

Summary
Quorum sensing is a process of chemical communication that bacteria use to track cell density and coordinate gene expression across a population. Bacteria-infecting viruses, called phages, can encode quorum-sensing components that enable them to integrate host cell density information into the lysis-lysogeny decision. Vibriophage VP882 is one such phage, and activation of its quorum-sensing pathway leads to the production of an antirepressor called Qtip. Qtip interferes with the prophage repressor (cIVP882), leading to host-cell lysis. Here, we show that Qtip interacts with the N-terminus of cIVP882, inhibiting both cIVP882 DNA binding and cIVP882 autoproteolysis. Qtip also sequesters cIVP882, localizing it to the poles. Qtip can localize to the poles independently of cIVP882. Alanine-scanning mutagenesis of Qtip shows that its localization and interference with cIVP882 activities are separable. Comparison of Qtip to a canonical phage antirepressor reveals that, despite both proteins interacting with their partner repressors, only Qtip drives polar localization.
Keywords: phage, quorum sensing, antirepressor, Qtip, cI repressor, lysis-lysogeny
Graphical Abstract
eTOC Blurb
Qtip is a 79-amino acid antirepressor that connects phage quorum-sensing to host-cell lysis. Silpe and Bridges, et al demonstrate Qtip binds, inactivates, and sequesters the phage cI repressor at the cell pole. Qtip mutagenesis reveals discrete sites required for repressor recognition and polar localization, showing the two functions are separable.
Introduction
Quorum sensing (QS) is a bacterial cell-cell communication process that controls collective behaviors. QS bacteria produce, release, and detect accumulated signaling molecules called autoinducers (AIs) [reviewed in (Papenfort and Bassler, 2016)]. QS-controlled behaviors include biofilm formation (Hammer and Bassler, 2003), virulence factor production (Miller et al., 2002; Zhu et al., 2002), competence (Meibom, 2005), and deployment of CRISPR-Cas defense systems that eliminate incoming foreign DNA from plasmids and bacteria-specific viruses called phages (Høyland-Kroghsbo et al., 2017). Phages also engage in chemical dialogs. For example, some Bacillus phages encode a phage-phage communication module termed the arbitrium system (Erez et al., 2017; Stokar-Avihail et al., 2019). In this case, phage-encoded proteins drive the production and release of signaling peptides, that, when detected by other prophages in the bacterial community, launch a lysogeny-activating program across the infected bacterial population. Phage-encoded QS components resembling host bacterial QS components have been identified using bioinformatics (Hargreaves et al., 2014; Silpe and Bassler, 2019a, 2019b), and in systems that have been tested, the phage-encoded QS receptors bind and respond to the AI molecule produced by the host bacterium. In one such instance, vibriophage VP882 encodes a QS receptor, called VqmAPhage, which is homologous to the bacterial VqmA QS receptor in Vibrio cholerae (Silpe and Bassler, 2019a). Both the host- and phage-encoded proteins bind to the host-produced AI, 3,5-dimethyl-pyrazin-2-ol (DPO), however, the AI-bound VqmA proteins control different pathways. Host VqmA binding to DPO regulates V. cholerae QS behaviors (Herzog et al., 2019; Papenfort et al., 2017), while binding of DPO by the phage-encoded VqmA activates lytic development, resulting in killing of the V. cholerae host (Silpe and Bassler, 2019a). In a similar vein, recently, the QS AI called 4,5-dihydroxy-2,3-pentanedione (DPD), that is produced by many species of bacteria (Chen et al., 2002; Schauder et al., 2001), has been shown to promote lytic behavior in phage T1 of Escherichia coli (Laganenka et al., 2019). How phage T1 detects the DPD AI is not yet understood.
Phage lysis-lysogeny decisions are often controlled by phage proteins that repress expression of genes encoding activators of phage lytic genes. Relief of repression launches the phage lytic programs. Thus, the transition from lysogeny to lysis requires a mechanism to inactivate the phage repressor [reviewed in (Ptashne, 2004)]. Commonly, phage repressor proteins are subject to post-translational regulation. In the case of phage lambda, the cI repressor protein (cILambda) is auto-cleaved in an SOS-activated, RecA-dependent manner (Roberts and Roberts, 1975; Sauer et al., 1982). Cleavage abolishes cILambda repressor activity. Repressors of other phages, for example, that of coliphage 186, are, by contrast, inactivated via protein-protein interactions with small antirepressor proteins (Shearwin et al., 1998). In these cases, production of the antirepressor is frequently controlled by a LexA-regulated promoter, and thus, despite different mechanisms, as in phage lambda, repressor inactivation is induced by the host SOS response (Heinzel et al., 1992; Kim et al., 2016; Lemire et al., 2011; Mardanov and Ravin, 2007; Quinones et al., 2005; Shearwin et al., 1998). Unlike autoproteolytic repressors, however, the mechanisms underlying antirepressor-mediated inactivation of phage repressors have seldom been fully characterized and it is possible that a given antirepressor can function by more than one mechanism. Known mechanisms include functioning as a competitive inhibitor of repressor DNA binding [reviewed in (Wang et al., 2014)], binding an allosteric site on a partner repressor protein and forcing it to adopt a DNA-incompetent confirmation (Kim et al., 2016), and sequestering the repressor rendering it insoluble (Davis et al., 2002).
The repressor encoded by vibriophage VP882, called cIVP882, can be inactivated by two mechanisms: RecA-dependent cleavage of the cIVP882 repressor occurs, similarly to lambda, following activation of the SOS response (Silpe and Bassler, 2019a). Additionally, antirepressor-mediated inactivation of the cIVP882 repressor is triggered by the phage-encoded QS pathway. Specifically, when VqmAPhage binds the host-produced DPO AI, the complex activates transcription of a phage gene encoding an antirepressor, Qtip, that inactivates cIVP882 [(Silpe and Bassler, 2019a) and Figure 1A]. By tuning into the SOS-response, VP882 prophages can connect their lysis-lysogeny decisions to the viability of the host and, by monitoring local bacterial population density, VP882 prophages can gauge the prospect of encountering future hosts.
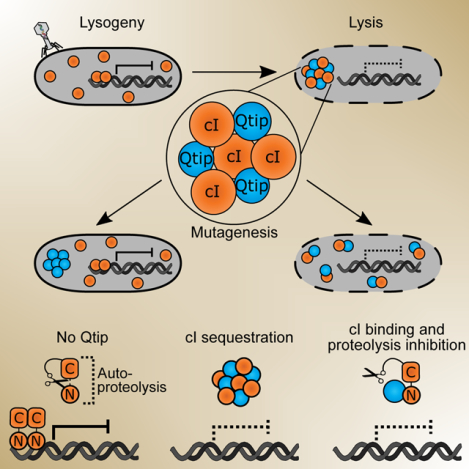
Figure 1. cIVP882 DNA binding is blocked by Qtip but a cIVP882 mutant lacking DNA-binding capability does not prevent Qtip recognition.
(A) Schematic depicting the phage VP882-encoded QS pathway and the reporter system used in this work. Top: VqmAPhage, when bound to DPO (white hexagon), activates expression of the qtip gene encoding the Qtip antirepressor. Qtip inactivates the cIVP882 repressor, enabling expression of Pq and subsequent Q-mediated host cell lysis. Bottom: The reporter system used in this work to monitor Qtip and cIVP882 activity. Pq is fused to the luciferase operon (lux). Light production is low when the cIVP882 repressor is functional and light production is high when Qtip is active and/or the cIVP882 repressor is non-functional. (B) Upper panel: EMSA analysis of Pq DNA retarded by cIVP882-HALO protein purified alone or in complex with HIS-Qtip. The relative amount of cIVP882-HALO in each lane is indicated (1x = ~12 nM). Lower panel: The reactions from the upper panel were subjected to SDS-PAGE analysis and imaged using a Cy5 filter set to visualize HALO-Alexa660, to which the cIVP882-HALO protein had been conjugated. The molecular weight marker is designated M. (C) Light production from E. coli harboring the Pq-lux reporter (no plasmid) or the Pq-lux reporter and a plasmid encoding WT cIVP882 or the cIVP882DBD*-HALO allele. Relative light units (RLU) were calculated by dividing bioluminescence by OD600. Data represented as mean ± SD with n = 3 biological replicates. (D) Upper panel: Average individual cell images of recombinant E. coli producing Qtip and either, cIVP882-HALO, cIVP882DBD*-HALO, or the HALO tag. HALO-TMR fluorescence intensity is displayed as a red heat map (black and white reflect the lowest and highest intensity, respectively). Dashed lines denote the average cell outlines. Scale bar = 1 μm. Upper left inset: Schematic showing aligned individual cells averaged to produce composite images in the upper panel (n = 20–25 cells per condition, see Methods). In the schematic, the HALO-TMR fluorescence intensity from three representative individual cells harboring Qtip and cIVP882DBD*-HALO is shown in inverted greyscale. Lower panel: Line plots of HALO-TMR fluorescence intensity extracted from individual cell images used to generate the composite images displayed in the upper panel. The distance along the x-axis is relative to the left most edge of each cell. Shaded regions represent ± 1 SD from the mean. See also Figure S1.
Qtip sequesters cIVP882, and Qtip-cIVP882 aggregates are localized at the cell poles (Silpe and Bassler, 2019a). However, the mechanism by which Qtip inactivates the cIVP882 repressor remains unknown. Qtip is a small protein (~8 kDa) with no predicted domains and no close homologs, preventing structure-function and homology-based predictions. To probe the Qtip antirepressor mechanism, here, we developed in vitro assays to show that Qtip inhibits cIVP882 DNA binding and autoproteolysis. We demonstrate that, while the C-terminal domain of cIVP882 possesses the autocleavage activity, Qtip recognizes the N-terminal domain of cIVP882, which harbors the cIVP882 DNA-binding activity. Mutation of some of the amino acid residues in cIVP882 that eliminate DNA binding do not interfere with Qtip binding to cIVP882, while other mutations eliminate both activities. Qtip inactivates repressors similar to cIVP882 but not those of lambda or phage P22. Likewise, Ant, the antirepressor from phage P22, inactivates repressors from lambda and P22 (Susskind and Botstein, 1975) but not from phage VP882. We show that, unlike Qtip, Ant does not alter the localization of its partner repressors. We mutagenized Qtip to identify residues essential for disabling cIVP882 DNA binding and residues required for polar localization. We show that these two Qtip properties are separable.
Results
Qtip Prevents cIVP882 from Binding to DNA
Previous work on phage VP882 showed that production of Qtip counteracts repression by cIVP882 at the target q promoter, leading to production of the Q antiterminator, launch of the lytic program, and host-cell lysis [Figure 1A and (Silpe and Bassler, 2019a)]. Moreover, microscopy demonstrated that Qtip alters the localization of cIVP882. In the absence of Qtip, cIVP882 is diffuse in the cytoplasm, and when Qtip is present, cIVP882 forms foci colocalized with Qtip at the cell poles (Silpe and Bassler, 2019a).
We sought to determine how Qtip binding alters the ability of cIVP882 to function as a repressor. We first examined whether Qtip interferes with cIVP882 binding to DNA. To do this, we co-expressed N-terminal-hexahistidine-tagged Qtip (HIS-Qtip) with C-terminal-HALO-tagged cIVP882 (cIVP882-HALO) and using Ni-NTA resin, purified HIS-Qtip in complex with cIVP882-HALO. In parallel, we purified the cIVP882-HALO protein using heparin-affinity chromatography. We measured cIVP882-HALO binding and binding of the cIVP882-HALO-His-Qtip complex to the cIVP882 target promoter, called Pq. Electromobility shift assays (EMSA) showed that the cIVP882-HALO protein shifted the Pq DNA, however, the cIVP882-HALO protein purified in complex with HIS-Qtip failed to do so (Figure 1B). These results indicate that Qtip binding to cIVP882 prevents cIVP882 from binding target DNA, possibly providing the mechanism by which, in phage VP882 lysogens, Qtip initiates the lytic program.
We wondered whether the amino acid residues required for cIVP882 to bind DNA are also required for cIVP882 to interact with Qtip. We used domain analysis to identify 6 residues in the cIVP882 N-terminus that are predicted to participate in sequence-specific DNA binding (InterProScan): L18, Q19, G32, S35, R39, and G40. Pairing these sites with our knowledge of cI homologs that Qtip does and does not sequester (Silpe and Bassler, 2019a), we aligned the Qtip-interacting cI proteins and found that 2 of these 6 sites (Q19 and S35) are shared between all of the susceptible proteins and are absent in the analogous positions in cILambda (Figure S1A), which is not sequestered by Qtip (Silpe and Bassler, 2019a). We mutated cIVP882 to make cIVP882Q19A/S35A and fused the double mutant protein to HALO. We call this construct cIVP882DBD*HALO. We tested the ability of cIVP882DBD*-HALO to repress its target Pq promoter using recombinant E. coli harboring a plasmid carrying the q promoter fused to the luciferase operon (Pq-lux). WT cIVP882-HALO repressed Pq-lux expression while cIVP882DBD*-HALO did not (Figure 1C), indicating that the Q19A/S35A double mutation leads to a DNA-binding defective cIVP882. We also assessed whether Qtip could sequester cIVP882DBD*-HALO by performing confocal microscopy on recombinant E. coli carrying Qtip and either WT cIVP882-HALO, cIVP882DBD*-HALO, or only HALO (no cIVP882), each labeled with HALO-TMR. The upper panel in Figure 1D shows composite images of the average HALO-TMR fluorescence signals from aligned individual cells expressing the specified constructs. We also measured the fluorescence intensity across the long axis of the aligned individual cells, displayed as an average line profile of fluorescence intensity as a function of the distance from the left cell pole as depicted in the figure (lower panel). We found that Qtip sequestration of cIVP882DBD*-HALO was indistinguishable from Qtip sequestration of WT cIVP882-HALO (Figure 1D), indicating that the residues required for the cIVP882 repressor to bind DNA are not required for Qtip to bind to the cIVP882 repressor.
RecA* Activates and Qtip Inhibits cIVP882 Autoproteolysis
In addition to binding DNA and Qtip, the cIVP882 repressor is subject to RecA-dependent autocleavage (Silpe and Bassler, 2019a). Based on domain prediction and homology modeling using the cILambda repressor and other LexA-like repressor proteins as references, we predicted that the cIVP882 N-terminal domain is responsible for DNA binding while cleavage is enabled by a catalytic domain (S24 peptidase-like) residing in the C-terminus. To assess autoproteolysis, we established in vitro conditions under which cIVP882 cleavage could be monitored. We incubated purified cIVP882-HALO protein with single stranded DNA (ssDNA), RecA, and ATP-γ-S. ssDNA and ATP-γ-S are known to stimulate RecA to adopt an activated state (termed RecA*), which, in the presence of other repressor proteins (e.g., LexA and cILambda) promotes their autoproteolytic activity (Giese et al., 2008; Ndjonka and Bell, 2006). Figure 2A shows that under these conditions, cIVP882-HALO cleavage occurred within 10 min at 37°C, while in reactions lacking ATP-γ-S or RecA, cIVP882-HALO did not undergo cleavage. The data in Figure 1B show that Qtip inhibits cIVP882 DNA binding. We examined whether Qtip could also affect cIVP882 autoproteolysis. Figure 2B shows that, in contrast to when cIVP882-HALO was purified alone, cIVP882-HALO purified in complex with Qtip did not undergo cleavage, suggesting that Qtip binding inhibits cIVP882 autoproteolytic activity.
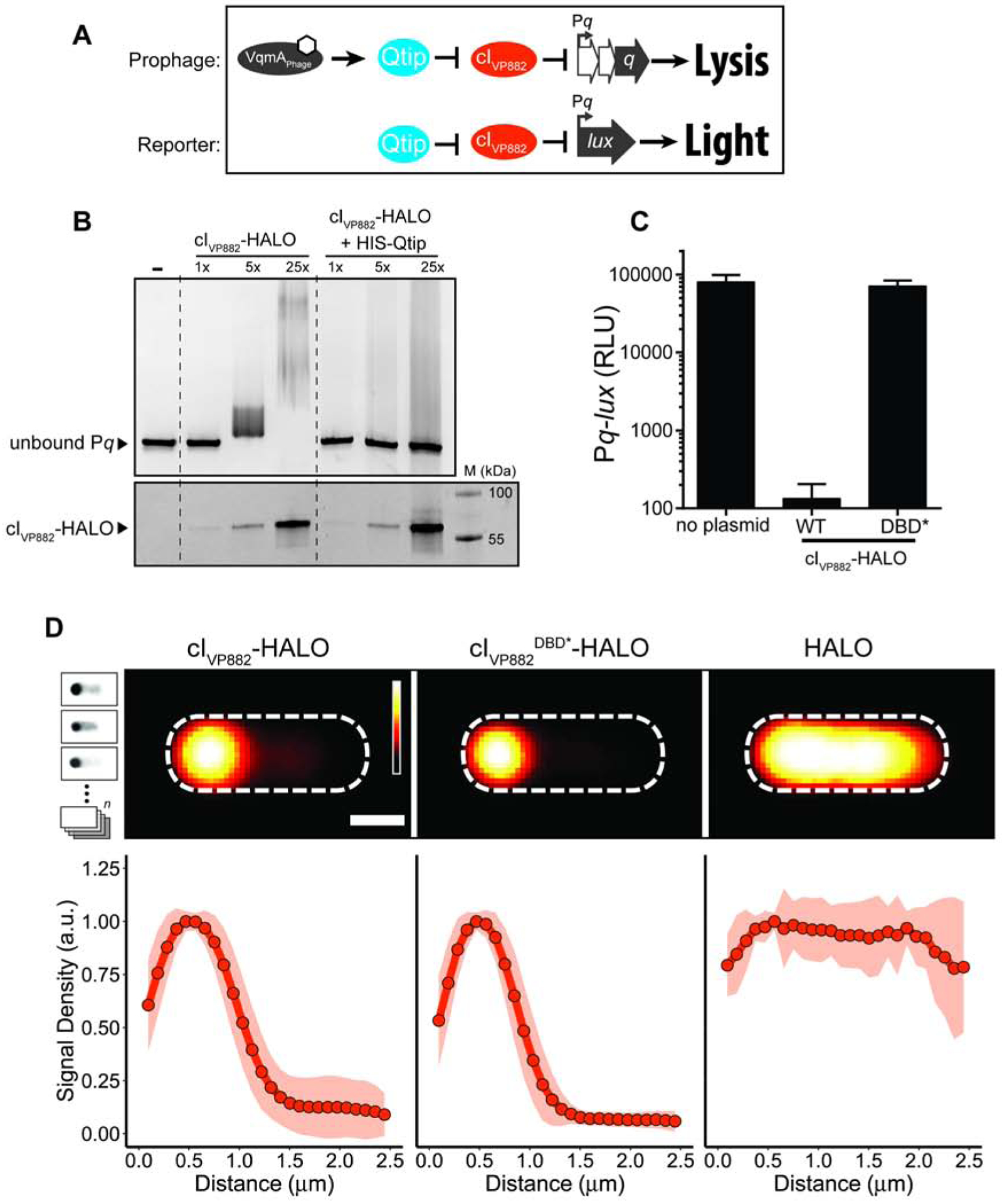
Figure 2. Qtip prevents in vitro cleavage of cIVP882 but cIVP882 mutants lacking cleavage or catalytic sites do not prevent Qitp recognition.

(A) In vitro cleavage of cIVP882-HALO monitored by SDS-PAGE analysis. The presence of RecA and/or ATP-γ-S in the reactions is indicated above each lane. (B) In vitro cleavage of cIVP882-HALO purified alone or in complex with HIS-Qtip. Samples were concentration matched according to the cIVP882-HALO Alexa660 signal. (C) In vitro cleavage of WT cIVP882-HALO and the indicated catalytic-site (K172A and S130A) and cleavage-site (G92D) variants. For (A-C): all cIVP882-HALO proteins were labeled with HALO-Alexa660. Upper panels, gels imaged using a Cy5 filter set for HALO-Alexa660 detection; lower panels, the same gels stained for total protein. Incubation times noted above each lane. Marker, M. In (B) and (C), all samples contained ATP-γ-S and RecA. (D) Composite images from individual cell analyses of E. coli producing Qtip and either WT cIVP882-HALO or the cleavage-site (G92D) or catalytic-site (S130A and K172A) cIVP882-HALO variant. HALO-TMR fluorescence intensity and scale bar represented as in Figure 1D. See also Figure S1.
We investigated the requirements for Qtip inhibition of cIVP882 autoproteolytic activity. Based on homology to cILambda, we predicted that cleavage of cIVP882 occurs between residues A91 and G92 (Figure S1A), analogous to the cleavage site between A111 and G112 in cILambda (Sauer et al., 1982), and that the two active site residues are S130 and K172 (Figure S1A), which match the known catalytic residues S149 and K192 in cILambda (Slilaty and Little, 1987). Consistent with these predictions, Figure 2C shows that the cIVP882S130A-HALO and cIVP882K172AHALO catalytic site mutants and the cIVP882G92D-HALO cleavage site mutant did not undergo autoproteolysis. Importantly, however, Figure 2D shows that, like WT cIVP882-HALO, the cIVP882HALO cleavage site and catalytic site mutants could all still be sequestered by Qtip. Thus, Qtip binding prevents cIVP882 autoproteolysis, but the residues involved in cIVP882 cleavage and catalysis are not required for Qtip to do so.
cIVP882 Autoproteolysis Occurs by an Intramolecular Mechanism
To probe the mechanism of cIVP882 autoproteolysis, we relied on established mechanisms underlying self-cleavage of related repressor proteins. Specifically, cleavage can occur via an intramolecular mechanism, as in the case of LexA and cILambda (Slilaty et al., 1986), or an intermolecular mechanism, as in the case of UmuD of E. coli (McDonald et al., 1998). To classify the catalytic behavior of cIVP882, we combined WT cIVP882-HALO with each of the catalytic-site variants, cIVP882S130A-HALO or cIVP882K172A-HALO. Our rationale was that if cIVP882 is cleaved by an intermolecular mechanism, then WT cIVP882-HALO, in addition to cleaving other WT cIVP882-HALO proteins, would cleave the catalytically dead cIVP882S130A-HALO and cIVP882K172AHALO mutants in trans. By contrast, if intramolecular cleavage is the mechanism, only the WT cIVP882-HALO protein would be cleaved.
To distinguish WT cIVP882-HALO from the cIVP882-HALO catalytic site mutants in our mixed reactions, we took advantage of the fact that the HALO tag can be labeled with different fluorophores and the fluorophores remain covalently bound and continue to fluoresce when excited under denaturing SDS-PAGE conditions (Figure S1B). Thus, we employed a red-fluorescent ligand, HALO-TMR, to label each of the catalytic site mutants (cIVP882S130A-HALO and cIVP882K172A-HALO) and a far-red/IR ligand, HALO-Alexa660, to label WT cIVP882-HALO. In our mixed reactions, the differentially tagged proteins could be resolved using filter sets that are specific to the excitation-emission spectrum of each fluorophore. The overlaid emission output of cIVP882-HALO conjugated to different fluorophores, prior to- and after mixing, is shown in Figure S1B. Mixing of the far-red labeled WT cIVP882-HALO with either of the red-fluorescent-labeled catalytic site mutants shows that only the WT (Alexa660-labeled) protein was cleaved (Figure 3A), indicating that the cIVP882 cleavage mechanism is intramolecular. To eliminate the possibility that the red-fluorescent HALO ligand prevents cleavage of cIVP882-HALO, we exchanged the fluorophores used to label the different proteins. Again, consistent with an intramolecular cleavage mechanism, only the WT (TMR-labeled) cIVP882-HALO protein was cleaved (Figure 3A). We conclude that cIVP882 is cleaved likely by the same intramolecular mechanism as cILambda, and cleavage relies on conserved catalytic residues and the identical alanyl-glycyl cleavage sequence.
Figure 3. cIVP882 is cleaved by an intramolecular mechanism and Qtip recognition occurs at the N-terminus of cIVP882.
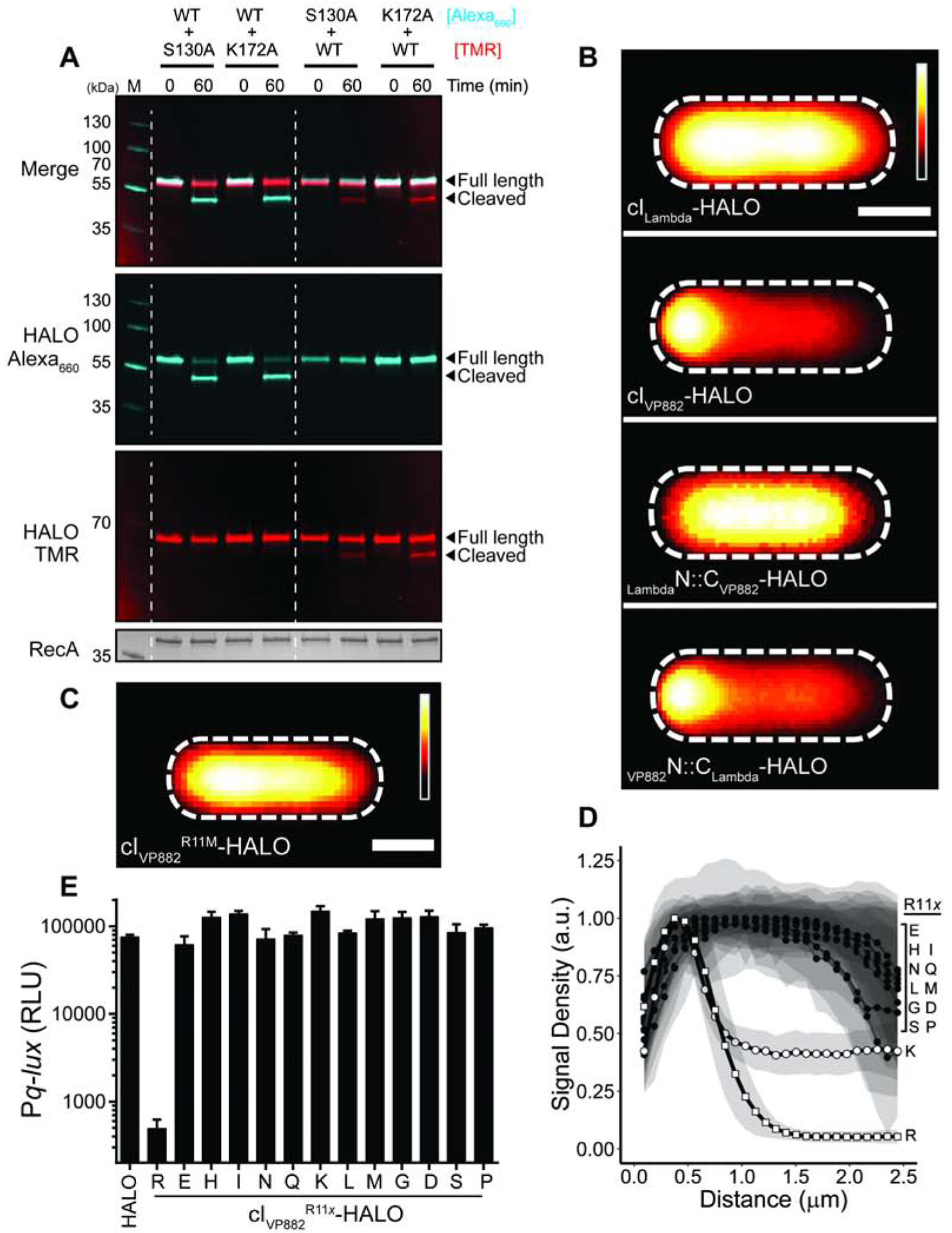
(A) In vitro cleavage of WT cIVP882-HALO mixed with cIVP882 catalytic-site variants monitored by SDS-PAGE analysis and differential labeling. The first lane has the marker, M. The HALO dye combinations used are designated to the right of the proteins. The lanes between the dashed white lines show WT cIVP882-HALO conjugated to HALO-Alexa660 (cyan) and catalytic-site variants conjugated to HALO-TMR (red). The lanes to the right of the second dashed white line show WT cIVP882-HALO conjugated to HALO-TMR (red) and the catalytic-site variants conjugated to HALO-Alexa660 (cyan). The gel was imaged under Cy5 and Cy3 filter sets to detect HALO-Alexa660 and HALO-TMR, respectively, as designated on the left (see also Figure S1B), prior to being stained for total protein (bottom panel). The composite of the HALO-Alexa660 and HALO-TMR channels is shown in the upper-most panel, designated Merge. (B) Composite images from individual cell analyses of E. coli producing Qtip and either cILambda, cIVP882, or the indicated chimeras, each fused to HALO and labeled with HALO-TMR. (C) As in (B) with Qtip and cIVP882R11M-HALO. In (B) and (C), HALO-TMR fluorescence intensity and scale bar represented as in Figure 1D. (D) Line plot of HALO-TMR fluorescence intensity extracted from individual cell images of E. coli producing Qtip and either WT cIVP882-HALO (designated R) or another cIVP882R11x-HALO variant (designated by the letters on the right). WT cIVP882-HALO (open squares) and cIVP882R11K-HALO (open circles) and the cIVP882R11x-HALO variants (closed circles). Distance along the x-axis as in Figure 1D. Shaded regions represent ± 1 SD from the mean. (E) Light production by E. coli harboring the Pq-lux reporter and a control plasmid (HALO), WT cIVP882-HALO (denoted R), or the designated cIVP882R11x-HALO variant. The x indicates the amino acid residue at position 11. RLU as in Figure 1. Data represented as mean ± SD with n = 3 biological replicates. See also Figures S1 and S2.
Qtip Recognizes the DNA-Binding Domain of cIVP882 but not that of cILambda
Our above results show that Qtip prevents cIVP882 autoproteolysis, however, Qtip does not depend on the cleavage or catalytic site residues in cIVP882 to sequester the protein (see Figure 2D). Moreover, cIVP882 has functionally analogous domains to those in cILambda, and yet, Qtip sequesters cIVP882 but not cILambda (Silpe and Bassler, 2019a). We thus wondered what determinants are required in a cI protein for recognition by Qtip. We took advantage of the conserved domain architecture between cIVP882 and cILambda and the similarities in their cleavage mechanisms to engineer two chimeric cI repressors for assessment of interaction with Qtip. Specifically, we exchanged the N-terminal (DNA-binding) and C-terminal (catalytic) domains of cIVP882 and cILambda, leading to VP882N::CLambda-HALO and LambdaN::CVP882-HALO proteins (Figure S1A). Both chimeras retained the ability to bind DNA (Figure S2A) and to undergo autoproteolysis (Figure S2B). Moreover, the DNA-binding specificity of each chimera is set by the protein from which the N-terminal DNA-binding portion was derived, i.e., the VP882N::CLambda-HALO chimera, like full length cIVP882-HALO, bound phage VP882 DNA but not lambda DNA (Figure S2A). Conversely, the LambdaN::CVP882-HALO chimera, while not as stable as the VP882N::CLambda-HALO chimera (see 0’ timepoint in Figure S2B, and Figure S2C), bound lambda DNA, but not VP882 DNA (Figure S2A). We assayed whether Qtip could bind to these chimeras. As controls, and consistent with our earlier findings (Silpe and Bassler, 2019a), we show that the cILambda-HALO protein was not sequestered by Qtip, whereas localization to the poles occurred for cIVP882-HALO (Figure 3B). LambdaN::CVP882-HALO, like cILambda-HALO, remained cytoplasmic in the presence of Qtip, suggesting that Qtip does not interact with cILambda-HALO or the LambdaN::CVP882-HALO chimera. By contrast, the VP882N::CLambda-HALO fusion was sequestered to the poles by Qtip similar to what occurred between Qtip and WT cIVP882-HALO (Figure 3B). These results suggest that Qtip recognizes the N-terminal region of cIVP882.
A Subset of the Amino Acid Residues Required for cIVP882 to Bind DNA are also Required for Qtip to Bind cIVP882
We have shown that Qtip binds the cIVP882 N-terminal, DNA-binding domain (Figure 3B) and yet the DNA-binding deficient variant, cIVP882DBD*-HALO, continues to be sequestered by Qtip (Figure 1D). These results suggest that DNA binding by cIVP882 and Qtip binding by cIVP882 could be separable functions. To examine this possibility, we sought to identify a cIVP882 mutant that was resistant to Qtip-directed localization. We randomly mutagenized WT cIVP882-HALO on a plasmid and transformed the pool of mutant plasmids into E. coli harboring qtip under an anhydrotetracyline (aTc)-inducible promoter. When co-produced with Qtip, cIVP882R11M-HALO remained cytoplasmic, suggesting that mutation of the arginine at position 11 of the cIVP882 repressor leads to a loss in Qtip recognition (Figures 3C and S1A). However, a trivial explanation for this phenotype is that the arginine to methionine change at position 11 provides an alternative cIVP882-HALO translation start site, and the absence of the first 10 amino acid residues prevents Qtip from binding. To eliminate this possibility, we used semi-arbitrary PCR to engineer random codons at position 11 in cIVP882-HALO. In addition to R11M, we recovered cIVP882R11x-HALO [where x = D, E, G, H, I, L, N, P, Q, S, K]. All of these alleles remained cytoplasmic in the presence of Qtip except for cIVP882R11K, which showed partial localization (Figure 3D). These results demonstrate that, in cIVP882, the amino acid at position 11 is critical for susceptibility to Qtip. To determine if resistance to Qtip is separable from the DNA-binding function of cIVP882, we tested cIVP882R11M-HALO and the 11 other R11 variants for their ability to repress Pq-lux. Despite the above cIVP882 domain analysis, which did not implicate R11 as being involved in DNA binding (Figure S1A), Figure 3E shows that none of our cIVP882R11x-HALO variants, including cIVP882R11K-HALO, repressed Pq-lux. We interpret these data to mean that, at least with respect to amino acid residue R11, elimination of Qtip recognition by cIVP882 also eliminates cIVP882 DNA-binding activity. Additionally, our findings that cIVP882R11K-HALO and cIVP882DBD*-HALO fail to bind DNA but can still be bound by Qtip (Figures 1D and 3D) suggest that Qtip binding to cIVP882 requires only a subset of the residues cIVP882 uses to bind DNA.
Qtip Localizes to Cell Poles in the Absence of its Partner Repressor
Having identified the residues in cIVP882 that confer its functions and drive its interaction with Qtip, we next sought to pinpoint the amino acids in Qtip that are crucial for its activities. To monitor Qtip localization in vivo, we employed a SNAP-Qtip fusion, which enables microscopic visualization and tracking using fluorescent SNAP ligands. We previously showed that SNAP-Qtip produced from an aTc-inducible promoter sequesters cIVP882-HALO identically to aTc-induced untagged Qtip (Silpe and Bassler, 2019a). Figures S3A and S3B demonstrate that, also like native Qtip, SNAP-Qtip derepressed cIVP882-mediated Pq-lux expression and induced lysis in VP882 lysogens. These results show that the SNAP tag does not interfere with known Qtip functions and the construct can be used as a fluorescent tool to probe Qtip properties.
The first question we addressed was whether Qtip could transit to the pole in the absence of cIVP882 or whether complex formation between Qtip and cIVP882 is required. Figure 4A shows that SNAP-Qtip localized to the cell pole irrespective of whether or not cIVP882-HALO was present. One possibility is that the observed Qtip polar localization occurred as a consequence of high-level production from the aTc-inducible promoter. We titrated down the inducer and we could find no concentration of aTc capable of generating measurable SNAP-Qtip signal that did not also cause polar localization (Figure S3C). Natural Qtip levels have not been established so we do not have a comparison. However, since the SNAP tag and cIVP882-HALO are both cytoplasmic [Figure 4A and (Silpe and Bassler, 2019a)], our data suggest that polar localization of the Qtip-cIVP882 complex is a property conferred by Qtip.
Figure 4. SNAP-Qtip localizes to the poles in the absence of its partner repressor cIVP882.
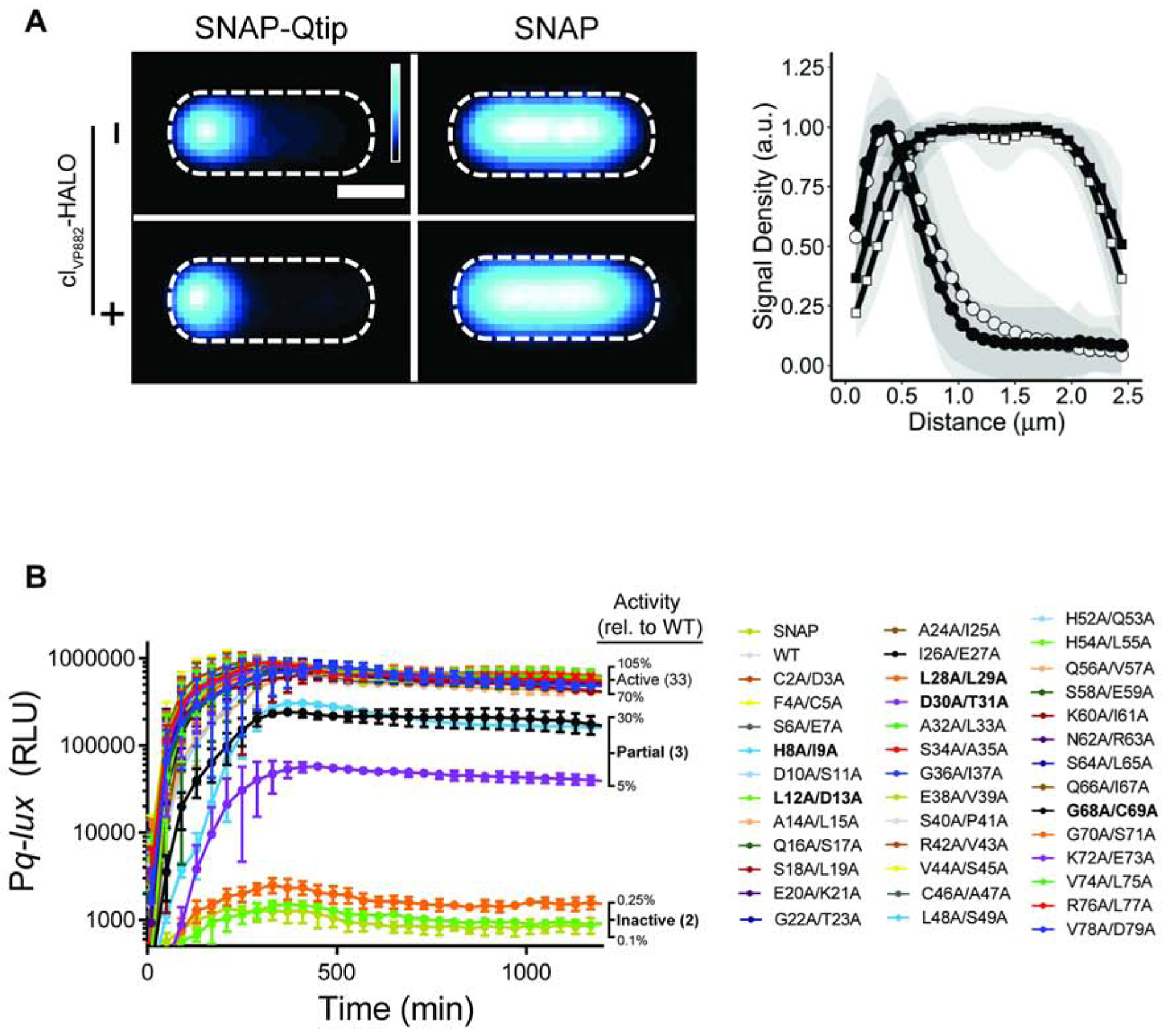
(A) Left: Composite images from individual cell analyses of E. coli producing SNAP-Qtip or SNAP, in the absence or presence of cIVP882-HALO (designated as − or + on the left side of the images) labeled with SNAP-JF503. SNAP-JF503 fluorescence intensity is displayed as a cyan heat map. Black and white reflect the lowest and highest intensity, respectively. Scale bar, as in Figure 1D. Right: Line plot of SNAP-JF503 fluorescence intensity extracted from individual cell images used in the left panel. Distance along the x-axis as in Figure 1D. Symbols: SNAP-Qtip (circles) and SNAP (squares), each in the absence (open) or presence (closed) of cIVP882-HALO. Shaded regions represent ± 1 SD from the mean. (B) Time-course of light production from the Pq-lux reporter in E. coli producing cIVP882 and either SNAP, WT SNAP-Qtip, or the designated SNAP-Qtip double-alanine variant. Variants exhibiting partial activity or that are inactive are shown in bold text in the key. RLU as in Figure 1. Data represented as mean ± SD with n = 3 biological replicates. See also Figures S3, S4, and S5.
Alanine-Scanning Mutagenesis of Qtip Decouples its Localization Function from its Function as an Inhibitor of cIVP882 Repressor Activity
Our finding that Qtip sequesters and inhibits cIVP882 autocleavage and DNA binding led us to wonder whether the ability of Qtip to localize cIVP882 is separable from its ability to inactivate cIVP882. To explore this issue, we performed alanine-scanning mutagenesis on Qtip. We altered two consecutive codons at a time. Qtip is a 79 amino acid protein. Excluding the start and stop codons and one existing Ala-Ala pair, this strategy required generation of 38 Qtip Ala-Ala double mutants, each fused to SNAP and cloned onto a plasmid under the aTc-inducible promoter. We assessed each Qtip allele for inactivation of cIVP882 DNA-binding activity and for co-localization with cIVP882-HALO.
Regarding cIVP882 DNA-binding and repressor function: we transformed the plasmids carrying the mutant SNAP-qtip alleles into E. coli harboring a second plasmid containing cIVP882 and Pq-lux. The logic was: cIVP882 represses Pq-lux, and SNAP-Qtip inactivates cIVP882, thus, introduction of a functional SNAP-Qtip mutant will induce light production, while introduction of a non-functional Qtip mutant will not (Figure 1A). Thirty three of the 38 SNAP-Qtip Ala-Ala mutants showed at least ~70% of wild-type (WT) SNAP-Qtip activity in the Pq-lux assay (Figure 4B). Of the 5 remaining SNAP-Qtip mutants, 3 (SNAP-QtipH8A/I9A, SNAP-QtipD30A/T31A, and SNAP-QtipG68A/C69A) exhibited partial activity (5%−30% of WT) and 2 (SNAP-QtipL12A/D13A and SNAP-QtipL28A/L29A) were incapable of driving light production (<0.3% of WT). Figure S4A shows that 4 of the 5 defective SNAP-Qtip mutant proteins were produced at approximately the same level as WT SNAP-Qtip. Only SNAP-QtipL28A/L29A was produced at lower than WT levels. Thus, differences in protein production for SNAP-QtipH8A/I9A, SNAP-QtipD30A/T31A, SNAP-QtipG68A/C69A, and SNAP-QtipL12A/D13A cannot be responsible for their attenuated activity.
Regarding co-localization with cIVP882: we used confocal microscopy to assess the localization patterns of the five defective SNAP-Qtip variants and cIVP882-HALO by labeling cells in which the proteins had been co-expressed with the red-fluorescent HALO-ligand HALO-TMR and the green fluorescent SNAP-ligand SNAP-JF503 (Grimm et al., 2017). Figure S4B shows that the 3 partially-inactive SNAP-Qtip mutants (SNAP-QtipH8A/I9A, SNAP-QtipD30A/T31A, and SNAP-QtipG68A/C69A) were also partially defective in localizing cIVP882-HALO to the pole. The 2 SNAP-Qtip mutants that failed to inactivate cIVP882-HALO (SNAP-QtipL12A/D13A and SNAP-QtipL28A/L29A) also failed to localize cIVP882-HALO (Figure S5A and B, left panels). Remarkably, however, SNAP-QtipL12A/D13A, while unable to inactivate the cIVP882 repressor or to localize cIVP882HALO to the pole, did itself localize to the cell pole (Figure S5A, left panel).
We next investigated whether, in the cases of the 2 Qtip mutants that failed to inactivate cIVP882 (L12A/D13A and L28A/L29A), both Ala substitutions or only a single Ala residue were required to confer the mutant phenotype. To do this, we made all of the corresponding single Ala substitutions. In the case of the SNAP-QtipL12A/D13A variant, the D13A alteration was sufficient for the mutant phenotype: SNAP-QtipD13A localized to the pole and it did not drive polar localization or inactivation of cIVP882 (Figure 5A and 5B). By contrast, SNAP-QtipL12A exhibited partial polar localization (Figure S5A, right panel) and it had measurable inhibitory activity against cIVP882 (Figure 5A).
Figure 5. Qtip polar localization and inhibitory activity against cIVP882 are separable.
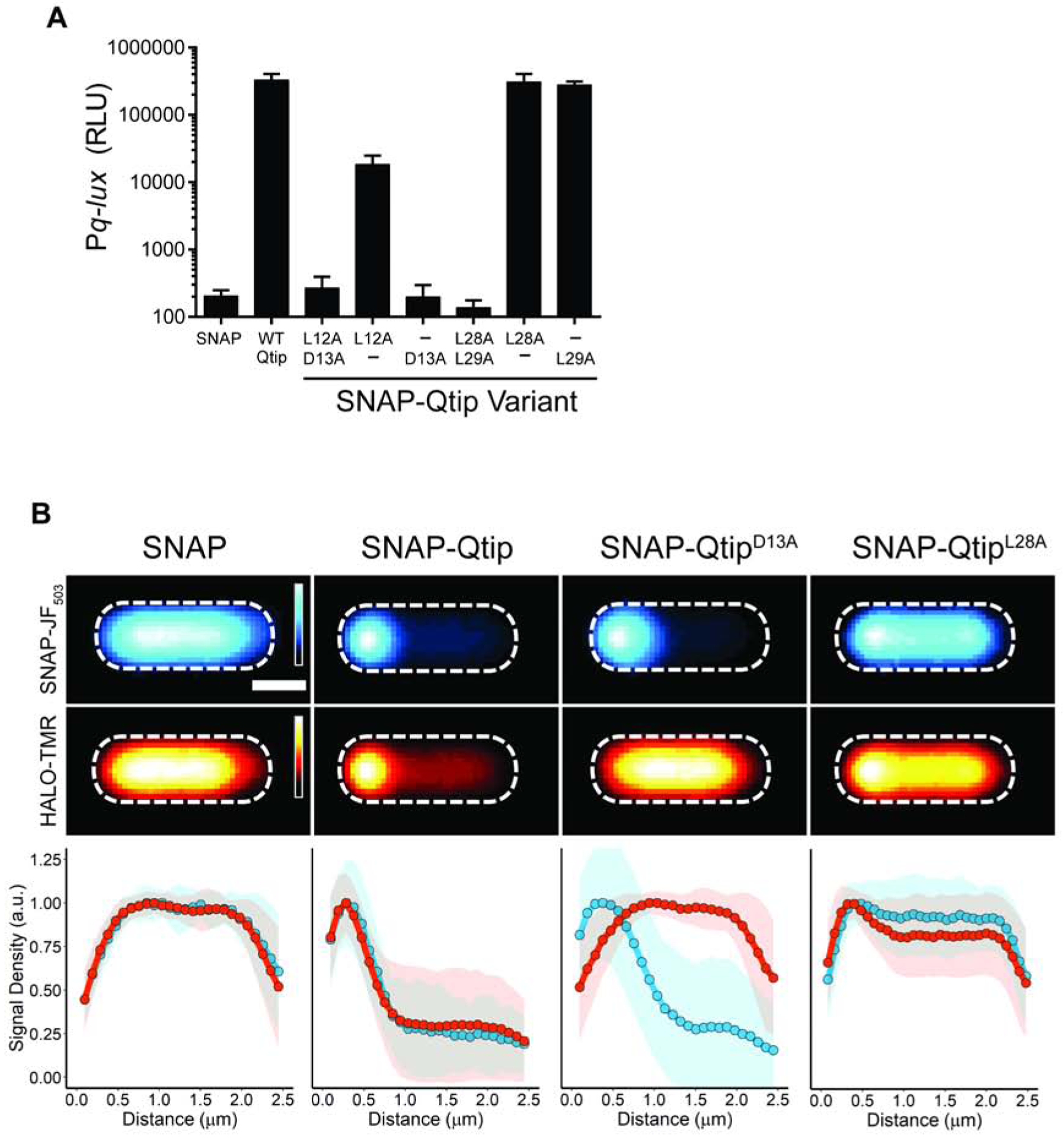
(A) Light production from the Pq-lux reporter in E. coli producing cIVP882 and SNAP, WT SNAP-Qtip, or the indicated SNAP-Qtip variant. RLU as in Figure 1. Data represented as mean ± SD with n = 3 biological replicates. (B) Upper two rows: Composite images from individual cell analyses of E. coli producing cIVP882-HALO and either SNAP, SNAP-Qtip, or the indicated SNAP-Qtip variant. Samples labeled with SNAP-JF503 (top row) and HALO-TMR (bottom row). Fluorescence intensity displayed as cyan (SNAP-JF503) and red (HALO-TMR) heat maps. Scale bar, as in Figure 1D. Lower: Line plots of the SNAP-JF503 (blue) and HALO-TMR (red) fluorescence intensities extracted from the single cells. Distance along the x-axis as in Figure 1D. Shaded regions represent ± 1 SD from the mean. See also Figures S4 and S5.
The case of SNAP-QtipL28A/L29A, which was cytoplasmic and failed to inactivate cIVP882, was less straightforward (see Figure S5B). The localization defect of SNAP-QtipL28A/L29A can be explained by mutation of only the first codon: SNAP-QtipL28A was cytoplasmic (Figure 5B), whereas SNAP-QtipL29A was partially localized at the pole (Figure S5B). However, the defect in inhibition of cIVP882 repressor activity by SNAP-QtipL28A/L29A cannot be explained by either of the single site variants: both SNAP-QtipL28A and SNAP-QtipL29A were active against cIVP882-HALO (Figure 5A). In addition, the poor production of SNAP-QtipL28A/L29A could not be ascribed to either single site variant as both SNAP-QtipL28A and SNAP-QtipL29A were produced at the levels of WT SNAP-Qtip (Figure S4A). Our interpretation is that Qtip residue L28 is required for Qtip localization but dispensable for inhibitory activity against cIVP882, whereas Qtip residue L29 is dispensable for both traits. In the SNAP-QtipL28A/L29A double mutant, there is a synthetic defect that causes reduced protein levels, which underpin the SNAP-QtipL28A/L29A defects against cIVP882-HALO. Collectively, our results with the defective Qtip mutants reveal that a single site substitution, D13A, abolishes the ability of Qtip to inactivate cIVP882 while not affecting the ability of Qtip to localize to the cell pole, and a single substitution at a different site, L28A, disrupts localization but not inhibitory activity against cIVP882.
The Phage P22 Antirepressor, Ant, Inactivates cILambda without Sequestration
Among the first antirepressors discovered was Ant of phage P22 (Botstein et al., 1975; Levine et al., 1975; Susskind and Botstein, 1975). While Ant is approximately four times the size of Qtip (34.6 kDa vs 8.4 kDa) and the proteins have no homology, the P22-Ant system does share similarities with the VP882-Qtip system: in both phages, the repressor protein can both be inactivated by cleavage and can be inactivated by an antirepressor, rather than one or the other. Ant, in addition to inactivating the P22 repressor, called C2, and inducing lysis, also binds to and interferes with DNA binding of cILambda (Susskind and Botstein, 1975), suggesting cross-reactivity analogous to what we observe between Qtip and other phage repressors (Silpe and Bassler, 2019a). We thus considered how features of Qtip-mediated repressor inactivation might parallel those of Ant. Specifically, we wondered whether polar localization occurs when Ant inactivates its partner repressors, which to our knowledge, has not yet been investigated.
To verify that Ant affects cILambda-directed lysis-lysogeny, we cloned the ant gene under the aTc-inducible promoter and introduced it into E. coli lysogenized by the temperaturesensitive lambda cI857 phage (Sussman and Jacob, 1962). Lambda cI857 only lysogenizes E. coli at temperatures at or below 30°C. As a control, and consistent with our above result showing that Qtip does not sequester cILambda [see Figure 3B and (Silpe and Bassler, 2019a)], we show that induction of Qtip or SNAP-Qtip production in the lambda cI857 lysogen did not induce lysis, whereas lysis occurred following induction in E. coli harboring the VP882 phage as an episome (Figure 6A and 6B). These results demonstrate that Qtip is a functional antirepressor for phage VP882 but not for phage lambda. In contrast, aTc-induced production of Ant inhibited growth of E. coli carrying the lambda cI857 lysogen, showing that the cILambda protein is inactivated by Ant (Figure 6A). Production of Ant in E. coli harboring the VP882 episome did not affect growth (Figure 6B) nor did Ant derepress Pq-lux expression in the E. coli reporter system (Figure S6A). Taken together, these results indicate that Ant is a functional antirepressor for phage lambda but not for phage VP882. Like Qtip, Ant remained functional when fused to SNAP (Ant-3xFLAG-SNAP; Figure 6A and 6B), however, unlike SNAP-Qtip, Ant3xFLAG-SNAP was cytoplasmic, and its localization did not change in the presence or absence of cIVP882-HALO, cILambda-HALO, or its natural partner, C2-HALO (Figure 6C). Similarly, the presence of Ant-3xFLAG-SNAP did not change the localization of any of the three repressor proteins (cIVP882-, cILambda-, and C2-HALO), and SNAP-Qtip did not change the localization of C2- or cILambda-HALO (Figure 6C). We conclude that while both Ant and Qtip are capable of inactivating their target and related repressors, Qtip induces polar localization, while Ant does not.
Figure 6. Ant is cytoplasmic and induces lysis in phage lambda but not in phage VP882, and Qtip localizes at the pole and induces lysis in phage VP882 but not in phage lambda.
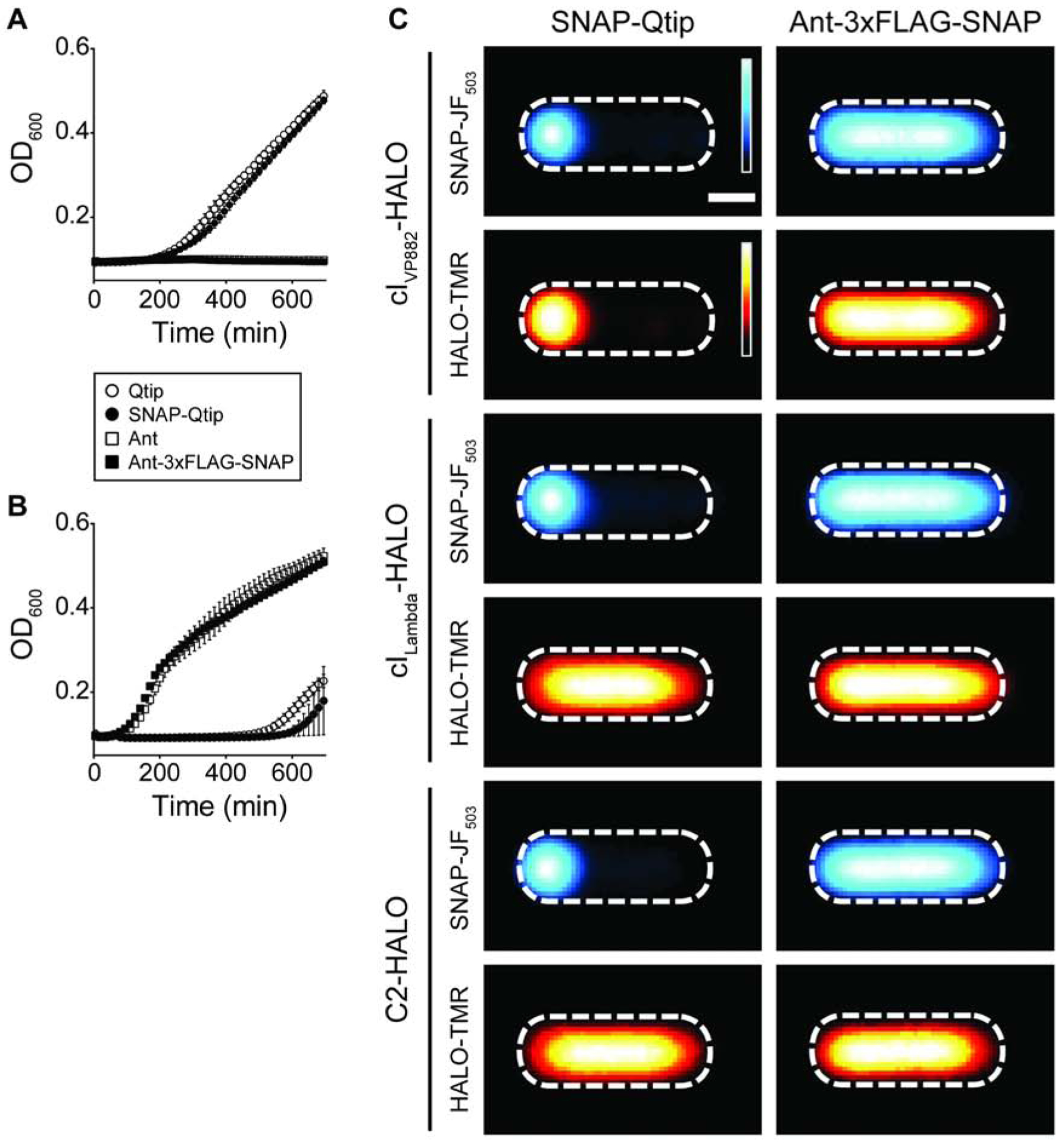
Growth curve of E. coli lysogenized by phage lambda cI857 (A) or phage VP882 CmR::Tn5 (B), each expressing the antirepressor constructs indicated in the key. (A) was performed at 30°C, (B) at 37°C. Data represented as mean ± SD with n = 3 biological replicates. (C) Composite images from individual cell analyses of E. coli producing SNAP-Qtip (left column) or Ant-3xFLAG-SNAP (right column) and cIVP882-HALO (first two rows), cILambda-HALO (third and fourth rows), or C2-HALO (fifth and sixth rows). SNAP and HALO labeled with SNAP-JF503 and HALO-TMR, respectively, and displayed using the same heat map color schemes as in Figure 5B. Scale bar, as in Figure 1D. See also Figure S6.
Discussion
Our investigation of the interactions between Qtip and cIVP882 revealed that, in some respects, cIVP882 behaves much like cILambda. Both repressors block lytic development in their respective phages and have SOS-responsive, RecA-activated catalytic domains located at their C-termini that promote intramolecular cleavage reactions. Both cIVP882 and cILambda also encode N-terminal DNA-binding domains, however, a feature present in cIVP882 that enables Qtip binding is absent from cILambda. Susceptibility to Qtip provides the VP882 phage with a QS-dependent trigger. Specifically, Qtip, which is produced in response to VqmAPhage binding to the host-produced QS AI DPO, prevents cIVP882 from binding DNA.
Temperate phages encoding antirepressors typically possess repressor proteins that lack catalytic C-terminal domains, presumably because the antirepressor fulfills that function. Thus, repressors in these systems are often half the size of cILambda (Lemire et al., 2011). cIVP882 and P22 C2 are curious in this regard because both possess autoproteolytic activity and are subject to inactivation by antirepressors (Qtip and Ant, respectively). Strikingly, early work on phage P22 showed that an Ant-overproducing P22 mutant that was exposed to UV light to induce RecA-dependent autoproteolysis did not undergo autoproteolysis, which is unlike a similarly treated non-Ant-overproducing P22 prophage (Prell and Harvey, 1983). This experiment suggested that Ant production “protects” C2 from autoproteolysis (Prell and Harvey, 1983). The experiment did not address whether the effect was direct or indirect. Our result showing that Qtip binding inhibits cIVP882 autoproteolysis in vitro is consistent with the Ant-C2 system, and furthermore demonstrates that antirepressors can directly alter the proteolytic susceptibility of the proteins they bind. We wonder whether antirepressor-directed inhibition of proteolytic degradation of phage repressors has implications for phage-host biology. While untested, we imagine that if antirepressor binding blocks access of RecA* to the repressor, there could be a larger pool of RecA* available to function in other crucial host SOS activities during stress (e.g., LexA cleavage and homologous DNA recombination). In support of this notion, production of the C-terminal domain of cILambda inhibits SOS induction of the LexAcontrolled regulon, suggesting the phage repressor titrates out the available RecA*, prohibiting it from performing its other functions (Ghodke et al., 2019).
In addition to inhibition of cIVP882, Qtip localizes cIVP882 to the cell poles. We show that inhibition and polar localization can be separated by single site mutations in Qtip that allow inhibition without localization (L28A) and vice-versa (D13A). Our objective in the mutant screen was to identify sites on Qtip that were absolutely required for Qtip function. Thus, we induced SNAP-qtip expression with a saturating amount of aTc to drive high level SNAP-Qtip production. Excluding the three double mutants with intermediate phenotypes (Figures 4B and S4B), this strategy revealed SNAP-QtipL12A/D13A, SNAP-QtipD13A, and SNAP-QtipL28A/L29A. Other SNAP-Qtip point mutants might exhibit interesting but more subtle defects if we performed the experiment using sub-maximal levels of inducer to lower the concentration of SNAP-Qtip produced. Going forward, it will be important to define how Qtip and cIVP882 interact. Currently, we are focused on solving the co-crystal structure of the Qtip-cIVP882 complex, which should reveal the interaction interface.
We also consider how the properties we discovered for Qtip compare to other antirepressors. Despite our data showing that the prototypical antirepressor, Ant, does not exhibit polar localization, to our knowledge, only one other phage antirepressor has been investigated at the single cell level and it, like Qtip, does localize to the poles (Davis et al., 2002). Specifically, the RS1 satellite phage to CTXφ of V. cholerae encodes the antirepressor, RstC, that binds to the CTXφ prophage repressor, RstR, rendering RstR insoluble. The consequence is production of cholera toxin and the transmission of the satellite and CTXφ phages to new cells (Davis et al., 2002). While RstC and Qtip have no homology, RstC is also a small protein (8.3 kDa) and it also induces aggregation of RstR (Davis et al., 2002). Unlike cIVP882, however, RstR does not have an obvious catalytic domain or cleavage site. Also similar to the phage VP882 system, CTXφ is induced by DNA damage via the host SOS response. However, unlike phage VP882, in CTXφ, regulation occurs via LexA binding to phage DNA to repress the expression of lytic genes (Quinones et al., 2005). Derepression occurs upon cleavage of LexA.
The biological significance of localization of Qtip and other antirepressors and the Qtip-directed sequestration of the repressor are not yet clear with respect to the host or the phage. One possibility is that localization ensures a high concentration of Qtip at the poles, which is where phage attachment and injection often occur. Specifically, tracking of quantum dot labeled bacteriophages, including one infecting V. cholerae, showed that phage adsorption occurs predominantly at the poles of the host cell (Edgar et al., 2008). Moreover, the FtsH protein of E. coli, which is involved in the regulation of lambda cII and cIII stability, is also localized to the poles (Edgar et al., 2008). One model proposed is that localization of FtsH at the site where lambda DNA first appears positions FtsH to regulate lysis-lysogeny of newly injected phage genomes (Edgar et al., 2008). Our work, showing that Qtip is also localized to the poles could implicate it in the superinfection process: the high local concentration of Qtip at the poles could enable it to rapidly act on repressors produced by newly infecting phages. While our current work shows that Ant of P22 does not share the same localization behavior as Qtip, it is worth noting that Ant is thought to be expressed early after infection, and the notion is that Ant plays a role during superinfection (Wu et al., 1987).
V. cholerae and Pseudomonas aeruginosa strains harboring QS-null mutations are frequently isolated (Feltner et al., 2016; Hammer and Bassler, 2003; Stutzmann and Blokesch, 2016) suggesting that there is pressure to avoid producing or to cheat on QS-produced public goods. As phage VP882 has the capacity to activate lysis in response to host DPO production, we wondered whether mutations in the phage-encoded QS pathway similarly arise. As one example, we identified a curious 8.5 kb contig (GenBank: NNHH01000051.1) in a recent dataset from mixed V. parahaemolyticus populations (Yang et al., 2019) (Figure S6B). The contig spans the region where vqmAPhage and qtip are located in phage VP882, between two conserved genes, repA and telN (Silpe and Bassler, 2019a). Aligned across its entire length, the contig is largely identical to VP882: the contig-derived RepA and TelN products have 99.2% (1229/1239) and 99.3% (534/538) amino acid identity, respectively, to the proteins encoded in phage VP882 (Figure S6B). However, between repA and telN, there is an 819 bp deletion on the contig that eliminates vqmAPhage, qtip, and the promoters and ribosome binding sites for both ORFs, leaving only the DNA encoding the first 36 amino acids of Qtip and the first 201 amino acids of VqmAPhage (Figure S6B). If this contig corresponds to a functional VP882-like phage, it suggests that the phage would be unresponsive to DPO and cannot execute QS-induced lysis. One can imagine scenarios in which inactivation of phage QS could benefit the host and/or the phage. For example, lack of the ability to induce host cell-density-dependent lysis could promote longer-term lysogeny, as has been shown to be prevalent for phages in gut ecosystems (Kim and Bae, 2018).
It is also worth noting that phage VP882 is a plasmid-like phage, which we previously showed allows it to be transformed and maintained in bacteria that likely fall outside of the natural host range of the phage (e.g., Salmonella typhimurium and E. coli) (Silpe and Bassler, 2019a). Perhaps existing as a plasmid enables multiple mechanisms of transfer to new cells. We suspect that bacterial species within the natural phage VP882 host range can contract VP882 as a phage, via infection, or they can be transformed, while species outside of the natural host range can take up VP882 as a plasmid, only via transformation. Interestingly, a recently deposited NCBI entry (GenBank: AAEKWQ010000030.1) of Salmonella paratyphi B variant L(+) tartrate(+) isolated from a human stool sample (CDC, National Salmonella Reference Laboratory) revealed a ~37.5 kb contig with ~90% identity (35,422 of 37,472 sites) on a nucleotide basis across its entire length to the genome of phage VP882 (Figure S6C). The Salmonella-derived contig has intact vqmAPhage and qtip genes, each with 99.1% (233/235 sites) and 100% (79/79 site) identity at the protein level, respectively, to those of phage VP882. Vibrios encode the DPO receptor and effector, VqmA and VqmR, respectively, and, as far as we are aware, Salmonella do not. Members from both bacterial families produce DPO (Papenfort et al., 2017). It is therefore possible that VP882-like phages, by encoding a receptor to a widely produced AI, can use the information encoded in DPO even in bacteria that, themselves, cannot.
In terms of the origin of the Salmonella-derived VP882-like contig, we imagine that since one area in which Salmonella and Vibrios come into contact is in the human host, it is plausible that the phage is transferred between species as a plasmid during human infection by the bacteria. In support of such a model, recent work showed that within the human gut, Salmonella persister cells promote the spread of broad-range resistance plasmids even in the absence of their selection (Bakkeren et al., 2019).
It is reported that as many as 80% of phage-encoded gene products are unrelated to proteins of known functions (Hatfull and Hendrix, 2011). Discovering the functions of uncharacterized phage products reveals the rich diversity of processes that phages can affect, including bacterial physiology, phage-phage interactions, and, in phages carried by endosymbiotic bacteria, the eukaryotic host (Perlmutter et al., 2019). Our results provide insight into one mechanism by which a newly discovered phage-encoded protein, Qtip, can function to control phage-bacterial interactions. More generally, based on our findings with phage VP882, we propose that plasmid-like phages have greater host flexibility than integrating phages, and we speculate that their transfer may occur between pathogens in the human host setting.
STAR Methods
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bonnie L. Bassler (bbassler@princeton.edu). All unique reagents generated in this study are available from the Lead Contact without restriction.
Experimental Model and Subject Details
Bacterial Strains and Growth Conditions
All strains were grown with aeration in Luria-Bertani (LB-Miller, BD-Difco) broth at 37°C except for the E. coli lambda cI857 lysogen which was grown at 30°C. Stra ins used in this study are listed in Table S1. Unless otherwise noted, antibiotics and inducers were used at: 100 μg mL−1 ampicillin (Amp, Sigma), 100 μg mL−1 kanamycin (Kan, GoldBio), 5 μg mL−1 chloramphenicol (Cm, Sigma), 100 ng mL−1 anhydrotetracycline (aTc, Clontech), and 0.4 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG, Fisher). For microscopy, HALO-TMR (Promega) and SNAP-JF503 (Lavis Lab) were used at concentrations of 1 and 2 μM, respectively. AB solid agar consisted of 1.5% agar, 0.3 M NaCl, 50 mM MgSO4, 0.2% casamino acids, 1 mM arginine, 1% glycerol, and 10 mM potassium phosphate at pH 7.5.
Cloning Techniques
Primers and dsDNA (gene blocks) used for plasmid construction are listed in Table S2, and all were obtained from Integrated DNA Technologies. Plasmids are listed in Table S3. Plasmid combinations are organized according to the figures and strains in which they appear in Table S4. Gibson assembly, intramolecular reclosure, and traditional cloning methods were employed for all cloning, as indicated in Table S2. PCR with Q5 High Fidelity Polymerase (NEB) was used to generate insert and backbone DNA. Gibson assembly relied on the HiFi DNA assembly mix (NEB). All enzymes used in cloning were obtained from NEB. Construction of the random cIVP882-HALO variants was carried out by GeneMorph II EZClone Domain Mutagenesis Kit (Agilent) using 250 ng of template DNA (pJES-183), primers JSO-1649/1650, and 32 cycles. Transfer of plasmids into the V. parahaemolyticus VP882 lysogen was carried out by conjugation followed by selective plating on 50 U mL−1 polymyxin B (Sigma) and Kan. In V. parahaemolyticus, 10 ng mL−1 aTc was used for induction rather than the 100 ng mL−1 inducer concentration used in E. coli.
Method Details
Growth, Lysis, and Reporter Assays
Unless otherwise noted, overnight cultures were back diluted 1:200 into fresh medium with appropriate antibiotics prior to being dispensed (200 μL) into 96 well plates (Corning Costar 3904). aTc was added as specified. Plates were shaken at 37°C or 30°C and a BioTek Synergy Neo2 Multi-Mode reader was used to measure OD600 and bioluminescence. Relative light units (RLU) were calculated by dividing the bioluminescence by the OD600 at that time. Testing the panel of SNAP-Qtip mutants (Figure 4B) made additional use of a BioSpa 8 automated incubator to read multiple plates at regular intervals. Unless otherwise noted, fixed time-point reporter assays were measured ~8 h after inoculation of the cells into the wells.
Protein Expression and Purification
For production and purification of cILambda-HALO-HIS, cIVP882-HALO-HIS, the chimeras, and cIVP882-HALO-HIS variants used for microscopy, cleavage assays, and DNA-binding (Pq-lux) assays, plasmids harboring the indicated constructs were expressed in E. coli T7Express lysY/Iq using 0.4 mM IPTG induction and 18°C incubation ove rnight. Cells were pelleted at 16,100 × g for 10 min and resuspended in lysis buffer (25 mM Tris-HCl pH 8, 150 mM NaCl, 1 mM DTT). Cells were lysed using sonication and subjected to centrifugation at 32,000 × g for 40 min. The supernatants were applied to Ni-NTA Superflow resin (Qiagen). Resin was washed with 5 column volumes (CVs) of 25 mM Tris-HCl pH 8, 150 mM NaCl, 20 mM imidazole, and 1 mM DTT, and eluted in 2 CVs of 25 mM Tris-HCl pH 8, 150 mM NaCl, 300 mM imidazole, and 1 mM DTT. Eluates were concentrated, incubated with 7 μM (final conc.) HALO-Alexa660 ligand on ice for 30 min before being loaded onto a Superdex-200 size exclusion column (GE Healthcare) in gel filtration buffer (25 mM Tris-HCl pH 8, 150 mM NaCl, and 1 mM DTT). For differential labeling experiments requiring the use of both HALO-Alexa660 and HALO-TMR labeled WT cIVP882-HALO, cIVP882S130A-HALO, and cIVP882K172A-HALO proteins, a second batch of lysates prepared from the relevant strains was purified using the identical procedure as for the HALO-Alexa660 labeled proteins except that the label used was HALO-TMR (5 μM final conc.). All proteins were concentrated, flash frozen, and stored at −80°C prior to use. Differentially labeled proteins were stored separately and mixed immediately before use.
For production and purification of cIVP882-HALO (no HIS) used for comparative analysis to cIVP882-HALO in complex with HIS-Qtip, cIVP882-HALO (no HIS) (pJES-190) was overexpressed in E. coli T7Express lysY/Iq as above. The clarified lysate was loaded onto a heparin column (GE Healthcare) and eluted by a linear gradient from buffer A (25 mM Tris-HCl pH 8, 1 mM DTT) to buffer B (25 mM Tris-HCl pH 8, 1 M NaCl, 1 mM DTT). Peak fractions were pooled, concentrated, treated with 7 μM HALO-Alexa660 ligand (final conc.), subjected to Superdex-200 size exclusion chromatography in gel filtration buffer, and stored at −80°C.
Production and purification of HIS-Qtip in complex with cIVP882-HALO was carried out as above with the HIS-tagged proteins, except that the E. coli T7Express lysY/Iq co-expressed HIS-Qtip (pJES-147) and cIVP882-HALO (pJES-190). The eluted complex was concentrated, treated with 7 μM HALO-Alexa660 ligand (final conc.), subjected to Superdex-200 size exclusion chromatography in gel filtration buffer, and stored at −80°C.
Electrophoretic Mobility Shift Assay (EMSA)
Primers JSO-956/957 (Table S2) and purified VP882 DNA were used to make the probe (257 bp). 20 ng of probe was used in each EMSA reaction. The highest concentration of cIVP882-HALO, designated 25x (Figure 1B), was ~300 nM. The concentration of the HIS-Qtip-cIVP882-HALO complex was matched to that of free cIVP882-HALO (no HIS-Qtip) based on the fluorescence from the cIVP882-HALO-conjuaged Alexa660 dye present in both samples. A 5-fold serial dilution of protein was applied to indicated lanes. The protein and probe were combined in binding buffer (25 mM Tris-HCl pH 8, 50 mM NaCl, 1 mM DTT), and incubated at RT for 15 min. The samples were subsequently loaded onto a Novex 6% DNA retardation gel (Thermo) at 4°C and electrophoresed in 1x TBE at 130 V for 2 h. The gel was stained for DNA by SYBR Green I Nucleic Acid Gel Stain (Thermo) and washed in 1x TBE for 20 min. The gel was subsequently imaged using an ImageQuant LAS 4000 imager under the SYBR Green setting. A duplicate batch of the samples was also subjected to electrophoresis in a 4–20% SDS-PAGE gel (Bio-Rad) to verify the correct loading of the HALO-Alexa660-labeled cIVP882-HALO in the EMSA. The gel was imaged using an ImageQuant LAS 4000 and detection of HALO and SNAP as outlined below.
in vitro Cleavage of Phage Repressor Proteins
10 μL of the indicated HALO-tagged repressor protein (0.5 mg mL−1) was mixed with 0.5 μL JSO-1538 ssDNA (1 mM, Table S2), 1 μL ATP-γ-S (50 mM, Sigma), and 10 μL 10x RecA buffer (NEB) in a 100 μL preparation. 2.5 μL RecA (2 mg mL−1, NEB) was added to initiate the reaction. All cleavage reactions were carried out at 37°C. At each time point, 10 μL aliquots were taken from the reactions and mixed with 5 μL 4x Laemmli Sample Buffer (BioRad). The samples were loaded onto a 4–20% SDS-PAGE gel for electrophoresis. The gel was imaged using an ImageQuant LAS 4000 imager under the Cy5 or the Cy3 and Cy5 settings before being stained with Coomassie Brilliant Blue. The RecA band revealed by Coomassie was used as the loading control (see below for additional details on in-gel imaging). In Figure 2A, RecA and ATP-γ-S were individually withheld from the reactions, as indicated in the figure. For the mixed reactions (Figure 3A), the total amount of cIVP882-HALO protein was doubled (20 μL per 100 μL reaction). Due to the extremely low yield of soluble HIS-Qtip-cIVP882-HALO complex that could be obtained using our approach, in Figure 2B, approximately 20-fold less cIVP882-HALO protein was used in the reactions relative to those in the other cleavage assays reported here. To ensure that equal amounts of free cIVP882-HALO and cIVP882-HALO in complex with HIS-Qtip were loaded onto gels, the amount of free cIVP882-HALO, as judged by HALO-Alexa660 fluorescence, was adjusted to match that of the labeled cIVP882-HALO in complex with HIS-Qtip.
In-Gel (Western) Detection of HALO and SNAP Constructs
SDS-PAGE gels were imaged for labeled HALO. HALO-TMR (excitation/emission: 555/585 nm) and HALO-Alexa660 (excitation/emission: 663/690 nm) were detected in a LAS 4000 imager under the Cy3 and Cy5 settings, respectively. Gels were subsequently stained for total protein using Coomassie Brilliant Blue. For assaying SNAP-Qtip production levels (Figure S4A), overnight cultures of the indicated strains were back diluted 1:500 in fresh medium containing 100 ng mL−1 aTc, 1 μM HALO-TMR, and 2 μM SNAP-Cell 647-SiR (NEB). Cultures were grown for 8 h at 37°C, prior to cell harvest, lysis (BugBuster, Millipore), and electrophoresis by SDS-PAGE. SNAP-Cell 647-SiR (excitation/emission: 647/661 nm) was detected by the LAS 4000 imager under the Cy5 setting making it compatible to use with HALO-TMR. Exposure times for all fluorophores never exceeded 30 sec.
Confocal Microscopy
Overnight cultures of E. coli T7Express lysY/Iq carrying the indicated HALO and/or SNAP fusions were diluted 1:500 in medium containing 100 ng mL−1 aTc, 1 μM HALO-TMR, and/or 2 μM SNAP-JF503. Cultures were returned to growth for 4–6 h, subjected to centrifugation, washed with PBS, and resuspended in fresh PBS to a final OD600 = 0.1–0.3. 8 μL of each sample was spotted onto a glass coverslip and overlaid with an AB agar pad. Samples were imaged approximately 40 min after preparation on a Leica SP8 Confocal microscope under photon counting mode. HALO-TMR was excited with 561 nm light and detected within the range 569–625 nm. SNAP was excited at 499 nm and detected between 513–542 nm. When imaged together, the HALO and SNAP channels were collected in sequential mode. Images for each sample were acquired as a tile-scan (3×3 or 4×4) and stitched in LASX (Leica Microsystems).
Image Analysis
Image analyses were performed in FIJI software (Version 1.52p). To generate intensity line traces and composite images of fluorescent SNAP and HALO fusions, individual cells were first segmented in the brightfield channel via an intensity-based thresholding procedure after image smoothing. Only cells of a defined size range were retained in further analysis for consistency. Individual segmented cells were manually rotated using the rotated rectangle function such that the long axis of the cell was oriented in the x-dimension. In cases where one cell poll contained a punctate signal, this pole was aligned to the left side of the image. For each x-aligned cell, the intensity profile was measured through a 0.5 μm thick (y-dimension) line traced through the long axis of the cell. Data were exported for quantitation and graphing in R software using gglplot2 (https://ggplot2.tidyverse.org). To account for cell-to-cell intensity differences, each line trace was normalized to the peak pixel intensity for that cell. For each condition (strain, fluorophore), the intensity traces of the individual cells were averaged, and the first standard deviation calculated to generate plots. To produce a composite image for each condition, the aligned individual cell images were concatenated and subsequently averaged. Resulting composite images were normalized to the highest pixel value within that image. For display, enhanced lookup tables were used (Red Hot for HALO-TMR fusions and Cyan Hot for SNAP-JF503 fusions). In all cases, n > 20 cells were analyzed.
Quantification and Statistical Analysis
Data are presented as the mean ± std unless otherwise indicated in the figure legends. The number of independent biological replicates for each experiment is indicated in the figure legends or methods. For imaging analysis, 20–25 single cells were used in compiling each composite. Cells were excluded from analysis if they were abnormal in size, contacting another cell, and/or exhibiting poor fluorescence from the SNAP- or HALO- channel.
Data and Software Availability
Unprocessed gels and micrographs that were used to in this study are deposited on Zenodo (https://doi.org/10.5281/zenodo.3576389). Other experimental data that support the findings of this study are available by reasonable request from the corresponding author.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| V. parahaemolyticus VP882 lysogen | BCRC | BCRC 80155 |
| E. coli T7Express lysY/Iq | NEB | Cat# C3013I |
| E. coli TOP10 | Invitrogen | Cat# C404010 |
| E. coli cI857 lysogen | (Sussman and Jacob, 1962); gift of Tom Silhavy | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SNAP-JF503 | Lavis Lab | N/A |
| SNAP-Cell 647-SiR | NEB | Cat# S9102S |
| HALO-TMR | Promega | Cat# G8251 |
| HALO-Alexa660 | Promega | Cat# G8471 |
| RecA and RecA Buffer | NEB | Cat# M0249L |
| ATP-γ-S | Sigma | Cat# A1388–5MG |
| Novex 6% DNA Retardation Gel | Thermo | Cat# EC63655BOX |
| SYBR Green I Nucleic Acid Gel Stain | Thermo | Cat# S7567 |
| 4–20% Mini-PROTEAN TGX Protein Gels | Bio-Rad | Cat# 4561096 |
| GeneMorph II EZClone Domain Mutagenesis Kit | Agilent | Cat# 200552 |
| Anhydrotetracycline | Takara | Cat# 631310 |
| Isopropyl β-D-1-thiogalactopyranoside | Fisher Scientific | Cat# BP1755–10 |
| Q5 High Fidelity Polymerase | NEB | Cat# M0491L |
| HiFi DNA assembly Mix | NEB | Cat# E2621L |
| Ni-NTA Superflow Resin | Qiagen | Cat# 30430 |
| Critical Commercial Assays | ||
| Synergy Neo2 Multi-Mode Reader | BioTek | N/A |
| BioSpa8 Automated Incubator | BioTek | N/A |
| ImageQuant LAS4000 | GE | N/A |
| Leica White Light Laser Confocal Microscope TCS SP8 | Leica Microsystems | N/A |
| Superdex 200 Increase 10/300 GL | GE Healthcare | Cat# 28990944 |
| HiTrap 5 ml Heparin HP | GE Healthcare | Cat# 17040701 |
| AKTA FPLC System | GE Healthcare | N/A |
| Deposited Data | ||
| Raw data | This paper, Zenodo (10.5281/zenodo.3576389) | https://doi.org/10.5281/zenodo.3576389 |
| Oligonucleotides | ||
| Primers used for plasmid construction | This study, IDT | Table S2 |
| Primers used for construction of EMSA probe | This study, IDT | Table S2 |
| dsDNA fragments (gBlocks) | This study, IDT | Table S2 |
| Recombinant DNA | ||
| Reporter and cI expression constructs | This study | Table S3 |
| cIVP882R11x-HALO-HIS mutants | This study | Table S3 |
| SNAP-Qtip mutants and Ant constructs | This study | Table S3 |
| Software and Algorithms | ||
| Prism v6 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| FIJI v1.52p | FIJI | http://fiji.sc/ |
| LASX v3.5.5.19976 | Leica Microsystems | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| RStudio Desktop 1.2.5033 | RStudio / The R Foundation | https://rstudio.com |
Highlights.
The phage antirepressor Qtip sequesters its partner cI repressor to the cell poles
Qtip bound repressor is inhibited for DNA-binding and autoproteolysis
Qtip recognizes the N-terminal domain of the cI repressor
Repressor recognition and polar localization are separable properties of Qtip
Acknowledgments
This work was supported by the Howard Hughes Medical Institute, NIH Grant R37GM065859, National Science Foundation Grant MCB-1713731 (to B.L.B.), NIGMS grant T32GM007388 (O.P.D), the Damon Runyon Fellowship Award, DRG-2302-17 (to A.A.B), a Charlotte Elizabeth Procter Fellowship provided by Princeton University (to J.E.S.), and a National Defense Science and Engineering Graduate Fellowship supported by the Department of Defense (to J.E.S). We are grateful to Dr. Luke Lavis for providing ample SNAP-JF503 ligand. We thank members of PulseNet and the National Salmonella Reference Laboratory for information on the reported Salmonella-derived contig. We thank Tom Silhavy for helpful discussions and Betsy Hart for the cI857 strain. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental Information
Supplemental information includes 4 tables and 6 figures and can be found with this article online.
Declaration of Interests
The authors declare no competing financial interests.
References
- Bakkeren E, Huisman JS, Fattinger SA, Hausmann A, Furter M, Egli A, Slack E, Sellin ME, Bonhoeffer S, Regoes RR, et al. (2019). Salmonella persisters promote the spread of antibiotic resistance plasmids in the gut. Nature 573, 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D, Lew KK, Jarvik V, and Swanson CA (1975). Role of antirepressor in the bipartite control of repression and immunity by bacteriophage P22. J. Mol. Biol 91, 439–462. [DOI] [PubMed] [Google Scholar]
- Chen X, Schauder S, Potier N, Dorsselaer AV, Pelczer I, Bassler BL, and Hughson FM (2002). Structural identification of a bacterial quorum-sensing signal containing boron. Nature 415, 545–549. [DOI] [PubMed] [Google Scholar]
- Davis BM, Kimsey HH, Kane AV, and Waldor MK (2002). A satellite phage-encoded antirepressor induces repressor aggregation and cholera toxin gene transfer. EMBO J. 21, 4240–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Rokney A, Feeney M, Semsey S, Kessel M, Goldberg MB, Adhya S, and Oppenheim AB (2008). Bacteriophage infection is targeted to cellular poles. Mol. Microbiol 68, 1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erez Z, Steinberger-Levy I, Shamir M, Doron S, Stokar-Avihail A, Peleg Y, Melamed S, Leavitt A, Savidor A, Albeck S, et al. (2017). Communication between viruses guides lysis–lysogeny decisions. Nature 541, 488–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltner JB, Wolter DJ, Pope CE, Groleau M-C, Smalley NE, Greenberg EP, Mayer-Hamblett N, Burns J, Déziel E, Hoffman LR, et al. (2016). LasR Variant Cystic Fibrosis Isolates Reveal an Adaptable Quorum-Sensing Hierarchy in Pseudomonas aeruginosa. MBio 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodke H, Paudel BP, Lewis JS, Jergic S, Gopal K, Romero ZJ, Wood EA, Woodgate R, Cox MM, and van Oijen AM (2019). Spatial and temporal organization of RecA in the Escherichia coli DNA-damage response. ELife 8, e42761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese KC, Michalowski CB, and Little JW (2008). RecA-Dependent Cleavage of LexA Dimers. J. Mol. Biol 377, 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, Muthusamy AK, Liang Y, Brown TA, Lemon WC, Patel R, Lu R, Macklin JJ, Keller PJ, Ji N, et al. (2017). A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat. Methods 14, 987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer BK, and Bassler BL (2003). Quorum sensing controls biofilm formation in Vibrio cholerae. Mol. Microbiol 50, 101–104. [DOI] [PubMed] [Google Scholar]
- Hargreaves KR, Kropinski AM, and Clokie MRJ (2014). What Does the Talking?: Quorum Sensing Signalling Genes Discovered in a Bacteriophage Genome. PLoS ONE 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfull GF, and Hendrix RW (2011). Bacteriophages and their Genomes. Curr. Opin. Virol 1, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel T, Velleman M, and Schuster H (1992). C1 repressor of phage P1 is inactivated by noncovalent binding of P1 Coi protein. J. Biol. Chem 267, 4183–4188. [PubMed] [Google Scholar]
- Herzog R, Peschek N, Fröhlich KS, Schumacher K, and Papenfort K (2019). Three autoinducer molecules act in concert to control virulence gene expression in Vibrio cholerae. Nucleic Acids Res. 47, 3171–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Høyland-Kroghsbo NM, Paczkowski J, Mukherjee S, Broniewski J, Westra E, BondyDenomy J, and Bassler BL (2017). Quorum sensing controls the Pseudomonas aeruginosa CRISPR-Cas adaptive immune system. Proc. Natl. Acad. Sci 114, 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M-S, and Bae J-W (2018). Lysogeny is prevalent and widely distributed in the murine gut microbiota. ISME J. 12, 1127–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Kim HJ, Son SH, Yoon HJ, Lim Y, Lee JW, Seok Y-J, Jin KS, Yu YG, Kim SK, et al. (2016). Noncanonical DNA-binding mode of repressor and its disassembly by antirepressor. Proc. Natl. Acad. Sci 113, E2480–E2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganenka L, Sander T, Lagonenko A, Chen Y, Link H, and Sourjik V (2019). Quorum Sensing and Metabolic State of the Host Control Lysogeny-Lysis Switch of Bacteriophage T1. MBio 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemire S, Figueroa-Bossi N, and Bossi L (2011). Bacteriophage Crosstalk: Coordination of Prophage Induction by Trans-Acting Antirepressors. PLOS Genet. 7, e1002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Truesdell S, Ramakrishnan T, and Bronson MJ (1975). Dual control of lysogeny by bacteriophage P22: An antirepressor locus and its controlling elements. J. Mol. Biol 91, 421–438. [DOI] [PubMed] [Google Scholar]
- Mardanov AV, and Ravin NV (2007). The Antirepressor Needed for Induction of Linear Plasmid-Prophage N15 Belongs to the SOS Regulon. J. Bacteriol 189, 6333–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JP, Frank EG, Levine AS, and Woodgate R (1998). Intermolecular cleavage by UmuD-like mutagenesis proteins. Proc. Natl. Acad. Sci 95, 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meibom KL (2005). Chitin Induces Natural Competence in Vibrio cholerae. Science 310, 1824–1827. [DOI] [PubMed] [Google Scholar]
- Miller MB, Skorupski K, Lenz DH, Taylor RK, and Bassler BL (2002). Parallel Quorum Sensing Systems Converge to Regulate Virulence in Vibrio cholerae. Cell 110, 303–314. [DOI] [PubMed] [Google Scholar]
- Ndjonka D, and Bell CE (2006). Structure of a hyper-cleavable monomeric fragment of phage lambda repressor containing the cleavage site region. J. Mol. Biol 362, 479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfort K, and Bassler BL (2016). Quorum sensing signal–response systems in Gramnegative bacteria. Nat. Rev. Microbiol 14, 576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfort K, Silpe JE, Schramma KR, Cong J-P, Seyedsayamdost MR, and Bassler BL (2017). A Vibrio cholerae autoinducer–receptor pair that controls biofilm formation. Nat. Chem. Biol 13, 551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmutter JI, Bordenstein SR, Unckless RL, LePage DP, Metcalf JA, Hill T, Martinez J, Jiggins FM, and Bordenstein SR (2019). The phage gene wmk is a candidate for male killing by a bacterial endosymbiont. PLOS Pathog. 15, e1007936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prell HH, and Harvey AM (1983). P22 antirepressor protein prevents in vivo recA-dependent proteolysis of P22 repressor. MGG Mol. Gen. Genet 190, 427–431. [DOI] [PubMed] [Google Scholar]
- Ptashne M (2004). A Genetic Switch, Third Edition: Phage Lambda Revisited (Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- Quinones M, Kimsey HH, and Waldor MK (2005). LexA Cleavage Is Required for CTX Prophage Induction. Mol. Cell 17, 291–300. [DOI] [PubMed] [Google Scholar]
- Roberts JW, and Roberts CW (1975). Proteolytic cleavage of bacteriophage lambda repressor in induction. Proc. Natl. Acad. Sci 72, 147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer RT, Ross MJ, and Ptashne M (1982). Cleavage of the lambda and P22 repressors by recA protein. J. Biol. Chem 257, 4458–4462. [PubMed] [Google Scholar]
- Schauder S, Shokat K, Surette MG, and Bassler BL (2001). The LuxS family of bacterial autoinducers: biosynthesis of a novel quorum-sensing signal molecule. Mol. Microbiol 41, 463–476. [DOI] [PubMed] [Google Scholar]
- Shearwin KE, Brumby AM, and Egan JB (1998). The Tum Protein of Coliphage 186 Is an Antirepressor. J. Biol. Chem 273, 5708–5715. [DOI] [PubMed] [Google Scholar]
- Silpe JE, and Bassler BL (2019a). A Host-Produced Quorum-Sensing Autoinducer Controls a Phage Lysis-Lysogeny Decision. Cell 176, 268–280.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silpe JE, and Bassler BL (2019b). Phage-Encoded LuxR-Type Receptors Responsive to Host-Produced Bacterial Quorum-Sensing Autoinducers. MBio 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slilaty SN, and Little JW (1987). Lysine-156 and serine-119 are required for LexA repressor cleavage: a possible mechanism. Proc. Natl. Acad. Sci 84, 3987–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slilaty SN, Rupley JA, and Little JW (1986). Intramolecular cleavage of lexA and phage lambda repressors: dependence of kinetics on repressor concentration, pH, temperature, and solvent. Biochemistry 25, 6866–6875. [DOI] [PubMed] [Google Scholar]
- Stokar-Avihail A, Tal N, Erez Z, Lopatina A, and Sorek R (2019). Widespread Utilization of Peptide Communication in Phages Infecting Soil and Pathogenic Bacteria. Cell Host Microbe 25, 746–755.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann S, and Blokesch M (2016). Circulation of a Quorum-Sensing-Impaired Variant of Vibrio cholerae Strain C6706 Masks Important Phenotypes. MSphere 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susskind MM, and Botstein D (1975). Mechanism of action of Salmonella phage P22 antirepressor. J. Mol. Biol 98, 413–424. [DOI] [PubMed] [Google Scholar]
- Sussman R, and Jacob F (1962). Sur un système de répression thermosensible chez le bactériophage λ d’Escherichia coli. C R Acad Sci 254, 1517–1519. [PubMed] [Google Scholar]
- Wang H-C, Ho C-H, Hsu K-C, Yang J-M, and Wang AH-J (2014). DNA mimic proteins: functions, structures, and bioinformatic analysis. Biochemistry 53, 2865–2874. [DOI] [PubMed] [Google Scholar]
- Wu TH, Liao SM, McClure WR, and Susskind MM (1987). Control of gene expression in bacteriophage P22 by a small antisense RNA. II. Characterization of mutants defective in repression. Genes Dev. 1, 204–212. [DOI] [PubMed] [Google Scholar]
- Yang C, Pei X, Wu Y, Yan L, Yan Y, Song Y, Coyle NM, Martinez-Urtaza J, Quince C, Hu Q, et al. (2019). Recent mixing of Vibrio parahaemolyticus populations. ISME J. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Miller MB, Vance RE, Dziejman M, Bassler BL, and Mekalanos JJ (2002). Quorum-sensing regulators control virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. U. S. A 99, 3129–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Unprocessed gels and micrographs that were used to in this study are deposited on Zenodo (https://doi.org/10.5281/zenodo.3576389). Other experimental data that support the findings of this study are available by reasonable request from the corresponding author.