

Publisher Summary
This chapter discusses the ways in which viral replication damages tissues and organs and the ways in which the body's own responses may cause damage. It presents examples illustrating the pathogenesis of four basic kinds of acute viral infection: respiratory, intestinal, generalized, and neurological. As the immune system plays a key role in protection against infections, viral damage to its components can exacerbate the severity of disease or predispose to superinfection with other viral or nonviral agents. Both specific acquired immunodeficiency and generalized immunosuppression can occur in viral infections. The immune response to viral infection may itself frequently contribute to the pathology of the disease. Inflammation with accompanying cellular infiltration is a regular feature of viral infection. Such common signs as erythema, edema, and enlargement of lymph nodes have an immunological basis. There are viral diseases in which the cardinal manifestations are caused by the body's immune response. Immunopathological (hypersensitivity) reactions are traditionally classified into types 1, 2, 3, and 4. For most viral infections, it is not known whether immunopathological effects make a significant contribution to disease and if so then as to which of the four classical hypersensitivity reactions is implicated. It is instructive to speculate about the possible involvement of different kinds of hypersensitivity reactions in viral diseases.
In the previous four chapters we have analyzed various aspects of viral infections of animals, exploring how viruses affect cells, how infection of animals occurs, how viruses spread to various parts of the body, and how the host animal responds to infection by immunological and other mechanisms. The clinical end product of these interactions, namely disease, is the subject of this chapter and Chapter 11 (persistent infections) and Chapter 12 (virus-induced tumors). The first part of this chapter covers the ways in which viral replication damages tissues and organs and the ways in which the body's own responses may cause damage. The second part of the chapter comprises examples illustrating the pathogenesis of four basic kinds of acute viral infection: respiratory, intestinal, generalized, and neurological.
DIRECT DAMAGE AT THE CELLULAR LEVEL
The mechanisms by which viruses damage cells have been discussed at the cellular and subcellular levels in Chapter 6. Here we apply these concepts at the level of tissues and organs. The severity of disease in the animal is not necessarily correlated with the degree of cytotoxicity of the virus in vitro. Many viruses that are cytocidal in cultured cells are harmless in vivo (e.g., many enteroviruses), whereas some that are noncytocidal in vitro cause a lethal disease in vivo (e.g., rabies virus). A great deal of cell and tissue damage can occur without producing signs of disease, e.g., a large number of intestinal or liver cells can be destroyed without significant clinical signs. Even when damage to cells impairs the function of an organ or tissue, this may be of minor importance, e.g., in muscle or subcutaneous tissue, but of great importance in the heart or the brain. Likewise, edema of a tissue may be unimportant in most sites in the body but may have serious consequences in the brain, because of the resulting increase in intracranial pressure, or in the lung, where it may interfere with gaseous exchange, or in the heart, where it may interfere with conduction.
Sometimes the whole pathological process in an animal may be explained by the direct damage to cells caused by a highly cytocidal virus. Mice infected intravenously with a large dose of Rift Valley fever virus, for example, develop overwhelming hepatic necrosis within 4 hours of injection, because the virions pass quickly through the Kupffer cells to infect the hepatic cells, which are rapidly lysed. In this experimental model, the defense mechanisms of the host, both nonspecific and specific, are quite unable to cope with the rapid lethal damage to a vital organ.
Epithelial Damage Predisposing to Secondary Bacterial Infection
As well as having direct adverse effects, viral infections of the respiratory or digestive tracts often predispose animals to secondary bacterial infections. Viral infection increases the susceptibility of the respiratory tract to bacteria that are normal commensals in the nose and throat. For example, in cattle, parainfluenza 3 virus or other viruses may destroy ciliated epithelia and cause exudation, allowing Pasteurella haemolytica and other bacteria to invade the lungs and cause secondary bacterial pneumonia, known in many countries as “shipping fever.” Rhinoviruses and respiratory syncytial viruses damage the mucosa in the nasopharynx and sinuses, predisposing to bacterial superinfection which commonly leads to purulent rhinitis, pharyngitis, and sinusitis. Similarly, in the intestinal tract, rotavirus and coronavirus infections may lead to an increase in susceptibility to enteropathogenic E. coli, and the synergistic effect leads to severe diarrhea.
Veterinary virologists are particularly conscious of the potentiating effect on viral diseases of coinfection with parasites. Animals are almost universally infected with protozoa and helminths, and a high parasitic load generally lowers their resistance to viruses and bacteria.
DAMAGE TO THE IMMUNE SYSTEM
Since the immune system plays a key role in protection against infections, viral damage to its components can exacerbate the severity of disease or predispose to superinfection with other viral or nonviral agents. Both specific acquired immunodeficiency and generalized immunosuppression can occur in viral infections.
Infection of the bursa of Fabricius in chickens (the site of B cell differentiation) with infectious bursal disease virus (a birnavirus) leads to atrophy of the bursa and a severe deficiency of B lymphocytes, with an increase in susceptibility to Marek's disease, Newcastle disease, infectious bronchitis, and infectious laryngotracheitis viruses. B and T cell deficiencies are produced in feline leukemia, thereby allowing intercurrent opportunistic infections to cause death.
Recently, a comparable example of great public health significance has emerged in humans. The agent that causes acquired immunodeficiency syndrome (AIDS) in humans, now designated human immunodeficiency virus (HIV), is a retrovirus, of the subfamily Lentivirinae. The virus destroys helper T (Th) cells, causing profound immunosuppression, which, after a prolonged clinical course, leads to death from opportunistic infections. This tropism for T cells is shared by other lentiviruses; e.g., visna–maedi virus of sheep, as well as by simian T-lymphotropic virus-III (STLV-III), which causes a similar disease in some species of monkeys.
Infections with certain other viruses (e.g., hog cholera, bovine virus diarrhea, canine distemper viruses, feline and canine parvoviruses) may temporarily suppress humoral and/or cell-mediated immune responses, but the results are not usually so catastrophic. The immune response to unrelated antigens is reduced or abrogated in such animals, and the situation is thus distinct from suppression of the immune response to the specific virus in question, which is called immune tolerance. The mechanisms involved in such general immunosuppression are not fully understood, but may result from the replication of virus in lymphocytes and/or macrophages, or from abnormal induction of suppressor T cells. Many viruses are capable of replication in macrophages (see Chapter 11), and several have been shown to grow in T cells, especially activated T cells. Some herpesviruses replicate nonproductively in B cells, transforming them and altering their function.
While viral infections can induce immunosuppression, conversely immunosuppression allows enhanced viral replication. When the immune system is suppressed by endogenous or exogenous factors, latent herpesvirus, adenovirus, or papovavirus infections can be reactivated. Such situations are frequently encountered following the use of cytotoxic drugs or irradiation for organ transplantation in humans. No doubt immunosuppression, usually of unknown origin, is often responsible, at least in part, for reactivation of herpesviruses in animals (see Chapter 11 and Chapter 19).
IMMUNOPATHOLOGICAL EFFECTS AND MECHANISMS
The immune response to viral infection may itself frequently contribute to the pathology of the disease. Inflammation with accompanying cellular infiltration is a regular feature of viral infection. Such common signs as erythema, edema, and enlargement of lymph nodes have an immunological basis. But there are viral diseases in which the cardinal manifestations are caused by the body's immune response. In the extreme case, the disease may be prevented by suppressing the immune response.
Immunopathological (hypersensitivity) reactions are traditionally classified into types 1, 2, 3, and 4. Although advances in cellular immunology have now blurred some of the distinctions, the classification is still convenient (Table 10-1 ). For most viral infections it is not known whether immunopathological effects make a significant contribution to disease, and if so, which of the four classical “hypersensitivity reactions” is implicated. Nevertheless, it is instructive to speculate about the possible involvement of different kinds of hypersensitivity reactions in viral diseases.
TABLE 10-1.
Basic Types of Hypersensitivity Reactionsa
| Hypersensitivity type | ||||
|---|---|---|---|---|
| item | 1 | 2 | 3 | 4 |
| Designation | Anaphylactic | Cytotoxic | Immune complex | Delayed—cell mediated |
| Time course | ||||
| Initiation | Minutes | Minutes | 3–6 hours | 18–24 hours |
| Persistence | Minutes | Dependent on antigen | Dependent on antigen | Weeks |
| and antibody | and antibody | |||
| Transfer with | IgE | IgM, IgG | IgG | T lymphocytes |
| Complement required | No | Usually | Yes | No |
| Histamine dependent | Yes | No | No | No |
| Histology | Edema, congestion, | Cell destruction, | Necrosis, neutrophils, | Lymphocytes, |
| eosinophils | phagocytosis | later plasma cells | macrophages, necrosis | |
| Viral immunopathological | Minor | Minor | Major | Major in brain, lung |
| effects | ?some rashes | ?some rashes | acute: fever | |
| chronic: immune | ||||
| complex disease | ||||
As categorized by Gell and Coombs.
Hypersensitivity Reactions—Type 1
These are anaphylactic reactions, which depend on the interaction of antigens with IgE antibodies on the surface of mast cells, resulting in the release of histamine and heparin and the activation of serotonin and plasma kinins. Except for its possible contribution to some types of rash and possibly in some acute respiratory infections, anaphylaxis is probably not important in viral immunopathology, but is responsible for some adverse reactions to viral vaccines.
Hypersensitivity Reactions—Type 2
These cytolytic reactions occur when antibody, having combined with viral antigen on the cell surface, activates the complement system, leading to cell lysis. Alternatively, antibodies can sensitize virus-infected cells to destruction by K cells, polymorphonuclear leukocytes, or macrophages, via antibody-dependent cell-mediated cytotoxicity. While it has been clearly demonstrated that virus-infected cells are readily lysed by all of these mechanisms in vitro, their significance in viral diseases in vivo is unclear, although there is some evidence that they may be operative in certain herpesvirus infections.
Hypersensitivity Reactions—Type 3
Antigen–antibody complexes cause inflammation and cell damage by a variety of mechanisms. If the reaction occurs in extravascular tissues there is edema, inflammation, and infiltration of polymorphonuclear leukocytes, which may later be replaced by mononuclear cells. This is a common cause of mild inflammatory reactions. These “immune complex” reactions constitute the classical Arthus response, and are of major importance, especially in persistent viral infections. If they occur in the blood, they produce circulating immune complexes, which are found in many viral infections, both acute and persistent. The fate of the immune complexes depends on the ratio of antibody to antigen. If there is a large excess of antibody, each antigen molecule is covered with antibody and removed by macrophages, which have receptors for the Fc fragment of the antibody molecule. If the amounts of antigen and antibody are about equal, lattice structures which develop into large aggregates are formed and removed rapidly by the reticuloendothelial system.
However, in some persistent infections viral antigens or virions themselves are continuously released into the blood, but the antibody response is weak and antibodies are of low avidity. Complexes continue to be deposited in glomeruli over periods of weeks, months, or even years, leading to impairment of glomerular filtration and eventually to chronic glomerulonephritis (see Chapter 11). A classic example is lymphocytic choriomeningitis infection in mice infected in utero or as neonates. Viral antigens are present in the blood and small amounts of nonneutralizing antibody are formed, giving rise to immune complexes which are progressively deposited upon renal glomerular membranes. Depending on the strain of mouse, the end result may be glomerulonephritis, uremia, and death. Circulating immune complexes may also be deposited in the walls of the small blood vessels in skin, joints, and choroid plexus, where they attract macrophages and activate complement. Prodromal rashes, seen commonly in exanthems in humans but rarely in domestic animals, are probably caused in this way.
In addition to these local effects, antigen–antibody complexes generate systemic reactions, such as the fever which marks the end of the incubation period in generalized viral infections. Fever is mediated by interleukin-1, which is liberated from macrophages and polymorphonuclear leukocytes.
Rarely, systemic immune complex reactions may activate the enzymes of the blood coagulation cascade, leading to histamine release and increased vascular permeability. Fibrin is deposited in the kidneys, lungs, adrenals, and pituitary gland, causing multiple thromboses with infarcts and scattered hemorrhages—a condition known as disseminated intravascular coagulation. This is seen in hemorrhagic fevers in humans, many of which are zoonoses caused by arenaviruses, bunyaviruses, filoviruses, or flaviviruses. Kittens infected with feline infectious peritonitis virus, a coronavirus, also display this phenomenon, and it probably occurs in other severe systemic diseases.
Hypersensitivity Reactions—Type 4
Unlike all the previous types, these “delayed hypersensitivity” reactions are mediated by cells rather than antibody. They are T lymphocyte-mediated immune reactions, involving inflammation, lymphocytic infiltration, macrophage accumulation, and activation by lymphokines secreted by Td cells. Once again, the classic model is lymphocytic choriomeningitis virus infection, this time primary infection of adult mice. After intracerebral inoculation this noncytocidal virus replicates harmlessly in the meninges, ependyma, and choroid plexus epithelium until about the seventh day, when a Tc lymphocyte-mediated immune response occurs, causing severe meningitis, cerebral edema, and death. Elsewhere than in the central nervous system, Tc cells help to control infection; within the rigid confines of the skull these changes are fatal. The death of mice infected in this way can be prevented by chemical immunosuppression, by X irradiation, or by antilymphocyte serum. Type 4 hypersensitivity reactions may also contribute to consolidation of the lung, probably mediated by Td cells, in various severe lower respiratory tract diseases.
Although occasionally the cause of immunopathology, cell-mediated immune responses are generally an important component of the process of recovery from viral infections (see Chapter 9), as becomes evident if they are abrogated by cytotoxic drugs, or are absent, as in some immunodeficiency diseases.
OTHER PHYSIOLOGICAL DISTURBANCES
Some pathological changes found in viral infections cannot be attributed to direct cell destruction by the virus, nor to inflammation, nor to immunopathology. Perhaps the most important of these effects relate to alterations in the function of various endocrine glands, notably the adrenals, in response to the “stress” of the infectious disease. Sometimes endocrine epithelial cells affected by noncytocidal viruses are not killed, but their secretory functions are damaged; e.g., infection of mice with encephalomyocarditis virus (a picornavirus) may lead to diabetes because of the action of the virus on the β cells of the islets of Langerhans. Lymphocytic choriomeningitis virus, by infecting somatotrophic cells of the anterior lobe of the pituitary gland of mice, may reduce the output of growth hormone, leading to dwarfism. These examples come from well-studied experimental animal models; similar changes probably occur in natural infections, but they have not yet been documented.
Most viral diseases are accompanied by a number of vague general clinical signs, such as fever, malaise, anorexia, and lassitude. Little is known about the causes of these signs, which collectively can significantly reduce the animal's performance and impede recovery. As discussed in Chapter 8, fever can be attributed to interleukin-1 and possibly to interferons. These and other soluble mediators produced by leukocytes, or released from virus-infected cells, may be responsible for the other clinical signs also.
REPRESENTATIVE DISEASE MODELS
We are now in a position to examine in some detail the pathogenesis of selected examples of each of the four main categories of acute infections: respiratory, intestinal, generalized, and those affecting the central nervous system. We begin with two prototypes of viral respiratory diseases: influenza, about which a great deal is known from experimental studies, and the complex respiratory infections of cattle known as shipping fever, as an example of synergistic virus–bacteria infection.
Influenza
The sequence of events in respiratory infections is rather similar no matter what kind of virus is involved. Acute respiratory infections are exemplified by influenza in horses and swine. Virus particles in aerosolized droplets or on fomites are inhaled and alight on the film of mucus that covers the epithelium of the upper respiratory tract. Droplets of different sizes alight at different levels of the respiratory tree, and infection may accordingly be initiated at particular levels, but in general the upper respiratory tract—i.e., the nasal passages, the pharynx, and the trachea—are the sites of initial infection. Immediately upon alighting, the virus is met by host defense mechanisms; if the animal has previously been infected with the same or a very similar strain of influenza virus, antibody (mainly IgA) present in the mucus may neutralize the virus. Mucus also contains glycoproteins similar to the receptor molecules on respiratory epithelial cells; these glycoproteins may combine with virions and prevent them from attaching to epithelial cells. In turn, the viral neuraminidase may destroy enough glycoprotein to allow virions to attach to and infect an epithelial cell.
If these host defenses fail, the virus moves deeper into the airways, where it faces another physiological barrier mechanism. This is the cleansing action of beating cilia; inhaled particles, including virions, are normally carried via the mucous flow generated by cilial beating action to the pharynx, where they are swallowed. However, it has been shown with influenza viruses and other respiratory viruses that initial invasion and destruction of just a few epithelial cells can initiate a lesion which can progressively damage the mucus protective layer and lay bare more and more epithelial cells.
Infection of epithelial cells of the respiratory tract follows attachment of the virus via its hemagglutinin to the specific receptor, N-acetylneuraminic acid, the terminal sugar of the oligosaccharide side-chains of common cellular glycoproteins and glycolipids. Once attached, the virus is taken up by endocytosis and, in the acid conditions of the phagolysosome, the viral envelope fuses with the phagolysosome membrane, releasing the viral nucleocapsid into the cytoplasm of the epithelial cell. Viral replication progresses and large numbers of progeny virions are budded from the plasma membrane into the lumen of the airway. Early in infection, cilial beating helps to move released progeny virus along the airway, thereby spreading the infection. As secretions become more profuse and viscous, the cilial beating becomes less effective, and later cilial beating is stopped as epithelial cells are destroyed.
In studies in experimental animals the spread of the infection via contiguous expansion from initial foci often does not stop until virtually every columnar epithelial cell at that airway level is infected. The result is complete denuding of large areas of epithelial surface (Plate 10-1 ) and the accumulation of large amounts of transudates, exudates, inflammatory infiltrates, and necrotic epithelial debris in the airways. The consequent respiratory distress is made worse by forced movement of animals and by coughing and sneezing. Where infection of the epithelium of the nasal passages, trachea, and bronchi proceeds to a fatal outcome, there is usually one or more of three complications: (1) bacterial superinfection, nurtured by the accumulation of fluid and necrotic debris, “growth medium,” in the airways, (2) infection and destruction of the lung parenchyma, the alveolar epithelium, and/or (3) blockage of airways that are so small in diameter that mucus plugs cannot be opened by forced air movements. Blockage of the airways is of most significance in the newborn. In all of these complications there is hypoxia, and a pathophysiological cascade that leads to acidosis and uncontrollable fluid exudation into airways.
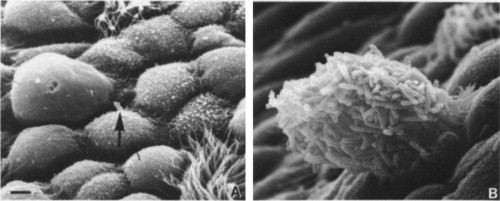
PLATE 10-1.
Scanning electron micrographs showing the adherence of Pseudomonas aeruginosa to the mouse trachea (bar = 2 µm). (A) Normal mouse trachea, showing a single bacterium (arrow) on a serous cell. (B) Microcolony adhering to desquamating cells in an influenza virus-infected trachea. [From R. Ramphal et al., Infect. Immun. 27, 614 (1980); courtesy Dr. P. A. Small, Jr.]
Degeneration of respiratory tract epithelial surfaces during influenza infection is extremely rapid, but so is regeneration. In studies of influenza in ferrets, for example, it has been shown that the development of a complete new columnar epithelial surface via hyperplasia of remaining transitional cells may be complete in a few days. The transitional epithelium and the newly differentiated columnar epithelium that arises from it are resistant to infection, probably by virtue of interferon production and a lack of virus receptors. Other host defenses, including antibody and cell-mediated immune mechanisms, also play a part in terminating the infection.
Influenza viruses, like most other respiratory viruses, have evolved a rapid transmission cycle, so as to outrace the host immune response. Influenza viruses, particularly human influenza viruses, have also evolved systems of genetic drift and shift (see Chapter 5) to circumvent host immunity, but they also depend on management programs that favor continual exposure of new susceptible horses and swine, as well as poultry—hence our need for vaccine.
Whereas influenza virus infection in mammals, as described, is generally restricted to the cells of the respiratory tract, in birds it is primarily an inapparent infection of the digestive tract; viremia and spread to other organs, leading to severe disease, occur with virulent strains of avian influenza virus.
Viral-Bacterial Synergism in Respiratory Infections (Shipping Fever)
In cattle there is a seasonal incidence of bronchopneumonia that corresponds with the extra stress of harsh climatic conditions and husbandry practices in fall and early winter, as well as the extra activity of respiratory viruses and mycoplasma at this time of year. This syndrome of bronchopneumonia, often extending to a true fibrinous pleuropneumonia, is called shipping fever in many countries. In the United States and Canada the syndrome represents the most economically important health problem in cattle, especially feedlot cattle. The syndrome, despite having diverse initial causes, has a common final pathway and etiology, the terminal manifestations being caused by overwhelming infection by Pasteurella haemolytica and to a lesser extent P. multocida.
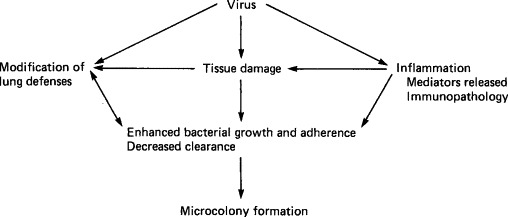
Respiratory virus infections, augmented by the pathophysiological effects of stress, alter the susceptibility of cattle to Pasteurella species that are normally present in the upper respiratory tract by a number of independent and interdependent mechanisms. The influence of respiratory epithelial tissue damage and fluid exudation into airways, as described above for influenza, is a major factor favoring bacterial growth following all respiratory virus infections. Viral infections can also alter bovine host defense mechanisms in other ways: (1) they can be directly immunosuppressive or can damage reticuloendothelial (macrophage and neutrophil) function in the lungs and airways, (2) they may induce such exuberant inflammatory response that the delicate epithelial surfaces of the alveoli are destroyed and the structure of the lungs collapsed and consolidated, or (3) they can alter the surface properties of respiratory epithelial cells so as to favor bacterial adherence, and the growth of bacterial microcolonies. These microcolonies in turn resist phagocytosis and the effects of antibodies and antibiotics, and can more readily enter the lower respiratory tract. Another effect of viral damage to epithelial cells is the release of iron, which enhances bacterial growth and colonization. The complex of interactions between various factors during the development of respiratory disease is represented diagrammatically in Fig. 10-1 .
FIG. 10-1.
Interactions between events associated with viral and bacterial infections in the development of respiratory disease (“shipping fever”) in cattle. [From L. A. Babiuk, In “Applied Virology” (E. Kurstak, ed.), p. 431. Academic Press, Orlando, 1984.]
Control of the viral infections that initiate shipping fever appears to be more important than control of the terminal bacterial pneumonia. The Pasteurella species involved are always present in the environment and in the upper respiratory tract of cattle, and it is not likely that they could be eliminated. Likewise, these bacterial species have proved most difficult to affect with vaccines. The speed with which antibiotic therapy is initiated does influence the eventual outcome, but in many cases irreparable damage has already been done by the time of diagnosis. If microcolonies have been established and have progressed to abscess formation, there is added difficulty in delivering effective antibiotic levels so as to reach the bacteria. Even when the animal does not die, it generally does not grow well, and it is prone to further debilitating diseases. Viral vaccine programs, aimed at preventing this pathogenetic pattern, must be measured and justified indirectly, by the effect they have on pneumonia prevalence. Producers and veterinarians must therefore collaborate in developing overall strategies for dealing with this syndrome.
Pathogenesis of Viral Diarrheas
Diarrhea in animals is often multifactorial, and interactions of infectious agents with immunological, environmental, and nutritional factors can often exacerbate the disease. The principal causes of viral diarrhea in domestic animals are rotaviruses and coronaviruses; other viruses involved are caliciviruses, astroviruses, and parvoviruses. Infection occurs by ingestion of virus, except with parvoviruses, in which infection of the intestinal tract is part of a systemic infection. The incubation period is very short.
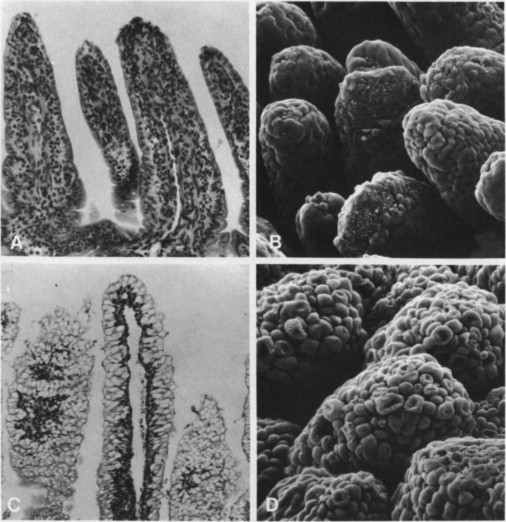
Different viruses characteristically infect different parts of the villi of the intestinal tract, with rotaviruses and coronaviruses infecting cells at the tip and parvoviruses cells in the crypts (Fig. 10-2 ). All cause marked shortening and occasional fusion of adjacent villi (Plate 10-2 ), so that the absorptive surface of the intestine is reduced, resulting in fluid accumulation and diarrhea. Infection generally begins in the proximal part of the small intestine and spreads progressively to the jejunum and ileum and sometimes to the colon. The extent of such spread depends on the initial dose, the virulence of the virus, and the host's immunological status. In the presence of maternal antibody, infection can occur, but the degree of replication is limited and diarrhea is mild or does not occur.
FIG. 10-2.
Preferred locations of replication of enteropathogenic viruses in intestinal villi and the effects on the villi: coronaviruses and rotaviruses in cells at the tips, parvoviruses in the multiplying cells in the crypts. [Based on L. A. Babiuk, In “Applied Virology” (E. Kurstak, ed.), p. 349. Academic Press, Orlando, 1984.]
PLATE 10-2.
Scanning electron and light micrographs of intestinal tissues from a gnotobiotic calf sacrificed 0.5 hour after onset of rotavirus diarrhea. (A) Proximal small intestine with shortened villi and a denuded villus tip (second from right) (H and E, ×112). (B) Appearance of same level of intestine as in (A) depicting denuded villi by scanning electron microscopy (×170). (C) Distal small intestine with normal vacuolated epithelial cells and normal villi (H and E, × 70). (D) Same area as in (C) seen by scanning electron microscopy. Epithelial cells appear round and protruding (×200).
[From C. A. Mebus et al., Vet. Pathol. 14, 273 (1977); and A. Z. Kapikian and R. M. Chanock, In “Virology” (B. N. Fields et al., eds.), Raven Press, New York; Courtesy A. Z. Kapikian.]
© 1987
With rotaviruses and coronaviruses, which infect cells at the tips of the villi, as infection progresses the absorptive cells are replaced by immature cuboidal epithelial cells whose absorptive capacity and enzymatic activity is greatly reduced. These cells are relatively resistant to viral infection, so that the disease is often self-limiting if dehydration is not so severe as to be lethal. The rate of recovery is rapid, since the crypt cells are not damaged. In contrast, recovery is slow after infections with parvoviruses, which infect cells of the crypts.
The mechanism of fluid loss in viral infections is different from that in bacterial infections, but the net loss may be of the same magnitude. In viral infections fluid loss is mainly a loss of extracellular fluid due to impaired absorption, and osmotic loss due primarily to the presence of undigested lactose in the lumen (in sucking animals), rather than active secretion. As virus destroys the absorptive cells there is a loss of those enzymes responsible for the digestion of disaccharides, and the loss of differentiated cells diminishes glucose, sodium carrier, and Na+,K+-ATPase activities. This leads to a loss of sodium, potassium, chloride, bicarbonate, and water, and the development of acidosis. Another cause of acidosis is the result of increased microbial activity associated with the fermentation of undigested milk. Acidosis can create a K+ ion exchange across the cellular membrane, affecting cellular functions that maintain the normal potassium concentration. Hypoglycemia due to decreased intestinal absorption, inhibited glyconeogenesis, and increased glycolysis follow, completing a complex of pathophysiological changes that if not promptly corrected results in death of the animal.
Effective management of viral diarrheas in young animals requires prompt action to prevent continued loss of fluids and electrolytes. This is most readily achieved on the farm by removal of milk from the diet. This reduces the amount of undigested lactose in the lumen of the intestine, and therefore reduces fluid loss and acidosis. Therapy also includes administration of balanced electrolyte solutions orally or parenterally. The use of intravenous fluid replacement and careful monitoring of dehydration status could save a large percentage of the most severely affected animals, but in many settings this is impractical.
In many cases of diarrhea in animals more than one virus is active; if two viruses have different sites of replication their combined effect may be severe. Furthermore, many bacterial infections, e.g., with enterotoxic E. coli, are more severe if combined with a viral infection.
Susceptibility decreases rapidly with increasing age; the viral diarrheas are essentially diseases of the first few weeks of life. To prevent infection of newborn animals, antibody must be present continuously in the lumen of the gut. This does not continue for more than about 7 days unless the dam is hyperimmunized against the common etiological agents. Since local immunization of the newborn is often not practical, this strategy of prevention should be actively promoted (see Chapter 14).
Pathogenesis of a Generalized Infection, Canine Distemper
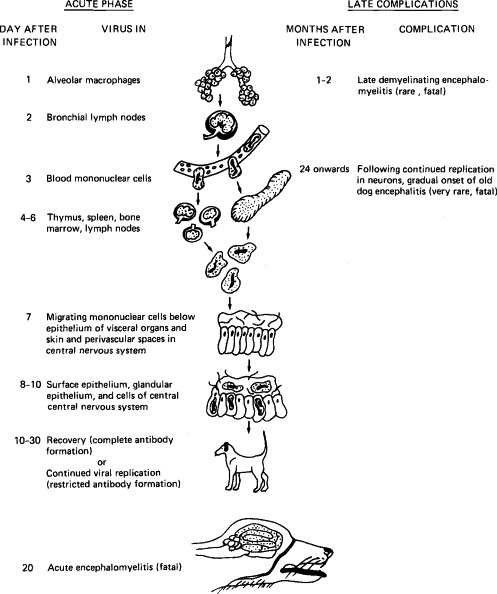
Canine distemper is caused by a virus of the genus Morbillivirus (family Paramyxoviridae), which is closely related to the viruses of measles and rinderpest. It usually causes an acute, self-limited systemic disease, but in some dogs virus invades the central nervous system and causes encephalitis. In a minority of dogs the virus persists in the brain and may cause the late neurological disease, old dog encephalitis (Fig. 10-3 ).
FIG. 10-3.
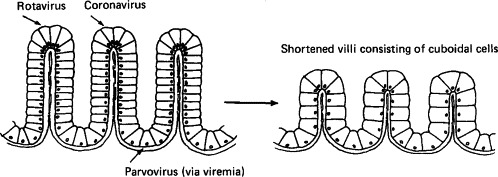
Diagram illustrating the pathogenesis of canine distemper. (Based on the work of Dr. M. J. G. Appel.)
Infection occurs via virus inhalation into the respiratory tract. Studies of the course of infection by fluorescent-antibody staining of tissues and organs have revealed that following initial infection of the respiratory epithelium and alveolar macrophages, virus is transferred within 2 days into mononuclear cells in the bronchial lymph nodes and tonsils. During the first week, before the onset of symptoms, cell-associated virus spreads through the bloodstream to the bone marrow, spleen, thymus, cervical and mesenteric lymph nodes, and macrophages in the lamina propria of the stomach and small intestine.
The rate of spread and distribution of virus after the eighth to ninth day varies, and appears to depend on the rate of development of neutralizing antibody, although the role of cell-mediated immunity has not been adequately studied. No antibody is found on the seventh day, but in some dogs the titer reaches 1:100 or higher on the eighth or ninth day. In such animals there is no further spread of virus, and virus disappears rapidly from the lymphatic tissues; the infection remains subclinical. If measurable antibody is not present on the ninth day or a titer of 1:100 has not been attained by the fourteenth day, virus spreads throughout the body. As well as continued infection of mononuclear cells in the lymphatic system, extensive infection of the epithelium in the intestinal, respiratory, and urogenital tracts, skin, and exocrine and endocrine glands occurs. Infection of the gastrointestinal tract causes vomiting and diarrhea, infection of the respiratory tract causes bronchitis and sometimes pneumonia, and infection of the skin is associated with dermatitis.
The brain is sometimes infected, usually when infection in visceral organs is subsiding. The virus appears first in meningeal macrophages and mononuclear cells in perivascular adventitia and later in ependymal cells, glial cells, and neurons. The infection of neurons is associated with behavioral changes, local myoclony, tonic–clonic spasms, and paresis, which often persist after recovery. Forty to sixty days after apparent recovery, some dogs suffer from postinfection encephalitis, with characteristic demyelination, often leading to death. In these dogs, high titers of neutralizing antibody occur in both blood and cerebrospinal fluid. In addition, very occasionally dogs that have recovered from distemper suffer from encephalitis years later—”old dog encephalitis.” Like subacute sclerosing panencephalitis in humans who have recovered from measles, this appears to be due to the very slow replication and spread of distemper virus in the brain. This complication, like acute canine distemper itself, has become more rare as distemper vaccination has become more general.
The course of acute canine distemper is affected by the extent of secondary bacterial infection, but this factor does not affect the central nervous system diseases. Recovery from canine distemper is followed by prolonged immunity, probably lifelong.
Pathogenesis of a Neurological Disease, Rabies
The pathogenesis of rabies is remarkable in that invasion, spread to the central nervous system, and the development of signs occur with mimimal immunological responses, yet even after infection has occurred, early administration of antibody and/or vaccine can often prevent disease.
Infection by the bite of a rabid animal usually results in deposition of rabies-infected saliva deep in the striated muscles, but rabies can occur, albeit with less certainty, after superficial abrasion of the skin. Initially virus replicates in the muscle cells or cells of the subepithelial tissues until it has reached a sufficient concentration to reach motor or sensory nerve endings in the muscle or skin. Here it appears to bind specifically to the acetylcholine receptor or other receptors and enters nerve endings. This begins the second phase of infection, in which neuronal infection and centripetal passive movement of viral nucleic acid within axons leads to involvement of the central nervous system. The incubation period—i.e., the time between the infective bite and the development of signs of central nervous system involvement—is usually between 14 and 90 days, but may occasionally be much longer, possibly because virus remains sequestered in striated muscle cells before entering peripheral nerves and ascending to the brain.
Although rabies proteins are highly antigenic, neither humoral nor cell-mediated responses occur during the stage of movement of virus from the site of the bite to the central nervous system, probably because very little antigen is delivered to the immune system; most is sequestered in muscle cells or within nerve axons. However, this early stage of infection is accessible to antibody and the classical Pasteurian postinfection vaccination (especially with the additional administration of hyperimmune immunoglobulin). Immunological intervention is effective during the long incubation period because of a delay between the initial viral replication in muscle cells and the entry of virus into the protected environment of the nervous system.
Movement along the nerves eventually delivers virus to the central nervous system, usually the spinal cord initially. An ascending wave of neuronal infection and neuronal dysfunction then occurs. Virus reaches the limbic system, where it replicates extensively, and the release of cortical control of behavior leads to “furious” rabies. Spread within the central nervous system continues, and when replication occurs in the neocortex the clinical picture changes to “dumb” rabies. Depression, coma, and death from respiratory arrest follow.
However, before this—and, indeed, coincidentally with its replication in the limbic system that leads to fury—virus moves centrifugally from the central nervous system down peripheral nerves to a variety of organs: adrenal cortex, pancreas, and, most importantly, the salivary glands. In the nervous system most virus is assembled upon cytoplasmic membranes within cells; the cells are not lysed, so that little viral antigen is released to stimulate host defense mechanisms. In the salivary gland, however, virions bud almost exclusively from plasma membranes at the luminal surface of mucous cells and are released in high concentrations into the saliva (Plate 10-3 ). Thus at the time when viral replication within the central nervous system causes the infected animal to become furious and bite indiscriminately, the saliva is highly infectious.
PLATE 10-3.
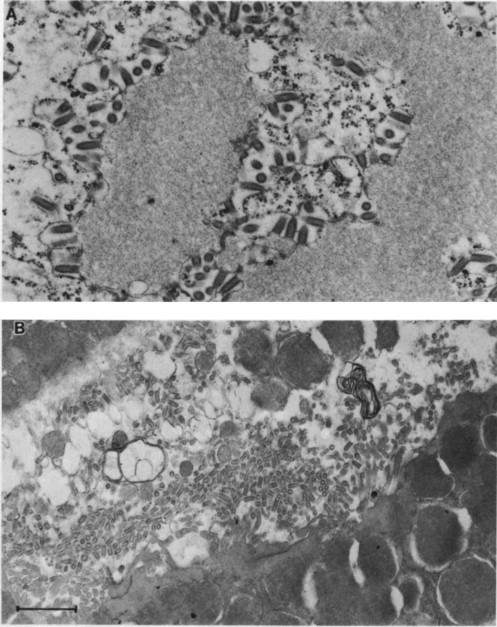
Electron micrographs of rabies virus infection in the brain (A) and salivary gland (B) (bar = 500 nm). In both organs the infection is noncytopathic, but in the brain nearly all virus is formed by budding upon internal membranes of neurons and so is trapped, while in the salivary gland nearly all virus is formed by budding upon the apical plasma membrane of mucous epithelial cells, where it is free to enter the salivary duct. Some reservoir host species can have 106 ID50 of rabies virus per milliliter of saliva at the time of peak transmissibility. (A) Street rabies virus from a dog in the cytoplasm of a neuron of a mouse 10 days after infection. Bullet-shaped virions are budding upon internal cellular membranes; the granular material is excess viral nucleocapsids forming an inclusion body which by light microscopy is seen as a Negri body. (B) Street rabies virus in the saliva of a fox. Bullet-shaped virions, having budded from mucous epithelial cells, are accumulating in the salivary duct where they are free to be transmitted in saliva injected during a bite.
On histopathological examination there is little evidence of brain damage, yet electron microscopic or fluorescent-antibody studies show that almost all neurons are infected. There is minimal cellular destruction to match the extensive neurological dysfunction seen in this disease.
FURTHER READING
- Appel M.J.G., Gillespie J.H. Canine distemper virus. Virol. Monogr. 1972;11(1) [Google Scholar]
- Babiuk L.A. Viral-bacterial synergistic interactions in respiratory infections. In: Kurstak E., editor. In “Applied Virology”. Academic Press; Orlando: 1984. p. 431. [Google Scholar]
- Babiuk L.A. Virus-induced gastroenteritis in animals. In: Kurstak E., editor. In “Applied Virology”. Academic Press; Orlando: 1984. p. 349. [Google Scholar]
- Lehmann-Grube F., Peralta L.M., Bruns M., Lohler J. Persistent infection of mice with the lymphocytic choriomeningitis virus. In: Fraenkel-Conrat H.C., Wagner R.R., editors. Vol. 18. Plenum Press; New York: 1984. p. 43. (In “Comprehensive Virology”). [Google Scholar]
- Mims C.A., White D.O. MRI principles. Blackwell Scientific Publications; Oxford: 1984. [Google Scholar]
- Murphy F.A. The pathogenesis of rabies virus infection. In: Koprowski H., Plotkin A., editors. In “The World's Debt to Pasteur”. Alan R. Liss; New York: 1980. p. 153. [Google Scholar]
- Koprowski H., Notkins A.L., Oldstone M.B.A., editors. “Concepts in Viral Pathogenesis.”. Springer-Verlag; New York: 1984. p. 153. [Google Scholar]
- Yates W.D.G. A review of infectious bovine rhinotracheitis, shipping fever pneumonia and viral-bacterial synergism in respiratory disease of cattle. Can. J. Comp. Med. 1990;46(225) [PMC free article] [PubMed] [Google Scholar]