

Staphylococcus aureus is an important pathogenic bacterium causing various diseases in humans. In the bacterium, transcription is typically regulated by the transcription factors located in the cytoplasm. In this study, we report an atypical transcription factor identified in S. aureus. Unlike most other transcription factors, the newly identified transcription factor is located in the cytoplasmic membrane, and its activity is proteolytically controlled by the membrane-bound AAA+ protease FtsH. The newly identified MTF, named MbtS, has the potential to regulate the transcription of over 200 genes. This study provides a molecular mechanism by which a protease affects bacterial transcription and illustrates the diversity of the bacterial transcriptional regulation.
KEYWORDS: Staphylococcus aureus, membrane localization, proteases, transcription factors
ABSTRACT
In bacteria, chromosomal DNA resides in the cytoplasm, and most transcription factors are also found in the cytoplasm. However, some transcription factors, called membrane-bound transcription factors (MTFs), reside in the cytoplasmic membrane. Here, we report the identification of a new MTF in the Gram-positive pathogen Staphylococcus aureus and its regulation by the protease FtsH. The MTF, named MbtS (membrane-bound transcription factor of Staphylococcus aureus), is encoded by SAUSA300_2640 and predicted to have an N-terminal DNA binding domain and three transmembrane helices. The MbtS protein was degraded by membrane vesicles containing FtsH or by the purified FtsH. MbtS bound to an inverted repeat sequence in its promoter region, and the DNA binding was essential for its transcription. Transcriptional comparison between the ftsH deletion mutant and the ftsH mbtS double mutant showed that MbtS could alter the transcription of over 200 genes. Although the MbtS protein was not detected in wild-type (WT) cells grown in a liquid medium, the protein was detected in some isolated colonies on an agar plate. In a murine model of a skin infection, the disruption of mbtS increased the lesion size. Based on these results, we concluded that MbtS is a new S. aureus MTF whose activity is proteolytically regulated by FtsH.
IMPORTANCE Staphylococcus aureus is an important pathogenic bacterium causing various diseases in humans. In the bacterium, transcription is typically regulated by the transcription factors located in the cytoplasm. In this study, we report an atypical transcription factor identified in S. aureus. Unlike most other transcription factors, the newly identified transcription factor is located in the cytoplasmic membrane, and its activity is proteolytically controlled by the membrane-bound AAA+ protease FtsH. The newly identified MTF, named MbtS, has the potential to regulate the transcription of over 200 genes. This study provides a molecular mechanism by which a protease affects bacterial transcription and illustrates the diversity of the bacterial transcriptional regulation.
INTRODUCTION
A typical bacterial chromosome carries several thousand genes, many of which are subjected to regulation of their expression by a class of proteins called transcription factors. Transcription factors are not a component of the RNA polymerase complex and regulate the transcription of the target genes by directly binding to their binding sequences (1). In bacteria, transcription factors typically reside in the cytoplasm, where the chromosome is located. On the other hand, in eukaryotic cells, transcription factors are often found in the membranes, and the release of the transcription factors from the membranes and subsequent nuclear transport are regulated by membrane-bound proteases or proteasomes (2, 3).
Although rare, membrane-bound transcription factors (MTFs) have been reported in Gram-negative bacteria. As integral membrane proteins with a DNA binding domain, their activities are often proteolytically regulated by intramembrane proteases. For example, the Gram-negative bacterium Vibrio cholerae is known to produce three MTFs: ToxR, TcpP, and TfoS. Of those, ToxR and TcpP have a cytoplasmic DNA binding domain and a periplasmic domain connected by a transmembrane helix (4, 5). Both transcription factors are involved in the transcriptional activation of toxT, the gene encoding the master regulator for the production of cholera toxin and toxin-coregulated pili (6, 7). For their function, ToxR and TcpP require an association with the membrane proteins ToxS and TcpH, respectively (7–10). In particular, TcpH protects TcpP from sequential degradation by Tsp and YaeL (also known as RseP) (8, 9). The third MTF, TfoS, is comprised of a periplasmic domain flanked by two transmembrane helices and a C-terminal DNA binding domain (11). In response to the chitin signal, TfoS positively regulates the production of the small RNA TfoR, which in turn increases the translation of the major competence regulator TfoX (11–13). The Gram-negative bacteria Escherichia coli and Salmonella enterica also produce an MTF called CadC (14, 15). Like ToxR and TcpP, CadC has a cytoplasmic DNA binding domain and a periplasmic domain connected by a transmembrane helix (16) and plays critical roles in stress responses (14, 15). To the best of our knowledge, an MTF has not yet been found in Gram-positive bacteria.
Staphylococcus aureus is a Gram-positive pathogenic bacterium causing a wide range of diseases in humans, including skin infections and sepsis (17, 18). In the bacterium, the AAA+ type metalloprotease FtsH is required for stress resistance and virulence (19, 20). FtsH is tethered to the membrane via two transmembrane helices located at the N terminus, and its active sites (i.e., ATPase and protease) reside in the C-terminal cytoplasmic domain (21, 22). Due to its active-site location, FtsH attacks target proteins from the cytoplasm and degrades only cytoplasmic and membrane proteins. For degradation initiation, FtsH does not recognize a specific sequence, and with weak unfoldase activity, it degrades loosely folded or unstructured proteins (21, 23). In S. aureus, FtsH degrades 11 proteins and indirectly represses the transcription of five genes (i.e., hslO, hrtAB, SAUSA300_2637, and SAUSA300_2640) (20). Since FtsH is a protease, not a transcription factor, we speculated that a transcription factor mediates the transcriptional effect of FtsH. Indeed, of the five genes, SAUSA300_2640 encodes a putative transcription factor with a DNA binding domain at the N terminus. Intriguingly, the putative transcription factor has three transmembrane helices and is predicted to reside in the cytoplasmic membrane. In this study, we named this putative MTF MbtS (membrane-bound transcription factor in S. aureus) and characterized its roles in staphylococcal transcription and pathogenesis.
RESULTS
MbtS is a putative transcription factor located in the cytoplasmic membrane.
The mbtS gene is located downstream of an operon consisting of SAUSA300_2641 and _2642 (Fig. 1A). According to the S. aureus transcriptome analysis by Mader et al. (24), the mbtS gene is cotranscribed with SAUSA300_2641 and SAUSA300_2642 from the promoter in front of SAUSA300_2642 (here designated P2642) (Fig. 1A). Therefore, it is likely that the mbtS gene forms an operon with SAUSA300_2641 and SAUSA300_2642 (Fig. 1A). However, a 259-bp noncoding sequence is located upstream of mbtS, raising the possibility that the sequence contains a promoter specific for mbtS (PmbtS in Fig. 1A). Protein sequence analysis predicted that the MbtS protein has a cytoplasmic DNA binding domain at the N terminus, three transmembrane helices, and another, 41-amino-acid (aa) cytoplasmic domain between the second and third transmembrane helices (Fig. 1B).
FIG 1.
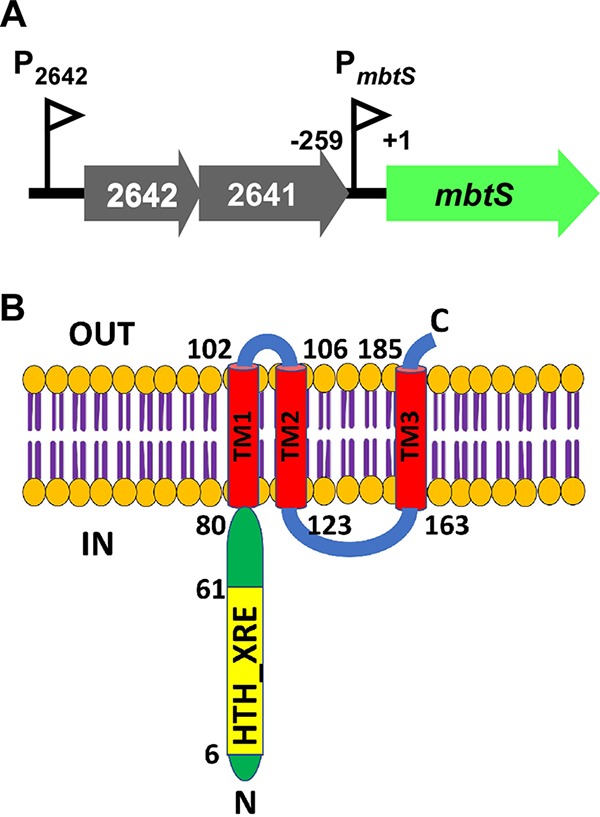
MbtS is a putative membrane-bound transcription factor. (A) Schematic presentation of the operon containing mbtS. Two promoters, PmbtS and P2642, are shown as triangular flags. The numbers indicate the relative nucleotide positions, where the start codon of mbtS is set to +1. (B) Topology of the MbtS protein predicted with SMART (http://smart.embl-heidelberg.de/). The pertinent amino acid positions are indicated. TM, transmembrane helix; N, N terminus; C, C terminus; IN, cytoplasm; OUT, extracytoplasmic region.
The expression of MbtS is negatively regulated by FtsH.
Previously, we have shown that the transcription of mbtS is drastically increased by the deletion of ftsH (20). To confirm the results, we carried out real-time quantitative reverse transcription-PCR (qRT-PCR) analysis for mbtS in the wild-type (WT) and the ftsH deletion mutant (ΔftsH) strains. The transcript level of mbtS was increased more than 40-fold in the ΔftsH mutant compared with the WT, confirming the previous results (WT versus ΔftsH in Fig. 2A). Further transposon insertion in mbtS abolished the transcription of mbtS (ΔftsH::mbtS in Fig. 2A). To detect MbtS, we generated an antibody with a peptide sequence (i.e., R29QSISNWENDKSLP42) in the N-terminal DNA binding domain of MbtS. In a Western blot analysis with the MbtS antibody, MbtS was detected only in the ftsH deletion mutant (Fig. 2B), corroborating the qRT-PCR results. As expected, the level of SaeQ, an FtsH target protein, was increased by the ftsH deletion, whereas the level of SrtA, a non-FtsH target, was not (Fig. 2B).
FIG 2.
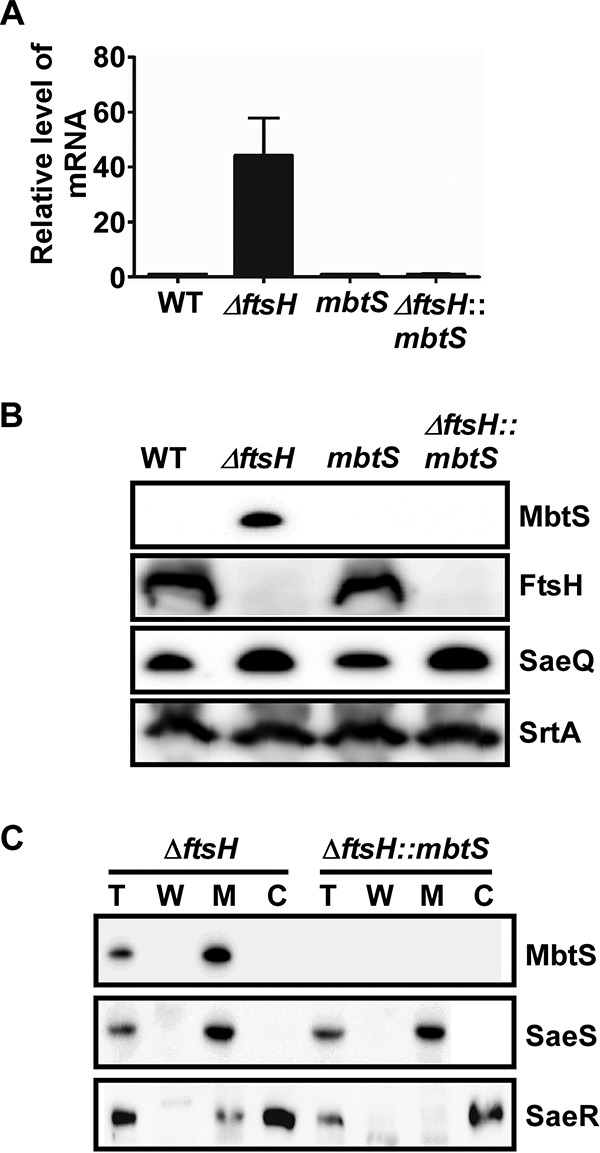
MbtS is a membrane-bound transcription factor regulated by FtsH. (A and B) Effect of the ftsH deletion on the expression of mbtS at the levels of the transcript (A) and the protein (B). The test strains were grown in TSB until the exponential growth phase, and then total RNA and proteins were prepared from whole-cell lysates as detailed in Materials and Methods. The transcript levels were determined by qRT-PCR, whereas the protein levels were determined by Western blotting. In the Western blot analysis, the membrane protein SaeQ, an FtsH substrate, was used to monitor the FtsH activity, whereas the membrane protein SrtA (sortase A), a non-FtsH substrate, was used as a negative control. WT, wild-type USA300; ΔftsH, ftsH deletion mutant; mbtS, USA300 with a transposon insertion at mbtS; ΔftsH::mbtS, ftsH deletion mutant with a transposon insertion at mbtS. The original Western blots are provided in Fig. S1 in the supplemental material. (C) Membrane localization of MbtS. Cells were grown until exponential growth phase, collected by centrifugation, and converted into protoplasts by lysostaphin treatment in the presence of 0.5 M sucrose. The supernatant was designated the cell wall fraction (W). The protoplasts were further lysed and subjected to ultracentrifugation, and the supernatant was designated the cytoplasm (C), whereas the pellet was washed and designated the membrane (M). The proteins were detected by Western blotting with cognate antibodies. The detected proteins are indicated on the right side of the blot. The sensor histidine kinase SaeS, a membrane protein, and the response regulator SaeR, a cytoplasmic protein, were used as fractionation controls. Sometimes a low level of SaeR is detected in the membrane fraction, possibly due to its interaction with SaeS. T, total protein.
To confirm the membrane localization of MbtS, we carried out cellular fractionation and Western blot analysis with the MbtS antibody. As predicted by protein sequence analysis (Fig. 1B), MbtS was detected only in the membrane (MbtS in Fig. 2C). Based on these results, we concluded that MbtS is a putative MTF whose expression is negatively regulated by FtsH.
MbtS is degraded by FtsH.
FtsH is a protease, not a transcription factor. To explain the transcriptional repression of mbtS by FtsH (Fig. 2A), we hypothesized that MbtS is an autoregulator directly degraded by FtsH. To examine the hypothesis, first we tested whether FtsH directly degrades MbtS. In the test, the membranes from the FtsH-overexpressing strain were mixed with the membranes from the ΔftsH mutant, which contains a high level of MbtS (Fig. 2B). In the presence of nonionic detergents and ATP, the full-length MbtS protein gradually degraded into two smaller fragments with apparent molecular weights of 16.6 kDa and 13.6 kDa, respectively (+ FtsH in Fig. 3A; see Fig. S2A in the supplemental material). In contrast, the membrane protein SrtA, which is not an FtsH substrate, was not affected. When only the MbtS-containing membranes were incubated, MbtS was not degraded (− FtsH in Fig. 3A), demonstrating the essential role of FtsH in the degradation of MbtS.
FIG 3.
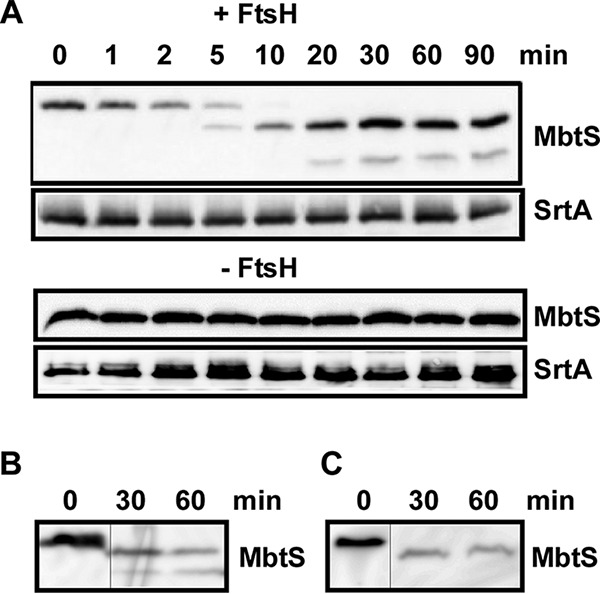
MbtS is degraded by FtsH. (A) Degradation of MbtS by membranes containing FtsH. Membranes containing MbtS were mixed with membranes containing FtsH (+ FtsH) or reaction buffer (− FtsH) in the presence of nonionic detergents. The degradation of MbtS was monitored by Western blotting. Sortase A (SrtA), a non-FtsH substrate located in the membrane, was used as a negative control. (B and C) Degradation of MbtS by purified FtsH. Strep-tagged FtsH was purified from E. coli and mixed with membranes (B) or cell lysate (C) containing MbtS. The degradation of MbtS was monitored by Western blotting. The original blots are provided in Fig. S2 in the supplemental material. The thin lines indicate the splicing point.
To provide FtsH in the protein degradation analysis, we used membranes containing FtsH, not purified FtsH. Therefore, we cannot rule out the possibility that other proteases embedded in the membrane are responsible for the MbtS degradation. To rule out the possibility, we overexpressed Strep-tagged FtsH (FtsH-Strep) in E. coli and purified the protein in the presence of nonionic detergent (see Materials and Methods). When the purified FtsH-Strep was mixed with the membranes containing MbtS from the ΔftsH mutant, MbtS was again degraded into two smaller fragments (Fig. 3B). Intriguingly, when FtsH-Strep was mixed with cell lysate of the ΔftsH mutant, the 13.6-kDa fragment was not observed (Fig. 3C), implying that it might be further degraded by proteases in the cell lysate. Nonetheless, these results demonstrate that MbtS is a new substrate of FtsH and is degraded by the protease.
MbtS recognizes an inverted repeat in its own promoter.
Since FtsH appears to degrade MbtS directly, next we examined whether MbtS autoregulates its transcription. Previously, we showed that the ftsH deletion increases the transcription of mbtS but not the transcription of either SAUSA300_2641 or SAUSA300_2642 (20). Therefore, it is likely that the PmbtS region contains a cis element that responds specifically to MbtS (Fig. 1A). To test this possibility, we made a PmbtS-lacZ fusion in pOS1 and inserted the fusion plasmid into the WT and ΔftsH strains. In the LacZ assay, the enzyme activity was significantly higher in the ΔftsH mutant than in the WT (Fig. 4A). More importantly, when the fusion plasmid was inserted into the ΔftsH::mbtS double mutant, the LacZ activity was completely abolished (ΔftsH::mbtS in Fig. 4A), confirming that MbtS positively autoregulates its own transcription.
FIG 4.

MbtS recognizes an inverted repeat in its own promoter. (A) Requirement of MbtS for the promoter activity of PmbtS. The PmbtS-lacZ fusion plasmid pOS1-PmbtS-lacZ was inserted into the test strains. Overnight cultures of the test strains were used for the LacZ assay. WT, wild-type strain USA300; ΔftsH, ftsH deletion mutant; ΔftsH::mbtS, ΔftsH with a transposon insertion in mbtS. MU, Miller units. The experiment was carried out in triplicate and repeated twice with similar results. (B) Mutational analyses of PmbtS. The PmbtS sequence in pOS1-PmbtS-lacZ was randomly mutated by error-prone PCR. The 105 mutations identified in 43 white colonies were plotted in PmbtS. On the x axis, −1 indicates the upstream nucleotide right next to the ATG start codon. The y axis shows the number of mutations identified. Two regions, −50/−40 and −100/−80, contained a noticeably higher number of mutations than other regions. Two red bars indicate the locations of two single mutations that abolished the PmbtS activity. The bars under the graph show the PmbtS sequence remaining in the deletion mutant plasmids (Δ1 to Δ5). The putative promoter elements (P−35 and P−10) and the 18-bp sequence containing an inverted repeat are indicated in green. The red Ts are the nucleotides corresponding to the red bar in the graph above. (C) LacZ activities of the five deletion mutants of PmbtS. WT, wild-type USA300; ΔftsH, ftsH deletion mutant of USA300; −, no mutation. (D) LacZ activities of the strains carrying the Δ5 plasmid with a point mutation in the putative MbtS binding sequence. WT, wild-type strain RN4220; ftsH, RN4220 carrying a transposon insertion in ftsH (RN4220::ftsH). The nucleotides in the inverted repeat are shown in red. The mutants indicated by m1 to m5 were used for further experiments (Fig. 5). ND, not determined.
To identify the cis element required for the positive autoregulation, we carried out a random mutagenesis analysis. In the analysis, the PmbtS sequence was mutated by error-prone DNA polymerases and fused with lacZ in the multicopy plasmid pOS1. The resulting pOS1-PmbtS-lacZ plasmids were inserted into the ftsH deletion mutant of USA300, which was subsequently spread on an agar plate containing X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside). From 43 white colonies on the plates, the mutant plasmids were purified, and the PmbtS region was sequenced to identify 105 mutations (∼2.4 mutations/colony). Although most plasmids contained multiple mutations (2 to 7), four plasmids had only one mutation, either in position −41 (three mutants, T/C transition) or in position −85 (one mutant, deletion of T) (red bars and red T nucleotides in Fig. 4B). When all mutations were mapped and graphed in the PmbtS region, a high number of mutations were identified in two regions (the −50/−40 and −100/−80 regions in Fig. 4B).
To narrow down the cis element further, we shortened the PmbtS sequence by 60 nucleotides (nt) (Δ1), 120 nt (Δ2), or 180 nt (Δ3) and fused the DNA fragments to lacZ in pOS1 (Fig. 4B). The mutant plasmids were inserted into the ftsH deletion mutant of USA300, and the LacZ activities of the mutant strains were measured. Although the strains containing the Δ1 and Δ2 mutant plasmids showed a wild-type LacZ activity, the strain carrying the Δ3 plasmid showed almost no activity (Fig. 4C), suggesting that the PmbtS sequence missing in Δ3, compared with Δ2, contains the cis element responding to MbtS.
To identify the cis element, we extended the PmbtS sequence in the Δ3 plasmid by 17 nt (Δ4) or 27 nt (Δ5) (Fig. 4B). In the ftsH deletion mutant of USA300, the 17-bp extension restored 10% of the LacZ activity, compared with that of the full-length PmbtS (Δ4 in Fig. 4C). Intriguingly, the restored LacZ activity of the Δ4 mutant was decreased by further disruption of mbtS (ΔftsH versus ΔftsH::mbtS in Fig. S3 in the supplemental material), indicating that the PmbtS region in the Δ4 mutant plasmid responds to the MbtS protein, albeit weakly. On the other hand, the 27-bp extension restored 80% of the LacZ activity (Δ5 in Fig. 4C). Inspection of the PmbtS sequence in the Δ5 plasmid identified not only putative −10 and −35 promoter sequences (between positions −41 and −69) but also an 18-bp sequence containing an inverted repeat (between positions −80 and −97) (green sequences in Fig. 4B). Notably, the T at position −97 is missing in the Δ4 mutant, explaining the low PmbtS activity of the mutant.
The identified 18-bp sequence, in particular, the inverted repeat, is likely the cis element serving as a binding site for the MbtS protein. To examine this possibility, we carried out site-directed mutagenesis. Using the Δ5 plasmid, we changed the 18-bp sequence and an additional two nucleotides to their complementary nucleotides (e.g., A to T and C to G). The mutated Δ5 plasmids were inserted into S. aureus RN4220 containing a transposon insertion in ftsH. It should be noted that, despite our multiple trials, we failed to insert one of the mutant plasmids (m3 in Fig. 4D) into the strain, suggesting that the plasmid might be toxic to the strain. As shown, when the nucleotides in the inverted repeat sequence were changed, 62% (8/13) showed markedly lower LacZ activity (red nucleotides in Fig. 4D). In contrast, when other nucleotides, either outside or between the inverted repeat, were mutated, such a marked reduction was not observed (the black TA and ACAT in Fig. 4D). Based on these results, we concluded that the 18-bp sequence, in particular, the inverted repeat sequence, is the cis element required for the positive autoregulation by MbtS, possibly by serving as the binding site for the protein.
MbtS binds to the DNA containing the inverted repeat sequence.
To examine whether the identified inverted repeat sequence is the binding site for MbtS, we carried out biolayer interferometry (BLI) analysis. We biotinylated the 5′ end of the 92-bp DNA (from −48 to −139 in Fig. 4B), which includes the inverted repeat sequence, and used it for a probe. For ligands, cytoplasmic membranes were purified from two different strains: the ΔftsH (for the MbtS protein) and ΔftsH::mbtS (a negative control) strains. The biotin-labeled DNA probe was attached to the streptavidin-coated sensor, and the probe then was immersed in membranes solubilized with nonionic detergents (see Materials and Methods). As shown in Fig. 5A, the DNA probe showed a binding activity only for the MbtS-containing membrane (i.e., ΔftsH), supporting the idea that the inverted repeat sequence is the binding site for MbtS.
FIG 5.
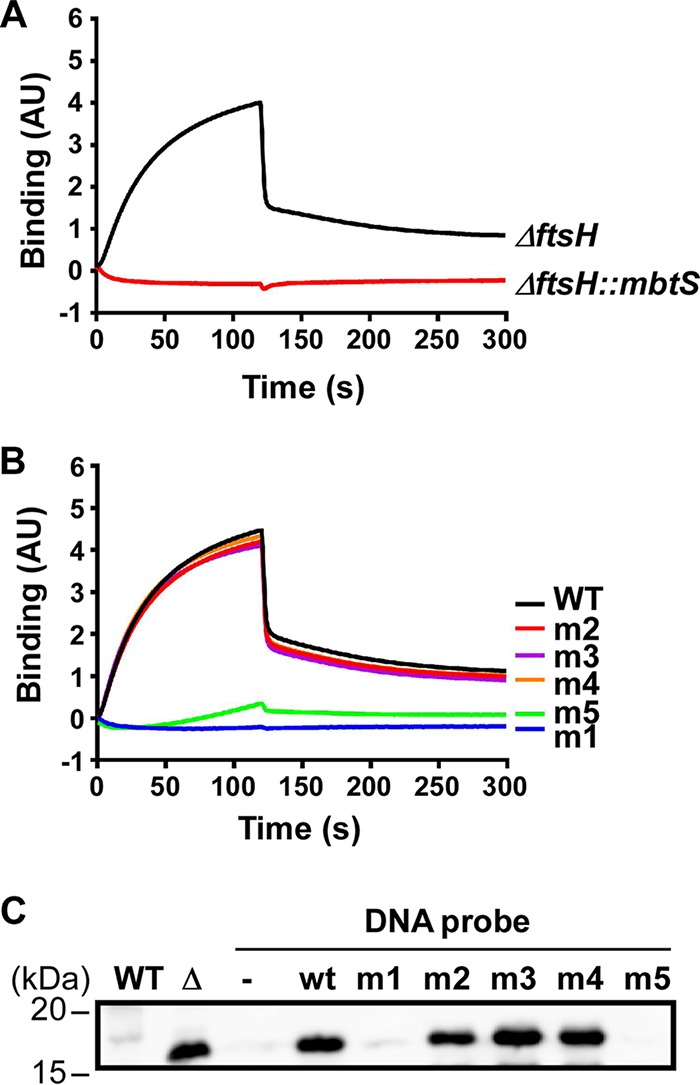
MbtS binds the DNA containing the inverted repeat sequence. (A) Biolayer interferometry (BLI) analysis with a DNA probe containing the inverted repeat sequence and solubilized membranes purified either from the ftsH deletion mutant of USA300 (ΔftsH) or from the ΔftsH mutant with a transposon insertion in mbtS (ΔftsH::mbtS). (B) BLI analysis with membranes containing MbtS and six DNA probes (1 wild-type [WT] and 5 mutants [m1 to m5]). The mutated nucleotides in the mutant probes are shown in Fig. 4D. (C) DNA affinity chromatography to examine MbtS binding to the WT and mutant DNA probes. WT, WT strain RN4220; Δ, RN4220 with a transposon insertion in ftsH; −, no DNA probe; wt, wild-type DNA probe; m1 to m5; mutant DNA probes.
If the inverted repeat sequence is the MbtS binding site, the mutations that abolished the PmbtS activity should abrogate the probe’s binding to MbtS. To test this hypothesis, we prepared one WT DNA probe and five mutant DNA probes. Of the five mutant probes, two contain the mutations that abolished the promoter activity (i.e., m1 and m5), and the other two probes have innocuous mutations (i.e., m2 and m4). The m3 mutant DNA probe was included because we failed to insert the m3 mutant plasmid into the ftsH deletion mutant of RN4220. Each of these six probes was attached to the sensor, and then the sensor was immersed into the solubilized membranes containing the MbtS protein. As can be seen, the two probes with an innocuous mutation (i.e., m2 and m4) and the m3 probe showed WT binding (Fig. 5B). In contrast, probes m1 and m5, whose mutations abolished the PmbtS activity, showed no binding activity (Fig. 5B), supporting the idea that the inverted repeat sequence is the MbtS binding site.
To confirm the MbtS binding to the DNA probes further, we carried out DNA affinity chromatography. We attached the biotin-labeled WT and mutant DNA probes to streptavidin-Sepharose resin and mixed the resin with solubilized membrane containing the MbtS protein. After washing the resins, we eluted the binding proteins with SDS-PAGE sample buffer and examined the presence of MbtS by Western blotting. The MbtS protein was eluted only from the DNA probes that showed binding activity in the BLI assay (i.e., WT, m2, m3, and m4) (Fig. 5C). In contrast, almost no MbtS was eluted from the probes containing the m1 or m5 mutation, which abolished both the PmbtS promoter activity and the binding activity in the BLI assay (Fig. 4D and 5B). Based on these results, we concluded that MbtS binds to the inverted repeat sequence in PmbtS.
MbtS can potentially regulate over 200 genes.
To identify the genes regulated by MbtS, first we compared the transcription profiles of the USA300 WT and an mbtS transposon insertion mutant by transcriptome sequencing (RNA-seq) analysis. However, no significant difference was found, indicating that, under the growth conditions used in our study, the MbtS concentration in the WT strain is too low to affect the gene transcription. Therefore, we repeated the RNA-seq analysis for the ΔftsH and ΔftsH::mbtS strains. With an arbitrary threshold of 2-fold changes, the disruption of mbtS altered the transcription of 17 genes (8 increased and 9 decreased) (Table 1). Notably, of the eight genes whose transcription was increased by the disruption of mtsB, seven encode proteases (Table 1). To confirm the RNA-seq results, we carried out qRT-PCR analysis for five genes (two increased and three decreased). As shown, all five genes showed transcriptional patterns similar to those determined by RNA-seq (splA, 1890, qcvT, pstS, and 2637 in Fig. 6A).
TABLE 1.
Genes whose transcription was affected by disruption of mbtS
| Category and gene ID | Gene name | Product | Fold change | P value |
|---|---|---|---|---|
| Increased | ||||
| SAUSA300_1525 | glyS | Glycyl-tRNA synthetase | 2.2 | 3.4E−97 |
| SAUSA300_1753 | splF | Serine protease SplF | 2.8 | 1.6E−65 |
| SAUSA300_1754 | splE | Serine protease SplE | 3.0 | 4.2E−47 |
| SAUSA300_1755 | splD | Serine protease SplD | 2.7 | 3.3E−40 |
| SAUSA300_1756 | splC | Serine protease SplC | 2.8 | 3.5E−53 |
| SAUSA300_1757 | splB | Serine protease SplB | 2.7 | 2.4E−69 |
| SAUSA300_1758 | splA | Serine protease SplA | 2.7 | 6.5E−53 |
| SAUSA300_1890 | Staphopain A | 2.1 | 4.0E−17 | |
| Decreased | ||||
| SAUSA300_0500 | tRNA Ile | −2.3 | 5.0E−03 | |
| SAUSA300_1283 | pstS | Phosphate ABC transporter, phosphate binding protein PstS | −2.7 | 1.7E−22 |
| SAUSA300_1496 | Glycine dehydrogenase, subunit 2 | −2.2 | 1.0E−137 | |
| SAUSA300_1497 | Glycine dehydrogenase, subunit 1 | −2.4 | 1.2E−118 | |
| SAUSA300_1498 | gcvT | Aminomethyltransferase | −2.5 | 9.7E−90 |
| SAUSA300_1839 | tRNA Ala | −2.8 | 1.7E−04 | |
| SAUSA300_2476 | ptsG | Phosphotransferase system, glucose-specific IIABC component | −2.3 | 2.5E−37 |
| SAUSA300_2637 | Conserved hypothetical protein | −2.6 | 4.1E−12 | |
| SAUSA300_2640 | mbtS | Putative transcriptional regulator | −105.4 | 0.0E+00 |
FIG 6.

Confirmation of the effect of MbtS on the transcription of staphylococcal genes. (A) qRT-PCR analysis for the genes whose transcription was affected by MbtS in RNA-seq. The experiment was carried out in quintuplicate. The analyzed genes are indicated. (B) Effect of the disruption of mbtS on the promoter activity of P2642. The reporter plasmid pOS1-P2642-lacZ was inserted into the test strains, and then the cells were grown overnight and the LacZ activity was measured with ONPG. The experiment was carried out in triplicate and repeated twice with similar results. (C and D) Effect of disruptions of SAUSA300_2641 and SAUSA300_2642 on PmbtS activity (C) and MbtS expression level (D). The reporter plasmid pOS1-PmbtS-lacZ was inserted into the test strains, and the LacZ activity was measured as described above. The proteins were detected by Western blotting. The LacZ assay was carried out in triplicate and repeated twice with similar results. The statistical significance was measured by an unpaired, two-tailed Student t test. **, P < 0.005; ***, P < 0.001; ****, P < 0.0001. WT, wild-type strain USA300; mbtS, USA300 with a transposon insertion in mbtS; ΔftsH, ftsH deletion mutant of USA300; ΔftsH::mbtS; ΔftsH mutant with a transposon insertion in mbtS; 2641, USA300 with a transposon insertion at SAUSA300_2641; 2642, USA300 with a transposon insertion at SAUSA300_2642.
Intriguingly, in the RNA-seq analysis, the transcription of SAUSA300_2641, a gene located right next to mbtS, was increased 1.9-fold (Fig. 1A; see Table S1 in the supplemental material). The qRT-PCR analysis also showed an approximately 5-fold transcription increase of SAUSA300_2641 in the ΔftsH::mbtS strain compared with the ΔftsH strain (2641 in Fig. 6A), raising the possibility that the P2642 promoter is negatively regulated by MbtS (Fig. 1A). Indeed, the promoter activity of P2642 was decreased by the ftsH deletion (i.e., an MbtS-replete condition) but was restored to 77% of the WT level by further disruption of mbtS (ΔftsH versus ΔftsH::mbtS in Fig. 6B), supporting the idea that P2642 is negatively regulated by MbtS. Conversely, the disruption of either SAUSA300_2641 or SAUSA300_2642 reduced the PmbtS promoter activity by approximately 4-fold without affecting the level of the MbtS protein (Fig. 6C and D; see Fig. S4 in the supplemental material), indicating that both the 2641 and 2642 proteins are required for full activity of PmbtS.
In the RNA-seq analysis, SAUSA300_2641 and 2642 were upregulated 1.9- and 1.4-fold, respectively (Table S1). Since P2642 appeared to be negatively regulated by MbtS (Fig. 6B), the 2-fold threshold was likely too stringent to identify all the genes affected by MbtS. When the threshold was lowered to 1.4-fold, the number of genes putatively affected by MbtS was increased to 232 (140 increased and 92 decreased) (see Tables S1 and S2 in the supplemental material). To examine whether those genes are affected by MbtS, we compared the transcript levels of five newly identified genes (two increased and three decreased) in the ΔftsH and ΔftsH::mbtS strains by qRT-PCR. All five genes showed a transcription pattern similar to those in RNA-seq (hla, cap5A, spa, clpC, and 0929 in Fig. 6A). Based on these results, we concluded that in the absence of FtsH, MbtS can potentially affect the transcription of over 200 genes.
Growth conditions can alter MbtS expression.
MbtS was not detected in the WT strain under the growth conditions we used (i.e., aerobic growth in tryptic soy broth [TSB]). To examine whether the MbtS expression can be modulated by nutritional conditions, we grew the WT strain of USA300 at 37°C in the following growth media: TSB, TSB without glucose, Todd-Hewitt broth, heart infusion broth, lysogeny broth (LB), chemically defined medium (CDM), CDM with lactose, and RPMI medium. Cells were collected at either the exponential or stationary growth phase and subjected to Western blot analysis. No MbtS was detected in any of the samples (data not shown), indicating that MbtS expression is not affected by the nutritional changes we tested.
Serendipitously, however, we found that on a tryptic soy agar plate containing X-Gal, some isolated colonies of the USA300 WT strain carrying pOS1-PmbtS-lacZ showed blue color, an indicator of increased PmbtS activity (WT in Fig. 7A). On the other hand, no blue colonies were observed in the mbtS transposon mutant (mbtS in Fig. 7A). To examine the expression of MbtS in those colonies, we collected two blue colonies (no. 1 and 2) and cells in the nonblue region (no. 3) and carried out Western blot analysis for MbtS. MbtS was detected in the blue colonies but not in the cells collected from the nonblue region (lanes 1 to 3 under WT in Fig. 7B). In contrast, all ΔftsH colonies showed MbtS expression at similar levels (lanes 1 to 3 under ΔftsH in Fig. 7B). Notably, the expression of SaeQ, an FtsH substrate, showed a pattern similar to that for MbtS (i.e., higher expression in isolated colonies), whereas the nonsubstrate protein SrtA did not (SaeQ versus SrtA in Fig. 7B). On the other hand, the expression levels of FtsH were equivalent regardless of the colony color (FtsH in Fig. 7B). These results indicate that the elevated production of MbtS in the blue colonies is probably due to the reduced activity of FtsH.
FIG 7.
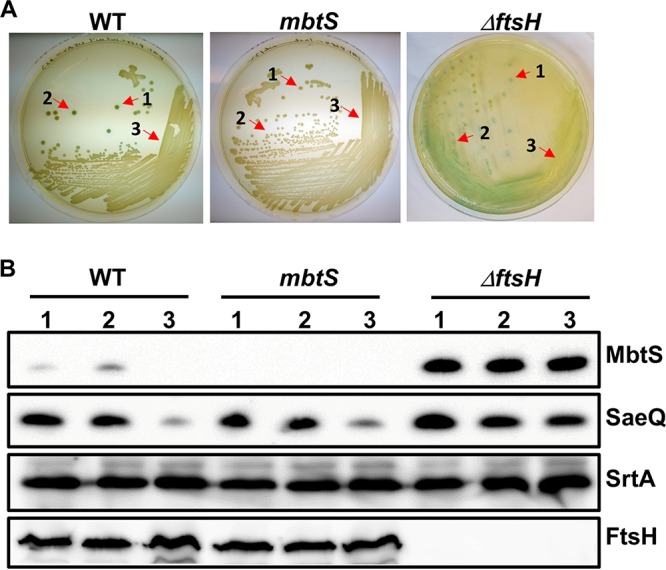
Growth conditions can alter MbtS expression. The test strains containing pOS1-PmbtS-lacZ were streaked on TSA containing X-Gal and incubated until some of the WT colonies turned blue. Cells were collected from two blue colonies (1 and 2) and a nonblue region of bacterial lawn (3) and lysed. An equal amount of proteins was subjected to Western blot analysis. SaeQ is an FtsH substrate protein and was used to monitor the FtsH proteolytic activity. Sortase A (SrtA) is not an FtsH substrate and used as a loading control. WT, wild-type strain USA300; mbtS, USA300 with a transposon insertion in mbtS; ΔftsH, ftsH deletion mutant of USA300.
MbtS decreases the size of skin lesions after infection with S. aureus.
Next, we examined whether MbtS contributes to the staphylococcal virulence in a murine model of blood infection. The four test strains, WT USA300, the mbtS transposon mutant, and the ΔftsH and ΔftsH::mbtS mutants, were administered to mice via retro-orbital injection, and the mice were watched for 2 weeks. Although the mbtS mutant showed a trend of reduced virulence compared with the WT strain, the reduction was not statistically significant (P = 0.0685) (WT versus mbtS in Fig. 8A). On the other hand, as reported previously, the ΔftsH mutant showed reduced virulence (20), and the virulence was not further reduced by additional disruption of mbtS (ΔftsH versus ΔftsH::mbtS in Fig. 8A).
FIG 8.
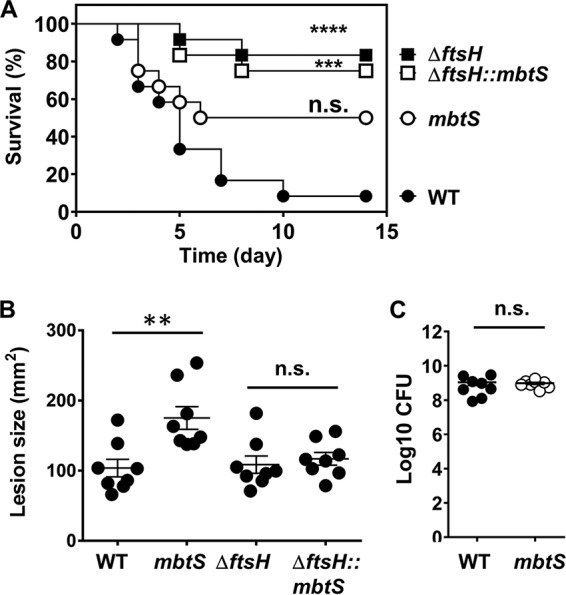
Effect of the disruption of mbtS on the staphylococcal virulence in a murine model of blood infection (A) and skin infection (B and C). (A) An equal amount of the test strains was administered to 10 C57BL/6J mice via the retro-orbital route, and then the mice were watched for 2 weeks. The statistical significance was measured by the log rank (Mantel-Cox) test. ***, P < 0.001; ****, P < 0.0001. (B and C) An equal amount of the test strains was injected s.c. into the right flanks of eight C57BL/6J mice. At day 2 postinfection, the lesion size was measured (B), and the bacterial CFU in the infected skin tissue was determined (C). The statistical significance was measured by an unpaired, two-tailed Student t test. **, P < 0.005; n.s., not significant. WT, wild-type strain USA300; mbtS, USA300 with a transposon insertion in mbtS; ΔftsH, ftsH deletion mutant of USA300; ΔftsH::mbtS, ΔftsH mutant with a transposon insertion in mbtS.
Since the skin is the primary infection site of S. aureus, we also examined the virulence contribution of MbtS in a skin infection model. In this model, the four test strains were administered subcutaneously, and the size of lesions was measured for 7 days. At day 2 postinfection, the lesion size in the mice infected with the mbtS mutant was significantly larger than that in the mice infected with the WT strain (Fig. 8B; see Fig. S5 in the supplemental material). However, when the bacterial loads in the lesions were measured, no significant differences were observed (Fig. 8B).
We examined whether the increased lesion size was caused by the recruitment of a higher number of innate immune cells such as neutrophils and macrophages to the infection site. The WT and the mbtS mutant were administered to mice via subcutaneous injection, and the infected skin was examined for recruitment of the innate immune cells at day 2 postinfection. However, at this time point, the skin tissues at the lesion sites were extensively damaged, and it was difficult to make the comparison (data not shown). Therefore, we examined the infected skin on day 1 postinfection. No substantial differences were observed in the infiltration of neutrophils (see Fig. S6A in the supplemental material) or macrophages (Fig. S6B) to the injection site. Therefore, we concluded that the larger lesion size was not caused by increased infiltration of the innate immune cells.
DISCUSSION
In this study, we identified MbtS as an MTF regulated by FtsH in S. aureus. Our conclusion is based on the following evidence. First, as a protein with both a DNA binding domain and three transmembrane helices, MbtS was found in the membrane (Fig. 1B and 2C). Second, the mbtS gene was essential for the transcription from PmbtS (Fig. 2A and 4A). Third, the PmbtS region contains an MbtS binding sequence (i.e., the inverted repeat sequence shown in Fig. 4B). Fourth, most of the mutations in the inverted repeat sequence abolished not only the promoter activity of PmbtS but also the DNA binding of MbtS (Fig. 4D and 5B and C). Finally, MbtS was degraded by the purified FtsH-Strep (Fig. 3B and C), and the increase of MbtS expression coincided with the decrease in the FtsH proteolytic activity, which was measured by the expression level of SaeQ (Fig. 7). Although, without the evidence from a DNA binding assay using purified MbtS, we cannot completely rule out the possibility that MbtS binds the DNA indirectly via a mediator, the presence of the N-terminal DNA binding domain strongly suggests that MbtS binds DNA directly.
In the protein degradation assay, FtsH cleaved MbtS into two smaller fragments of 16.6 kDa and 13.6 kDa (Fig. 3; see Fig. S2 in the supplemental material). The 13.6-kDa fragment appeared after the 16.6-kDa fragment was generated (Fig. 3A), suggesting that the 13.6-kDa fragment might be derived from the 16.6-kDa fragment. Since the MbtS antibody used in the experiment detects a peptide sequence in the N-terminal DNA binding domain, these MbtS fragments are likely generated by cleavage at the second cytoplasmic domain, located between the second and the third transmembrane helices (aa 123 to 163 in Fig. 1B). This also fits with the fact that FtsH attacks membrane protein from the cytoplasmic side. Intriguingly, no such fragments were detected in the WT cells; instead, the entire MbtS disappeared (Fig. 2B). These results might imply that the smaller fragments are further degraded in the WT cells by another protease(s). Alternatively, the conditions in the protein degradation assay might not be optimal for FtsH to degrade MbtS completely.
In the PmbtS region, the distance between the inverted repeat sequence and the putative −35 hexamer is 10 bp, which allows nearly one turn of the DNA helix (25). Therefore, upon binding to the DNA, MbtS is expected to be on the same side as the RNA polymerase and possibly directly interacts with the polymerase. Compared with the consensus sequence for bacterial σ70 binding (−35 TTGACA/−10 TATAAT), the putative promoter sequence of PmbtS (i.e., −35 TGACTT/−10 TAGTCT), in particular, the −35 hexamer, is poorly conserved, which probably explains the requirement of MbtS for the promoter activity. The inverted repeat in the binding sequence also suggests that MbtS binds to DNA as a dimer or a higher multimer. These hypotheses, i.e., a poorly conserved −35 hexamer and binding as a dimer, are currently being tested in our laboratory.
The full promoter activity of PmbtS required both the 2641 and 2642 proteins, which are encoded by the upstream genes SAUSA300_2641 and _2642 (Fig. 1A and 6C). Both 2641 and 2642 are small proteins of 131 aa and 117 aa, respectively. With four transmembrane helices, they are predicted to reside in the membrane. Therefore, it is not a far-fetched idea that these proteins form a complex with MbtS and activate the transcriptional activity of MbtS. Intriguingly, although the disruption of SAUSA300_2641 and _2642 reduced the promoter activity of PmbtS, the overall level of MbtS was not noticeably reduced (Fig. 6D; see Fig. S4 in the supplemental material). This might suggest that 2641 and 2642 negatively affect the stability of MbtS. However, this is pure speculation, and at this time we cannot explain this apparent disconnect between the decreased activity of PmbtS and the unchanged level of MbtS.
The deletion of ftsH increases the transcription of hslO, hrtAB, SAUSA300_2637, and mbtS (20). Of the five, along with mbtS, SAUSA300_2637 showed decreased transcription upon disruption of mbtS (Fig. 6A and Table 1), implying that MbtS mediates the transcription regulation of SAUSA300_2637 by FtsH. Inspection of the promoter region of SAUSA300_2637 identified an 18-bp sequence (i.e., TGAGAACTCCTTACCACA) similar to the MbtS binding sequence in PmbtS (i.e., TGTCAAAACATTTAGTCA). When a 251-bp DNA probe containing the 18-bp sequence was generated by PCR amplification with the primer set P3696/P3697 (Table 2) and used for DNA affinity chromatography, it did not bind to MbtS (data not shown), suggesting that SAUSA300_2637 is probably not a direct target of MbtS. A BLAST search with the 18-bp MbtS binding sequence or with the degenerate sequence TGNCNAANNNNNTNNNCA, where N is any nucleotide, did not identify any other genes containing the MbtS binding sequence in their promoter regions. Therefore, it appears that the mbtS gene is the only direct target of MbtS and that other genes are likely affected by MbtS indirectly. For example, the upregulation of the splABCDEF operon in the mbtS mutant might be explained by the increased transcription of the SaeRS two-component system (Table S1), a known positive regulator of the splABCDEF operon (26, 27). Further work is needed to determine how MbtS, with only one direct target, can affect the transcription of over 200 genes.
TABLE 2.
Oligonucleotides used in this study
| Category and name | Sequence (5′→3′)a | Target |
|---|---|---|
| pOS1-PmbtS-lacZ construction | ||
| P3302 | GTTCACCACCTTTTCCCTATGAATAACCATCTCGTTCCCTATACTTTTTAATTTGT | PmbtS |
| P3303 | AGCCTTAAAGACGATCCGGGAAAAAGATTAGAAAAGCATAGTTGGAAAGCT | PmbtS |
| P3304 | AGGGAACGAGATGGTTATTCATAGGGAAAAGGTGGTGAACTACTGT | lacZ |
| P3305 | GCGGGCAGTGAGCGCAACGCACGCCAAGCTTGCATGC | lacZ |
| P3306 | CAGGCATGCAAGCTTGGCGTGCGTTGCGCTCACTGC | pOS1 |
| P3307 | TATGCTTTTCTAATCTTTTTCCCGGATCGTCTTTAAGGCT | pOS1 |
| pOS1-PmbtS-lacZ deletion mutagenesis | ||
| P3330 | GCATCATTAATAAGTGAAATTCAAGTTGGCATTG | PmbtS Δ1 |
| P3331 | AGGTCTGTCTTAAGGGAGTCTTCGAAC | PmbtS Δ2 |
| P3332 | ATCATAAAGGTGACTTGATTTAACTTTATCTGATAG | PmbtS Δ3 |
| P3372 | GTCAAAACATTTAGTCAATCATAAAGG | PmbtS Δ4 |
| P3463 | GTTGTCGTATGTCAAAACATTTAGTCAATC | PmbtS Δ5 |
| pOS1-PmbtS-lacZ Δ5 point mutagenesis | ||
| P3481 | CGACAACTATGCTTTTCTAATCTTTTTCCCGG | PmbtS Δ5 |
| P3482 | TAAGTCAAAACATTTAGTCAATCATAAAGG | PmbtS Δ5 |
| P3483 | TTTGTCAAAACATTTAGTCAATCATAAAGG | PmbtS Δ5 |
| P3484 | AATGTCAAAACATTTAGTCAATCATAAAGG | PmbtS Δ5 |
| P3466 | ATACGACAACTATGCTTTTCTAATCTTTTTCCC | PmbtS Δ5 |
| P3431 | CTCAAAACATTTAGTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3432 | GACAAAACATTTAGTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3433 | GTGAAAACATTTAGTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3434 | GTCTAAACATTTAGTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3435 | GTCATAACATTTAGTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3436 | GTCAATACATTTAGTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3437 | GTCAAATCATTTAGTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3438 | GTCAAAAGATTTAGTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3439 | GTCAAAACTTTTAGTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3440 | GTCAAAACAATTAGTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3441 | GTCAAAACATATAGTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3442 | GTCAAAACATTAAGTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3443 | GTCAAAACATTTTGTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3444 | GTCAAAACATTTACTCAATCATAAAGGTGAC | PmbtS Δ5 |
| P3445 | GTCAAAACATTTAGACAATCATAAAGGTGAC | PmbtS Δ5 |
| P3446 | GTCAAAACATTTAGTGAATCATAAAGGTGAC | PmbtS Δ5 |
| P3447 | GTCAAAACATTTAGTCTATCATAAAGGTGAC | PmbtS Δ5 |
| BLI assay | ||
| P3556 | Biotin-AGGTCTGTCTTAAGGGAGTCTTCGAAC | PmbtS |
| P3557 | CAGATAAAGTTAAATCAAGTCACCTTTATG | PmbtS |
| P3670 | CAGATAAAGTTAAATCAAGTCACCTTTATGATTGACTAAATGTTTTGAGATACGACAAC | PmbtS m1 |
| P3671 | CAGATAAAGTTAAATCAAGTCACCTTTATGATTGACTAAATCTTTTGACATACGACAAC | PmbtS m2 |
| P3672 | CAGATAAAGTTAAATCAAGTCACCTTTATGATTGACAAAATGTTTTGACATACGACAAC | PmbtS m3 |
| P3673 | CAGATAAAGTTAAATCAAGTCACCTTTATGATTGTCTAAATGTTTTGACATACGACAAC | PmbtS m4 |
| P3674 | CAGATAAAGTTAAATCAAGTCACCTTTATGATTCACTAAATGTTTTGACATACGACAAC | PmbtS m5 |
| P3696 | Biotin-CTTTATCTAATCCACTGCGTCTAATTATTCCG | P2637 |
| P3697 | GGTTAGCATCGTATGTACCACTTCTTG | P2637 |
| pYJ-ftsH-His6 construction | ||
| P3764 | TAAAATAAGCTTGATATCGATGGGAAGTAGGAGGAAATG | ftsH |
| P3765 | TGACATTAGAAAACCGACTGCGTACTTCCAATCCAATGC | ftsH |
| P3766 | AGCATTGGATTGGAAGTACGCAGTCGGTTTTCTAATGTCAC | pYJ335 |
| P3767 | TCATTTCCTCCTACTTCCCATCGATATCAAGC | pYJ335 |
| pET28a-ftsH-Strep construction | ||
| P4218 | CTTTAAGAAGGAGATATACCATGCAGAAAGCTTTTCGCAATGTG | ftsH |
| P4219 | CGGATCTCATTTTTCGAACTGCGGGTGGCTCCATTTATTGTCTGGGTGATTTGGATC | ftsH |
| P4220 | TAAATGGAGCCACCCGCAGTTCGAAAAATGAGATCCGGCTGCTAACAAAGCCC | pET28a |
| P4221 | TTGCGAAAAGCTTTCTGCATGGTATATCTCCTTCTTAAAGTTAAACA | pET28a |
| qRT-PCR | ||
| P3081 | GATGATTTAGTAAAAGGGACC | mbtS |
| P3082 | GATTGCCACCGTTACACCCC | mbtS |
| P1395 | GGCGGCCTTATTGGTGCAAATG | hla |
| P1396 | CCATATACCGGGTTCCAAGA | hla |
| P2902 | CCAAGGGAACTCGTTTTACG | saeR |
| P2903 | ACGCATAGGGACTTCGTGAC | saeR |
| P3093 | GTATGGCGAAGCACAGTGATC | 2637 |
| P3094 | GGTTTAGTGGAGCCATTTATTG | 2637 |
| P3099 | CAAATGATCACAGCATTTGGTACAG | gyrB |
| P3100 | CGGCATCAGTCATAATGACGAT | gyrB |
| P4710 | TGCACCAAGAAGAACAGTGG | qcvT |
| P4711 | CGCTTTTAATTGACGCTTACG | qcvT |
| P4716 | CATTCAATTGCCAAAGCAGA | splA |
| P4717 | TTTCCTCCGCCTTTACCTTT | splA |
| P4718 | CATGCAGAAGCTGTGATGAGA | 1890 |
| P4719 | GCGTGCATACCATTTCTTGA | 1890 |
| P4720 | TGGTGATAAAGTCGCTGTGC | 2641 |
| P4721 | CGATTTTCCAGCTTTCCAAC | 2641 |
| P4727 | GCTGGTACAGGTGCTGGTTT | pstS |
| P4728 | CAACCGTTACACCATCTTGC | pstS |
| P4731 | CGGCACTACTGCTGACAAAA | spa |
| P4732 | AACGCTGCACCTAAGGCTAA | spa |
| P4733 | AGCGTTTGTCACAACTGTCAAT | 0929 |
| P4734 | ACCGAGAAATGCCTCCAAAT | 0929 |
| P4739 | GGTCATGATGATGGTGGACA | clpC |
| P4740 | ACTTGAACCACCGAATCCAG | clpC |
| P4741 | CAGTTTATGGCGCAAGAGGT | cap5A |
| P4742 | GCGAAGCTATTCGCAATTTT | cap5A |
Mutated nucleotides are underlined.
In the murine infection experiments, the mbtS disruption increased the size of skin lesions caused by staphylococcal infection (Fig. 8B). However, it did not affect either the virulence or the survival of the bacterium (Fig. 8A and C). The increased lesion size was not due to altered recruitment of proinflammatory cells such as neutrophils and macrophages; therefore, the molecular basis behind the phenotype remains unexplained. Intriguingly, despite the minor role it plays in bacterial pathogenesis, the mbtS gene is strongly conserved in S. aureus genomes. When 473 complete S. aureus genome sequences were searched with the MbtS protein sequence in PATRIC (https://www.patricbrc.org/), all genomes contained at least one gene whose product shows 85% to 100% identity to MbtS. This strong conservation of the gene implies that the protein might be needed for the survival of the bacterium under certain circumstances.
In summary, we identified a new MTF, MbtS, in S. aureus. Compared with other bacterial MTFs, MbtS is unique in that its degradation is initiated by an AAA+ protease, FtsH. As a result, the MbtS activity is induced by the reduction of the FtsH proteolytic activity. At this time, it is unknown how the FtsH proteolytic activity is regulated and why S. aureus links the FtsH proteolytic activity to the expression of MbtS. Therefore, future research is warranted to answer these questions. Finally, the identification of MbtS demonstrates the diversity of the bacterial transcriptional regulation and may suggest that MTFs are distributed in bacteria more widely than previously thought.
MATERIALS AND METHODS
Ethics statement.
All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal protocol was approved by the ethics committee of Renji Hospital, School of Medicine, Shanghai Jiaotong University (protocol number RJ-M-2014-0305), and the IUSM-NW IACUC (protocol number NW-43). Every effort was made to minimize the suffering of the animals.
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 3. E. coli and S. aureus were grown in lysogeny broth and tryptic soy broth (TSB), respectively. For transduction of mutations and plasmids, heart infusion broth (HIB) supplemented with 5 mM CaCl2 was used. When necessary, antibiotics were added to the growth media at the following concentrations: ampicillin, 100 μg/ml; erythromycin, 10 μg/ml; and chloramphenicol, 5 μg/ml.
TABLE 3.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics | Source or reference |
|---|---|---|
| E. coli strains | ||
| DH5α | Plasmid free, Lac− | New England Biolabs |
| BL21(DE3) | IPTG-inducible T7 RNA polymerase | Invitrogen |
| S. aureus strains | ||
| RN4220 | Restriction deficient, prophage cured | 36 |
| RN4220::ftsH | Transposon insertion of ΦΝΞ 4818 in ftsH was transduced into RN4220 | This study |
| USA300-0114 | Clinical isolate | NARSAa |
| USA300-P23 | USA300-0114 without plasmids 2 and 3 | 37 |
| NE132 | SAUSA300_2640 (= mbtS) mutant from Nebraska Transposon Mutant Library | BEI Resources |
| NE1755 | SAUSA300_2641 mutant from Nebraska Transposon Mutant Library | BEI Resources |
| NE 373 | SAUSA300_2642 mutant from Nebraska Transposon Mutant Library | BEI Resources |
| USA300::mbtS | Transposon insertion of NE132 in mbtS was transduced into USA300-P23 | This study |
| USA300 ΔftsH | ftsH deletion mutant of USA300-P23 | 20 |
| USA300 ΔftsH::mbtS | Transposon insertion in mbtS in NE132 was transduced into USA300ΔftsH | This study |
| USA300 ΔftsH::2641 | Transposon insertion in SAUSA300_2641 in NE1755 was transduced into USA300 ΔftsH | This study |
| USA300 ΔftsH::2642 | Transposon insertion in SAUSA300_2642 in NE373 was transduced into USA300 ΔftsH | This study |
| Plasmids | ||
| pET28a | Protein expression vector | Novagen |
| pET28a-ftsH-Strep | pET28a containing the ftsH gene with Strep tag sequence at the C terminus | This study |
| pOS1 | E. coli-S. aureus shuttle vector, Cmr | 38 |
| pOS1-PmbtS-lacZ | lacZ gene fused to the promoter sequence of mbtS | This study |
| pYJ335 | E. coli-S. aureus shuttle vector with anhydrotetracycline-inducible promoter | 39 |
| pCL-ftsH-His6 | ftsH-His6 cloned in pCL55 | 20 |
| pYJ-ftsH-His6 | pYJ335 containing the ftsH gene with His tag sequence at the C terminus | This study |
NARSA, Network on Antimicrobial Resistance in Staphylococcus aureus.
Plasmid constructions.
pOS1-PmbtS-lacZ was constructed by Gibson assembly (28). The PmbtS region (Fig. 1A) was PCR amplified with the primer set P3302/P3303, whereas the lacZ gene and the pOS1 plasmid were amplified with P3304/P3305 and P3306/P3307, respectively. For the DNA amplification, Phusion high-fidelity DNA polymerase (NEB) was used. The amplified PCR products were purified, subjected to Gibson assembly, and inserted into E. coli DH5α. After confirming the integrity of the plasmid by DNA sequencing of the PmbtS region, the resulting plasmid was electroporated into S. aureus RN4220, from which the plasmid was further moved to other strains of S. aureus by Φ85-mediated transduction. The deletion mutants of pOS1-PmbtS-lacZ shown in Fig. 4B and C were constructed by PCR amplification and self-ligation. The mutant pOS1-PmbtS-lacZ plasmids (Δ1 to Δ5) were amplified with the following sets of primers: P3307/P3330 (Δ1), P3307/P3331 (Δ2), P3307/P3332 (Δ3), P3307/P3372 (Δ4), and P3307/P3463 (Δ5) (Table 2). The PCR products were treated with T4 polynucleotide kinase (NEB) and self-ligated with T4 DNA ligase (NEB). The ligated plasmids were inserted into E. coli. Once their integrity was confirmed by DNA sequencing, the mutant plasmids were electroporated into S. aureus RN4220, from which the plasmids were further transduced into S. aureus USA300 ΔftsH with Φ85.
Point mutations in Fig. 4D were generated in pOS1-PmbtS-lacZ Δ5 by PCR amplification and self-ligation. To mutate positions 1 through 3 (i.e., TAT in Fig. 4D), pOS1-PmbtS-lacZ Δ5 was amplified by the following sets of primers: P3481/P3482 (position 1), P3481/P3483 (position 2), and P3481/P3484 (position 3) (Table 2). Mutation of the rest of the nucleotide positions shown in Fig. 3D was carried out by amplifying the same plasmid with the primer P3466 and one of primers P3431 to P3447 (Table 2). The resulting PCR products were processed as described above for the deletion mutants. All mutations were confirmed by DNA sequencing. The resulting plasmids were electroporated into strain RN4220 containing a transposon insertion in ftsH (i.e., RN4220::ftsH).
To construct pYJ-ftsH-His6, the vector and the insert DNA were PCR amplified with the primer sets P3766/P3767 (with pYJ335 as a template) and P3764/P3765 (with pCL-ftsH-His6 as a template), respectively (Table 2). The amplified PCR products were subjected to Gibson assembly and inserted into E. coli DH5α. The resulting plasmid, pYJ-ftsH-His6, was further electroporated into S. aureus RN4220 and subsequently transduced into USA300ΔftsH.
To construct pET28a-ftsH-Strep, pET28a and ftsH-Strep were PCR amplified with the primer sets P4220/P4221 (with pET28a as a template) and P4218/P4219 (with USA300 chromosomal DNA as a template), respectively (Table 2). After purification, the amplified PCR products were subjected to Gibson assembly and inserted into E. coli DH5α. The resulting plasmid was finally inserted into E. coli BL21(DE3) for protein expression and purification.
Purification of FtsH-Strep.
The FtsH-Strep protein was purified as described before with minor modifications (29). Briefly, overnight cultures of E. coli BL21(DE3) harboring pET28a-ftsH-Strep were transferred into fresh LB medium containing 50 μg/ml ampicillin. Cells were grown at 37°C to exponential growth phase (optical density at 600 nm [OD600] ≈ 0.5), 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added, and the cells were further incubated at 37°C for 5 h. Cells were collected, washed once with 10 mM Tris HCl (pH 8.0), and suspended in binding buffer (50 mM Tris HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA). After the addition of lysozyme (150 μg/ml), cells were subjected to a brief sonication, an equal volume of 1× binding buffer containing 0.2% n-dodecyl β-d-maltoside (Sigma) was added, and the cells were incubated on ice for 1 h. Cell debris was removed by ultracentrifugation at 45,000 × g at 4°C for 30 min. The soluble fraction containing the solubilized FtsH-Strep was applied to Strep-Tactin resin (IBA) per the manufacturer’s protocol. Finally, the buffer of the purified protein was exchanged first with TBS buffer (10 mM Tris HCl [pH 7.5], 138 mM NaCl, 2.7 mM KCl) and then with TBS buffer containing 25% glycerol. The protein solution was concentrated using an Amicon Ultra-30 instrument (MW 30,000; Millipore). Protein concentration was determined by the bicinchoninic acid (BCA) assay (Pierce).
Cell fractionation.
Cellular components were fractionated into cell wall, cell membrane, and cytoplasm, as described previously (30). Briefly, the test strains were grown until exponential growth phase and collected by centrifugation. Cells were lysed with lysostaphin (50 μg/ml final concentration) in TSM (50 mM Tris HCl, 0.5 M sucrose, 10 mM MgCl2, pH 7.5). The protoplasts were collected by centrifugation (4,600 × g, 5 min), and the obtained supernatant was designated the cell wall fraction. The collected protoplasts were suspended in membrane buffer (100 mM Tris HCl, 100 mM NaCl, 10 mM MgCl2, pH 7.5), broken by sonication, and subjected to ultracentrifugation (48,000 rpm, 4°C, 90 min). The collected supernatant was designated the cytoplasm fraction, whereas the pellet was washed with the membrane buffer and designated the membrane fraction.
Purification of cell membranes.
RN4220::ftsH, USA300 ΔftsH and USA300 ΔftsH::mbtS were grown in 100 ml TSB and TSB containing 10 μg/ml of erythromycin with shaking (200 rpm) at 37°C overnight. Cells were collected by centrifugation (9,000 rpm, 4°C, 15 min), and the cell pellet was washed with deionized water once, suspended in 10 ml TSM, treated with lysostaphin (50 μg/ml final concentration) at 37°C for 1 h, and sonicated (20 s, 3 times) on ice. The resulting cell lysate was centrifuged (12,000 rpm, 4°C, 30 min), and the supernatant was collected. The supernatant was further centrifuged (48,000 rpm, 4°C, 90 min), and the membrane pellet was washed twice with 1 ml phosphate-buffered saline (PBS), suspended in 1 ml membrane storage buffer (50 mM Tris HCl, 50 mM KCl, 5 mM MgCl2, 1 mM CaCl2, 20% glycerol, 0.1% Triton X-100, 0.01% Tween 20, pH 7.4), and sonicated (10 s, 3 times) on ice. After further centrifugation (13,000 rpm, 4°C, 5 min), the supernatant was collected and stored at −80°C until use. Protein concentrations (typically 7 to 12 mg/ml) in the purified membranes were measured by the BCA assay (Pierce).
The membrane vesicles for protein degradation assay were prepared as described before (31). Briefly, overnight cultures of USA300 ΔftsH and USA300 ΔftsH harboring pYJ335 or pYJ-ftsH-His6 were transferred into fresh TSB containing 10 μg/ml of erythromycin and incubated for 2 h. Anhydrotetracycline (100 ng/ml final concentration) was then added to induce the expression of FtsH, and the culture was further incubated for 6 h. Cells were collected by centrifugation, suspended in TSM buffer containing lysostaphin (50 μg/ml), and incubated at 37°C for 30 min. After brief sonication of the cell lysates, cell debris was removed by centrifugation at 7,000 rpm and 4°C for 10 min. The membrane fraction was collected by ultracentrifugation (45,000 × g for 45 min; Beckman Optima Max-XP ultracentrifuge). The membranes were washed twice in TKM buffer (50 mM Tris HCl, 50 mM KCl, 1 mM MgCl2) and collected by ultracentrifugation (45,000 × g, 4°C, 30 min). Finally, the membranes were suspended in TKMG buffer (50 mM Tris HCl, 50 mM KCl, 1 mM MgCl2, 25% glycerol, pH 8.0) and stored at −80°C until use.
Western blot analysis.
Western blot analysis was carried out as described previously (31). Briefly, test strains were grown in TSB containing appropriate antibiotics until exponential growth phase. Cells were collected by centrifugation, lysed with lysostaphin (50 μg/ml), and subjected to SDS-PAGE. The proteins were transferred to Protran BA nitrocellulose membranes (Whatman) and probed with cognate antibodies.
For the Western blot analysis of the bacterial colonies shown in Fig. 7B, the three strains USA300, USA300 ΔftsH, and USA300 ΔftsH::mbtS carrying pOS1-PmbtS-lacZ were streaked on tryptic soy agar (TSA) containing X-Gal (20 μg/ml), grown at 37°C overnight, and further incubated at room temperature until blue colonies appeared. From two isolated colonies and one region of bacterial lawn, bacterial cells were collected and lysed in 50 mM Tris HCl buffer (pH 8.0) with lysostaphin (50 μg/ml) at 37°C for 20 min. The protein concentration in the total cell extracts was quantified using the BCA assay (Pierce), and an equal amount of the cell extracts (50 μg) was subjected to SDS-PAGE, followed by Western blotting with anti-MbtS and anti-SaeQ antibodies. As a control for a non-FtsH substrate, SrtA was detected with the anti-SrtA antibody.
All primary antibodies were polyclonal and were generated in either mouse (FtsH antibody) or rabbit (MbtS, SaeQ, SaeS, and SrtA antibodies). In particular, the MbtS antibody was generated by immunizing rabbits with a 14-aa peptide (RQSISNWENDKSLP) located in the DNA binding domain of MbtS. For secondary antibodies, either horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Cell Signaling) or HRP-conjugated anti-rabbit IgG (Cell Signaling) was used. The proteins were visualized with the SuperSignal West Pico Plus chemiluminescent substrate (Thermo Scientific), and the images were taken and processed with LAS-4000 (GE Healthcare). The molecular weights of the target proteins are as follows: FtsH, 77.8 kDa; SaeS, 39.7 kDa; SrtA, 23.5 kDa; MbtS, 21.8 kDa; and SaeQ, 17.7 kDa. In the Western blot analysis, although the Spa protein (staphylococcal protein A, 52 kDa without signal peptide) was also detected, due to the significant difference in the molecular weight, the Spa signal did not interfere with the test protein signals.
Protein degradation assay.
MbtS degradation by FtsH was assayed as described by Liu et al. (20), with minor modifications. The membrane vesicles harboring FtsH (500 μg in total protein content) and MbtS (300 μg in total protein content) were mixed and incubated in degradation buffer {50 mM Tris HCl (pH 8.0), 5 mM MgCl2, 12.5 μM zinc acetate [Zn(OAc)2], 20 mM KCl, 100 mM NaCl, 1 mM dithiothreitol (DTT), 10% (wt/vol) glycerol, 12.5% (wt/vol) polyethylene glycol (PEG) 3350, and 0.1% n-dodecyl β-d-maltoside} at 42°C for 2 min. When purified FtsH-Strep was used, FtsH-Strep (3 μg) was mixed with either the membrane vesicles containing MbtS (300 μg in total protein content) or the cell lysate of ΔftsH (300 μg in total protein content). To initiate the protein degradation, 8 mM ATP (final volume of 300 μl) was added. An aliquot (25 μl) was removed at the indicated times, mixed with SDS-PAGE sample buffer to stop the reaction, and placed on ice until SDS-PAGE was performed. Samples were heated at 100°C for 5 min, and 7 μl of each sample was subjected to 13% SDS-PAGE, followed by Western blotting with the anti-MbtS antibody (GL Biochem, Shanghai, China). As a control, SrtA and FtsH were also detected with anti-SrtA and anti-FtsH antibodies, respectively.
BLI assay.
For the biolayer interferometer (BLI) assay, biotin-labeled probes (92 nt) were PCR amplified with P3556 and one of the following primers: P3557 (WT), P3670 (m1), P3671 (m2), P3672 (m3), P3673 (m4), and P3674 (m5). The biotin-labeled DNA probes were diluted to 0.2 μM with PBS. The membranes were diluted to 1 mg/ml protein with DNA binding buffer (50 mM Tris HCl [pH 7.4], 50 mM KCl, 5 mM MgCl2, 1 mM CaCl2, 10% glycerol, 0.1% Triton X-100, and 0.01% Tween 20). The assay was carried out with Octet Red 384 (Pall FortéBio) at Bindley Bioscience Center, Biophysical Analysis Lab, Purdue University, by following the manufacturer’s recommendations. Briefly, the streptavidin-coated biosensor was hydrated in DNA binding buffer for 10 min, and then it was exposed to DNA binding buffer containing the biotinylated DNA probes for 120 s to load the DNA probes. The loaded biosensor was exposed to DNA binding buffer for 30 s to establish the baseline, and then the biosensor was exposed to the solubilized membranes for 120 s to acquire the binding curve. Finally, the biosensor was exposed to DNA buffer for 180 s to acquire the dissociation curve.
DNA affinity chromatography.
Streptavidin-Sepharose resin (20 μl per sample; BioVision) was mixed with 100 μl PBS. After brief centrifugation, the pellet was washed with 100 μl PBS and mixed with 10 μl of the DNA probe (∼50 ng/μl). After incubation at room temperature for 10 min, 100 μl of DNA binding buffer was added to the sample and briefly centrifuged. The resulting pellet was further washed twice with 200 μl DNA binding buffer. The washed resin was mixed with the purified membranes (70 μl; protein concentration, ≈1 mg/ml). After 30 min of incubation at room temperature, the resin was washed five times with 200 μl of DNA binding buffer. Finally, the resin pellet was mixed with 10 μl of 2× SDS-PAGE sample buffer, boiled for 10 min, and subjected to 12.5% SDS-PAGE and Western blotting with the anti-MbtS antibody.
RNA-seq analysis.
The test strains, i.e., WT USA300, USA300 mbtS, USA300 ΔftsH, and USA300 ΔftsH::mbtS, were grown in TSB at 37°C overnight. The next day, the cultures were diluted 100-fold in TSB and incubated in a shaking incubator at 37°C for 4 h. Samples were harvested using TRIzol solution, suspended in TE buffer (10 mM Tris HCl [pH 8.0], 1 mM EDTA), and treated with lysostaphin (50 μg/ml final concentration) at 37°C for 5 min. Cells were homogenized with a Precellys 24 homogenizer (Bertin Technologies) (three 30-s pulses at 6,500 rpm). From the cell homogenates, total RNA was purified with the RNeasy kit (Qiagen) according to the manufacturer’s recommendations. Triplicates of the isolated RNA were sent to the Center for Medical Genomics at Indiana University School of Medicine. After the depletion of rRNA with the QIAseq FastSelect rRNA removal kit (Qiagen), cDNA library preparation was carried out by following the KAPA RNA Hyper Prep kit technical data sheet KR0961, v3.15 (Roche Corporate). A Phred quality score (Q score) was used to measure the quality of sequencing.
qRT-PCR.
From the same RNA used for RNA-seq, cDNA was synthesized with a high-capacity cDNA reverse transcription kit (Applied Biosystems) according to the manufacturer’s instructions. The cDNA was used for quantitative PCR (qPCR) with SYBR green PCR master mix (Applied Biosystems) in a QuantStudio 6 Flex real-time PCR system (Applied Biosystems). The relative amount of cDNA was determined by using a standard curve obtained from PCR with serially diluted genomic DNA, and results were normalized to the levels of gyrB, used as an internal control. Data shown are averages from at least three independent experiments. The statistical analysis was carried out with Prism 8 (GraphPad). The primers used in this assay are presented in Table 2.
β-Galactosidase (LacZ) assay.
The β-galactosidase assays were carried out as described previously (32). Test strains were grown in TSB at 37°C overnight. The resulting cultures were used for the assay. The LacZ activity was measured with ortho-nitrophenyl-β-galactoside (ONPG) as a substrate and normalized by optical density at 600 nm.
Animal experiment.
The murine blood infection experiment was carried out as described previously (33). In the experiment, 10 sex-matched C57BL/6J mice (8 weeks old; Jackson Laboratory) were used. The skin infection experiment was carried out as described previously (34). After subcutaneous (s.c.) injection of the bacterial strains into the right flank, the lesion size was measured every day for 7 days. The skin lesion size was quantitated by taking a digital image of the lesion and processing the image with ImageJ (35). To determine the CFU in the infected skin tissues, the harvested skin tissue was suspended in PBS and homogenized with a TissuRuptor II instrument (Qiagen). The tissue homogenates were serially diluted and spread on a TSA plate. The agar plates were incubated at 37°C overnight, and the colonies on each plate were counted.
Immunohistochemical analysis.
PBS, WT USA300, and USA300::mbtS were injected s.c. into three female mice per strain, as described for the animal experiment. At day 1 or 2 postinfection, the infected skin at the site of injection was dissected and fixed in 4% paraformaldehyde in PBS at room temperature for 3 h. Samples were washed 3 times in PBS for 5 min, placed in TBS medium (Triangle Biological Sciences, Durham, NC), frozen in dry-ice-cooled isopentane, and stored at −70°C until needed. Samples were sliced with a cryostat at a thickness of 12 μm and adhered to Superfrost Plus microscope slides. The tissue sections were dried at room temperature for 10 min, treated with 0.2% Triton X-100 in PBS for 5 min, and rinsed 3 times with PBS. The tissue sections were blocked with PBS with 0.05% Tween 20 and 20% calf serum (PBST-S) at room temperature for 30 min and incubated at 4°C overnight with the primary antibodies in PBST-S. The following primary antibodies were used: phycoerythrin (PE)-conjugated rat IgG against lymphocyte antigen 6 complex (catalog number 127607; BioLegend) to detect murine neutrophils and polyclonal goat IgG against mouse MMR/CD206 (catalog number AF2535; R&D Systems) to detect murine macrophages. For the visualization of CD206, tissue sections treated with the CD206 antibody were further incubated with a Cy3-conjugated anti-goat antibody (Jackson ImmunoResearch Laboratory, West Grove, PA). To visualize the connective tissue, the samples were incubated with fluorescein-conjugated wheat germ agglutinin (WGA-fluorescein) (1 μg/ml; Molecular Probes, Eugene, OR). Nuclei were visualized by 5 min of incubation with 4′,6′-diamidino-2-phenylindole (DAPI) solution (Sigma, St. Louis, MO) in PBST. The samples were examined and photographed with a Leica microscope.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ji Ma, Purdue University (currently at Columbia University), for his assistance with the BLI assay.
This study was supported in part by the National Natural Science Foundation of China (grant 81772139), the Innovative Research Team of High-Level Local Universities in Shanghai, and the Cultivation Fund from Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University (grant PYIII-17-001) (Q.L.) and by a Research Enhancement Grant from Indiana University and the Indiana Clinical and Translational Sciences Institute (T.B.), which was funded in part by grant UL1TR002529 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award.
The funders had no role in study design, data collection and interpretation, or the decision to submit this work for publication.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Balleza E, López-Bojorquez LN, Martínez-Antonio A, Resendis-Antonio O, Lozada-Chávez I, Balderas-Martínez YI, Encarnación S, Collado-Vides J. 2009. Regulation by transcription factors in bacteria: beyond description. FEMS Microbiol Rev 33:133–151. doi: 10.1111/j.1574-6976.2008.00145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoppe T, Rape M, Jentsch S. 2001. Membrane-bound transcription factors: regulated release by RIP or RUP. Curr Opin Cell Biol 13:344–348. doi: 10.1016/s0955-0674(00)00218-0. [DOI] [PubMed] [Google Scholar]
- 3.Liu Y, Li P, Fan L, Wu M. 2018. The nuclear transportation routes of membrane-bound transcription factors. Cell Commun Signal 16:12. doi: 10.1186/s12964-018-0224-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller VL, Taylor RK, Mekalanos JJ. 1987. Cholera toxin transcriptional activator toxR is a transmembrane DNA binding protein. Cell 48:271–279. doi: 10.1016/0092-8674(87)90430-2. [DOI] [PubMed] [Google Scholar]
- 5.Hase CC, Mekalanos JJ. 1998. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A 95:730–734. doi: 10.1073/pnas.95.2.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crawford JA, Krukonis ES, DiRita VJ. 2003. Membrane localization of the ToxR winged-helix domain is required for TcpP-mediated virulence gene activation in Vibrio cholerae. Mol Microbiol 47:1459–1473. doi: 10.1046/j.1365-2958.2003.03398.x. [DOI] [PubMed] [Google Scholar]
- 7.DiRita VJ, Mekalanos JJ. 1991. Periplasmic interaction between two membrane regulatory proteins, ToxR and ToxS, results in signal transduction and transcriptional activation. Cell 64:29–37. doi: 10.1016/0092-8674(91)90206-e. [DOI] [PubMed] [Google Scholar]
- 8.Teoh WP, Matson JS, DiRita VJ. 2015. Regulated intramembrane proteolysis of the virulence activator TcpP in Vibrio cholerae is initiated by the tail-specific protease (Tsp). Mol Microbiol 97:822–831. doi: 10.1111/mmi.13069. [DOI] [PubMed] [Google Scholar]
- 9.Beck NA, Krukonis ES, DiRita VJ. 2004. TcpH influences virulence gene expression in Vibrio cholerae by inhibiting degradation of the transcription activator TcpP. J Bacteriol 186:8309–8316. doi: 10.1128/JB.186.24.8309-8316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carroll PA, Tashima KT, Rogers MB, DiRita VJ, Calderwood SB. 1997. Phase variation in tcpH modulates expression of the ToxR regulon in Vibrio cholerae. Mol Microbiol 25:1099–1111. doi: 10.1046/j.1365-2958.1997.5371901.x. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto S, Mitobe J, Ishikawa T, Wai SN, Ohnishi M, Watanabe H, Izumiya H. 2014. Regulation of natural competence by the orphan two-component system sensor kinase ChiS involves a non-canonical transmembrane regulator in Vibrio cholerae. Mol Microbiol 91:326–347. doi: 10.1111/mmi.12462. [DOI] [PubMed] [Google Scholar]
- 12.Dalia AB, Lazinski DW, Camilli A. 2014. Identification of a membrane-bound transcriptional regulator that links chitin and natural competence in Vibrio cholerae. mBio 5:e01028-13. doi: 10.1128/mBio.01028-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lo Scrudato M, Blokesch M. 2012. The regulatory network of natural competence and transformation of Vibrio cholerae. PLoS Genet 8:e1002778. doi: 10.1371/journal.pgen.1002778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuper C, Jung K. 2005. CadC-mediated activation of the cadBA promoter in Escherichia coli. J Mol Microbiol Biotechnol 10:26–39. doi: 10.1159/000090346. [DOI] [PubMed] [Google Scholar]
- 15.Lee YH, Kim BH, Kim JH, Yoon WS, Bang SH, Park YK. 2007. CadC has a global translational effect during acid adaptation in Salmonella enterica serovar Typhimurium. J Bacteriol 189:2417–2425. doi: 10.1128/JB.01277-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindner E, White SH. 2014. Topology, dimerization, and stability of the single-span membrane protein CadC. J Mol Biol 426:2942–2957. doi: 10.1016/j.jmb.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 18.Gordon RJ, Lowy FD. 2008. Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis 46(Suppl 5):S350–S359. doi: 10.1086/533591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lithgow JK, Ingham E, Foster SJ. 2004. Role of the hprT-ftsH locus in Staphylococcus aureus. Microbiology 150:373–381. doi: 10.1099/mic.0.26674-0. [DOI] [PubMed] [Google Scholar]
- 20.Liu Q, Hu M, Yeo WS, He L, Li T, Zhu Y, Meng H, Wang Y, Lee H, Liu X, Li M, Bae T. 2017. Rewiring of the FtsH regulatory network by a single nucleotide change in saeS of Staphylococcus aureus. Sci Rep 7:8456. doi: 10.1038/s41598-017-08774-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ito K, Akiyama Y. 2005. Cellular functions, mechanism of action, and regulation of FtsH protease. Annu Rev Microbiol 59:211–231. doi: 10.1146/annurev.micro.59.030804.121316. [DOI] [PubMed] [Google Scholar]
- 22.Langklotz S, Baumann U, Narberhaus F. 2012. Structure and function of the bacterial AAA protease FtsH. Biochim Biophys Acta 1823:40–48. doi: 10.1016/j.bbamcr.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 23.Herman C, Prakash S, Lu CZ, Matouschek A, Gross CA. 2003. Lack of a robust unfoldase activity confers a unique level of substrate specificity to the universal AAA protease FtsH. Mol Cell 11:659–669. doi: 10.1016/s1097-2765(03)00068-6. [DOI] [PubMed] [Google Scholar]
- 24.Mäder U, Nicolas P, Depke M, Pané-Farré J, Debarbouille M, van der Kooi-Pol MM, Guérin C, Dérozier S, Hiron A, Jarmer H, Leduc A, Michalik S, Reilman E, Schaffer M, Schmidt F, Bessières P, Noirot P, Hecker M, Msadek T, Völker U, van Dijl JM. 2016. Staphylococcus aureus transcriptome architecture: from laboratory to infection-mimicking conditions. PLoS Genet 12:e1005962. doi: 10.1371/journal.pgen.1005962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levitt M. 1978. How many base-pairs per turn does DNA have in solution and in chromatin? Some theoretical calculations. Proc Natl Acad Sci U S A 75:640–644. doi: 10.1073/pnas.75.2.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rogasch K, Ruhmling V, Pane-Farre J, Hoper D, Weinberg C, Fuchs S, Schmudde M, Broker BM, Wolz C, Hecker M, Engelmann S. 2006. Influence of the two-component system SaeRS on global gene expression in two different Staphylococcus aureus strains. J Bacteriol 188:7742–7758. doi: 10.1128/JB.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Q, Yeo WS, Bae T. 2016. The SaeRS two-component system of Staphylococcus aureus. Genes (Basel) 7:81. doi: 10.3390/genes7100081. [DOI] [Google Scholar]
- 28.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 29.Yeo WS, Zwir I, Huang HV, Shin D, Kato A, Groisman EA. 2012. Intrinsic negative feedback governs activation surge in two-component regulatory systems. Mol Cell 45:409–421. doi: 10.1016/j.molcel.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeong DW, Cho H, Jones MB, Shatzkes K, Sun F, Ji Q, Liu Q, Peterson SN, He C, Bae T. 2012. The auxiliary protein complex SaePQ activates the phosphatase activity of sensor kinase SaeS in the SaeRS two-component system of Staphylococcus aureus. Mol Microbiol 86:331–348. doi: 10.1111/j.1365-2958.2012.08198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Q, Cho H, Yeo WS, Bae T. 2015. The extracytoplasmic linker peptide of the sensor protein SaeS tunes the kinase activity required for staphylococcal virulence in response to host signals. PLoS Pathog 11:e1004799. doi: 10.1371/journal.ppat.1004799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun F, Cho H, Jeong DW, Li C, He C, Bae T. 2010. Aureusimines in Staphylococcus aureus are not involved in virulence. PLoS One 5:e15703. doi: 10.1371/journal.pone.0015703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeo WS, Arya R, Kim KK, Jeong H, Cho KH, Bae T. 2018. The FDA-approved anti-cancer drugs, streptozotocin and floxuridine, reduce the virulence of Staphylococcus aureus. Sci Rep 8:2521. doi: 10.1038/s41598-018-20617-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho H, Jeong DW, Liu Q, Yeo WS, Vogl T, Skaar EP, Chazin WJ, Bae T. 2015. Calprotectin increases the activity of the SaeRS two component system and murine mortality during Staphylococcus aureus infections. PLoS Pathog 11:e1005026. doi: 10.1371/journal.ppat.1005026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kreiswirth BN, Löfdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 37.Jeong DW, Cho H, Lee H, Li C, Garza J, Fried M, Bae T. 2011. Identification of P3 promoter and distinct roles of the two promoters of the SaeRS two-component system in Staphylococcus aureus. J Bacteriol 193:4672–4684. doi: 10.1128/JB.00353-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bubeck Wardenburg J, Williams WA, Missiakas D. 2006. Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc Natl Acad Sci U S A 103:13831–13836. doi: 10.1073/pnas.0603072103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji Y, Marra A, Rosenberg M, Woodnutt G. 1999. Regulated antisense RNA eliminates alpha-toxin virulence in Staphylococcus aureus infection. J Bacteriol 181:6585–6590. doi: 10.1128/JB.181.21.6585-6590.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.