

Abstract
Antiviral therapy is one of the most exciting aspects of virology, since it has successfully employed basic science to generate very effective treatments for serious viral infections. Table 1 lists selected examples of those human viral diseases for which there are established antiviral drugs. Therapy for human immunodeficiency virus (HIV) infection has demonstrated that the potential impact antivirals can have on a lethal, chronic infection with lifesaving therapy administered to more than 12 million individuals by 2015. This dramatic advance is about to be recapitulated for the treatment of hepatitis C virus (HCV) infection. The development of new antiviral drugs is very much a work in progress, with active drug discovery programs for filoviruses, coronaviruses, dengue, and others.
Keywords: Antiviral drugs, Drug resistance, Entry inhibitors, Integrase inhibitors, Interferons, Neuraminidase inhibitors, Nucleoside and nucleotide analogues, Polymerase inhibitors, Protease inhibitors
Antiviral therapy is one of the most exciting aspects of virology, since it has successfully employed basic science to generate very effective treatments for serious viral infections. Table 1 lists selected examples of those human viral diseases for which there are established antiviral drugs. Therapy for human immunodeficiency virus (HIV) infection has demonstrated the potential impact antivirals can have on a lethal, chronic infection with lifesaving therapy administered to more than 12 million individuals by 2015. This dramatic advance is about to be recapitulated for the treatment of hepatitis C virus (HCV) infection. The development of new antiviral drugs is very much a work in progress, with active drug discovery programs for filoviruses, coronaviruses, dengue, and others.
Table 1.
Viral Diseases for Which There Are Established Antiviral Drugs: Some Examples
| Virus Family | Specific Virus and (Disease) | Example of Drug | Mechanism of Action |
|---|---|---|---|
| Orthomyxovirus | Influenza virus (influenza) | Amantadine | Binds and blocks the H+ ion channel formed by the viral M2 proteins, prevents RNA uncoating; type A viruses only |
| Oseltamivir | Binds the enzymatic site on the viral neuraminidase, prevents cleavage of terminal sialic acid residues, and release of virions from infected cells; all influenza type A and B viruses | ||
| Retrovirus | HIV (AIDS) | Zidovudine (AZT) | Reverse transcriptase inhibitor; nucleoside analogue; prevents synthesis of DNA transcripts |
| Nevirapine | Reverse transcriptase inhibitor; nonnucleoside analogue; prevents synthesis of DNA transcripts | ||
| Atazanavir | Protease inhibitor; blocks processing of viral proteins | ||
| Maraviroc | Entry inhibitor; binds host cell CCR5 to inhibit binding of R5-tropic HIV to this coreceptor | ||
| Raltegravir | Integrase strand transfer inhibitor; blocks integration of linear dsDNA reverse transcript | ||
| Hepadnavirus | Hepatitis B virus (chronic hepatitis) | Tenofovir, emtricitabine | HBV DNA polymerase inhibitor as well as HIV reverse transcriptase inhibitor; nucleotide analogue; prevents synthesis of viral DNA |
| Hepacivirus | Hepatitis C virus (chronic hepatitis) | Sobosfuvir | Nucleoside analogue inhibitor of viral RNA polymerase (NS5) |
| Simeprevir | Protease NS3 inhibitor—blocks processing of viral polypeptide | ||
| Ledipasvir | Viral NS5A inhibitor—targets viral protein essential for replication but whose function is incompletely characterized | ||
| Herpesvirus | Herpes simplex (encephalitis) | Acyclovir | Viral DNA polymerase inhibitor; guanine derivative; prevents synthesis of DNA transcripts |
| Cytomegalovirus (retinitis) | Ganciclovir, valganciclovir | Viral DNA polymerase inhibitor; acyclovir derivative; prevents synthesis of DNA transcripts | |
| Poxvirus Adenovirus Polyoma virus |
Variola (smallpox) Adenovirus viremia BK virus in renal transplant patients |
Brincidofovir | Viral polymerase inhibitor; Cytosine derivative; prevents synthesis of DNA transcripts |
The conceptual approach to drug development is in flux. In the past, the primary focus has been upon virus targets, and this continues to be a very productive strategy. It is now being complemented by a wider set of approaches, so that present strategies include: compounds that target generic viral targets such as RNA or DNA synthesis and could be active against a range of different viruses and compounds that are directed against host cellular activities necessary for virus replication, which might target one or a spectrum of viruses (Figure 1 ). Furthermore, established methodologies for drug discovery are now supplemented with the use of large databases and evolving methods in computational biology. Finally, there is an increased emphasis on the repurposing of drugs already approved for human use, driven by the inordinate time and cost of drug development. This chapter explores all of these issues.
Figure 1.

The various scientific strategies for development of antiviral drugs. Left panel: direct-acting antivirals that target a specific viral protein and are aimed at a single virus target or a target for multiple viruses. This cartoon shows inhibitors of viral polymerases or proteases but other viral proteins may also be targeted. Right panel: drugs that target cellular processes that are essential for replication of one of several viruses. The cartoon shows several classes of inhibitors but there are many other cellular functions that could be targets.
Reconceived after Bekerman and Einav (2015). CypA: cyclophilin A.
We begin by discussing the mechanisms by which antiviral agents act, illustrated by selected examples. The presentation attempts to highlight the importance of viral pathogenesis for designing different therapeutic strategies for individual viral diseases. We continue with a brief discussion of pharmacodynamics and toxicity, critical hurdles that a safe and effective drug must pass. This section closes with a discussion of the new horizons in drug development. The pathway to drug development with all its challenges is next described, followed by an overview of those virus infections for which the most effective therapy is available. We conclude with a section on the future of antiviral therapy.
1. Principles of Antiviral Therapy
1.1. Virus Targets
Viral proteins. Current understanding of the molecular replication of individual viruses provides a detailed elucidation of the role of individual viral proteins. It is possible to map functional domains within viral proteins and to image their structures. These data can be used for “rational” drug design, either to synthesize small molecules that will bind to active sites on viral proteins, or to develop high-throughput screening procedures to test a very large battery of small molecules for those that block a specific activity.
HIV serves as a useful example, since there has been an exhaustive effort to develop antiviral drugs exploiting many of the viral proteins. Most of the anti-HIV drugs target one of the viral enzymes, either the reverse transcriptase, the protease, or the integrase (Table 2 ). In particular, there are many drugs that block reverse transcription, an enzymatic activity not expressed in normal cells. There are two classes of reverse transcriptase inhibitors: nucleoside and nonnucleoside (NRTIs and NNRTIs, respectively). NRTIs are compounds that are incorporated in to the nascent DNA chain and block its elongation. NNRTIs bind directly to the enzyme itself, inhibit its function, and may lead to its degradation. The other major enzymatic drug target is the viral protease that cuts gag and pol viral polypeptides to produce mature proteins. Protease inhibitors usually bind to the catalytic site on the protease molecule.
Table 2.
HIV Drugs Approved As of Early 2015
| Year Approved | Generic Name | Manufacturer |
|---|---|---|
| NRTIs (nucleoside reverse transcriptase inhibitors) | ||
| 1987 | Zidovudine (AZT) | GSK |
| 1991 | Didanosine (ddI) | BMS |
| 1992 | Zalcitabine (ddC)a | Roche |
| 1994 | Stavudine (d4T) | BMS |
| 1995 | Lamivudine (3TC) | GSK |
| 1998 | Abacavir | GSK |
| 2001 | Tenofovir | Gilead |
| 2003 | Emtricitabine (FTC) | Gilead |
| NNRTIs (nonnucleoside reverse transcriptase inhibitors) | ||
| 1996 | Nevirapine | Boehringer Ingelheim |
| 1997 | Delavirdine | Pfizer |
| 1998 | Efavirenz | BMS, Merck |
| 2002 | Etravirine | Tibotec, J&J |
| 2011 | Rilpivirine | Tibotec, J&J |
| PIs (protease inhibitors) | ||
| 1995 | Saquinavir | Roche |
| 1996 | Ritonavir | Abbott |
| 1996 | Indinavir | Merck |
| 1997 | Nelfinavir | Pfizer |
| 1999 | Amprenavira | GSK |
| 2000 | Lopinavir and ritonavir | Abbott |
| 2003 | Atazanavir | BMS |
| 2003 | Fosamprenavir | GSK |
| 2005 | Timpranavir | Boehringer Ingelheim |
| 2006 | Darunavir | Tibotec, J&J |
| Entry inhibitor | ||
| 2003 | Enfuvirtide | Roche |
| CCR5 antagonist | ||
| 2007 | Maraviroc | Pfizer |
| Integrase strand transfer inhibitors | ||
| 2007 | Raltegravir | Merck |
| 2012 | Elvitegravir (as combination, see below) | Gilead |
| 2013 | Dolutegravir | GSK |
| Nucleoside reverse transcriptase inhibitor combinations | ||
| 1997 | Zidovudine and lamivudine | GSK |
| 2000 | Abacavir, zidovudine, and Lamivudine | GSK |
| 2004 | Abacavir and lamivudine | GSK |
| 2004 | Tenofovir and emtricitabine | Gilead |
| Multiclass combinations | ||
| 2006 | Efavirenz, emtricitabine, and tenofovir | BMS and Gilead |
| 2011 | Rilpivirine, emtricitabine, and tenofovir | Gilead |
| 2012 | Elvitegravir, eobicistat, emtricitabine, and tenofovir | Gilead |
Manufacturer: GSK, GlaxoSmithKline; BMS, Bristol-Myers Squibb; J&J, Johnson & Johnson.
Drug has been withdrawn.
In addition, there are drugs that inhibit other steps in the HIV replication cycle. The initial step in HIV cellular entry is binding of the viral gp120 to the cellular coreceptor (see Chapter 9, HIV/AIDS). HIV gp41 then undergoes a conformational change that exposes its N-terminal fusion domain, which inserts into the plasma membrane of the host cell. Close to the N terminus of gp41 is a heptad repeat (HR1) that forms a three-helix bundle. HR1 associates with another three-helix bundle (HR2) at the C terminus of gp41, which forces the molecule into a hairpin configuration. A synthetic oligopeptide analogue of HR2, enfuvirtide (originally called T20), can bind to HR1 and prevent this hairpin formation, thereby blocking HIV-1 cellular entry. Enfuvirtide has been shown to be active in HIV-1-infected patients who have “failed” other anti-HIV drug therapy (Lazzarin et al., 2003).
Viral mutagens. The survival of viruses depends in part on their ability to evolve in response to antiviral pressures, such as host immune responses. From this perspective, the rapid mutational rate of RNA viruses, in particular, facilitates the selection of fitness mutants among an ever-present swarm of genetic variants. In theory, the polymerases of RNA viruses have evolved to an optimal balance of processivity and fidelity (mutational rate), which permits the generation of large numbers of progeny with many genetic variants. These variants facilitate rapid adaptation to selective pressures, such as immune responses and drug treatment.
A counterintuitive approach to antiviral drugs is the use of mutagens that can increase viral mutational rate, so that fit variants are overwhelmed by less fit or nonsense mutants leading to a lethal accumulation of errors or “error catastrophe” (discussed in Chapter 17, Virus Evolution). At least one antiviral drug, ribavirin, a nucleoside analogue, has been proposed to act through this mechanism. It has been shown to both increase mutation rate and decrease the production of infectious particles in several viruses including poliovirus and Hantaan virus.
RNA interference (RNAi). In the mid-1990s, RNA interference was discovered serendipitously when an attempt to overexpress specific plant genes, using viral vectors, instead resulted in the knockout or silencing of those genes. RNAi is described in more detail in Chapter 3, Concepts of viral pathogenesis. Its potential as an antiviral therapeutic has been shown in a proof-of-concept clinical trial for hepatitis C (Figure 2 ).
Figure 2.
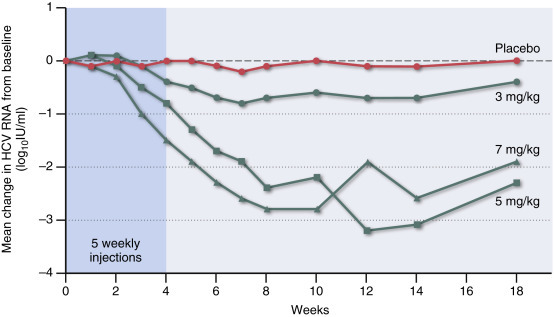
MicroRNA (miRNA) treatment has the potential as a treatment modality for selected human viral infections. This example is a trial in which microRNA-122 was targeted for the treatment of HCV. This miRNA is expressed by liver cells and the replication of HCV is dependent upon a functional interaction of its genome with miRNA-122. Shown are the mean changes in HCV RNA levels from baseline for patients receiving 3, 5, or 7 mg of miravirsen (an antisense oligonucleotide that sequesters miRNA-122) per kilogram of body weight, as compared with placebo. Miravirsen was administered in 5 weekly subcutaneous injections during the first 29 days of the study (shading). The dashed line indicates no change from baseline.
Redrawn from data in Janssen et al. (2013).
1.2. Cellular Targets
Cellular targets have a theoretical advantage over viral targets since they will not undergo escape mutations. On the other hand, targeting cellular molecules can interfere with vital host functions. This could be a problem for long-term treatment, but might not be a critical impediment for acute infections. Cellular targets have not been a major focus in the past but are now receiving increasing attention (Table 3 ). Techniques for identifying potential cellular targets are covered in Chapter 11, Systems Virology and Chapter 12, The Virus–Host Interactome.
Table 3.
Antiviral Drugs Directed against Cellular Targets
| Drug | Virus | Mechanism of Action | Clinical Status | References |
|---|---|---|---|---|
| Maraviroc | HIV | Blocks CCR5 coreceptor | Approved | Fatkenheuer et al. (2005) |
| Cyclosporin A and related compounds | HCV | Binds and inactivates cyclophilin A, a cis–trans isomerase | Phase III trials | Lin and Gallay (2013) |
| Deoxynojirimycin, Castanospermine | Enveloped RNA viruses | Binds and blocks ER-resident glucosidases essential for virus maturation | Approved for other indications Phase II trials |
Chang et al. (2013) |
| Erlotinib Sunitinib |
HCV | Binds and blocks AP2M1, a subunit of adaptor protein complex, required for maturation of HCV virions | Research | Neveu et al. (2012) |
| Nilotinib | Ebola | Blocks c-Abl/1 tyrosine kinase, required to phosphorylate Ebola virus proteins | Research | Garcia et al. (2012) |
CCR5 (chemokine receptor 5) is a coreceptor for HIV (see Chapter 9, HIV/AIDS). Some individuals are homozygous for a mutation in CCR5 (the “delta 32” mutation) that prevents the expression of this host gene. These persons do not show any ill effect from this mutation and are very resistant to HIV infection. These observations suggest that a small molecule that blocks the receptor domain on CCR5 might safely be used to treat HIV infection, without interfering with any essential cellular functions (Fatkenheuer et al., 2005). Maraviroc is such an inhibitor and has been proven effective for HIV treatment.
Cyclosporin A (CsA) is an approved drug that binds to—and inactivates—a cellular molecule, cyclophilin A. Cyclophilins are a family of cis–trans isomerases that convert prolines from the trans to the cis form. As a family, cyclophilins play an essential role in many cellular processes that require protein folding and trafficking, and this activity is essential for the replication of HCV. Interestingly, CsA, which is approved as an immunosuppressive drug, was discovered serendipitously to have strong activity against HCV (Lin and Gallay, 2013). Of note, mice with cyclophilin A knockouts are generally healthy, so this molecule does not seem to be essential for life (at least in a shoebox).
Another focus of cellular targets has been the attempt to develop a pan-virus strategy for antiviral therapeutics. One example was directed at phosphatidylserine, an anionic phospholipid that is located on the inner leaflet of the plasma membrane but exposed in virus-infected cells. An antibody directed against anionic phospholipids interfered with the replication of arenaviruses (Soares et al., 2008). Antibody treatment of infected guinea pigs spared them from a potentially lethal infection with Pichinde, an arenavirus.
1.3. Viral Pathogenesis and Antiviral Strategy
The pathogenesis, transmission, and epidemiological characteristics of individual viruses are important determinants of the potential efficacy of antiviral drugs. Viruses that have a very short incubation period and generation time, and spread very rapidly, tend to be poor candidates for antiviral treatment because it is difficult to complete diagnosis and initiate therapy in a timely fashion. Influenza is a good example of a serious illness with a short incubation period (18–72 h). Neuraminidase inhibitors are quite effective anti-influenza drugs but need to be given prior to infection or very soon after symptoms appear. This drawback may be overcome under certain circumstances; in the presence of a pandemic wave of influenza that is spreading across a community, antiviral drugs could be widely administered as a short-term prophylactic, thereby anticipating potential infection.
Persistent viral infections that cause significant chronic illness are attractive targets for antiviral treatment. Their slow course permits an accurate diagnosis and evaluation prior to initiating therapy. Furthermore, there are a few persistent infections, such as hepatitis B and hepatitis C, which carry a long-term risk of liver failure or hepatocellular cancer. With an estimated >150 million cases of hepatitis C and >350 million cases of hepatitis B globally, these diseases constitute significant opportunities for treatment. Effective therapeutic intervention, particularly long-lasting viral suppression or even viral clearance (a “cure”), would significantly reduce the disease burden.
In many persistent infections, there is a dynamic balance between the persistent virus and host defenses. Thus, some persons infected with hepatitis B virus (HBV) are able to clear the infection even after years of persistence. This pattern has two implications. First, it suggests that antiviral therapy might tip the balance in favor of the host and lead to viral clearance, and second, it suggests that antiviral antibodies might be used in synergy with antiviral drugs to improve the therapeutic outcome.
Therapeutic antibody. Neutralizing antibodies are a major mediator of the preexposure protection conferred by many established viral vaccines (see Chapter 19, Viral Vaccines). In addition, antibodies induced during primary infection play a role in clearance and recovery from certain acute viral diseases. Therefore, it is plausible that passive antibody administered during acute viral infection might be therapeutic.
Primary infection with West Nile virus (WNV), a flavivirus, is one example. Following transmission by mosquito bite, WNV initiates a plasma viremia followed by invasion of the central nervous system, resulting in potentially fatal encephalitis. Experiments in immunologically deficient mice have shown that both antibody and cellular immunity play a role in the outcome of infection. Surprisingly, antibody also plays a role in the clearance of virus from the central nervous system, even when infection has been well established in neurons. When administered to mice undergoing acute WNV encephalitis, passive antibody can markedly improve survival, at least under experimental conditions. Recently, it has been found that neutralizing antibodies are very effective when used for treatment of acute infection with Ebola virus in an animal model (see below).
Interferons (IFNs) and interferon inducers. Type 1 interferons (IFN-α and -β) are an important component of the innate immune response (discussed in Chapter 4, Innate Immunity). IFNs induce a complex pleiotropic response that inhibits viral replication in several different ways, in addition to activating antigen-specific adaptive immune responses. Use of exogenous IFN as an antiviral therapy has been tried for many viral infections but—to date—has been used mainly for the treatment of two persistent infections, HCV and HBV (discussed below). However, IFN therapy has substantial unwanted side effects in humans, which limit its practical utilization.
Cytokine storm. Several serious viral infections of humans produce disease via an excessive or imbalanced host response, leading to intense dysregulation of proinflammatory cytokines and chemokines. This cytokine storm can cause a range of serious disease manifestations. These include acute lung injury in association with respiratory viruses such as highly pathogenic influenza viruses (avian H5N1 and 1918 H1N1) and SARS, or a shock syndrome in fatal cases of Ebola virus infection. Recent studies of the host–virus interactome (see Chapter 12, The Virus–Host Interactome) have made it possible to identify some of the components of this pathological host response. Such insights may be useful in formulating new therapies to ameliorate an excessive deleterious innate response.
1.4. Drug-Resistance Mutations
Drug-resistant viral mutants constitute a major problem in antiviral therapy. The frequency of resistant mutants varies widely (see Chapter 17, Virus Evolution) and is determined by a number of factors (Sidebar 1 ).
-
1.
RNA viruses have a mutation rate estimated at 10−4 (1 mutation in 10,000 base replications) that is much higher than the rate for DNA viruses (10−8); the difference reflects poor fidelity, as well as the absence of cellular proofreading mechanisms for RNA polymerases.
-
2.
The replication rate of the virus during a specific infection will vary widely and influence the rate at which mutant virions are produced. For instance, it has been estimated that during an HIV-1 infection, 108–1011 virions are produced daily; this would yield 104–107 virions with single point mutations (or an average 1–1000 mutants for each of the 10,000 bases) each day. At the other end of the scale, human papillomavirus (HPV, a DNA virus) replicates very slowly in vivo, so that very few mutant virions would be synthesized daily. These differences are reflected in the observation that individual primary HIV isolates consist of a “swarm” of viruses the sequences of which—after several years of infection—vary from 5% to 10%, for different genes. In contrast, primary isolates of DNA viruses show much less variation.
-
3.
Different classes of drugs target diverse viral functions that vary in their importance for viral replication, and individual drugs vary in the degree to which they can block their targeted function. Furthermore, resistant mutants vary in their ability to replicate in the presence of the drug and also—absent drug—in their replicative capacity or fitness. These nuances are reflected in the observation that different HIV-1 NRTIs—which are directed against the same viral function—select for different escape mutations.
-
4.
The in vivo selective pressure of a specific drug will depend upon both its intrinsic ability to block an essential virus function and its pharmacodynamics, which will determine its actual concentration at sites of viral replication. As the selective pressure increases, the relative advantages of mutants increase, but the rate of replication of wild-type virus decreases. As replication diminishes, the probability that resistant viruses will emerge diminishes. The selection of escape mutants is maximized when the drug concentration is high enough to select for resistant mutants, but not so high that it substantially inhibits virus replication (see Chapter 17, Viral Evolution).
Sidebar 1. Determinants of antiviral drug resistance.
-
•
Variation in viral mutation rate, which is about 10,000-fold greater for RNA than DNA viruses.
-
•
Variation in in vivo viral replication, which determines the rate at which mutants are generated.
-
•
Variation in the structural mechanism of drug-mediated viral inactivation, which determines the frequency and fitness of resistant mutants.
-
•
Variation in drug-mediated selective pressure in vivo, which determines the relative replication rates of wild-type and mutant viruses.
-
•
Concurrent use of several drugs that act upon different viral functions will markedly reduce the frequency of resistant virions, since these must possess multiple mutations.
One important implication of the foregoing considerations is the potential advantage of multidrug therapy. If a virus has to replicate in the presence of three diverse drugs each of which select for different resistance mutations at a frequency of 10−4, then triple mutants (assuming no interaction between various mutations) would occur at 10−12, which might be a very rare phenomenon. In the case of HIV-1, there has been a comparison of multiple drug therapy as new compounds have been introduced (Tang and Shafer, 2012). There is a dramatic stepwise increase in efficacy with each additional drug (Figure 3 ). For HIV-1, triple drug therapy is usually required to suppress viral replication and minimize escape mutations.
Figure 3.

Virus mutants may escape a single or even two drugs, but are much less likely to escape from three or more drugs administered simultaneously. HIV-infected treatment-naïve patients were started in regimens of two nucleosides (azidothymidine (ZDV) and lamivudine (3TC)), the protease inhibitors indinavir (IDV), or the combination of all three drugs. The decrease in the level of blood HIV RNA from baseline is shown. The rises in plasma HIV RNA after several months in the nucleoside and protease only arms was associated with the emergence of resistance mutations in the genes of the respective target enzymes.
Drawn from data in Gulick et al. (1997).
1.5. Pharmacodynamics
A critical aspect of drug efficacy is its behavior in vivo. Many compounds that appear active in cell culture systems fail when tested in animals. The pharmacodynamics of a drug depends on at least the following parameters: (1) Is the compound soluble? (2) Can it be absorbed if given by mouth or does it require injection or even intravenous administration? (3) Is the drug active in its administered formulation or does it require biochemical processing in the liver to be activated? (4) How fast is the compound released into the blood? Does it circulate as a free molecule in plasma or does it bind to albumin or other plasma proteins? (5) Where does the drug act, in blood or in specific target tissues? How fast does it enter target tissues? (6) What is the half-life of the compound in blood and in tissues? (7) Is the drug inactivated in the liver or excreted in the urine or intestinal tract? (8) Does the drug achieve therapeutic levels in blood or target tissues? What is the dosage regimen required to maintain therapeutic levels? Thus, a complex set of experiments must be conducted to determine whether a candidate compound meets pharmacodynamic criteria that make it practical for use.
Related to the pharmacodynamics and often studied in concert is toxicity. Usually, a standard battery of tests is conducted to detect unwanted side effects of a candidate drug. Toxic effects may be unpredictable from the mode of action of an antiviral compound. Both pharmacodynamic and toxicity studies are required by the Food and Drug Administration as part of the application for an IND (Investigational New Drug) which must be obtained prior to Phase I trials in humans (see below).
1.6. New Horizons for Drug Development
A new era for drug development is dawning, based on advances in computational biology and large public databases. Also, there is a broadened conceptual view, to include the host–pathogen interactome, cellular targets, and drug repurposing (Casadevall and Pirofski, 2015). Major incentives are the inefficiencies in established development pathways, the many years required, the inordinate cost, and the low yield, discussed below. Table 4 lists some recent publications where diverse applications of computational biology are proposed for drug discovery. Several examples will illustrate the approaches being taken.
Table 4.
Computational and Big Data Strategies for Drug Discovery: Some Recent Publications
| Category | Message | References |
|---|---|---|
| General | Big data: hype versus utility | Hu and Bajorath (2014) |
| General | Integration diverse data on human diseases: network-based models | Berg (2014) |
| General | Mathematical modeling: network-based multiscale strategy | Wangand Diesbock (2014) |
| General | Broad spectrum antiviral drugs | Beker et al. (2015) |
| General | Using systems biology to find therapeutic targets | Dopazo (2013) |
| Platform | Open innovative drug discovery platform for data mining | Alvim-Gaston et al. (2014) |
| Platform | Activity-based protein profiling to identify antiviral targets | Blais et al (2013) |
| Platform | Using host–pathogen interactome to identify antiviral targets | Brown et al. (2011) |
| Cellular antiviral | Genomics screen to identify antiviral proteins | Brass et al. (2009) |
| Cellular antiviral | Used siRNA and other omics to identify cellular antiviral factors | Munk et al. (2011) |
| Cellular antiviral | Genomic screen to identify cellular factors critical for virus replication | Schwegman et al. (2008) |
| Repurposing | Repurposing: computational methods | Jin and Wong (2014) |
| Repurposing | Computational data mining of publicly available databases | Law et al. (2013b) |
| Repurposing | Computational methods for data mining FDA-approved drugs | Ekins et al. (2011) |
| Repurposing | Used tissue culture of MERS coronavirus to screen FDA-approved drugs | Dyall et al. (2014) |
| Metabolomics | Inventory of drugs that alter metabolism | Fillet and Frederich (2014) |
| Toxicity | Used gene expression profiling to detect side effects | Verbist et al. (2015) |
| Structural | Example of computational designed inhibitor of a viral anti-host cell protein | Procko et al. (2014) |
| Structural | CANDO: inventory of protein–protein interactions | Minie et al. (2014) |
One such computational approach was used by Josset et al. (2014) when studying the transcriptome response to virulent influenza A viruses, H7N9, H5N1, and seasonal H3N2 viruses, in a human lung epithelial cell system. To identify potential antiviral compounds, a data-based approach was used which relies on the assumption that an effective drug would have the inverse effect on cellular transcriptional response to that of the targeted virus. Using a publically available database, Connectivity Map, which contains thousands of gene expression profiles from over a thousand compounds, several drugs were identified as potential antivirals against the H7N9 virus. These included cellular kinase inhibitors as well as some FDA-approved drugs, such as troglitazone and minocycline. A similar approach has been taken for a number of other viruses, such as dengue, HIV-1, and hepatitis C (Munk et al., 2011; Brass et al., 2009).
The CANDO (computational analysis of novel drug opportunities) platform focuses on potential interactions between small molecules and proteins of interest (Minie et al., 2014). Input comes from a variety of sources, including structural homologies, curated databases, and other information in the scientific literature. The goal is to identify small molecules that might interact with specific proteins and thereby interfere with their function. This could be applied to blocking viral functions or host proteins critical to viral replication.
A set of cell lines were used by Verbist et al. (2015) to test the effect of over 700 drug candidates on gene transcripts using a microarray readout. The focus was on a predetermined set of genes, some of which—if upregulated—could be toxic, while others—if upregulated—could be therapeutic. The data were used to make some go/no-go decisions to aid in selection of compounds for further investigation.
Broad-spectrum antiviral drugs. Some viruses share steps in their replication strategies so it would be possible to design direct-acting antiviral compounds that would inhibit families of viruses in contrast to a single target. Several compounds that inhibit RNA polymerases are being investigated for their potential to treat a number of viruses (Warren et al., 2014; Furuta et al., 2013). Brincidofovir, a nucleotide analogue that can block the action of DNA polymerases, has been proposed as a candidate treatment for several dsDNA viruses (Florescu et al., 2014).
Antiviral cellular targets. As noted above, there are examples of cellular targets that—if compromised—will inhibit individual or classes of viruses (Table 3). How does a system’s approach exploit this potential opportunity for drug discovery? First, it offers a systematic approach to identifying cellular enzymes, proteins, or processes that are essential for the replication of a virus. Second, it provides a broad-based approach—using siRNA, knockouts, or other methods—to determining which potential cellular targets may be “expendable.” Third, it will enable a search for potential inhibitors of these cellular targets. Fourth, it can be used for screens to select anticellular compounds for their ability to inhibit virus replication in vitro or in vivo.
Although these methods have not yet produced approved therapeutics, the various examples cited in Table 3, Table 4 demonstrate the potential for these newer approaches to drug discovery.
2. Examples of Antiviral Therapy
How are new drugs developed? We begin with a short discussion of the process of drug discovery. To give a sense of the scope and diversity of approved drugs, and the variables that influence their efficacy, this section presents some examples of approved antiviral therapy. More detailed information is available in clinical texts and reviews (see Further Reading).
2.1. Challenges of Drug Development and Utility of Drug Repurposing
New antiviral drugs follow a well-worn developmental pathway that is usually required before drugs are approved for use in human subjects. Overall this is a very slow, cumbersome, and expensive process, which can take at least 10 years and cost more than $1 billion. Figure 4 shows a typical pathway for drug development in the United States. What the figure does not show is the cost of all the candidate compounds that never make it to market, because they fail to pass one of the successive hurdles shown in Figure 4. It is guesstimated that only 1 in 20 compounds makes it from the beginning to the end of this process.
Figure 4.
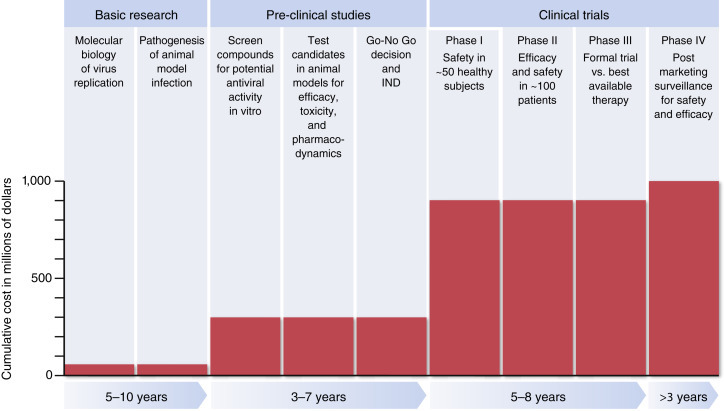
The pathway to development of a new antiviral drug is slow and expensive. The figure shows typical parameters for drug development.
Based on data in Tufts (2014).
The drug development process can be divided into three phases: basic research, preclinical studies, and clinical trials. Basic research involves identification of a druggable target molecule or step in viral replication. Preclinical studies require an informed or high-throughput search for compounds that will inactivate the target molecule, and evidence that it will work in an animal model of the viral disease under study. In animal models, there are a complex set of pharmacological parameters that must meet practical standards, including the dose and route of administration, the pharmacodynamics and frequency of administration, the concentration in blood or key target organs, and possible requirement for activation in the liver. Care must be taken to search for possible toxic effects at therapeutic drug levels.
Once past these steps, the compound becomes eligible for testing in humans, assuming that an IND approval can be obtained from the USA Food and Drug Administration. Typically, there are three sequential phases of clinical trials: Phase I focuses on safety; Phase II on some parameter that can serve as a surrogate for efficacy; and Phase III-controlled trials in human populations to determine efficacy and safety.
There are several consequences of this slow and expensive process. In most countries, the early basic research that identifies potential druggable targets takes place at not-for-profit research institutions and is funded by both government and private organizations. Further drug development is usually conducted by for-profit pharmaceutical or biotechnology companies. Considering the cost, there is an understandable reluctance to develop drugs unless there is a remunerative potential market. AIDS and hepatitis C are good examples of diseases for which there is an attractive market, while Ebola is an example of an orphan disease for which there was a very limited potential market—at least until the pandemic of 2013–2015.
Drug repurposing. The impediments to new drug development have activated the field of drug repurposing. There are large numbers of drugs, directed against cellular pathways, which have been approved by the U.S. Food and Drug Administration for human use. It is possible that some of them could also have antiviral activity, either by blocking a cellular pathway critical for virus replication or by ameliorating the innate response to a virus which—in some instances—contributes to disease pathogenesis (Ekins et al., 2011). If a previously approved drug can be identified as an effective antiviral, this “shortcut” in the drug development pathway could be quicker and less expensive (Dyall et al., 2014).
There are a number of strategies for drug repurposing, some of which are noted in Table 4. One example is cyclosporin A, described above, as a compound that inhibits the maturation of HCV. A non-immunosuppressive derivative of cyclosporin A, that maintained activity against HCV was included in a trial of patients with HCV-induced inflammatory liver disease. It induced an antiviral response due to its anti-cyclophilin A activity.
2.2. Influenza Virus
Influenza is one of the most prevalent viral diseases, affecting an estimated 10–20% of the population annually, with 3–5 million cases of severe respiratory illness and 0.5–1 million deaths. In the United States alone, there is an annual excess mortality of about 35,000 attributed to influenza. As explained above, the pathogenesis of influenza—its very short incubation period of 18–72 h and its acute course—makes it a difficult target for antiviral therapy.
Inhibitors of viral entry (M2 inhibitors). As one step in its cellular entry pathway, influenza virus is endocytosed into an acidic vacuole; an H+ ion channel formed by the viral M2 proteins then facilitates acidification of the interior of the virion, which in turn permits dissociation of the matrix protein from the ribonucleoprotein viral core that enters the cytosol and initiates replication. Amantadine and rimantadine, related drugs, bind in the ion channel of influenza A viruses and prevent the final step in viral entry. These drugs have been proven effective for both prophylaxis and therapy; however, drug-resistant mutants are frequently isolated from patients after a few days of therapy. Moreover, all circulating influenza viruses over the past decade have maintained resistance to this class of drugs.
Inhibitors of virus release (neuraminidase inhibitors). As influenza virus buds from a host cell, the viral hemagglutinin binds to receptors on the cell surface, which contain N-acetylneuraminic (sialic acid) residues. Release of virions is accomplished by action of the neuraminidase on the viral surface, which cleaves terminal cellular sialic acid residues and frees virions to spread to adjacent uninfected cells. The neuraminidase inhibitors, oseltamivir and zanamivir, which are sialic acid analogues, bind to the catalytic site on the neuraminidase, thereby inhibiting viral release and spread. The active domain of the neuraminidase is highly conserved in order to maintain this enzymatic function; however, escape mutants emerge after monotherapy in patients with prolonged virus shedding (most often children and immunosuppressed individuals). Some influenza virus strains carrying resistance mutations to this class of drugs have circulated successfully.
Not surprisingly, influenza drugs have limited efficacy in clinical application. If administered within 48 h of the onset of symptoms, each of these drugs reduced the duration of symptoms by about 1 day and reduced the time to return to normal activity by about 1 day also.
2.3. Human Immunodeficiency Virus
There has been a greater research investment in antiviral drugs for HIV than for any other virus, because the large number of infected persons, the persistent nature of infection, and the 100% fatality rate among untreated patients created an ethical imperative and offered a very lucrative market for effective therapies (Gunthard et al., 2014). As a result, there is a panoply of FDA-approved products (Table 2). The action of these drugs has been described in the foregoing section on antiviral compounds. The ability to effectively treat this inevitably fatal infection is one of the great triumphs of scientific medicine. It is now guesstimated that a subject age 20 who is recently infected with HIV and is on optimal HAART (highly active antiretroviral therapy) has a life expectancy of 50 years (contrasted with 5 years absent treatment). However, present regimens that control HIV replication do not provide a “cure” since they fail to eradicate latent HIV genomes (see Chapter 9, HIV/AIDS).
HIV drug resistance. The loss of susceptibility of isolates from subjects treated with the first antiretroviral as monotherapy, AZT, was associated with the cumulative acquisition of mutations in the target gene of this NRTI, the reverse transcriptase. As explained above, a combination of three different drugs is needed (Figure 3). Protease or integrase inhibitors are usually included in combination with two NRTIs in antiretroviral regimens for initial therapy. The development of fixed dose combinations of three drugs in a single capsule is an important practical advance, which has enhanced patient compliance and become a standard regimen. At present, over 10 million people receive a single pill once daily to treat HIV (Table 2).
For HIV prevention, another important application of antiretroviral treatment is preexposure prophylaxis (PreP). In several trials, PreP appears to offer a potentially powerful adjunct to treatment of infected patients, particularly in a high exposure context (Cohen, 2015b).
Persistence of HIV genomes. When HIV-infected patients initiate potent combination therapy, the titers of viral RNA in plasma drop in a biphasic manner (see Figure 7 in Chapter 9, HIV/AIDS). There is an early and rapid decline lasting about 1 week and a slower decline lasting several weeks, which then plateaus often below the limits of detection. However, if treatment is stopped, viremia inevitably reappears, usually in a few weeks, indicating that viral eradication has not been achieved. Even if continuing low-level HIV replication is completely suppressed, persistence of HIV, in the form of latent proviral genomes in resting CD4+ T cells, precludes eradication with present regimens. Substantial investigative efforts have been initiated to identify approaches to eradicate the latent reservoir, which would require activating latent HIV genomes and killing the infected cells (Cohen, 2015a). However, this is at best a long-term initiative.
2.4. Hepatitis C Virus
HCV is an important cause of human disease, since it is estimated that there are >150 million persistently infected humans worldwide, many of whom develop chronic hepatitis, liver failure, or hepatocellular cancer. HCV is a hepacivirus, a positive-stranded RNA virus, in the flavivirus family. HCV has presented a difficult experimental challenge because there are limited cell culture systems (recently improved) and the only animal model is the chimpanzee.
The natural history of HCV is clouded by the insidious, asymptomatic nature of infection. About 25% of patients eliminate the virus within 3–24 months after infection. Of persistently infected patients, about 50% experience little chemical or clinical evidence of disease, while 50% have chronic hepatitis that can progress to end-stage cirrhosis or hepatocellular cancer over a period of 5–40 years. These data indicate that in many patients there is a delicate balance between HCV and host defenses, supporting the notion that antiviral therapy could lead to viral clearance.
Prior to 2014, treatment with HCV focused on a combination of ribavirin and pegylated (polyethylene glycol-conjugated) interferon. IFN-α2 produces a virological response that is sustained for many months after ending a course of treatment, with a substantial number of “cures” (about 75% of patients infected with genotype 1 HCV). However, this treatment strategy caused serious toxic side effects associated with long-term administration of interferon.
The recent discovery and development of drugs against HCV has produced a revolution in therapy (De Clercq, 2014; Rice and Saeed, 2014). These compounds are called direct-acting agents (DAA) to differentiate them from interferons. Interferon-free regimens of drugs that target HCV polymerase, protease, and the NS5A and NS5B proteins can result in viral clearance without relapse. This is effectively a cure in 90–100% of those treated (Petta and Craxi, 2015). These recent successes are energizing the development of other antiviral drugs.
2.5. Hepatitis B Virus
As mentioned above, the course of HBV infection is variable and a high proportion of patients spontaneously clear the virus (see Chapter 7, Patterns of Infection). Persistent HBV infections can be divided into several categories. Patients with lower virus titers (<105 HBV DNA copies per ml serum) and normal liver function tests (serum alanine aminotransferase) are not usually treated since they are at relatively low risk of end-stage liver disease (cirrhosis or hepatocellular carcinoma). Treatments, with an IFN and either a nucleoside or a nucleotide reverse transcriptase inhibitor, are used for patients with more severe disease. IFN-α administered parenterally for months to years induces a therapeutic response in about one-third of patients. However, present regimens do not clear the virus and replication resumes after interruption of drug therapy. In view of the toxic effects of prolonged interferon treatment and the low response rate of present therapies, there was a need for improved treatments. The development of potent nucleoside (entecavir) and nucleotide (tenofovir) HBV polymerase inhibitors has provided more tolerable and effective long-term treatments.
A major obstacle to a cure of HBV is the existence in infected hepatocytes of covalently closed circular (ccc) DNA, which maintains the viral genome as a nuclear episome. One approach is the use of the CRISPr/Cas9 system to introduce DNA nucleases specific for cccDNA (Kennedy et al., 2015; Dong et al., 2015; Ahmed et al., 2015), although the delivery of this system in vivo is a challenge. A number of drugs directed against HBV enzymes are in use or under development; they suppress HBV replication and reduce the level of cccDNA episomes (Shi et al., 2015; Tavis et al., 2015; Liu et al., 2015). Finally, there are newly developed systems for screening anti-HBV compounds (Suresh et al., 2015; Ishida et al., 2015). The estimated >350 million HBV infections worldwide provide a major incentive for research and development, and the current investment in the development of HBV drugs holds promise for the introduction of curative regimens.
2.6. Herpesviruses
There are several human herpesviruses that cause considerable morbidity (and rare mortality), including herpes simplex virus (HSV, cold sores and genital lesions), varicella zoster virus (VZV, chicken pox and herpes zoster), and cytomegalovirus (CMV, retinitis and other complications in immunosuppressed subjects). Herpesvirus infections offer special challenges for treatment because they cause persistent lifelong latent infections with intermittent activation of active replication and accompanying disease. During the latent phase viral DNA is maintained in the nucleus, making it unlikely that effective therapy can eradicate the virus. Therefore, treatment is directed at the symptomatic phase of infection.
Herpesviruses are large DNA viruses that encode their own DNA polymerases, which are required for transcription of their genomes. Acyclovir, the first highly effective compound for the treatment of HSV and VZV, is a nucleoside analogue DNA chain terminator. Acyclovir undergoes in vivo activation by addition of three phosphates to its side chain. The first phosphate is added by a virus-encoded thymidine kinase so that the drug is converted to its active triphosphate only in HSV- and VZV-infected cells. The second and third phosphates are added by cellular kinases. Acyclovir triphosphate is incorporated into nascent DNA chains but—absent a ribose—the DNA polymerase cannot add further nucleotides and the chain is terminated. This dual viral target (thymidine kinase and DNA polymerase) of acyclovir confers great specificity (and hence safety) to this class of drugs.
Several other closely related compounds with improved clinical activity have also been developed. The lipid-conjugated nucleotide, brincidofovir, is a DNA polymerase inhibitor active against HSV, VZV, and CMV. There are number of compounds directed against the viral polymerase or other viral proteins that are now under test as potential anti-herpesvirus agents. Among the nonnucleoside compounds are helicase–primase inhibitors, that target other aspects of viral DNA synthesis.
Absent exogenous therapy, the host’s virus-specific immune response plays an important role in the control of the active phase of herpesvirus infection and represents an important complementary arm of therapy. In fact, reimmunization of VZV-immune subjects reduces the incidence of herpes zoster in the elderly.
2.7. Ebola Virus
Ebola is a zoonotic filovirus that has caused multiple small outbreaks in Africa since 1976, and one large pandemic in West Africa that began in 2013 but finally waned in May, 2015 (see Chapter 16, Emerging Viral Diseases). From the viewpoint of treatment or prevention, it is an “orphan” or neglected disease. In response to the ongoing pandemic, a crash program to develop Ebola treatments has been initiated, and a few compounds are in early clinical trials (Table 5 ).
Table 5.
Drugs in Ebola Virus Clinical Trials. This Table, Which is Updated on a Continuous Basis, Summarizes the Data on Drugs That Are Either Being Tested or Considered for Testing in Patients with Ebola Virus Disease. This Truncated Version is Limited to Those Drugs That Look Promising Based on High Efficacy in Nonhuman Primates and Low Toxicity in Nonhuman Primates or in Phase I Human Trials in April, 2015. A Full Version of This Table is Posted on the WHO Website, http://who.int/medicines/ebola-treatment/2015-0116_TablesofEbolaDrugs.pdf, accessed April, 2015
| Drug/Company | Drug Type | Ebola Preclinical Data | Known Safety Issues | Availability And Logistical Considerations | Comments |
|---|---|---|---|---|---|
| Category A: drugs already under evaluation in formal clinical trials in West Africa | |||||
| Favipiravir (Fuji/Toyama Japan) | Small molecule antiviral with activity against many RNA viruses. Functions through inhibiting viral RNA-dependent RNA polymerase. Approved in Japan for treating novel/pandemic influenza. |
In vitro inhibition IC50 64 μM; higher than that needed for influenza. Mice: protected at 300 mg/kg. Nonhuman primate (NHP): antiviral effect seen; 2 log reduction in viremia. Model limitation due to frequent need to anesthetize NHP to administer drug orally. |
Clinical use in healthy volunteers up to 3.6 g on first day followed by 800 mg twice daily (BID). No safety issues identified. Increased drug exposure in setting of hepatic dysfunction |
200 mg tablets; dosing at 6 g/first day requires 30 tablets—potentially difficult to swallow. 1.6 million tablets available free (10,000 treatment courses). Thermostable. |
4 patients received drug under compassionate use. No conclusions possible from these patients, but no obvious safety concerns identified. Clinical efficacy trial began in guinea in December 2014. Target 6 g dosing (day 1) followed by 2.4 g per day (day 2–10). Preliminary data presented in early February by investigators do not permit a firm conclusion regarding efficacy and more data are required. |
| Category B: drugs that have been prioritized for testing in human efficacy trials but for which such trials are not yet underway | |||||
| Zmapp (MappBio, USA) | Cocktail of three monoclonal antibodies produced in tobacco plants. | NHP: 100% survival when administered 5 days after virus challenge. | No formal safety studies in humans yet. Phase I safety study initiated in January 2015. | Supply reported to be 15 treatment courses every 6 weeks. | 8 patients treated on compassionate grounds to date. No conclusion regarding safety or efficacy possible. Some adverse reactions noted—possibly due to immune complex formation with virus. Phase I safety/PK study started in January 2015. Efficacy study due to start in early 2015. |
| AVI-7537 (Sarepta, USA) | Antisense polymorpholino oligonucleotide. Inhibits Ebola virus replication by binding to RNA in sequence-specific manner to VP24 gene. Specific to this strain of Ebola. | NHP: 100% survival for Marburg virus (using Marburg sequence) and 50–60% survival for Ebola using Ebola sequence. | Phase I safety study completed. Tolerability demonstrated. |
Limited no. of doses available. | No clinical trials planned at this time. |
Recent experimental data have shown that a cocktail of neutralizing monoclonal antibodies (ZMapp) against Ebola virus can have a dramatic therapeutic effect in a nonhuman primate model that closely simulates Ebola disease in humans (Qui et al., 2014; Murin et al., 2014). Monkeys were challenged with a 100% lethal dose of Ebola virus that kills animals in 4–8 days. Antibody treatment was only initiated 3–5 days after infection and—amazingly—it protected all animals, which were reported to recover to normal health. A crash program to scale up the production of these antibodies has been initiated but it is not known—at this writing—when these will be available for use in humans. ZMapp or a similar antibody cocktail is a very promising therapy and appears to be the most efficacious of the interventions so far developed. A wide variety of compounds with different modes of action are under investigation as potential drugs to treat Ebola. One proposed strategy to develop algorithms to screen FDA-approved drugs for their anti-Ebola activity (Ekins and Coffee, 2015; Vejlkovic et al., 2015; Litterman et al., 2015). This field is moving rapidly and the data in Table 5 will be outdated by the time of publication.
An important advance for the treatment of Ebola infection is a rapid point-of-care diagnostic test kit that does not rely on electric power, refrigeration, or complex equipment and highly trained technicians. Such a test was approved in 2015 by the World Health Organization (First antigen rapid test for Ebola, 2015). The ability to determine whether patients presenting with fevers of unknown origin in an Ebola-endemic area need anti-Ebola treatment is a critical adjunct to effective therapeutic interventions.
It is also relevant to note that there are several promising Ebola vaccine candidates in human trials in West Africa in 2015 (see Chapter 19, Viral Vaccines). If approved for human use, they will also serve as a critical tool for control of Ebola outbreaks (Rampling et al., 2015; Agnandji et al., 2015; Lipsitch et al., 2015; Marzi et al., 2015).
On a practical note, one of the ironies in the development of Ebola treatments is that the pandemic was waning—May, 2015—just as new compounds were readied for clinical trials (Kuehn, 2015; Fleck, 2015; Kupferschmidt, 2015). Hopefully, it will be possible to test the safety of those interventions that appear most promising, so that they could be used on a compassionate basis when the next outbreak occurs.
3. The Future of Drug Development
There is an exciting future for development of new antiviral drugs, which has been energized by the recent success with HCV. What are the considerations that will guide this field in the near future? Both scientific and public health issues are in play.
In the scientific arena, as set forth in Figure 1, there are a number of pathways for drug development: direct-acting antiviral compounds that are virus-specific; direct-acting compounds that are somewhat generic; cellular targets that are virus-specific; and broad cellular targets. The pathogenesis of different infections will have an important role in the selection and efficacy of such putative drugs. There is great potential for drugs to treat persistent infections with serious long-term consequences, while drugs may be less practical for acute infections that resolve before a virus-specific diagnosis can be made. The molecular nature of the virus–host infection is important, since it can determine whether drugs that control an infection will also lead to a permanent cure. A comparison of HIV (control no cure) and HCV (control and cure) illustrates that point.
What will guide the search for effective antiviral compounds? The recent successes for HCV and HIV suggest that direct-acting antiviral molecules still remain the gold standard that will inform many initiatives. One Achilles heel of such compounds is viral escape, but extensive experience with HIV has shown that the use of triple combinations can overcome this problem. Recent experience with HCV has provided several lessons: understanding the virus replication cycle at a molecular level paves the way for identification of candidate compounds; these insights can be used to construct high-throughput screening assays; and this strategy can lead to products that are clinically effective and safe (Scheel and Rice, 2013; Kim et al., 2013; Rice and Saeed, 2014).
The burgeoning field of systems biology offers significant new strategies for identifying antiviral compounds, particularly in the intertwined fields of cellular targets and drug repurposing (Table 3). Drugs that target host functions always carry the risk of unwanted side effects, particularly for long-term drug treatment. However, toxicity may be less of an issue for drugs used to treat acute infections such as Ebola hemorrhagic fever. The utility of a drug directed against the CCR5 molecule (for HIV treatment) demonstrates that this is a viable strategy.
The era of personalized (or precision) medicine is in its infancy (see Gary Gilliland commentary in the Chapter 22, What Lies Ahead?). In fact, personalization of treatment regimens is already a standard practice for certain persistent viral infections such as hepatitis B and C (Lok, 2015). Future trials of candidate drugs will more and more include genetic data on the participants to determine if drug efficacy or toxicity is localized to subpopulations (Insel et al., 2015; Ramamoothry et al., 2015; Johannessen et al., 2015). This may “rescue” certain candidate compounds for use in preselected patients.
Given these considerations, which viral diseases may be targets for drug development in the near future? A list of persistent infections would include: HBV; HPV; and some of the herpesviruses. In each instance, there is a large global population of infected persons who would benefit from a cure. In addition, cure or control of large numbers of virus carriers should have an impact on prevalence in the population. Among acute infections, those that carry a high mortality would be candidates, particularly viruses with epidemic potential, such as Ebola and other hemorrhagic fevers (Table 4). Dengue and influenza type A viruses are also high on this list, and experience with existing influenza antiviral compounds indicates that viral escape is common, mandating the need for multiple drug therapy.
Using this information, pharmaceutical companies have the research tools that enable them to develop or select compounds that have significant antiviral activity. However, drug development has become a daunting challenge, due both to scientific hurdles and financial requirements. In vivo bioavailability, pharmacodynamics, and potential toxicity are additional major obstacles that must be overcome during the development of safe and effective antiviral chemotherapeutics. Development of a new drug from discovery to licensure often takes as long as 10 years and costs more than $1 billion dollars. Repurposing of drugs already approved for human use could help to address these obstacles.
Clearly, there is an exciting future for antiviral drug development (De Clercq, 2013). This field demonstrates the practical yield of research in basic virology, viral pathogenesis, and viral epidemiology. However, support for “orphan” drugs, particularly those for the great neglected diseases, remains a major ethical and strategic challenge for the high-income countries, both for government and private sectors alike (Karan and Pogge, 2015). To date, we have failed to implement an effective strategy for the development and distribution of therapeutics for the neglected infectious diseases that continue to plague low-income countries.
Further Reading
Reviews, Chapters, and Books
- http://www.hcvguidelines.org/ (regularly updated expert guidelines in this rapidly changing field).
- Ahmed M, Wang F, Levin A, et al. Targeting the Achilles heel of the hepatitis B virus: a review of current tratments against covalently closed cirulcar DNA. Drug discovery today 2015, doi: 10.1016/j.drudis.2015.01.008. [DOI] [PubMed]
- Bekerman E, Einav S. Combating emerging viral threats. Science 2015, 348: 282–283. [DOI] [PMC free article] [PubMed]
- Brown JR, Magid-Slav M, Sanseau P, Rajpal DK. Computational biology approaches for selecting host-pathogen drug targets. Drug Discovery Today 2011, 16: 229–236. [DOI] [PubMed]
- Casadevall A, Pirofski L-A. Ditch the term pathogen. Nature 2014, 516: 165–166. [DOI] [PubMed]
- De Clercq E. Antivirals: past, present and future. Biochemical Pharmacology 2013, 85: 727–744. [DOI] [PubMed]
- De Clercq E. Current race in the development of DAAs (direct-acting antivirals) against HCV. Biochemical Pharmacology 2014, 89: 441–452. [DOI] [PubMed]
- Günthard HF, Aberg JA, Eron JJ, Hoy JF, Telenti A, Benson CA, Burger DM, Cahn P, Gallant JE, Glesby MJ, Reiss P, Saag MS, Thomas DL, Jacobsen DM, Volberding PA. Antiretroviral Treatment of Adult HIV-1 Infection: 2014 Recommendations of the International Antiviral Society–USA Panel. JAMA. 2014, 312: 410–25. [DOI] [PubMed]
- Insel PA, Amara SG, Blaschke TF. Introduction to the theme “precision medicine and prediction in pharmacology”. Annual review of Pharmacology and toxicology 2015, 55: 11–14. [DOI] [PubMed]
- Johannessen CM, Clemons PA, Wagner BK. Integrating phenotypic small-molecule profiling and human genetics: the next pahse in drug discovery. Trends in Genetics 2015, 31: 16–23. [DOI] [PMC free article] [PubMed]
- Karan A, Pogge T. Ebola and the need for restructuring pharmaceutical incentives. journal of global health 2015, 5: 1–4. [DOI] [PMC free article] [PubMed]
- Law GL, Korth MJ, Benecke AG, et al. Systems virology: host-directed approaches to viral pathogenesis and drug targeting. Nature reviews microbiology 2013a, 11: 455–466. [DOI] [PMC free article] [PubMed]
- Law GL, Tisoncik-Go J, Korth MJ, et al. Drug repurposing: a better approach for infectious disease drug discovery? Current Opinion in Immunology 2013b, 25: 588–592. [DOI] [PMC free article] [PubMed]
- Menendez Arias L, Richman D, et al. Antivirals and Resistance. Current Opinion in Virology October 2014, 8: 1–116. [DOI] [PMC free article] [PubMed]
- Ramamoothry A, Pacanowski MA, Zhang I. Racial/ethnic differences in drug disposition and response: review of recently approved drugs. Clinical pharmacology and therapeutics 2015, 97: 263–273. [DOI] [PubMed]
- Scheel TKH, Rice CM. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nature Medicine 2013, 19: 837–849. [DOI] [PMC free article] [PubMed]
- Schwegman A, Brombacher F. Host-directed drug targeting of factors hijacked by pathogens. Science Signaling 2008, 1: 1–7 (re8). [DOI] [PubMed]
- Tang MW, Shafer RW. HIV-1 antiretroviral resistance. Drugs 2012, 72: e1–e25. [DOI] [PMC free article] [PubMed]
- Wilson RC, Doudna JA. Molecular Mechanisms of RNA Interference. Annual Review of Biophysics 2013, 42: 217–239. [DOI] [PMC free article] [PubMed]
Original Contributions
- Agnandji ST, Huttner A, Zinser ME, et al. Phase 1 trials of rVSV Ebola vaccine in Africa and Europe – preliminary report. NEJM 2015, doi: 10.1056/NEJMoa502924. [DOI] [PMC free article] [PubMed]
- Alvim-Gaston M, Grese T, Mahoui A, et al. Open innovation drug discovery (OIDD): a potential path to novel therapeutic chemical space. Current topics in medicinal chemistry 2014, 14: 294–303. [DOI] [PubMed]
- Berg E. Systems biology in drug discovery and development. Drug discovery today 19: 113–125. [DOI] [PubMed]
- Blais DR, Nasher N, McKay CS, et al. Activity-based protein profiling of host-virus interactions. Cell 2012, 30: 89–97. [DOI] [PMC free article] [PubMed]
- Brass AL, Huang I-C, Beita Y, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1virus, West Nile virus, and dengue virus. Cell 2009, 139: 1243–1254. [DOI] [PMC free article] [PubMed]
- Chang J Block TM, Guo J-T. Antiviral therapies targeting host ER alpha-glucosidases: current status and future directions. Antiviral research 2013, 99: 251–260. [DOI] [PMC free article] [PubMed]
- Cohen J. Drug flushes out hidden AIDS virus. Science 2015a, 347: 1056. [DOI] [PubMed]
- Cohen J. Doubts dispelled about HIV prevention. Science 2015b, 347: 1055–1056. [DOI] [PubMed]
- Dong C, Qu L, Want H, et al. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antiviral research 2015, 118: 110–117. [DOI] [PubMed]
- Dopoazo J. Genomics and transcriptomics in drug discovery. Drug discovery today 2014, 19: 126–132. [DOI] [PubMed]
- Dyall J, Coleman CM, Hart BJ, et al. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrobial agents and chemotherapy 2014, 58: 4885–4893. [DOI] [PMC free article] [PubMed]
- Ekins S, Coffee M. FDA approved drugs as potential Ebola treatments. F1000Research 2015, 4: 48. [DOI] [PMC free article] [PubMed]
- Fatkenheuer G, Poznia AL, Johnson MA, et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV. Nature medicine 2005, 11: 1170–1172. [DOI] [PubMed]
- Fillet M, Frederich M. Ethe emergence of metabolomics as a keh disciploine the the drug discovery process. Drug discovery today 2015, doi: org/10.1016/j.ddtec.2015.01.006. [DOI] [PubMed]
- First Antigen Rapid Test for Ebola through Emergency Assessment and Eligible for Procurement. Accessed at http://www.who.int/medicines/ebola-treatment/1st_antigen_RT_Ebola/en/, April, 2015.
- Fleck F. Tough challenges for testing Ebola therapeutics. Bulletin of the WHO 2015 93: 70–71. [DOI] [PMC free article] [PubMed]
- Florescu DF, Keck MA. Development of CMX001 (Brincidofovir) for the treatment of serious conditions caused by dsDNA viruses. Expert review of anti-infective therapy 2014, 12: 1171–1178. [DOI] [PubMed]
- Furuta Y, Gowen BB, Takahashi K, et al. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral research 2013, 100: 446–454. [DOI] [PMC free article] [PubMed]
- Garcia M, Cooper A, Shi W, et al. Productive replication of Ebola virus is regulated by the c-Abl1 tyrosine kinase. Science translational medicine 2012, 4: 123ra24. [DOI] [PMC free article] [PubMed]
- Guidotti LG, Ando K, Hobbs MV, Ishikawa T, Rundel L, Schreiber RD, Chisari FV. Cytotoxic T lymphocytes inhibit hepatitis B virus gene expression by a noncytolytic mechanism in transgenic mice. Proceedings of the National Academy of Sciences, 1994, 91: 3764–3768. [DOI] [PMC free article] [PubMed]
- Gulick RM, Mellors JW, Havlir D. Eron JJ, Gonzalez C, McMahon D, Richman DD, Valentine FT, Jonas L, Meibohm A, Emini EA, Chodakewitz JA. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. New England Journal of Medicine 1997, 337:734–739. [DOI] [PubMed]
- Hu Y, Bajorath J. Learning from “big data”: compounds and targets. Drug Discovery today 2014, 19: 357–360. [DOI] [PubMed]
- Ishida Y, Yamasaki C, Yanagi A, et al. Novel robust in vitro hepatitis B virus infection model using fresh human hepatocytes isolated from humanized mice. American journal of pathology 2015, 185: 1275–1285. [DOI] [PubMed]
- Janssen H, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, Persson R, King BD, Kauppinen S, Levin AA, Hodges MR. Treatment of HCV Infection by Targeting MicroRNA. New England J Medicine 2013, 368: 1685–1694. [DOI] [PubMed]
- Jin G, Wong STC. Toward better drug repositioning: prioritizing and integrating exisint metods n efficient pipelines. Drug discovery today 2014, 19: 637–644. [DOI] [PMC free article] [PubMed]
- Josset L, Zeng H, Kelly SM, et al. Transcriptomic characterization of the novel avian-origin influenza A (H7N9) virus: specific host response and responses intermediate between avian (H5N1 and H7N7) and human (H3N2) viruses and implication for treatment options. mbio 2014, 5: 1-12 e01102–13. [DOI] [PMC free article] [PubMed]
- Kennedy EM, Bassit LC, Mueller H, et al. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA nuclease. Virology 2015, 476: 196–205. [DOI] [PMC free article] [PubMed]
- Kim H-Y, Li X, Jones CT, et al. Development of a multiplex phenotypic cell-based high throughput screening assay to identify novel hepatitis C antivirals. Antiviral Research 2013, 99: 6–11. [DOI] [PubMed]
- Kuehn BM. As Ebola epidemic begins to slow, trials of drugs and vaccines speed up. JAMA 2015, 313: 10001002. [DOI] [PubMed]
- Kupferschmidt K. As Ebola wanes, trials jockey for patients. Science 2015, 348: 20. [DOI] [PubMed]
- Lazzarin A, Clotet B, Cooper D, Reynes J, Arasteh K, Nelson M, Katlama C, Stellbrink H-J, Delfraissy J-F, Lange J, Huson L, DeMasi R, Wat C, Delehanty J, Drobnes C, Salgo M. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. New England J Medicine 2003, 348: 2186–2185. [DOI] [PubMed]
- Lin K, Gallay P. Curing a viral infection by targeting the host: the example of cyclophilin inhibitors. Antiviral research 2013, 99: 68–77. [DOI] [PMC free article] [PubMed]
- Lipsitch M, Eyal N, Halloran ME, et al. Ebola and beyond. Science 2015, 348: 46–48. [DOI] [PMC free article] [PubMed]
- Litterman N, Lipinski C, Ekins S. Small molecules with antiviral activity against Ebola virus. F1000Research 2015, 4: 38. [DOI] [PMC free article] [PubMed]
- Liu Y, Sheng J, Fokine A, et al. Structure and inhibition of EV-D68, a virus that causes respiratory illness in children. Science 2015, 347: 71–74. [DOI] [PMC free article] [PubMed]
- Liu N, Zhao F, Jia H, et al. Non-nucleoside anti-HBV agents: adnvances in structural optimization and mechanism of action investigations. Medchemcomm 2015, 6: 521–535.
- Lok AS. Personalized treatment of hepatitis B. Clinical and molecular hepatology 2015, 21: 1–6. [DOI] [PMC free article] [PubMed]
- Marzi A, Halfmann P, Hill-Batorski L, et al. An Ebola whole-virus vaccine is protective in nonhuman primates. Science 2015, 348: 439–442. [DOI] [PMC free article] [PubMed]
- Minie M, Chopra G, Sethi G, et al. CANDO and the infinite drug discovery frontier. Drug Discovery Today 2014, 19: 1353–1363. [DOI] [PMC free article] [PubMed]
- Neveu G, Barouch-Bentov R, Ziv-Av A, et al. Identification and targeting of an interaction between a tyrosine motif within hepatitis C virus core protein and AP2M1 essential for viral assembly. PLOS pathogens 2012, 8: e1002845. [DOI] [PMC free article] [PubMed]
- Murin CD, Fusco ML, Bornholdt ZA, et al. Structures of protective antibodies reveal sites of vulnerability on Ebola virus. PNAS 2014, 111: 17182–17187. [DOI] [PMC free article] [PubMed]
- Petta S, Craxi A. Current and future HCV therapy: do we still need other anti-HCV drugs? Liver International 2015, 35: S1: 4–10. [DOI] [PubMed]
- Procko E, Berguig GY, Shen BW, et al. A computationally designed inhibitor of an Epstein-Barr viral Bcl-2 protein induces apoptosis in infected cells. Cell 2014, 157: 1644–1656. [DOI] [PMC free article] [PubMed]
- Qui X, Wong C, Audit J, et al. Reversion of advanced Ebola disease in nonhuman primates with ZMapp. Nature 2014; 514: 47–53. [DOI] [PMC free article] [PubMed]
- Rampling T, Ewer K, Bowyer G, et al. A monovalent chimpanzee adenovirus Ebola vaccine – preliminary report. NEJM 2015, doi: 10.1056/NEJMoa1411627. [DOI] [PMC free article] [PubMed]
- Rice CM, Saeed M. Treatment triumphs. Nature 2014, 510: 43–44. [DOI] [PubMed]
- Shi M, Sun WL, Hua YY, et al. Effects of entecavir on hepatitis B virus covalently closed circular DNA in hepatitis B e Antigen-positive patients with hepatitis B. PLOSone 2015, doi: 10.1371/journal.pone.0117741. [DOI] [PMC free article] [PubMed]
- Soares MM, King SW, Thorpe PE. Targeting inside-out phosphatidylserine as a therapeutic strategy for viral diseases. Nature Medicine 2008, 14: 1357–1362. [DOI] [PMC free article] [PubMed]
- Suresh V, Krishnakumar KA, Asha VV. A new fluorescent based screening system for high throughput screening of drugs targing HBV-core and HBsAg interaction. Biomedicine and pharmacology 2015, 70: 305–316. [DOI] [PubMed]
- TablesofEbolaDrugs.pdf. http://who.int/medicines/ebola-treatment/2015-0116, accessed April, 2015.
- Tavis JE, Lomonosova E. The hepatitis B virus ribonuclease H as a drug target. Antiviral research 2015, 118: 132–138. [DOI] [PMC free article] [PubMed]
- Tufts Center for the Study of Drug Development (2014), Cost to Develop and Win Marketing Approval for a New Drug Is $2.6 Billion, http://csdd.tufts.edu/news/complete_story/pr_tufts_csdd_2014_cost_study.
- Veljkovic V, Loiseau PM, Figadere B, et al. Virtual screen fo repurposing approved and experimental drugs for candidate inhibitors of Ebola virus infection. F1000Research 2015, 4: 34. [DOI] [PMC free article] [PubMed]
- Verbist B, Klambauer G, Vervoort L, et al. Using transcriptomics to guide lead optimization in drug discovery projects: lesson learned from the QSTAR project. Drug discovery today 2015, doi: org/10.1016/j.drudis.2014.12.014. [DOI] [PubMed]
- Wang Z, Deisboeck TS. Mathematical modeling in cancer drug discovery. Drug discovery today 2014, 19: 145–150. [DOI] [PubMed]
- Warren TK, Wells J, Panchal RG, et al. Protection against figovirus disesaee by a nove broad spectrum nucleoside analogue BCX4430. Nature 2014, 508: 402–405. [DOI] [PMC free article] [PubMed]