

Abstract
Owing to its rarity and severe nature, the treatment for generalized pseudohypoaldosteronism type 1 (PHA1), a genetic disorder in the epithelial sodium channel (ENaC), is exclusively experience-based. In particular, the usefulness of dietary potassium restriction in PHA1 remains unclear with the absence of theoretical background to elucidate its utility. First, we demonstrated the effect of potassium restriction in a 13-month-old patient with ENaC γ-subunit gene mutations via a retrospective chart review; reduction of daily dietary potassium intake from 40 to 20 mEq induced rapid restoration of volume depletion, as evidenced by weight gain, elevation of the serum sodium level from 133 to 141 mEq/L, decreased urinary sodium excretion, and normalized renin activity. The serum potassium level decreased from 5.6 to 4.5 mEq/L. Next, we attempted to elucidate the pathophysiological basis of the usefulness of potassium restriction, leveraged by the increased knowledge regarding the roles of with-no-lysine kinases (WNKs) in the distal nephron. When potassium is restricted, the WNK signal will turn “on” in the distal nephron via reduction in the intracellular chloride level. Consequently, the sodium reabsorption from the Na+Cl− cotransporter (NCC) in the distal convoluted tubule and possibly from pendrin in the β-intercalated cell will increase. Thus, potassium restriction causes NCC and pendrin to compensate for the non-functional ENaC in the collecting duct. In conclusion, dietary potassium restriction is one of the indispensable treatments for generalized PHA1.
Keywords: Aldosterone, ENaC, NCC, ROMK, With-no-lysine kinase
Introduction
Pseudohypoaldosteronism type 1 (PHA1) is a rare genetic disorder characterized by aldosterone unresponsiveness in the distal nephron, which results in urinary sodium wasting [1–4]. According to the corresponding genetic background, PHA1 is subdivided into the renal (autosomal dominant, OMIM #177735) and generalized (autosomal recessive, OMIM #264350) forms [1, 2, 4]; the former is caused by heterozygous mutations in the aldosterone receptor gene (NR3C2) and the latter by homozygous or compound heterozygous mutations in the epithelial sodium channel (ENaC-α, -β, -γ) genes [1–5]. Patients with generalized PHA1 are at a risk of developing life-threatening salt-wasting crisis with severe hyperkalemia from the neonatal period to adulthood; this is because sodium loss occurs not only at the distal nephron but also at other target tissues of aldosterone, such as the sweat gland [6] and lung epithelium [7].
To avoid salt-wasting crisis and to maintain sodium/potassium homeostasis, the mainstay treatment for generalized PHA1 is supplementation of salt at a high dose [1–4]. Regular use of sodium bicarbonate and/or cation-exchange resins is also recommended [1, 2, 8]. However, the usefulness of potassium restriction in diets has only been described anecdotally [1, 8] and has not been evaluated in detail. In addition, the pathophysiology as to the effect of potassium restriction in generalized PHA1 has not been elucidated.
Herein, we describe our experience in treating a patient with generalized PHA1, where the usefulness of potassium restriction was illustrated. In addition, we attempted to clarify the mechanism of potassium restriction in attenuating sodium loss.
Patient report and methods
The case of the male patient of Japanese ancestry assessed here has been described twice elsewhere [5, 9]. In brief, he was born in 1989 and collapsed at 7 days of age with marked hyponatremia (116 mEq/L), hypochloremia (84 mEq/L), hyperkalemia (8.6 mEq/L), and metabolic acidosis (HCO3− 14.7 mmol/L). Molecular investigation revealed compound heterozygous mutations in the ENaC γ-subunit gene (1627delG and 1570-1 G > A) [5]. Following recovery via intensive treatment, including peritoneal dialysis, he was treated with salt supplementation (up to 7 g/day) with oral sodium polystyrene sulfate (Kayexalate®). A high-sodium and low-potassium (HSLP) formula was used until 12 months of age. Despite the above-mentioned treatment, he experienced additional episodes of salt-wasting crisis (n = 14, the lowest sodium level 121 mEq/L, the highest potassium level 10.6 mEq/L, the lowest HCO3− level 13.2 mmol/L) during the initial 13 months.
When he was 13 months, we noticed that his potassium intake had substantially increased because of the cessation of the HSLP formula use and the increased intake of cow milk and solid food. At that time, his weight gain had been poor, showing only a 160-g increment during the precedent 4 weeks, although his body mass index was at the 53.2nd percentile. Therefore, potassium restriction was empirically introduced in 1990 (almost 30 years ago), hoping that it may ameliorate his clinical condition. It was achieved by total substitution of cow milk with the HSLP formula.
We now rechecked his clinical course by utilizing past medical records covering 9 days prior to potassium restriction and 2 weeks thereafter to illustrate its therapeutic usefulness. We assessed the changes in his body weight, electrolyte levels, fractional excretion of sodium (FENa), and hormonal levels, such as plasma renin activity (PRA) and aldosterone levels. The total daily intake of both sodium and potassium was calculated using the nursing records of consumed food/milk and supplemented salt.
Results
The changes in body weight and laboratory data around the introduction of dietary potassium restriction are shown in Fig. 1. Substitution of cow milk with the HSLP formula reduced the daily potassium intake level from roughly 40 to 20 mEq; the sodium intake level from the diet and salt supplementation was stable at approximately 170 mEq/day. For an unclear reason, the Kayexalate dose was increased from 2.5 to 7 g/day during this period.
Fig. 1.
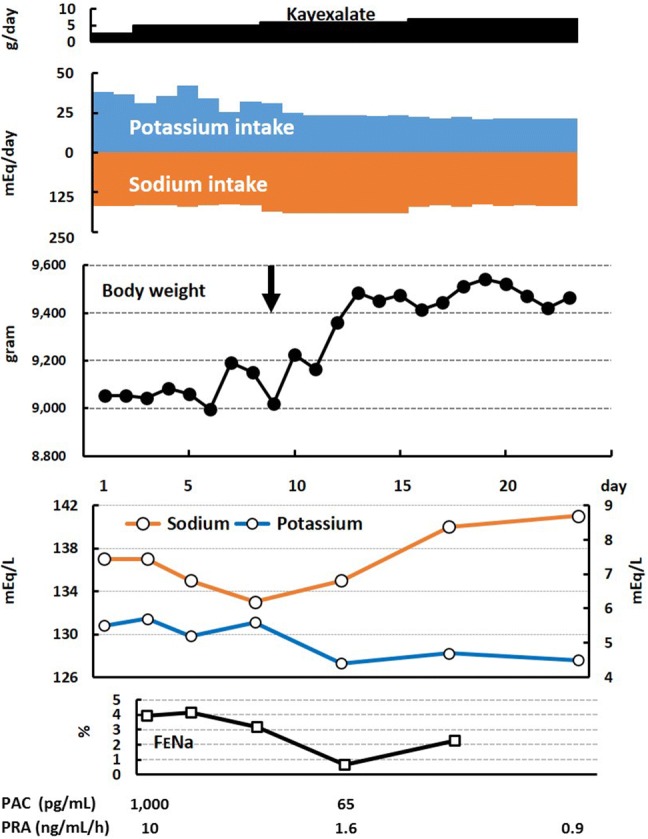
Clinical course around the introduction of dietary potassium restriction. The vertical arrow indicates the initiation of the restriction. FENa fractional excretion of sodium, PAC plasma aldosterone concentration, PRA plasma renin activity
Soon after potassium restriction, a weight increment of 260 g (5.1%) in 4 days was observed, with maintenance of the weight thereafter. Along with weight increment, an increase in the serum sodium level was observed, from 133 to 141 mEq/L. Conversely, the serum potassium level showed a reciprocal change, from 5.6 to 4.5 mEq/L. The FENa and PRA also decreased. The plasma aldosterone level was measured only twice. However, its level after potassium restriction (65 pg/mL) was below the normal range for the corresponding age: 101–789 pg/mL [10].
With the continuation of potassium restriction thereafter, he was free from salt-wasting crisis during the next 6 weeks and was discharged after a 15-month-long hospitalization since birth.
Discussion
The usefulness of dietary potassium restriction in generalized PHA1 is not self-explanatory but warrants verification. Although well-designed clinical trials are necessary, they are impractical considering the rarity and severe nature of the disease. Therefore, our retrospective evaluation before and after potassium restriction may be of significance and is the first in-depth report regarding this issue.
As shown in Fig. 1, potassium restriction yielded acutely beneficial effects, including weight gain and reciprocal changes in the serum sodium/potassium levels. It not only simply decreased the serum potassium level, but seemed to reduce urinary sodium loss, as evidenced by the decrease in the FENa and PRA. A rapid increase in body weight reflects a decrease in sodium loss, leading to restoration of volume depletion. Unfortunately, we did not measure the aldosterone levels more frequently; nevertheless, the aldosterone level, measured once during potassium restriction, substantially decreased. Dismayingly, the Kayexalate dose was increased along with potassium restriction. However, Kayexalate had been used in the patient from 1 month of age; at 4–7 months of age, he was administered with 8–10 g of Kayexalate per day. Considering that he experienced repetitive salt-wasting crises during administration of high amounts of Kayexalate, we can conclude that administration of 5–7 g of Kayexalate during potassium restriction seems to play a minimal role in reducing urinary sodium loss.
Therefore, we assume that potassium restriction was effective. In fact, at 30 years ago, we strongly believed that potassium restriction is one of the indispensable treatments for generalized PHA1. However, the responsible pathophysiology has been lacking. At present, we can elucidate the pathophysiological basis of the beneficial effect of potassium restriction, leveraged by the increased knowledge regarding the roles of with-no-lysine kinases (WNKs) for electrolyte regulation in the distal nephron [11–17]. Interestingly, investigation of pseudohypoaldosteronism type 2 (PHA2), i.e., Gordon syndrome, allowed the understanding of WNKs and relevant kinases, such as sterile20-related proline-alanine-rich kinase (SPAK) and oxidative stress response kinase 1 (OSR1). Therefore, the knowledge obtained from investigations of PHA2 helped us to understand the pathophysiology of PHA1.
Figure 2a depicts the physiological adaptation of the distal nephron to a high potassium intake [11–16]. WNK4 is regarded as a molecular switch that positively or negatively regulates the Na+Cl− cotransporter (NCC) in the distal convoluted tubule (DCT). A high potassium intake will turn the WNK signal “off” via the increased intracellular chloride level. The reduced WNK signal will inactivate the downstream SPAK/OSR1, which will consequently inhibit the NCC in the DCT. The resultant diminished sodium reabsorption via the NCC will enhance the sodium delivery to the cortical collecting duct (CCD), which will facilitate sodium reabsorption from the ENaC and potassium secretion via the flow-dependent large Ca2+-activated K+ channel (BK). Potassium secretion through the renal outer medullary K+ channel (ROMK) will also increase because of the greater lumen negative voltage generated by the ENaC. Hyperkalemia-stimulated aldosterone secretion will further enhance the activity of both the ENaC and ROMK. In addition, the reduced WNK signal also plays a role in enhancing ROMK activity. Therefore, in response to potassium intake, WNK4 will induce activation of the kaliuretic mode in the distal nephron.
Fig. 2.

Regulation of sodium/potassium handling at the distal nephron in normal conditions (a, d) and in generalized PHA1 (b, c). a Potassium load will turn the WNK signal “off” via increased intracellular chloride levels in DCT-2. The reduced WNK signal will inactivate the downstream SPAK/OSR1, which will consequently inhibit the activity and density of the NCC in the DCT-2 apical membrane. The stimulatory effect of ALD toward the NCC is overridden by the inhibitory effect of the WNK signal [13]. The resultant diminished sodium reabsorption via the NCC will enhance the sodium delivery to the CCD, which will facilitate sodium reabsorption from the ENaC and potassium secretion via BK. Hyperkalemia-stimulated ALD secretion will further enhance the activity of the ENaC via sgk1. Potassium secretion through the ROMK will also increase because of the greater lumen negative voltage generated by the increased sodium entry from the ENaC, increased sgk1 signal, and reduced WNK signal. The effect of the WNK signal on the ENaC is less understood and is thus omitted here. b In patients with PHA1 with a high potassium intake, sodium reabsorption from both the NCC and ENaC will be reduced. Because decreased sodium entry from the ENaC will result in a diminished lumen negative voltage, potassium secretion through the ROMK will be impaired at the CCD. c When potassium intake is restricted in PHA1, sodium reabsorption from the NCC will recover, because the turned-“on” WNK signal will activate the downstream SPAK/OSR1/NCC cascade. In addition, in mouse model of PHA2 with disease-causing WNK4 mutation, Cl−/HCO3− exchanger pendrin, located in β-intercalated cell in CCD, is shown to be upregulated. Because pendrin-dependent apical chloride influx can be coupled to apical sodium influx by the Na+-driven Cl−/HCO3− exchanger (NDCBE), WNK signal “on” state may enhance sodium reabsorption via β-intercalated cell [17]. In this situation, the plasma ALD level will decrease, as long as potassium restriction is sufficient and volume depletion is negligible. d Physiological adaptation to volume depletion. AnGII will turn the WNK signal “on” while also stimulating ALD secretion. Both WNK and ALD will enhance the NCC activity, while the latter will also up-regulate the ENaC. Although the ROMK will be up-regulated by ALD, the WNK signal may cancel it to prevent excessive potassium excretion. If the ENaC is inactive (as in PHA1) in this situation, the NCC and possibly the pendrin/NDCBE system may compensate for the ENaC. The possible direct action of AnGII to the NCC is omitted here. PHA1 pseudohypoaldosteronism type 1, PHA2 pseudohypoaldosteronism type 2, ENaC epithelial sodium channel, WNK with-no-lysine kinase, SPAK sterile20-related proline-alanine-rich kinase, OSR1 oxidative stress response kinase 1, DCT-2 distal convoluted tubule late segment, CCD cortical collecting duct, BK flow-dependent large Ca2+-activated K+ channel, ROMK renal outer medullary K+ channel, ALD aldosterone, sgk1 serum and glucocorticoid regulated kinase 1, NDCBE Na+-driven Cl−/HCO3− exchanger, AnGII angiotensin II
When the above-mentioned situation occurs in PHA1 (Fig. 2b), both the NCC and ENaC will fail to reabsorb sodium, whereas potassium excretion via the ROMK will be impaired. This scenario will lead to chronic volume depletion and/or repetitive salt-wasting crises in PHA1, despite supplementation of salt at high amounts. In contrast, potassium restriction in PHA1 (Fig. 2c) will turn the WNK signal “on” and activate SPAK/OSR1 and the NCC [11–13, 15]. Therefore, in this situation, the impaired sodium reabsorption from the ENaC can be compensated by the NCC. In addition, sodium reabsorption via pendrin/Na+-driven Cl−/HCO3− exchanger (NDCBE) system located in β-intercalated cell is shown to be upregulated in WNK signal “on” state [17]. The aldosterone level may decrease in this situation, as long as hyperkalemia diminishes during potassium restriction, as in the case of our patient. Therefore, the aldosterone level may be used as an indicator of adequacy of treatments, including salt supplementation and potassium restriction.
Apart from potassium restriction, the WNK “on” state is also induced by volume depletion, which is mediated by angiotensin II (Fig. 2d) [11, 12, 16]. Although speculative, the NCC activity, and possibly the pendrin/NDCBE system, in PHA1 may be up-regulated by persistent stimulation by angiotensin II, considering that PHA1 is characterized by chronic volume depletion. This assumption is in accordance with our previous finding that the urinary NCC levels were increased in our patient aged 20 years [9]. To elucidate the pathophysiology of PHA1, consideration of the compensative role exerted by the NCC and pendrin/NDCBE system is essential.
Conclusion
When dietary potassium is restricted in generalized PHA1 with the non-functional ENaC, the compensatory mechanisms will be exerted by the WNK signal “on” state. Potassium restriction is one of the indispensable treatments for PHA1.
Funding
None.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
All procedures performed in this study were in accordance with the ethical standards of national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. This study was in accordance with the ethical standards of Kanagawa Children’s Medical Center.
Informed consent
Informed consent was obtained from the patient.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
4/29/2020
In the Original publication of the article, there are two minor errors in Fig.��2 and these include one missing arrow in Fig.��2d and appears as an incorrectly drawn solid lines as dashed line in Fig.��2d.
References
- 1.Zennaro MC, Hubert EL, Fernandes-Rosa FL. Aldosterone resistance: structural and functional considerations and new perspectives. Mol Cell Endocrinol. 2012;350:206–215. doi: 10.1016/j.mce.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 2.Riepe FG. Pseudohypoaldosteronism. Endocr Dev. 2013;24:86–95. doi: 10.1159/000342508. [DOI] [PubMed] [Google Scholar]
- 3.Hanukoglu I, Hanukoglu A. Epithelial sodium channel (ENaC) family: phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene. 2016;579:95–132. doi: 10.1016/j.gene.2015.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tajima T, Morikawa S, Nakamura A. Clinical features and molecular basis of pseudohypoaldosteronism type 1. Clin Pediatr Endocrinol. 2017;26:109–117. doi: 10.1297/cpe.26.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adachi M, Tachibana K, Asakura Y, Abe S, Nakae J, Tajima T, Fujieda K. Compound heterozygous mutations in the gamma subunit gene of ENaC (1627delG and 1570-1G %3e A) in one sporadic Japanese patient with a systemic form of pseudohypoaldosteronism type 1. J Clin Endocrinol Metab. 2001;86:9–12. doi: 10.1210/jcem.86.1.7116. [DOI] [PubMed] [Google Scholar]
- 6.Hanukoglu I, Boggula VR, Vaknine H, Sharma S, Kleyman T, Hanukoglu A. Expression of epithelial sodium channel (ENaC) and CFTR in the human epidermis and epidermal appendages. Histochem Cell Biol. 2017;147:733–748. doi: 10.1007/s00418-016-1535-3. [DOI] [PubMed] [Google Scholar]
- 7.Kerem E, Bistritzer T, Hanukoglu A, Hofmann T, Zhou Z, Bennett W, MacLaughlin E, Barker P, Nash M, Quittell L, Boucher R, Knowles MR. Pulmonary epithelial sodium-channel dysfunction and excess airway liquid in pseudohypoaldosteronism. N Engl J Med. 1999;341:156–162. doi: 10.1056/NEJM199907153410304. [DOI] [PubMed] [Google Scholar]
- 8.Güran T, Değirmenci S, Bulut İK, Say A, Riepe FG, Güran Ö. Critical points in the management of pseudohypoaldosteronism type 1. J Clin Res Pediatr Endocrinol. 2011;3:98–100. doi: 10.4274/jcrpe.v3i2.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adachi M, Asakura Y, Muroya K, Tajima T, Fujieda K, Kuribayashi E, Uchida S. Increased Na reabsorption via the Na-Cl cotransporter in autosomal recessive pseudohypoaldosteronism. Clin Exp Nephrol. 2010;14:228–232. doi: 10.1007/s10157-010-0277-0. [DOI] [PubMed] [Google Scholar]
- 10.Fanelli F, Baronio F, Ortolano R, Mezzullo M, Cassio A, Pagotto U, Balsamo A. Normative basal values of hormones and proteins of gonadal and adrenal functions from birth to adulthood. Sex Dev. 2018;12:50–94. doi: 10.1159/000486840. [DOI] [PubMed] [Google Scholar]
- 11.Argaiz ER, Gamba G. The regulation of Na+Cl− cotransporter by with-no-lysine kinase 4. Curr Opin Nephrol Hypertens. 2016;25:417–423. doi: 10.1097/MNH.0000000000000247. [DOI] [PubMed] [Google Scholar]
- 12.Hadchouel J, Ellison DH, Gamba G. Regulation of renal electrolyte transport by WNK and SPAK-OSR1 kinases. Annu Rev Physiol. 2016;78:367–389. doi: 10.1146/annurev-physiol-021115-105431. [DOI] [PubMed] [Google Scholar]
- 13.van der Lubbe N, Moes AD, Rosenbaek LL, Schoep S, Meima ME, Danser AH, Fenton RA, Zietse R, Hoorn EJ. K+-induced natriuresis is preserved during Na+ depletion and accompanied by inhibition of the Na+-Cl- cotransporter. Am J Physiol Renal Physiol. 2013;305:F1177–F1188. doi: 10.1152/ajprenal.00201.2013. [DOI] [PubMed] [Google Scholar]
- 14.Welling PA. Regulation of renal potassium secretion: molecular mechanisms. Semin Nephrol. 2013;33:215–228. doi: 10.1016/j.semnephrol.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Naito S, Ohta A, Sohara E, Ohta E, Rai T, Sasaki S, Uchida S. Regulation of WNK1 kinase by extracellular potassium. Clin Exp Nephrol. 2011;15:195–202. doi: 10.1007/s10157-010-0378-9. [DOI] [PubMed] [Google Scholar]
- 16.Arroyo JP, Ronzaud C, Lagnaz D, Staub O, Gamba G. Aldosterone paradox: differential regulation of ion transport in distal nephron. Physiology (Bethesda) 2011;26:115–123. doi: 10.1152/physiol.00049.2010. [DOI] [PubMed] [Google Scholar]
- 17.López-Cayuqueo KI, Chavez-Canales M, Pillot A, Houillier P, Jayat M, Baraka-Vidot J, Trepiccione F, Baudrie V, Büsst C, Soukaseum C, Kumai Y, Jeunemaître X, Hadchouel J, Eladari D, Chambrey R. A mouse model of pseudohypoaldosteronism type II reveals a novel mechanism of renal tubular acidosis. Kidney Int. 2018;94:514–523. doi: 10.1016/j.kint.2018.05.001. [DOI] [PubMed] [Google Scholar]