mononucleotide protects against beta-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Res 1643,1–9. [DOI] [PubMed] [Google Scholar]
- Ward JM, Stoyas CA, Switonski PM, Ichou F, Fan W, Collins B, Wall CE, Adanyeguh I, Niu C, Sopher BL, et al. (2019). Metabolic and Organelle Morphology Defects in Mice and Human Patients Define Spinocerebellar Ataxia Type 7 as a Mitochondrial Disease. Cell Rep 26,1189–1202 ell86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PA, Harder JM, Foxworth NE, Cochran KE, Philip VM, Porciatti V, Smithies 0, and John SW (2017). Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 355, 756–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, and Sauve AA (2006). NAD metabolism and sirtuins: metabolic regulation of protein deacetylation in stress and toxicity. AAPS J 8, E632–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SY, Pennesi ME, Weeber EJ, Xu B, Atkinson R, Chen S, Armstrong DL, Wu SM, Sweatt JD, and Zoghbi HY (2003). SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron 37, 383–401. [DOI] [PubMed] [Google Scholar]
- Young JE, Gouw L, Propp S, Sopher BL, Taylor J, Lin A, Hermel E, Logvinova A, Chen SF, Chen S, et al. (2007). Proteolytic cleavage of ataxin-7 by caspase-7 modulates cellular toxicity and transcriptional dysregulation. J Biol Chem 282, 30150–30160. [DOI] [PubMed] [Google Scholar]
- Zagha E, Lang EJ, and Rudy B (2008). Kv3.3 channels at the Purkinje cell soma are necessary for generation of the classical complex spike waveform. J Neurosci 28,1291–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R, et al. (2016). NAD(+] repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352,1436–1443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Differentially expressed genes from cerebellar transcriptome analysis. [related to Figure 1; comprehensive table of all transcriptome changes, which are highlighted in Figure 1]
Table S2. Transcription Factor Binding Site analysis of differentially expressed genes. [related to Figure 3; comprehensive set of results of oPOSSUM analysis, the key result of which is highlighted in Figure 3]
Table S3. Nanostring analysis of SCA7 cerebellum differentially expressed genes. ]related to Figure 4; raw data for digital qPCR analysis of the 100 differentially expressed genes performed on cerebellar RNAs from fxSCA7 92Q, Sirt1 uOE, Sirt1 uOE – fxSCA7 92Q, and WT control mice, and comparative analysis, the results of which are presented in Figure 4]
Table S4. Post-mortem sample information. [related to Figure 7; provides descriptive information for the seven human cerebellum samples used in the poly(ADP-ribose) immunostaining experiment, the results of which are shown in Figure 7]
Data Availability Statement
The transcriptome data set obtained from RNA-Seq analysis of cerebellum RNAs isolated from 12 week-old and 29 week-old SCA7 mice and littermate control mice is available at the NCBI GEO website, accession #GSE139090.