

Abstract
Over the past 20 years, antiviral therapy has undergone a revolution and is now making a significant impact on the treatment of major virus diseases, for example HIV and viral hepatitis B and C. Targets include viral reverse transcriptases and proteases. The use of interferon to boost host immunity has also proved effective. Our understanding of chemotherapeutic strategies and drug resistance has become considerably more sophisticated since the widespread introduction of routine genome sequencing of virus isolates.
Keywords: Antiviral agents, clinical application, clinical strategies, antiviral drugs, disinfectants, newer approaches
Over the past two decades, antiviral therapy has undergone a revolution. After a long era of hope tempered by disappointment when there were only one or two encouragingly successful agents, many viral infections can now be treated by one or more antiviral regimens, and our understanding of chemotherapeutic strategies and drug resistance has become considerably more sophisticated. A majority of the agents in Table 12.1 have been developed in the past 20 years (Box 12.1 ).
Table 12.1.
Antiviral Drugs in Clinical or Experimental Use
| Process | Target | Viruses | Agent |
|---|---|---|---|
| Virus attachment to receptor | Ligand on virus capsid | Picornaviruses | Receptor analogs, disoxaril, pleconaril |
| Virus interaction with co-receptor | CCR5 co-receptor | HIV | Maraviroc |
| Conformation change during fusion of virion with cell | HR1 region of gp41 | HIV | Enfuvirtide (Fuzeon) |
| Uncoating and release | M2 ion channel | Influenza A | Amantadine, rimantadine |
| Replication of viral DNA | Viral DNA polymerase | Herpesviruses | Nucleosides; acyclovir, valacyclovir, famciclovir |
| Herpesviruses (CMV) | Ganciclovir, valganciclovir, cidofovir | ||
| Herpesviruses (CMV) | Pyrophosphate analog phosphonoformic acid (foscarnet) | ||
| Hepatitis B | Adefovir, entecavir, lamivudine, telbivudine, emtricitabine, tenofovir disoproxil fumarate | ||
| Replication of viral RNA | NS-5B polymerase | HCV | Sofosbuvir (nucleoside) dasabuvir (non-nucleoside) |
| Reverse transcription | Reverse transcriptase (RT) | HIV | Nucleoside RT inhibitors; zidovudine, didanosine, zalcitabine, stavudine, lamivudine, abacavir, emtricitabine (FTC) |
| Reverse transcription | Reverse transcriptase (RT) | HIV | Non-nucleoside RT inhibitors; nevirapine, delavirdine, efavirenz |
| Reverse transcription | RT, DNA polymerase | HIV, HBV | Tenofovir disoproxil fumarate |
| Integration of viral genome | Viral integrase | HIV | Raltegravir, elvitegravir |
| Cleavage of viral polyprotein precursors | Viral protease | HIV | Saquinavir, ritonavir, indinavir, nelfinavir, amprenavir, lopinavir |
| NS3-NS4A serine protease | HCV | Boceprevir, telaprevir, simeprevir, asunaprevir, paritaprevir | |
| RNA virus replication | Multiple targets | HCV (with interferon) Respiratory syncytial virus | Ribavirin |
| NS5A | HCV | Daclatasvir, ledipasvir | |
| Release of progeny virions from cell | Viral neuraminidase | Influenza A and B | Zanamavir, oseltamivir |
| General antiviral and immunomodulatory effects | Multiple IFN-stimulated genes | HBV, HCV (with ribavirin), AIDS-related Kaposi’s sarcoma | Interferon-α |
Box 12.1. Major Landmarks in the History of Antiviral Chemotherapy.
-
●
1962—idoxuridine, adenine arabinoside; these were relatively toxic, and their efficacy and value limited to specific situations, for example, topical use (for idoxuridine)
-
●
1983—acyclovir was a major breakthrough. It provided proof that highly effective, safe antiviral drugs were possible, and showed an example of how to do it
-
●
1980s—interferon-α became used more widely following its cloning, allowing its benefits and toxicity to be defined better
-
●
1990—AZT (zidovudine); the first RT inhibitor of major benefit in HIV
-
●
Mid-1990s—protease inhibitors and Highly Active Anti-retroviral Therapy (HAART) for HIV infection—interferon/ribavirin for hepatitis C
-
●
Early 2000s—lamivudine and other RT inhibitors for HBV, neuraminidase inhibitors for influenza
-
●
2011—directly acting antivirals against the HCV protease and RNA-dependent RNA polymerase
In broad terms, there are three classes of chemical agents used to combat infection: (1) Antiviral drugs act by suppressing or preventing viral replication in infected cells, and are sufficiently non-toxic that they can be used to treat infected patients, (2) Disinfectants are designed to destroy the infectivity of free virus particles, but are mostly toxic for the host cell and have no significant clinical effect against established infection, (3) The many antibiotics now available to fight bacteria have no activity against viruses. The only circumstances when it may be appropriate to prescribe antibiotics in viral infections are (1) to prevent or treat serious bacterial superinfection of a viral disease (e.g. bacterial pneumonia complicating influenza), or (2) to play safe where there is a real possibility of a serious bacterial illness, as in meningitis or pneumonia, until laboratory identification of the etiological agent is available. However, the widespread practice of prescribing antibiotics as a knee-jerk response to any infection leads to a number of undesirable consequences and is irresponsible medical practice.
The development of antiviral drugs has lagged behind the success with antibacterial agents for a number of reasons (Box 12.2 ). However, three major advances in the 1980s and 1990s provided the pharmaceutical industry with considerable optimism. These were (1) the discovery and increasing use of acyclovir, (2) the production of interferon by recombinant DNA technology, and (3) the introduction of an increasing array of effective drugs against HIV coupled with a new understanding as to how these weapons may be applied in clinical practice. We are now seeing a plethora of new agents against many significant human viral diseases reaching the stage of clinical trials, combined with a greatly expanding knowledge about how to manage such treatments and associated side effects. It is noteworthy that many of the currently available antiviral drugs are for treatment of chronic infections, where the timing of drug administration is not as critical as for the treatment of acute infections. Also most drugs only suppress replication but do not by themselves eradicate infection, and true therapeutic success and eradication of infection also requires an effective immune response, otherwise rebound of replication may be seen when the administration of the drug ceases.
Box 12.2. Why Has Development of Antiviral Drugs Lagged Behind the Introduction of Antibiotics?
-
1.
Unlike bacteria, replication of viruses is carried out within host cells and largely by host biochemical processes. Hence it is more difficult to find inhibitors of virus replication that are not also damaging to host cells or their functions, leading to side effects
-
2.
With many acute viral infections, the stage of peak virus replication is finished by the time the patient seeks treatment
-
3.
For many agents, there are problems in attaining adequate intracellular drug concentrations, or adequate distribution of drug to the site of infection
-
4.
Screening and testing candidate drugs can be more difficult, because, for example, there are no certified animal models
-
5.
Until recently, accurate laboratory diagnosis has often taken longer for viral than for bacterial infections
-
6.
Drugs shown to be effective in vitro are often ineffective in vivo for a variety of other reasons
Strategies for the Development of AntiViral Agents
In theory, any of the steps in the viral replication cycle represent possible targets for selective chemotherapeutic attack. Steps unique to virus replication are potential targets, as are those for which an inhibitor can be devised that has a greater selective activity against a virus-specific process than against a corresponding host process. Examples are shown in Table 12.1. A further refinement of this approach is illustrated by the nucleoside analog acycloguanosine (acyclovir), which preferentially inhibits the herpesvirus DNA polymerase essential for replication of viral DNA, rather than host cell polymerases. Acyclovir is in fact an inactive prodrug that requires another herpesvirus-coded enzyme, thymidine kinase, to phosphorylate it to its active form. As the viral enzyme occurs only in infected cells, such prodrugs are non-toxic for uninfected cells; the specificity of its action against viral rather than host replication is based on two separate selective steps.
New antiviral chemotherapeutic agents can come from a number of different sources:
-
1.
Large pharmaceutical companies house large banks of compounds with possible biological activities. These are screened for a desired function, for example, inhibition of the in vitro replication of a particular virus, either by direct testing of a candidate drug, or as part of sophisticated very high-throughput screening protocols. An example is AZT (zidovudine), the first successful nucleoside reverse transcriptase inhibitor (NRTI) used against HIV. This was synthesized in 1964 as a possible anti-cancer drug but was found to be ineffective. Much later it was reported as having anti-retrovirus activity in 1974, and then screened and found to be active against HIV in the mid-1980s (Box 12.3 ).
-
2.
Knowledge of the detailed structure of an active site critical for virus replication, for example, a viral enzyme, ligand for a receptor, or a regulatory target, allows for the creation of specific inhibitors through rational design. The neuraminidase inhibitors, a class of potent inhibitors of influenza replication, were developed by Mark von Itzstein after the fine structure of the neuraminidase protein was solved by Peter Colman using x-ray crystallography; this knowledge allowed the rational synthesis of chemical derivatives that bind tightly to the active pocket of the enzyme. A similar approach can be used to develop drugs that act by binding directly to the capsid (or envelope) of the virion itself, thereby blocking early steps in the replication cycle, for example, attachment, penetration, or uncoating. Once the three-dimensional structure of the whole surface of isometric virions such as picornaviruses is known, the receptor-binding site (the ligand) on the critical capsid protein can be characterized in atomic detail. Complexes of viral proteins with purified cell receptors or receptor mimics can be crystallized and examined directly. The receptor-binding site on the virion has generally turned out to be a “canyon,” cleft, or depression on the external surface of the protein. Further steps are taken to analyze the structure of the viral protein after binding to a compound known to neutralize infectivity, to map the particular amino acid residues found to be substituted in resistant mutants of virus, or to use site-specific mutagenesis in order to identify critical residues. This information can then be exploited to design better synthetic drugs, using computer modeling to optimize the fit and the binding energy of the drug–virus interaction.
-
3.
Once an agent is found which displays a degree of specific inhibition for a viral enzyme or other process integral to viral replication (“lead” molecule), related analogs (congeners) of the prototype are then synthesized with a view to enhancing its activity, solubility, and bioavailability, or reducing its toxicity.
-
4.
It is often proposed that natural products (e.g., “folk medicines”) that are reported to be of benefit against particular diseases may contain active ingredients from which new classes of potent antivirals can be developed. This area of discovery continues to hold hope, but all too often these products turn out to be complex mixtures and the apparent therapeutic benefit seems to wane as the material is fractionated into its components.
Box 12.3. Sources for New Candidate Antiviral Drugs.
-
1.
Blind screening of banks of randomly synthesized compounds
-
2.
Selection or synthesis of compounds known or predicted to inhibit a target process in virus replication, for example, by binding to an active site or intermediate
-
3.
Modification of compounds already known to have antiviral effects (“leads”), to improve efficacy or overcome drawbacks
-
4.
Naturally occurring substances or products reported to be of therapeutic benefit
Other approaches based on similar principles include the targeting of regulatory genes crucial for optimal virus replication, such as the TAR region of nascent HIV RNA transcripts, to which the product of the HIV tat gene binds. Potentially, the replication of such viruses could be blocked by agents that bind either to the protein product of such a viral regulatory gene, or to the recognition site in the regulatory region of the viral genome with which that protein normally interacts. Antisense RNAs, micro-RNAs (miRNAs), and small interfering RNAs (siRNA) are other approaches that are yet to be fully exploited.
A successful new antiviral drug typically goes through three stages of evaluation—initial in vitro screening in cell culture to determine its therapeutic index against a particular virus, followed by animal studies to determine pharmacokinetics, toxicity, and (if a suitable model exists) efficacy, followed finally by successive human phase I, II, and III studies. For any one successful drug this process can take 10 years or longer and can cost many millions of dollars. Many thousands of other potential candidates usually need to be discarded along this route, having failed one of the successive stages listed in Box 12.4 .
Box 12.4. Sequential Stages in the Development of New Antiviral Drugs.
-
I.Screening in vitro for antiviral inhibitory activity
- Infected and control cell cultures are incubated with dilutions of the compound to be tested.
- Chemotherapeutic index=(1) the minimum drug concentration toxic to cells (e.g., measured by trypan blue staining, reduced plating efficiency, or rate of cell division), divided by (2) the minimum drug concentration that inhibits virus replication (e.g., measured by virus yield or cytopathic effect). A promising drug would have a C.I.>100
-
II.Animal studies
-
●Pharmacokinetics; includes mechanism and routes of absorption; tissue distribution; metabolism, detoxification, excretion; half-life (t½) in different tissue compartments
-
●Toxicity—acute (biochemistry, hematology, immunosuppression)—chronic (allergenicity, mutagenicity, carcinogenicity, teratogenicity)
-
●Efficacy in experimental infection in vivo, if such a model is available
-
●
-
III.Human studies—Phase 1, 2, and 3
-
●Route; oral versus parenteral (intramuscular, intravenous, subcutaneous), topical (e.g., nasal, aerosol, skin, eye) versus systemic
-
●Dose and timing
-
●Efficacy; this requires
-
1.careful selection of population to be studied (e.g., enlist individuals already being exposed or infected)
-
2.comparison with (usually) a control group receiving current standard treatment (withholding treatment from a placebo control group is not ethically acceptable)
-
1.
-
●Toxicity; recording and evaluation of major and minor adverse events in control and treated group
-
●
The classical first test of any putative chemotherapeutic agent is, of course, inhibition of viral replication. In the presence of dilutions of the agent, the multiplication of suitable indicator viruses in cultured cells is measured by a reduction in the yield of virions or of some convenient viral marker. The toxicity of the drug for uninfected human cells may be measured crudely by the cytopathic effect or, more sensitively, by reduction in cell plating efficiency or cell doubling time. In general, only those agents displaying a therapeutic index of at least 10 and preferably 100 to 1000 are worth pursuing further (Box 12.4).
Ideally, the drug should be water-soluble, chemically and metabolically stable, moderately apolar, and satisfactorily taken up into cells. Pharmacokinetic studies, first in animals and then in humans, address such questions as the mechanism and rate of absorption following various routes of administration, tissue distribution, metabolism, detoxification, and excretion of the drug. Tests for acute toxicity encompass comprehensive clinical surveillance of all the body systems, biochemical tests (e.g., for liver and kidney function), hematology, tests for immunosuppression, and so on. Longer-term investigations screen for chronic toxicity, allergenicity, mutagenicity, carcinogenicity, and teratogenicity.
Clinical Application
Formulation and Methods of Delivery
The route of administration of an antiviral agent is a prime consideration in assessing its general acceptability. The oral route is, naturally, by far the most convenient for the patient. Nasal drops or sprays may be acceptable for upper respiratory infections but can be irritating, whereas continuous delivery of aerosols through a facemask or oxygen tent is generally appropriate only for very sick and hospitalized patients. Topical preparations (creams, ointments, etc.) are satisfactory for superficial infections of skin, genitalia, or eye, provided the infection is relatively localized; penetration of drugs through the skin can be enhanced by mixing with substances such as polyethylene glycol. Parenteral administration is the only option in the case of some drugs and may, in any case, be required for serious systemic infections; intravenous infusion usually necessitates hospitalization.
Some drugs have to be used at very high, potentially toxic concentrations because of poor solubility or poor penetration into cells. Delivery of antiviral concentrations of compounds into cells can sometimes be achieved either by incorporating the drug into liposomes or by conjugating the compound to a hydrophobic membrane anchor. Sophisticated chemistry may also be required to modify potential antivirals, such as synthetic peptides or oligonucleotides, which are otherwise rapidly degraded intra- or extracellularly. Some experimental antiviral drugs are being conjugated to antiviral antibodies, or incorporated into liposomes coated with such antibody, to direct these to virus-infected cells.
Emergence of Drug-Resistant Mutants
With almost every new antiviral agent, drug-resistant mutants soon emerge in vitro and in vivo, especially during long-term therapy of chronic infections and among immunocompromised patients (Box 12.5 ). In its simplest form, resistance may be due to a single point mutation in the gene encoding the particular viral protein that is the target of the compound. Stepwise increases in the degree of resistance may occur as further nucleotide substitutions accumulate, often in a particular order. Drug-resistant strains often show less replication fitness and/or less virulence, and may be replaced by wild-type virus in cell culture or in a patient unless maintained by the selection pressure of the continuing presence of the drug. However, as a demonstration of the power of selection, such resistant strains may also develop further compensatory mutations that enhance replication fitness.
BOX 12.5. Resistance to Antiviral Drugs.
-
●
During replication of virus nucleic acids, errors (base changes) continually occur. Error rates range from 1 in 10−4 or less. Selection then occurs, and any base change with a replication advantage will be amplified. We will then see this as a “mutation”
-
●
Drug resistance arises by single or multiple base changes, that may affect a key viral protein or nucleic acid motif or sequences affecting this, for example, folding, leading to reduced susceptibility to the action of the drug
-
●
Resistant strains are often less virulent and/or replication competent than wild-type virus, but are given a selective advantage and maintained by the selection pressure of drug presence
-
●
The drug may still be clinically useful even after resistant strains appear
-
●
May get rebound of wild-type virus replication when the drug is withdrawn
-
●
Compensatory mutations may develop that enhance the replication competence of virus containing a drug resistance mutation
-
●Assays for drug resistance may be
-
(1)Clinical, that is, the reappearance of virus, or an increase in the virus level in the patient during therapy
-
(2)Phenotypic in vitro, that is, virus replication occurring in cell culture in the presence of higher concentrations of drug
-
(3)Genotypic, that is, detection of base changes in the virus sequence that are known to be associated with drug resistance
-
(1)
Clinical virus isolates may be tested for drug sensitivity by growth in cultured cells in the presence of serial dilutions of the agent (a “phenotypic” assay). For virus/drug combinations where resistance is regularly associated with particular mutations, the genome sequence of the patient’s isolate is matched against a library of known sequences; this allows prediction of the resistance pattern of the new isolate and choice of optimal antiviral therapy. This approach is most highly developed in the case of HIV. Of course, often the first indication during long-term treatment of a persistent infection that resistance has developed may be clinical, that is, a deterioration in the patient’s symptoms or an increase in viral load.
Resistance mutations can only develop while a virus is replicating, a situation most likely to develop during monotherapy where the drug concentration is intermediate, that is, sufficient to create selection pressure but not high enough to prevent replication completely. The principles needed to minimize this problem are well understood from experience with antibiotic resistance in bacteria and cancer chemotherapy. Antiviral agents should be administered in sufficiently high dosage and without interruption, so that inhibitory drug levels are maintained. Combination therapy with two or more agents (preferably with distinct modes of action) is theoretically attractive and has been shown with HIV to greatly delay the appearance of resistance and improve the therapeutic response. Furthermore, if this allows one or both drugs to be given at a lower dose, combination therapy can reduce the incidence of toxic side effects. Patient compliance became a major difficulty for the treatment of HIV infections requiring the maintenance of strict drug regimens, as patients eventually grow tired of maintaining strict drug regimens necessitating the taking of 20 pills at different times each day, particularly when they start to feel better! It was a major advance with the licensing in 1997 of a single tablet containing two drugs (Combivir) and later (2006) three drugs, taken once a day (e.g., Atripla, containing emtricitabine, tenofovir, and efavirenz); six more combination treatments have been approved by the United States Federal Drug Agency in the past 4 years.
Clinical Strategies
Antiviral drugs are used successfully with a number of different strategies in mind, including:
-
1.
Long-term therapy in chronic infection, particularly HIV, HBV, and HCV. It is important to realize that different possible therapeutic goals may be targeted, for example (a) eradication of virus from the body, (b) suppression of virus replication so that viral load in the plasma is greatly reduced or undetectable, (c) delaying or preventing clinical disease, disease progression, or death, or (d) improving laboratory markers of disease, for example, liver function tests (hepatitis), and CD4 cell count (HIV). These options should be clearly understood both in the planning of clinical treatment and in the design of drug trials. The outcome of a long-term treatment course is designated as either IR (initial response), ETR (end-of-treatment response, i.e., possibly followed by rebound), or SR (sustained response, i.e., no rebound occurring).
-
2.
Immediate treatment of acute infection, for example, neuraminidase inhibitors for influenza, acyclovir or a derivative for acute herpes simplex infection. As in many of these infections the virus replication peaks early in the clinical disease and wanes thereafter: a rapid initiation of treatment has been shown to be very important to achieve optimal benefit.
-
3.
Prophylaxis against disease progression or recrudescence, or to prevent viral shedding. Patients suffering from frequent recrudescence of genital herpes can have their lives transformed by long-term daily maintenance therapy with acyclovir, famciclovir, or valaciclovir. Similarly, use of valganciclovir in transplant patients, either prophylactically to all those with pre-existing CMV antibody (i.e., persistent infection), or pre-emptively for those found to have laboratory evidence of CMV replication, has led to a major reduction in CMV disease among these patients.
-
4.
Prevention of virus transmission. Without intervention, approximately 25% of infants born to HIV-infected mothers become infected. This can be reduced to approximately 2%, a dramatically important result, by an appropriate combination of three antiviral drugs given to the mother in late pregnancy and to the infant after birth, combined with Caesarean section if viral suppression has not been achieved by the time of labor, and combined with avoidance of breastfeeding. Other regimens include intravenous zidovudine, or nevirapine as a short course or single dose to mother and infant at delivery.
Antiviral drug regimens are also used for post-exposure protection against HIV transmission to individuals following known sexual exposure to HIV. Recently, daily pre-exposure prophylaxis has been approved for individuals thought to be at on-going high risk of acquiring HIV, using the combination tablet Truvada (tenofovir and emtricitabine). Wider adoption of this approach in the future could even bring some similarity between HIV prevention and malaria prophylaxis.
Mechanisms of Action and Role of Individual AntiViral Drugs
Interferons
The interferons (IFNs) have offered potential advantages as ideal antiviral agents since being discovered in 1957. These are natural cellular products of viral infection and display a broad spectrum of activity against essentially all viruses (see Chapter 5: Innate Immunity, Chapter 7: Pathogenesis of Virus Infections, and Chapter 8: Patterns of Infection). Early clinical trials were conducted with inadequate amounts of semi-purified interferons produced either by treating cultured human leukocytes or fibroblasts with a paramyxovirus, or with a synthetic double-stranded RNA. However, the cloning and expression of the gene for human interferon α (IFN-α) in Escherichia coli in 1980 allowed the production of therapeutic quantities of IFN-α at a greatly reduced cost, allowing its true clinical role to be explored. Since then, the genes for all known subtypes of human IFN-α as well as IFN-β and IFN-γ have been cloned in prokaryotic and/or eukaryotic cells.
Interferons are not effective by mouth and must therefore be administered by injection. IFN-α is much more active in vivo than IFN-β or IFN-γ, probably because the latter do not achieve or maintain the required blood levels after intramuscular administration. Toxic side effects are regularly observed and may be significant with doses in excess of 107 units per day, even when highly purified cloned IFN subtypes are employed. Fever regularly accompanies high doses but lasts only a day or so. Severe fatigue is the most debilitating symptom and may be accompanied by malaise, anorexia, myalgia, headache, nausea, vomiting, weight loss, erythema, and tenderness at the injection site, partial alopecia (reversible), dry mouth, reversible peripheral sensory neuropathy, or signs referable to the central nervous system. Depression is a common and sometimes severe side effect. Various indicators of myelosuppression (granulocytopenia, thrombocytopenia, and leukopenia) and abnormal liver function tests, both reversible on cessation of therapy, are regularly observed should high-dose interferon administration be prolonged.
Despite its promise, there are only a limited number of situations where interferon-α has become standard therapy. These include hepatitis C (in combination with ribavirin), hepatitis B, and AIDS-related Kaposi’s sarcoma. Genital warts have been successfully treated, and juvenile laryngeal papillomatosis, a severe condition calling for repeated surgical removal following recurrences, can be arrested by local injection of interferon; however, the tumors reappear when therapy is withdrawn (Chapter 19: Papillomaviruses). Other non-viral conditions that may undergo temporary remission or partial regression with vigorous interferon therapy include hairy cell leukemia, malignant melanoma, chronic myelocytic leukemia, and in the case of multiple sclerosis, interferon-β. In these situations interferons may be acting not as antivirals but as cytokines exerting immunomodulatory effects. Because of the above pattern of side effects, long-term systemic treatment with interferon is often uncomfortable or unpleasant, and it is not uncommon for patients undergoing long-term treatment for hepatitis C to withdraw from their treatment course. The recent development of very effective interferon-free antiviral regimens for treatment of hepatitis C is a major advance that has been hailed by patients and clinicians alike.
Blocking Attachment or Fusion
One theoretical option for antiviral therapy is to inhibit the first step in the viral replication cycle, namely, attachment of the virion to its specific receptor on the host cell membrane. Substances designed to mimic either the cell receptor or the viral ligand should theoretically accomplish this. For example, attachment of HIV to its receptor (CD4) can be blocked by soluble CD4 (which binds to the virion), or by a synthetic peptide corresponding to the ligand on the HIV envelope glycoprotein gp120 that binds to the cell receptor. Another example of the latter is the blockage of the HIV co-receptor CCR-5, using either a ligand mimic or an antibody that binds to the site. Maraviroc is a CCR5 co-receptor antagonist now approved for treatment of HIV; when co-administered with standard treatment it has been shown to lead to an improved outcome. A major problem with ligand mimics is that saturation of cell receptors may occur, and therefore interfere with the normal physiological function of that membrane glycoprotein. Maraviroc has been reported to cause allergic reactions and hepatotoxicity. Receptor mimics may be safe but would need to be confirmed as not eliciting an autoimmune response.
X-ray crystallography has provided detailed information about the binding site of a wide range of antiviral agents that block the uncoating of picornaviruses. Many of the studies to date have used human rhinovirus type 14 (HRV-14) as a model. Most of the drugs, in spite of a diversity in chemical structure, bind to the same site on HRV-14, namely, a hydrophobic pocket that lies immediately beneath the floor of the canyon that comprises the ligand (receptor-binding site) on the viral capsid protein VP1. Following binding of the drug, hydrophobic interactions result in deformation of the canyon floor; this inhibits virion attachment to cell receptors, but, more importantly, also inhibits uncoating of the virion. This is thought to occur by locking VP1 into a conformation, thus preventing the disassembly of the virion that normally occurs in the acidic environment of the endosome. When administered prophylactically rather than therapeutically, antivirals of this nature have been claimed to reduce the symptoms of the common colds induced by certain rhinovirus serotypes but not others. A promising example of such a drug was Pleconaril, which was shown in randomized double-blind clinical trials to reduce the symptoms in patients with self-diagnosed colds: resistant virus strains have been reported. However, its efficacy was not fully established, and as a consequence the Food and Drug Administration of the United States declined to approve its use in 2003. While this approach has been slow to bear fruit, it does engender some optimism that antiviral drugs may eventually provide a better answer to the virologists’ age-old challenge, “when will you produce a cure for the common cold?”
The anti-HIV drug enfuvirtide (T-20, Fuzeon) represents the first example of the novel approach of fusion inhibitors. During fusion between the HIV envelope and the host cell membrane, a necessary step is binding between the HR1 and HR2 regions of the gp41 envelope protein which leads to gp41 refolding. The short peptide enfuvirtide mimics the HR2 segment of the gp41 envelope protein and binds to the HR1 region of gp41, thereby preventing HR1–HR2 binding and virus–cell fusion. Being a 36-amino acid peptide, enfuvirtide has presented a special challenge in drug synthesis and delivery. It is administered subcutaneously, being used in combination with other drugs where other treatments have failed. Local side effects at the injection site, and a range of systemic side effects, are not uncommon.
Blocking Uncoating—Ion Channel Blockers
In the 1960s the simple three-ringed symmetrical amine amantadine was synthesized and shown to inhibit the replication of influenza A viruses (but not influenza B viruses). The principal target of amantadine is the protein M2, which is a minor component of the influenza viral envelope. Influenza B lacks an M2 protein, and thus amantadine’s specificity for influenza A can be understood. M2 forms a tetrameric transmembrane ion channel that reduces the pH gradient across the envelope of incoming virions within acidic endosomes. It is also thought to play a role in assisting the transport of newly synthesized HA to the plasma membrane by reducing transmembrane gradients across the trans-Golgi cisternae. Thus, amantadine acts at these two distinct steps in the replication cycle as an ion channel blocker. First, by raising the pH of the endosome, amantadine prevents the pH 5-mediated conformational change in the HA molecule required for fusion of viral envelope with endosomal membrane. Second, later in the replication cycle, by disturbing the ionic environment within the exocytic pathway, amantadine prevents newly synthesized HA from assuming the correct conformation for incorporation into the envelope of budding virions.
Therapeutically, amantadine has been reported to reduce the severity of symptoms in about 50% of cases, but only if given both within the first 24 to 48 hours and at high doses. Administered prophylactically, it can significantly reduce the incidence of clinical influenza (50% to 90% in various trials). However, in practice, its use as a prophylactic demands the ingestion of 200 mg daily for 1 to 2 months from the commencement of an influenza epidemic in the community, and it is clear that vaccination presents a safer and cheaper alternative.
Amantadine has a narrow therapeutic window, with side effects commonly occurring at doses that do not greatly exceed the therapeutic dose. Such side effects relate mainly to the central nervous system (loss of concentration, insomnia, nervousness, light-headedness, drowsiness, anxiety, confusion) but are generally reversible and mild. In 1969 the drug was found to relieve symptoms of Parkinson’s disease and other extrapyramidal syndromes, and today is more widely used for this indication than as an antiviral.
Rimantadine (Fig. 12.4), is a methylated derivative that shows less central nervous system side effects and is the drug of choice in most cases. Both drugs are given orally, but either may be delivered to hospitalized patients by aerosol spray. As a majority of recent isolates of H3N2 and pandemic H1N1 show resistance to adamantanes, the newer neuraminidase inhibitors have largely replaced the use of both drugs.
Figure 12.4.

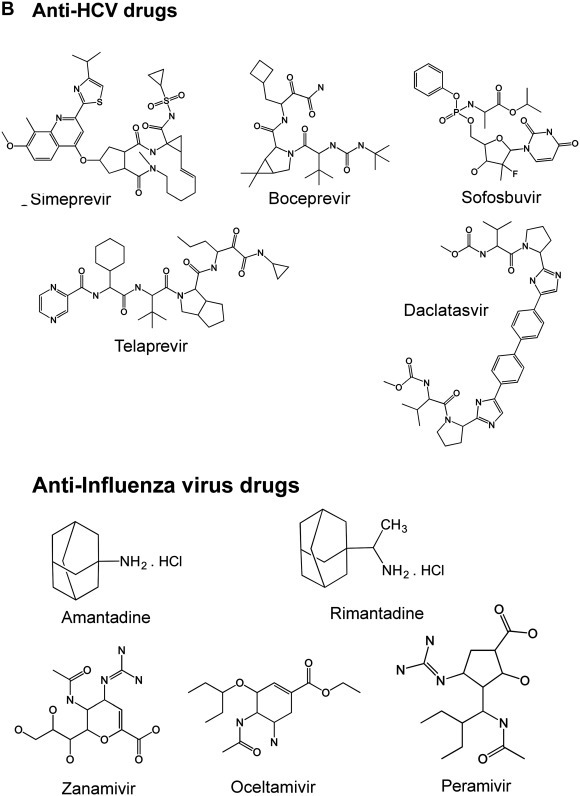
Chemical structures of some antiviral agents used against hepatitis B, hepatitis C and influenza viruses. (A) Anti-HBV shopped; (B) Anti-HCV and Influenza shopped.
Courtesy of Steven Polyak.
Although the above compounds are relatively specific for influenza A virus, different short hydrophobic transmembrane proteins that function as ion channels are encoded by a number of different viruses; these include the vpu protein of HIV, the E protein of SARS-CoV, and the p7 protein of HCV. Drugs that act as ion channel blockers against some of these are under active development, and may represent the prototype of a new class of compounds active against many other viruses.
Inhibitors of Viral DNA Polymerase
Many of the successful inhibitors of viral replication are nucleoside analogs, with antiviral activity particularly against the herpesviruses or HIV. The early prototypes, such as adenine arabinoside, are relatively undiscriminating inhibitors of both cellular and viral DNA synthesis. Understandably, these early compounds often produced toxic side effects, directed especially at dividing cells in the bone marrow and gastrointestinal tract.
Acycloguanosine (Acyclovir) and Homologs
A major breakthrough in antiviral chemotherapy occurred in 1977 when Elion and colleagues developed a prodrug that depends on a viral enzyme to convert it to its active form. Acycloguanosine, now commonly known as acyclovir, is a guanine derivative with an acyclic side chain, the full chemical name being 9-(2-hydroxyethoxymethyl) guanine (Fig. 12.1 ). Its unique advantage over earlier nucleoside derivatives is that the herpesvirus-encoded enzyme, thymidine kinase (TK), which has broader specificity than cellular TK, is required to phosphorylate acycloguanosine intracellularly to acycloguanosine monophosphate (ACG-P); a cellular GMP kinase then completes the phosphorylation to the active agent, acycloguanosine triphosphate (ACG-PPP) (Fig. 12.2 ). Further, ACG-PPP inhibits the herpesvirus-encoded DNA polymerase at least 10 times more effectively than it does cellular DNA polymerase α. It acts as both inhibitor and substrate of the viral enzyme, competing with GTP and being incorporated into DNA: chain termination is the result as acyclovir lacks the 3′-hydroxyl group required for chain elongation. Since activation of the prodrug needs the viral TK, acyclovir is essentially non-toxic to uninfected cells but is powerfully inhibitory to viral DNA synthesis in infected cells, giving it much greater selectivity than is the case with the earlier nucleoside analogs.
Figure 12.1.
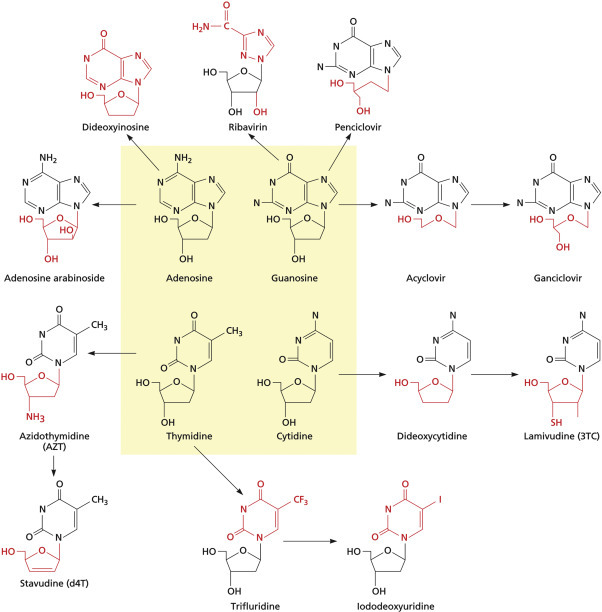
Many successful inhibitors of viral replication are nucleoside or nucleotide analogs. The four natural deoxynucleotides are highlighted in the yellow central box, and arrows connect these to related antiviral drugs. Chemical modifications giving rise to each antiviral drug are highlighted in red.
Reproduced from Flint, S.J. et al., 2009. Principles of Virology, third ed., vol. II, p. 292, ASM Press, Washington, DC, with permission.
Figure 12.2.
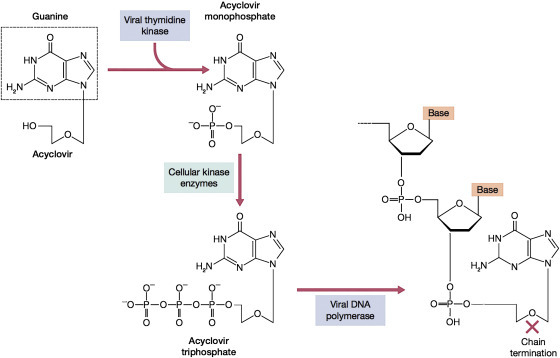
Mechanism of selective inhibition of herpesvirus replication by acyclovir. Acyclovir is an inactive prodrug, and must be phosphorylated to the active triphosphate compound within the cell. The first step, to the monophosphate is carried out by the viral thymidine kinase but not the host cell kinases, so this step can only proceed in infected cells. The monophosphate is then converted to the active triphosphate by cellular kinases. ACV-TTP then inhibits the viral DNA polymerase 10 times more than the host DNA polymerase α; furthermore, when it is incorporated into the growing DNA, it acts as a chain terminator because it has no 3′ hydroxyl group of the sugar ring.
Reproduced from Flint, S.J. et al., 2009. Principles of Virology, third ed., p. 694, ASM Press, Washington, DC, with permission.
Herpes simplex viruses types 1 and 2 (HSV-1 and -2) are both highly sensitive to acyclovir; varicella-zoster virus (VZV) is susceptible at somewhat higher concentrations of the drug. Other human herpesviruses, lacking a gene coding for TK, are susceptible only at much greater doses; this results from the limited production of ACG-P by cellular GMP kinase. The relative sensitivity of different herpesviruses seems to depend on a rather complex interplay of at least three variables: (1) the efficiency of the virus-coded TK (if any) in converting acyclovir to ACG-P, (2) the efficiency of cellular kinases in converting this intermediate to ACG-PPP, and (3) the susceptibility of the viral DNA polymerase to ACG-PPP. The use of acyclovir for treatment of various herpesvirus diseases is discussed in Chapter 17: Herpesviruses. Acyclovir (Zovirax) may be delivered orally, by slow intravenous infusion, or topically as an aqueous cream. As anticipated from in vitro studies, the drug is essentially non-toxic. Acyclovir levels must be carefully monitored in patients with dehydration or renal impairment, as the drug, which is excreted unchanged through the kidneys, is relatively insoluble, and crystalluria may occur. Nevertheless, its use has revolutionized treatment and suppression of severe muco-cutaneous herpes simplex infections, herpes simplex encephalitis, and disseminated infections with HSV, varicella-zoster, and CMV.
Acyclovir-resistant mutants of HSV can be recovered in vivo and in cell culture. The mutation is usually located in the gene coding for the viral thymidine kinase, but more rarely is seen in the DNA polymerase gene. There are two kinds of TK mutants: (1) those failing to produce appreciable levels of TK (TK−), (2) those in which the enzyme is produced but has an altered substrate specificity such that it can no longer satisfactorily phosphorylate acyclovir (TKa). TK− mutants, the most common, may contain a mutation, deletion, or insertion leading to premature termination of translation or the production of a non-functional enzyme, whereas TKa mutants result from a point mutation causing a more subtle alteration in substrate specificity so that the enzyme no longer phosphorylates acyclovir. Nearly all TK− mutants, while able to establish latent infection of ganglia, have reduced virulence and reduced ability to reactivate; these strains most commonly arise, and cause severe disease, in immunocompromised hosts.
Acyclovir has the limitations of low oral absorption and a short half-life, typically needing to be taken five times a day. More recent derivatives of acyclovir are valacyclovir (a valine ester of acyclovir), which is absorbed three to five times better than acyclovir after oral administration, and famciclovir, which is also well absorbed and is rapidly metabolized to the active compound penciclovir. Both these latter drugs have longer half-lives and require less frequent dosage than acyclovir. As expected, TK− mutants resistant to acyclovir have a cross-resistance to related nucleoside analogs, but not to foscarnet or the nucleotide analog cidofovir which therefore can be used to treat infected patients (Fig. 12.3 ).
Figure 12.3.

Structure of valacyclovir. Valacyclovir, the valine ester of acyclovir, is absorbed from the gut into the circulation three to five times faster than acyclovir. Once taken up by the cell, host enzymes cleave off the valine side chain. Its longer half-life in the body means that doses do not need to be as frequent as for the parent compound acyclovir.
Reproduced from Flint, S.J. et al., 2009. Principles of Virology, third ed., vol. II, p. 289, ASM Press, Washington, DC, with permission.
Ganciclovir
Acyclovir is much less potent against CMV as much less phosphorylated ACV accumulates in CMV-infected cells than in HSV- or VZV-infected cells. A derivative of acyclovir, 9-(1,3-dihydroxy-2-propoxy) methylguanine (DHPG), known as ganciclovir (GCV) (Fig. 12.1), has much greater inhibitory activity against CMV than does ACV, and was the first drug approved for use against CMV. GCV is phosphorylated in CMV-infected cells, not by TK but by an unusual virus-coded protease, the product of the UL97 gene; further phosphorylation by cellular kinases yields the active triphosphate, which inhibits the viral DNA polymerase. Resistance in some mutants maps to the phosphorylation gene, in others to the DNA polymerase gene.
Ganciclovir has been used principally to treat severe CMV infections such as retinitis, colitis, and pneumonia in AIDS patients and in transplant recipients. Given intravenously for some weeks, ganciclovir may produce a temporary remission in a proportion of cases, but unfortunately the condition generally recurs following its withdrawal. The antiviral activity of GCV is not as selective as ACV against HSV, and accordingly the drug is relatively toxic. Severe neutropenia, anemia, and thrombocytopenia are common side effects, and the drug also is associated with gastrointestinal symptoms including diarrhea, renal problems, and hypersensitivity. The valine ester valganciclovir is a prodrug that is rapidly converted to GCV in the body. It is more available orally, and can be used as daily oral prophylaxis against CMV in HIV and transplant patients.
Acyclic Nucleoside Phosphonates
Cidofovir is an acyclic cytosine analog that contains a phosphonate group. This group mimics a monophosphate group, and accordingly cidofovir is poorly taken up into cells, but has a long intracellular half-life. Its activity does not require a viral TK or protein kinase to synthesize the monophosphate, and it therefore has a broad antiviral spectrum against herpesviruses, adenoviruses, papillomaviruses, and poxviruses. It is used to treat CMV retinitis in HIV patients by intravenous administration. Drugs with similar structure include adefovir (used against hepatitis B) and tenofovir (used in HIV and hepatitis B).
Trisodium Phosphonoformate (PFA, Foscarnet)
Trisodium phosphonoformate, known also as phosphonoformic acid (PFA) or foscarnet (Fig. 12.1), is a non-nucleoside inhibitor of the DNA polymerases of herpesviruses and hepatitis B, as well as the reverse transcriptase of HIV. It acts through a non-competitive inhibition of the pyrophosphate-binding site on the enzyme. Resistance maps to the DNA polymerase gene. PFA may be given intravenously to treat CMV retinitis and severe herpesvirus infections, particularly those resistant to other antiviral drugs. The drug also displays some activity against hepatitis B in vitro but has been ineffective in vivo.
Although foscarnet displays some selectivity in that it inhibits cellular DNA polymerase α only at higher concentrations than the level required to inhibit viral DNA polymerase, it accumulates in bone and can lead to renal toxicity, electrolyte disturbances, and genital ulceration. Accordingly, it tends to be reserved for life-threatening conditions.
Inhibitors of RNA Virus Replication
A rather unusual nucleoside analog, 1-β-d-ribofuranosyl-l,2,4-triazole-3-carboxamide, known as ribavirin (Fig. 12.1), was first synthesized in 1972. Despite the fact that it inhibits the growth of a wide spectrum of RNA and DNA viruses in cultured cells and experimental animals, it is still only approved in most countries for a limited range of indications. These are chronic HCV infections in combination with interferon (although this role has now been largely superseded by the use of direct acting antiviral drugs); and severe respiratory syncytial virus infections, where it is delivered using a nebulizer to generate a small-particle aerosol administered via a mask or oxygen tent for 3 to 6 days. When given by oral administration at the usual dosage of about 1 gram per day, a substantial minority of recipients develop a reversible anemia with increased reticulocyte numbers and elevated serum bilirubin levels: immunosuppression and teratogenic effects have been demonstrated in animals. Its mechanism of action is not clear. The fact that ribavirin monophosphate inhibits the cellular enzyme IMP dehydrogenase, decreasing the pool of GTP, as well as inhibiting guanylyltransferase-mediated 5′-capping of mRNA, suggests that it may be acting on cellular pathways that are somewhat more critical to the virus than to the cell. It has also been proposed that it may inhibit viral RNA polymerases, and/or increase the mutation rate of the viral polymerase to a “catastrophic” level where few functional genomes are produced. The 3-carboxamidine derivative viramidine (taribavirin), a prodrug of ribavirin, has a similar spectrum of activity but may have slightly less toxicity.
Oral or intravenous ribavirin has been used in viral hemorrhagic fevers including Lassa, Crimean-Congo hemorrhagic fever, and Hantavirus infection, and there is some evidence of benefit.
Inhibitors of Reverse Transcriptase
Nucleoside RT Inhibitors
The advent of AIDS in the early 1980s stimulated a major search for new approaches to antiviral therapy. Few in the medical and scientific community had experience with lentiviruses and there were no effective anti-retroviral agents. Enormous strides were thenceforth made rapidly, and a new era of viral chemotherapy was born.
The first compound to display sufficient antiviral activity in vivo to be licensed for human use was 3′-azido-2′,3′-dideoxythymidine, otherwise known as azidothymidine, AZT, or zidovudine (Fig. 12.1), an inhibitor of reverse transcriptase. Unlike acyclovir, AZT is phosphorylated by cellular kinases to AZT triphosphate (AZT-PPP), which exerts its antiviral effect against HIV by inhibiting HIV reverse transcriptase, being as it is accepted by the enzyme in preference to TTP. AZT-PPP binds to the reverse transcriptase approximately 100 times more efficiently than it does to the cellular DNA polymerase α. AZT-PPP is incorporated into the growing HIV DNA chain, leading to premature chain termination. In addition, AZT monophosphate (AZT-P) competes successfully for the enzyme thymidylate kinase, resulting in depletion of the intracellular pool of TTP. Since AZT’s activity does not depend on phosphorylation by viral enzymes, it is less selective and more toxic than acyclovir. Clearly, AZT suppresses replication but does not eliminate proviral DNA from infected cells. The ultimate goal of eradication of HIV from the body can only be achieved if recruitment of infection to new cells is prevented and if cells already infected (the HIV “reservoir”) eventually die.
Toxicity of AZT is a serious issue, including marrow suppression (neutropenia, macrocytic anemia), reversible wasting of proximal muscles, nausea, and headaches. The newer NRTIs (see Table 12.1) have different patterns of side effects, and resistance to many of these agents maps at different sites in the reverse transcriptase molecule from that of AZT (see also Chapter 23: Retroviruses).
Non-nucleoside Reverse Transcriptase Inhibitors
In the late 1980s several classes of inhibitors (“TIBO” and “HEPT”) were developed and found to be highly specific and potent inhibitors of the reverse transcriptase of HIV-1. These drugs bind non-competitively to a hydrophobic pocket 10 Å distant from the enzyme catalytic site, leading to conformational changes in the p66 thumb domain of the reverse transcriptase, so impairing the function of the reverse transcriptase catalytic site. The first non-nucleoside reverse transcriptase inhibitor to be approved (in 1997) was nevirapine, followed by delavirdine and efavirenz, while etravirine (approved by the United States Food and Drug Agency in 2008) has a slightly different structure and is of value in patients with virus resistant to the earlier non-nucleoside reverse transcriptase inhibitors. These are well absorbed and the associated long half-lives in the body means once or twice daily dosage is effective. Side effects include hepatotoxicity and psychiatric manifestations. Since non-nucleoside reverse transcriptase inhibitors have a different molecular target and mechanism of action from non-nucleoside reverse transcriptase inhibitors, these are of value in combination with non-nucleoside reverse transcriptase inhibitors in delaying the emergence of drug resistance, and indeed the two together along with protease inhibitors are a mainstay of current therapy.
Emergence of resistance to all of the above drugs is a continuing issue that requires careful management. Further details can be found in Chapter 23: Retroviruses.
Nucleoside Analogs in Hepatitis B
In addition to tenofovir and adefovir, several other analogs have become useful for the treatment of hepatitis B virus infection. The first of these, lamivudine (3TC), was first synthesized in 1988 and licensed for use against HIV in 1995. Later it was found to be active against the HBV DNA polymerase at lower dosages, and has since been used widely for the treatment of chronic hepatitis B infection. It has a low toxicity and a long half-life, but drug-resistant HBV variants regularly develop when used alone, and its preferred use is in combination therapy. Emtricitabine (FTC) is a similar drug, and is often used in combination with tenofovir in patients co-infected with HIV and hepatitis B virus. Entecavir is a more recently introduced nucleoside analog that is particularly active against the DNA polymerase of hepatitis B virus but not against the HIV reverse transcriptase. A 48-week course of entecavir treatment in chronic hepatitis B has been shown to give suppression of serum hepatitis B virus DNA and normalization of liver function in a greater proportion of patients than with lamivudine treatment, although the safety profiles of the two drugs are similar. Other drugs to which hepatitis B virus responds in a proportion of patients include telbivudine and pegylated interferon.
The efficacy and accompanying drawbacks in treating hepatitis B patients have been documented for each of these drugs as monotherapy, but evidence for or against their use in combination therapy is only now being explored, despite the obvious theoretical appeal of combination therapy.
Inhibitors of Viral Proteases
Cleavage of viral proteins by proteases is essential at several stages in many viral replication cycles: examples include activation of envelope fusion glycoproteins, activation of some viral enzymes, post-translational cleavage of the polyprotein product of polycistronic mRNA, and maturation of the virion. As many of the proteases are virus-coded, it should be possible to find agents that specifically inhibit a viral protease without interfering with essential cellular proteases.
The development of HIV protease inhibitors is the first example of this approach. The three-dimensional structure of the HIV protease was solved by X-ray crystallography. The active site was identified and found to accommodate seven amino acids, using enzyme produced in large quantities by recombinant DNA technology. The enzyme was found to cleave sequences containing the dipeptides Tyr-Pro or Phe-Pro (Fig. 12.4 ). When a hydroxyethylene linkage replaces the peptide bond, cleavage does not occur; viral proteins continue to be made, but the viral particles budding from cells are immature and non-infectious. A range of different inhibitors that bind to, and block, the active site are now in routine use, usually as part of combination therapy (Table 12.1; Fig. 12.5 ). As would be expected, resistant strains of HIV may arise, many of which are unique to a particular inhibitor. Side effects, which can be troublesome, include nausea, vomiting, diarrhea, hyperlipidemia, insulin resistance, redistribution of body fat to the trunk regions, and abnormal liver function.
Figure 12.5.

Comparison between one of the sites cleaved by the HIV protease and the protease inhibitor saquinavir. (A) Amino acid sequence within the Gag-Pol protein showing one cleavage site (between tyrosine and proline, shown by the red arrow). (B and C) The corresponding structures of the protease inhibitor saquinavir (Ro 31-8959) and darunavir (WHO/ATC code J05AE10).
Reproduced from Flint, S.J. et al., 2009. Principles of Virology, third ed., vol. II, p. 303, ASM Press, Washington, DC, with permission.
Direct-acting Antiviral Drugs for Hepatitis C Virus Infection
Antiviral treatment of hepatitis C infection has undergone a radical change since 2011, with the progressive introduction of three classes of direct-acting antiviral drugs (DAAs). This development has been possible due to the synthesis of specific inhibitors of the active sites of three key viral gene products: the NS3-4A protease, the NS5A RNA-binding protein, and the NS5B RNA-dependent RNA polymerase. For example, sofosbuvir is a nucleotide monophosphate analog that, after conversion to the active triphosphate within the cell, becomes incorporated into the growing RNA chain and acts as a chain terminator. The NS3-NS4A protease of hepatitis C virus complex offers an attractive target. The peptidomimetic drugs boceprevir and telaprevir were licensed for use in the United States in 2011, but have since been largely replaced by second-generation protease inhibitors (simeprevir, asunaprevir, paritaprevir) with improved safety and dosage profiles. When used in combination with pegylated interferon and ribavirin the rate of sustained virological response nearly doubled and they have been of particular benefit in more difficult-to-treat patients such as those with cirrhosis or those infected with HCV genotype 1. Sustained viral responses were achieved with shorter courses of treatment, and more recent trials have demonstrated success with interferon-free regimens in most patients. These drugs represent the first of a large number of direct-acting antivirals at different stages of development, ushering in an exciting new phase in the treatment of hepatitis C virus. Six different combinations of these drugs are now recommended for use against different HCV genotypes, and permanent cure can be achieved for most patients using a shorter and completely interferon-free regimen. A truly remarkable advance.
Inhibitors of Viral Integration
Integration of the HIV genome through the action of the virus-coded enzyme integrase is an essential stage in HIV replication. The development of an assay for the DNA strand transfer step in the integration process allowed screening of compounds for inhibition of this step. The first compound to reach clinical use was raltegravir in 2008, followed by elvitegravir and dolutegravir. A number of clinical trials have shown that combination regimens inclusive of an integrase inhibitor were equally effective or superior to standard regimens, and more detailed information about toxicity and guidelines for use are being developed in the light of further experience.
Neuraminidase Inhibitors
The development of highly effective neuraminidase inhibitors became feasible when analysis of the three-dimensional structure of influenza neuraminidase revealed the location and structure of the catalytic site (see above). Two successful agents zanamavir (delivered intranasally) and oseltamivir (delivered orally) have become important in influenza treatment and prophylaxis. Both bind strongly to the enzyme active site and prevent release of progeny virions, thereby preventing infection of new cells and the further spread of infection. When used within 24 to 30 hours of the onset of symptoms, either drug can shorten the duration of symptoms by one to three days, and these are also useful as prophylactic treatment. In contrast to the adamantanes, zanamavir and oseltamivir cause very little toxicity and are associated with less problems of drug resistance. Each has a broad spectrum of action across influenza strains of both type A and type B, and a low rate of drug resistance makes each an important component in planning for future pandemics. Stockpiles of drugs are maintained to provide rapid prophylaxis and treatment, if required in the lead-up period before a specific vaccine against a new pandemic strain becomes available. However, cost and logistics will of course limit how extensively and for how long such treatment can be sustained in the face of a new pandemic. More recently developed neuraminidase inhibitors include peramivir, an intravenous agent authorized for emergency use in hospitalized patients suffering from 2009 H1N1 influenza, and laninamivir, which is active against oseltamivir-resistant viruses and has such a long-lasting effect that a single inhalation on the first day of treatment appears to be effective.
Newer Approaches Under Development
Virus-Specific Oligonucleotides
Theoretically, short synthetic antisense oligodeoxynucleotides, complementary in sequence to viral mRNA, could inhibit viral gene expression with high specificity. For example, hybridization to viral mRNA may prevent the splicing, transport, or translation of that mRNA, or render it susceptible to degradation by RNase H. On the other hand, hybridization to either viral DNA or cDNA might block transcription or replication, or block the attachment of DNA-binding regulatory proteins. Short single-stranded DNA sequences of this nature have displayed antiviral activity against HIV, herpes simplex virus, and influenza viruses in cultured cells. Moreover, the ability of naturally occurring micro-RNAs (miRNAs) to regulate gene function has provided encouragement for such an approach against viruses. In practice, major problems with the specificity, stability, and uptake of oligodeoxynucleotides have hindered this work. Oligonucleotides are rapidly degraded by extracellular and intracellular nucleases, unless the backbone of the molecule is modified, for example, to phosphorothioate linkages, to circumvent this problem. Moreover, the uptake of oligonucleotides into cells is very inefficient; attempts are being made to facilitate entry, such as the coupling of miRNAs to a hydrophobic peptide or lipid.
Inhibitors of Regulatory Proteins
Many of the genes of HIV are regulatory genes, the sole or principal function of which is to control the expression of other genes. For example, the Tat protein binds to a specific responsive element called TAR which is present in both the integrated HIV cDNA and all HIV mRNAs. This binding results in enhanced expression of all the HIV genes, including tat itself, thereby constituting a positive feedback loop that enables production of large numbers of progeny virions. Clearly, an agent capable of binding either to the Tat protein or to the TAR nucleotide sequence should be a most effective inhibitor of HIV replication. Screening based on inhibition of the Tat–TAR interaction has identified a number of compounds active in cell culture, but none of these is currently used clinically, partly because these agents are not easily delivered. As other regulatory elements are being discovered for many other viruses, this novel approach to antiviral chemotherapy has considerable appeal.
Microbicides
Efforts have increased recently to develop effective topical microbicides—ointments or creams designed to prevent virus uptake at the site of entry into the body. Such agents might act by inactivating extracellular virus, by blocking attachment to cells, or by preventing the first round of virus replication at the entry site. Prevention of HIV and genital herpes simplex virus infection has received most attention, and the attraction of this approach includes the fact that it can be applied by the patient at the time when it is most needed. Ideally it should prevent infection before it is initiated, and the agents are likely to be relatively cheap. Agents that have been investigated include nonoxynol-9, a commercially available spermicide that unfortunately was found to damage epithelia and possibly enhance HIV transmission; various polymers, for example, polystyrene sulfonate that binds to herpes simplex virus glycoprotein B and inhibits virus uptake and spread; specific blockers of virus entry; and classical antiviral drugs, for example, reverse transcriptase inhibitors, applied locally. This area is under active research.
Further Reading
- Mousseau G., Valente S. Strategies to block HIV transcription: Focus on small molecule tat inhibitors. Biology. 2012;2012(1):668–697. doi: 10.3390/biology1030668. http://dx.doi.org/10.3390/biology1030668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman D.D., Whitley R.J., Hayden F.G., editors. Clinical virology. third ed. ASM Press; 2009. [Google Scholar]