

Abstract
Previous reports suggest that type I and type II Interferon can co-operatively inhibit some virus replication, e.g. HCV, SARS-CoV, HSV-1. To find out the molecular mechanism underlying this phenomenon, we analyzed the transcription profile stimulated by IFN-α and IFN-γ in Huh-7 cells and found that the transcription of a subset of IFN stimulated genes (ISGs) including BclG, XAF1, TRAIL and TAP1 was enhanced when IFN-α and γ were both present. Promoter analysis of BclG revealed that IRF-1 and STAT1 were both required in this process. Enhanced IRF-1/DNA complex formation was observed in interferon co-treatment group by gel shift analysis. Furthermore, IRF-1 activation was found to be generally required in this cluster of ISGs. STAT1 tyrosine phosphorylation was elevated by IFN combination treatment, however, only the hyper-transactivation of GAS but not ISRE was observed. In conclusion, hyper-activation of IRF-1 and elevated STAT1 dimer formation may be two general switches which contribute to a much more robust antiviral symphony against virus replication when type I and type II IFNs are co-administered.
Keywords: Interferon regulatory factor-1, Interferon-α, Interferon-γ
1. Introduction
Interferons (IFNs) are pleiotropic cytokines that mediate antiviral, anti-proliferative, anti-tumor and immuno-modulatory activities [1]. Interferons are categorized into two major classes, i.e. type I (predominantly IFN-α and β) and type II (IFN-γ) interferon. These two classes of interferon bind to different pairs of receptors on the cell membrane but have overlapping route of signaling downstream [1]. Type I interferon preferentially triggers the formation of the ISG factor 3 (ISGF3) complex composed of phosphorylated STAT1 and STAT2 together with IRF-9 which binds to IFN-stimulated response elements (ISRE) within promoters of certain ISGs. Type II interferon induces the tyrosine phosphorylation of STAT1 and results in the formation of STAT1 homodimers which bind to IFN-γ activated site (GAS) elements [2]. In addition, type II interferon can potently induce the expression of IRF-1, which has been shown to stimulate various genes with anti-viral, anti-proliferative or immuno-modulatory activities [3].
Human type I IFNs have long been recognized clinically for their antiviral and immuno-modulatory effects. IFN-α has been widely used to treat hepatitis B and hepatitis C virus infection although with limited percentage of sustained response [4], [5]. Similar to IFN-α, IFN-γ has potent antiviral activity in many in vitro and in vivo virus replication models [6], [7], however its application in clinical viral infection turned out to be unsuccessful and toxicity was also frequently observed [8].
Interestingly, in recent years, a growing body of evidence has shown that when type I and type II IFN are co-administered, the replication of many viruses is inhibited even more strikingly. Sainz et al. demonstrated in a series of articles that type I and type II IFN could synergize to inhibit herpes simplex virus type 1 (HSV-1), human cytomegalovirus and severe acute respiratory syndrome coronavirus(SARS-CoV) replication [9], [10], [11], Fuchizaki et al. also reported that combination of mouse IFN-α and IFN-γ can prolong the survival period of mice infected with mouse hepatitis virus type 2. This is consistent with the lower levels of heptocellular necrosis and serum alanine aminotransferase (ALT) and decreased titers of MHV-2 virus load [12]. It was also the case in an in vitro HCV replicon system in which IFN-α or IFN-β is co-administered with IFN-γ [13], [14]. Furthermore, an IFN-γ priming IFN-α boost protocol in a clinical trial successfully invoked the response against HCV in six of the nineteen patients who all failed in a previous 6-month IFN-α mono-therapy [15].
In the light of all the experimental and clinical evidence, it is timely to probe the molecular process underlying these phenomena. A small cluster of genes were identified whose expression was enhanced after co-treatment including BclG (Bcl-2 family protein G), XAF1 (X-linked inhibitor of apoptosis associated factor-1), TRAIL (TNF-related apoptosis inducing ligand) and TAP1 (transporter 1). Subsequent promoter analysis of BclG showed that IRF-1 (interferon regulatory factor 1) and STAT1 (signal transducer and activator of transcription 1) were both essential for full enhancement of gene expression. Moreover, enhanced IRF-1/DNA complex formation was also observed in EMSA analysis. Knock down of IRF-1 expression by specific small interfering RNA (siRNA) not only suppressed the enhancement of BclG but also other members of this cluster suggesting the general role of IRF-1 in this process. STAT1 tyrosine phosphorylation was enhanced, while only the activation of GAS (IFN-gamma-activated site) but not ISRE (interferon-stimulated response element) was observed. Taken together, we postulate that IRF-1 hyper-activation and elevated STAT1 dimer formation associate with enhanced expression of a subset of interferon stimulated genes (ISGs) which may lead to even greater antiviral activity.
2. Materials and methods
2.1. Cell culture and transfection
Huh-7 cells were maintained in DMEM supplemented with 10% fetal calf serum, 2 mmol/ml l-glutamine, penicillin and streptomycin (Gibco, BRL). Plasmid DNA was transfected using FuGENE 6 (Roche), siRNA was transfected with Oligofectamine (Invitrogen).
2.2. Reagents
The antibodies to IRF-1 (sc-497), IRF-2(sc-498) and IRF-9 (sc-496) were obtained from Santa Cruz, antibodies to STAT1 (#9172) and Phospho-STAT1 (Tyr 701) (#9171) were from Cell Signaling, BclG antibody (PAB10209) was obtained from Orbigen. Antibody to β-actin was from Sigma. Human IFN-α and IFN-γ were purchased from Calbiochem and Peprotech respectively.
2.3. Quantitative RT-PCR
Total RNA of Huh-7 cells after various stimulations was extracted using TRIzol reagent (Gibco BRL) followed by 30 min of DNaseI digestion of remaining genomic DNA. Pure RNA was reverse transcribed using SuperScript II (Invitrogen) and random hexamer primer according to the manufacturer's instructions. Real-time PCR (iCyler, Bio-Rad) was performed as instructed in iCycler resource guide. Briefly, reactions were carried out in 25 μl volume containing 2 μl cDNA template, 400 nM of each forward and reverse primer, SYBR GreenI and 1.25 unit hot-start ExTaq (TaKaRa). A linearized plasmid containing 188 bp GAPDH cDNA was quantified and diluted to 4 × 102–4 × 105 copy/μl as standard. Cycle numbers of the logarithmic linear phase were plotted against the logarithm of the concentration of template DNA. The primers used are listed in Table 1 (Upper panel).
Table 1.
Primers, DNA probes and siRNAs used in this study
| Target | Sequence (5′–3′) |
|---|---|
| Real time PCR primers | |
| BclG | (+)-TGCACAGTTTATCTTTTCACTC |
| (−)-TGAGAACCTGAGGAAATCTGT | |
| XAF1 | (+)-AACCCTCAACAAACCAGGC |
| (−)-TCTCTTGCCTGATTGCTGTG, | |
| TRAIL [35] | (+)-CCCAATGACGAAGAGAGTATGAACA |
| (−)-CTCAAAATCATCTTTCTAACGAGCTGA | |
| TAP1 | (+)-TTATCCTGGATGATGCCACCAG |
| (−)-GAGAAGCACTGAGCG GGAGTA | |
| MxA | (+)-GCTACACACCGTGACGGATATGG |
| (−)-CGAGCTGGATTGGAAAGCCC | |
| GAPDH | (+)-GGTATCGTGGAAGGACTCATGAC |
| (−)-ATGCCAGTGAGCTTCCCGTTCAGC | |
| EMSA probes | |
| IRF-E | (+)-GGTTATTTAGGTTTCTCTTTCATTTC |
| (−)-GAAATGAAAGAGAACCTAAATAACCTTT | |
| IRF-E mutant | (+)-AAAGGTTATTTAGGTTTCTCAATCATTTC |
| (−)-GAAATGATTGAGAAACCTAAATAACCTTT | |
| siRNAs | |
| Control | (+)-UUCUCCGAACGUGUCACGUdtdt |
| (−)-ACGUGACACGUUCGGAGAAdtdt | |
| IRF-1 [36] | (+)-CCAAGAAC CAGAGAAAAGAdtdt |
| (−)-UCUUUUCUCUGGUUCUUGGdtdt | |
The table shows the sequence of primers used in the real-time PCR assays, probes in EMSA and siRNAs in gene silencing. Pimers or siRNAs that were previous reported was cited after the target name. (+), forward primers; (−), reverse primers.
2.4. Reporter assay
Dual luciferase assays were performed according to the manufacturer's protocols (Promega).
2.5. Western blotting
Huh-7 cells were extracted by using SDS sample buffer (50 mM Tris–HCl, pH 6.8, 2% w/v SDS, 10% glycerol 100 mM DTT and 0.1% w/v bromophenol blue). Equal amount of protein extracts were loaded on a 7.5 or 10% SDS-PAGE gel. After electrophoresis, protein was transferred onto a 0.2 μm nitrocellulose membrane (Protran™, Perkin Elmer) in transfer buffer (25 mM Tris, 180 mM glycine and 20% methanol, pH 8.3). The membranes were incubated with various antibodies followed by the addition of horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse IgG (Santa Cruz). Proteins were visualized by an ECL western blotting system (Western lightning™, Perkin Elmer).
2.6. Plasmids
BclG promoter (− 1000 to + 146) was amplified from human genomic DNA of a healthy donor by PCR and inserted into pGL3-Basic (Promega). Subsequent truncation or deletion mutants were done by further sub-cloning or fusion PCR. The sequences of primers used for cloning and mutation are available upon request. pRC/CMV/STAT1 Y701F was a gift from Dr. Darnell (Rockefeller University). pcDNA3-IRF1 was generously provided by Dr. Sakamoto (Tokyo Medical and Dental University). pISRE-luc, pGAS-luc were purchased from Stratagene, pRL-TK was from Promega.
2.7. Electrophoretic mobility shift assay (EMSA)
Huh-7 nuclear extracts were prepared using a nuclear extraction kit (Active Motif). 32P labeling of DNA probe and binding reaction was done using a gel shift kit from Promega (E3050) according to the instructions. Briefly, 10 μg/reaction nuclear extract was incubated at room temperature with 32P labeled IRF-E in BclG promoter in 1× binding buffer. The sequences of the probes are listed in Table 1 (middle panel). The antibodies to IRF-1, IRF-2, IRF-9, cold IRF-E or mutant IRF-E were added 20 min before 32P labeled probe was included in supershift or competition reactions. After 20 min of incubation with probe at room temperature, the complex was loaded on a 6% polyacrylamide gel in 0.5× TBE and subjected to electrophoresis. The gel was dried and analyzed using a phosphor-imaging instrument (Fuji Medical Systems).
2.8. siRNA
The sequences of siRNAs are listed in Table 1 (lower panel). All the siRNAs were synthesized by Genechem Co. Ltd. Transfection of siRNA was performed as described in the Oligofectamine™ user manual (Invitrogen).
3. Results
3.1. Promoter analysis of BclG identified IRF-1 and STAT1 binding sites
In a microarray analysis, a class of genes was shown to be hyper-activated when IFN-α/γ was co-administered (unpublished data). Subsequent real-time PCR confirmed that, at least, the transcription of BclG long form, TAP1, XAF1 and TRAIL was indeed enhanced (Table 2 ). BclG is a novel member of Bcl-2 family with Bcl-2 homology domains and is pro-apoptotic when over-expressed. BclG encodes two proteins by alternative splicing; the long form of BclG is expressed widely in various tissues whereas the short form is restricted in testis [16]. Since this gene had not been reported to be an ISG at the time of analysis, it was selected as a prototype for further examination. We cloned − 1000 to + 146 region of BclG promoter and inserted it into pGL3-Basic. In silico analysis of this region identified an IFN-gamma-activated site (GAS) and an Interferon regulatory factor element (IRF-E) close together between − 204 and − 139 (http: //mbs.cbrc.jp/research/db/TFSEARCH. html). A cAMP responsive element (CRE) was also identified downstream (Fig. 1A).
Table 2.
Expression profiles of BclG, XAF1, TRAIL and TAP1
| Gene | Copy number per 106 GAPDH |
|||
|---|---|---|---|---|
| Untreated | IFN-α | IFN-γ | IFN-α/γ | |
| BclG | 4.54 × 102 | 4.60 × 102 | 7.52 × 102 | 2.34 × 103 |
| XAF1 | 4.67 × 102 | 1.50 × 104 | 2.15 × 103 | 5.54 × 104 |
| TRAIL | 1.43 × 103 | 6.96 × 103 | 3.42 × 103 | 2.93 × 104 |
| TAP1 | 3.46 | 7.94 | 4.96 × 101 | 1.23 × 102 |
Fig. 1.
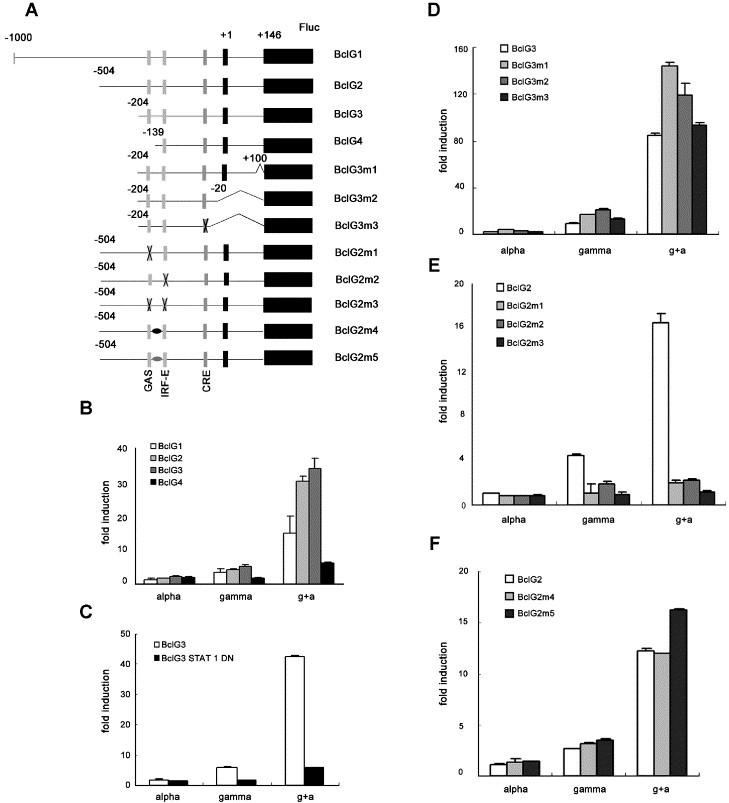
Mapping of DNA elements responsible for synergistic induction of BclG. (A) Schematic illustration of pGL3 reporter constructs containing BclG promoter and various mutants. (B–F) Huh-7 cells were transfected with one of the above plasmids and pRL-TK, 36 or 48 h later, IFN-α (50 U/ml) or/and IFN-γ (200 U/ml) was added. Luciferase activity was measured 6 h after stimulation. Relative fold induction of full length BclG promoter and 5′ proximal truncation mutants (B), BclG3 co-transfected with vector or STAT1 Y701F (C), 3′ truncation mutants (D), GAS and/or IRF-E deleted promoter (E) and two intervening sequenced changed mutants (F) was shown compared with untreated. The various columns are indicated on the graph.
The expression of full-length reporter construct showed clear synergism in IFN-α/γ co-treatment (Fig. 1B). The activation was totally dependent upon tyrosine phosphorylation of STAT1 as over-expression of a Y701F mutant made BclG3 unresponsive to interferon co-treatment (Fig. 1C). Truncation of 5′ proximal sequence did not alleviate but increased the synergism until the GAS element was deleted (BclG4, Fig. 1B). However, we could still detect about three fold induction in co-treatment suggesting that another factor is involved in − 139 to + 146 region. Further internal deletions on the downstream sequence until − 20 did not alter the expression pattern (Fig. 1D), deletion of CRE also had no effect. We therefore focused on the GAS and IRF-E elements. Deletions of GAS or IRF-E both significantly altered the responsiveness to IFN-α/γ treatment. Double deletion led to complete unresponsiveness to either cytokine (Fig. 1E). Since the GAS and IRF-E are very close with only 20 bp in between, we changed the intervening sequence into other two random sequences, however, no significant difference was observed (Fig. 1F). Thus, we conclude that IRF-E and GAS are both important for synergistic activation of BclG transcription.
3.2. Enhanced IRF-1 binding activity with IRF-E
Since previous results (Fig. 1) demonstrated indispensable role of IRF-E, we sought to find out which IRF binds to this element. Neither IRF-3 nor IRF-7 seemed to participate in BclG transcription as co-transfection of their dominant negative forms did not significantly alter its expression pattern (data not shown). However, when IRF-1 was co-expressed, the basal expression was greatly increased. Besides, a dose dependent phenomenon was also observed (Fig. 2A). Gel shift analysis suggested that over-expressed IRF-1 could bind to the IRF-E of BclG promoter (arrow A), addition of IRF-1 specific antibody effectively eliminated this complex (Fig. 2B).
Fig. 2.
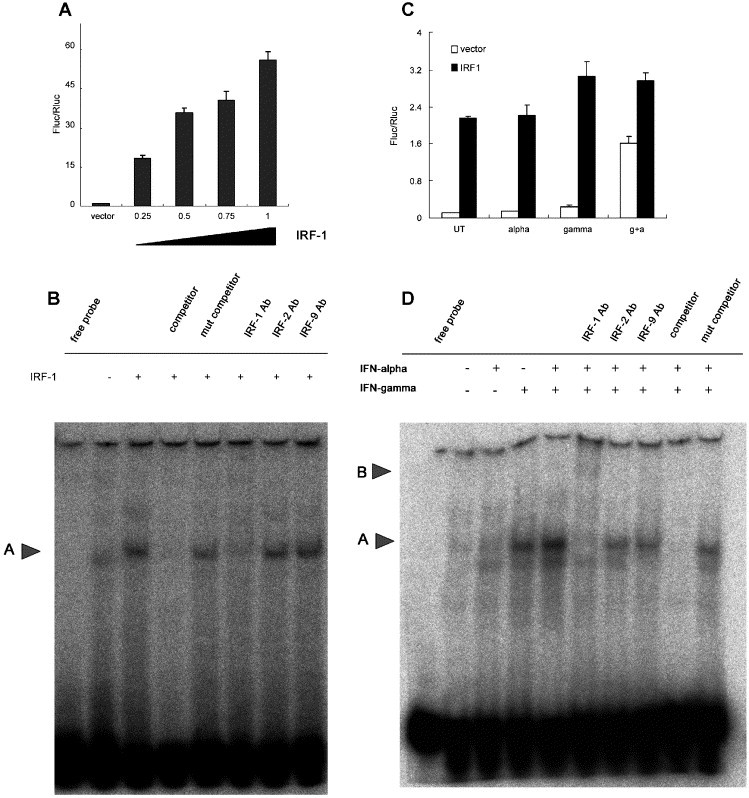
The binding activity of IRF-1 to IRF-E is enhanced after combined IFN treatment. (A) Increasing amount of wild type IRF-1 was co-transfected with BclG2 promoter reporter without stimuli, relative luciferase activity (Fluc/Rluc) was measured 48 h after. (B) Huh-7 cells were transfected with vector or pcDNA3-IRF-1, nuclear protein was extracted 48 h later. Gel shift analysis was performed as described in Materials and methods. Arrow A indicates shifted complex. (C) BclG2 were co-transfected with vector or pcDNA3-IRF-1, IFN-α (50 U/ml) or/and IFN-γ (200 U/ml) was added 48 h after transfection, luciferase activity was measured 6 h after. (D) Huh-7 nuclear extract was prepared 3 h after IFN-α (200 U/ml) and/or IFN-γ (1000 U/ml) stimulation and subjected to EMSA analysis. Arrow A indicates shifted complex and arrow B shows super-shifted complex.
To investigate whether IRF-1 was indeed responsible for the synergistic activation of BclG in physiological settings, we co-transfected BclG2 and IRF-1 and then treated the cells with IFN-α/γ (Fig. 2C). IFN-α did not significantly alter the reporter expression, whereas IFN-γ could slightly increase luciferase activity which supported the idea that other factors (e.g. STAT1) may be involved in the activation of BclG transcription in IFN-γ treatment. Co-expression of IRF-1 also abolished the enhanced reporter expression after IFN co-treatment which confirmed the critical role of IRF-1 in this process. To further verify that IRF-1 is the nuclear factor associated with IRF-E, we performed EMSA analysis by using IRF-E corresponding to − 132 to − 109 of BclG promoter as the probe. As shown in Fig. 2D, IFN-α/γ co-treatment led to an enhanced band shift (arrow A) compared with IFN-γ treatment. While addition of a specific IRF-1 antibody super-shifted the complex (arrow B), addition of antibody to IRF-2 and IRF-9 had no effect. The shift band was effectively inhibited by adding 100 fold cold probe but not by mutant oligo.
Western blot analysis of BclG confirmed the synergistic effect although slightly different compared with the transcription level (Fig. 3 ). When IFN-α was added, a marked decrease of BclG was observed and it is possible that BclG degradation was activated in IFN-α treatment since BclG was found to be linked to MNSF (monoclonal nonspecific suppressor factor), a ubiquitin homology protein [17]. Although IFN-γ treatment did not result in a significant increase of BclG protein, co-treatment clearly up-regulated its expression (Fig. 3). The different induction profiles in protein and mRNA level are presumably caused by the half-life of BclG and possibly by the different degradation efficiency in various stimulation contexts. As to IRF-1 level, minimal expression was observed in untreated or IFN-α treated cells, but after IFN-γ treatment, an obvious induction was seen as described in previous reports [18]. In contrast to the enhanced binding activity in EMSA experiment, no significant increase of IRF-1 was observed in IFN-α/γ co-treatment which is still not well understood. IRF-2 level remained constant in all treatment groups.
Fig. 3.
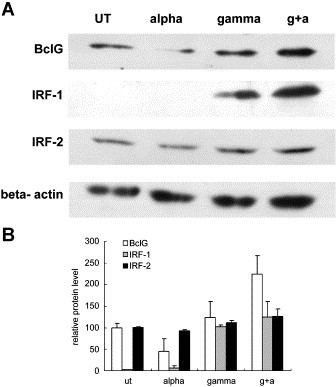
The expression of BclG is enhanced in IFN-α and IFN-γ co-treatment. (A) Western blot analysis of BclG, IRF-1, IRF-2 and β-actin was performed on Huh-7 cell stimulated by IFN-α (200 U/ml) or/and IFN-γ (1000 U/ml) for 6 h. (B) Densitometric quantification and statistical analysis of protein expression.
3.3. Knock down of IRF-1 expression by siRNA efficiently suppressed gene enhancement by IFN-α/γ co-treatment
To further elucidate the role of IRF-1 in altered transcription profile and synergistic antiviral activity after IFN co-treatment, synthetic siRNA directed against IRF-1 was used to abrogate its activation. IRF-1 induction after IFN-γ treatment was significantly inhibited when IRF-1 siRNA was transfected (Fig. 4 ) while IRF-2 expression unchanged.
Fig. 4.
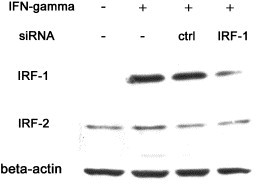
Knock down of IRF-1 protein expression by short interfering RNA. Control or IRF-1 siRNA (200 nM) was transfected into Huh-7 cells, 24 h later, culture medium was refreshed with or without IFN-γ (200 U/ml). Cell extracts were prepared 6 h after stimulation. Western blot analysis was done on IRF-1, IRF-2 and β-actin.
The effect of IRF-1 siRNA on synergistically enhanced genes was further assessed by quantitative RT-PCR. As expected, enhanced mRNA level of BclG was significantly reduced (3.82 × 103 copy/106 GAPDH versus 9.19 × 102 after IFN-α/γ treatment, p < 0.05). Similar expression profile changes were observed in TAP1 (1.23 × 102 versus 4.34 × 101, p < 0.05), XAF1 (3.67 × 104 versus 1.42 × 104 , p < 0.05) and TRAIL (9.04 × 103 versus 6.70 × 103, p < 0.05) (Fig. 5A). In silico transcription factor binding site analysis on the promoter of XAF1 and TRAIL also identified putative IRF-1 binding sites (Fig. 5B). To ascertain the specificity of our IRF-1 siRNA, the expression of MxA, a well-known IRF-1 independent ISG, was also quantified. Indeed, the transfection of IRF-1 siRNA did not affect the mRNA level of MxA (1.30 × 105 versus 1.57 × 105, p > 0.05) after IFN combination treatment (Fig. 5A). These data strongly support the idea that IRF-1 activation is a general switch leading to enhanced gene expression in a subset of ISGs. Interestingly, a previous report has shown that IRF-1 is required for TRAIL induction by both retinoic acid and IFN-γ [19]. As to TAP1, a GAS and IRF-E arrangement in its promoter similar to that of BclG has been indicated and that both elements were crucial for its induction after IFN-γ treatment [20].
Fig. 5.
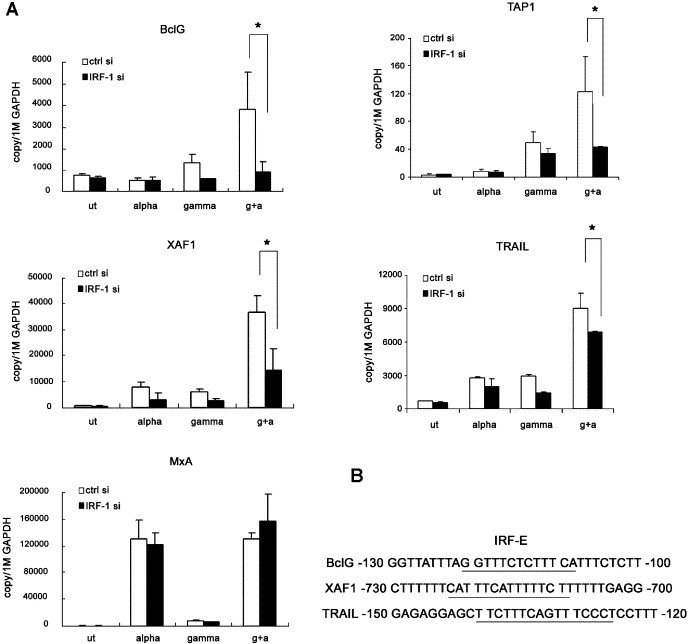
Inhibition of ISG hyper-activation by IRF-1 siRNA. (A) Huh-7 cells were transfected with control or IRF-1 siRNA, 24 h later IFN-α (200 U/ml) and/or IFN-γ (1000 U/ml) was added for 6 h, mRNA level of BclG, XAF1, TRAIL, TAP1 and MxA was quantified by Real time PCR. * indicates p < 0.05 by a Student's t test. (B) In silico analysis of promoter of BclG, XAF1 and TRAIL reveals the existence of IRF-E.
3.4. The phosphorylation status and transcriptional potency of STAT1 after IFN-α/γ co-treatment
Since there has been evidence showing that IFN-γ priming could amplify IFN-α induced STAT1 activation in human macrophage [21], we checked STAT1 phosphorylation in Huh-7 cells after 30 min of IFN-α/γ treatment. Tyrosine phosphorylation of STAT1 could be detected in either IFN-α or IFN-γ mono-treatment although with differing intensities, in accordance with published data, STAT1 hyper-phosphorylation was readily observed after co-treatment (Fig. 6A). As STAT1 plays a pivotal role in transcription activation in both IFN-α and IFN-γ signaling, we monitored the induction of downstream interferon-stimulated response element (ISRE) and GAS reporter expression. IFN-γ treatment led to a lagged ISRE activation compared with IFN-α, however, the combined cytokines did not enhance ISRE activity (Fig. 6B) suggesting that interferon stimulated gene factor 3 (ISGF3) complex formation was not elevated. In the GAS assay, IFN-α/γ co-treatment resulted in about two fold activation compared with IFN-γ treatment which suggested increased STAT1 homo-dimer formation (Fig. 6C).
Fig. 6.
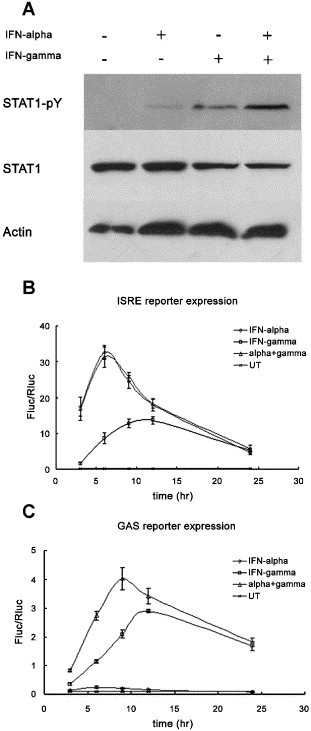
Tyrosine phosphorylation status and trans-activation potency of STAT1. Huh-7 cells were treated with IFN-α (100 U/ml) and/or IFN-γ (200 U/ml), cell extracts were prepared 30 min after. Western blotting of tyrosine phosphorylated STAT1, total STAT1 and β-actin was shown (A). (B) And (C) Huh-7 cells were co-transfected with ISRE or GAS reporter and pRL-TK, 24 h later, IFN-α (200 U/ml) and/or IFN-γ (1000 U/ml) was added. Cells were lysed at different time points after stimulation and relative luciferase activity was measured (Fluc/Rluc).
4. Discussion
Basic research and clinical trials have repeatedly demonstrated that type I and type II IFN are inter-dependent. Signaling by IFN-γ is shown to be dependent on the IFN-α/β receptor components in knock-out mice models [22]. A recent clinical trial using IFN-alfacon and IFN-γ 1b to treat HCV patients unresponsive to PEG-IFN and ribavirin treatment has shown about 35% sustained virological response which was remarkably higher than that in prolonged standard treatment [23]. The underlying mechanism is believed to be two fold: (1) The innate antiviral effects of IFN-α may be enhanced by addition of type II IFN, (2) The adaptive immunity is re-tuned to Th1 response thus facilitating virus eradication by specific CTLs (cytotoxic T lymphocyte).
In this study, we focused on the immediate antiviral activity after IFN combination treatment in a host cell-line which supports HCV replication. A cluster of genes (BclG, TRAIL, XAF1 and TAP1) was shown to be transcriptionally enhanced after IFN-α/γ co-treatment, subsequent promoter analysis of BclG revealed IRF-1 and STAT1 as crucial regulators. Interestingly, a similar GAS and IRF-E arrangement in the promoter of gp91(phox) has also been documented, which allowed rapid induction of gp91 expression by IFN-γ treatment in U937 cells [24]. Nevertheless, due to its strict tissue specific (myeloid) expression pattern [25], gp91(phox) could not be detected in Huh-7 cells (unpublished data).
Although increased STAT1 phosphorylation was demonstrated, we found that only the GAS but not ISRE mediated transcription was enhanced suggesting that STAT1 homo-dimer rather than ISGF3 formation was preferably induced. As to IRF-1, it is dramatically upregulated upon some virus infection or IFN stimulation [18]. Other observations suggest that IRF-1 functions as a regulator of cellular response to IFNs by affecting a set of ISGs [18]. Interestingly, in this study, the DNA binding activity of IRF-1 was shown to be enhanced after IFN-α/γ co-treatment. Further analysis supports the general role of IRF-1 in regulating this cluster of genes. Indeed, basal expression level of IRF-1 was found to be significantly lower in cells harboring the replicon [26] indicating that IRF-1 could greatly hamper the replication of HCV, and a variant clone of HCV replicon could block the DNA binding activity of IRF-1 and suppress the expression of some IRF-1 dependent ISGs [27].
Intriguingly, based on previous reports, BclG, XAF1 and TRAIL are all thought to be associated with apoptosis [16], [28], [29], [30]. BclG can be conjugated with MNSF, a 14.5 kDa protein with 36% identity with ubiquitin, the post-translational modification of BclG might be implicated in T-cell activation [17]. In our preliminary study, BclG long form did not show significant pro-apoptotic effect, nor was there any direct anti-HCV activity at least in Huh-7 cells (data not shown). Given that its molecular function is still not well defined, BclG merits further investigations in anti-viral or pro-apoptotic activities. TRAIL, a member of the TNF family, has selective apoptosis activity in cancer cells while normal cells are largely insensitive [19]. TRAIL−/− mice display no overt phenotype but an increased susceptibility to tumor initiation and metastasis [28]. X-linked inhibitor of apoptosis associated factor-1 (XAF1) was shown to be associated with XIAP; expression of XAF1 triggers a redistribution of XIAP from cytosol to the nucleus and antagonizes XIAP activities [29]. XAF1 protein induction is observed predominantly in melanoma cell lines sensitive to pro-apoptotic effects of IFN-β. Cells constitutively expressing XAF1 were more sensitive to TRAIL induced apoptosis [30].
The similar expression kinetics and co-operative pro-apoptotic activity indicated that this cluster of enhanced genes is finely regulated to fulfill a specific task, e.g. immuno-surveillance of cancer cells as reported [31]. However, their roles in control of virus replication cannot be excluded. Indeed, it has been proposed that a novel antiviral function of type I IFN is the selective induction of apoptosis in virally infected cells [32], [33], [34]. On the other hand, viruses also encode various genes that can antagonize the pro-apoptotic signaling. For example, NS5A protein of HCV is well known for its anti-apoptotic activity although the exact mechanism is still controversial [35], [36]; EBV (Epstein–Barr virus)-encoded poly(A) RNAs (EBERs) exhibit striking resistance of IFN-α induced apoptosis in Burkitt's lymphoma cell lines [37].
Overall, our data provide the evidence that hyper-induction of subclass of ISGs is mainly controlled by hyper-activated IRF-1 and elevated STAT1 dimer formation when IFN-α and IFN-γ are co-administered. Although only four enhanced genes were unequivocally identified in this study, we believe that many other genes of this cluster are yet to be found. Further investigations are needed to expand this cluster and elucidate the functions of its members as some of them may play important roles in the innate antiviral activity of interferon. We are also aware of the fact that the enhanced IRF-1 trans-activation capacity, as shown in EMSA, does not correlate well with its protein expression level. Intensive analysis is being done to elucidate the mechanism underlying this phenomenon.
Acknowledgements
We thank Dr. Reena (Shanghai public health center) for helpful discussions on our manuscript. This work was supported by Chinese State Basic Research Foundation Grant (2005CB522902), National Natural Science Fund for distinguished scholars (30425041) and Shanghai Sci & Tech Research Program (04dz19208,05xd14001).
References
- 1.Stark G.R., Kerr I.M., Williams B.R., Silverman R.H., Schreiber R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 2.Platanias L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev., Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 3.Kroger A., Koster M., Schroeder K., Hauser H., Mueller P.P. Activities of IRF-1. J. Interferon Cytokine Res. 2002;22:5–14. doi: 10.1089/107999002753452610. [DOI] [PubMed] [Google Scholar]
- 4.Feld J.J., Hoofnagle J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature. 2005;436:967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- 5.Lee W.M. Hepatitis B virus infection. N. Engl. J. Med. 1997;337:1733–1745. doi: 10.1056/NEJM199712113372406. [DOI] [PubMed] [Google Scholar]
- 6.Guidotti L.G., Chisari F.V. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu. Rev. Immunol. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- 7.Chesler D.A., Reiss C.S. The role of IFN-gamma in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev. 2002;13:441–454. doi: 10.1016/s1359-6101(02)00044-8. [DOI] [PubMed] [Google Scholar]
- 8.Kakumu S., Ishikawa T., Mizokami M., Orido E., Yoshioka K., Wakita T., Yamamoto M. Treatment with human gamma interferon of chronic hepatitis B: comparative study with alpha interferon. J. Med. Virol. 1991;35:32–37. doi: 10.1002/jmv.1890350108. [DOI] [PubMed] [Google Scholar]
- 9.Sainz B., Jr., Halford W.P. Alpha/Beta interferon and gamma interferon synergize to inhibit the replication of herpes simplex virus type 1. J. Virol. 2002;76:11541–11550. doi: 10.1128/JVI.76.22.11541-11550.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sainz B., Jr., Mossel E.C., Peters C.J., Garry R.F. Interferon-beta and interferon-gamma synergistically inhibit the replication of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) Virology. 2004;329:11–17. doi: 10.1016/j.virol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sainz B., Jr., Lamarca H.L., Garry R.F., Morris C.A. Synergistic inhibition of human cytomegalovirus replication by interferon-alpha/beta and interferon-gamma. Virol. J. 2005;2:14. doi: 10.1186/1743-422X-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuchizaki U., Kaneko S., Nakamoto Y., Sugiyama Y., Imagawa K., Kikuchi M., Kobayashi K. Synergistic antiviral effect of a combination of mouse interferon-alpha and interferon-gamma on mouse hepatitis virus. J. Med. Virol. 2003;69:188–194. doi: 10.1002/jmv.10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larkin J., Jin L., Farmen M., Venable D., Huang Y., Tan S.L., Glass J.I. Synergistic antiviral activity of human interferon combinations in the hepatitis C virus replicon system. J. Interferon Cytokine Res. 2003;23:247–257. doi: 10.1089/107999003321829962. [DOI] [PubMed] [Google Scholar]
- 14.Okuse C., Rinaudo J.A., Farrar K., Wells F., Korba B.E. Enhancement of antiviral activity against hepatitis C virus in vitro by interferon combination therapy. Antivir. Res. 2005;65:23–34. doi: 10.1016/j.antiviral.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 15.Katayama K., Kasahara A., Sasaki Y., Kashiwagi T., Naito M., Masuzawa M., Katoh M., Yoshihara H., Kamada T., Mukuda T., Hijioka T., Hori M., Hayashi N. Immunological response to interferon-gamma priming prior to interferon-alpha treatment in refractory chronic hepatitis C in relation to viral clearance. J. Viral Hepatitis. 2001;8:180–185. doi: 10.1046/j.1365-2893.2001.00274.x. [DOI] [PubMed] [Google Scholar]
- 16.Guo B., Godzik A., Reed J.C. Bcl-G, a novel pro-apoptotic member of the Bcl-2 family. J. Biol. Chem. 2001;276:2780–2785. doi: 10.1074/jbc.M005889200. [DOI] [PubMed] [Google Scholar]
- 17.Nakamura M., Tanigawa Y. Characterization of ubiquitin-like polypeptide acceptor protein, a novel pro-apoptotic member of the Bcl2 family. Eur. J. Biochem. 2003;270:4052–4058. doi: 10.1046/j.1432-1033.2003.03790.x. [DOI] [PubMed] [Google Scholar]
- 18.Taniguchi T., Ogasawara K., Takaoka A., Tanaka N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 2001;19:623–655. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- 19.Clarke N., Jimenez-Lara A.M., Voltz E., Gronemeyer H. Tumor suppressor IRF-1 mediates retinoid and interferon anticancer signaling to death ligand TRAIL. EMBO J. 2004;23:3051–3060. doi: 10.1038/sj.emboj.7600302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brucet M., Marques L., Sebastian C., Lloberas J., Celada A. Regulation of murine Tap1 and Lmp2 genes in macrophages by interferon gamma is mediated by STAT1 and IRF-1. Genes Immun. 2004;5:26–35. doi: 10.1038/sj.gene.6364035. [DOI] [PubMed] [Google Scholar]
- 21.Tassiulas I., Hu X., Ho H., Kashyap Y., Paik P., Hu Y., Lowell C.A., Ivashkiv L.B. Amplification of IFN-alpha-induced STAT1 activation and inflammatory function by Syk and ITAM-containing adaptors. Nat. Immunol. 2004;5:1181–1189. doi: 10.1038/ni1126. [DOI] [PubMed] [Google Scholar]
- 22.Takaoka A., Mitani Y., Suemori H., Sato M., Yokochi T., Noguchi S., Tanaka N., Taniguchi T. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science. 2000;288:2357–2360. doi: 10.1126/science.288.5475.2357. [DOI] [PubMed] [Google Scholar]
- 23.Blatt L. Combination Interferon (Type I and Type II) therapy for chronic hepatitis C. The Annual Meeting of International Society for Interferon and Cytokine Research. 2005;15:111. (Suppl.) [Google Scholar]
- 24.Kumatori A., Yang D., Suzuki S., Nakamura M. Cooperation of STAT-1 and IRF-1 in interferon-gamma-induced transcription of the gp91(phox) gene. J. Biol. Chem. 2002;277:9103–9111. doi: 10.1074/jbc.M109803200. [DOI] [PubMed] [Google Scholar]
- 25.Royer-Pokora B., Kunkel L.M., Monaco A.P., Goff S.C., Newburger P.E., Baehner R.L., Cole F.S., Curnutte J.T., Orkin S.H. Cloning the gene for an inherited human disorder–chronic granulomatous disease–on the basis of its chromosomal location. Nature. 1986;322:32–38. doi: 10.1038/322032a0. [DOI] [PubMed] [Google Scholar]
- 26.Kanazawa N., Kurosaki M., Sakamoto N., Enomoto N., Itsui Y., Yamashiro T., Tanabe Y., Maekawa S., Nakagawa M., Chen C.H., Kakinuma S., Oshima S., Nakamura T., Kato T., Wakita T., Watanabe M. Regulation of hepatitis C virus replication by interferon regulatory factor 1. J. Virol. 2004;78:9713–9720. doi: 10.1128/JVI.78.18.9713-9720.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pflugheber J., Fredericksen B., Sumpter R., Jr., Wang C., Ware F., Sodora D.L., Gale M., Jr. Regulation of PKR and IRF-1 during hepatitis C virus RNA replication. Proc. Natl. Acad. Sci. U. S. A. 2002;99:4650–4655. doi: 10.1073/pnas.062055699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cretney E., Takeda K., Yagita H., Glaccum M., Peschon J.J., Smyth M.J. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J. Immunol. 2002;168:1356–1361. doi: 10.4049/jimmunol.168.3.1356. [DOI] [PubMed] [Google Scholar]
- 29.Liston P., Fong W.G., Kelly N.L., Toji S., Miyazaki T., Conte D., Tamai K., Craig C.G., McBurney M.W., Korneluk R.G. Identification of XAF1 as an antagonist of XIAP anti-Caspase activity. Nat. Cell Biol. 2001;3:128–133. doi: 10.1038/35055027. [DOI] [PubMed] [Google Scholar]
- 30.Leaman D.W., Chawla-Sarkar M., Vyas K., Reheman M., Tamai K., Toji S., Borden E.C. Identification of X-linked inhibitor of apoptosis-associated factor-1 as an interferon-stimulated gene that augments TRAIL Apo2L-induced apoptosis. J. Biol. Chem. 2002;277:28504–28511. doi: 10.1074/jbc.M204851200. [DOI] [PubMed] [Google Scholar]
- 31.Chawla-Sarkar M., Lindner D.J., Liu Y.F., Williams B.R., Sen G.C., Silverman R.H., Borden E.C. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237–249. doi: 10.1023/a:1023668705040. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka N., Sato M., Lamphier M.S., Nozawa H., Oda E., Noguchi S., Schreiber R.D., Tsujimoto Y., Taniguchi T. Type I interferons are essential mediators of apoptotic death in virally infected cells. Genes Cells. 1998;3:29–37. doi: 10.1046/j.1365-2443.1998.00164.x. [DOI] [PubMed] [Google Scholar]
- 33.Balachandran S., Roberts P.C., Kipperman T., Bhalla K.N., Compans R.W., Archer D.R., Barber G.N. Alpha/beta interferons potentiate virus-induced apoptosis through activation of the FADD/Caspase-8 death signaling pathway. J. Virol. 2000;74:1513–1523. doi: 10.1128/jvi.74.3.1513-1523.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barber G.N. Host defense, viruses and apoptosis. Cell Death Differ. 2001;8:113–126. doi: 10.1038/sj.cdd.4400823. [DOI] [PubMed] [Google Scholar]
- 35.Gale M., Jr., Kwieciszewski B., Dossett M., Nakao H., Katze M.G. Antiapoptotic and oncogenic potentials of hepatitis C virus are linked to interferon resistance by viral repression of the PKR protein kinase. J. Virol. 1999;73:6506–6516. doi: 10.1128/jvi.73.8.6506-6516.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Street A., Macdonald A., Crowder K., Harris M. The Hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J. Biol. Chem. 2004;279:12232–12241. doi: 10.1074/jbc.M312245200. [DOI] [PubMed] [Google Scholar]
- 37.Nanbo A., Inoue K., Adachi-Takasawa K., Takada K. Epstein–Barr virus RNA confers resistance to interferon-alpha-induced apoptosis in Burkitt's lymphoma. EMBO J. 2002;21:954–965. doi: 10.1093/emboj/21.5.954. [DOI] [PMC free article] [PubMed] [Google Scholar]