

Cryptic transcription antagonizes OXPHOS and cardiogenesis.
Abstract
Cryptic transcription occurs widely across the eukaryotic genome; however, its regulation during vertebrate development is not understood. Here, we show that two class I histone deacetylases, Hdac1 and Hdac2, silence cryptic transcription to promote mitochondrial function in developing murine hearts. Mice lacking Hdac1 and Hdac2 in heart exhibit defective developmental switch from anaerobic to mitochondrial oxidative phosphorylation (OXPHOS), severe defects in mitochondrial mass, mitochondrial function, and complete embryonic lethality. Hdac1/Hdac2 promotes the transition to OXPHOS by enforcing transcriptional fidelity of metabolic gene programs. Mechanistically, Hdac1/Hdac2 deacetylates histone residues including H3K23, H3K14, and H4K16 to suppress cryptic transcriptional initiation within the coding regions of actively transcribed metabolic genes. Thus, Hdac1/2-mediated epigenetic silencing of cryptic transcription is essential for mitochondrial function during early vertebrate development.
INTRODUCTION
The mammalian heart forms early during development, requiring constant energy to maintain its critical pumping action. During early cardiac development, cardiomyocytes switch energy production from anaerobic pathways to mitochondrial oxidative phosphorylation (OXPHOS) (1). Multiple studies link defective developmental energy generation and congenital heart disease (CHD), with almost every metabolic disruption associated with cardiac symptoms (2). Although recent advances identified the transition to OXPHOS as a checkpoint and potential origin for metabolic CHD, the molecular basis for this developmental metabolic transition remains elusive (1, 3, 4). In adult cardiomyopathies and metabolic syndrome, pathologic reversal of the anaerobic to OXPHOS switch occurs, suggesting epigenetic regulation (5). Concordantly, intracellular metabolic transitions, in both development and disease, alter a myriad of lysine residues on histone tails, influencing gene expression (6). Emerging data highlight that intracellular metabolites modulate activity of histone deacetylases (HDACs) and histone acetylation in cultured cells (7, 8). Despite this, the roles of HDACs during developmental metabolic transitions remain undefined.
RESULTS
Among mammalian HDACs, paralogous Hdac1 and Hdac2 have the highest activity toward nucleosome substrates, enabling chromatin remodeling (9). To investigate the role of Hdac1 and Hdac2 in early cardiac metabolism, we genetically ablated murine Hdac1/Hdac2 using Nkx2.5IRES-Cre (10), Hdac1Flox, and Hdac2Flox (11) (Nkx2.5;1KO2KO) alleles. Complete loss of Hdac1/Hdac2 was observed in the developing heart starting at embryonic day 10.5 (E10.5) (fig. S1). Nkx2.5;1KO2KO embryos were comparable in size at E10.5; however, these embryos exhibited complete embryonic lethality by E11.5 (Fig. 1, A to C). We observed increased light transmission through the primitive heart tube (PHT) in E10.5 Nkx2.5;1KO2KO embryos, suggesting defective cardiogenesis (Fig. 1C). Morphologic staining revealed a thinner myocardium and reduced eosinophilic cytoplasm in E10.5 Nkx2.5;1KO2KO cardiomyocytes (Fig. 1, D and E) without major changes in proliferation, apoptosis, or cell number (not shown). Transmission electron micrographs (TEMs) of E10.5 PHTs showed Nkx2.5;1KO2KO cardiomyocytes had accumulations of cytoplasmic lipid droplets, a fragmented contractile network, and abnormal mitochondria consistent with failure to initiate OXPHOS (Fig. 1, F and G).
Fig. 1. Loss of Hdac1/Hdac2 within Nkx2.5IRES-Cre+ cells causes defective cardiogenesis and complete embryonic lethality.
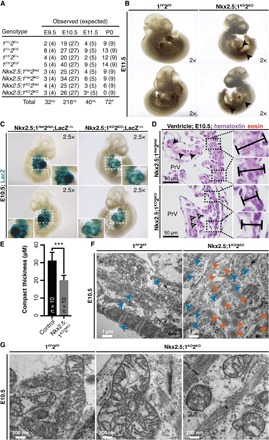
(A) Nkx2.5IRES-Cre;1F+2F+ was crossed with 1FF2FF, and samples were recovered at various time points. aThree embryos dying. *P < 0.05; ns, not significant. (B) Nkx2.5;1KO2KO and 1FF2FF embryos at embryonic day 11.5 (E11.5) (arrows, pooled blood). (C) Nkx2.5;1Het2Het;R26R-LacZ−/+ and Nkx2.5;1KO2KO;R26R-LacZ−/+ E10.5 embryos. (D) Hematoxylin and eosin–stained Nkx2.5;1Het2Het and Nkx2.5;1KO2KO E10.5 sagittal sections at atrioventricular canal (AVC) level (arrows, eosinophilic cytoplasm; bars, compact thickness). (E) Compact myocardial thickness in control and Nkx2.5;1KO2KO E10.5 primitive ventricles (PrVs). (F) Transmission electron micrographs (TEMs) of Nkx2.5;1KO2KO and 1FF2FF E10.5 cardiomyocytes (blue, contractile fibers; orange, cytoplasmic lipid droplets). (G) TEM of Nkx2.5;1KO2KO and 1FF2FF E10.5 cardiomyocyte mitochondrial structure, density, and size.
To define transcriptional changes, we performed Affymetrix Clariom D Pico assays on E10.5 Nkx2.5;1KO2KO PHTs. Among 8127 differentially regulated transcripts with NCBI (National Center for Biotechnology Information) identifiers, 52% were reduced in Nkx2.5;1KO2KO, while 48% were increased relative to controls (Fig. 2, A and B). Protein ANalysis THrough Evolutionary Relationships (PANTHER) analysis showed enrichment in mitochondrial metabolism categories, such as tricarboxylic acid cycle (TCA), adenosine 5′-triphosphate (ATP) formation, cristae formation, and complex I biogenesis without corollary upstream changes (Fig. 2C and fig. S2) (12). Despite relatively equal numbers of up-/down-regulated transcripts, enriched categories contained almost exclusively down-regulated genes, suggesting that Hdac1/Hdac2 facilitates metabolic gene expression (Fig. 2C). We found corresponding reductions in mitochondrial DNA content and protein expression in E10.5 Nkx2.5;1KO2KO PHTs (Fig. 2, D and E). Similarly, reduced protein expression of critical mitochondrial proteins in neonatal 1KO2KO hearts with normal contractile protein network and reduced oxygen consumption in 1KO2KO cardiomyocytes confirm requirement of Hdac1 and Hdac2 for mitochondrial function (fig. S3, A to C). There is a strong interrelationship between cardiac bioenergetics and structural development. Transition from early anaerobic fetal metabolism to OXPHOS, facilitated by mitochondrial permeability transition pore (mPTP) closure between E9.5 and E11.5, is necessary and sufficient to drive cardiomyocyte differentiation (13). E10.5 is a metabolic checkpoint with disruptions of cardiac bioenergetics associated with early embryonic lethality (12, 14). Consequently, we propose that loss of Hdac1/Hdac2 disrupts the anaerobic to OXPHOS switch, arresting cardiogenesis at E10.5.
Fig. 2. Primitive heart tubes lacking Hdac1 and Hdac2 exhibit down-regulation of metabolic gene programs.
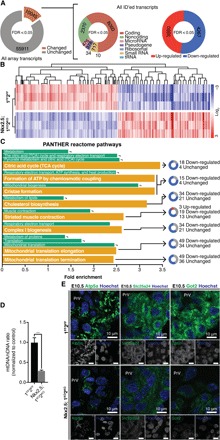
(A) Analysis of differentially regulated transcripts at various levels. (B) Heatmap of identified, differentially regulated transcripts grouped by Spearman clustering. (C) Top enriched PANTHER reactome terms (gold) with parent categories (green). Doughnuts show distribution of transcript changes. (D) Mitochondrial DNA (mtDNA) quantification relative to nuclear DNA (nDNA) in Nkx2.5;1KO2KO and 1FF2FF E10.5 PHTs. (E) Atp5a, Slc25a24, and Got2 staining on Nkx2.5;1KO2KO and 1FF2FF E10.5 sagittal sections at AVC level with Hoechst nuclear counterstain. Grayscale images are unedited. tRNA, transfer RNA.
We next determined how Hdac1/Hdac2 cooperates to facilitate metabolic transcriptional programs during cardiogenesis. Examination of exon-level expression data revealed a transcriptional signature of lowered initiation at canonical transcription start sites (TSSCs) but increased initiation from downstream, alternative transcription start sites (TSSAs) specifically in down-regulated metabolic transcripts (Fig. 3, A to G, and fig. S4, A and B). Highlighting two critical transcripts, citrate synthase (CS) and NADH:ubiquinone oxioreductase subunit B9 (Ndufb9), we looked for evidence of noncanonical transcripts using 5′ rapid amplification of complementary DNA (cDNA) ends (5′RACE). We identified strong control bands corresponding to canonical transcripts for both CS and Ndufb9 (Fig. 3, H and I). No canonical transcript was found for CS following Hdac1/Hdac2 ablation, instead a truncated transcript originating from a distal TSSA was identified (Fig. 3H). Two alternative Ndufb9 transcripts were identified, originating from two distinct TSSAs. We observed decreased intensity of the canonical and second alternative Ndufb9 transcripts in E10.5 Nkx2.5;1KO2KO hearts (Fig. 3I). To confirm our findings, we quantified abundance of early exon junctions relative to late exon junctions, finding a significantly reduced ratio of early to late exon junction expression in Nkx2.5;1KO2KOs consistent with reduced canonical and increased alternative transcription (fig. S4C). Together, these results suggest that Hdac1/Hdac2 represses cryptic initiation of transcription from intragenic TSSAs to facilitate the anaerobic to OXPHOS switch during early cardiac development.
Fig. 3. Hdac1 and Hdac2 suppress cryptic transcription during cardiogenesis.
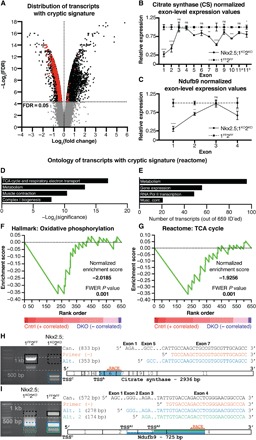
(A) Distribution of differentially regulated transcripts (black), differentially regulated transcripts with a cryptic signature (red), and unchanged transcripts (gray) in Nkx2.5;1KO2KO compared with 1FF2FF E10.5 PHTs. (B and C) Normalized exon-level expression values for citrate synthase (CS; B) and Ndufb9 (C) in Nkx2.5;1KO2KO and 1FF2FF E10.5 PHTs. (D to G) PANTHER analysis (D and E) and Gene Set Enrichment Analysis (GSEA) (F and G) of differentially regulated, identified cryptic transcripts. (H and I) 5′ rapid amplification of complementary DNA (cDNA) ends (5′RACE) for CS (F) and Ndufb9 (E). Left: 5′RACE products (black, canonical; blue/green, alternative). Right: Alignment of sequenced products. TSSC, canonical TSS; TSSA#, alternative TSS; ns, not significant; FWER, Family-wise error rate; Pol II, polymerase II.
Transcription-induced histone modifications at an alternative transcription start sites can antagonize the permissive environment of proximal promoter and enhancer elements, attenuating gene expression from canonical transcription start site (15, 16). Supporting this model, several covalent histone modifications have been associated with cryptic transcription including H3K14ac, H3K23ac, H4K16ac, and H3K36me3 (17). Embryonic and postnatal hearts lacking Hdac1 and Hdac2 exhibited increased numbers of H3K23ac and H4K16ac foci in cardiomyocyte nuclei (Fig. 4, A and B, and fig. S5, A and B). Similarly, we observed enrichment of H3K36me3, H3K23ac, H3K14ac, and H4K16ac at the TSSAs of CS and Ndufb9, consistent with cryptic initiation of transcription (Fig. 4, C to F, and fig. S5C). Hdac1 and Hdac2 occupancy at the TSSA of CS, TSSA1 of Ndufb9, and TSSA2 of Ndufb9 in E10.5 PHTs (Fig. 4, G to I) suggests that Hdac1/Hdac2 cooperatively and directly suppresses activating histone marks throughout actively transcribed gene bodies, restricting transcription to defined TSSCs (Fig. 4J).
Fig. 4. Hdac1 and Hdac2 cooperate to suppress cryptic transcription through deacetylation of key histone residues at alternative transcription start sites during mammalian cardiogenesis.
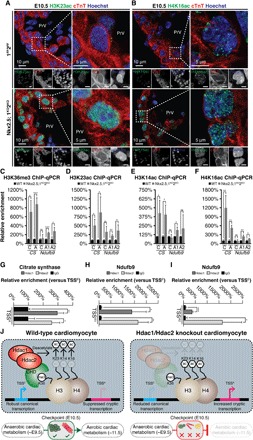
(A and B) H3K23ac (A) and H4K16 (B) staining on Nkx2.5;1KO2KO and 1FF2FF E10.5 sagittal sections at AVC level with cardiac troponin (cTnT) and Hoechst counterstains. Grayscale images are unedited. (C to E) ChIP-qPCR showing relative enrichment (TSSA versus TSSC) of Hdac1and Hdac2 at CS TSSA (C) and Ndufb9 TSSA1 (D), TSSA2 (E), normalized to immunoglobulin G (IgG). (F to I) H3K36me3 (E), H3K23ac (F), H3K14ac (G), and H4K16ac (H) ChIP-qPCR showing relative enrichment over TSSC/TSSAs for CS and Ndufb9 in Nkx2.5;1KO2KO and 1FF2FF E10.5 PHTs. (J) Hdac1/Hdac2 controls cardiomyocyte development at a critical metabolic checkpoint. CHD, chromodomain containing subunit. WT, wild type.
DISCUSSION
To our knowledge, this report is the first link between HDACs and cryptic transcription during vertebrate development and the first to link cryptic transcription and mitochondrial function. Cryptic transcription is observed in lower organisms and mammalian cell lines, yet no reports describe cryptic transcription in a vertebrate system (15, 17). In yeast, cryptic transcription is regulated by Set2 and Rpd3s (18–20). Set2, an H3K36 methyltransferase, is recruited to active loci by RNA polymerase II, causing accumulation of H3K36 trimethylation. H3K36me3 recruits Rpd3s (homologous to the Sin3 complex) through Eaf3’s chromodomain to deacetylate residues including H3K23, H3K14, and H4K16 (15, 17, 18, 21). This axis restores basal chromatin conformations following transcription, preventing cryptic initiation of transcription. HDAC or DNA methyl transferase inhibition in cultured cells increases cryptic transcription across the genome, suggesting, much like inappropriately condensed chromatin can disrupt initiation, cryptic transcription results from inappropriately open chromatin (16, 17). Open conformations expose noncanonical promoter and enhancer regions, driving the synthesis of noncoding mRNA transcripts and disrupting canonical transcription. Cumulatively, these results suggest that chromatin homeostasis is crucial for ordered transcription.
Here, we demonstrate that mammalian Rpd3 homologs, Hdac1/Hdac2, redundantly suppress cryptic initiation of transcription during mammalian cardiogenesis. Loss of Hdac1/Hdac2 in developing murine hearts promotes cryptic transcription at metabolic loci, including CS and Ndufb9, during a critical period of bioenergetic maturation. Mechanistically, we identified accumulation of H3K36 trimethylation and H3K23/H3K14/H4K16 acetylation, consistent with inappropriately open chromatin at the TSSAs of CS and Ndufb9. This suggests that Hdac1/Hdac2, much like Rpd3, suppresses cryptic transcription by restoring basal chromatin accessibility following transcription (Fig. 4J).
Our study reconciles prior observations that HDACs, classically viewed as chromatin condensers and transcriptional repressors, exhibit residency at actively transcribed loci during development (22). In addition, transcriptome analysis following Hdac1/Hdac2 inhibition/inactivation reveals differential gene expression consistent with HDACs having both permissive and repressive roles. We show that Hdac1/Hdac2 restores basal chromatin conformation of active loci by pruning activating histone marks deposited during transcription. Further, Hdac1/Hdac2 cooperatively regulates genome-wide levels of H3K23 and H4K16 acetylation, two marks associated with cryptic transcription, with loss of Hdac1/Hdac2 increasing cryptic transcription. These data, and previous literature reports, establish Hdac1/Hdac2 with duals roles, first their classically defined repressive role and now novel function as repressors of cryptic transcription.
Our study highlights the integration of epigenetics, transcriptional fidelity, and metabolism during murine cardiogenesis. Early in development, the PHT sustains contraction through anaerobic metabolism (1, 12). Between E9.5 and E11.5, the mPTP closes, establishing an electrochemical gradient for oxidative energy production (3, 13). Failure is associated with lethality, positioning E10.5 as a checkpoint of cardiac bioenergetic maturation (12, 14). Abnormally thinned myocardium, down-regulated metabolic genes, and lethality between E10.5 and E11.5 in Nkx2.5;1KO2KO suggest a failure of this checkpoint. In addition, cytoplasmic accumulations of lipid droplets and abnormal mitochondrial morphology are consistent with a failed transition. Mechanistically, Hdac1/Hdac2 maintains transcriptional fidelity of gene programs required for the mitochondrial biogenesis, including all complexes of the electron transport chain. Overall, this study identifies Hdac1/Hdac2 as regulators of cardiac bioenergetic maturity, canonical initiation, and cryptic transcription during mammalian cardiogenesis.
MATERIALS AND METHODS
Animals
Nkx2.5IRES-Cre (stock#: 024637), ROSAmT/mG (stock#: 007576), αMHCMerCreMer (stock#: 005657), and R26RLacZ (stock#: 003474) were obtained from the Jackson laboratory. Hdac1Flox, Hdac2Flox, and CMVCre(ERT2) mice used in this study have been previously described (11). All animal protocols were approved by the University of Massachusetts Medical School Institutional Animal Care and Use Committee. Embryonic samples were generated via timed mating. Following the identification of a vaginal plug, pregnant females were isolated and euthanized, and embryos were dissected at the appropriate embryonic time point (ranging from 9.5 to 11.5 days after fertilization). Mice were then separated into double knockout and control groups according to their genotype and developmental stage.
Antibodies and reagents
The following antibodies were used in this study: Hdac1 (Abcam), Hdac2 (Thermo-Fisher), cardiac troponin [cTnT; Developmental Studies Hybridoma Bank (DSHB)], MYH (Santa Cruz), ATP5A (Abcam), SLC25A24 (DSHB), GOT2 (DSHB), H3K36me3 (Abcam), H3K23ac (Abcam), H3K14ac (Active Motif), H4K16ac (Abcam), RNA polymerase II (Epigentek), and nonimmune immunoglobulin G (IgG; Epigentek). Eosin Y and Harris modified hematoxylin were obtained from Fisher, while X-gal was purchased from 5-Prime. Vectashield permanent mounting medium, Vectashield fluorescent mounting medium, Vectashield Elite ABC reagent kit, Vector TrueVIEW Autofluorescence Quenching Kit, and DAB Peroxidase Substrate kit were purchased from Vector Labs. ProLong Glass Antifade Mounting Medium was purchased from Thermo Fisher Scientific. Donkey serum and horse serum were obtained from Sigma. RNeasy Plus Micro, QIAquick Gel Extraction, and DNeasy Blood and Tissue kits were obtained from Qiagen. Chromaflash High Sensitivity ChIP Kit was from Epigentek. SMARTer RACE 5′/3′ kit was from Takara Bio.
Histology
Tissue samples were collected, fixed in 4% paraformaldehyde in 1× phosphate-buffered saline (PBS) at 4°C overnight, dehydrated through an ethanol gradient, embedded in paraffin, and sectioned at 8-μm thickness on a Leica fully motorized rotary microtome.
Imaging
Images of dissected embryos were captured on a Leica Mz10 F fluorescence stereomicroscopy via a 0.7× C-mount, Achromat 1.0 x 90–mm objective, SOLA light engine (Lumencore), and DS-Fi1 color camera (Nikon) through NIS-Elements Basic Research Software (Nikon). Images of stained sections were captures using a Nikon Eclipse 80i microscope via CFI Plan Fluro 4×/10×/20×/40× objective lenses, SOLA light engine, DS-Fi1 color camera, and NIS-Elements Basic Research Software. Confocal microscopy images were acquired using a Leica TCS SP5 II Laser Scanning Confocal microscope. Alexa Fluor 488 and 568 were simultaneously excited at 488 and 561 nm with confocal lasers, respectively. Emissions were split by an Main Beam Splitter 488/561/633 and captured with two detection ranges (ch1, 493 to 536 nm; ch2, 576 to 685 nm). For nuclear staining, Hoechst was excited using a Chameleon Ti:sapphire pulse laser (755 nm) (Coherent Inc.) and was emission detected at 387 to 486 nm. Image stacks of vertical projections were assembled using ImageJ software (National Institutes of Health). Additional confocal images were acquired with a Zeiss LSM800 airyscan confocal scanning microscope equipped with a solid state laser module with 405-, 488-, 561-, and 640-nm beam splitter, Plan-Apochromat objectives with DIC (63×/1.4 oil, 20×/0.8, and 10×/0.45), and Zeiss’s Zen 2.5 imaging software. All analyses were performed on at least three biologically independent samples (n = 3).
Hematoxylin and eosin staining
Staining was performed via deparafanizing sections in xylenes, rehydrating with an ethanol gradient, a 30-s stain with 30% Harris modified hematoxylin (in diH2O) followed by a 30-s counterstain with eosin Y. Stained slides were rinsed and then dehydrated through an ethanol gradient, cleared with xylenes, and mounted using Vectashield permanent mounting medium. All analyses were performed on at least three biologically independent samples (n = 3).
LacZ (β-gal) staining
Samples for LacZ (β-gal) staining were quickly dissected in cold PBS and fixed in 2% paraformaldehyde for 30 min at 4°C. Samples were then washed in PBS at room temperature and stained overnight in staining solution (5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM MgCl2, 0.01% deoxycholic acid, 0.04% NP-40, 0.1% X-gal, in 1× PBS) in the dark at 37°C or until staining developed. Samples were then washed in 1× PBS and fixed overnight in 4% paraformaldehyde in PBS at 4°C. All analyses were performed on at least three biologically independent samples (n = 3).
Immunohistochemistry
Selected sections were deparaffinized in xylenes, rehydrated through an ethanol gradient, pretreated using the Aptum Antigen Retriever 2100 and Aptum R-buffer A or sodium citrate buffer (pH 6.0). Sections for immunofluorescence staining were blocked with 10% donkey serum/0.3% Triton X-100 in PBS for 1 hour at room temperature. Sections were then washed with PBS and probed with primary antibody (in 10% donkey serum in PBS) overnight at 4°C. Subsequently, slides were washed with PBS and incubated with Alexa Fluor 488/546/568–conjugated secondary antibodies (1:500) and Hoechst nuclear counterstain (1:1000) at room temperature for 1 hour. Following incubation, slides were rinsed, quenched with Vector TrueVIEW Autofluroescence quenching kit according to the manufacturer’s guidelines, rinsed again, and mounted with either Vectashield fluorescent mounting medium or ProLong Glass Antifade mountant. All analyses were performed on at least three biologically independent samples (n = 3).
Morphometric analysis
Quantitative morphologic analysis was performed using ImageJ software to measure and record anatomical features of interest. Primitive ventricular thickness was estimated by comparing the average of four serial measurements on each section of interest across 10 independent biologic samples from both genotypes of interest (n = 10). Raw pixel values were converted to a micron scale according to a conversion factor related to the microscope objective/imaging parameters used.
Quantification of immunohistochemistry/immunofluorescence
Cell counting was performed using ImageJ software to measure and record staining points of interest. The ImageJ Cell Counter plugin was used to record the total number of cardiomyocyte nuclei (cTnT+ or MYH+) in each section and the number of double-positive nuclei (cTnT+ or MYH+ AND CC3+ or PHH3+). The number of double-positive nuclei per 100 cardiomyocyte nuclei is presented as either apoptotic index (CC3) or proliferative index (PHH3). All analyses were performed with 13 to 16 biologically independent samples (n = 13 to 16). Control genotypes include 1++2++, 1FF2FF, Nkx2.5;1Het2Het, and Nkx2.5;1Het2KO.
Transcriptome analysis
PHTs were isolated from E10.5 mouse embryos and snap frozen in liquid nitrogen before being stored at −80°C. RNA was extracted from pools of 20 1FF2FF and Nkx1.5;1KO2KO hearts using a Qiagen RNeasy Plus Micro kit (n = 20). The Clariom D Pico Assay (Affymetrix) was performed in triplicate (n = 3) from pooled samples of 20 E10.5 PHTs by the University of Massachusetts Genomics Core Facility. Raw data were analyzed using the Affymetrix Transcriptome Analysis Console with a significant cutoff of false discovery rate (FDR) <0.05. Data were further analyzed using Database for Annotation, Visualization and Integrated Discovery (DAVID) Functional Annotations Bioinformatics Microarray Analysis, PANTHER Gene List Analysis, and Broad Institute Gene Set Enrichment Analysis (GSEA) using default settings. Spearman hierarchical clustering was performed to visualize interrelationships within our data. Lists of enriched ontologic categories were generated and used to create heatmaps, enrichment plots, and doughnut charts in GraphPad Prism. Heatmaps were generated using GraphPad Prism. Relative transcript expression is reported as the log-transformed expression value normalized to the median across all six samples.
Transmission electron microscopy
PHTs from E10.5 mouse embryos were dissected in cold 1× PBS and immediately immersed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2) and fixed overnight at 4°C. Samples were processed and analyzed at the University of Massachusetts Medical School Core Electron Microscopy Facility according to standard procedures. Briefly, after fixation, the samples were rinsed three times in the same fixation buffer and postfixed with 1% osmium tetroxide for 1 hour at room temperature. Samples were then washed three times with diH2O for 20 min and dehydrated through a graded ethanol series of 20% increments before two changes in 100% ethanol. The samples were then infiltrated first with two changes of 100% propylene oxide and then with 50/50% propylene oxide/SPI-Pon 812 resin. The following day, six changes of fresh 100% SPI-Pon 812 resin were performed before samples were polymerized at 68°C in flat embedding molds. The samples were then reoriented as to obtain cross sections of the heart tubes, and 1-μm sections were cut and stained with toluidine blue to confirm orientation. Approximately 70-nm sections of this area were collected and placed on copper support grids and contrasted with lead citrate and uranyl acetate. Sections were then examined using a FEI Tecnai 12 TEM with 120-kV accelerating voltage, and images were captured using a Gatan TEM charge-coupled device camera. All analyses were performed in triplicate (n = 3).
Mitochondrial DNA/nuclear DNA Quantification
PHTs were isolated from E10.5 mouse embryos and snap frozen in liquid nitrogen before being stored at −80°C. Total DNA was extracted from a pool of five biologically independent 1FF2FF and Nkx2.5;1KO2KO hearts (n = 5) using a Qiagen DNeasy Blood and Tissue kit (n = 5). Relative levels of mitochondrial (mtDNA) and nuclear DNA (nDNA) were quantified by quantitative polymerase chain reaction (PCR) using SYBR Green Master Mix and previously reported primers (below). mtDNA levels were normalized to nDNA levels and then compared between 1FF2FF and Nkx2.5;1KO2KO samples. Primer sequences are included in the Supplementary Materials.
Rapid amplification of cDNA ends
5′RACE experiments were performed using a Takara Bio SMARTer 5′RACE and 3′RACE Kit according to manufacturer guidelines. Briefly, PHTs were isolated from E10.5 mouse embryos and snap frozen in liquid nitrogen before being stored at −80°C. 1FF2FF and Nkx2.5;1KO2KO total RNA was isolated using a Qiagen RNeasy Plus Micro Kit from pools of three biologically independent samples (n = 3), and first-strand cDNA was synthesized using reagents and protocols provided by the kit. Using primers complementary to sequences in distal exons of key metabolic genes (below) and reagents provided in the kit, 5′RACE was performed. Products were resolved in a 1% tris-acetate-EDTA (TAE)/ethidium bromide gel, extracted using a QIAquick Gel Extraction Kit (Qiagen), and sequenced (Eurofins Genomics). Specificity and alignment of sequences were determined using MacVector. Primer sequences are included in the Supplementary Materials.
Chromatin immunoprecipitation quantitative PCR
Chromatin immunoprecipitation quantitative PCR (ChIP-qPCR) experiments were performed using an Epigentek ChromaFlash High Sensitivity ChIP Kit (Epigentek) according to the manufacturer’s guidelines. Briefly, 1+/+2+/+ (WT) and Nkx2.5;1KO2KO (DKO) PHTs were isolated from E10.5 mouse embryos and snap frozen in liquid nitrogen before storage at −80°C. Biologically independent samples were pooled (n = 7 to 10), briefly fixed in 1% formaldehyde in 1× PBS at room temperature for 30 min before being quenched with glycine for 5 min, and washed with 1× PBS. Chromatin fragmentation was performed in lysis buffer (Epigentek) using a Covaris M220 (two cycles of the following: peak power, 50; duty factor, 20; cycles/burst, 200; duration, 3 min). Proteins were immunoprecipitated from 0.7 to 2 μg of chromatin in ChIP buffer using Hdac1, Hdac2, H3K36me3, H3K23ac, H3K14ac, H4K16ac, RNA polymerase II, and nonimmune IgG antibodies on an orbital shaker (100 rpm) at room temperature for 1 hour before being transferred to 4°C overnight. Immunoprecipitated antibody-chromatin complexes were washed four times with the provided wash buffer, treated with RNase A, proteinase K, and eluted using elution buffer. DNA was further purified using the included purification columns and protocol. Using purified immunoprecipitated DNA, enrichment of the targets at various locations were measured using qPCR with SYBR Green Master Mix and the primers. Primer sequences are included in the Supplementary Materials.
Quantitative reverse transcription PCR
PHTs were isolated from E10.5 mouse embryos and snap frozen in liquid nitrogen before being stored at −80°C. Total RNA was isolated from pools of 10 biologically independent 1FF2FF and Nkx2.5;1KO2KO samples (n = 10) using a Qiagen RNeasy Plus Micro kit, quantified, and identical amounts of cDNA were generated via reverse transcription using iScript reverse transcription supermix (Bio-Rad) according to the manufacturer’s guidelines. Transcription expression was measured by qPCR using SYBR Green Master Mix. Relative expression levels were calculated using the ∆∆Ct method using glyceraldehyde-3-phosphate dehydrogenase (Gapdh) (unchanged in our transcriptome analysis) as a reference transcript. Primer sequences are included in the Supplementary Materials.
Cardiomyocyte isolation
Cardiomyocytes were prepared from P0 1FF;2FF hearts of approximately 10 mouse pups. Ventricular tissues were predigested in trypsin (0.5 mg/ml; Sigma)–Hank’s balanced salt solution without Ca2+ at 4°C for 30 min with constant shaking. Cells were dissociated by four rounds of 4-min digestion with purified type II collagenase (Gibco) in Hank’s balanced salt solution at 37°C. Fibroblasts were removed from the cell suspension by two rounds of 1-hour differential plating at 37°C in Dulbecco’s modified Eagle medium/M199 (4:1 ratio) medium containing 10% horse serum, 5% fetal bovine serum, penicillin (100 U/ml), streptomycin (100 μg/ml), and Hepes (25 mM) in the presence of 5% CO2. Myocytes were cultured on fibronectin-coated (Sigma) tissue culture plates.
Oxygen consumption rate
Oxygen consumption was measured using a Seahorse XFe96 Analyzer at 25°C according to the manufacturer’s protocol. In brief, isolated cardiomyocytes from P0 1FF2FF hearts were equally plated into each well of a 96-well microplate containing 200 μl of M9 buffer and infected with control or Cre-expressing viral media supplemented with polybrene reagent (10 μg/ml). After 48 hours, basal respiration was measured for a total of 30 min, in 6-min intervals that included a 2-min mix, a 2-min time delay, and a 2-min measurement.
CUT&RUN
CUT&RUN was performed as previously described with the following modifications (23–25). Nuclei were isolated from 24 control and Nkx2.5; 1FF;2FF E10.5 heart tubes. Briefly, frozen tissues were dissociated with a Dounce homogenizer using swelling buffer (10 mM Hepes, 2 mM MgCl2, 3 mM CaCl2) followed by lysis buffer (10 mM Hepes, 2 mM MgCl2, 3 mM CaCl2, 10% glycerol, 1% IGEPAL). The isolated nuclei were washed twice in PBS and resuspended in wash buffer (20 mM Hepes, pH 7.5, 150 mM NaCl, 0.5 mM spermidine, protease inhibitors). Concanavalin A–coated beads were washed twice in 1 ml of binding buffer (20 mM Hepes-KOH, pH 7.9, 10 mM KCl, 1 mM CaCl2, 1 mM MnCl2) and resuspended in binding buffer equal to the volume of bead slurry (10 μl per sample) before they were added to the cells. Bead-bound cells were incubated with 6 μg of H4K16ac antibody (Abcam) and IgG (Abcam) in 150 μl of antibody buffer (wash buffer, 2 mM EDTA) overnight at 4°C. Protein A–micrococcal nuclease was added at a concentration of 700 ng/ml in wash buffer and incubated at 4°C for 1 hour. Chromatin cleavage was initiated after incubation with 1 ml of low-salt rinse buffer (0.5 mM spermidine, 20 mM Hepes, pH 7.5) using 10 mM CaCl2-containing incubation buffer [10 mM CaCl2, 3.5 mM Hepes, pH 7.5) at 0°C for 30 min. Digestion was stopped with the addition of 200 μl of stop buffer [200 mM NaCl, 20 mM EDTA, 4 mM EGTA, RNase A (50 μg/ml), glycogen (40 μg/ml), yeast spike-in DNA (10 pg/ml)]. Digested fragments were released by incubating at 37°C for 30 min. DNA was purified using phenol/chloroform extraction followed by ethanol precipitation. Cut&Run library preparation included Illumina paired-end adaptor ligation and amplification. In all cases, library quality and concentration were determined using Fragment Analyzer (UMASS Medical School Deep Sequencing Core). Deep sequencing of H4K16ac libraries were performed by UMASS Medical School Deep Sequencing Core.
Cryptic event identification and volcano plot
We considered all transcripts with a fold change FDR <0.05 and determined the presence of alternative transcripts from intragenic transcription start sites (cryptic event) by comparing the exon-level probe signals in DKO and Cntrl transcripts. A cryptic event was called when a single transcript had a significantly decreased expression of the first probe and no difference between a later probe in DKO compared to Cntrl. We hypothesized that this transcriptional signature of distal normalization was consistent with intragenic transcript generation. Using this list of transcripts with cryptic events, we created a volcano plot of log2(fold change) versus −log2(FDR) and performed gene ontology using PANTHER Gene List Analysis.
Statistical analysis
Data are shown as means with error bars representing the SD [mean(SD)]. Significant differences between means were determined using an unpaired two-tailed Student’s t test, the Holm-Sidak method, or χ2 test. The level of significance (α) was set at 0.05 for all analyses. Per GraphPad Prism standard annotation: ns, P > 0.05, *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Analysis was performed using either Microsoft Excel or GraphPad Prism 8.
Supplementary Material
Acknowledgments
Funding: This work was supported by R01HL118100 (to C.M.T.) and R01AG047182 (to C.M.H.). Author contributions: C.M.T. conceived the study. Z.J.M. and C.M.T. designed experiments and wrote the manuscript. Z.J.M., S.S., L.M.B., and C.M.T. performed experiments and acquired and analyzed the data. T.S. and C.M.H. contributed reagents, methodology, and analysis tools. All authors reviewed results and approved the final version of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/15/eaax5150/DC1
REFERENCES AND NOTES
- 1.Lopaschuk G. D., Jaswal J. S., Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharmacol. 56, 130–140 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Teekakirikul P., Kelly M. A., Rehm H. L., Lakdawala N. K., Funke B. H., Inherited cardiomyopathies: Molecular genetics and clinical genetic testing in the postgenomic era. J. Mol. Diagn. 15, 158–170 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Porter G. A. Jr., Hom J., Hoffman D., Quintanilla R., Bentley K. d. M., Sheu S.-S., Bioenergetics, mitochondria, and cardiac myocyte differentiation. Prog. Pediatr. Cardiol. 31, 75–81 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scaglia F., Towbin J. A., Craigen W. J., Belmont J. W., Smith E. O., Neish S. R., Ware S. M., Hunter J. V., Fernbach S. D., Vladutiu G. D., Wong L.-J. C., Vogel H., Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics 114, 925–931 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Dorn G. W. II, Vega R. B., Kelly D. P., Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 29, 1981–1991 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berger S. L., Sassone-Corsi P., Metabolic signaling to chromatin. Cold Spring Harb. Perspect. Biol. 8, a019463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wellen K. E., Hatzivassiliou G., Sachdeva U. M., Bui T. V., Cross J. R., Thompson C. B., ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nakahata Y., Sahar S., Astarita G., Kaluzova M., Sassone-Corsi P., Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 324, 654–657 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moser M. A., Hagelkruys A., Seiser C., Transcription and beyond: The role of mammalian class I lysine deacetylases. Chromosoma 123, 67–78 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stanley E. G., Biben C., Elefanty A., Barnett L., Koentgen F., Robb L., Harvey R. P., Efficient Cre-mediated deletion in cardiac progenitor cells conferred by a 3'UTR-ires-Cre allele of the homeobox gene Nkx2-5. Int. J. Dev. Biol. 46, 431–439 (2002). [PubMed] [Google Scholar]
- 11.Milstone Z. J., Lawson G., Trivedi C. M., Histone deacetylase 1 and 2 are essential for murine neural crest proliferation, pharyngeal arch development, and craniofacial morphogenesis. Dev. Dyn. 246, 1015–1026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vega R. B., Horton J. L., Kelly D. P., Maintaining ancient organelles: Mitochondrial biogenesis and maturation. Circ. Res. 116, 1820–1834 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hom J. R., Quintanilla R. A., Hoffman D. L., de Mesy Bentley K. L., Molkentin J. D., Sheu S.-S., Porter G. A. Jr., The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Dev. Cell 21, 469–478 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gómez-Del Arco P., Perdiguero E., Yunes-Leites P. S., Acín-Pérez R., Zeini M., Garcia-Gomez A., Sreenivasan K., Jiménez-Alcázar M., Segalés J., López-Maderuelo D., Ornés B., Jiménez-Borreguero L. J., D’Amato G., Enshell-Seijffers D., Morgan B., Georgopoulos K., Islam A. B. M. M. K., Braun T., de la Pompa J. L., Kim J., Enriquez J. A., Ballestar E., Muñoz-Cánoves P., Redondo J. M., The chromatin remodeling complex Chd4/NuRD controls striated muscle identity and metabolic homeostasis. Cell Metab. 23, 881–892 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Smolle M., Workman J. L., Transcription-associated histone modifications and cryptic transcription. Biochim. Biophys. Acta 1829, 84–97 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berretta J., Morillon A., Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep. 10, 973–982 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brocks D., Schmidt C. R., Daskalakis M., Jang H. S., Shah N. M., Li D., Li J., Zhang B., Hou Y., Laudato S., Lipka D. B., Schott J., Bierhoff H., Assenov Y., Helf M., Ressnerova A., Islam M. S., Lindroth A. M., Haas S., Essers M., Imbusch C. D., Brors B., Oehme I., Witt O., Lübbert M., Mallm J.-P., Rippe K., Will R., Weichenhan D., Stoecklin G., Gerhäuser C., Oakes C. C., Wang T., Plass C., DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat. Genet. 49, 1052–1060 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li B., Gogol M., Carey M., Lee D., Seidel C., Workman J. L., Combined action of PHD and chromo domains directs the Rpd3S HDAC to transcribed chromatin. Science 316, 1050–1054 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Keogh M.-C., Kurdistani S. K., Morris S. A., Ahn S. H., Podolny V., Collins S. R., Schuldiner M., Chin K., Punna T., Thompson N. J., Boone C., Emili A., Weissman J. S., Hughes T. R., Strahl B. D., Grunstein M., Greenblatt J. F., Buratowski S., Krogan N. J., Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 123, 593–605 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Carrozza M. J., Li B., Florens L., Suganuma T., Swanson S. K., Lee K. K., Shia W.-J., Anderson S., Yates J., Washburn M. P., Workman J. L., Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123, 581–592 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Sen P., Dang W., Donahue G., Dai J., Dorsey J., Cao X., Liu W., Cao K., Perry R., Lee J. Y., Wasko B. M., Carr D. T., He C., Robison B., Wagner J., Gregory B. D., Kaeberlein M., Kennedy B. K., Boeke J. D., Berger S. L., H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 29, 1362–1376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z., Zang C., Cui K., Schones D. E., Barski A., Peng W., Zhao K., Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138, 1019–1031 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meers M. P., Bryson T. D., Henikoff J. G., Henikoff S., Improved CUT&RUN chromatin profiling tools. eLife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meers M. P., Tenenbaum D., Henikoff S., Peak calling by sparse enrichment analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin 12, 42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skene P. J., Henikoff S., An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, e21856 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/15/eaax5150/DC1