

Abstract
High throughput screening, increased accuracy and the coupling of real-time quantitative PCR (Q-PCR) to robotic set-up systems are beginning to revolutionise biotechnology. Applications of Q-PCR within biotechnology are discussed with particular emphasis on the following areas of biosafety and genetic stability testing: (a) determination of the biodistribution of gene therapy vectors in animals; (b) quantification of the residual DNA in final product therapeutics; (c) detection of viral and bacterial nucleic acid in contaminated cell banks and final products; (d) quantification of the level of virus removal in process validation viral clearance studies; (e) specific detection of retroviral RT activity in vaccines with high sensitivity; and (f) transgene copy number determination for monitoring genetic stability during production. Methods employed for Q-PCR assay validation as required in ICH Topic Q2A Validation of Analytical Methods: Definitions and Terminology (1st June 1995) are also reviewed.
Keywords: Real-time, Q-PCR, Biosafety, Genetic stability, Testing, Biotechnology
1. Introduction
Real-time quantitative polymerase chain reaction (Q-PCR) has revolutionised the detection and quantification of nucleic acid and retroviral reverse transcriptase because of fast throughput capability (Morris et al., 1996, Lovatt et al., 1999, Nitsche et al., 1999). The incorporation of this technology into gene expression, copy number determination, gene/cell therapy biodistribution and pathogen detection studies is of benefit to the discovery and marketing of novel therapeutics and biopharmaceutical products.
Q-PCR studies can be performed using an ABI PRISM™ sequence detector 7700, 7900HT, or 5700 machine. This technology can precisely quantify the amount of nucleic acid target sequence in DNA or RNA extracted from a wide variety of samples including: animal tissues; final products; cell banks; chromatography eluate; and bulk harvest material. During amplification with target-specific oligonucleotide primers, a fluorogenic oligonucleotide probe with both reporter dye (FAM, VIC, TET, JOE, or HEX) and quencher dye (TAMRA) attached, anneals specifically to the amplified product between the amplimers. The detection reaction utilises the 5′ exonuclease activity of AmpliTaq Gold™ DNA polymerase to cleave the reporter dye from the probe, which results in an increased fluorescence when target is present (Fig. 1 a). A sequence detector instrument calculates a reporter dye value (R n) for each sample during each cycle of amplification. The value of R n, or the normalised reporter signal, represents the fluorescence of the reporter dye divided by a passive reference dye. During PCR, R n increases as the amplicon copy number increases until the reaction approaches a plateau. The value ΔR n represents the normalised reporter signal (R n) minus the baseline signal established in the first few cycles of PCR. Like R n, ΔR n increases during PCR as the amplicon copy number increases until the reaction reaches a plateau (Fig. 1b). The use of a sequence detector allows measurement of the amplified product in direct proportion to the increase in fluorescence emission continuously during PCR amplification. The threshold cycle (C T) value is calculated in real time and is defined as the PCR cycle number at which an increase in reporter fluorescence above the baseline signal can first be detected. Therefore the cycling point of a given amplification where a significant increase in the fluorescence signal associated with exponential growth of the PCR product is first detected, represents the C T value of that amplification (Fig. 1b). A linear relationship exists between the C T value and starting quantity of target molecule, allowing the construction of a standard curve. The greater the amount of target copies in the reaction, the lower the C T value (Fig. 2 ) (Heid et al., 1996, Livak and Perkin Elmer, 1998).
Fig. 1.
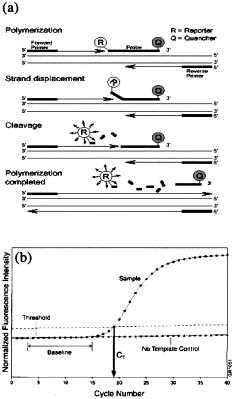
The 5′ Exonuclease Assay and the Real-Time Q-PCR Amplification Plot. (a) Diagram showing cleavage of the FAM reporter dye on the TaqMan probe during PCR amplification. (b) Amplification plot showing exponential increase in FAM reporter dye and the calculation of the CT value.
Fig. 2.
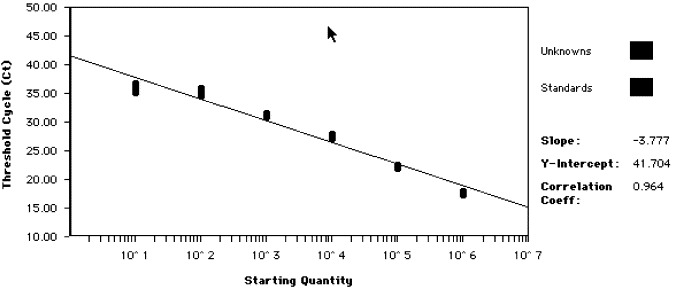
Standard curve showing linear relationship between CT and starting copy number. Serial 10-fold dilutions in the range of 106–10 copies of viral target DNA were analysed as standards. The slope, y-intercept and correlation co-efficient of the generated standard curve are shown on the right hand side.
There are many advantages of real time Q-PCR over conventional end-point PCR. For example, real-time Q-PCR is performed in a closed reaction system and eliminates the post-PCR processing of PCR products, thereby increasing throughput and reducing the chances of carryover contamination. Moreover, the TaqMan Q-PCR system incorporates a chemical system termed AmpErase to avoid amplicon contamination. Like conventional qualitative PCR assays, Q-PCR assays should operate under a strict contamination control system (i.e. separate air spaces for all reactions) to give increased assurance of re-avoiding cross-contamination. Because of the increased throughput of real time Q-PCR, a higher assay sensitivity can be achieved with relative ease. Multiple replicated reactions can be used to increase the chance of observing a low level virus below the limit of detection (Heid et al., 1996, Livak and Perkin Elmer, 1998). In addition, coupling real-time Q-PCR to robotic extraction and PCR set-up systems will increase throughput further and minimise human error.
C T values are also less sensitive than end-point reactions to the effects of inhibitors. Compared to endpoint PCR, real time Q-PCR offers streamline assay development, reproducible results and a large dynamic range. More importantly, if the sample contains viral sequences, then the amount of target nucleic acid molecule present is given. This will provide an indication of the level of viral contamination present, which could be useful in deciding what further action to take. If, for example, the PCR-positive sample was from bulk harvest material, further process validation could be used to determine if the sample still gave a positive result (Heid et al., 1996, Livak and Perkin Elmer, 1998).
2. Assay development and validation
2.1. Primer design, optimisation and specificity
Primer design for real-time TaqMan PCR can be performed using the Primer Express™ Software on the ABI 5700, 7700, or 7900HT following PE Applied Biosystems recommendations. One of the first steps in the identification of the nucleic acid target is comparison of the target sequence or amplicon using multiple sequence alignments with DNA sequence databanks. This allows an assessment of potential cross-reaction with other related sequences that may produce problems in the Q-PCR testing. Determination of primer specificity is of utmost importance when performing pathogen detection studies. False positive reactions, although uncommon, can lead to major problems in the release of biotechnology products for clinical trial. Therefore, assay specificity studies are critical during validation to ensure confidence in reporting positive results. The Q-PCR assay should be able to discriminate between the target and nucleic acids that are likely to be present in test material.
For detection of viruses such as Human immunodeficiency virus (HIV), and other viruses which show sequence variation, it is extremely important to ensure the primers and probe are targeted to a conserved region of the viral genome. Selection of a region with highly conserved sequences facilitates the design of degenerate amplimers and probes, allowing detection of the maximum amount of variants and strains possible (Shown in Fig. 3 for Bovine/Porcine circovirus). In addition, the stability of the target sequence should be considered, in an attempt to ensure that the amplicon does not reside on an unstable genetic element that may be a hot spot for deletions, such as those that occur in the Polyoma virus (Bendig and Folk, 1979, Persing, 1993).
Fig. 3.
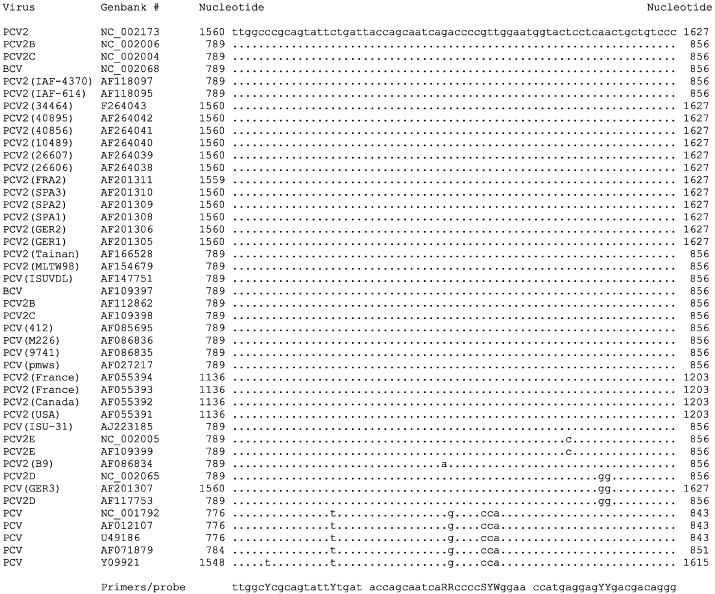
5′–3′ multiple sequence alignment of a conserved region of the Bovine circovirus and Porcine circovirus rep region. TaqMan primers and probe are designed as degenerated oligonucleotides. Degenerated nucleotides are: Y=CT; R=A+G; S=G+C; W=A+T.
To ensure technical reliability, assay optimisation studies are essential, together with validation to ICH guidelines (ICH Topic Q2A, 1995, ICH Topic Q2B, 1997, Cope, 2000). A minimum of two operators should perform specificity, linearity, detection limit, accuracy and repeatability studies. Each operator should determine these assay parameters at least once, using a minimum of five concentrations over the range of the assay. The importance of optimisation and validation is highlighted when working with non-validated rapidly developed Q-PCR assays. High background fluorescent signals leading to reduced assay sensitivity and abnormal amplification signals have been observed in assays before assay optimisation and validation, further supporting the need for assay validation. More importantly, a less experienced operator may not be able to distinguish between genuine and abnormal amplification signals during testing with a non-optimised, non-validated assay.
Primer optimisation is carried out using an array of primer concentrations with a constant amount of probe and target template. Because high ΔR n values reflect increased levels of amplification product and efficient reactions, the combination of forward and reverse primer showing the maximum ΔR n, is selected. Probe optimisation is performed using the optimised primer conditions, whereby the concentration of probe is varied. The probe concentration showing the lowest C T value is chosen as optimal, and reflects the most efficient reaction conditions.
2.2. Linearity, detection limit, precision and reproducibility
Assay linearity, detection limit and precision are assessed using eight replicated reactions over a range of five concentrations. To assess reproducibility, the correlation coefficient (r 2), y-intercept and slope of the regression line should be calculated using a minimum of two independent operators on the ABI 7700, 7900HT, or 5700 systems. The minimum accepted value for the linear correlation coefficient at the P=0.05 statistical level when using four standards is 0.88 (Wardlaw, 1987), and Q-PCR assays will normally show values of r 2>0.90.
Two major complicating factors in establishing the detection limit for a real-time PCR assay using biological material are: (1) the number of target nucleic molecules present in infected cells and clinical samples, or electron microscopy counted viral stocks, may be variable; (2) there may be no culture methods available for the target organism. Therefore, the limit of detection is firstly validated using purified target molecules in the form of recombinant plasmid DNA or recombinant RNA. The assay limit of detection with 95% confidence, is defined as the lowest amount of target producing a confidence interval that is significantly lower than that obtained from eight negative controls and no template controls. Within the group of eight replicates containing target levels at the limit of detection, there should be no failed reactions, and all replicates should produce amplification signals with a C T value less than 40. Assay limit of detection is routinely determined as 10–100 nucleic acid copies in a background of 200 000 cellular genomes (for DNA targets) or 100 ng of cellular RNA (for RNA targets). Ultimate assessment of assay reproducibility requires the positive cut-off point to be determined. The positive cut-off level is the minimum number of target molecules that are detected in 95% of test runs. The positive cut-off point of an assay can be measured over a period of 12 months using different batches of reagents and several different operators.
2.3. Nucleic acid extraction validation
The extraction of viral nucleic acids is widely used in diagnostic and research laboratories. However, the suitability or yield of nucleic acid from extraction procedures can vary depending on the nucleic acid and the biological material (Kok et al., 2000). Therefore, to ensure efficient extraction of target nucleic acid, the procedure should be qualified for the particular target and biological material to be used. Since Q-PCR allows the quantification of a nucleic acid target molecule, it can be used to determine the percentage recovery of the viral or cellular DNA target achieved during the extraction procedure (Table 1 ). To assess further the performance of the extraction procedures, the extracted test nucleic acid can be spiked with target molecules to determine the percentage of PCR inhibition. The calculated copy number of the test sample is subsequently adjusted, taking into account the amount lost in extraction and the estimated level of inhibition. PCR inhibition is particularly important to assess when analysing many different biological materials that may contain various levels of inhibitory substances.
Table 1.
Recovery of viral and cellular nucleic acid using Qiagen extraction methodology
| DNA or RNA spike | Recovery |
|---|---|
| BVDV 1000 RNA copies | 11% |
| Enterovirus (10 000 TCID50) | 14% |
| MLV 100 copies | 41% |
| Porcine CMV 549 copies | 43% |
| Polyoma virus 5000 copies | 40% |
| HPV-18 DNA 1000 copies | 87% |
| Host cell DNA 78 pg per | 22% |
| 0.5 ml Therapeutic dose |
The Qiagen RNeasy mini kit was used to extract mammalian cells spiked with 10 000 TCID50 Poliovirus RNA equivalents. The QIAmp viral RNA kit was used to extract cell free systems containing 1000 and 100 RNA copies of BVDV and MLV, respectively. The Qiagen DNA mini kit was used to extract mammalian cells spiked with Porcine CMV, HPV-18 and Polyoma DNA. Approximately 15 copies (78 pg) of mammalian host cell DNA were extracted using the Qiagen mini kit containing carrier molecule.
When extracting nucleic acid from cell free samples, especially final products, it is important to include a carrier molecule in the procedure. This allows maximum recovery of low levels of nucleic acid and is critical for ensuring maximum sensitivity of the PCR assay. This is essential when screening vaccines for contaminating host cell DNA. As determined by Q-PCR, typical extraction recoveries of 22% and greater can be achieved in 78 pg of human cell line DNA per 0.5-ml therapeutic dose, using the Qiagen DNA mini kit (Table 1). Similar results are obtained when using Q-PCR assays for primate, rodent, chicken and porcine cellular DNA.
Inhibitory factors which interfere with PCR are most apparent when performing extraction of viral or plasmid nucleic acid from animal tissues and secretions. Substances such as: heparin; urea and haemoglobin; ethanol; and iso-propanol will inhibit PCR and affect extraction efficiency (Qiagen, 1999). The use of methods which remove these molecules are of utmost importance when screening tissue during gene and cell therapy biodistribution studies. Therefore in these studies, the use of optimised high throughput nucleic acid extraction systems and the examination of the recovery of 10–100 copies of target nucleic acid from the highest amount of tissue possible should be determined. Recovery in percentage values of a plasmid DNA per 100 μg of animal tissue are shown (Table 2 ).
Table 2.
Recovery of 10–100 copies of viral nucleic acid target per 100 microgram of animal tissue extracted with the Qiagen DNA mini kit (tissue and blood protocol)
| Animal tissue | Recovery |
|---|---|
| Brain | 60–90% |
| Kidney | 70–95% |
| Spleen | 68–80% |
| Lung | 50–75% |
| Gonads | 60–90% |
| Muscle | 10–40% |
| Blood | 10–20% |
| Heart | 70–100% |
| Lymph node | 30–90% |
| Liver | 65–100% |
2.4. Data analysis
After cycling is complete, data are analysed in real time and the C T values of each replicate observed and recorded. The standard deviations and confidence limits of C T values of the replicates are calculated to detect similarities and significant differences between groups of replicates.
2.4.1. Positive test
Firstly, it is not impossible that a positive result may derive from nucleic acid of previously unrecognised origin that shows non-specific interactions, due to sequence homology with the amplification target used in detection. In a valid assay, a positive result for the test sample is considered to have occurred when the following four conditions are met when testing the sample with two independent amplimer sets to the specified target. (1) There are significant amplification signals present in the test sample reactions. (2) There are no amplification signals present in the sentinel controls. (3) There are no amplification signals present in the negative controls. (4) There are significant amplification signals present in the test article spiked at the limit of detection.
Detection of the target sequence by PCR in any of the sentinel or negative controls invalidates the assay and is indicative of contamination. In a positive test with confidence, analysis of data results in the C T values of the test sample, spiked test sample and positive control samples being significantly less than the negative/sentinel reactions. When the level of target molecules is below the limit of detection, positive reactions can occur in one of three or two of three replicates. Because negative replicates produce a C T=40, the confidence interval of the test sample is not significantly different from the negative control reactions. In this case, a confidence interval that contains positive amplification signals in test sample reactions is produced, but overlaps with the negative controls. The result could therefore be considered ‘equivocal with signals detected below the validated assay sensitivity’. Increases in sample volume can be analysed, or if appropriate, testing carried out by an independent procedure, such as cell infectivity with Q-PCR end point analysis
2.4.2. Negative test
In a valid assay, a negative result for the test sample is considered to have occurred when the following four conditions are all met when testing the sample. (1) There are no amplification signals present in the test sample reactions. (2) There are no amplification signals present in the sentinel controls. (3) There are no amplification signals present in the negative controls. (4) There are amplification signals detected in the all spiked positive control reactions.
In a negative test with confidence, analysis of the data should result in the C T value of the sentinel controls, negative controls and the test article being significantly greater than those from the test article spiked at the limit of detection. Occasionally, one in three, or two in three replicates of the test sample spiked at the limit of detection may contain C T=40. If no amplifications are detected in the test article, the confidence intervals show that the spiked data is not significantly different from the test article and negative control data. In this case, the result would be considered negative below confidence, since the test was not able to detect amplification signals in all of the spiked replicates.
2.5. Validation of quantitative data
The accuracy of the quantitative data for a number of Q-PCR assays can be determined using a minimum of eight replicates containing identical amounts of DNA or viral RNA spread across a range of concentrations. During the analysis, four of the replicates are termed standards and the other four replicates designated unknowns. After generation of the standard curve and quantification of the unknowns, the accuracy of the quantitative software in combination with an assessment of the Q-PCR assay was determined. The results show that Q-PCR assays are able to quantify accurately the correct amount to within one standard deviation for both DNA and RNA targets (Table 3 ). In more detail, the value X=1000 on the standard curve (which is the actual amount in the Enterovirus RNA in the standard), corresponds to a calculated value Y=994±97 [which is the mean (± standard deviation) amount calculated in the unknown replicates containing identical amounts of nucleic acid as the standard]. Because the sensitivity of the ABI 7700 and 7900HT is a two- to fourfold copy number difference, a mean value of Y that varies by more than fourfold could be calculated as being different from the actual value of Y, if working at the two- to fourfold copy number difference level. Therefore, for an actual value (X) of 1000 copies the acceptable criteria could be a one- to twofold variation on either side of the 1000 copies (e.g. from 500–750 to 1500–2000 copies); a total of two- to fourfold variation of the actual value (X). Therefore, the value of Y=994±97 would be within the acceptable criteria. Any value outside of the acceptable limit could be considered as too highly variable for accurate quantification. This approach may allow one to assess the performance of the standards and the overall Q-PCR assay in terms of quantification accuracy.
Table 3.
Assessment of copy number accuracy
| Q-PCR assay | Actual | Calculated |
|---|---|---|
| (X) | (Y) | |
| Host cell DNA | 300 pg | 324 pg±25 |
| (pg) | 30 pg | 31 pg±14 |
| 3 pg | 5 pg±2 | |
| Enterovirus | 10 000 | 9702±1434 |
| (viral RNA copies) | 1000 | 994±97 |
Actual values (X) are the known amount in the standard reaction. Values (Y) are the amounts calculated in the ‘standardised unknowns’ from the generated standard curve, and are expressed as mean copy number ±S.D.
3. Pathogen detection and quantification
Adventitious agents pose significant risk if present in vaccines and therapeutics. Therefore, highly sensitive PCR assays for the detection of these agents are crucial for safety testing. Numerous viral nucleic acid detection assays are available, and the available number of fully validated Q-PCR pathogen detection assays is increasing at a considerable rate. Besides viral-specific nucleic acid detection, one very important use of Q-PCR technology in safety testing is the detection of contaminating retroviruses by the identification of the presence of retroviral reverse transcriptase activity (Lovatt et al., 1999).
3.1. F-PERT for the detection of retroviral reverse transcriptase
The fluorescent product enhanced reverse transcriptase (F-PERT) assay, is an extremely sensitive tests for the detection of reverse transcriptase (RT). F-PERT is up to 105-fold more sensitive than conventional RT assays for detecting the presence of retroviruses (Maudru and Peden, 1997, Lovatt et al., 1999). This increased sensitivity, prompted The Center for Biologics Evaluation and Research (CBER) to request PERT testing on all viral vaccines being investigated for humans. Consequently, F-PERT is the first choice for detection of RT in live viral vaccines, gene therapy preparations and the screening of animals and patients for Porcine endogenous retrovirus (PERV) in xenotransplantation trials. F-PERT is a RT-dependent Q-PCR assay and therefore combines the broad specificity of conventional RT assays with the high sensitivity and fast throughput of Q-PCR. As with conventional RT assays, F-PERT is utilised to detect the RT activity packaged into extracellular retrovirus particles. The assay involves converting RNA template to cDNA and then amplifying the cDNA using product-specific primers. As no exogenous RT activity is added to the reaction, cDNA will only be generated if the sample itself contains RT activity. When RT activity is not present, the product will not be detected (Silver et al., 1993, Pyra et al., 1994, Heneine et al., 1995, Arnold et al., 1998).
One major advantage of F-PERT assays over other similar types of assays, is the capability to discriminate between the amplification signals generated by retroviral RT activity and those which are a result of the RT-like activity from DNA polymerases. F-PERT uses activated calf thymus DNA to suppress the RT-like activity from cellular and Taq DNA polymerases with no reduction in the RT activity (Table 4 ). A linear relationship between threshold cycle (C T) and the number of MLV retrovirus particles or RT molecules exists, allowing quantification of MLV retrovirus, in terms of a number retroviral RT equivalents in unknown samples (Lovatt et al., 1999, Brorson et al., 2001). The F-PERT assay is able to detect a wide range of retroviral RT activities, including that from Porcine endogenous retrovirus (PERV), Murine leukaemia virus (MLV), Simian foamy virus (SFV), Simian immunodeficiency virus (SIVmac) and Squirrel monkey retrovirus (SMRV). The detection limit of F-PERT with 95% confidence for SMRV, MLV and PERV is approximately 1000 virion particles and 1000–10 000 molecules of purified AMV RT enzyme, however, the assay can detect lower levels of retrovirus with decreased frequency. An overview of the F-PERT assay is described in a flow diagram in Fig. 4 .
Table 4.
Suppression of DNA polymerase activity with activated calf thymus (aCT) DNA
| Sample | α | β | δ | Taq | 1000 |
|---|---|---|---|---|---|
| SMRV | |||||
| Minus | 37–40 | 40–40 | 26–28 | 22–31 | 22–29 |
| aCT | (−) | (−) | (+) | (+) | (+) |
| Plus | 40–40 | 40–40 | 39–40 | 40–40 | 26–28 |
| aCT | (−) | (−) | (−) | (−) | (+) |
CT values obtained from 1 to 5 units (U) of enzyme which gave the highest activity in F-PERT reactions without aCT DNA and suppression of that activity by aCT DNA (BMV RNA to aCT ratio of 1:104). Abbreviations:Taq: 5 U native Taq DNA polymerase; α: 1 U calf thymus DNA polymerase α; β: 1 U human DNA polymerase β; δ: 0.1 U DNA polymerase δ and 75 ng of δ-PCNA cofactor; TdT: 1 U terminal deoxynucleotide transferase; SMRV: 103 Squirrel monkey retrovirus particles; activated calf thymus DNA: aCT (Lovatt et al., 1999).
Fig. 4.
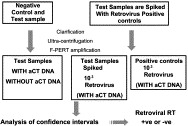
Overview of the F-PERT assay.
The high sensitivity of F-PERT has ensured that this Q-PCR assay plays a significant role in the evaluation of the retroviral status of vaccines and other biological material from the biotechnology industry, where safety of products is paramount. More specifically, when performed in combination with co-cultivation, induction and infectivity assays, together with electron microscopy (EM) and pathogen-specific PCR detection assays, F-PERT assists in providing a detailed risk assessment for the presence of adventitious agents in therapeutics.
3.2. Detection and quantification of viral nucleic acid
Viruses may contaminate cell lines from a number of sources including: (1) those present in the individual from whom the cell line is derived; (2) viruses of animal origin in media and tissue culture reagents; and (3) those from operators or other sources that could have contaminated the cells during handling in non-GMP conditions. For these reasons, all cell lines, primary cells, or tissue products used for therapeutic purposes require testing for the presence of a range of viruses. The selection of viruses to be tested depends upon the origin of the cell line and raw material used in manufacture. The development and validation of PCR assays to meet the requirements of regulatory authorities is a key element in the production and marketing of final products. An increasing number of published or validated Q-PCR assays are becoming available, and these tests can be utilised for cell line, raw material, or final product testing (Table 5, Table 6 ). Testing for the presence of human viruses is required when cells of human origin or products obtained from human blood or tissues are involved. Examples include mouse–human and human–human hybridomas, and cell lines of human origin such as MRC-5, and PER.C6 used in the production of Adenovirus vectors for therapy. In broad terms, the viruses that have to be screened for in a human cell line are those associated with severe or oncogenic diseases, and those that are likely to infect the cell type, particularly those that might establish latent or abortive replication in cells.
Table 5.
Q-PCR assays for human viruses
| Human virus | Target | Comment/references |
|---|---|---|
| gene/region | ||
| Human immuno- | gag gene (HIV-1) | Modified from |
| deficiency virus | env gene (HIV-2) | Vet et al., 1999a |
| Enteroviruses | 5′ Untranslated | Designed to detect over 20 |
| region | enterovirus typesa | |
| Human | MIE gene | Nitsche et al., 1999 |
| cytomegalovirus | HXFL4 gene | Najioullah et al., 2001 |
| Coronaviruses | pol gene | Detects human, bovine, porcine |
| and murine coronavirusa | ||
| Human papillomavirus | E1 gene | Josefsson et al., 1999 |
| Human polyomavirus | VP1 gene (BK) | Biel et al., 2001a |
| VP1 gene (JC) | ||
| Epstein–Barr virus | BALF5 gene | Kimura et al., 1999 |
| Validation at Q-One Biotech Ltd. | ||
| (in progress) | ||
| Hepatitis type A | 5′ NCR | No known published assaya |
| Hepatitis type B | core gene | Mercier et al., 1999a |
| Hepatitis type C | 5′ NCR | Mercier et al., 1999a |
| Measles virus | F and N gene | No known published assaya |
| Human T-cell | tat gene | Modified from |
| leukaemia virus | Vet et al., 1999a | |
| Influenza A | Matrix protein gene | van Elden et al., 2001 |
| Influenza B | Schweiger et al., 2000 | |
| Influenza Ca | Hemagglutinin gene | No known published assaya |
| Seg4 | ||
| Reovirus type 3 | S4 | No known published assaya |
| Human adenovirus | E1 | No known published assaya |
| Adeno-associated virus | cap | No known published assaya |
| B19 parvovirus | VP1 gene | Aberham et al., 2001a |
| Varicella zoster virus | gene 38 | Hawrami and Breuer, 1999 |
| Herpes viruses | gB gene (HSV 1 | Ryncarz et al., 1999 |
| and 2) | ||
| KS330 Bam | Kennedy et al., 1998 | |
| (HHV-8) | ||
| TTV | 5′ UTR ORF2 | Kato et al., 2000 |
Q-PCR assay is also developed and validated at Q-One Biotech Ltd.
Table 6.
Q-PCR assays for animal viruses
| Animal virus | Target | Comment/References |
|---|---|---|
| gene/region | ||
| Xenotropic murine | U3 LTR region | No known published assaya |
| leukemia virus | ||
| Bornavirus | Gp18 gene | No known published assaya |
| Simian adenovirus (SA7) | E1 gene | No known published assaya |
| Bovine papillomavirus | Early region | No known published assaya |
| Porcine endogenous | Gag gene | No known published assaya |
| retrovirus | ||
| Mouse mammary tumour | Sag gene | No known published assaya |
| virus | ||
| Bovine rotavirus G6 and | VP7 gene | No known published assaya |
| G10 | ||
| Bovine viral diarrhoea virus | 5′ UTR | No known published assaya |
| (BVDV) | ||
| Simian immuno- | Gag gene | Suryanarayana et al., 1998 |
| deficiency virus | ||
| Leutenegger et al., 2001 | ||
| Hofmann-Lehmann et al., 2000 | ||
| SV40 | Large T-antigen | Shi et al., 1999a |
| Shi et al., 1999b | ||
| Hamster polyomavirus | Small T-antigen | No known published assaya |
| Coronaviruses | Pol gene | No known published assaya |
| Bunyavirus | M-segment | No known published assaya |
| Porcine cytomegalovirus | OF-1 gene | No known published assaya |
| Bovine polyomavirus | VP1 gene | No known published assaya |
| Mouse parvovirus (MMV) | NS1 gene | No known published assaya |
| Rat endogenous retrovirus | Gag gene | No known published assaya |
| Porcine/bovine circovirus | Rep gene | No known published assaya |
| Bovine herpes 1 and 4 | TK gene | No known published assaya |
Q-PCR assay is also developed and validated at Q-One Biotech Ltd.
Animal viruses pose significant risk if the cell line or therapeutic is of animal origin and/or if animal products such as foetal calf serum, trypsin, or insulin are used in the manufacturing cultures. All new animal reagents used in manufacture should be screened by appropriate techniques. Bovine serum and porcine trypsin are usually screened using an appropriate Title 9 Code of Federal Regulation (9CFR) assay. However, some caution is required as 9CFR assays using minimal volumes may miss critical viruses. In addition, some viruses are not detected by these assays, therefore PCR should be employed. Many viruses are of concern in the manufacturing process and are not the subject of this review. Instead, this section focuses on examples of viruses that have recently emerged as agents of concern, namely Bovine polyomavirus, Bunyavirus and Porcine circovirus.
3.2.1. Bovine polyomavirus
Recent concern has been expressed about the contamination of bovine products with the Bovine polyomavirus (BPyV). This virus belongs to a family, some of which are oncogenic in their own host or in heterologous hosts. Infection of cattle is widespread, and up to 70% of foetal calf serum batches contain BPyV sequences by PCR and infectious virus can be isolated from some batches (Schuurman et al., 1991). The most worrying feature of this virus is that it is probably zoonotic. It replicates in primate cells, and while antibody to the virus is lacking in the general population, 71% of veterinarians, 50% of cattle farmers and 40% of abattoir workers are sero-positive (Parry and Gardner, 1986). Detection of BPyV by quantitative PCR is fully validated to ICH guidelines and the assay is capable of detecting 100 viral genome copies on a routine basis.
3.2.2. Bunyavirus
The Bunyaviridae family contains over 300 viruses grouped into five genera. Unlike most other enveloped viruses, they bud into intracytoplasmic vesicles associated with the golgi apparatus forming virions 80–120 nm in diameter. The genome of the virus is segmented into three strands permitting complex reassortants, as occurs with Influenza virus. Consequently, the taxonomy of these viruses can be complex (Lundstrom, 1999, Borucki et al., 1999, Mahy, 1998). Within the Bunyavirus genus, the most relevant serogroups or genetic complexes are the Californian serogroup and the Bunyamwera serogroup. Within the Bunyamwera group, the Cache valley virus (CVV) is a recognised infection of sheep and cattle. CVV can be transmitted congenitally and infection of foetal calf serum is possible (McLean et al., 1996, Blackmore and Grimstad, 1998, Edwards et al., 1997, Chung et al., 1991), emphasising that CVV has the potential to be problematic in large scale fermentation culture during manufacturing. CVV is lytic to cell lines including, CHO-K1 and Vero, and therefore can also be detected by culture-based bio-assays (Edwards et al., 1997). Since CVV is suspected to cause foetal malformations in cattle the presence of this virus in serum represents a major safety concern (Edwards et al., 1997). Detection of CVV by quantitative RT-PCR is fully validated to ICH guidelines and the assay is capable of detecting 100 viral RNA copies on a routine basis. The 95% confidence interval of C T values produced by 100 viral copies per 100 ng of non-infected cellular RNA was significantly different from the negative control RNA using two operators (Table 7 ). The CVV assay is able to detect the following strains of CVV: Ar66-2126 (Genbank no. AF231115); MN550 (Genbank no. AF231114); 90P686 (Genbank no. AF231113); RU68 (Genbank no. AF187824); 807270 (Genbank no. AF186243); CK102 (Genbank no. AF186242); MI80-1-450 (Genbank no. AF186241); and 6V633 (Genbank no. AF082739, AF82576).
Table 7.
Detection limit, reproducibility and linearity of CVV Q-PCR assay using two operators
| Negative | 100 copies | Linearity | CV |
|---|---|---|---|
| control RNA | of viral RNA | (r2) | |
| 40–40 | 34.5–34.9 | 0.998 | 1.90% |
| (−) | (+) | ||
| 40–40 | 33.3–33.5 | 0.964 | 1.07% |
| (−) | (+) | ||
3.2.3. Bovine and Porcine circovirus
Circoviruses are amongst the smallest mammalian viruses at approximately 17 nm, and like parvoviruses, are highly resistant in the environment. Porcine circovirus type 2 (PCV2) has a nucleotide sequence homology to Bovine circovirus (BCV) and has been shown to be involved in the formations of organ lesions in pigs. The close relationship between these two viruses has led to suggestions that this group of viruses may cross species barriers. In some cases, circoviruses can remain as latent and persist as non-cytopathic infections in vitro (Allan and Ellis, 2000). Techniques currently available for detection of all forms of circovirus are limited, and there is no current test that differentiates between infectious virus and nucleic acids. Consequently, specific Q-PCR assays are required to detect these viruses. As shown in Fig. 3, multiple sequence alignment of the Bovine circovirus and Porcine circovirus genomes has identified the Q-PCR primers and probe that will detect a range of strains isolated from many global locations. The Bovine circovirus and Porcine circovirus Q-PCR is fully validated to ICH guidelines and can routinely detect 100 genome copies.
In summary, CVV, BPyV, Porcine circovirus and Bovine circovirus represent a new threat to biotechnology products and steps need to be incorporated to monitor these viruses. Appropriate sub-pool testing of foetal calf serum and the testing of other raw materials should be conducted. Since it is possible that effects on cell viability may not be noted, appropriate bulk harvest screening of cultures should be undertaken to exclude the presence of these viruses, thereby avoiding expensive contamination of the down-stream purification train.
4. Clearance studies
Process validation studies are usually performed using a downscale of the manufacturer's purification process. This allows the assessment of the capacity and efficiency of the process to remove and inactivate viruses, DNA and other contaminants. All manufacturers producing recombinant proteins from rodent cell lines, e.g. Hybridoma and CHO, are required to perform Murine leukemia virus clearance studies for Phase I/II clinical trials. Generally, processes for phase III studies require clearance of three or more other viruses to be demonstrated (Federal Register, 1998). The federal guidelines specify that infectivity should be used, but where this is not technically feasible alternative technologies may be justified. However, an increasing number of manufacturers are using both infectivity and Q-PCR to demonstrate viral removal.
4.1. Xenotropic MLV and viral clearance
Murine leukemia viruses are endogenous proviruses that are divided into two main groups, ecotropic and non-ecotropic viruses. Ecotropic viruses can infect only mouse cells and non-ecotropic viruses are sub-divided into three groups: xenotropic; polytropic; and modified polytropic. Xenotropic MLV infect human cells, but not their natural rodent host cells. Polytropic MLVs, also termed mink-cell focus (MCF) forming MLV can infect both rodent and non-rodent cells, and have a broader host range than xenotropic MLV. Unlike xenotropic and ecotropic MLV, infectious polytropic MLV do not pre-exist in the mouse germ line, but can arise by recombination between infectious ecotropic MLV and endogenous MLV-related polytropic sequences. In certain laboratory mouse strains, the progeny of several non-ecotropic MLV can recombine with exogenous or endogenous MLV sequences to result in oncogenic variants such as MCF virus (Tomonaga and Coffin, 1998). Proviral xenotropic MLV that are capable of producing infectious virus are present in inbred mice populations, such as Balb-C. An endogenous xenotropic MLV, termed bxv-1 appears to be the LTR donor for in leukemogenic MCF forming MLV. In addition, xenotropic MLV derived from the Bxv-1 locus, first described in BALB/c mice are infectious (Kozak and Ruscetti, 1992).
Murine hybridoma cells used for pharmaceutical monocolonal antibody production express xenotropic MLV, and regulatory agencies recommend that retrovirus should be inactivated or removed during manufacturing. In addition, MLV is used as a model virus to evaluate the elimination of non-infectious CHO cell retrovirus particles. Such retroviral clearance studies can use virus inactivation treatments such as low pH of elution buffers during manufacturing. Viral clearance studies in scaled down conditions, involve spiking the load sample with a known amount of infectious virus to allow assessment of retroviral removal by examination of the eluate by infectivity assay or Q-PCR. However, when buffers that inactivate virus are used in the chromatography process, the reduction of infectious virus titre in the eluate is due to the combined result of chromatography removal and buffer inactivation (Lau et al., 1999). In these cases, infectivity assays will not provide clearance information, whereas Q-PCR resolves this problem.
Initially, it is essential to qualify the viral clearance Q-PCR assay using quantitative results on both the spiked load and unspiked load control. For cell lines other than murine hybrodoma, this ensures the absence of non-specific interactions that obscure the clearance data. For retroviral nucleic acid clearance, it is essential to perform DNA and RNA copy number quantification on the viral spike and any spiked column elutions. Analysis of the X-MLV nucleic acid copy number in the absence of RT enzyme allows an estimation of the percentage of proviral DNA targets which can result from the background residual host cell DNA of the viral production cell line. By recalculating the RNA copy number, this ensures that detection of viral RNA genome is achieved and the results take into account any background integrated provirus present in residual production cell genomic DNA. The difference between two RNA copy numbers obtained from the viral spike and spiked column elutions allows calculation of viral removal (Fig. 5 ).
Fig. 5.
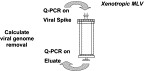
Q-PCR for viral clearance studies. For example, X-MLV stock of 109 RNA genome copies per ml (approx. 106 TCID50) is diluted 10% in the load to give a 108 X-MLV genome per ml spiked load. If the background signal from the bulk harvest material is negligible and the quantification limit of the X-MLV Q-PCR is 104 RNA copies per ml, we have a 4-log window to calculate viral RNA removal.
For the column elution samples that already contain xenotropic MLV, during RNA extraction a different virus can be spiked as an internal extraction/amplification control in order to distinguish it from the MLV already present. The internal control spike should not cross-react with the actual xenotropic MLV to simplify calculations. An unrelated RNA virus of known TCID50 or naked viral RNA can be used to control for PCR/extraction interference and viral recovery during extraction. Viral removal can be obtained by calculating the mean RNA copy number of the load, hold control and eluate from column runs. In addition to X-MLV quantification, Q-PCR assays to measure clearance of the endogenous retrovirus from Chinese hamster ovary (CHO) cells and SV40 are now available (Shi et al., 1999a, Shi et al., 1999b, de Wit et al., 2000). Taken together these approaches will allow the manufacturer's clearance process to be assessed for the ability to remove logs of viral genomes and thereby pave the way for optimisation of procedures to generate maximum viral clearance.
4.2. Contaminating host cell DNA assays
Manufacturers of biopharmaceuticals must ensure that final products produced from starting cell substrates contain minimum levels of residual host cell DNA. Current regulatory guidelines established by the FDA and WHO recommend that products manufactured from continuous cell lines contain less than 100 pg of cellular DNA per dose (Lewis et al., 1999a). The dangers of residual DNA include: malignant transformation by activated cellular and/or viral oncogenes; aberrant gene expression by insertion of sequences in sensitive control regions of genes; as well as the production of infectious viruses from viral DNA. In response to these concerns, a number of Q-PCR assays have been designed to detect and quantify residual DNA (Table 8 ). The quantification limit of residual DNA Q-PCR assays are usually within the 5 pg per reaction or 50–100 pg (10–20 genome equivalents) per ml. This sensitivity is expected when using a target such as the beta actin gene as the target. Sensitivity can sometimes be increased if a target with a higher copy number is used, for example a sequence repeat region within the genome. We are developing Q-PCR assays for these repeat regions for rodent and primate residual DNA to obtain sensitivity in the fg range, however, one advantage of using a gene such as β-actin is gene stability, as this target should not undergo copy number changes within a stable cell line. Repeat sequences are known to be less stable (Lewis et al., 1999b, Lobachev et al., 2000) and may undergo significant changes in copy number on the genome, and therefore have the potential to obscure the residual DNA data if a shift in target copy number occurs during production. For these reasons, the choice of PCR target is important and the use of repeat regions may not be necessary, when sensitivity to pg levels is required.
Table 8.
Contaminating DNA Q-PCR assays validated to ICH guidelines
| Host cell | Target gene | Sensitivity | Sensitivity |
|---|---|---|---|
| (Q-PCR) | Q-PCR | hybridisationb | |
| 293 | Adenovirus E1 | 500 fg–5 pg | 50 pg |
| E. coli | 23S RNA | 500 fga | 100 pg |
| HeLa | HPV-18 E7 | 5–50 pg | 50 pg |
| Primate | β-actin | 5–50 pg | 50 pg |
| Rodent | GAPDH | 500 fg–5 pg | 20 pg |
| (CHO) | |||
| Rodent | GAPDH | 500 fg–5 pg | 20 pg |
| (mouse) | |||
| Pichia | β-actin | 500 fg–5 pg | ND |
| Chicken | β-actin | 5 pg | ND |
| Porcine | PERV | 500 fg–5 pg | ND |
Abbreviations: ND: not determined; PERV: porcine endogenous retrovirus; GAPDH: glutaraldehyde-6-phosphate dehydrogenase.
Assay has background signals equivalent to that below 50 fg of E. coli DNA. These signals are due to residual DNA present in AmpliTaq DNA polymerase.
Slot-blot technique using 32P-radiolabeled probe (genomic DNA), hybridisation and autoradiography.
Besides the qualification of final products for batch release, residual DNA assays are used to demonstrate clearance of host cell DNA during process validation. In addition, since some test samples are most likely to contain detectable levels of residual DNA, another appropriate internal control DNA that is unrelated to the host cell and test sample is spiked into the sample. The recovery of the internal control is quantified using a PCR reaction specific for the unrelated target DNA. It is important that the internal control target and test sample target do not cross-react in terms of sequence specificity. This simplifies DNA recovery calculations for each individual test sample.
Q-PCR for residual DNA is far more robust than conventional hybridisation methods. For example, Q-PCR is more precise and has a larger dymanic range than conventional hybridisation methods (Smith et al., 1999). Problems with standard curves can be encountered when performing linear regression with conventional hybridisation autoradiography. This is due to the fact that X-ray films have a threshold for both response and sensitivity, resulting in a non-linear response to the hybridisation signal (Cornett et al., 1999). Q-PCR on the other hand, combines a highly linear response coupled with sensitivity comparable to hybridisation assays. In addition, turnaround time for the Q-PCR technique is more rapid, less labour intensive and can be automated with robotic systems to remove operator error during sample manipulation. Q-PCR can still estimate the amount of DNA in benzonase treated products, for example, if DNA is sheared down to sizes of 50–100 bp.
5. Gene and cell therapy biodistribution studies
Q-PCR is essential in determining the efficacy of gene and cell therapeutics. Moreover, the biodistribution analysis of such novel therapeutics is critical for assessment of their bio-safety and for meeting the increasingly stringent regulatory guidelines. In this way, regulatory issues concerning infection or the transfer of the gene to normal or distal tissues, as well as the target site, are addressed. In addition, investigations of the possibility of integration or expression in the germ line are performed. If possible, whole organs should be homogenised, an aliquot extracted and the remaining homogenate archived for further analysis. Homogenisation of whole organs gives insurance that patches or localisation of infection in the organ can be detected. If the therapeutic is viral or cellular based, then detection of viral or cellular RNA expression in tissues of concern may be indicative of virus or cellular replication in these tissues. Therefore, the assay target, conditions and controls used in the biodistribution study are critical for generating the highest quality data to satisfy regulatory authorities. A pre-study will be most important, to assess the best conditions that can be applied in the overall biodistribution assay.
5.1. Pre-studies for biodistribution assays
Problems that can occur during a bio-distribution study are dependant on the complexity of the study design. Pre-studies can be widely variable depending on the complexity of the therapeutic vector. However, in simplistic terms, the pre-study can be used to demonstrate that optimal PCR, nucleic acid extraction and maximum assay sensitivity are all addressed. For high quality data, it may also be necessary for the preparation of an inactivated vector to provide the material for administration into the negative control animals. This will help control for the presence of any residual vector nucleic acid that becomes lodged in animal organs and therefore provide information to distinguish between signals that can be generated from the replicating vector and those from the residual nucleic acid. In addition, if the vector is a DNA virus, the use of Q-RT-PCR with the incorporation of DNase during extraction and a minus RT reaction control will help distinguish between viral expression and residual DNA in the animal tissue. The method for inactivation of the viral vector will depend on the virus, and may be either UV or B-propriolactone treatment. The inactivated preparation should then be verified by amplification using cell culture and the replicating virus vector examined by an appropriate detection method, for example cytopathic effect or Q-PCR. Healthy cells that have been treated with inactivated vector can be passed through several times to dilute any residual inactivated target in the culture. Samples of cells taken at each passage time point can be quantified by Q-PCR with and without RT enzyme to demonstrate reduction of residual viral nucleic acid and the absence of low level infection.
Detection limit, linearity and reproducibility should all be determined during assay development and validation. Once the reaction conditions are optimal, spiked negative control organs are analysed to determine the lowest amount of target that can be detected in the maximum amount of test sample tissue. The optimal extraction conditions can be applied in the biodistribution study.
5.2. Study design
The animal species used in the biodistribution study is extremely important. The species must be acceptable to the regulatory authorities for use in safety evaluation studies and extensive background data should be available. The chosen animal species should be susceptible to infection. The route of administration can be the intended human route for the vector, however, the intravenous route allows better assessment of the vector biodistribution. Both animal sexes should be used in the study and the administrative dose should be calculated using maximum human dose based on equivalent unit per kilogram of body weight. An example of an animal study design to examine vector biodistribution could be as follows: group A: high dose (50–100×human maximum); group B: low dose (1×human dose); and group C: high dose inactivated. Each group contains three female and three male animals. At time points 8, 48 and 168 h, one animal is killed and blood, lymph nodes, lung, spleen, brain, kidneys, gonads, heart and liver harvested under carefully controlled conditions to prevent cross-contamination of vector between different organs. Organs should be either snap frozen or stored at −80 °C in individual vials for Q-PCR analysis.
5.2.1. Number of Q-PCR reaction replicates
To generate confidence intervals, a minimum of three replicated wells are required, but more are preferred. This ensures that results are expressed as positive or negative with confidence. The use of only duplicate or single wells for the controls does not allow comparison of groups to be carried out accurately, due to the significant high value of the t-statistic when using one degree of freedom. However, if working to a limited budget, performing the study using duplicate reaction replicates can allow the data to be expressed as the mean copy number within a specified standard deviation. The use of duplicate wells with a mean and standard deviation allows the generation of a 68.2% confidence interval. However, the use of single wells gives no information on well-to-well reproducibility and should only be used for research purposes rather than bio-safety testing, where confidence in results is essential.
To produce an accurate set of data covering the dynamic range of the assay a standard curve with a minimum of four or five standards should be included. A minimum of four or five different 10-fold dilutions should in most cases achieve an assessment of the C T values over the range of the assay on every PCR run.
5.2.2. Sentinel control extractions
These controls are essential since some of the animals dosed with a gene therapy vectors will most likely contain some PCR-positive tissues. If sentinel extractions are not included while handling positive samples, there will be no control for the level of cross-contamination between samples during extraction. The importance of the sentinel extraction is highlighted since the Q-PCR reaction sentinel can be negative and the extraction sentinels positive.
5.2.3. Internal control for test sample DNA
This control ensures recovery of test animal nucleic acid. This is necessary since any validation of the extraction procedure will have been performed in another time point than the actual test sample assay. This control can be replaced with the pre-extraction spike recovery control mentioned below, if the target is not integrated into the cellular DNA.
5.2.4. Spike recovery controls
If the integration of vector DNA into the host genome is not being assessed, both pre-extraction and post-extraction spike controls to quantify the percent of recovery during extraction and any inhibition during PCR should be performed. Even if a pre-study nucleic acid extraction validation already gives this information, the test sample biodistribution assay is performed at a time point other than that of the pre-study. Possible variables between those time points are, for example, variation in solution batches, different operators, errors, or problems with extraction reagents. The spike recovery controls performed at the time of testing ensure that these potential problems are detected.
5.2.5. Internal positive controls (IPC)
IPC, also known as TaqMan exogenous internal positive control reagents, can be included in all Q-PCR master mixes. This control can replace post-extraction spike controls, since PCR inhibition is detected with IPC reagents. IPC is a set of primers and probe targeted to a non-biological synthetic template. In the multiplexed PCR reaction, IPC is detected using a VIC-labelled probe and the viral/cell target nucleic acid is detected using a FAM-labelled probe. IPC reagents are spiked into the reactions to distinguish true target negatives from PCR inhibition. In addition, data generated from PCR-negative wells in the presence of IPC ensure that they are truly negative for the viral target, and are not due to a failed amplifications, thereby giving extra confidence in the results.
6. Gene copy number and genetic stability
In addition to viral detection and quantification, Q-PCR is widely used for the detection of changes in gene copy number, mRNA expression and plasmid DNA levels. Q-PCR combined with DNA sequencing and restriction mapping are commonly used in genetic stability testing. Examples of common uses of Q-PCR in research, development and quality control include: (1) detection of changes in plasmid DNA copy number in E. coli and yeast cell banks from development projects to large scale production; (2) detection of changes in transgene copy number of chromosomal-integrated expression cassettes (yeast and mammalian cell banks); (3) copy number of transgene mRNA levels to assess expression and (4) changes in gene expression in response to the therapeutic product.
6.1. Plasmid DNA copy number
Recent approaches to vaccination involve the direct introduction of recombinant plasmid DNA gene therapy vectors into appropriate tissue (Robertson and Griffiths, 1998, Mahato et al., 1999). This includes the administration of plasmid DNA complexed with cationic lipids for the treatment of genetic and acquired diseases (Gao and Haung, 1995). Since, plasmids are receiving increasing attention for use as vectors in gene therapy preparations, methods to monitor their stability and yield is of fundamental importance during manufacturing. Monitoring changes in transgene copy number and levels of mRNA expression is critical for the isolation of stable cell lines and in the streamlining of constructs for production. However, production and purification of large amounts of the therapeutic plasmid vector can be hindered by a number of factors, including bacterial host strain, plasmid DNA instability and copy number per cell. Together with restriction endonuclease mapping, nucleotide sequence analysis and phenotypic testing, routine monitoring of plasmid copy number is of utmost importance in large scale cultures where genetic instability can cause significant problems (Smith et al., 1996). Conventional procedures for the determination of plasmid DNA copy number are laborious, requiring DNA extraction, restriction endonuclease digestion, agarose electrophoreseis and ethidium bromide staining. Therefore, any high throughput methods which can calculate the copy number of plasmid vector constructs during fermentation are paramount in obtaining maximum efficiency in any downstream process.
Using Q-PCR assays targeted to regions such as: the plasmid origin of replication; the antibiotic resistance gene; the human CMV promoter; or the therapeutic transgene, it is possible to identify specific changes in plasmid copy number of different microbial constructs. By assessing the relationship between cell number and C T values, linearity was demonstrated allowing the direct determination of plasmid DNA copy number from culture samples relative to the reference plasmid containing E. coli strain. A fourfold difference in plasmid DNA copy number was detected, therefore the procedure can be used to monitor microbial fermentation or optimise production methods (Table 9, Table 10 ).
Table 9.
Detection of changes in plasmid DNA copy number in vitro
| Plasmid copies | 1 | 4 | 16 | 64 |
|---|---|---|---|---|
| per genome | ||||
| equivalent | ||||
| CT | ||||
| (95% CI) | 26.1–28.6 | 23.4–25.8 | 20.3–22.6 | 16.6–19.7 |
Plasmid DNA was analysed in a background of E. coli genomic DNA. Confidence intervals show statistical differences. Abbreviation: CI: confidence interval.
Table 10.
Differences between high and low copy number plasmids in vivo
| E. coli strain | Number of cells |
||
|---|---|---|---|
| 5×103 | 1.25×103 | 3.12×102 | |
| JM109 | 18.2–19.6 | 20.5–22.8 | 24.8–28.4 |
| (pUC18) | |||
| JM109 | 21.0–24.3 | 26.7–37.2 | 40.0–40.0 |
| (pBR322) | |||
A fourfold difference in cell number was detected. Confidence interval data show statistical differences.
The two methods for quantifying target molecules by real-time Q-PCR are termed relative and absolute quantification. Relative quantification can use an endogenous control to normalise the amount of plasmid molecules in a sample. The normalised value can assess significant increases or decreases in the copy number of plasmid DNA and is independent of conventional measurement of cell number. The incorporation of relative quantification could provide the potential to screen colonies or volumes of culture directly. The relative quantification will calculate numerical difference between the endogenous chromosomal gene and the number of plasmid molecules, in a microbial sample. In this way, significant differences between plasmid target and endogenous control, are detected to provide information on plasmid DNA molecules per cell. Using this approach, the assay could be independent of OD600 standardisation and still be able to detect a two- to fourfold difference in copy number. Taken together, Q-PCR has severe implications in monitoring the genetic stability of plasmid DNA in microbial constructs selected for production.
6.2. Stability of chromosomally-integrated transgenes
Q-PCR can be used to measure the copy number over a wide range of different cell numbers or genome equivalents, and is able to assess copy number stability in master cell bank (MCB), working cell bank (WCB) and post production cell bank (PCB). Significant changes are demonstrated by performing statistical analysis on the C T value obtained from the MCB, WCB and PCB. The copy number for each cell bank is expressed in a confidence interval and if the numerical C T values of the confidence intervals from each cell bank overlap, this demonstrates that the data derived from each stage of the manufacturing process are derived from the same statistical populations. The statistical data at the P=0.05 level, provides a means of monitoring significant changes in the copy number during the manufacturing process.
Q-PCR is more accurate than the Southern blot technique for the detection of alterations in the gene copy number, since estimation by Southern blotting relies on comparison of the banding intensity on an autoradiograph. The Southern blot analysis generates useful information about the structural integrity of the integrated construct but provides only an approximate evaluation of the copy number. In terms of stability, an increase in copy number of the transgene in the cell line can occur by chromosomal duplication. If significant increases in copy number are detected in the manufacturing process, Fluorescent In Situ Hybridisation (FISH) analysis can be performed to investigate further the stability of the construct. Direct chromosomal duplication, with no associated structural alterations, would be missed by southern analysis, if the integrated expression cassette remains unaltered when duplicated. Structural analysis will only detect changes in the expression cassette, such as those caused by deletions, insertions and recombination/duplication of the integrated construct to another chromosomal location. Therefore, rapid screening with Q-PCR assays will aid in the detection of any chromosomal duplication, which results in an increase in copy number but no associated structural alterations detectable by Southern blot analysis.
7. Conclusions
Because of the increasing rate of discovery of previously unrecognised viruses, safety is becoming of the utmost concern in biotechnology manufacturing. In addition, there is additional pressure for fast assay development, optimisation and validation of testing methods. Now that validated tests are available, Q-PCR is now beginning to revolutionise the production and batch release of products. The high versatility of Q-PCR assays coupled with the high reproducibility and fast throughput ability will no doubt assist in decreasing the time for the development, safety testing and final marketing of novel biotechnology products.
Acknowledgements
I would like to thank David Onions, Malcolm Brattle, Gillian Lees, Ken Smith, Iain Doherty, Danny Galbraith and Steve Gibson for their advice throughout the Q-PCR assay development program. I also thank Cliff Schorr, Christine McLean and Phillipe Grimm for constructive discussions. I also thank John Black and Donna McMutrie, Mike Moran, Angela Griffen, Sharron Kinnon and Terry Collins for all the hard work that they have carried out for the development program, and for significantly contributing towards solving the problems encountered.
References
- Aberham C., Pendl C., Gros P., Zerlauth G., Gessner M. A quantitative, internally controlled real-time PCR assay for the detection of parvovirus B19 DNA. J. Virol. Methods. 2001;92:183–192. doi: 10.1016/s0166-0934(00)00292-5. [DOI] [PubMed] [Google Scholar]
- Allan G.M., Ellis J.A. Porcine circoviruses: a review. J. Vet. Diagn. Invest. 2000;12:3–14. doi: 10.1177/104063870001200102. [DOI] [PubMed] [Google Scholar]
- Arnold B.A., Hepler R.W., Keller P.M. One step fluorescent probe product enhanced reverse transcriptase assay. Biotechniques. 1998;25:98–106. doi: 10.2144/98251st06. [DOI] [PubMed] [Google Scholar]
- Bendig M.M., Folk W.R. Deletion mutants of polyoma virus defining a nonessential region between the origin of replication and the initiation codon for early proteins. J. Virol. 1979;32:530–535. doi: 10.1128/jvi.32.2.530-535.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel S.S., Held K.H., Landt O. Rapid quantification and differentiation of human polyomavirus DNA in undiluted urine from patients after bone marrow transplantation. J. Clin. Microbiol. 2001;38:3689–3695. doi: 10.1128/jcm.38.10.3689-3695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmore C.G., Grimstad P.R. Cache valley and Potosi viruses (Bunyaviridae) in white tailed deer (Odocoileus virginianus): experimental infections and antibody prevalence in natural populations. Am. J. Trop. Med. Hyg. 1998;59:704–709. doi: 10.4269/ajtmh.1998.59.704. [DOI] [PubMed] [Google Scholar]
- Borucki M.K., Chandler L.J., Parker B.M., Blair C.D., Beaty B.J. Bunyavirus superinfection and segment reassortment in transovarially infected mosquitos. J. Gen. Virol. 1999;80:3173–3179. doi: 10.1099/0022-1317-80-12-3173. [DOI] [PubMed] [Google Scholar]
- Brorson K., Swann P.G., Lizzio E., Maudru T., Peden K., Stein K.E. Use of quantitative product enhanced reverse transcriptase assay to monitor retrovirus levels in mAb cell-culture and downstream processing. Biotechnol. Prog. 2001;17:188–196. doi: 10.1021/bp000153q. [DOI] [PubMed] [Google Scholar]
- Chung S.I., Livingston C.W., Jr., Jones C.W., Collisson E.W. Cache valley virus infection in Texas sheep flocks. J. Am. Vet. Med. Assoc. 1991;199:337–340. [PubMed] [Google Scholar]
- Cope M. Analytical method validation. Eur. J. Parenteral Sci. 2000;5:93–96. [Google Scholar]
- Cornett J.H., Sawyer J., Shanahan D. Membrane hybridisation. In: Saunders G.C., Parkes H.C., editors. Analytical Molecular Biology. Quality and Validation. Royal Society of Chemistry; Cambridge: 1999. [Google Scholar]; chapter 9, p. 135.
- de Wit C., Fautz C., Xu Y. Real time quantitative PCR for retrovirus-like particle quantification in CHO cell culture. Biologicals. 2000;28:137–148. doi: 10.1006/biol.2000.0250. [DOI] [PubMed] [Google Scholar]
- Edwards J.F., Karabatsos N., Collisson E.W., de la Concha Bermejillo A. Ovine fetal malformations induced by utero inoculation of Main Drain, San Angelo and La Crosse viruses. Am. J. Trop. Med. Hyg. 1997;56:171–176. doi: 10.4269/ajtmh.1997.56.171. [DOI] [PubMed] [Google Scholar]
- Federal Register 1998. Q5A Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin. Vol. 63, no 185, 51075–51084. [PubMed]
- Gao X., Haung I. Cationic liposome-mediated gene transfer. Gene Therapy. 1995;2:710–722. [PubMed] [Google Scholar]
- Hawrami K., Breuer J. Development of a fluorogenic polymerase chain reaction assay (TaqMan) for the detection and quantification of varcella zoster virus. J. Virol. Methods. 1999;79:33–40. doi: 10.1016/s0166-0934(98)00176-1. [DOI] [PubMed] [Google Scholar]
- Heid C.A., Stevens J., Livak K.J., Williams P.M. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Heneine W., Yamamoto S., Switzer W.M., Spira T.J., Folks T.M. Detection of reverse transcriptase by a highly sensitive assay in sera from persons infected with human immunodeficiency virus type 1. J. Infectious Dis. 1995;171:1210–1216. doi: 10.1093/infdis/171.5.1210. [DOI] [PubMed] [Google Scholar]
- Hofmann-Lehmann R., Swenerton R.K., Liska V. Sensitive and robust one-tube real-time reverse transcriptase-polymerase chain reaction to quantify SIV RNA load: comparison of one-versus two-enzyme systems. AIDS Res. Hum. Retroviruses. 2000;16:1247–1257. doi: 10.1089/08892220050117014. [DOI] [PubMed] [Google Scholar]
- ICH Topic Q2A, 1st June 1995. Validation of Analytical Methods. Definitions and Terminology. The European Agency for the Evaluation of Medicinal Products, CPMP/ICH/381/95.
- ICH Topic Q2B, 18th June 1997. Validation of Analytical Methods. Methodolgy. The European Agency for the Evaluation of Medicinal Products, CPMP/ICH/281/95.
- Josefsson A., Livak K., Gyllensten U. Detection and quantitation of human papillomavirus by using the fluorescent 5′ exonuclease assay. J. Clin. Microbiol. 1999;37:490–496. doi: 10.1128/jcm.37.3.490-496.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T., Mizokami M., Mukaide M. Development of a TT virus DNA quantification system using real-time detection PCR. J Clin. Microbiol. 2000;38:94–98. doi: 10.1128/jcm.38.1.94-98.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M.M., O'Leary J.J., Oates J.L., Lucas S.B., Howells D.D., Picton S., McGee J.O. Human herpes virus 8 (HHV-8) in Kaposi's sarcoma: lack of association with bcl-2 and p53 expression. Mol. Pathol. 1998;51:155–159. doi: 10.1136/mp.51.3.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H., Morita M., Yabuta Y. Quantitative analysis of Epstein–Barr virus load by a using real time PCR assay. J. Clin. Micriobiol. 1999;37:132–136. doi: 10.1128/jcm.37.1.132-136.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok T., Wati S., Bayly B., Devonshire-Gill D., Higgins G. Comparison of six nucleic acid extraction methods for the detection of viral DNA or RNA sequences in four different non-serum specimen types. J. Clin. Virol. 2000;16:59–63. doi: 10.1016/s1386-6532(99)00066-9. [DOI] [PubMed] [Google Scholar]
- Kozak C.A., Ruscetti S. Retroviruses in rodents. In: Levy J.A., editor. vol. 1. Plenum Press; New York: 1992. (The Retroviridae). [Google Scholar]
- Lau A.S.L., Lie Y.S., Norling L.A. Quantitative competitive reverse transcription-PCR as a method to evaluate retrovirus removal during chromatography procedures. J. Biotechnol. 1999;75:105–115. doi: 10.1016/s0168-1656(99)00152-2. [DOI] [PubMed] [Google Scholar]
- Leutenegger C.M., Higgins J., Mathews T.B. Real-time TaqMan PCR as a specific and more sensitive alternative to the branched-chain DNA assay for quantitation of Simian immunodeficiency virus RNA. AIDS Res. Hum. Retroviruses. 2001;17:243–251. doi: 10.1089/088922201750063160. [DOI] [PubMed] [Google Scholar]
- Lewis A.M. Jr., Krause P., Peden. K., 1999a. A defined approach to the regulatory assessment of the use of neoplastic cells as substrates for viral vaccine manufacture. A Draft. For the cell–substrate–adventitious agent working/interest group. Center for Biologics Evaluation and Research, http://www.fda.gov/cber/minutes/brief091499.pdf.
- Lewis S., Akgun E., Jasin M. Palindromic DNA and genome stability. Further studies. Ann. N.Y. Acad. Sci. 1999;870:45–57. doi: 10.1111/j.1749-6632.1999.tb08864.x. [DOI] [PubMed] [Google Scholar]
- Livak, K.J., Perkin Elmer, 1998. Quantitation of DNA/RNA using real time PCR detection. PE Applied Biosystems technical literature.
- Lobachev K.S., Stenger J.E., Kozyreva O.G., Jurka J., Gordenin D.A., Resnick M.A. Inverted Alu repeats unstable in yeast are excluded from the human genome. EMBO J. 2000;19:3822–3830. doi: 10.1093/emboj/19.14.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovatt A., Black J., Galbraith D. High throughput detection of retrovirus-associated reverse transcriptase using an improved Fluorescent Product Enhanced Reverse Transcriptase Assay (F-PERT) and its comparison to conventional detection methods. J. Virol. Methods. 1999;82:185–200. doi: 10.1016/s0166-0934(99)00111-1. [DOI] [PubMed] [Google Scholar]
- Lundstrom J.O. Mosquito-borne viruses in Western Europe: a review. J. Vector Ecol. 1999;24:1–39. [PubMed] [Google Scholar]
- Mahato, R.I., Smith, L.C., Rolland, 1999. Pharmaceutical perspectives of nonviral gene therapy. Adv. Genetics 41, 95–156. [DOI] [PubMed]
- Mahy B.W. Zoonoses and haemorrhagic fever. Dev. Biol. Stand. 1998;93:31–36. [PubMed] [Google Scholar]
- Maudru T., Peden K.W. Elimination of background signals in a modified polymerase chain reaction-based reverse transcriptase assay. J. Virol. Methods. 1997;66:247–261. doi: 10.1016/s0166-0934(97)00067-0. [DOI] [PubMed] [Google Scholar]
- McLean R.G., Kirk L.J., Shriner R.B. The role of deer as a possible reservoir host of Potosi virus recognised arbovirus in the United States. J. Wildl. Dis. 1996;32:444–452. doi: 10.7589/0090-3558-32.3.444. [DOI] [PubMed] [Google Scholar]
- Mercier B., Burlot L., Feree C. Simultaneous screening for HBV DNA and HCV RNA genomes in blood donations using a novel TaqMan PCR. J. Virol. Methods. 1999;77:1–9. doi: 10.1016/s0166-0934(98)00075-5. [DOI] [PubMed] [Google Scholar]
- Morris T., Robertson B., Gallagher M. Rapid reverse transcription-PCR detection of hepatitis C virus RNA in serum using TaqMan fluorogenic detection system. J. Clin. Microbiol. 1996;34:2933–2936. doi: 10.1128/jcm.34.12.2933-2936.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najioullah F., Thouvenot D., Bruno L. Development of a real-time PCR procedure including an internal control for the measurement of HCMV viral load. J. Virol. Methods. 2001;92:55–64. doi: 10.1016/s0166-0934(00)00273-1. [DOI] [PubMed] [Google Scholar]
- Nitsche A., Steuer N., Schmidt C.A., Landt O., Siegert W. Different real-time PCR formats compared for the quantitative detection of human cytomegalovirus DNA. Clin. Chem. 1999;45:1932–1937. [PubMed] [Google Scholar]
- Parry J.V., Gardner S.D. Human exposure to bovine polyomavirus: a zoonosis? Arch. Virol. 1986;87:287–296. doi: 10.1007/BF01315306. [DOI] [PubMed] [Google Scholar]
- Persing D.H. Target selection and optimisation of amplification reactions. In: Persing D.H., Smith T.F., Tenover F.C., White T.J., editors. Diagnostic Molecular Microbiology, Principles and Applications. American Society for Microbiology; Washington D.C: 1993. [Google Scholar]
- Pyra H., Böni J., Schüpbach J. Ultra-sensitive retrovirus detection by a reverse transcriptase assay based on product enhancement. Proc. Natl. Acad. Sci. U.S.A. 1994;91:1544–1548. doi: 10.1073/pnas.91.4.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiagen, 1999. Critical factors for successful PCR. Practical Guidelines Technical Literature.
- Robertson J., Griffiths E. WHO guidelines for assuring the quality of DNA vaccines. Biologicals. 1998;26:205–212. doi: 10.1006/biol.1998.0155. [DOI] [PubMed] [Google Scholar]
- Ryncarz A.J., Goddard J., Wald A., Huang M.L., Roizman B., Corey L. Development of a high-throughput quantitative assay for detecting Herpes simplex virus DNA in clinical samples. J. Clin. Microbiol. 1999;37:1941–1947. doi: 10.1128/jcm.37.6.1941-1947.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweiger B., Zadow I., Heckler R., Timm H., Pauli G. Application of a fluorogenic PCR assay for typing and subtyping of influenza viruses in respiratory samples. J. Clin. Microbiol. 2000;38:1552–1558. doi: 10.1128/jcm.38.4.1552-1558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurman R., van Steenis B., van Strien A. Frequent detection of Bovine polyomavirus in commercial batches of calf serum by using the polymerase chain reaction. J. Gen. Virol. 1991;72:2739–2745. doi: 10.1099/0022-1317-72-11-2739. [DOI] [PubMed] [Google Scholar]
- Shi L., Ho J., Norling L.A., Roy M., Xu Y. A real time quantitative PCR-based method for the detection and quantification of Simian virus 40. Biologicals. 1999;27:241–252. doi: 10.1006/biol.1999.0212. [DOI] [PubMed] [Google Scholar]
- Shi L., Norling L.A., Lau A.S., Krejci S., Laney A.J., Xu Y. Real time quantitative PCR as a method to evaluate Simian virus 40 removal during pharmaceutical protein purification. Biologicals. 1999;27:253–262. doi: 10.1006/biol.1999.0213. [DOI] [PubMed] [Google Scholar]
- Silver J., Maudru T., Fujita K., Repaske R. An RT PCR assay for the enzyme activity of reverse transcriptase capable of detecting single virions. Nucleic Acid Res. 1993;21:3593–3594. doi: 10.1093/nar/21.15.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K.T., Shepherd A.J., Boyd J.E., Lees G.M. Gene delivery systems for use in gene therapy: An overview of quality assurance and safety issues. Gene Therapy. 1996;3:190–200. [PubMed] [Google Scholar]
- Smith G.J., Helf M., Nesbit C., Betita H.A., Meek J., Ferre F. Fast and accurate method for quantitating E. coli host–cell DNA contamination in plasmid DNA preparations. Biotechniques. 1999;26:518–526. doi: 10.2144/99263rr03. [DOI] [PubMed] [Google Scholar]
- Suryanarayana K., Wiltrout T.A., Vasquez G.M., Hrsch V.M., Lifson J.D. Plasma SIV RNA viral load determination by real time quantification of product generation in reverse transcriptase polymerase chain reaction. AIDS Res. Hum. Retroviruses. 1998;14:183–189. doi: 10.1089/aid.1998.14.183. [DOI] [PubMed] [Google Scholar]
- Tomonaga K., Coffin J.M. Structure and distribution of endogenous non-ecotropic Murine leukemia virus in wild mice. J. Virol. 1998;72:8289–8300. doi: 10.1128/jvi.72.10.8289-8300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Elden L.J., Nijhuis M., Schipper P., Schuurman R., van Loon A.M. Simultaneous detection of influenza viruses A and B using real-time quantitative PCR. J. Clin. Microbiol. 2001;39:196–200. doi: 10.1128/JCM.39.1.196-200.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vet J.A.M., Majithia A.R., Marras S.A., Tyagi S., Dube S., Poise B.J., Kramier F.R. Multiplex detection of four pathogenic retroviruses using molecular beacons. Proc. Natl. Acad. Sci. USA. 1999;96:6394–6399. doi: 10.1073/pnas.96.11.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw, A.C., 1987. Correlation, regression and line fitting through graph points: standard curves. Practical Statistics for Experimental Biologists. John Wiley and Sons Ltd., 1985, Chapter 9, p. 188.