

Key concepts
-
■New tools are available to aid vaccine manufacturers in meeting challenges for new vaccine development
-
□Many technologies that are already available continue to be improved, including adjuvants and novel vaccine delivery platforms
-
□New methods are yet to be fully exploited, including reverse engineering (going from pathogen gene sequences to immunogenic epitopes), screening of peptide libraries, and methods to increase antigen purity, cross-protection and thermostability
-
□
-
■
Persistent, highly variable and/or novel pathogens, and complex (eg polymicrobial) infections present challenges to vaccine designers. Targets include new prophylactic vaccines that prevent the emergence of disease and new therapeutic vaccines which augment or redirect the existing immune response to control persistent infections, malignancies, autoimmune diseases and addiction
-
■
State-of-the-art technology combined with a more comprehensive understanding of disease aetiology, pathogen biology and host immunity will assist future vaccine developments to combat elusive pathogens and protect populations with special needs
Introduction
The advances made in vaccine technology since Edward Jenner vaccinated the young James Phipps against smallpox have had a spectacular impact on human health over the last two centuries (see Chapter 1 – Vaccine evolution). Vaccines have been fundamental in the control and elimination of many debilitating and lethal diseases, and more diseases are currently targeted for eradication by vaccination. Recent major breakthroughs in immunology, molecular biology, genomics, proteomics, biochemistry and computing sciences have driven vaccine technology forward, and will continue to do so.
Many challenges remain, however, including persistent or latent infections, pathogens with complex life cycles, antigenic drift and shift in pathogens subject to selective pressures, challenging populations and emerging infections. To address these challenges researchers are exploring many avenues: novel adjuvants are being developed that enhance the immune response elicited by a vaccine while maintaining high levels of tolerability; methods of protective antigen identification are iterated with every success; vaccine storage and transport systems are improving (including optimising the cold chain and developing temperature-stable vaccines); and new and potentially more convenient methods of vaccine administration are being pursued.
High priority targets include life-threatening diseases, such as malaria, tuberculosis (TB) and human immunodeficiency virus (HIV), as well as problematic infections caused by ubiquitous agents, such as respiratory syncytial virus (RSV), cytomegalovirus (CMV) and Staphylococcus aureus. Non-traditional vaccines are also likely to become available for the management of addiction, and the prevention, treatment and cure of malignancies.
This chapter is not meant as a compendium of all new-generation vaccines, but rather as an outline of the modern principles that will likely facilitate the development of future vaccines. As shown in Figure 6.1 , there are several key elements that are likely to be the foundation for the development of future vaccines. This chapter will illustrate these elements and provide examples that show promise.
Figure 6.1.

Modern elements of vaccine development.
Strategies to improve the prevention and/or treatment of diseases through the use of vaccination are multifaceted. Methods include improved antigen identification, purification and presentation; use of new adjuvants; and more targeted vaccine administration for use against elusive pathogens and in special populations.
New adjuvants
Achievements of adjuvants
Since the first use of an adjuvant in a human vaccine over 80 years ago, adjuvant technology has improved significantly with respect to improving vaccine immunogenicity and efficacy. Over 30 currently licensed vaccines have an adjuvant component in their formulation (see Chapter 4 – Vaccine adjuvants; Figure 4.1). The advances in adjuvant design have been driven by parallel advances in vaccine technology as many modern vaccines consist of highly purified antigens – with low non-specific reactogenicity which require combination with adjuvants to enhance the immune response. Future developments in adjuvant technology are expected to provide stronger immune priming, enhance immune responses in specific populations, and lead to antigen sparing. Adjuvants to date have demonstrated an ability to increase and broaden the immune response – examples include MF59™ or AS03 adjuvants used in various influenza vaccines, and aluminium or AS04 used in human papillomavirus (HPV) vaccines. The impact of new adjuvant technology on vaccine efficacy has been demonstrated in influenza vaccines and in candidate malaria vaccines currently undergoing clinical testing, and has opened the door to antigen-specific immunotherapy for cancer.
The role of adjuvants in future vaccines
New adjuvants must aim to drive the immune response that is associated with lifelong protection. New adjuvants and adjuvant combinations will play many roles in future vaccines as illustrated in Figure 6.2 . Adjuvants will need to be individually selected for specific vaccine targets in order to achieve the desired goal (ie enhanced immunogenicity, induction of specific immune profile etc). To deliver this aim, some adjuvants will be mixed with free antigens, while others will need to be covalently linked to the antigenic moiety as part of a complex molecule.
Figure 6.2.
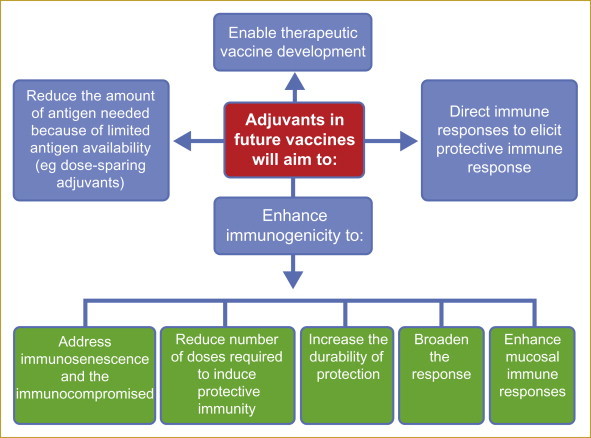
Roles of adjuvants in future vaccines.
New and future adjuvants (discussed in Chapter 4 – Vaccine adjuvants) will be employed to fulfil various roles aimed at maximising vaccine efficacy and durability, thus reducing the amount of antigen needed when in short supply in an effort to increase the impact of vaccination programmes.
Some examples of new adjuvants that have been evaluated in humans or that are in clinical trials are listed in Table 6.1 (also see Chapter 4 – Vaccine adjuvants).
Examples of new adjuvants are the nanoemulsions developed by NanoBio Corporation. Nanoemulsions are oil-in-water emulsions manufactured in various sizes ≤400 nm and stabilised by a surfactant. These technologies are amenable to topical and mucosal administration and can be used to deliver antigens or used alone to physically disrupt the outer membrane of pathogenic organisms. When administered as a vaccine, the nanoemulsion enhances vaccine antigen uptake by antigen-presenting cells, which then carry the antigen to the lymph nodes – the site of adaptive immune response initiation. Nanoemulsion vaccines administered intranasally elicit both mucosal immunity and a systemic immune response.
Table 6.1.
Adjuvants contained in vaccines currently in development
| Adjuvant name | Description | Route of delivery | Current vaccine targets | Manufacturer |
|---|---|---|---|---|
| Early-stage development (Phase I) | ||||
| NanoStat™ | Nanoemulsion | Intranasal | Influenza Hepatitis B∗ |
NanoBio Corporation |
| CpG | TLR9 agonist | Intramuscular | Malaria | Coley |
| LT | Escherichia coli heat-labile toxin | Intranasal | TB | Novartis |
| Montanide™ ISA720 | Water-in-oil emulsion | Intramuscular | Malaria Cancer |
Seppic |
| Resiquimod | TLR7/8 agonist | Intramuscular | Melanoma | 3M |
|
ISCOM™ (with antigen included) ISCOMATRIX™ (without antigen) |
Lipids, cholesterol, saponin in a cage-like structure | Intramuscular | Alzheimer's disease | CSL Behring |
| IC31™ | TLR9 agonist and targets antigen to APC | Intramuscular | Influenza | Intercell |
| Mid-stage development (Phase II) | ||||
|
ISCOM™ (with antigen included) ISCOMATRIX™ (without antigen) |
Lipids, cholesterol, saponin in a cage-like structure | Intramuscular | Influenza Melanoma HCV |
CSL Behring |
| Flagellin (conjugated with antigen) | TLR5 agonist | Intramuscular | Influenza | VaxInnate |
| IC31™ | TLR9 agonist and targets antigen to APC | Intramuscular | TB | Intercell |
| MF59™ | Squalene-based oil-in-water emulsion | Intramuscular | CMV | Novartis |
| AS01 | Liposome, lipopolysaccharide derivative (MPL) and saponin (QS21) | Intramuscular | TB HIV |
GSK Biologicals |
| Late-stage development (Phase III) | ||||
| ISS | TLR9 agonist | Intramuscular | HBV | Dynavax |
| AS01 | Liposome, lipopolysaccharide derivative (MPL) and saponin (QS21) | Intramuscular | Malaria | GSK Biologicals |
| AS03 | α-tocopherol and squalene in an oil-in-water emulsion | Intramuscular | Influenza | GSK Biologicals |
| AS04 | Lipopolysaccharide derivative (MPL) and aluminium salt | Intramuscular | HSV | GSK Biologicals |
| AS15 | Liposome, lipopolysaccharide derivative (MPL), saponin (QS21) and TLR9 agonist (CpG) | Intramuscular | Lung cancer Melanoma |
GSK Biologicals |
| Montanide™ ISA51 | Water-in-oil emulsion | Intramuscular | Cancer | Seppic |
| MF59™∗∗ | Squalene-based oil-in-water emulsion | Intramuscular | Influenza | Novartis |
| MPL | Lipopolysaccharide derivative | Subcutaneous | Non-small-cell lung cancer | Merck |
TLR, Toll-like receptor; TB, tuberculosis; APC, antigen-presenting cell; HCV, hepatitis C virus; CMV, cytomegalovirus; MPL, monophosphoryl lipid A; HIV, human immunodeficiency virus; HBV, hepatitis B virus; HSV, herpes simplex virus; FDA, Food and Drug Administration; EU, European Union. Every effort has been made to verify the information in the above table. The information included is not meant to be exhaustive but is intended to provide an overview of the subject matter.
National Institutes of Health funding received for Phase I trials – FDA approval pending as of April 2010.
MF59™ adjuvanted influenza vaccines has been approved in the EU and not in the USA
New approaches to antigen selection and stability
Modern approaches to antigen design tend to eschew classical trial and error techniques in favour of identifying the type of pathogenic structures (ie antigens) that are most likely to be important immunogens based on their structural signature or physical location within the pathogen (Table 6.2 ) (see Chapter 3 – Vaccine antigens).
Table 6.2.
Key areas of modern vaccine-related antigen research and new approaches to antigen discovery
|
Novel approaches to antigen identification and purification
The T or B cell immune responses to an antigen are targeted to precise regions of the antigen (ie epitopes – either linear or three-dimensional conformational structures; in the case of protein antigens these are specific peptide epitopes). Historically, simple, linear, synthetic peptide epitope vaccines have been poorly immunogenic because they lack a specific conformation and are easily degraded by a variety of extracellular and cell-surface proteases that serve to limit epitope presentation to T cells and/or result in destruction of the B-cell epitope. Peptide vaccines need to survive this environment in order to participate in successful major histocompatibility complex (MHC) class II presentation (see Chapter 2 – Vaccine immunology).
Subunit and individual epitope vaccines need to be optimised to ensure adequate immunogenicity. Novel strategies are being developed and exploited in order to identify antigens recognised by T and B cells, thus facilitating a more knowledge-based vaccine design. One of the most common ways to identify these antigens is to measure cellular proliferation (T or B cells) upon in vitro stimulation with antigen. High-throughput screening assays of candidate synthetic peptides that drive cellular proliferation help speed the rate of antigen discovery.
Reverse vaccinology
Reverse vaccinology combines knowledge of the pathogen's genome sequence with known protein sequences via computer analysis, to predict protein expression and post-translational modifications and identify likely vaccine candidates (see Chapter 3 – Vaccine antigens; Figure 3.5).
The development of epitope-based vaccines is one example of reverse vaccinology where computer software combines prediction algorithms to suggest sequences similar to those for pathogenic components. Epitope mapping, combined with the creation of more stable poly-epitope vaccines, may lead to the successful translation of this technology into products.
Poly-epitope vaccines and MHC restriction
MHC molecules exhibit widely varying binding specificities; a vaccine expressing a single peptide antigen would therefore only target a few MHC molecules and thus only be recognised by the T cells of individuals carrying a specific MHC phenotype. Poly-epitope technology could be used to generate a synthetic protein carrying antigenic epitopes from multiple strains or pathogens. This would overcome the MHC restriction and afford protection in individuals carrying different MHC types.
Pathogen libraries
The screening of pathogen peptide libraries is another example of new approaches to antigen discovery. Screening methods are used to identify antigens that can stimulate CD4+ or CD8 + T cells, or which bind to antibodies from humans known to have been infected with the relevant pathogen. Where peptide screening uses antibodies, an additional consideration is the synthesis of antigens that contain the tertiary (folding/three-dimensional) structure of the native immunogen, since vaccine efficacy can be impacted by infidelities in the structure of the final product. Incorrect protein folding may result in a less immunogenic antigen or an antigen that induces an immune response that differs from that of the native immunogen. The mimicking of the three-dimensional structure of the native immunogen is important during the synthesis of antigens that are being used to target B-cell responses. Conversely, the requirement for folding is reduced for T cells since T cells bind only processed peptides, from degraded proteins. Likewise, DNA expression libraries using the pathogen genomic DNA have been screened using animal model systems to identify genes encoding proteins that afford protection against infection or disease caused by the pathogen.
One example is Genocea's vaccine development programmes that are built around a broad platform for the rapid discovery of T-cell antigens. The process is explained in Figure 6.3 . T-cell antigens, specifically antigens that stimulate CD4+ and CD8+ T cells, are critical to generating disease-specific cellular immune responses and long-term T-cell memory.
Figure 6.3.

Genocea's T-cell antigen discovery technology.
Genocea's vaccine development programmes are built around a transformational platform for the rapid discovery of T-cell antigens. T-cell antigens, specifically antigens that stimulate CD4+ and CD8+ T cells, are critical to generating disease-specific cellular immune responses and long-term T-cell memory (Genocea website, 2011).
Antigen stability
Stability of the final product is another important consideration. Adverse environmental conditions can result in degradation of the vaccine, rendering it non-immunogenic. In order to maintain product integrity many vaccines (particularly live vaccines) must be stored at cold temperatures (≤4°C). The maintenance of the vaccine at this temperature from production site to distribution site, and medical office or clinic, is referred to as the ‘cold chain’. Maintaining the cold chain is much less of a challenge in resource-rich countries, but can be a major barrier to vaccine implementation in resource-limited areas. Ongoing research designed to increase our understanding of vaccine degradation may address the problems associated with cold chain management and lead to the development of thermostable vaccines.
Modifying vaccine formulations to increase tolerance to temperature fluctuations is likely to increase the shelf-life of the product and reduce transport and wastage issues.
New approaches to antigen delivery
Issues affecting antigen presentation
The level of antigen presentation which occurs with some current vaccines may sometimes be insufficient to drive long-lasting immune responses of high quality (see Chapter 3 – Vaccine antigens). This may be due to inadequate exposure of the antigen to immature antigen-presenting cells (APCs) rapid or subimmunogenic degradation or sequestration of antigens, or lack of immunogenicity due to the physical presentation of the antigen. The discovery and refinement of new and varied options for antigen presentation is expected to allow the design of vaccines to produce specific immune profiles. Some of these technologies have been shown to facilitate oral delivery to target mucosal immune responses and also trigger both innate and adaptive immune systems, including T- and B-cell effector and memory responses.
Viral vector vaccines
Candidate viral vector vaccines utilise a non-pathogenic virus to carry and subsequently induce expression of genes that produce immunogenic foreign proteins at high levels in the host. These are taken up by immature APCs, and have been shown to lead to a robust, long-lasting immune response to the target antigen (Figure 6.4 ). Viral vector vaccines, eg recombinant poxvirus vaccines, can be administered mucosally to stimulate mucosal immune responses. The attenuated modified vaccinia virus Ankara (rMVA) vectors are showing promise as mucosal delivery vectors.
Figure 6.4.

Viral vectors for vaccines.
Viral vector vaccines exploit the natural ability of viruses to infect or otherwise enter (in the case of disabled viral vectors) host cells, and then deliver pathogen-specific antigens. Antigen-encoding genes are isolated from the pathogen and inserted into the viral vector genome. The viral vector can then be used as a factory for production of large quantities of pathogen antigen in vivo, following introduction of the vector into the vaccine recipient, with the pathogen antigen then expressed on the surface of the infected/transduced host cells or exported out of the producer cell.
MHC, major histocompatibility complex.
Pre-existing immunity to the viral vaccine vector is an impediment to successful use of this approach. As ways to avoid anti-vector immunity, viruses can be attenuated or inactivated, by deleting or replacing pathogenic genes. Figure 6.4 demonstrates how viral vaccine vectors are made. DNA expressing an immunogenic transgene (the vaccine antigen) is inserted into the viral vector genome for expression following administration into the recipient; expression of the vaccine antigen can be boosted by using a variety of DNA promoters. If the viral vector is no longer able to grow and replicate, the virus is grown using a cell line (a so-called complementing cell line) that has been engineered to produce the missing viral product.
Often, viral genes are removed in an effort to reduce or eliminate the pathogenicity of the vector and in some cases viral genes are removed to make the vector itself less immunogenic; an anti-vector immune response would greatly reduce the ability of the vector to induce an antigen-specific response. Examples of viral vector candidate vaccines in clinical development are listed in Table 6.3 .
Table 6.3.
Examples of viral vector candidate vaccines in various stages of clinical development
| Vector | Pathogen target | Manufacturer |
|---|---|---|
| Early-stage development (Phase I) | ||
| Adenovirus | HIV Ebola-Marburg HCV |
Crucell |
| Adenovirus | Pandemic influenza | Paxvax |
| Adenovirus | Pandemic influenza | Vaxin |
| Attenuated influenza virus | Influenza HIV TB |
Polymun Scientific |
| Human or bovine PIV3 | PIV3/RSV | Medimmune AstraZeneca |
| Recombinant modified vaccinia Ankara | Measles | Bavarian Nordic |
| Replication incompetent adenovector | HSV | GenVec |
| Replication incompetent adenovector | HCV | Okairos |
| Vaccinia virus | HCV | Transgene |
| Mid-stage development (Phase II) | ||
| Adenovirus | Malaria TB |
Crucell |
| Adenovirus | HIV | Paxvax |
| Adeno-associated virus | HIV | Targeted Genetics |
| rMVA | Cancer | Oxford Biomedica |
| rMVA | TB | Emergent Biosolutions |
| rMVA | Smallpox Cancer HIV |
Bavarian Nordic |
| Replication incompetent adenovector | HIV Malaria |
GenVec |
| Replication incompetent adenovector | Malaria | Okairos |
| rMVA | Cancer HPV |
Transgene/Novartis Transgene/Roche |
| Vaccinia and fowlpox | Cancer | Bavarian Nordic |
| Yellow fever virus | Dengue West Nile |
Sanofi Pasteur |
| Late-stage development (Phase III) | ||
| rMVA | Cancer | Oxford Biomedica |
| Canarypox virus | HIV | Sanofi Pasteur |
HIV, human immunodeficiency virus; HCV, hepatitis C virus; TB, tuberculosis; PIV3, human parainfluenza virus type 3; RSV, respiratory syncytial virus; HSV, herpes simplex virus; rMVA, modified vaccinia virus Ankara; HPV, human papillomavirus. Every effort has been made to verify the information in this table. The information included is not meant to be exhaustive but is intended to provide an overview of the subject matter.
Bacterial vector vaccines
Non-pathogenic bacterial vectors have many features that make them an attractive vaccine platform. Bacterial vectors can be engineered for maximum safety (eg deletion of two or more genes from the same metabolic pathway), and to express large numbers of foreign antigens (Figure 6.5 ).
Figure 6.5.

Bacterial vectors for vaccines.
Antigen-encoding genes are isolated from the pathogen and inserted into endogenous bacterial plasmid DNA. The bacterial vector can then be used as a factory for production of large quantities of pathogen antigen in vivo, following administration of the bacterial vector to the vaccine recipient, with the pathogen antigen then displayed on the surface of the bacteria or secreted.
Two key issues affecting bacterial vaccine vectors are: a) to decide whether the optimal platform should be a bacterial vaccine in its own right or a bacterial vector system to deliver exogenous antigens; and b) to determine whether re-administration of the vector, either with the same or different target antigens, will fail because of the immune response to the bacterial vector vaccine at the time of its initial administration.
Initial assessments of the feasibility of using attenuated bacterial vectors for the delivery of foreign antigens have focused on Salmonella species. Bacterial vaccine vectors for humans, however, have been disappointing so far. It may be necessary to develop unique bacterial vaccine vectors for delivering exogenous antigens, in which case the vectors can be modified to allow for re-use. For example, if immunity against the vector, which is a major impediment to vaccine re-use, is determined by antibodies against the surface structures of the bacterium (such as lipopolysaccharide [LPS]), the dedicated vaccine vector could be developed to lack expression of LPS or to express truncated/different forms of LPS to the target, thereby avoiding priming of the immune response and allowing for re-use of the vector and/or vaccine. Some potential options for live, attenuated bacterial vectors are shown in Table 6.4 .
Table 6.4.
Bacterial vector vaccines in clinical development
| Vector | Pathogen target | Manufacturer/developer |
|---|---|---|
| Early-stage development (Phase I) | ||
| Salmonella typhi | HIV Helicobacter pylori Streptococcus pneumoniae |
Vaccine platform being developed under Gates Foundation support at Arizona State University |
| Vibrio cholerae | ETEC/cholera | Celldex |
| Listeria monocytogenes | HPV | Advaxis |
| Mid-stage development (Phase II) | ||
| Salmonella typhi | Typhoid | Celldex |
| Salmonella typhi | HBV Typhoid |
Emergent Biosolutions |
| Vibrio cholerae | Cholera | Celldex |
HIV, human immunodeficiency virus; ETEC, enterotoxigenic Escherichia coli; HPV, human papillomavirus; HBV, hepatitis B virus. Every effort has been made to verify the information in this table. The information included is not meant to be exhaustive but is intended to provide an overview of the subject matter.
DNA vaccines
DNA vaccines are the result of the discovery in the early 1990s that the gene, rather than the encoded protein, if delivered in an ‘expressible’ form, could induce an immune response (see Chapter 1 – Vaccine evolution). The principle behind DNA vaccines is that the antigenic molecule is produced within the host from the DNA or RNA that is injected, in contrast to more traditional vaccination where the antigen is supplied in the vaccine formulation. The gene(s) for target antigen(s) is/are usually encoded in a circular plasmid expression vector under the control of promoter sequences that direct gene expression in mammalian cells, which is achieved after injection into mammals.
The DNA vaccine process can circumvent some of the major issues resulting from recombinant protein administration. The construction and production of the plasmids carrying the gene of interest together with the promoter sequences is relatively simple; antigens expressed from plasmids retain their native conformation, the gene can be readily modified to produce tailored antigens, and bacterial plasmid DNA is intrinsically immunogenic (subsequently shown to result from the pathogen-associated molecular patterns [PAMPs] it carries). Additional desirable features include the ability to engineer and deliver genetic adjuvants in tandem or parallel with the antigen, the potential to deliver multiple antigen genes in one construct or within other constructs that encode adjuvanting protein(s), and the ability to induce both cellular and humoral immune responses. Despite promising data in pre-clinical testing, DNA vaccine candidates have shown only limited success in clinical settings so far. One of the current drawbacks of DNA vaccines is the inefficiency of conventional delivery methods for the plasmid DNA; however, emerging proprietary particle-mediated delivery technology or electroporation technology seeks to improve this situation. With the electroporation method, brief electrical pulses are applied at the site of immunisation which causes a transient disruption of cell membranes. This results in an enhancement in uptake of the DNA vaccine between 10–100-fold. Examples of DNA candidate vaccines in clinical development are presented in Table 6.5 .
Table 6.5.
DNA vaccines in clinical development
| Target |
Manufacturer |
|---|---|
| Early-stage development (Phase I) | |
| HIV Cancer |
Ichor |
| HPV Cancer |
Inovio |
| Influenza HBV |
PowderMed/Pfizer Vaccines |
| Melanoma | Memorial Sloan-Kettering Cancer Center |
| Cancer Pandemic influenza SARS Ebola West Nile Anthrax |
Vical |
| Mid-stage development (Phase II) | |
| HIV | FIT Biotech |
| HBV HIV |
Genexine |
| Acute myelocytic leukaemia | Immunomic Therapeutics |
| Cancer HCV |
Inovio |
| CMV HIV |
Vical |
| Late-stage development (Phase III) | |
| Melanoma | Vical |
HIV, human immunodeficiency virus; HPV, human papillomavirus; HBV, hepatitis B virus; SARS, severe acute respiratory syndrome; HCV, hepatitis C virus; CMV, cytomegalovirus. Every effort has been made to verify the information in this table. The information included is not meant to be exhaustive but is intended to provide an overview of the subject matter.
Dendritic cell vaccines
Dendritic cell (DC) vaccines typically use monocytes harvested from the blood (in most cases from the individual who will receive the vaccine) to produce immature DCs in vitro. The monocytes are antigen-loaded and treated to induce their maturation into APCs and infused back into the patient. The first Food and Drug Administration (FDA)-approved DC vaccine, designed for the treatment of prostate cancer, was licensed in 2010 (Sipuleucel-T); examples of other targets for DC vaccine therapy are presented in Table 6.6 . DC vaccines offer an individualised approach to therapeutic vaccine development, but represent a specialised method of vaccination that is currently limited to aggressive cancers, and the treatment of serious, intractable infections.
DC vaccines hold great promise for the treatment of cancer, HIV and other chronic infections. Utilising the patient's own DCs, this is truly an individualised biomedical intervention.
Table 6.6.
DC vaccines and candidate vaccines licensed and in clinical development
| Target | Stage of development | Manufacturer |
|---|---|---|
| Melanoma | Phase I (therapeutic) | Immutep |
| HIV | Phase I (therapeutic) | Baylor Research Institute |
| Melanoma | Phase II (therapeutic) | Immutep |
| Prostate cancer | FDA licensed 2010 | Dendreon |
HIV, human immunodeficiency virus; FDA, Food and Drug Administration. Every effort has been made to verify the information in this table. The information included is not meant to be exhaustive but is intended to provide an overview of the subject matter.
A comparison between the strengths and weaknesses of selected new vaccine platforms is presented in Table 6.7 .
Table 6.7.
Advantages and disadvantages of viral, bacterial, DNA and DC-based vaccines
| Advantages | Disadvantages |
|---|---|
| Viral vectors | |
| Use pathways that induce robust and durable cellular and humoral immunity May be applied to the mucosal surface (facilitating oral delivery) Easily engineered |
Potential for lack of attenuation of the vector Potential for over-attenuation with associated poor immunogenicity Risk of recombination resulting in restored virulence Efficacy decreased by pre-existing vector immunity Vector-specific responses may limit subsequent doses Potential for some vectors to establish persistent/latent infection |
| Bacterial vectors | |
| Directed delivery of target antigen to specific cells including macrophages Large antigen-carrying capacity Safety maximised by removing several genes Potential for mucosal immunity Oral delivery possible |
Possible genomic instability at the site of insertion giving low antigen expression levels Expression of bacterial antigens may further reduce vaccine-specific immunogenicity Efficacy decreased by existing vector immunity Difficult to optimise engineering without conducting a number of clinical trials |
| DNA vaccines | |
| Defined composition Non-replicating platform capable of inducing T-cell immunity Potential application in development of therapeutic vaccines Construct may code for multiple epitopes and also inducers of innate immune responses |
Poor immunogenicity in humans Concerns/issues regarding potential for construct to integrate with host genome |
| DC vaccines | |
| Individualised approach to therapeutic vaccine development Can induce potent T-cell responses Clinical responses can be achieved |
Expensive (these vaccines typically use autologous DCs) with sophisticated logistics Mature DCs rapidly lose viability and function after injection Difficult to achieve clinically sufficient levels of circulating antigen-specific T cells |
DC, dendritic cell.
New approaches to vaccine administration
Developing administration techniques that place the vaccine directly at the site(s) where pathogens are most likely to initiate an infection (eg mucosal or respiratory sites) is likely to improve vaccine efficacy and safety.
Traditional methods of vaccine administration can potentially pose a number of limitations with respect to reactogenicity, immunogenicity, convenience, efficacy, safety and cost-effectiveness. Most vaccines are administered by intramuscular (IM) injection, while selected vaccines are delivered intranasally (eg via droplet for live attenuated influenza vaccine and via aerosol for measles vaccine); by the oral route (eg polio and rotavirus vaccines); or subcutaneously (particularly live viral vaccines such as the measles-mumps-rubella [MMR] vaccine and varicella zoster-containing vaccines). Ongoing research on alternative experimental administration strategies includes ballistic delivery to skin (the gene gun), the transdermal patch and other intradermal methods, plus sublingual, aerosol, rectal and vaginal mucosal vaccines. The main advantages of alternative delivery strategies are the potential to induce immune responses at the common portals of pathogen entry (eg oral polio vaccine replicating in the gut), potential convenience (eg ease of use of the transdermal patch), potential combination of vaccines to reduce or simplify the vaccination schedule, and reduction or elimination of administration via standard hypodermic needle injection. Despite the intuitive value of these approaches, few vaccines today are administered via non-IM routes. This is for several reasons including feasibility, lack of proven efficacy and limited safety data. Some problems have been observed with new routes of delivery, for example, after the 2000 launch of an inactivated intranasal influenza vaccine (a virosome formulation adjuvanted by heat labile enterotoxoid of Escherichia coli), post-licensure data indicated a significantly increased risk of Bell's palsy in vaccinees and forced its withdrawal from the market. This experience led to a higher level of caution in the development of intranasal vaccines.
Transdermal microneedle patch vaccine administration utilises an array of microneedles (Figure 6.6) to deliver the vaccine to the epidermis, which is rich in innate and adaptive immune response elements.
Aerosol delivery: ‘Mass immunization of almost all susceptible children in a short period of time, has the potential of rapidly eliminating measles as a public health problem. Immunization by inhalation of aerosolised measles vaccine provides a procedure that could make such a mass programme possible, especially in parts of the world where measles continues to be a serious problem…’ (Sabin et al., 1983).
Administering the measles vaccine as an aerosol, either as nebulised vaccine or as a dry powder, provides a promising alternative to subcutaneous administration, particularly in countries with concerns over inadequately safe injection practices. Numerous clinical trials with aerosolised measles vaccine have been performed in populations of various ages and appear to be equally or more immunogenic than subcutaneous vaccination in adults and children over 9 months old (data from younger children are inconclusive, possibly because of administration difficulties). Measles vaccine is not currently licensed for respiratory administration, though this route is being comprehensively studied by the World Health Organization's (WHO) Measles Aerosol Project and a Phase II/III study is underway in India to confirm that the efficacy of inhaled measles vaccine is equivalent to that of existing routes of administration. In parallel, various delivery devices are in development.
New vaccines for complex and challenging targets
In the future, new vaccines will target not only important established infectious diseases (eg malaria, TB, HIV, Lassa fever, severe acute respiratory syndrome [SARS]), but also emerging or yet to be discovered infectious diseases. New vaccines may also target diseases that result in illnesses manifesting as autoimmune disease (eg diabetes mellitus, multiple sclerosis) or chronic inflammation. New vaccines are likely to address the problem of immunosenescence in the elderly; and therapeutic vaccines may offer new treatments for the control of persistent infectious diseases, cancer and illnesses such as Alzheimer's disease.
Figure 6.6.

Microneedles.
An alternative to IM injection for vaccine administration is the use of a transdermal patch containing an array of microneedles, typically <1 mm long; much shorter and less intrusive than the needles typically used for IM injections (20–30 mm in length) (A). Microneedles pierce the epidermis of the skin, a site rich in innate and adaptive immune response elements, and deliver the vaccine antigen painlessly. Adhesive, needle-free transdermal patches are also in development (B).
IM, intramuscular.
Vaccines addressing persistent infection and malignancy
Persistent infections include both chronic infections, characterised by ongoing replication of the pathogen (eg chronic hepatitis B virus [HBV] and hepatitis C virus [HCV] infections, malaria, Helicobacter pylori infections); and latent infections, where the pathogen, after the first infection, remains dormant in the host until triggered to reactivate (eg recurrent herpes simplex virus [HSV] infection, herpes zoster, reactivation TB). Since the natural immune responses in persistently infected hosts fail to clear the infection, mimicking the immune response to natural infection with immunisation may not be sufficient.
Today, the only example of a licensed vaccine against a latent infection is the zoster vaccine; the vaccine formulation is the high potency (about 15-fold) version of the live, attenuated varicella zoster virus (VZV) vaccine. This vaccine has been used to boost the anti-VZV cell-mediated immune response in older subjects and has been shown to reduce the overall incidence of zoster by 50% in subjects aged 60 years or older (Oxman et al., 2005).
The issue of whether a vaccine protects against infection or disease is critical with regard to pathogens capable of establishing persistent infection. While a vaccine that protects against disease may afford some benefit, if the vaccine fails to prevent initial infection, the pathogen may establish a persistent infection with long-term disease consequences, such as recurrent infection, organ damage or malignancy.
Future vaccines may control persistent infections either by preventing the initial infection or disease (ie prophylactic vaccines) or by augmenting or redirecting immune responses in the persistently infected host in order to eliminate or control the chronic infection (ie therapeutic vaccines).
Therapeutic vaccines are designed to stimulate an immune response that can control or cure persistent infections, malignancies, autoimmune diseases, degenerative diseases or addiction. This approach may enhance existing responses, engender new responses, or alter the existing balance of immune responses. It is important to make the distinction between this immunotherapeutic application of vaccine technology and prophylactic vaccines that prevent infection/disease from occurring, although similar immune mechanisms are involved – see Prophylactic and therapeutic vaccines for cancer.
An increased understanding of human immunology and of host–pathogen interactions should enable the identification of the type(s) of immunity required to effectively prevent or control persistent infections (see Chapter 2 – Vaccine immunology). Some examples of persistent infections are shown in Table 6.8 .
Table 6.8.
Pathogens capable of establishing persistent infections
| Pathogen | Type of persistent infection | Disease |
|---|---|---|
| Hepatitis viruses B, C, D and E (in immunocompromised patients) | Chronic | Chronic hepatitis, cirrhosis, malignancy |
| HIV | Chronic | HIV/AIDS |
| Papovaviruses | Chronic | Progressive multifocal leukoencephalopathy |
| Prions | Chronic | Spongiform encephalopathies |
| Helicobacter pylori | Chronic | Chronic gastritis, gastric and duodenal ulcers, and gastric and duodenal cancer |
| HPV | Chronic | Cervical cancer, other genital and oral cancers |
| HSV | Latent | Recurrent HSV infections, HSV encephalitis |
| VZV | Latent | Zoster |
| EBV | Latent | Reactivation diseases including glandular fever and nasopharyngeal carcinoma |
| Mycobacterium tuberculosis | Latent | Reactivation TB |
| CMV | Latent | Severe multisystem reactivation disease in immunocompromised patients |
HIV, human immunodeficiency virus; AIDS, acquired immunodeficiency syndrome; HPV, human papillomavirus; HSV, herpes simplex virus; VZV, varicella zoster virus; EBV, Epstein–Barr virus; TB, tuberculosis; CMV, cytomegalovirus. Every effort has been made to verify the information in the above table. The information included is not meant to be exhaustive but is intended to provide an overview of the subject matter.
Tuberculosis
Mycobacterium tuberculosis can persist in a latent state within the human host for years without causing disease (latent TB). Protection against miliary (disseminated) TB in children is provided by the bacille Calmette–Guérin (BCG) vaccine, developed through culture attenuation of Mycobacterium bovis early in the 20th century, which is routinely given in many countries. The vaccine, however, provides only modest and often temporary protection against pulmonary TB, and provides lower efficacy in resource-limited regions closer to the equator. In addition, vaccination with live, attenuated Mycobacterium bovis is a particular concern in HIV-positive individuals, especially those with advanced immune suppression; this population would particularly benefit from TB vaccination as TB is a leading cause of death worldwide for people with HIV/acquired immunodeficiency syndrome (AIDS). However, a recent Phase III trial demonstrated that protection against TB can be provided to individuals with HIV by using an inactivated whole-cell mycobacterial vaccine (von Reyn et al., 2010). The current state of TB vaccine development has been summarised in reviews by Walker et al. (2010) and Lambert et al. (2009) and examples of vaccines in development are shown in Table 6.9 .
Table 6.9.
Examples of Mycobacterium tuberculosis vaccines in development
| Approach | Vaccine name | Comment | Manufacturer |
|---|---|---|---|
| Early-stage development (Phase I) | |||
| TLR9 agonist, fusion protein H4 | SSI HyVac4 (AERAS-404) | Adjuvanted with IC31™ | Statens Serum Institut, Sanofi Pasteur, Intercell and Aeras |
| Mid-stage development (Phase II) | |||
| Fusion protein | GSK M72 | Adjuvanted with AS01 | GSK Biologicals |
| Adenovirus vector 35 | Crucell Ad35 (AERAS-402) | Viral vector | Crucell |
| Modified vaccinia Ankara virus | MVA85A (AERAS-485) | Viral vector | The Oxford-Emergent TB Consortium Ltd |
TLR, Toll-like receptor; SSI, Statens Serum Institut; AERAS, The Aeras Global TB Vaccine Foundation; GSK, GlaxoSmithKline; AS, Adjuvant Systems; MVA, modified vaccinia Ankara. Every effort has been made to verify the information in this table. The information included is not meant to be exhaustive but is intended to provide an overview of the subject matter.
Cytomegalovirus
Cytomegalovirus, a herpes virus, establishes latent infection in cells in the bone marrow and peripheral blood. Primary infection during pregnancy is associated with congenital infection that frequently causes a well-characterised spectrum of abnormalities and disabilities, which may be severe or fatal. Reactivation in pregnancy is common, but is unlikely to cause severe congenital infection, although some manifestations, especially hearing loss, remain common. Reactivation of CMV is of special concern in immunocompromised individuals, where severe and fatal pulmonary, hepatic and central nervous system infections are common. Gastrointestinal disease and retinitis are common in association with HIV. A successful CMV vaccine has proved elusive for more than 30 years. Based upon the observation that antibodies to the CMV envelope glycoprotein B (gB) could neutralise the virus, and with the advent of genetically engineered viral proteins, new research began in the late 1980s on a CMV gB subunit vaccine. This included the use of a novel adjuvant, MF59™ (Pass et al., 1999). A recent Phase II clinical trial in CMV-seronegative women ≤1 year post-partum has shown the potential of gB/MF59 in decreasing incident cases of maternal and congenital CMV infection (Pass et al., 2009). This is the first evidence that a CMV vaccine can protect against infection.
An alternative approach to the development of a CMV vaccine has been to utilise DNA vaccination to induce host responses to CMV gB and phosphoprotein 65 (pp65 is another viral target). Recent studies have shown that injection of combinations of plasmids, formulated with an adjuvant, can induce vaccine-specific immune responses, and can prime for effective memory responses.
Herpes simplex virus
The hallmark of herpes simplex virus types 1 and 2 (HSV-1 and HSV-2) is their ability to establish and maintain latent infection in sensory ganglion neurons. Periodic reactivation of the latent infection results in recurrent infections. Both HSV-1 and HSV-2 can cause myriad diseases but the greatest public health problem is genital herpes. Genital HSV-2 infection increases the risk of HIV acquisition and transmission, and control of genital herpes has been predicted to significantly impact the HIV epidemic. Given the complex natural history of HSV infections, vaccines could have a variety of possible risks and benefits (Table 6.10 ).
Table 6.10.
Possible effects and consequences of genital HSV vaccines
| Effect | Positive consequences | Negative consequences |
|---|---|---|
| Prevent mucosal infection | No clinical disease No latency No recurrences |
None |
| Prevent development of clinical disease but not mucosal infection | No symptomatic disease | Latency established Recurrent infections can develop with possible transmission to susceptible partners |
| Prevent latent infection | No latency and no recurrences | Without preventing mucosal infection there could be an initial episode of symptomatic genital herpes |
| Reduce latent infection | Possibly fewer recurrences or less severe outbreaks, and less risk of transmission | Still a risk of initial and recurrent infections and hence transmission |
HSV, herpes simplex virus.
An effective HSV vaccine has been sought for more than 80 years. Recently, an HSV-2 glycoprotein D (gD2) candidate vaccine containing the AS04 adjuvant (see Chapter 4 – Vaccine adjuvants), was tested in three large, double-blind, Phase III controlled trials. The first two studies recruited volunteers with a partner with genital herpes disease and found the candidate vaccine was 73% effective against genital herpes disease in women seronegative for both HSV-1 and HSV-2 (Stanberry et al., 2002). Trends towards protection against infection were also observed, but were not statistically significant. The candidate vaccine was not effective in HSV-1 seropositive women; or in men, regardless of their HSV seropositivity status. These were the first studies to report a significant difference in vaccine efficacy between men and women. This finding could have important implications for other vaccines targeting sexually transmitted diseases. The basis for this difference could relate to differences in how men and women respond to novel adjuvants or may reflect differences in the acquisition and natural history of genital herpes in men and women. A third Phase III efficacy trial of the gD2 candidate vaccine in HSV-1 and HSV-2 negative women who thought themselves possibly at risk of acquiring genital herpes (a different risk population than in the original two trials) has been completed and is being analysed. An initial assessment of the results of the third trial showed that the vaccine had an acceptable safety profile but the primary trial endpoint, prevention of genital herpes disease, was not met (NIAID, 2010). Although the development of the vaccine has been stopped, further analyses and comparison of the trials may guide researchers as they continue seeking vaccines to control HSV infections.
Vaccines for pathogens with a complex life cycle
As discussed in Chapter 2 – Vaccine immunology, some pathogens have complex life cycles. One specific example is parasites, sometimes using more than a single host, where each development phase is marked by differential expression of major proteins, meaning that possible antigen targets are host- and development-phase specific. Taenid worms aside, vaccines against parasites have been extremely difficult to develop and only a limited number have performed well in later-stage clinical trials.
Plasmodium falciparum
The protozoan parasite Plasmodium falciparum, the most common cause of malaria, has a complex life cycle, as shown in Figure 6.7 . The Plasmodium parasite has a genome encoding more than 5000 proteins, and presents different allelic and immunogenic structures at each stage of the life cycle. Many of the key antigens are subject to antigenic variation. The complexity of the Plasmodia has made the development of an effective malaria vaccine extremely challenging. Over the past 30 years there have been more than 90 candidate vaccines that have not reached advanced stages of development.
Figure 6.7.

The life cycle of Plasmodium falciparum with stages identified for vaccine development.
The parasite responsible for malaria is delivered as sporozoites to the host during a blood feed by a carrier Anopheles spp mosquito. Sporozoites travel to the liver, and develop into merozoites. Infected hepatocytes rupture, releasing merozoites into the bloodstream. The infection becomes symptomatic as increasing numbers of red blood cells are invaded by merozoites. The merozoites can also develop into male and female gametocytes, lysing the blood cells upon maturation. A blood feed by another mosquito allows uptake of the gametocytes. Sexual reproduction within the mosquito generates oocysts that eventually rupture and release sporozoites that subsequently migrate to the mosquito's salivary gland to restart the life cycle when the mosquito feeds. Candidate vaccines have been designed to target the pre-erythrocytic, blood and transmission stages of the life cycle.
A number of new malaria candidate vaccines that utilise adjuvants or viral vectors are presently in clinical trials (see Appendices, Supplementary Table 7). One of the furthest advanced of these new candidate vaccines is RTS,S/AS01. The vaccine targets the pre-erythrocytic stage of the parasite (Figure 6.7). To be protective, a vaccine targeted at this phase needs to induce humoral immunity, to prevent parasites from invading the liver, and cell-mediated immunity to destroy hepatocytes that become infected in the face of the humoral immune response. The RTS,S antigen, produced in Saccharomyces cerevisiae, contains sequences of the P. falciparum circumsporozoite protein, linked to the hepatitis B surface antigen (HBsAg). This chimeric protein spontaneously assembles into mixed polymeric particulate structures. In Phase II studies, the RTS,S/AS01 candidate vaccine induced a strong neutralising antibody response and cell-mediated immunity, and afforded protection against malaria (Bejon et al., 2008, Abdulla et al., 2008). RTS,S/AS01 has been selected to proceed to Phase III clinical testing due to its higher efficacy compared with alternative formulations. If successful, the RTS,S/AS01 candidate vaccine could be the first licensed human vaccine against a parasite. Other malaria candidate vaccines in development are shown in Appendices, Supplementary Table 7.
Vaccines against pathogens that exhibit antigenic variability
Pathogens may mutate or recombine to change their antigenic profile. Antigenic drift refers to a gradual process whereby point mutations in genes encoding antigenic proteins change the antigen sufficiently so that over time previously effective antibodies and vaccines no longer effectively control the pathogen and hence new vaccines need to be created. Antigenic shift is a more dramatic event where there is a recombination of genes between different pathogen strains that gives rise to a new strain with a unique antigenic profile.
In theory, pathogens are susceptible to selective pressure and an immunological environment that provides strong selective pressures should provide the ‘bottleneck’ that drives selection. This occurs with influenza viruses, where the high mutation frequency allows for the selection of mutants that are not neutralised. The risk of vaccine-mediated immune selection of pathogens, though certainly present, is difficult to demonstrate. Moreover, peptide vaccines only use the antigenic epitope so the risk of pathogen evolution is theoretically increased. However, this phenomenon has not been regularly observed in experimental studies and may reflect the complex nature of most vaccine antigens and the presence of immune responses against multiple antigens and multiple epitopes within antigens.
Serotype replacement, where the distribution of specific microbial serotypes within communities changes after the introduction of vaccines, has occurred for some bacterial pathogens and may be a consequence of the use of capsular vaccines that address only a limited number of serotypes.
Similarly, since their introduction in the 1940s, the use of antibiotics has exerted a selective pressure on bacterial strains leading to selection for common resistance alleles (eg the extended-spectrum beta-lactamase [ESBL] resistance of enteric bacteria and beta-lactamase resistance in gonococci). To date, there has been no requirement to remodel a vaccine because of vaccine-mediated immune escape; however, new vaccines against the pneumococcus have been licensed, including additional capsular types, to expand the geographical coverage of most frequent types and, in part, to counter the observed phenomenon of serotype replacement. Annual seasonal influenza infections are subject to natural antigenic drift which requires the reformulation of the vaccine when drifts occur, but there is no evidence that the deployment of the vaccine accelerates this drift.
Antigenic shift, while not the result of selective pressure, gives rise to viral strains containing a mixture of the surface antigens from the parent strains. Pathogens that can undergo antigenic shift, including influenza viruses (Figure 6.8 ), present major challenges for vaccine developers.
Figure 6.8.

Antigenic shift resulting in a new influenza virus.
When two (or more) influenza viruses infect the same host cell, genetic segments from both strains can become packaged into the same progeny virion. This results in a new virus strain that displays a distinct and varying array of surface antigens, relative to the parent strains. The shifted strains are all possible candidates for a pandemic infection due to their new features to which most of the population is naïve.
HA, haemagglutinin; NA, neuraminidase.
Nevertheless, as described in Chapter 3 – Vaccine antigens and Chapter 4 – Vaccine adjuvants, there has been progress in the development of influenza vaccines that target strains against which the vaccinee has limited or no pre-existing immunity, arising as the result of antigenic drift and shift (Table 6.11 ).
Table 6.11.
Research to develop new influenza vaccines continues
| Research area | Research goal |
|---|---|
| Alternative methods for antigen delivery | Virus-like particle vaccines Recombinant vector vaccines |
| Use of adjuvants | Enhance immunogenicity and reduce dose Overcome immunosenescence |
| Antigen presentation, concentration and use of conserved sequences | Re-explore the development of DNA vaccines Utilise conserved viral antigens in the development of a universal vaccine |
Another approach to the problem of influenza genome shifts has been to target weakly immunogenic conserved antigens such as the influenza M2e protein. One approach to addressing the weak immunogenicity of the antigen has been to link it to a potent Toll-like receptor adjuvant such as flagellin, an approach developed by VaxInnate Inc.
Human immunodeficiency virus
During primary infection of a single individual with HIV, mutations in surface proteins of the virus lead to selection of a ‘cloud’ of antigenic variants that can evade the cell-mediated immune responses complicating the development of broadly effective vaccines. This propensity for mutation has given rise to many strains of HIV (Figure 6.9 ). Two types of HIV, HIV-1 and HIV-2, have been identified, with HIV-1 being the most common. On a global scale, HIV-1 strains are differentiated according to their respective group and subtypes (or ‘clades’) within groups. The amino acid sequence of the viral envelope glycoprotein gp120 shows 25–35% divergence between clades and up to 20% divergence within any given clade, which constitutes a formidable hurdle to vaccine development. This is made worse by recombination between clades of HIV-1, which has produced circulating recombinant forms (CRFs) which differ in antigenicity depending on the geographical region.
A Phase III clinical trial combining Global Solutions for Infectious Diseases' AIDSVAX ™ with Sanofi Pasteur's recombinant canarypox vector vaccine (ALVAC ™) expressing CRF-AE gp120 and subtype B Gag, Pol and Nef antigens (surface glycoproteins, replication enzymes and non-structural accessory proteins, respectively) was modestly successful with an efficacy of 31.2% (95% confidence interval, 1.1−52.1; p=0.04) against HIV infection in 16,000 Thai volunteers. The vaccine purposely targeted HIV-1 strains specific to Thailand (Rerks-Ngarm et al., 2009). This trial was the first to show a degree of efficacy for an HIV candidate vaccine.
Figure 6.9.

Division of HIV-1 subtypes.
Mutations of HIV have given rise to many clades and region-specific combinations of clades, known as CRFs, of which some examples are shown. The genetic variation between HIV clades and CRFs has challenging implications for the development of HIV vaccines; future HIV vaccines may need to be clade- or region-specific, or cross-protective for all clades, groups and types.
HIV, human immunodeficiency virus; CRFs, circulating recombinant forms.
Since the initiation of HIV vaccine programmes, more than 30 candidate vaccines have been tested in over 80 Phase I/II clinical trials involving more than 10,000 healthy human volunteers. Regrettably, all attempts to date have failed to yield a licensed HIV vaccine.
Development of HIV vaccines: the toughest task?
Development of a vaccine for HIV/AIDS is exceptionally difficult due to HIV targeting of key immune cells during infection; the marked variability in HIV strain sequence and antigenicity between and within individuals; the lack of understanding of successful immune control; and the failure to reproduce in humans the results of successful vaccine trials in monkey (simian immunodeficiency virus [SIV]) models. Recent human trials aimed at inducing CD8+ lymphocyte cytotoxicity (the key immune effector in SIV trials) have failed, probably because the induced repertoire lacks sufficient potency and breadth of specificity. Vaccine development has been spurred however, by the ability of human ‘elite controllers’ and old world monkeys to control viral load and/or disease; by the recent demonstration that broad neutralising antibody to HIV can be induced in humans; and by the 2009 ‘Thai vaccine trial’ which demonstrated, for the first time, limited efficacy of an HIV vaccine.
Questions remain concerning the immune mechanisms behind vaccines that achieve partial protection. Regardless of the unknowns, the ability to prevent infection in at least some individuals still offers real hope that a globally effective HIV vaccine might be possible. Current research is comparing the immune responses of subjects who are naturally protected against HIV with those who were infected, seeking to find the elusive immunological mechanisms of protection to help guide the design of future T-cell vaccines against the virus.
Targets of HIV/AIDS vaccine candidates
Current vaccine candidates are aimed at inducing multiple types of immune effectors with a single vaccine. These effectors include CD4+ and CD8+ T-cell lymphocytes of increased potency and breadth, and broad neutralising and perhaps non-neutralising antibody, to handle the many circulating HIV strains. The 2009 ‘Thai vaccine trial’ suggested a need to examine the role of non-neutralising antibody and the possibility of preventing HIV acquisition, not just progressive immunodeficiency. A better understanding of the multiple subsets of CD4+ lymphocytes in HIV infection and the role of DCs as initial targets for infection are at the forefront of these new efforts. New hybrid viral vectors, synthetic antigens (developed with the aid of three-dimensional modelling), novel adjuvants that manipulate the immune system to induce desirable responses and more useful animal models are also being developed and tested.
Development of vaccines that induce broad neutralising antibodies to highly variable viruses, such as HIV and influenza, has proved to be extremely difficult. However, screening HIV-infected individuals for such antibodies has allowed the identification of previously undiscovered viral epitopes which can be incorporated into structure-based vaccine design.
Group A Streptococcus
Infections of group A streptococcal serotypes (ie Streptococcus pyogenes) account for approximately 85% of cases of uncomplicated bacterial pharyngitis and streptococcal invasive infections in North America. The M protein of group A streptococci is a major virulence determinant of these organisms and also functions as a major target for protective antibodies. One of several strategies for vaccine prevention of these infections is based on type-specific M protein epitopes. However, group A streptococcal vaccine development faces many obstacles: i) the widespread diversity of circulating M protein types; ii) immunological cross-reactivity between epitopes in the M protein and several human tissues introducing an autoimmune risk; and iii) animal models are of limited value because humans are the only hosts for group A streptococci. In an attempt to partially overcome some of these obstacles, a design strategy akin to that of the pneumococcal polysaccharide vaccines has been employed to generate a group A streptococci multivalent M protein-based vaccine containing type-specific determinants from 26 different M serotypes. This multivalent vaccine is currently in clinical development.
The ‘prime-boost’ approach
The term ‘prime boost’ (or heterologous boosting) describes an approach to vaccination where one type of vaccine, such as a live-vector vaccine, is administered followed by a second type of vaccine, such as a recombinant subunit vaccine. This is in contrast with the traditional method of homologous boosting in which two or more doses of the same vaccine are given successively. The intent of prime-boost vaccination is to induce different types of immune responses and enhance the overall immune response, a result that may not occur if only one type of vaccine were to be given for all doses. This approach has been employed in trials with, for example, TB, CMV, malaria and HIV candidate vaccines. For example, in studies on new TB vaccines, subjects already primed with the live, attenuated BCG vaccine have been boosted with a subunit adjuvanted vaccine (see Tuberculosis).
Undesirable immune responses elicited by vaccines
Respiratory syncytial virus
Respiratory syncytial virus is a common cause of bronchiolitis and pneumonia in infants, and exacerbations of chronic obstructive pulmonary disease in the elderly. The development of an effective vaccine has been challenging; natural immunity to RSV infection is incomplete and re-infections occur in all age groups. Moreover, the primary target population for vaccination is newborns and young infants, and they are a challenging population as they have relatively immature immune systems and the presence of maternal antibodies may interfere with vaccination of the young infant (see Chapter 2 – Vaccine immunology). The initial efforts to develop a formalin-inactivated cell culture-derived RSV vaccine resulted in an unanticipated enhancement of natural RSV disease in some of the RSV-naïve infants who received the vaccine in a clinical trial and subsequently were exposed to RSV. The exacerbated disease is thought to be due to an exaggerated T helper type 2 cell immune response (see Chapter 2 – Vaccine immunology). Safety concerns regarding the potential of vaccines to trigger or prime for immunopathological responses has resulted in a cautious approach to the development of RSV vaccines. The vaccine candidates most advanced in clinical development use two different approaches – one uses a live, attenuated virus with a gene deletion deliberately targeted to minimise immunopathological responses. The other approach uses a live viral vector to deliver only a key RSV surface antigen, thereby avoiding the risk of an immunopathological response arising from exposure to the RSV virus itself.
Vaccines for neglected tropical and non-tropical diseases
Infectious illnesses exert a major burden of disease in developing countries. The greatest burden is caused by diseases for which we currently have no vaccines, eg taeniid cestode parasites are associated with high human morbidity and losses in livestock. Global efforts to reduce these infections in humans are ongoing through the use of antihelminthics and the implementation of lifestyle changes, but this is having little effect. However, substantial progress has been made towards developing veterinary vaccines which encourages investigation of the potential use of similar vaccines in humans to prevent, for example, hydatid disease (arising from infection with Echinococcus granulosus) and cysticercosis (from infection with Taenia solium).
Relative to their burden on society, such diseases have a low priority for funding. Unless comprehensive measures are taken to address the gaps in funding, research and global immunisation coverage, developing countries will continue to be overwhelmed by some of the most devastating diseases. In order to improve the situation, collaborative schemes are underway that bring together academic institutions, industry and public/charitable financing organisations. Recent initiatives include the Novartis Vaccines Institute for Global Health, the MSD–Wellcome Trust Hilleman Laboratories and the Alliance for Case Studies for Global Health.
Human Hookworm Vaccine Initiative featured in Case Studies for Global Health
The Human Hookworm Vaccine Initiative (HHVI), an international product development partnership based at the Sabin Vaccine Institute, was established in 2000 to develop the world's first ever safe, affordable, multivalent recombinant vaccine against human hookworm infection. Such a vaccine could impact an estimated 3.2 billion at risk individuals. Sabin Vaccine's HHVI is one of 32 projects chosen for inclusion in Case Studies for Global Health released on 20 November 2009 by the Alliance for Case Studies for Global Health. Other diseases include HIV, TB and malaria, and lesser-known diseases such as dengue fever and Japanese encephalitis. The Alliance is a collaboration of The Bill & Melinda Gates Foundation, the World Health Organization's Special Programme for Research and Training in Tropical Diseases (TDR), Global Health Progress (GHP), the International AIDS Vaccine Initiative (IAVI) and the Association of University Technology Managers (AUTM).
Vaccines for novel and emerging pathogens
It is estimated that 99% of microbes are yet to be discovered. Using nucleic acid sequencing strategies, Ian Lipkin has discovered close to 200 new viruses including the LuJo virus, a new arenavirus that has caused several fatal cases of haemorrhagic fever in Zambia and South Africa. Behavioural and environmental changes may facilitate the emergence and spread of new pathogens, while novel methods of discovery may allow for the more rapid development of vaccines against emergent diseases, before the new pathogens become widespread public health problems, as was the case in the development of a Sanofi Pasteur vaccine against the SARS coronavirus infection.
The microbiome, a term coined by Joshua Lederberg, is defined as the totality of microbes within a defined environment. The human microbiota has co-evolved with their hosts and appears to play important roles in human health and disease. The Human Microbiome Project is a National Institutes of Health initiative that seeks to determine the relationship between human health and changes in the human microbiome. By using revolutionary sequencing technologies to characterise the microbiology of five body sites – oral cavity, skin, vagina, gut and nasal tract/lung – an association may be made between the microbiomes associated with either the healthy body state or disease. Characterising microbes associated with disease-related pathogens may allow for the development of new vaccines that preserve or protect the healthy microbiome and hence could protect human health. Some of the areas of current research are outlined in the box, right.
Diseases being explored in connection with the human microbiome include psoriasis and atopic dermatitis, inflammatory bowel disease, urethritis and sexually transmitted diseases, obesity, oesophageal adenocarcinoma, necrotising enterocolitis and paediatric abdominal pain.
Vaccines for conditions not generally associated with infectious diseases
Some conditions traditionally thought of as non-infectious may in fact have infectious origins (Table 6.12 ); therefore, vaccination could be a strategy to prevent these diseases. Other diseases may result from an interaction between the host’s genetic background and a particular microbe (a so-called gene-environment interaction). Some diseases have an established link with an identified infectious agent. For example, primary CMV infection is a known cause of congenital mental retardation; similarly the link between bacterial vaginosis and foetal prematurity is widely accepted. While some links have been established, others remain speculative (Table 6.12).
Table 6.12.
Examples of non-infectious diseases where infectious agents have been identified or postulated as contributory causes
| Disease | Proposed infectious agent | Vaccine status | Causal link |
|---|---|---|---|
| Cervical cancer | HPV | Licensed vaccine | Established |
| Gastric ulcer disease Gastric cancer |
Helicobacter pylori | In development | Established |
| Systemic lupus erythematosus | Trypanosoma cruzi | In development | Speculative and indirect (via the formation of anti-P and antiphospholipid antibodies) |
| Glandular fever Burkitt's lymphoma Nasopharyngeal carcinoma Multiple sclerosis |
EBV | In development | Established Strong Established Strong |
| Hepatocellular carcinoma | HBV HCV |
HBV – licensed vaccine HCV – in development |
Established Strong |
| Kaposi's sarcoma | HHV-8 (in patients with AIDS) | No candidate vaccine | Strong |
| Foetal prematurity | Bacterial vaginosis | No candidate vaccine | Established |
| Congenital mental retardation | CMV | In development | Established |
| Asthma exacerbations | Rhinovirus | No candidate vaccine | Established |
| Diabetes mellitus (type I) | Fungal infection/mycotoxins | No candidate vaccine | Speculative |
| Alzheimer's disease/dementia/cognitive impairment | HSV-1 Helicobacter pylori Picornavirus Borna disease virus Chlamydia pneumoniae |
HSV – in development Helicobacter pylori – in development Others – no candidate vaccine |
Speculative |
| Schizophrenia | CMV Toxoplasma gondii HHV-6 |
CMV – in development Others – no candidate vaccine |
Speculative |
| Atherosclerosis | Chlamydia pneumoniae | No candidate vaccine | Speculative |
| Hypertension | HHV-8 | No candidate vaccine | Weak/unlikely |
| Obesity/weight gain | Adenovirus-36 | No candidate vaccine | Speculative, possibly geographical |
| Rheumatoid arthritis | EBV HHV-6 |
EBV – in development HHV-6 – no candidate vaccine |
Speculative |
| Crohn's disease | Mycobacterium avium subspecies paratuberculosis | No candidate vaccine | Conflicting evidence |
HPV, human papillomavirus; EBV, Epstein–Barr virus; HBV, hepatitis B virus; HCV, hepatitis C virus; HHV, human herpesvirus; AIDS, acquired immunodeficiency syndrome; CMV, cytomegalovirus; HSV, herpes simplex virus. Every effort has been made to verify the information in this table. The information included is not meant to be exhaustive but is intended to provide an overview of the subject matter.
Prophylactic and therapeutic vaccines for addiction
Candidate vaccines are in development for the prevention and treatment of various types of addiction. The basic concept is to induce the production of antibodies which will bind the drug and impede its crossing the blood–brain barrier to exert its psychoactive effects. Several nicotine candidate vaccines have now entered clinical trials. A cocaine candidate vaccine has also shown some benefit in a Phase IIb clinical trial. The key issue to date for both nicotine and cocaine candidate vaccines has been to induce high immunoglobulin (Ig)G anti-drug antibody levels, which appear to be critical in achieving some degree of efficacy. Candidate vaccines against methamphetamine addiction are also in early development.
“It's easy to quit smoking. I've done it hundreds of times” Mark Twain
Prophylactic and therapeutic vaccines for cancer
To date, the approach to developing prophylactic cancer vaccines has been to target infectious diseases that cause or contribute to the development of cancer such as HPV (cervical cancer) and HBV (hepatocellular carcinoma). Examples of infectious diseases associated with cancer are shown in Table 6.13 . The successful development of a nicotine vaccine would be expected to reduce cigarette smoking-related lung cancer.
Infectious diseases cause approximately 17% of new cancers worldwide, about 1.5 million (26%) cancers in low-resource and middle-resource countries (where 84% of the world's population resides), and 360,000 (7.2%) cancers in high-resource countries (where 16% of the world's population resides).
Table 6.13.
Infectious diseases associated with cancer
| Pathogen | Type of organism | Associated cancer(s) |
|---|---|---|
| HBV | Virus | Hepatocellular carcinoma |
| HCV | Virus | Hepatocellular carcinoma |
| HPV (high-risk types) | Virus | Cervical cancer, vaginal cancer, vulvar cancer, oropharyngeal cancer, anal cancer, penile cancer |
| EBV | Virus | Burkitt's lymphoma, non-Hodgkin's lymphoma, Hodgkin's lymphoma, nasopharyngeal carcinoma |
| HHV | Virus | Kaposi's sarcoma, primary effusion lymphoma |
| Human T-cell lymphotropic virus 1 | Virus | Acute T-cell leukaemia |
| JC and BK polyomaviruses | Virus | Brain tumours |
| Helicobacter pylori | Bacterium | Stomach cancer |
| Schistosoma hematobium (schistosomiasis) | Parasite | Bladder cancer |
| Opisthorchis viverrini (liver flukes) | Parasite | Cholangiocarcinoma |
HBV, hepatitis B virus; HCV, hepatitis C virus; HPV, human papillomavirus; EBV, epstein–Barr virus; HHV, human herpesvirus 8; JC and BK, initials of patients from whom polyomavirus isolates were obtained. Every effort has been made to verify the information in this table. The information included is not meant to be exhaustive but is intended to provide an overview of the subject matter.
Some cancers express tissue-specific antigens that can be targeted by the immune system. Therapeutic cancer vaccines aim to target tumour-associated antigens (TAA) with T-cell mediated immune responses. TAA can be related to the genetic changes that drive the cancer (eg Ras oncogene), or inappropriate up-regulation/expression of genes (eg carcinoembryonic antigen). With such TAA targets, vaccines aim to maximally stimulate a cytotoxic T-cell response and their design often includes adjuvants to enhance antigen presentation. Tumours develop in a multistep process in the face of the host immune response and frequently evolve to escape immune control. Mechanisms of evasion include genetic changes (loss of human leukocyte antigen/TAA expression) and induction of immune regulatory systems (T-cell anergy due to the activity of Treg cells) which limit anti-tumour immunity. The key approach for therapeutic cancer vaccines is resetting the immune response to deliver anti-tumour immunity that alters or destroys cancer cells and hence eliminates or reduces the tumour. One strategy uses the patient's own tumour as the immunogen, thereby providing all the potential idiotypic changes that might act as TAA, in conjunction with antigen-presenting DCs harvested from the same patient and activated in vitro (see Dendritic cell vaccines). There are different types of therapeutic candidate vaccines currently undergoing clinical trials for numerous types of cancer (Table 6.14 ). The most advanced candidates currently in Phase III are described in Chapter 4 – Vaccine adjuvants.
Table 6.14.
Examples of therapeutic cancer vaccines in clinical trials
| Cancer type | Vaccine approach | Manufacturer/Developer |
|---|---|---|
| Bladder cancer | Multi-epitope cancer vaccine to the MUC1 tumour-associated antigen | Vaxil Bio Therapeutics Ltd |
| Brain tumours | Dendritic cell vaccine | Samuel Oschin Comprehensive Cancer Institute |
| Cervical cancer | RO5217790 consists of recombinant modified vaccinia Ankara viral vector encoding mutated forms of human papillomavirus type 16 genes for the viral oncoproteins E6 and E7 and the hIL2 | Roche |
| Melanoma | DNA vaccine (tyrosinase-related protein 2) – an immunobody adjuvant approach ASCI, MAGE-A3/AS15 Human anti-CTLA-4 antibody (MDX-010) in combination with a gp100 melanoma vaccine (MDX-1379) |
Scancell Ltd GSK Biologicals Bristol-Myers Squibb |
| Lung cancer | Modified mRNA vaccine encoding five antigens including NY-ESO-1 ASCI, MAGE-A3/AS15 |
CureVac GmbH GSK Biologicals |
| Colorectal cancer | Modified vaccinia Ankara encoding the tumour antigen 5T4 | Oxford BioMedica |
MUC1, Mucin 1 cell surface; hIL2, human cytokine interleukin-2; ASCI, antigen-specific cancer immunotherapeutics; MAGE, melanoma-associated antigen; CTL, cytotoxic T lymphocyte. Every effort has been made to verify the information in this table. The information included is not meant to be exhaustive but is intended to provide an overview of the subject matter.
There has been some success in the development of therapeutic cancer vaccines, with the FDA approval of the first DC vaccine designed for the treatment of prostate cancer in 2010 (see Dendritic cell vaccines). Other vaccines have been licensed in individual countries for treatment of cancers including non-small-cell lung cancer and melanoma.
New and more effective vaccines for specific populations
Developing vaccines that are effective in all populations is difficult because some populations do not respond adequately to traditional vaccine approaches. However, this presents opportunities for the application of novel technologies and adjuvants. Some of the considerations for vaccines designed for use in special populations include: immunosenescence in the elderly; the poor immunological response to traditional vaccines seen in immunocompromised individuals (patients with HIV, transplant recipients); the crossing of vaccine components into the foetal bloodstream when vaccines are administered to pregnant women; and the safety and immunogenicity concerns surrounding vaccines for neonates due to their naïve and immature immune system.
Vaccines for pregnant women
Cell-mediated immunity is depressed in pregnant women, leaving them at high risk of infection from pathogens, including those harmful to the foetus. Most live, attenuated vaccines are contraindicated during pregnancy because of the theoretical risk of foetal infection from the vaccine. However, inactivated viral or bacterial vaccines can be administered. Pregnant women can, therefore, be vaccinated against some infections, including several that pass from mother to foetus (such as hepatitis A and B), and against infections acquired by the infant in the first few months of life (often from close contact with the mother). In the latter case, the infant can be protected by transfer of maternal antibodies during late gestation. Examples of diseases that can be prevented in pregnant women include influenza, tetanus, diphtheria and probably pertussis. Other diseases, such as those caused by the so-called TORCH pathogens (toxoplasma, others including syphilis, CMV and HSV), are not yet preventable through vaccination though encouraging Phase II results have been presented for a vaccine to prevent Group B streptococcus carriage in pregnant women (Hillier et al., 2009, Smith, 2009).
The 2009 H1N1 pandemic influenza outbreak posed an increased risk to pregnant women and vaccination was specifically recommended in pregnant women as one of the high-risk groups. The pandemic example has emphasised once more the importance of protecting pregnant women against influenza. Seasonal influenza vaccination in pregnancy is well tolerated and the benefit–risk profile when administered to pregnant women supports its use during pregnancy. Many public health authorities worldwide recommend seasonal influenza vaccination in pregnant women and this recommendation is motivated not only by the potentially severe course of influenza during pregnancy, but also by the need to protect vulnerable infants against influenza during their first months of life. Boosting RSV immunity in pregnant women through vaccination may be another approach to protecting the newborn against RSV infection during the most vulnerable early months after birth.
Vaccines for neonatal infants
Neonatal immunisation is a strategy to protect infants against infections during a particularly vulnerable period. A recent study showed that immunisation with an acellular pertussis vaccine at birth and 1 month of age induces high IgG anti-pertussis antibody titres by 2 months of age (Wood et al., 2010). It is hoped that this approach may reduce death and morbidity from Bordetella pertussis infection in the first 3 months of life.
Vaccines for the elderly
The elderly respond poorly to vaccination as the immune system becomes more senescent with increasing age and, therefore, new vaccine technologies are needed to improve the response to vaccination in this population. In the late 1990s, an influenza vaccine adjuvanted with the oil-in-water emulsion, MF59™, was shown to be more effective at inducing high immune responses in the elderly (Minutello et al., 1999). Alternative vaccine administration techniques have also been studied in the elderly. Research showed that in subjects 60 years of age or older, an influenza vaccine administered with an intradermal microinjection system induced significantly higher antibody titres compared with IM vaccination (Arnou et al., 2009). Subsequently, a microinjection system influenza vaccine was licensed for use in Europe, and a high antigen dose formulation has been licensed for the elderly in the USA.
Vaccines for those who are immunocompromised
Individuals with cancer, HIV infection or who are asplenic can be immunocompromised as a result of their condition. Patients can also be immunocompromised as a result of therapy, eg when receiving an organ transplant, radiation therapy or immunosuppressive medication. Such patients are therefore at an elevated risk of infection from pathogens such as herpesviruses (particularly CMV and Epstein–Barr virus), HBV, HCV, pneumocystis and coinfections and represent a special population regarding immunisation. Despite a likely reduction in the efficacy of vaccinations in immunocompromised individuals, immunisation remains a frequent recommendation in the hope that at least partial immunity will be achieved. Eliciting a response from vaccination in immunocompromised patients may require an increase in the dose and/or number of doses; altering the dosing interval; selecting a different vaccine formulation; or administration via an alternative route. Evidence in this patient population is lacking and guidelines are often based on theoretical assumptions.
Live vaccines are generally contraindicated in immunocompromised or immunosuppressed individuals due to the risk of an active and symptomatic infection resulting from the vaccine itself (non-controlled replication process). Encouragingly, vaccine formulations with highly purified antigens and novel adjuvants or alternative deliveries have been shown to induce more effective immune responses than the classical inactivated vaccines in immunocompromised hosts, including patients with end-stage renal diseases in pre-haemodialysis and haemodialysis (see Chapter 4 – Vaccine Adjuvants), patients with HIV and those who have received haematological stem cell transplants.
Conclusion
The future of vaccine development can build on the knowledge and experience gained over the last 200 years, and at the same time can take advantage of the most cutting-edge technologies and research. New approaches to antigen selection and production, antigen delivery, adjuvantation and vaccine administration will allow us to target established and emerging diseases, and populations with complex needs. Vaccination has been one of the most successful and cost-effective health interventions ever conceived and is now expanding further into cancer and chronic diseases. This expansion of scope and the subsequent impact on human disease is likely to continue into the future in currently unforeseen ways, further increasing the importance of vaccine science and engineering in improving human health.
References
- Abdulla S., Oberholzer R., Juma O. Safety and immunogenicity of RTS,S/AS02D malaria vaccine in infants. N Engl J Med. 2008;359:2533–2544. doi: 10.1056/NEJMoa0807773. [DOI] [PubMed] [Google Scholar]
- Arnou R., Icardi G., De Decker M. Intradermal influenza vaccine for older adults: a randomized controlled multicenter phase III study. Vaccine. 2009;27:7304–7312. doi: 10.1016/j.vaccine.2009.10.033. [DOI] [PubMed] [Google Scholar]
- Bejon P., Lusingu J., Olotu A. Efficacy of RTS,S/AS01E vaccine against malaria in children 5 to 17 months of age. N Engl J Med. 2008;359:2521–2532. doi: 10.1056/NEJMoa0807381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier S, Ferris D, Fine P et al. Women receiving Group B Streptococcus serotype III tetanus toxoid (GBS III-TT) vaccine have reduced vaginal and rectal acquisition of GBS Type III. Infectious Diseases Society of America (IDSA) 47th Annual Meeting: Abstract 186. Presented 30 October 2009
- Lambert P.H., Hawkridge T., Hanekom W.A. New vaccines against tuberculosis. Clin Chest Med. 2009;30:811–826. doi: 10.1016/j.ccm.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Minutello M., Senatore F., Cecchinelli G. Safety and immunogenicity of an inactivated subunit influenza virus vaccine combined with MF59 adjuvant emulsion in elderly subjects, immunized for three consecutive influenza seasons. Vaccine. 1999;17:99–104. doi: 10.1016/s0264-410x(98)00185-6. [DOI] [PubMed] [Google Scholar]
- Oxman M.N., Levin M.J., Johnson G.R. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med. 2005;352:2271–2284. doi: 10.1056/NEJMoa051016. [DOI] [PubMed] [Google Scholar]
- Pass R.F., Duliegè A.M., Boppana S. A subunit cytomegalovirus vaccine based on recombinant envelope glycoprotein B and a new adjuvant. J Infect Dis. 1999;180:970–975. doi: 10.1086/315022. [DOI] [PubMed] [Google Scholar]
- Pass R.F., Zhang C., Evans A. Vaccine prevention of maternal cytomegalovirus infection. N Engl J Med. 2009;360:1191–1199. doi: 10.1056/NEJMoa0804749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rerks-Ngarm S., Pitisuttithum P., Nitayaphan S. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- Sabin A., Flores Areciga A., Fernandez de Castro J. Successful immunization of children with and without maternal antibody by aerosolized measles vaccine. I. Different results with undiluted human diploid cell and chick embryo fibroblast vaccines. JAMA. 1983;249:2651–2662. [PubMed] [Google Scholar]
- Smith M. IDSA: Strep Vaccine Safe, Effective in Early Trial MedPage Today 02 November 2009 http://www.medpagetoday.com/MeetingCoverage/IDSA/16739 Date accessed: 19 April 2011 (reviewed by Dori F Zaleznik, MD, Associate Clinical Professor of Medicine, Harvard Medical School, Boston and Dorothy Caputo, MA, RN, BC-ADM, CDE, Nurse Planner)
- Stanberry L.R., Spruance S.L., Cunningham A.L., GlaxoSmithKline Herpes Vaccine Efficacy Study Group Glycoprotein-D-adjuvant vaccine to prevent genital herpes. N Engl J Med. 2002;347:1652–1661. doi: 10.1056/NEJMoa011915. [DOI] [PubMed] [Google Scholar]
- von Reyn C.F., Mtei L., Arbeit R.D. Prevention of tuberculosis in Bacille Calmette-Guérin-primed, HIV-infected adults boosted with an inactivated whole-cell mycobacterial vaccine. AIDS. 2010;24:675–685. doi: 10.1097/QAD.0b013e3283350f1b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker K.B., Brennan M.J., Ho M.M. The second Geneva Consensus: recommendations for novel live TB vaccines. Vaccine. 2010;28:2259–2270. doi: 10.1016/j.vaccine.2009.12.083. [DOI] [PubMed] [Google Scholar]
- Wood N., McIntyre P., Marshall H. Acellular pertussis vaccine at birth and one month induces antibody responses by two months of age. Pediatr Infect Dis J. 2010;29:209–215. doi: 10.1097/INF.0b013e3181bc98d5. [DOI] [PubMed] [Google Scholar]
- Genocea technology. Available at http://genocea.com/technology/technology.html. Date accessed: 16 March 2011.
Further reading
- Barrett P.N., Portsmouth D., Ehrlich H.J. Developing cell culture-derived pandemic vaccines. Curr Opin Mol Ther. 2010;12:21–30. [PubMed] [Google Scholar]
- Flores J. Seeking new pathways for HIV vaccine discovery. Future Microbiol. 2009;4:1–7. doi: 10.2217/17460913.4.1.1. [DOI] [PubMed] [Google Scholar]
- Fontham E.T. Infectious diseases and global cancer control. CA Cancer J Clin. 2009;59:5–7. doi: 10.3322/caac.20000. [DOI] [PubMed] [Google Scholar]
- Goldenberg R.L., Hauth J.C., Andrews W.W. Intrauterine infection and preterm delivery. N Engl J Med. 2000;342:1500–1507. doi: 10.1056/NEJM200005183422007. [DOI] [PubMed] [Google Scholar]
- Lipkin I. Straight talk with…Ian Lipkin. Nat Med. 2009;15:982–983. doi: 10.1038/nm0909-982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HMP Working Group N.I.H., Peterson J., Garges S., Giovanni M. The NIH human microbiome project. Genome Res. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappuoli R. Reverse vaccinology, a genome-based approach to vaccine development. Vaccine. 2001;19:2688–2691. doi: 10.1016/s0264-410x(00)00554-5. [DOI] [PubMed] [Google Scholar]
Internet resources
- 3M: http://solutions.3m.com/wps/portal/3M/en_WW/DrugDeliverySystems/DDSD/
- Advaxis: http://www.advaxis.com/
- AERAS Global TB Vaccine Foundation: http://www.aeras.org/home/home.php
- Alliance for Case Studies for Global Health . 2009. Case studies for global health: Building relationships. Sharing knowledge.http://www.casestudiesforglobalhealth.org/case_study_PDFs/GlobalHealthCaseStudies.pdf ISBN #0-9778444-8-X. Date accessed: 8 April 2011. [Google Scholar]
- Alliance for Case Studies for Global Health: http://www.casestudiesforglobalhealth.org/
- Association of University Technology Managers: http://www.autm.net/Home.htm
- AstraZeneca: http://www.astrazeneca.com/Home
- Bavarian Nordic A/S: http://www.bavarian-nordic.com/
- Baylor Research Institute: http://www.baylorhealth.edu/Research/Default.htm
- Bill & Melinda Gates Foundation: http://www.gatesfoundation.org/Pages/home.aspx
- Bristol-Myers Squibb: http://www.bms.com/pages/default.aspx
- Celldex Therapeutics: http://www.celldextherapeutics.com/
- Coley Pharmaceutical Group: http://www.pfizer.com/coleypharma/index.html
- Crucell: http://www.crucell.com/
- CSL Behring: http://www.csl.com.au/businesses/csl-behring.htm
- CureVac GmbH: http://www.curevac.com/
- Dendreon: http://www.dendreon.com/
- Dynavax Technologies: http://www.dynavax.com/
- Emergent Biosolutions: http://www.emergentbiosolutions.com/
- FIT Biotech: http://www.fitbiotech.com/
- Genexine: http://www.genexine.com/
- Genocea Biosciences: http://www.genocea.com/
- GenVec Inc: http://www.genvec.com/
- GSK Biologicals: http://www.gsk.com/
- Global Health Progress: http://globalhealthprogress.org/
- Global Solutions for Infectious Diseases: http://www.isis-innovation.com/spinout/OETC.html
- Ichor Medical Systems: http://www.ichorms.com/
- Immunomic Therapeutics: http://www.immunomix.com/index.html
- Immutep SA: http://www.immutep.com/
- Inovio Pharmaceuticals: http://www.inovio.com/
- Intercell AG: http://www.intercell.com/
- International AIDS Vaccine Initiative: http://www.iavi.org/Pages/home.aspx
- MedImmune LLC: http://www.medimmune.com/
- Memorial Sloane-Kettering Cancer Center: http://www.mskcc.org/mskcc/html/44.cfm
- MSD-Wellcome Trust Hilleman Laboratories: http://www.hillemanlaboratories.in/
- NanoBio Corporation: http://www.nanobio.com/
- National Institute of Allergy and Infectious Diseases (NIAID) Statement: Study finds genital herpes vaccine ineffective in women. 30 Sep 2010. http://www.niaid.nih.gov/news/newsreleases/2010/Pages/Herpevac.aspx Date accessed: 8 April 2011.
- National Institute of Allergy and Infectious Diseases, Vaccine Research Center: http://www.niaid.nih.gov/about/organization/vrc/about/Pages/goals.aspx
- Novartis International AG: http://www.novartis.com/index.shtml
- Novartis Vaccines Institute for Global Health: http://www.corporatecitizenship.novartis.com/patients/access-medicines/nvgh.shtml
- Okairos: http://www.okairos.it/e/index.php
- Oxford Biomedica: http://www.oxfordbiomedica.co.uk/
- Oxford-Emergent Tuberculosis Consortium Ltd: http://www.isis-innovation.com/spinout/OETC.html [DOI] [PubMed]
- PaxVax Inc: http://paxvax.com/
- Pfizer: http://www.pfizer.com/home/
- Polymun Scientific: http://www.polymun.at/
- PowderMed: http://www.powdermed.com/
- Roche: http://www.roche.com/index.htm
- Sabin Vaccine Institute: http://www.sabin.org/
- Samuel Oschin Comprehensive Cancer Institute: http://www.cedars-sinai.edu/Patients/Programs-and-Services/Cancer-Center/
- Sanofi Pasteur: http://www.sanofipasteur.com/sanofi-pasteur2/front/index.jsp?siteCode=SP_CORP
- Scancell Ltd: http://www.scancell.co.uk/
- Seppic: http://www.seppic.com/
- Statens Serum Institut: http://www.ssi.dk/English.aspx
- Swiss Vaccine Research Institute: http://www.swissvaccineresearchinstitute.ch/
- Targeted Genetics: http://www.targen.com/about_us.htm
- Transgene SA: http://www.transgene.fr/
- University of Maryland School of Medicine, Center for Vaccine Development: http://medschool.umaryland.edu/cvd/
- University of Texas Medical Branch, Sealy Center for Vaccine Development: http://www.utmb.edu/scvd/
- Vacin Inc: http://www.vaxin.com/
- Vical: http://www.vical.com/
- Vaxil BioTherapeutics Ltd: http://www.vaxilbio.com/
- VaxInnate Inc: http://www.vaxinnate.com/
- World Health Organization's Special Programme for Research and Training in Tropical Diseases: http://apps.who.int/tdr/
Trademark disclaimers
- AIDSVAX™ is a trademark of Global Solutions for Infectious Diseases Corporation; ALVAC™ is a trademark of Connaught Technology Corporation; IC31™ is a trademark of Intercell; ISCOM™ and ISCOMATRIX™ are trademarks of Iscotec AB Corp; MF59™ is a trademark of Novartis; Montanide™ is a trademark of Seppic; NanoStat™ is a trademark of NanoBio Corporation. For details of trademarks not listed above please see the manufacturer's website (see section Internet resources).