

In the present study, we investigated a collection of Listeria monocytogenes strains that all carried sigB operon mutations. The mutants all had reduced σB activity and were found to have a growth advantage under conditions of mild heat stress (42°C). In mixed cultures, these mutants outcompeted the wild type when mild heat stress was present but not at an optimal growth temperature. An analysis of 22,340 published L. monocytogenes genome sequences found a high rate of premature stop codons present in genes positively regulating σB activity. Together, these findings suggest that the occurrence of mutations that attenuate σB activity can be favored under conditions of mild stress, probably highlighting the burden on cellular resources that stems from deploying the general stress response.
KEYWORDS: Listeria monocytogenes, competition, mutations, rsbS, rsbT, rsbU, sigB, sigma B, stress
ABSTRACT
In Listeria monocytogenes, the full details of how stress signals are integrated into the σB regulatory pathway are not yet available. To help shed light on this question, we investigated a collection of transposon mutants that were predicted to have compromised activity of the alternative sigma factor B (σB). These mutants were tested for acid tolerance, a trait that is known to be under σB regulation, and they were found to display increased acid sensitivity, similar to a mutant lacking σB (ΔsigB). The transposon insertions were confirmed by whole-genome sequencing, but in each case, the strains were also found to carry a frameshift mutation in the sigB operon. The changes were predicted to result in premature stop codons, with negative consequences for σB activation, independently of the transposon location. Reduced σB activation in these mutants was confirmed. Growth measurements under conditions similar to those used during the construction of the transposon library revealed that the frameshifted sigB operon alleles conferred a growth advantage at higher temperatures, during late exponential phase. Mixed-culture experiments at 42°C demonstrated that the loss of σB activity allowed mutants to take over a population of parental bacteria. Together, our results suggest that mutations affecting σB activity can arise during laboratory culture because of the growth advantage conferred by these mutations under mild stress conditions. The data highlight the significant cost of stress protection in this foodborne pathogen and emphasize the need for whole-genome sequence analysis of newly constructed strains to confirm the expected genotype.
IMPORTANCE In the present study, we investigated a collection of Listeria monocytogenes strains that all carried sigB operon mutations. The mutants all had reduced σB activity and were found to have a growth advantage under conditions of mild heat stress (42°C). In mixed cultures, these mutants outcompeted the wild type when mild heat stress was present but not at an optimal growth temperature. An analysis of 22,340 published L. monocytogenes genome sequences found a high rate of premature stop codons present in genes positively regulating σB activity. Together, these findings suggest that the occurrence of mutations that attenuate σB activity can be favored under conditions of mild stress, probably highlighting the burden on cellular resources that stems from deploying the general stress response.
INTRODUCTION
Listeria monocytogenes is the causative agent of listeriosis, which can sicken immunocompromised individuals and pregnant women and is associated with a high mortality rate (typically 25 to 30%) (1, 2). L. monocytogenes is ubiquitous in the environment (3), partly due to its ability to survive and grow under a wide range of harsh conditions, such as low pH, high osmolality (4), and elevated concentrations of bile salts (5). This robustness is partly under the control of the stress-inducible sigma factor sigma B (σB), which is responsible for the upregulation of a regulon composed of approximately 300 genes (6, 7). σB also plays a role in establishing infections, as it is necessary for L. monocytogenes survival in the gastrointestinal tract (8), and it contributes to the regulation of the internalin genes inlA and inlB, which are required for host cell invasion (9).
In Bacillus subtilis, σB is regulated by a signal transduction pathway that is primarily encoded in the polycistronic sigB operon, which comprises eight genes, rsbR, rsbS, rsbT, rsbU, rsbV, rsbW, sigB, and rsbX (10). In L. monocytogenes, two additional genes, mazE and mazF, are located upstream of rsbR and are also cotranscribed with this operon (11). The σB signal transduction pathway has been well studied in B. subtilis, where the main components of the system are all conserved and share a high degree of similarity with their L. monocytogenes counterparts (12). In the absence of stress, the anti-sigma factor RsbW sequesters σB, blocking its interaction with RNA polymerase (13, 14). Upon encountering environmental or starvation stress, an unknown signal is detected and integrated by the stressosome, a supramolecular complex composed of RsbR, RsbS, RsbT (15–18), and a number of RsbR paralogues (Lmo0161, RsbL, Lmo1642, and Lmo1842) (19, 20). This triggers the serine-threonine kinase activity of RsbT, resulting in the phosphorylation of RsbR and RsbS and the subsequent release of RsbT from the stressosome (21, 22). Once free, RsbT interacts with RsbU, activating its serine phosphatase activity, which in turn results in the dephosphorylation of the anti-anti-sigma factor RsbV. The anti-sigma factor RsbW, which is also a serine kinase, possesses a higher affinity for nonphosphorylated RsbV than σB, resulting in the sequestration of RsbW by RsbV and the release σB (23), allowing it to interact with RNA polymerase and instigate the transcription of the σB regulon.
Several studies, in both L. monocytogenes and B. subtilis, have shown that mutations constructed within the sigB operon result in reduced σB activity as a consequence of impaired signal transduction through this pathway (21, 24–28). Although the main elements of the signal transduction pathway from the stressosome to σB are well described, the nature of the signals detected and the molecular mechanisms involved in the transduction of these signals are still largely unknown. Paradoxically, a number of studies have reported that the loss of sigB can result in a higher growth rate under some culture conditions. In a chemically defined medium with limiting glucose, mutant strains lacking either sigB, rsbV, or rsbT grow faster at 37°C than the wild type (WT) (29). At 3°C, a growth advantage has also been reported for a sigB mutant strain in complex medium (30). In the presence of sublethal doses of blue light, mutant strains lacking σB show improved growth in both liquid and solid complex media (31). These findings suggest that deploying the σB-controlled general stress response can, under some conditions, impose a cost on cells that results in a reduced growth rate.
In an attempt to develop a better understanding of the factors that influence stress sensing via the stressosome, we focused on a set of transposon mutant strains in L. monocytogenes EGD-e that were previously suggested to have altered σB activity (32). In their study, Tiensuu and colleagues discovered that L. monocytogenes forms distinct rings in soft-agar plates when exposed to cycles of light and darkness, a phenotype that is mediated by σB and requires the action of RsbL (also known as Lmo0799), a stressosome-associated RsbR paralogue that acts as a blue-light sensor (19). Those authors described the isolation of several transposon mutant strains that failed to produce these rings in response to oscillating cycles of light (“ringless” phenotype) and suggested that the genes carrying the transposon insertions could be involved in modulating σB activity. The present study focused on this collection of mutant strains in the expectation that the mutated genes might give new insights into the mechanisms that lead to the activation of σB in response to stress and potentially into the nature of the stress signals detected.
During the preliminary stages of the present study, whole-genome sequencing (WGS) was used to confirm the location of the transposon insertions. While confirming the presence of the transposons, it also revealed the presence of distinct frameshift mutations in the sigB operon in each of the ringless strains. This suggested a simpler explanation for the ringless phenotype of the transposon mutant strains, namely, that the sigB operon alleles reduced σB activity, which in turn compromised “ring” formation. The emergence of mutations in the sigB operon of L. monocytogenes during laboratory culture has been reported in a number of other studies (28, 31, 33–35). It has been suggested that the occurrence of mutations impairing σB function might be particularly associated with the loss of surface proteins (36). However, the selective pressure driving the emergence of these alleles is unknown. Here, we investigated the properties of these sigB operon mutant strains to determine whether σB activity was affected and whether this affected their fitness. The frameshift alleles that arose in the sigB operon of these strains were associated with reduced acid tolerance as well as a marked reduction in σB activity. Furthermore, in the presence of mild heat stress, the mutations produced a fitness advantage in mixed populations with WT bacteria that was qualitatively similar to that seen in a sigB deletion mutant strain. Together, these observations suggest that the loss of σB activity can confer a growth advantage under conditions used routinely during laboratory culture of L. monocytogenes. We propose that this effect is responsible for the common emergence of mutations in the sigB operon under laboratory conditions and that this finding has important implications for researchers studying any phenotypic properties of this pathogen.
RESULTS
Ringless transposon mutants display acid-sensitive phenotypes.
Five “ringless” transposon mutant strains from the study by Tiensuu et al. [A4:E8, C10:A8, C14:C12, D9:B6, and D2:C10 (32), here renamed 1RsbS (H23R), 2RsbU (E103K), 3RsbS (H23R), 4RsbV (E42R), and 5RsbV (R47Y), respectively] were selected to investigate the possible effects of these insertions on the regulation of σB activity in L. monocytogenes EGD-e (Tables 1 and 2). These ringless mutant strains were first reconfirmed to be defective for ring formation (data not shown), a phenotype exhibited on soft-agar media in response to 12-h cycles of light and dark and known to be under σB control (32) (Table 1). Since σB plays an important role in acid tolerance, we reasoned that transposon mutations influencing σB activity would also lead to an altered acid resistance phenotype. To investigate this, the five ringless mutant strains were tested for their ability to withstand a challenge at pH 2.5 and compared to the WT (EGD-e) and two control strains that harbor a transposon insertion but still retain the ring phenotype (B14:A6 and B15:E2) (32). The WT and the two ring-forming strains behaved similarly in this assay, showing high-level resistance to lethal acidic conditions, while a mutant strain lacking σB (ΔsigB) was exquisitely sensitive to acid, showing significantly decreased viable counts at 30 min and no viable counts at 60 min (Fig. 1). The ringless transposon mutant strains all displayed a significant increase in acid sensitivity compared to the ring-forming WT and ring-forming control strains. These data strongly suggested that σB activity was compromised in these strains.
TABLE 1.
Strains and plasmids used for this study
| Plasmid or straina | Transposonb | Ring formatione | Reference or source |
|---|---|---|---|
| Plasmids | |||
| pKSV7-Plmo2230::egfp | NAf | NA | 24 |
| pMAD | NA | NA | 56 |
| pMAD::ΔsigB | NA | NA | 68 |
| pMAD::ΔrsbX | NA | NA | This study |
| pEX-K168::Δlmo0596 | NA | NA | Eurofins Genomics |
| pMAD::Δlmo0596 | NA | NA | This study |
| Strains | |||
| Escherichia coli One Shot TOP10 | NA | NA | Invitrogen |
| L. monocytogenes EGD-e WT | − | + | K. Boor |
| L. monocytogenes EGD-e ΔsigB | − | − | This study |
| L. monocytogenes EGD-e ΔrsbX | − | NA | This study |
| L. monocytogenes EGD-e Δlmo0596 | − | + | This study |
| L. monocytogenes EGD-e ΔsreB | − | + | 37 |
| L. monocytogenes EGD-e A4:E8c [1RsbS (H23R)]d | + | − | 32 |
| L. monocytogenes EGD-e A4:D7c | + | − | 32 |
| L. monocytogenes EGD-e A1:D10c | + | − | 32 |
| L. monocytogenes EGD-e A4:B1c | + | − | 32 |
| L. monocytogenes EGD-e A3:G10c | + | − | 32 |
| L. monocytogenes EGD-e C10:A8c [2RsbU (E103K)]d | + | − | 32 |
| L. monocytogenes EGD-e C14:C12c [3RsbS (H23R)]d | + | − | 32 |
| L. monocytogenes EGD-e C12:F3c | + | − | 32 |
| L. monocytogenes EGD-e C9:C1c | + | − | 32 |
| L. monocytogenes EGD-e D9:B6c [4RsbV (E42R)]d | + | − | 32 |
| L. monocytogenes EGD-e D2:C10c [5RsbV (R47Y)]d | + | − | 32 |
| L. monocytogenes EGD-e B14:A6c | + | + | 32 |
| L. monocytogenes EGD-e B15:E2c | + | + | 32 |
| eGFP reporter strains | |||
| L. monocytogenes EGD-e WT/pKSV7-Plmo2230::egfp | 24 | ||
| L. monocytogenes EGD-e ΔsigB/pKSV7-Plmo2230::egfp | This study | ||
| L. monocytogenes EGD-e A4:E8/pKSV7-Plmo2230::egfp | This study | ||
| L. monocytogenes EGD-e C10:A8/pKSV7-Plmo2230::egfp | This study | ||
| L. monocytogenes EGD-e C14:C12/pKSV7-Plmo2230::egfp | This study | ||
| L. monocytogenes EGD-e D9:B6/pKSV7-Plmo2230::egfp | This study | ||
| L. monocytogenes EGD-e D2:C10/pKSV7-Plmo2230::egfp | This study | ||
| L. monocytogenes EGD-e B14:A6/pKSV7-Plmo2230::egfp | This study | ||
| L. monocytogenes EGD-e B15:E2/pKSV7-Plmo2230::egfp | This study | ||
See Table 2 for full genotype descriptions.
Presence or absence of a chromosomal mariner-based transposon insertion.
Nomenclature consistent with that described previously by Tiensuu et al. (32).
New designation of the transposon mutant strain.
Ring formation on soft-agar plates in response to cycles of light/dark (ring formation requires σB).
NA, not applicable.
TABLE 2.
WGS results for L. monocytogenes EGD-e transposon mutant strains
| Strain codeh | Transposon locationh | Transposon location (by WGS) | SNP location in sigB operona | Frameshift resultf | New strain name |
|---|---|---|---|---|---|
| A4:E8 | lmo0040 | lmo0770c | IN(rsbS)67 (+GATC) | p.H23Rfs*16 | 1RsbS (H23R) |
| A4:D7 | lmo0101-lmo0102b | —e | IN(rsbS)67 (+GATC) | p.H23Rfs*16 | NAi |
| A1:D10 | lmo0842 | —e | IN(rsbS)67 (+GATC) | p.H23Rfs*16 | NA |
| A4:B1 | lmo2682 | —e | IN(rsbS)67 (+GATC) | p.H23Rfs*16 | NA |
| A3:G10 | lmo2777 | —e | IN(rsbS)67 (+GATC) | p.H23Rfs*16 | NA |
| C10:A8 | lmo0124 | lmo0125c | Δ(rsbU)306 (−G) | p.E103Kfs*7 | 2RsbU (E103K) |
| C14:C12 | lmo0595-lmo0596b | —e | IN(rsbS)67 (+GATC) | p.H23Rfs*16 | 3RsbS (H23R) |
| C12:F3 | lmo0774 | —e | Δ(rsbU)506 (−AT) | p.Y170Rfs*28 | NA |
| C9:C1 | lmo1736 | —e | Δ(rsbS)6 (−T) | p.D21Efs*6 | NA |
| D9:B6 | lmo0101 | lmo0101-lmo0102b,c,d | IN(rsbV)136 (+TGTAC) | p.R47Yfs*6 | 4RsbV (E42R) |
| D2:C10 | lmo2668 | —e | IN(rsbV)120 (+A) | p.E42Rfs*17 | 5RsbV (R47Y) |
| B14:A6 | —g | lmo1671 | None | NA | NA |
| B15:E2 | —g | lmo2287 | None | NA | NA |
SNP position in the L. monocytogenes chromosome identified by WGS.
Transposon insertion located in the intergenic region.
Different transposon position than initially reported.
Not in the same position as A4:D7.
Same position as reported previously (32).
Nomenclature was adapted from that recommended by the Human Genome Variations Society (79). For example, in “p.H23Rfs*16,” p.H23R refers to the first encoded residue affected in the new protein, resulting in a histidine-to-arginine substitution at codon 23, while fs*16 refers to the number of codons in the new reading frame that would be translated prior to encountering a stop codon.
Not previously analyzed by WGS (32).
See reference 32.
NA, not applicable.
FIG 1.
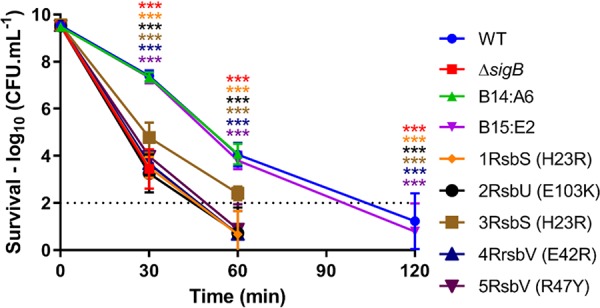
“Ringless” transposon mutant strains have acid-sensitive phenotypes. Stationary-phase cultures were grown in BHI medium at 37°C before being challenged in acidified BHI medium (pH 2.5) at 37°C. At 0, 30, 50, and 120 min, samples were taken for viable count measurements. The dotted line represents the detection threshold. Each marker represents the measurement average from three independent biological replicates performed, with the respective standard deviations. Statistical analysis was performed using a paired Student t test (***, P value of <0.001).
The ringless transposon mutants harbor secondary mutations in the sigB operon conferring reduced σB activity.
To assess whether additional mutations could be present in the transposon mutant strains displaying the ring and ringless phenotypes, we performed WGS on each of them. The locations of the transposon insertions were found to be identical to those described previously by Tiensuu et al. (32) for eight of the strains tested (Table 2). However, there were three exceptions: the loci reported to carry the transposon insertions in strains 1RsbS (H23R) (lmo0040), 2RsbU (E103K) (lmo0124), and 4RsbV (E42R) (lmo0101) were somewhat different in the WGS analysis that we performed (Table 2). This difference is likely due to the different methods used to determine their positions in the two studies.
An analysis of the genome sequences for single nucleotide polymorphisms (SNPs) revealed that all 11 of the ringless mutant strains had mutations in the sigB operon. Specifically, either the rsbS, rsbU, or rsbV gene carried frameshift mutations that were predicted to result in deeply truncated versions of the corresponding protein product (Fig. 2 and Table 2). Six of the transposon mutant strains carried the same mutant allele in the rsbS gene, a 4-nucleotide insertion resulting in a premature stop at codon 38 (H23R) (Table 2). The other five mutant strains all carried unique frameshift alleles in rsbU, rsbS, or rsbV (Table 2). The two ring-forming control strains selected for our study (B14:A6 and B15:E2) showed no mutation in any of the sigB operon genes. Based on the known functions of the proteins encoded by the sigB operon, it seemed likely that these mutations, rather than the transposon insertions, were responsible for both the ringless and acid-sensitive phenotypes.
FIG 2.

Locations of the frameshift mutations in the sigB operon. Shown is a layout of the sigB operon with the respective promoter regions and terminator. Each arrow represents the ORFs of mazE, mazF, rsbR, rsbS, rsbT, rsbU, rsbV, rsbW, sigB, and rsbX with the respective locations of the identified mutations. White sections of the ORFs represent the alternate reading frames produced by the frameshift mutations in rsbS, rsbU, and rsbV until a PMSC is encoded.
To investigate this further, we focused our attention on strain 3RsbS (H23R) (formerly C14:C12 [Table 1]), predicted to express a truncated RsbS protein, since the loss of ring formation in this transposon-carrying strain was partially complemented in the study by Tiensuu et al. by providing the gene affected by the transposon insertion (lmo0596) in trans (32). The complementation suggested that the transposon insertion was responsible for the ringless phenotype, rather than the SNP in the rsbS gene, which we now suspected to be the principal cause of the phenotype. The WGS analysis revealed that the transposon was located 186 bp upstream from the start codon of lmo0596 and 40 bp upstream from the divergently transcribed gene sreB, which encodes a small RNA (sRNA) S-adenosylmethionine (SAM) riboswitch (37). We reasoned that if a disruption of either lmo0596 or sreB was responsible for the ringless phenotype, then the deletion of either gene should produce a similar phenotype. The deletion of neither gene produced a ringless phenotype, whereas the loss of sigB produced the expected loss of ring formation (see Fig. S1A in the supplemental material). If the transposon impacted σB activity through effects on either lmo0596 or sreB, then the loss of these genes might be expected to affect acid tolerance, another highly σB-dependent phenotype. However, the deletion of neither gene produced any detectable effect on survival at pH 2.5, unlike the ΔsigB mutation, which produced an acid-sensitive phenotype (Fig. S1B and C). Similarly, if the transposon insertion in this strain was affecting σB activity via an effect on these genes, the transposon might be expected to affect the transcription of one or both genes. Reverse transcription-quantitative PCR (RT-qPCR) was used to measure the relative levels of lmo0596 and sreB transcription in the transposon-carrying strain 3RsbS compared to the wild-type, the ΔsigB mutant, and two other transposon-carrying strains (the ring-forming strain B12:A6 and the ringless strain 1RsbS). The transcription of lmo0596 was confirmed to be σB dependent, as previously suggested (38), but no difference in transcription was observed between the 3RsbS and 1RsbS strains, which carry transposon insertions in completely separate loci (Table 2; Fig. S1D), nor was sreB transcription affected by the presence of the transposon (Fig. S1D). Finally, the deletion of lmo0596 had no effect on the transcription of the σB-dependent gene lmo2230, suggesting that this gene does not play a role in regulating σB activity (Fig. S1E). Taken together, these results suggest that the transposon insertion in strain 3RsbS is not responsible for the observed ringless phenotype in 3RsbS (H23S), and we therefore conclude that the altered phenotype in the 3RsbS (H23R) transposon mutant is due solely to the frameshift mutation in rsbS and suggest that the sigB operon mutations in the other strains are most likely responsible for their σB-related phenotypes as well.
To investigate this further, σB activity was measured in these strains using a previously described transcriptional reporter that fuses the highly σB-dependent promoter of lmo2230 (annotated as a putative arsenate reductase) to the enhanced green fluorescent protein (eGFP) gene (egfp) (39). This reporter was integrated into the genomes, upstream of the original promoter of lmo2230, of the WT, the ΔsigB mutant strain, and seven of the transposon insertion strains, including five ringless and two ring-forming control strains (Table 1). Fluorescence was recorded by fluorescence microscopy following growth to stationary phase at 37°C, a condition under which σB is known to be highly active (24, 39). As expected, the ΔsigB mutant strain had almost no detectable fluorescence, while the WT and ring-forming transposon control strains (B14:A6 and B15:E2) produced a strong fluorescent signal (Fig. 3A and B). In contrast, the ringless strains that carried mutations in rsbS, rsbU, or rsbV all produced a greatly reduced fluorescence signal albeit not as low as that of the ΔsigB deletion mutant strain. Western blotting was used to determine the levels of eGFP protein in the reporter strains under the same growth conditions. The results confirmed what was observed microscopically: very low levels of eGFP expression in the ΔsigB mutant strain and in the strains harboring sigB operon mutations but not in the WT or ring-forming transposon mutant strains (Fig. 3C and D). These data confirmed that σB activity was reduced in the strains that displayed both an acid-sensitive and a ringless phenotype. The most parsimonious explanation for these observations is that the sigB operon alleles in the ringless mutant strains were directly responsible for the reduced σB activity and associated phenotypes, especially since defined mutations in these genes, in both L. monocytogenes and B. subtilis, have previously been shown to result in reduced σB activity (14, 27–29, 31, 36, 40, 41). Moreover, the ring-forming transposon mutant strains used as controls exhibited phenotypes similar to that of the WT strain. It is highly unlikely, therefore, that the transposons themselves were responsible for any of the phenotypes detected in these strains.
FIG 3.

Transposon mutant strains carrying sigB operon mutations all have decreased σB activity. Stationary-phase cultures of WT, ΔsigB, and transposon mutant strains transformed with pKSV7-Plmo2230::egfp were grown at 37°C. (A and B) Measurements of fluorescence were made using images obtained by fluorescence microscopy (A) and particle quantification from fluorescence microscopy images (B). A total of 15 images were taken across three biological replicates. Particle counts were normalized against the WT strain and converted to percentages. (C) Western blotting using anti-GFP antibodies. The arrow shows the eGFP protein with a size of 27 kDa. (D) Quantification of the percentage of eGFP normalized against the WT, obtained from Western blot images. Data were generated from three independent biological replicates. Statistical analysis was performed by a paired Student t test (***, P value of <0.001; NS, nonsignificant).
The rsbS frameshift results in a polar effect on rsbT.
The sigB operon has the gene order mazEF-rsbRSTUVW-sigB-rsbX, with a σA promoter located upstream from rsbR and a σB-dependent promoter upstream from rsbV (Fig. 2). Mutations in rsbS, rsbU, and rsbV could potentially produce polar effects on downstream genes, further impacting the pathway leading to σB activation. To address this, Western blot assays using polyclonal antibodies against RsbT and σB were performed on each of the strains. The levels of RsbT were markedly reduced in the ringless strains carrying the rsbS-H23R allele but were similar to those of the WT for the strains carrying either the rsbU or rsbV frameshift alleles (Fig. 4A). In contrast, the levels of σB protein were similar in all the strains (with the exception of the ΔsigB mutant strain) (Fig. 4B). These data show that the effects of the rsbU and rsbV frameshift alleles on σB activity are probably directly caused by the loss of these proteins. The rsbS frameshift allele affects the expression of RsbT, suggesting that these genes may be translationally coupled and that the loss of σB activity in this strain likely arises through the loss of both stressosome-associated proteins.
FIG 4.

Frameshift mutations in rsbS produce a polar effect resulting in the inhibition of RsbT translation. Western blot images were obtained from total protein extractions of stationary-phase WT, ΔsigB, and transposon mutant strains grown at 37°C. Western blots were probed with rabbit polyclonal anti-RsbT (A) and anti-σB (B) antibodies. Arrows point to the respective proteins, RsbT with a predicted size of 14.7 kDa and σB with a predicted size of 29.5 kDa.
Reduced σB activity confers an increased growth rate at higher temperatures.
Since the procedure for generating the transposon mutants involved an incubation step at a range of different temperatures (32, 42), we hypothesized that sigB operon mutations might arise if the mutant strains had a growth advantage under these conditions. To assess this, cultures of the WT, the ΔsigB mutant strain, one of the ringless mutant strain [designated 2RsbU (E103K)], and a ring-forming transposon mutant strain control (B14:A6) were grown at 30°C, 37°C, 40°C, and 42°C, and their growth rates were determined (Fig. 5). At 30°C, there was no significant effect of the genotype on the growth rates, whereas at higher temperatures, the ringless mutant strains lacking sigB or with an rsbU frameshift allele (E103K) exhibited a slight increase in the growth rate. At 42°C, these two mutant strains grew noticeably faster than the WT and ring-forming control B14:A6 strains, especially as the cultures approached stationary phase between 4 and 7 h after inoculation (Fig. 5D). This finding raised the possibility that mutations arising spontaneously in the sigB operon could be selected for at higher temperatures because of a growth advantage relative to WT cells, possibly due to the reduced σB activity.
FIG 5.

Loss of σB activity is associated with increased growth rates at elevated temperatures. (A to D) Measurements of OD600 values for the WT, ΔsigB, 2RsbU (E103K), and B14:A6 strains were performed every hour for 12 h in BHI medium at 30°C (A), 37°C (B), 40°C (C), and 42°C (D). (E) Growth rates in hours were calculated by using the log10 (OD600) value for the transition period from data points between 4 and 5 h (for 37°C, 40°C, and 42°C) and between 5 and 6 h (30°C). Data were generated from three biological replicates. Statistical analysis was performed using a paired Student t test (*, P value of <0.05; **, P value of <0.01; ***, P value of <0.001; NS, nonsignificant).
To further examine this possibility, we performed mixed-culture competition experiments to determine if the growth rate advantage would enable the ΔsigB and rsbU mutant (2RsbU [E103K]) to outcompete the WT during growth at elevated temperatures. Cultures were prepared by mixing the WT with the ΔsigB mutant strain, the ringless rsbU mutant strain [2RsbU (E103K)], or the ring-forming control strain B14:A6 at a ratio of 1,000:1 (WT to mutant). Cultures were grown over a period of 5 days, with daily passage into fresh medium, and dilutions were plated daily onto brain heart infusion (BHI) agar to determine the relative proportion of each strain. The erythromycin (Ery) resistance of the transposon-containing strains was used to differentiate the WT from mutant strains, while a difference in the colony color was used to discriminate the WT and the ΔsigB mutant strains, as described in Materials and Methods. When the mixed cultures were grown at 42°C, the ringless mutant strains carrying the rsbU frameshift allele accumulated steadily, reaching approximately 50% of the population after 5 days (Fig. 6A). In contrast, the ring-forming control strain did not accumulate in the culture over this period (Fig. 6B). At 42°C, the ΔsigB mutant strain accumulated within the population, dominating it (80:20) by the end of 5 days. However, at 30°C, where this growth advantage was absent (Fig. 5A), the ΔsigB mutant strain failed to fully dominate the population, reaching approximately 10% by the end of 5 days (Fig. 6D). When cultures were mixed 1:1 and grown at 42°C, the advantage was less evident for the ringless strain carrying the rsbU allele, but the ΔsigB mutant strain still dominated the WT under these conditions (Fig. S2). When the WT was started as the minority strain (1:1,000) in these competition assays, it failed to emerge as the dominant population when competed against any of three mutant strains tested (Fig. S2E to G). Overall, the data suggest that strains with reduced σB activity can accumulate in a mixed population when the growth temperature is elevated.
FIG 6.

Loss of σB activity is associated with a competitive advantage at 42°C. Shown are data for mixed cultures at ratios of 1,000:1 of the WT with 2RsbU (E103K) (A), the WT with B14:A6 (B), and the WT with the ΔsigB mutant (C) incubated at 42°C and the WT with the ΔsigB mutant incubated at 30°C (D). Passages were made every 24 h for 5 days. WT and transposon mutant strains were distinguished by erythromycin resistance. WT and ΔsigB mutant strains were distinguished by colony color. Results are depicted as relative abundances of CFU per milliliter. Data were generated from three independent biological replicates. Statistical analysis was performed using a paired Student t test (*, P value of <0.05; **, P value of <0.01; ***, P value of <0.001; NS, nonsignificant).
Loss of rsbX results in reduced competitiveness.
The observations described above suggest that a growth advantage arises in strains with reduced σB activity when mild heat stress is present. A corollary of this conclusion is that increased σB activity might be expected to have a negative effect on growth and competitiveness. In the current model of σB regulation in L. monocytogenes, RsbX is believed to act as phosphatase whose function is to reset the resting state of the stressosome following a stress signaling event (43, 44). Thus, RsbX plays a negative role in regulating σB activity, and the loss of RsbX is expected to have produced a strain with elevated σB activity. To investigate this, competition experiments were performed with a ΔrsbX mutant strain to test the competitiveness of this strain relative to the WT at both 30°C and 42°C (Fig. 7). When the ΔrsbX mutant was present as the minority strain (1,000-fold underrepresented at the start of the experiment), it failed to dominate the culture over 5 days of passage, regardless of the temperature (Fig. 7A and B), although it recovered its population somewhat over this period. When the WT was the minority strain, it outgrew the ΔrsbX strain over the same time period (Fig. 7C and F), showing that the WT had a competitive advantage over the mutant strain. When the populations were equal at the start of the experiment, the WT tended to dominate the mixed cultures (Fig. 7B and E). This result was observed at both incubation temperatures, probably because the growth of the ΔrsbX strain was inhibited regardless of the temperature. Overall, these results are consistent with the idea that mutations that increase σB activity produce a growth disadvantage.
FIG 7.

Competitive advantage of the WT strain when challenged against the ΔrsbX mutant strain at both 30°C and 42°C. Competition experiments with mixed cultures of the WT and ΔrsbX mutant strains were performed at 42°C and 30°C, and the data are shown as relative abundances in percentages of CFU per milliliter. (A to C) Cultures of the WT and ΔrsbX strains incubated at 42°C were mixed at ratios of 1,000:1 (A), 1:1 (B), and 1:1,000 (C). (D to E) Cultures at the same ratios were mixed and incubated at 30°C. Passages were made every 24 h for 5 days. Statistical analysis was performed using a paired Student t test (*, P value of <0.05; **, P value of <0.01; ***, P value of <0.001).
Premature stop codons occur with a higher-than-average frequency in the positive regulators of σB within the sigB operon.
The occurrence of sigB operon mutations in this study and in other studies (28, 31, 33–36) prompted us to investigate whether undocumented premature stop codons (PMSCs) might be present in the sigB operon in genome sequences deposited in public databases, so we searched for PMSCs in the sigB operon among 22,340 L. monocytogenes genome assemblies. The PMSC rate per 100 bp (expressed as a percentage per 100 bp to normalize for gene length) for genes in the sigB operon that positively affect σB activity (rsbT, rsbU, and rsbV) was considerably higher than for genes that act negatively (rsbW and rsbX). Indeed, of all the genes that we included in the analysis, rsbU was found to have the highest occurrence of PMSCs (Fig. 8A and B). Unexpectedly, mazF showed a high PMSC rate similar to those of rsbT, rsbU, and rsbV. The gene mazF encodes an endoribonuclease, a component of a toxin-antitoxin system along with mazE, which is speculated to be an additional regulator for σB in Staphylococcus aureus (45, 46) and to have a positive effect on σB-dependent genes in L. monocytogenes in the presence of norfloxacin (47). The high PMSC rate of mazF may be associated with its putative positive regulation of σB; thus, it may also be subjected to the same selective pressure as σB positive regulators. Interestingly, rsbW, which encodes the anti-sigma factor that negatively regulates σB activity, had no occurrence of PMSCs. As expected, essential genes such as sigA, which encodes the principal housekeeping sigma factor in L. monocytogenes, have a very low rate of PMSC occurrence, suggesting that this measure reflects biological significance rather than just sequencing errors in the database. Interestingly, we found that the gene inlA, encoding internalin A, was found to possess a very high PMSC rate per 100 bp (0.86% from a total of 4,576 PMSCs found [data not shown]). A previous study found that many L. monocytogenes strains in lineage II possess PMSCs in inlA (48); for this reason, we excluded this gene from Fig. 8. Interestingly, inlB, which shares the same operon as inlA, also possesses a high PMSC rate although not as high as that of inlA (Fig. 8). Taken together, these data indicate that within the published genome sequence data for L. monocytogenes, there is a high occurrence of mutations that are predicted to reduce σB activity and a low occurrence of mutations that would act to increase it.
FIG 8.

Occurrence of PMSCs is elevated in genes encoding positive effectors of σB activity. Shown are results for PMSC mutations retrieved from the in silico analysis of the 22,340 genomes of L. monocytogenes strains available. (A and B) The total number of identified PMSCs by ORF length (A) and the PMSC rate normalized by 100 bp of the ORF length (B) for genes comprising the sigB operon and characterized as putative positive regulators of σB activity (red), the putative σB negative regulators rsbX and rsbW (black), and the mazEF genes (orange). MLST genes are in blue, and rsbR paralogues are in purple. The housekeeping gene sigA and the sigma factor genes sigL and sigH are in gray. Two genes downstream of the sigB operon, lmo0897 and lmo0898, and genes encoding metabolic pathways of glutathione (lmo1702, lmo0983, lmo1433, lmo0906, and lmo2770), glycerol (lmo1293), threonine (thrC), isoleucine (ilvA and ilvB), and glucose (lmo2253) are in green, and prfA, inlB, hly, plcA, and plcB are in brown. (C) Expanded area indicated by the square in panel B.
DISCUSSION
In this study, we investigated the emergence and selection of spontaneous mutations inactivating σB within populations of L. monocytogenes. We first identified these mutations in a collection of L. monocytogenes “ringless” transposon mutant strains whose response to visible light was altered, a phenotype that requires both the blue-light sensor RsbL and the stress-inducible sigma factor σB (32). One interpretation of the mutants’ behavior was that the transposon insertions somehow influenced the signal transduction pathway leading to σB activation in response to light. Unexpectedly, we found that, in addition to the transposon insertions, all of the ringless mutant strains that we sequenced carried mutations in the sigB operon (rsbS, rsbU, or rsbV), which were predicted to produce premature stop codons in the corresponding coding sequences. In addition to the ringless phenotype, the transposon mutant strains also exhibit reduced acid tolerance and a marked decrease in σB activation compared to the WT strain. We conclude that the ringless and acid-sensitive phenotypes of these mutant strains are due to the presence of the sigB operon mutations rather than the transposon insertions (32). These mutations most likely interfere with signal transduction through the σB regulatory pathway since they are predicted to affect stressosome function (rsbS), dephosphorylation of the anti-anti-sigma factor RsbV (rsbU, which encodes a phosphatase), or partner switching with the anti-sigma factor RsbW (rsbV). Indeed, these data provide additional genetic support for the existing model for σB activation in L. monocytogenes (reviewed in references 49 and 50).
In previous studies, knockout deletions of the rsbS, rsbV, or rsbU gene in both L. monocytogenes and B. subtilis resulted in the impairment of signal transduction and reduced resistance against stress (14, 27–29, 31, 36, 40, 41). The premature stop codons identified in this study do not result in the full loss of σB activity since the σB-dependent Plmo2230::egfp reporter is still expressed in these transposon mutant strains (Fig. 3), and this correlates with an intermediate acid tolerance phenotype between that of the WT and that of the ΔsigB mutant strain (Fig. 1). It is noteworthy that no mutations were detected in sigB itself, which could suggest that a partial loss of function might be more advantageous than a complete loss of σB activity. The extent of the σB activity detected appears to depend on which sigB operon frameshift allele is present. There is significantly more σB activity present in strains carrying the frameshift allele encoding RsbS-H23R than there is in the strains carrying either of the two rsbV frameshift alleles (Fig. 3). This result suggests that the loss of a functional RsbV protein has a greater impact on σB activity than the loss of RsbS. RsbV serves as an anti-anti-sigma factor, whose role is to sequester the anti-sigma factor RsbW under stress conditions, thereby releasing σB for participation in transcription, while RsbS is an integral component of the stressosome stress-sensing apparatus (16, 51). It is possible that some stress signals can still be transduced through the pathway in the absence of RsbS but that this is less likely in the absence of RsbV. In B. subtilis, there is additional input into the pathway, which allows energy stress to be sensed via the RsbP and RsbQ proteins, independently of the stressosome (52). This pathway is not present in L. monocytogenes (29), but this does not preclude some other stressosome-independent means of transducing stress signals in this pathogen. Indeed, there is evidence that even in the absence of RsbV, some σB activation can occur under some growth conditions (40). In B. subtilis, RsbV-independent activation of σB occurs at elevated temperatures (53), and in L. monocytogenes, similar observations have been made during growth at low temperatures (40, 54). The possibility that RsbW could be regulated posttranslationally, for example, by proteolysis, allowing an additional layer of control over σB, is certainly worthy of further investigation.
Emergence of mutated alleles within the sigB operon.
The data presented here suggest an explanation for the common detection of mutations in the sigB operon of L. monocytogenes (28, 33–36, 55), including a previous study in our own laboratory where an rsbV missense mutation arose during routine subculture (31). Although the occurrence of sigB operon mutations has been reported by others, the mechanism(s) that drives the selection of these mutations has remained elusive. Here, we show that the loss of σB function confers a competitive advantage under conditions where sublethal stress (in this case, heat stress) prevails. The advantage is manifested as both an increased growth rate (Fig. 5) and increased competitiveness in mixed cultures (Fig. 6). Furthermore, the absence of rsbX, which is predicted to increase σB activity, confers a competitive disadvantage under the same conditions (Fig. 7). Since a variety of the protocols for genetically modifying L. monocytogenes include a step with prolonged incubation at an elevated temperature (typically 40°C to 44°C) to prevent the replication of plasmids possessing a heat-sensitive origin of replication, used in order to encourage allele integration into the chromosome (42, 56), it is possible that this provides the necessary selective pressure to select mutant strains that negatively affect σB activity. Previous studies have reported fast-growth phenotypes for L. monocytogenes mutant strains lacking σB under stress conditions. In the presence of inhibitory doses of blue light, sigB mutant strains grow faster than the WT in liquid and solid media (31). Under glucose-limited conditions, mutant strains lacking σB or the positively acting regulator RsbT or RsbV were found to grow faster than the WT parent (29). A similar phenotype was observed during growth at 3°C (30) and under conditions of osmotic stress (57). Indeed, there is evidence in B. subtilis that mutant strains lacking σB can dominate the population in nutrient-limited chemostats (58). Taken together with the findings presented in this study, where a competitive advantage is demonstrated for mutant strains with reduced σB activity in mixed populations, it seems likely that this phenotype is the reason for the common emergence of mutations affecting σB activity during routine laboratory culture of this pathogen.
An analysis of the over 22,000 L. monocytogenes publicly available genome assemblies revealed a high rate of premature stop codons within genes of the sigB operon that positively regulate σB activity (Fig. 8), suggesting that there is some selective pressure driving this process. It is not possible to determine precisely when these PMSCs occurred, and so it is unclear at present whether these mutations arose during laboratory culture or whether they were already present in the wild isolates. It is possible that conditions other than mild heat stress can confer a growth advantage on mutant strains with reduced σB activity, as has been observed previously with light stress and salt stress (57, 59). In Escherichia coli, prolonged starvation during stationary phase frequently results in mutant strains displaying a so-called GASP (growth advantage in stationary phase) phenotype, and these arise as a result of the reduced expression or activity of σS (RpoS), the sigma factor that controls the general stress response in this organism (60, 61). Indeed, mutations affecting rpoS frequently arise during laboratory domestication of E. coli strains (62). A similar explanation has been postulated to account for these observations in E. coli; namely, the loss of σS function arises when a fitness advantage accrues through the allocation of resources to growth rather than to the costly general stress response (63, 64). Thus, the phenomenon that we describe in this study is likely a reflection of a general biological principle where competition within populations occasionally favors the emergence of variants that have acquired a growth advantage at the expense of their ability to withstand potentially lethal stress.
σB deployment is a trade-off between growth and survival.
Our study raises the important questions of how and why σB negatively affects growth and competitiveness in mixed cultures at elevated temperatures. A number of possible, perhaps coexisting, models could account for these phenomena. First, it is possible that the expression of the large σB regulon (approximately 300 genes), many components of which are actively involved in homeostatic and repair functions, represents a significant energy burden on the cells. Freeing the cells from this energy burden could make more resources available for biosynthesis and growth. Second, it is possible that there is a limited transcriptional capacity in the cell and that the absence of σB allows the housekeeping sigma factor (σA) to have greater access to the RNA polymerase core enzyme, which leads to the more efficient transcription of genes involved in growth-related functions. This idea has been proposed previously to account for the emergence of sigma S (rpoS) mutations in E. coli (65–67). Third, it is possible that σB actively restricts growth, perhaps to ensure that protection and repair functions have sufficient time to mitigate the damaging effects of stress. We recently provided evidence for this model, as we identified an sRNA under σB transcriptional control (Rli47) that acts to restrict the biosynthesis of isoleucine, even when isoleucine is absent from the growth medium (68). This somewhat surprising finding could provide evidence of deliberate σB-controlled growth restriction. It is clear that further experiments will be needed to specifically test these possibilities and establish the basis for the fast-growth phenotype associated with the loss of σB activity.
Overall, this study highlights the frequent occurrence of secondary mutations negatively affecting σB activity in newly constructed strains of L. monocytogenes. It further suggests that caution needs to be exercised by researchers to ensure that this issue does not confound the interpretation of phenotypic data, especially where temperature selection has been used during strain construction. The availability and comparative affordability of whole-genome sequencing for bacteria make it possible to routinely sequence the genomes of newly constructed strains in order to avoid this issue, and indeed, the availability of these data will make it easier to compare the behaviors of strains between groups. Finally, this study clearly illustrates the cost to the cell of deploying the σB-mediated general stress response; protection against stress is a resource-intensive process, but presumably, the long-term survival benefits outweigh the short-term costs.
MATERIALS AND METHODS
Bacterial strains, plasmids, and primers.
Bacterial strains and plasmids used in this study are presented in Table 1. Oligonucleotide primer sequences are shown in Table 3. Strains were grown in brain heart infusion (BHI) broth or agar (LabM) at 37°C unless otherwise specified, with constant shaking at 150 rpm. Cells were grown for 16 h until stationary phase was reached. The following antibiotics were added to the media when required: chloramphenicol (Chl) at 10 μg · ml−1, erythromycin (Ery) at 5 μg · ml−1, tetracycline (Tet) at 2.5 μg · ml−1 for L. monocytogenes mutant strains, and ampicillin (Amp) at 100 μg · ml−1 for E. coli.
TABLE 3.
Primers used for this study
| Primer sequence (5′–3′) | Target |
|---|---|
| GCCTTGTCGCCATCTTTG | egfp-lmo2230-F integration |
| GGCCGTTTACATCTCCATC | egfp-lmo2230-R integration |
| ATAACGGCACAAGCTTCG | sigB-flank_F |
| TTATGGCGTCAACAGTCG | sigB-flank_R |
| GGAGTAAATGAACAAGGCAG | rsbX-flank_F |
| CGCTAGTTTAAAAGGTGTTATGG | rsbX-flank_R |
| CGGTCGACGTAGAGTCCATCGCCCGAA | rsbX-A |
| TTACTCCACTTCCTCATTCTGCAAC | rsbX-B |
| AATGAGGAAGTGGAGTAACCATAACACC | rsbX-C |
| CGAGATCTATCATTCCGGCAACAAGTAAATCTTGG | rsbX-D |
| CTAGCTAATGTTACGTTACA | pMAD-U |
| GCGAGAAGAATCATAATGGG | pMAD-L |
| GAAAATAAATTCCGGTTGCTAAGGC | lmo0596-A |
| AATCGAGTCGGATGGTTCTTGTT | lmo0596-B |
| CCACTCCTCTTTGATATGTATTTAT | lmo0596-flank_F |
| GGCAGATGAATGCACTTATG | lmo0596-flank_R |
| RT-qPCR | |
| TGGGGAGCAAACAGGATTAG | 16S_F |
| TAAGGTTCTTCGCGTTGCTT | 16S_R |
| CATATTCGAAGTGCCATTGC | lmo2230_F |
| CTGAACTAGGTGAATAAGACAAAC | lmo2230_R |
| CCTAAACTTGGATTTCCGACTTATCTT | sreB_F |
| TTCTTATCACGAAAGGTGGAGGG | sreB_R |
| GGGTACTAGCTGACGGAATTTTATC | lmo0596_F |
| CCCACATACCGAAAAGTAATACGAG | lmo0596_R |
Construction of genetically modified L. monocytogenes.
An L. monocytogenes transposon mutant strain library was previously constructed by Tiensuu et al. (32). Electrocompetent cells were created as previously described (69). The L. monocytogenes EGD-e ΔsigB mutant strain was constructed using a previously built shuttle vector, pMAD::ΔsigB (68). The shuttle vector pMAD::ΔrsbX was generated by amplifying by PCR the rsbX-flanking regions using primers rsbX-A and -B and primers rsbX-C and -D in separate reactions. Both flanks were joined together by splicing by overlap extension (SOE) PCR (69) using primers rsbX-A and -D. The resulting amplicon was digested with SalI and BglII and cloned into the pMAD vector, creating pMAD::ΔrsbX. Confirmation of the construct was carried out by PCR using primers pMAD-U and pMAD-L. pMAD::Δlmo0596 was constructed by digesting the artificially synthesized vector pEX-K168::Δlmo0596 (Eurofins Genomics) with BamHI and SalI. The digestion product, containing 300 bp both upstream and downstream of the lmo0596 open reading frame (ORF), was ligated into the pMAD vector, creating pMAD::Δlmo0596. Its construction was verified by PCR with primers lmo0596-A and lmo0596-B. The mutagenic vectors pMAD::ΔsigB, pMAD::ΔrsbX, and pMAD::Δlmo0596 were separately transformed into electrocompetent L. monocytogenes EGD-e cells. Transformants were selected from BHI agar supplemented with Ery for growth at 30°C. Chromosomal integration of the vector was achieved by growing transformants at 39°C overnight in BHI medium supplemented with Ery. Cultures were plated onto BHI agar containing the same antibiotic and X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) (50 μg · ml−1), and colonies were allowed to grow at 41°C overnight. Blue colonies were grown at 30°C overnight and at 39°C for 3 h. Serial dilutions were plated onto BHI–X-Gal (50 μg · ml−1) plates and grown at 30°C for 2 days. White colonies (indicating excision and loss of the plasmid) were screened for erythromycin sensitivity, and deletion knockouts of sigB, rsbX, and lmo0596 were checked by colony PCR using primers sigB-flank_F and sigB-flank_R, lmo0596-flank_F and lmo0596-flank_R, and rsbX-flank_F and rsbX-flank_R, respectively. The σB reporter vector pKSV7-Plmo2230::egfp (24) was transformed into electrocompetent L. monocytogenes transposon mutant strains. Electrotransformed colonies were selected from BHI agar plates containing Chl and incubated at 30°C. Plasmid integration in the chromosome was achieved as previously described (24), by incubating fluorescent cells at 42°C. The plasmid’s chromosomal integration occurred upstream of the original lmo2230 promoter region by homologous recombination. Integration was verified by PCR (using primers egfp-lmo2230-F integration and egfp-lmo2230-F integration).
Acid tolerance (pH 2.5).
Stationary-phase cultures were pelleted down by centrifugation and resuspended in BHI medium previously acidified with 5 M HCl until pH 2.5 was reached. Resuspensions were incubated in a water bath at 37°C for 120 min. Samples were taken at 0, 30, 60, and 120 min; serially diluted from 10−7 to 10−2 in phosphate-buffered saline (PBS); and plated onto BHI agar plates. Plates were then incubated at 37°C for 24 h, and colonies were counted. A minimum of three biological replicates were made.
Whole-genome sequencing.
Transposon mutant strain genomic DNA was extracted using a DNeasy blood and tissue kit (Qiagen) according to the manufacturer’s recommendations. Purified genomic DNA was sent to MicrobesNG for whole-genome sequencing (WGS). The obtained trimmed reads were used for SNP identification analysis performed with Breseq (70). The transposon sequence was located in each transposon mutant strain by using the contig reads on Mauve-Multiple Genome Alignment (71). The L. monocytogenes EGD-e (NCBI RefSeq accession no. NC_003210.1) genomic sequence was used as the reference genome in both analyses.
Microscopic quantification of eGFP fluorescence.
Cells containing the eGFP σB reporter (pKSV7-Plmo2230::egfp) integrated into the L. monocytogenes chromosome were grown for 16 h to stationary phase. Cultures were mixed in a 1:1 (vol/vol) mixture of ice-cold methanol–ethanol and placed at −20°C for 10 min. The mixtures were centrifuged afterward at 10,000 × g for 10 min at 4°C. The supernatant was removed, the pellet was resuspended in ice-cold PBS, and cells were further adjusted to an optical density at 600 nm (OD600) of 1. Fluorescence microscopy was performed with a Nikon Eclipse E600 microscope by using a B-2A filter covering the eGFP excitation and emission wavelengths. A total of 5 images per biological replicate were taken across different fields of the slide. Images were digitally captured using a charge-coupled-device (CCD) camera attached to the microscope. Relative fluorescence intensities were reported after automated image processing of multiple fields with ImageJ 1.44 software (72), with appropriate manipulations as described previously by others (73, 74).
SDS-PAGE and Western blotting.
The total protein fractions were extracted from stationary-phase cultures grown at 37°C. Tet was added to the cultures, and the cultures were centrifuged at 9,000 × g for 15 min at 4°C. Cells were resuspended in sonication buffer containing 13 mM Tris-HCl, 0.123 mM EDTA, and 10.67 mM MgCl2 adjusted to pH 8.0. Cell suspensions were digested with 1 mg · ml−1 lysozyme (Sigma-Aldrich) for 30 min and centrifuged again. Pellets were resuspended in sonication buffer containing 1% (vol/vol) protease inhibitor cocktail (catalog number P2714; Sigma-Aldrich). Resuspensions were then transferred into cryotubes containing zirconia-silica beads (Thistle Scientific) and bead beaten in a FastPrep-24 instrument at a speed of 6 m · s−1 for 40 s, twice. Lysates were centrifuged at 13,000 × g for 30 min at 4°C, and the supernatant was recovered. Total protein quantification was performed using a DC protein assay (Bio-Rad) according to the manufacturer’s recommendations. Protein extracts were normalized to a total protein concentration of 0.8 mg · ml−1, and 12 μl of each sample was separated by SDS-PAGE (15% acrylamide-bisacrylamide), along with the PageRuler Plus prestained protein ladder (Thermo Scientific). Separated protein was transferred onto a polyvinylidene difluoride (PVDF) membrane and blocked with Tris-buffered saline (TBS) supplemented with 3% (wt/vol) skim milk powder (Sigma). SDS-PAGE gels were further stained with GelCode blue staining reagent (Thermo Scientific) and destained with a destaining solution (20% acetic acid and 10% methanol) (see Fig. S3 in the supplemental material). For immunoblots, rabbit polyclonal IgG anti-GFP (Santa Cruz Biotechnology) and rabbit polyclonal IgG anti-σB (40) were diluted 1:500 and 1:1,500, respectively, in TBST (TBS supplemented with 0.5% [vol/vol] Tween 20; Sigma). Rabbit polyclonal IgG anti-RsbT was diluted 1:5,000 with a SignalBoost immunoreaction enhancer kit (Merck). The secondary antibody mouse anti-rabbit IgG–horseradish peroxidase (HRP) (Santa Cruz Biotechnology) was diluted 1:6,500 in TBS or with the SignalBoost immunoreaction enhancer kit (Merck) when required. Blots were visualized on an Odyssey Fc imaging system (Li-Cor Biosciences).
Growth kinetics.
Stationary-phase cells of the L. monocytogenes WT, ΔsigB, B14:A6, and 2RsbU (E103K) strains grown in BHI medium at 37°C were adjusted to an OD600 of 0.05 and grown in BHI medium at 30°C, 37°C, 40°C, and 42°C. The OD600 of each culture was measured every hour for 12 h. Growth rates were calculated by determining the slope during the transition into the stationary phase, between 4 and 5 h at 37°C, 40°C, and 42°C and between 6 and 7 h at 30°C. A minimum of 3 biological replicates were made.
Competition experiments.
L. monocytogenes WT, the ringless 2RsbU (E103K) strain, and the ring-forming control strain B14:A6 were grown in BHI medium at 37°C for 16 h until stationary phase was reached. Cultures were adjusted to an initial OD600 of 0.05 in fresh BHI medium at final ratios of 1:1,000, 1:1, and 1,000:1 of mixtures of the WT with 2RsbU (E103K) and of the WT with B14:A6, at the indicated temperatures. Passages were made every 24 h by diluting the cultures 1:100 into fresh BHI medium. Samples were taken, diluted to 10−7 in PBS in every passage, plated onto both BHI medium and BHI medium supplemented with erythromycin to distinguish the non-erythromycin-resistant WT from the resistant transposon mutant strain colonies, and incubated at 37°C. Colonies were counted after 24 h of incubation. Ratios were calculated by subtracting the number of erythromycin-resistant colonies from the total number of colonies. Competition cultures of the WT versus the ΔsigB mutant and of the WT versus the ΔrsbX mutant were performed as described above, with the following modifications. Cultures were incubated on BHI agar plates for 24 h at 37°C. Differences in colony morphology were used to distinguish the WT from both the ΔrsbX and ΔsigB strains. The colony size of ΔrsbX colonies was significantly smaller than that of the WT strain. For WT-versus-ΔsigB mutant competitions, BHI plates were further incubated at 30°C for 7 days, and strains were distinguished by differences in colony color. We observed that the WT colonies appeared whiter than the ΔsigB colonies. Confirmation of WT and ΔsigB mutant strain colonies was carried out by colony PCR by amplifying the flanking regions of sigB (using primers sigB-flank_F and sigB-flank_R). A total of 60 colonies, 30 of each color, were tested using a MyTaq DNA polymerase kit (Bioline). WT and ΔrsbX mutant strains were also confirmed via the same method by amplifying the flanking regions of rsbX (using primers rsbX-flank_F and rsbX-flank_R) from a total of 24 colonies, 12 of the WT and 12 of the ΔrsbX mutant. This method enables the WT to be distinguished from the ΔsigB mutant with a precision of 98.3% and enables the WT to be distinguished from the ΔrsbX mutant with 100% precision.
In silico analysis of premature stop codon occurrence rates.
A DNA BLAST database was constructed for 22,340 L. monocytogenes genome assemblies available from the NCBI database (182 complete genomes, 45 chromosomal genomes, and 22,113 assembly scaffolds or contigs [accessed 5 July 2019]). DNA coding sequences of 41 genes of interest, including all 10 genes from the sigB operon, 2 genes immediately downstream from the sigB operon, 7 housekeeping genes used for multilocus sequencing typing (MLST) (75), 4 rsbR paralogues, 3 sigma factor genes, 5 virulence factors, and 10 genes intervening in several different metabolic pathways, were extracted from the L. monocytogenes EGD-e strain and used as query sequences. BLASTn was performed with each query sequence against all 22,340 DNA databases, and the DNA sequences were then extracted for the closest match. Genes were considered absent or incomplete in a given genome assembly when either (i) the BLASTn top hit was empty, (ii) the start or end of the subject sequence was less than 20 bp from the end or start of the contig in which the subject sequence was found, or (iii) the subject sequence was shorter than the query sequence by ≥25 bp. Otherwise, the gene was considered present and complete in the genome assembly and then translated with Biopython (76) according to the bacterial translate table to determine the position of stop codons and identify premature stop codons (PMSCs) (defined as codons that truncate the gene length to less than 90% of the length of the gene in the reference strain EGD-e). For each gene analyzed, the rate of occurrence of PMSCs was calculated as a rate normalized to 100 bp to account for the different gene lengths in the analysis.
Ring formation phenotype.
Cultures were grown overnight at 30°C, and 2 μl was spotted onto BHI soft-agar plates (0.3% [wt/vol] agar no. 2) previously dried in a laminar flow hood for 10 min. Plates were incubated at 30°C for 4 days either in constant darkness or exposed to cycles of 12 h of light and dark in a blue-light array apparatus with an average intensity of 0.2 mW · cm−2. Photographs of the grown cultures were acquired using the Syngene GBox-Chemi 16 bioimaging system.
RNA extraction and RT-qPCR.
Cultures of L. monocytogenes WT and ΔsigB and Δlmo0596 mutant strains grown overnight in BHI medium were adjusted to an initial OD600 of 0.05 in fresh BHI medium and allowed to grow until mid-log phase was reached (OD600 = 0.8) or allowed to grow overnight until stationary phase was reached. Transcription was stopped by diluting the cultures in RNAlater (Sigma) at a 1:5 ratio. Total RNA was extracted using an RNeasy minikit (Qiagen) according to the manufacturer’s recommendations. Cell disruption was achieved by bead beating in a FastPrep instrument with the same parameters as the ones mentioned above. Total DNA was digested with Turbo DNA-free (Invitrogen) digestion according to the manufacturer’s recommendations. RNA integrity was verified by 0.7% agarose gel electrophoresis. cDNA was synthesized with the SuperScript IV first-strand synthesis system (Invitrogen) according to the manufacturer’s recommendations and further quantified using Qubit (Invitrogen). RT-qPCR was performed using a QuantiTect SYBR green PCR kit (Qiagen) and primers for the target genes (Table 3). Primer efficiencies for the target genes 16S, lmo2230, sreB, and lmo0596 were determined using purified L. monocytogenes genomic DNA. Samples were analyzed on a LightCycler 480 system (Roche) with the following parameters: 95°C for 15 min; 45 cycles of 15 s at 95°C, 15 s at 53°C, and 30 s at 72°C; a melting curve drawn for 5 s at 95°C and 1 min at 55°C, followed by increases of 0.11°C · s−1 until 95°C was reached; and cooling for 30 s at 40°C. Cycle quantification values were calculated by using LightCycler 480 software version 1.5.1 (Roche) and the Pfaffl relative expression formula (77, 78). The expression of the 16S rRNA gene was used as a reference. Three biological replicates, each in technical duplicates, were performed. Results are expressed as log2 relative expression ratios normalized against the WT strain.
Statistical analysis.
Student’s t test was performed using GraphPad Prism 8.
Supplementary Material
ACKNOWLEDGMENTS
This project has received funding from the European Union’s Horizon 2020 research-and-innovation program under Marie Skłodowska-Curie grant agreement no. 721456. Jialun Wu was funded by the Department of Agriculture, Food and the Marine (17/F/244).
We acknowledge Nicholas Waters for help in scripting.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Mylonakis E, Paliou M, Hohmann EL, Calderwood SB, Wing EJ. 2002. Listeriosis during pregnancy: a case series and review of 222 cases. Medicine (Baltimore) 81:260–269. doi: 10.1097/00005792-200207000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Farber J, Peterkin P. 1991. Listeria monocytogenes, a food-borne pathogen. Microbiol Mol Biol Rev 55:476–511. doi: 10.1128/MMBR.55.3.476-511.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbuddhe SB, Chakraborty T. 2009. Listeria as an enteroinvasive gastrointestinal pathogen, p 173–195. In Sasakawa C. (ed), Molecular mechanisms of bacterial infection via the gut. Springer-Verlag, Berlin, Germany. [DOI] [PubMed] [Google Scholar]
- 4.Gandhi M, Chikindas ML. 2007. Listeria: a foodborne pathogen that knows how to survive. Int J Food Microbiol 113:1–15. doi: 10.1016/j.ijfoodmicro.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 5.Jensen VB, Harty JT, Jones BD. 1998. Interactions of the invasive pathogens Salmonella typhimurium, Listeria monocytogenes, and Shigella flexneri with M cells and murine Peyer’s patches. Infect Immun 66:3758–3766. doi: 10.1128/IAI.66.8.3758-3766.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Y, Orsi RH, Boor KJ, Wiedmann M, Guariglia-Oropeza V. 2017. Home alone: elimination of all but one alternative sigma factor in Listeria monocytogenes allows prediction of new roles for σB. Front Microbiol 8:1910. doi: 10.3389/fmicb.2017.01910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaturongakul S, Raengpradub S, Palmer ME, Bergholz TM, Orsi RH, Hu Y, Ollinger J, Wiedmann M, Boor KJ. 2011. Transcriptomic and phenotypic analyses identify coregulated, overlapping regulons among PrfA, CtsR, HrcA, and the alternative sigma factors σB, σC, σH, and σL in Listeria monocytogenes. Appl Environ Microbiol 77:187–200. doi: 10.1128/AEM.00952-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sleator RD, Watson D, Hill C, Gahan CG. 2009. The interaction between Listeria monocytogenes and the host gastrointestinal tract. Microbiology 155:2463–2475. doi: 10.1099/mic.0.030205-0. [DOI] [PubMed] [Google Scholar]
- 9.Kim H, Marquis H, Boor KJ. 2005. σB contributes to Listeria monocytogenes invasion by controlling expression of inlA and inlB. Microbiology 151:3215–3222. doi: 10.1099/mic.0.28070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wise AA, Price CW. 1995. Four additional genes in the sigB operon of Bacillus subtilis that control activity of the general stress factor sigma B in response to environmental signals. J Bacteriol 177:123–133. doi: 10.1128/jb.177.1.123-133.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K, Barthelemy M, Vergassola M, Nahori M-A, Soubigou G, Régnault B, Coppée J-Y, Lecuit M, Johansson J, Cossart P. 2009. The Listeria transcriptional landscape from saprophytism to virulence. Nature 459:950–956. doi: 10.1038/nature08080. [DOI] [PubMed] [Google Scholar]
- 12.Ferreira A, Gray M, Wiedmann M, Boor KJ. 2004. Comparative genomic analysis of the sigB operon in Listeria monocytogenes and in other Gram-positive bacteria. Curr Microbiol 48:39–46. doi: 10.1007/s00284-003-4020-x. [DOI] [PubMed] [Google Scholar]
- 13.Boylan SA, Rutherford A, Thomas SM, Price CW. 1992. Activation of Bacillus subtilis transcription factor sigma B by a regulatory pathway responsive to stationary-phase signals. J Bacteriol 174:3695–3706. doi: 10.1128/jb.174.11.3695-3706.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benson AK, Haldenwang WG. 1992. Characterization of a regulatory network that controls sigma B expression in Bacillus subtilis. J Bacteriol 174:749–757. doi: 10.1128/jb.174.3.749-757.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marles-Wright J, Lewis RJ. 2010. The stressosome: molecular architecture of a signalling hub. Biochem Soc Trans 38:928–933. doi: 10.1042/BST0380928. [DOI] [PubMed] [Google Scholar]
- 16.Pané-Farré J, Quin MB, Lewis RJ, Marles-Wright J. 2017. Structure and function of the stressosome signalling hub, p 1–41. In Harris JR, Marles-Wright J (ed), Macromolecular protein complexes: structure and function. Springer, Cham, Switzerland. [DOI] [PubMed] [Google Scholar]
- 17.Impens F, Rolhion N, Radoshevich L, Bécavin C, Duval M, Mellin J, del Portillo FG, Pucciarelli MG, Williams AH, Cossart P. 2017. N-terminomics identifies Prli42 as a membrane miniprotein conserved in Firmicutes and critical for stressosome activation in Listeria monocytogenes. Nat Microbiol 2:17005. doi: 10.1038/nmicrobiol.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez L, Reeves A, Haldenwang W. 2010. Stressosomes formed in Bacillus subtilis from the RsbR protein of Listeria monocytogenes allow σB activation following exposure to either physical or nutritional stress. J Bacteriol 192:6279–6286. doi: 10.1128/JB.00467-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ondrusch N, Kreft J. 2011. Blue and red light modulates SigB-dependent gene transcription, swimming motility and invasiveness in Listeria monocytogenes. PLoS One 6:e16151. doi: 10.1371/journal.pone.0016151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delumeau O, Chen C-C, Murray JW, Yudkin MD, Lewis RJ. 2006. High-molecular-weight complexes of RsbR and paralogues in the environmental signaling pathway of Bacillus subtilis. J Bacteriol 188:7885–7892. doi: 10.1128/JB.00892-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaidenko TA, Yang X, Lee YM, Price CW. 1999. Threonine phosphorylation of modulator protein RsbR governs its ability to regulate a serine kinase in the environmental stress signaling pathway of Bacillus subtilis. J Mol Biol 288:29–39. doi: 10.1006/jmbi.1999.2665. [DOI] [PubMed] [Google Scholar]
- 22.Kim T-J, Gaidenko TA, Price CW. 2004. A multicomponent protein complex mediates environmental stress signaling in Bacillus subtilis. J Mol Biol 341:135–150. doi: 10.1016/j.jmb.2004.05.043. [DOI] [PubMed] [Google Scholar]
- 23.Yang X, Kang CM, Brody MS, Price CW. 1996. Opposing pairs of serine protein kinases and phosphatases transmit signals of environmental stress to activate a bacterial transcription factor. Genes Dev 10:2265–2275. doi: 10.1101/gad.10.18.2265. [DOI] [PubMed] [Google Scholar]
- 24.Utratna M, Cosgrave E, Baustian C, Ceredig R, O’Byrne C. 2012. Development and optimization of an EGFP-based reporter for measuring the general stress response in Listeria monocytogenes. Bioeng Bugs 3:93–103. doi: 10.4161/bbug.19476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang CM, Vijay K, Price CW. 1998. Serine kinase activity of a Bacillus subtilis switch protein is required to transduce environmental stress signals but not to activate its target PP2C phosphatase. Mol Microbiol 30:189–196. doi: 10.1046/j.1365-2958.1998.01052.x. [DOI] [PubMed] [Google Scholar]
- 26.Kim T-J, Gaidenko TA, Price CW. 2004. In vivo phosphorylation of partner switching regulators correlates with stress transmission in the environmental signaling pathway of Bacillus subtilis. J Bacteriol 186:6124–6132. doi: 10.1128/JB.186.18.6124-6132.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shin J-H, Brody MS, Price CW. 2010. Physical and antibiotic stresses require activation of the RsbU phosphatase to induce the general stress response in Listeria monocytogenes. Microbiology 156:2660–2669. doi: 10.1099/mic.0.041202-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tran V, Geraci K, Midili G, Satterwhite W, Wright R, Bonilla CY. 2018. Resilience to oxidative and nitrosative stress is mediated by the stressosome, RsbP and SigB in Bacillus subtilis. bioRxiv 460303. [DOI] [PubMed]
- 29.Chaturongakul S, Boor KJ. 2004. RsbT and RsbV contribute to σB-dependent survival under environmental, energy, and intracellular stress conditions in Listeria monocytogenes. Appl Environ Microbiol 70:5349–5356. doi: 10.1128/AEM.70.9.5349-5356.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brøndsted L, Kallipolitis BH, Ingmer H, Knöchel S. 2003. kdpE and a putative RsbQ homologue contribute to growth of Listeria monocytogenes at high osmolarity and low temperature. FEMS Microbiol Lett 219:233–239. doi: 10.1016/S0378-1097(03)00052-1. [DOI] [PubMed] [Google Scholar]
- 31.O’Donoghue B. 2016. A molecular genetic investigation into stress sensing in the food-borne pathogen Listeria monocytogenes: roles for RsbR and its paralogues. Doctoral thesis National University of Ireland, Galway, Ireland: http://hdl.handle.net/10379/6310. [Google Scholar]
- 32.Tiensuu T, Andersson C, Rydén P, Johansson J. 2013. Cycles of light and dark co‐ordinate reversible colony differentiation in Listeria monocytogenes. Mol Microbiol 87:909–924. doi: 10.1111/mmi.12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hauf S, Herrmann J, Miethke M, Gibhardt J, Commichau FM, Müller R, Fuchs S, Halbedel S. 2019. Aurantimycin resistance genes contribute to survival of Listeria monocytogenes during life in the environment. Mol Microbiol 111:1009–1024. doi: 10.1111/mmi.14205. [DOI] [PubMed] [Google Scholar]
- 34.Hingston P, Chen J, Dhillon BK, Laing C, Bertelli C, Gannon V, Tasara T, Allen K, Brinkman FSL, Truelstrup Hansen L, Wang S. 2017. Genotypes associated with Listeria monocytogenes isolates displaying impaired or enhanced tolerances to cold, salt, acid, or desiccation stress. Front Microbiol 8:369. doi: 10.3389/fmicb.2017.00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asakura H, Kawamoto K, Okada Y, Kasuga F, Makino S, Yamamoto S, Igimi S. 2012. Intrahost passage alters SigB-dependent acid resistance and host cell-associated kinetics of Listeria monocytogenes. Infect Genet Evol 12:94–101. doi: 10.1016/j.meegid.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 36.Quereda JJ, Pucciarelli MG, Botello-Morte L, Calvo E, Carvalho F, Bouchier C, Vieira A, Mariscotti JF, Chakraborty T, Cossart P, Hain T, Cabanes D, García-Del Portillo F. 2013. Occurrence of mutations impairing sigma factor B (SigB) function upon inactivation of Listeria monocytogenes genes encoding surface proteins. Microbiology 159:1328–1339. doi: 10.1099/mic.0.067744-0. [DOI] [PubMed] [Google Scholar]
- 37.Loh E, Dussurget O, Gripenland J, Vaitkevicius K, Tiensuu T, Mandin P, Repoila F, Buchrieser C, Cossart P, Johansson J. 2009. A trans-acting riboswitch controls expression of the virulence regulator PrfA in Listeria monocytogenes. Cell 139:770–779. doi: 10.1016/j.cell.2009.08.046. [DOI] [PubMed] [Google Scholar]
- 38.Rauch M, Luo Q, Müller-Altrock S, Goebel W. 2005. SigB-dependent in vitro transcription of prfA and some newly identified genes of Listeria monocytogenes whose expression is affected by PrfA in vivo. J Bacteriol 187:800–804. doi: 10.1128/JB.187.2.800-804.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Utratna M, Shaw I, Starr E, O’Byrne CP. 2011. Rapid, transient, and proportional activation of σB in response to osmotic stress in Listeria monocytogenes. Appl Environ Microbiol 77:7841–7845. doi: 10.1128/AEM.05732-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Utratna M, Cosgrave E, Baustian C, Ceredig RH, O’Byrne CP. 2014. Effects of growth phase and temperature on activity within a Listeria monocytogenes population: evidence for RsbV-independent activation of σB at refrigeration temperatures. Biomed Res Int 2014:641647. doi: 10.1155/2014/641647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Z, Meng Q, Qiao J, Yang L, Cai X, Wang G, Chen C, Zhang L. 2013. RsbV of Listeria monocytogenes contributes to regulation of environmental stress and virulence. Arch Microbiol 195:113–120. doi: 10.1007/s00203-012-0855-5. [DOI] [PubMed] [Google Scholar]
- 42.Cao M, Bitar AP, Marquis H. 2007. A mariner-based transposition system for Listeria monocytogenes. Appl Environ Microbiol 73:2758–2761. doi: 10.1128/AEM.02844-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen CC, Lewis RJ, Harris R, Yudkin MD, Delumeau O. 2003. A supramolecular complex in the environmental stress signalling pathway of Bacillus subtilis. Mol Microbiol 49:1657–1669. doi: 10.1046/j.1365-2958.2003.03663.x. [DOI] [PubMed] [Google Scholar]
- 44.Eymann C, Schulz S, Gronau K, Becher D, Hecker M, Price CW. 2011. In vivo phosphorylation patterns of key stressosome proteins define a second feedback loop that limits activation of Bacillus subtilis σB. Mol Microbiol 80:798–810. doi: 10.1111/j.1365-2958.2011.07609.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu Z, Tamber S, Memmi G, Donegan NP, Cheung AL. 2009. Overexpression of MazFsa in Staphylococcus aureus induces bacteriostasis by selectively targeting mRNAs for cleavage. J Bacteriol 191:2051–2059. doi: 10.1128/JB.00907-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schuster CF, Mechler L, Nolle N, Krismer B, Zelder M-E, Götz F, Bertram R. 2015. The MazEF toxin-antitoxin system alters the β-lactam susceptibility of Staphylococcus aureus. PLoS One 10:e0126118. doi: 10.1371/journal.pone.0126118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Curtis TD, Takeuchi I, Gram L, Knudsen GM. 2017. The influence of the toxin/antitoxin mazEF on growth and survival of Listeria monocytogenes under stress. Toxins (Basel) 9:31. doi: 10.3390/toxins9010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orsi R, Ripoll D, Yeung M, Nightingale K, Wiedmann M. 2007. Recombination and positive selection contribute to evolution of Listeria monocytogenes inlA. Microbiology 153:2666–2678. doi: 10.1099/mic.0.2007/007310-0. [DOI] [PubMed] [Google Scholar]
- 49.Dorey A, Marinho C, Piveteau P, O’Byrne C. 2019. Role and regulation of the stress activated sigma factor sigma B (σB) in the saprophytic and host-associated life stages of Listeria monocytogenes. Adv Appl Microbiol 106:1–48. doi: 10.1016/bs.aambs.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 50.Tiensuu T, Guerreiro DN, Oliveira AH, O’Byrne C, Johansson J. 2019. Flick of a switch: regulatory mechanisms allowing Listeria monocytogenes to transition from a saprophyte to a killer. Microbiology 165:819–833. doi: 10.1099/mic.0.000808. [DOI] [PubMed] [Google Scholar]
- 51.Marles-Wright J, Lewis RJ. 2008. The Bacillus subtilis stressosome: a signal integration and transduction hub. Commun Integr Biol 1:182–184. doi: 10.4161/cib.1.2.7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Voelker U, Voelker A, Haldenwang WG. 1996. Reactivation of the Bacillus subtilis anti-sigma B antagonist, RsbV, by stress- or starvation-induced phosphatase activities. J Bacteriol 178:5456–5463. doi: 10.1128/jb.178.18.5456-5463.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holtmann G, Brigulla M, Steil L, Schütz A, Barnekow K, Völker U, Bremer E. 2004. RsbV-independent induction of the SigB-dependent general stress regulon of Bacillus subtilis during growth at high temperature. J Bacteriol 186:6150–6158. doi: 10.1128/JB.186.18.6150-6158.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brigulla M, Hoffmann T, Krisp A, Völker A, Bremer E, Völker U. 2003. Chill induction of the SigB-dependent general stress response in Bacillus subtilis and its contribution to low-temperature adaptation. J Bacteriol 185:4305–4314. doi: 10.1128/jb.185.15.4305-4314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hsu C-Y, Cairns L, Hobley L, Abbott J, O’Byrne C, Stanley-Wall NR. 21 January 2020. Genomic differences between Listeria monocytogenes EGDe isolates reveals crucial roles for SigB and wall rhamnosylation in biofilm formation. J Bacteriol doi: 10.1128/JB.00692-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arnaud M, Chastanet A, Débarbouillé M. 2004. New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol 70:6887–6891. doi: 10.1128/AEM.70.11.6887-6891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abram F, Starr E, Karatzas KAG, Matlawska-Wasowska K, Boyd A, Wiedmann M, Boor KJ, Connally D, O’Byrne CP. 2008. Identification of components of the sigma B regulon in Listeria monocytogenes that contribute to acid and salt tolerance. Appl Environ Microbiol 74:6848–6858. doi: 10.1128/AEM.00442-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schweder T, Kolyschkow A, Völker U, Hecker M. 1999. Analysis of the expression and function of the σB-dependent general stress regulon of Bacillus subtilis during slow growth. Arch Microbiol 171:439–443. doi: 10.1007/s002030050731. [DOI] [PubMed] [Google Scholar]
- 59.O’Donoghue B, NicAogáin K, Bennett C, Conneely A, Tiensuu T, Johansson J, O’Byrne C. 2016. Blue-light inhibition of Listeria monocytogenes growth is mediated by reactive oxygen species and is influenced by σB and the blue-light sensor Lmo0799. Appl Environ Microbiol 82:4017–4027. doi: 10.1128/AEM.00685-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Finkel SE. 2006. Long-term survival during stationary phase: evolution and the GASP phenotype. Nat Rev Microbiol 4:113–120. doi: 10.1038/nrmicro1340. [DOI] [PubMed] [Google Scholar]
- 61.Zambrano MM, Kolter R. 1996. GASPing for life in stationary phase. Cell 86:181–184. doi: 10.1016/s0092-8674(00)80089-6. [DOI] [PubMed] [Google Scholar]
- 62.Liu B, Eydallin G, Maharjan RP, Feng L, Wang L, Ferenci T. 2017. Natural Escherichia coli isolates rapidly acquire genetic changes upon laboratory domestication. Microbiology 163:22–30. doi: 10.1099/mic.0.000405. [DOI] [PubMed] [Google Scholar]
- 63.Maharjan R, Nilsson S, Sung J, Haynes K, Beardmore RE, Hurst LD, Ferenci T, Gudelj I. 2013. The form of a trade‐off determines the response to competition. Ecol Lett 16:1267–1276. doi: 10.1111/ele.12159. [DOI] [PubMed] [Google Scholar]
- 64.Gudelj I, Weitz JS, Ferenci T, Horner-Devine MC, Marx CJ, Meyer JR, Forde SE. 2010. An integrative approach to understanding microbial diversity: from intracellular mechanisms to community structure. Ecol Lett 13:1073–1084. doi: 10.1111/j.1461-0248.2010.01507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Farewell A, Kvint K, Nyström T. 1998. Negative regulation by RpoS: a case of sigma factor competition. Mol Microbiol 29:1039–1051. doi: 10.1046/j.1365-2958.1998.00990.x. [DOI] [PubMed] [Google Scholar]
- 66.Notley-McRobb L, King T, Ferenci T. 2002. rpoS mutations and loss of general stress resistance in Escherichia coli populations as a consequence of conflict between competing stress responses. J Bacteriol 184:806–811. doi: 10.1128/jb.184.3.806-811.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nyström T. 2004. Growth versus maintenance: a trade‐off dictated by RNA polymerase availability and sigma factor competition? Mol Microbiol 54:855–862. doi: 10.1111/j.1365-2958.2004.04342.x. [DOI] [PubMed] [Google Scholar]
- 68.Marinho CM, Dos Santos PT, Kallipolitis BH, Johansson J, Ignatov D, Guerreiro DN, Piveteau P, O’Byrne CP. 2019. The σB-dependent regulatory sRNA Rli47 represses isoleucine biosynthesis in Listeria monocytogenes through a direct interaction with the ilvA transcript. RNA Biol 16:1424–1437. doi: 10.1080/15476286.2019.1632776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Monk IR, Gahan CG, Hill C. 2008. Tools for functional postgenomic analysis of Listeria monocytogenes. Appl Environ Microbiol 74:3921–3934. doi: 10.1128/AEM.00314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deatherage DE, Barrick JE. 2014. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq, p 165–188. In Sun L, Shou W (ed), Engineering and analyzing multicellular systems. Springer, New York, NY. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Darling AC, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Collins TJ. 2007. ImageJ for microscopy. Biotechniques 43:25–30. doi: 10.2144/000112517. [DOI] [PubMed] [Google Scholar]
- 73.Hamilton N. 2009. Quantification and its applications in fluorescent microscopy imaging. Traffic 10:951–961. doi: 10.1111/j.1600-0854.2009.00938.x. [DOI] [PubMed] [Google Scholar]
- 74.Selinummi J, Seppälä J, Yli-Harja O, Puhakka JA. 2005. Software for quantification of labeled bacteria from digital microscope images by automated image analysis. Biotechniques 39:859–863. doi: 10.2144/000112018. [DOI] [PubMed] [Google Scholar]
- 75.Salcedo C, Arreaza L, Alcalá B, de la Fuente L, Vázquez JA. 2003. Development of a multilocus sequence typing method for analysis of Listeria monocytogenes clones. J Clin Microbiol 41:757–762. doi: 10.1128/jcm.41.2.757-762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, de Hoon MJL. 2009. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pfaffl M, Georgieva TM, Georgiev IP, Ontsouka E, Hageleit M, Blum J. 2002. Real-time RT-PCR quantification of insulin-like growth factor (IGF)-1, IGF-1 receptor, IGF-2, IGF-2 receptor, insulin receptor, growth hormone receptor, IGF-binding proteins 1, 2 and 3 in the bovine species. Domest Anim Endocrinol 22:91–102. doi: 10.1016/s0739-7240(01)00128-x. [DOI] [PubMed] [Google Scholar]
- 79.den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T, Antonarakis SE, Taschner PE. 2016. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat 37:564–569. doi: 10.1002/humu.22981. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.