

Abstract
Cell culture is a very versatile tool in the investigation of basic scientific and translation research questions. The advantage of using cell lines in scientific research is their homogeneity and associated reproducibility in data generated. This chapter introduces the principles behind the setup of a cell culture lab and the guidelines that ensure safety of the lab personnel as well as the cultured cells. It also addresses potential microbiological contaminants and how they can be avoided but also detected early. Since the selection of a particular cell line and specific cell culture conditions depends on the readout of the desired assay, this chapter will present a generalized overview of common mammalian cell culture components and properties that contribute to a suitable cell culture microenvironment. Consequently, this chapter outlines several techniques that are crucial for cell propagation and can be easily adapted to a broad number of cell types and experimental procedures.
Keywords: Cell culture, asepsis, primary cell, contamination, medium, supplements, incubator, biosafety level, hazard group, maintenance
Introduction
Cell culture refers to laboratory methods that enable the growth of eukaryotic or prokaryotic cells in physiological conditions. Its origin can be found in the early 20th century when it was introduced to study tissue growth and maturation, virus biology and vaccine development, the role of genes in disease and health, and the use of large-scale hybrid cell lines to generate biopharmaceuticals. The experimental applications of cultured cells are as diverse as the cell types that can be grown in vitro. In a clinical context, however, cell culture is most commonly linked to creating model systems that study basic cell biology, replicate disease mechanisms, or investigate the toxicity of novel drug compounds. One of the advantages of using cell culture for these applications is the feasibility to manipulate genes and molecular pathways. Furthermore, the homogeneity of clonal cell populations or specific cell types and well-defined culture systems removes interfering genetic or environmental variables, and therefore allows for data generation of high reproducibility and consistency that cannot be warranted when studying whole organ systems.
In Principle
The Cell Culture Laboratory
Cell Culture Laboratory Safety
The exciting application of cell culture techniques in biomedical research requires the management of potential hazards linked to infectious agents harbored by cultured cells (e.g., HBV or HIV), but also the control of reagents that can be of toxic, corrosive, or mutagenic nature. These potential hazards can endanger the health of laboratory workers when introduced into the body (e.g., via contact of skin and mucous membranes with solids, liquids, or aerosols) and threaten the environment when handled improperly (Fig. 9.1 ).
Figure 9.1.
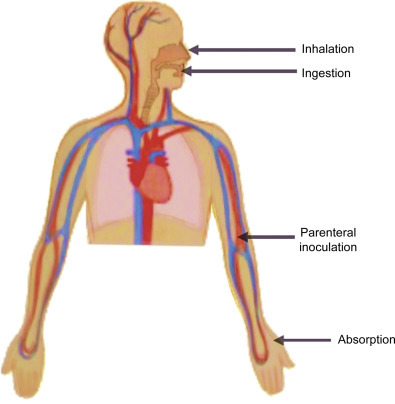
Routes of exposure to biohazards.
Biohazards in the lab can enter the body through contaminated needles (parenteral inoculation), the consumption of food or application of make-up in the lab (ingestion), the exposure of biohazardous aerosols (inhalation), and contact of skin and mucous membrane with contaminations. Personal protective equipment and biosafety cabinets are put in place to block exposure of researchers to biohazardous agents.
Before commencing any cell culture work, the reduced or eliminated exposure to potentially hazardous agents therefore needs to be ensured to minimize infection, pathogenicity, allergic reactions, and contact with released toxins. This can be achieved by stringent training of lab personnel and implementation of standard cell culture practices (Table 9.1 ), which should be reviewed and revised regularly by laboratory members and the institute’s safety committee. Additionally, when working with primary cells isolated directly from human tissue, it is important to screen donors from which cells were derived for disease-causing pathogens. Up-to-date immunizations against infectious diseases such as Hepatitis B are also highly recommended for laboratory staff working with primary cells.
Table 9.1.
Guidelines for Cellular Lab Safety
|
|
|
|
|
|
|
|
|
|
|
Safe Handling of Cell Lines
The Advisory Committee on Dangerous Pathogens (ACDP) is a national body managed by the Health and Safety Executive (HSE). It advises on hazards and risks to workers and others from exposure to pathogens and has published these recommendations [1]. Since some cell types are pathogenic or carry disease-causing agents, it is important to first determine their Hazard Group and implement appropriate safety measures. This includes a written risk assessment and review of the laboratory facilities. Microorganisms classified as Hazard Group 1 (e.g., Escherichia coli K-12) or 2 (e.g., Staphylococcus aureus) represent a low or moderate health risk to laboratory workers and the community, and relies on effective prophylaxis or treatment options. Cell culture work with biological agents of Hazard Group 3 (e.g., severe acute respiratory syndrome-associated coronavirus (SARS-CoV)), and 4 (e.g., Ebola viruses) involves biological agents that carry high health risks and may lack treatment options upon infection. Thus, laboratory spaces need to provide containment levels corresponding to the Hazard Group of the cultured cell types. These are referred to as biosafety levels (BSL) and carry the corresponding numbers (BSL1–4). As such, laboratories designated as BSL1 will follow standard microbiological practices, while BSL2 laboratories will need to be restricted to trained personnel, who are taught to take extreme precaution handling sharp items and to limit infectious aerosols by utilizing physical containment equipment as well as Class II biosafety cabinets.
Safe Experimental Procedures in the Cell Culture Laboratory
In order to ensure a safe working environment with cell lines and biohazardous agents, personal protective equipment (PPE) must be worn in the cell culture lab. Lab coats, gloves, and goggles create a barrier between the laboratory worker and potentially hazardous sources. Furthermore, biosafety cabinets rely on a steady, unidirectional flow of HEPA-filtered air and create an enclosed, ventilated workspace. This minimizes the exposure of researchers and the environment to hazardous material associated with the cultured cells, while simultaneously protecting the cell cultures from contaminations. While handling cell culture media and carrying out experiments in the cell culture lab, it is also recommended to review the Material Safety Data Sheet (MSDS) associated with laboratory reagents. It details the chemical and physical properties of the product, outlines suitable storage and disposal routes, informs about potential health hazards and toxicity, and advises on PPE that should be in place when handling this product.
Equipment for the cell culture laboratory
Despite the various techniques and assays carried out in different cell culture labs, the common theme of cell culture work is asepsis—the creation of a microenvironment free of unwanted pathogenic microorganisms, including bacteria, viruses, fungi, and parasites. Since asepsis is a crucial component of successful cell culture work, a separate room or designated area should be dedicated to this work and not be utilized for other purposes. Several pieces of equipment can aid in achieving such a sterile workspace and generally lead to higher efficiency, accuracy, and consistency of the cell culture performance (Table 9.2 ).
Table 9.2.
Recommended Equipment for the Cell Culture Laboratory
| Equipment | Purpose |
|---|---|
| Biosafety cabinet |
|
| Humid CO2 incubator |
|
| Inverted light microscope |
|
| Fridge, freezers (−20°C, −80°C), liquid nitrogen storage |
|
| Centrifuge |
|
| pH meter |
|
| Pipettes and pipettors |
|
| Cell media and supplementary components |
|
| Hemacytometer |
|
| Autoclave |
|
| Vacuum pump |
|
| Water bath (with adjustable temperature) |
|
| Cell culture dishes |
|
| Containers for waste (biohazardous) |
|
Aseptic Cell Culture Practices
While the previous section has explored methods aimed at decreasing the exposure of hazardous substances to the laboratory worker, this section will address the practices that should be put in place by laboratory workers to protect the cultured cells. Indeed, microbiological infections represent the main problem for the maintenance of cells in vitro. Infectious agents such as bacteria are toxic for eukaryotic cells and ultimately lead to cell death. Furthermore, even low levels of contamination can result in abnormal results and lead to wrong scientific interpretations. By adhering to several techniques that ensure asepsis in the cell culture lab, researchers can reduce the frequency and extent of contaminations and diminish loss of cells, resources, and time. This can be achieved by eliminating the entry of microorganisms into the cell culture through contaminated equipment, media, cell culture components, incubators, work surfaces, and defect or opened cell culture vessels.
Creating an Aseptic Work Environment
Given that atmospheric air is laden with microparticles of potentially infectious nature, the biosafety cabinet is the most crucial piece of equipment to restrict nonsterile aerosols and airborne components from contaminating cultured cells. The biosafety cabinet should be located in a laboratory space that does not interrupt its airflow through external sources of wind (e.g., drafts from windows or doors). Most biosafety cabinets require a warm-up time after which the work surface should be decontaminated with an antifungal detergent (e.g., 5% Trigene) followed by 70% ethanol. All equipment entering the biosafety cabinet also needs to be sprayed and wiped with 70% ethanol. The number of items used in the biosafety cabinet, however, should be kept at a minimum to avoid any obstruction of airflow. The biosafety cabinet should only be turned off after its daily use has been completed and the ultraviolet lamp may be turned on to sterilize the exposed surface areas over night. Regular maintenance also includes cleaning of the area under the work surface onto which media may spill through the grill. Furthermore, routine servicing through biosafety cabinet engineers can ensure correct airflow and full filter capacity of this important piece of cell culture equipment.
It is critical to keep all other surfaces in contact with the cell culture vessels or media components clean. This includes the incubator, centrifuge, microscope, water bath, fridge, and freezer. Stainless steel incubators allow for easy cleaning and protect the surfaces from corrosion of the humid environment. Treatment solutions can be added to water baths to prevent the growth of microbes. On a larger scale, the equipment stored in the cell culture space should be kept free from dust and regular cleaning of cell culture floors is advisable.
Laboratory staff can contribute to a clean work surface by washing hands with soap before and after working with cell cultures. Disposable gloves sprayed with 70% ethanol and lab coats can further reduce the introduction of contaminants carried by hair, skin cells, or dust. However, gloves need to be removed when leaving the cell culture space and lab coats should also be worn only within the confinement of the cell culture laboratory. Furthermore, lab coats need to be washed at hot temperatures on a regular basis.
Using Aseptic Reagents and Media for Cell Culture
The main sources of contamination are laboratory staff, the environment, and the culture medium. Commercially sourced media and supplementary cell culture products are generally supplied in sterile condition. In addition, filter-sterilizing allows for the generation of cell culture media that are based on nonsterile culture reagents, while autoclaving is conventionally used to sterilize equipment in contact with cultured cells. The filter-sterilization of liquids can be achieved by forcing the liquid through a 0.22 μM polyethersulfone low-binding filter system using a vacuum pump. The addition of antibiotics (e.g., Penicillin/Streptomycin) further limits the risk of bacterial growth in media bottles after opening and in cell culture vessels. However, some laboratories refrain from using antibiotics routinely since it can facilitate the emergence of resistant bacteria strains, allow for low-level background contaminations, and may lead to interference with cell metabolisms and experimental outcomes.
Contaminations
Since contaminations can generally not be avoided altogether, it is important to train cell culture laboratory staff to recognize early signs in order to prevent the spread of contaminants to other cells or cell culture products. Contaminants are most commonly of biological nature and can include bacteria, fungi, viruses, and parasites (Fig. 9.2 ). It is important to limit biological contaminants since they can alter the phenotype and genotype of the cultured cell line through competition for nutrients, synthesis of alkaline, acidic or toxic by-products, and the potential interference of viral components with the cell culture genome. Other contaminants may include the introduction of undesired chemicals impurities (e.g., plasticizers in cell culture vessels) or other cell types cocultured in the lab.
Figure 9.2.

Microbial contaminants in cell culture.
(A) Depending on the bacterial strain introduced, the morphology of bacterial contamination under the microscope can vary from rodlike shapes, cocci, flagellated to barely visible. (B) Yeast forms multicellular stringlike structures that appear ovoid in shape. (C) Mold growth is marked by the production of multicellular, highly connected, thin filaments (hyphae).
Bacterial Contamination
The bacteria kingdom includes highly ubiquitous, prokaryotic microorganisms characterized by the size of a few micrometers in diameter, wide diversity in their morphologies, and fast doubling times through asexual reproduction. While the latter property allows for ready detection in cell culture supernatants shortly after infection, it also facilitates quick spread. Cell cultures affected by bacterial contamination generally appear turbid in appearance. Furthermore, the high metabolic rates of bacteria can modify the pH of the culture media and thus change the color of phenol red to yellow. While bacteria may be detected as small particles at low microscope magnification, their distinct shapes are generally detected at higher magnification. While bacterial strains such as E. coli can therefore be uncovered quite easily due to their size (~2 μM) and flagella-induced mobility, other strains such as Mycoplasma are smaller in size (<1 μM), immobile, and therefore not as easily detectable. As a result, Mycoplasma infections can go unnoticed for a longer time and usually only become apparent through declining quality of the cultured cells. This can manifest as reduced cell proliferation and cell death. In order to monitor cell cultures for potential infections with Mycoplasma, it is advisable to routinely test cultures for their presence using polymerase chain reaction (PCR), enzyme-linked immunosorbent assay (ELISA), or immunostaining [2].
Fungal Contamination
Yeasts are unicellular eukaryotes that form multicellular string-like structures during asexual reproduction. These budding cells appear ovoid in shape, can grow to approximately 4 μM of size and are therefore easily detected at low microscope magnifications.
Molds are additional members of the fungi kingdom that can be found in cell cultures. Their growth is marked by the production of multicellular, highly connected, thin filaments (hyphae).
Cell culture supernatants contaminated with yeasts or molds appear turbid and although the pH remains stable during the initial stages of infection, it increases in high contaminant concentrations. Yeast contaminations may also be accompanied by a distinct smell. Since fungal species can spread via airborne spores, it is particularly important to identify and contain such contaminations quickly.
Viral Contamination
Viruses are infectious agents that rely on host cells for their own replication. Owing to their limited size of upto 300 nm and their intracellular lifecycle, they are not visible in generic light microscopy and very difficult to detect. While some viruses may induce morphological changes in the cultured cells (cytopathic effects), other species may integrate into the cellular genome and alter the phenotype of the investigated cell line. Viruses can enter cell cultures, for example, through the use of animal-derived cell culture products such as trypsin or fetal bovine serum and are a serious health concern for laboratory workers. The presence of viral contaminants can be challenging to confirm but generally relies on PCR, ELISA, immunocytochemistry, or electron microscopy [3].
Eliminating contaminations
Regardless of the type of contamination identified, affected cell cultures should be removed from the cell culture room and discarded to prevent the spread of infectious agents to other cultures. Furthermore, it is important to determine the source of contamination. It is advisable to dispose of culture media and other cell culture components that have been in contact with the contaminated cells and to clean the surfaces that have touched the contaminated vessel (e.g., incubator, biosafety cabinet, microscope, aspirator).
It is not recommended to treat or proceed culturing infected cells, since any handling of contaminated cultures will increase the potential spread of contaminants—especially airborne fungal spores. Furthermore, the use of antifungal compounds to contain an established infection can interfere with the metabolism of cultured cells. Similar consequences are expected if deciding to prolong the cultures using antibiotics such as 1% Ciprofloxacin to diminish the bacterial growth: the continued release of endotoxins from bacteria will impact the cellular metabolism and likely falsify the cellular readout.
The elimination of contaminants from the cell culture laboratory is a very tedious task and reinforces the importance of prophylactic, aseptic measures to prevent contaminants taking root in the first place.
The Cell Line
The choice of a cell line for cell culture depends heavily on the functional properties and specific readouts required of the cell model [4]. The selected cell lines will also need to align with the available equipments and requirements of their specific hazard group. Cells cultured in the lab can be classified into three different types: primary cells, transformed cells, and self-renewing cells. Primary cells, such as fibroblasts obtained from skin biopsies and hepatocytes isolated from liver explants, are directly isolated from human tissue. Biomedical and translational research oftentimes relies on using these cell types since they are good representatives of their tissue of origin. However, there are stringent biosafety restrictions associated with handling these cell types. Furthermore, primary cells are generally characterized as “finite” and therefore rely on a continuous supply of stocks since their proliferation ceases after a limited amount of cell divisions and cell expansion is oftentimes impossible. Transformed cells can be generated either naturally or by genetic manipulation. While the use of such immortalized cell lines leads to a cellular platform that generates fast growth rates and stable conditions for maintenance and cloning, their manipulated genotype may result in karyotypic abnormalities and nonphysiological phenotypes. On the other hand, standardized cell lines derived from human or nonhuman species or (e.g., Chinese hamster ovary (CHO), HeLa, human umbilical vein endothelial cells (HUVEC)) are oftentimes thoroughly characterized and may therefore be easier to set-up. Self-renewing cells include, for example, embryonic stem cells, induced pluripotent stem cells, neural and intestinal stem cells. These cells carry the capacity to differentiate into a diversity of other cells types, while their self-renewing property allows for long-term maintenance in vitro. Self-renewing cell types oftentimes act as physiologically relevant representatives of in vivo mechanisms.
Cell lines can be obtained commercially, where certain quality control measures are in place that guarantee genomic stability and absence of contaminants. Other places to source cell lines from can be cell banks or other cell culture laboratories. The introduction of new cell lines in a lab should always be accompanied by a Mycoplasma PCR test to ensure clean cultures.
The Cell Culture Microenvironment
Regardless of the cell line chosen, a common requirement will be the selection of suitable growth conditions. This also includes the format of cell growth. There are several advantages and disadvantages associated with culturing cells either in suspension or in plated forms. While fast-growing cells in suspension are more suitable for experiments that aim to isolate recombinant proteins, adherent cells are more appropriate for studies in which the polarity of cells is a crucial component of the cell’s functionality (e.g., epithelial cells). Cells grown in suspension generally adopt spherical shapes, while adherent cells display spiked or polygonal morphologies.
The Cell Culture Medium
The goal to create an environment that allows for maximum cell propagation is achieved primarily through the incubator (i.e., temperature, humidity, O2, and CO2 tensions) and the basal cell culture medium and its supplements. This includes not only the supply of nutrients such as carbohydrates, vitamins, amino acids, minerals, growth factors, hormones, but also components that control physicochemical properties such as the culture’s pH and cellular osmotic pressure. Additionally, the solid or semisolid growth substrate and the cell density allow for cell–matrix anchoring and cell–cell interactions respectively, which further govern the imitation of a physiologically relevant microenvironment.
A great variety of cell culture medium compositions have been created for the requirements of specific cell types and can be classified according to their level of supplemented serum. Serum in the form of fetal bovine serum (FBS) is most commonly added to basal media that already contain a standard formulation based on amino acids, vitamins, carbon sources (e.g., glucose), and inorganic salts. Serum provides cells with growth factors and hormones and acts as a carrier for lipids and enzymes, and the transportation of micronutrients and trace elements. Several labs, however, aim to reduce the supplementation of basal media with animal-derived factors such as serum since it is an undefined component that can highly vary between batches. It is also a costly cell culture product, carries the risk of causing undesired stimulatory or inhibitory effects on cellular growth and function, and may introduce contaminations if not sourced from reliable suppliers. Reduced-serum or serum-free media rely on formulations that reduce or replace serum with more defined components. This generally yields cell cultures characterized by greater consistency in growth and in downstream experimental applications. The concentration of supplements can also be adjusted according to the specific needs of the cell types.
Temperature, pH, CO2, and O2 Levels
The desired temperature for cell cultures depends on the body temperature of the species and the microenvironment from which the cultured cell types were isolated. While most human and mammalian cell lines are incubated at 36–37°C, cell lines originating from cold-blooded animals can be maintained at wider temperature ranges between 15°C and 26°C. The pH level for most human and mammalian cell lines cultured in the lab should be tightly controlled and kept at a physiological pH level of 7.2–7.4. In contrast, some fibroblast cell lines favor slightly more alkaline conditions between pH 7.4 and 7.7, while transformed cell lines prefer more acidic environments between pH 7.0 and 7.4 [5].
Stable temperatures for cell cultures can be achieved through incubators that tightly regulate and monitor the temperature of the cell culture environment. As the cells propagate, their growth requires energy supplied in the medium, for example in the form of glucose. When metabolized, its by-products include pyruvic acid, lactic acid, and CO2. Since the pH level is dependent on the balance of CO2 and HCO3 − (bicarbonate), the addition of bicarbonate-based buffers to cell culture media can equilibrate the CO2 concentrations. Other pH buffers can be of organic nature and include 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (10–25 mM) or 3-(N-morpholino)propanesulfonic acid (MOPS) (20 mM). Many cell culture media contain pH indicators (e.g., phenol red), which display a color range between acidic (yellow) and alkaline (pink) conditions. Furthermore, fluctuations in atmospheric CO2 concentrations can also alter the pH level. Cells should therefore be cultured in incubators that also allow for CO2 tensions to be adjusted to 5–7%. To delay shifts in pH, glucose may be substituted by another carbon source such as galactose or fructose. Although this will slow down the rate of cell growth, it will also reduce the accumulation of by-products such as lactic acid.
For specific cell cultures that mimic pathological or physiological conditions under low oxygen tension (hypoxia), it is recommended to use hypoxic incubators with adjustable 1–21% oxygen concentrations balanced with nitrogen.
Subculturing
When the available space in the cell culture vessel reaches ~80% confluency (coverage), cells need to be transferred to new vessels to continue their growth. This process, referred to as “passaging,” generates subcultures or subclones, and requires enzymatic digestion or mechanical disruption of the adherent cell monolayer to detach cells from their tissue-culture-treated substrate (Fig. 9.3 ). While the growth of adherent cells is limited or enabled by the available surface area, it is the concentration of cells in the medium that creates the rate-limiting step in suspension cultures. It is therefore essential to monitor the growth rates in suspension cultures over time.
Figure 9.3.
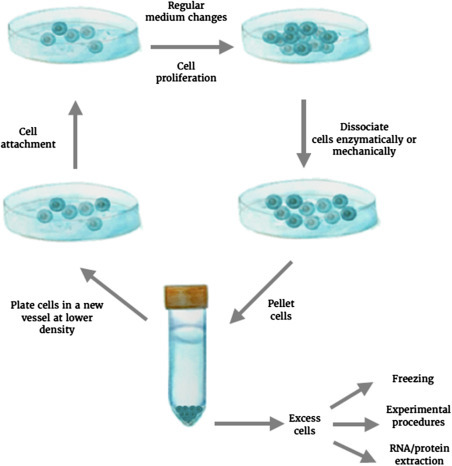
Cell maintenance.
Adherent cell lines are maintained in flasks or plates and regular medium changes ensure healthy cell propagation. Once ~80% confluency has been reached, cells are enzymatically or mechanically dissociated from their plating substrates. The detached cells can be collected in a Falcon tube and pelleted. With a subset of these cells new cell culture vessels can be seeded, while any remaining cells can be frozen or utilized for downstream experiments.
In Practice
This section explains the basic protocols required for the maintenance of cell cultures. Since some of these protocols may need to be amended to accommodate the specific requirements of various cell types, it is helpful to review the recommendations of the cell line supplier.
Dissociating Adherent Cells from Culture Vessels for Subculturing
Cells cultured in vitro over time will deplete nutrients supplied in the medium, release toxic metabolites and grow in number. In order to expand and/or maintain a healthy cell culture, it is therefore essential to produce a new culture with a subset of cells from the originating culture, removing toxic by-products, and replenishing nutrients with fresh medium. A suitable time for passaging is reached when the growth of adherent cells reaches ~80 % confluency. They can then be enzymatically digested or mechanically dissociated to lift off their substrate. In a biosafety cabinet, cells are washed with phosphate-buffered saline (PBS) free of Mg2+ and Ca2+ to remove dead cells and are incubated at 37°C with sufficient digestive enzymes or chelating agent to cover the monolayer (e.g., trypsin, dispase, collagenase, ethylenediaminetetraacetic acid (EDTA)). The time required to detach the anchored cells from their substrate and cell–cell interactions can take 1–60 minutes and depends on the cell type and the digestive enzymes used. The extent of dissociation can be monitored under a light microscope and once complete, tapping of the culture vessel should dislodge remaining adherent cells. The dissociated cells are collected in a sterile Falcon tube and the culture vessel should also be washed with a medium containing an inhibitor for the enzymatic digestion and dissociation of cells. Collected cells can then be concentrated and counted according to protocols 4.3 and 4.4 and seeded in new culture vessels at the desired concentrations. Lower cell concentrations (~104 cells/mL) are suitable for cell lines with fast proliferation rates, while higher cell concentrations (~105 cells/mL) are more adapted for cells with slower growth rates.
Note: It is good lab practice to record the number of passages that have taken place since the culture has been initiated. Some cell lines are not suitable for experimental work beyond a given passage number since chromosomal abnormalities tend to increase in mammalian lines with cell divisions over time.
Subculturing of Suspension Cultures
The subculturing of suspension cultures can be achieved by aseptically removing one-third of the cell suspension solution and replacing the volume with prewarmed complete medium.
Pelleting Cells
In order to concentrate cells for transfer to new cell culture vessels, freezing, or other experimental assays, the cell suspension is centrifuged at 300 ×g for 10 minutes. After removing the supernatant, the cell pellet is resuspended in the desired medium through gently pipetting cells up and down three times.
Note: Single cells can be quite fragile and it is therefore advisable to not centrifuge at higher speeds or to pipette them vigorously.
Quantification of Cells and Determining Cell Viability
Cells can die in the process of culturing or during handling and passaging. When relying on a specific concentration of live cells to start a culture or needing a specific number of live cells for an assay it is important to distinguish between live and dead cells. Cell counting is also helpful when assessing growth rates. Since cells are commonly cultured in the millions, the number of cells are first counted in a small volume and then extrapolated to the full cell volume. To achieve this, all cells are dissociated, pelleted, and evenly resuspended in a suitable medium volume. In a 1:1 dilution with 0.4% trypan Blue, a small volume of the cell suspension is mixed in an Eppendorf tube. Trypan Blue dye permeates only nonviable cells that can therefore be excluded from the subsequent quantification [6]. This occurs by loading 10 μL of the cell mixture in Trypan Blue onto a hemacytometer (Fig. 9.4 ). Using an inverted microscope, phase contrast, and a magnification of at least 10X, all cells located in the four outer squares are counted. Viable cells contain a darker “halo,” while nonviable cells stain blue/black. To determine the total number of viable cells, the number of cells found in all four squares is divided by 4 (to determine the average cell number in 1 mm2), multiplied by 104 (to obtain the cell number per mL), multiplied by 2 (to account for the dilution factor of Trypan Blue) and multiplied by the initial medium volume of the entire cell suspension. The percentage of viable cells can be determined by dividing the number of unstained cells by the total number of cells, and multiplying the ratio by 100. A healthy cell culture is characterized by 80–95% cell viability.
Figure 9.4.
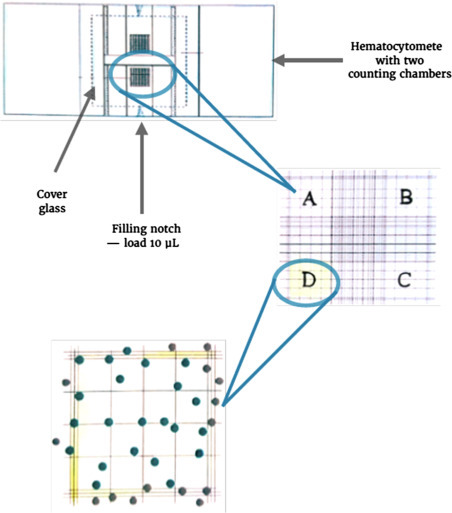
Cell quantification using Trypan Blue.
The hemacytometer is prepared by covering both counting chambers with a cover glass. Subsequently, 10 μL of a 1:1 cell suspension with 0.4 % Trypan Blue is loaded onto the filling notch of one of the counting chambers. Through capillary action, this volume will cover the grid that can be observed in an inverted microscope at magnifications of at least 10X. The average of cells covering squares A–D determines the number of cells per mm2. Viable cells in these squares are counted by excluding nonviable cells that appear black due to their absorption of Trypan Blue through their permeable cell membranes. Only cells overlapping with one of the outer horizontal and vertical borders should be included.
Freezing Cells
When a surplus of cells becomes available during subculturing, they can be preserved at that passage through freezing with cryoprotective agents (e.g., glycerol or dimethyl sulfoxide (DMSO)) that prevent the formation of harmful extra- or intracellular crystals [7]. To that end, cells are dissociated from the culture vessel and condensed as described in protocol 4.3. The cell pellet is resuspended in 1 mL of freezing medium (e.g., knockout serum replacement medium supplemented with 10% DMSO) and ~1 ×106 cells are transferred into each cryovial. After 20–30 minutes, the cryoprotectant will have penetrated the cells. Cooled down overnight at −80°C at a controlled freezing rate of 1–2°C/min, the vials are then transferred to liquid nitrogen for long-term storage.
Note: While glycerol and DMSO are both suitable cyroprotective agents, handling of DMSO needs to be carefully monitored. In high (stock) concentrations, DMSO is toxic to personnel and cultured cells and therefore cannot be added to cells without prior dilution. This toxicity also affects cells in freezing medium containing 10% DMSO when left for several hours at room temperature, highlighting the need to transfer cells to −80°C for storage within 30 minutes. In general, chemically protective gloves should be worn to safeguard personnel from the hazards of DMSO and its solutes to easily penetrate membranes, including the skin.
Thawing Cryopreserved Cells
Most mammalian cells can be preserved in liquid nitrogen (<130°C) for numerous years since all biological processes are halted at these temperatures. To recover cells, 10 mL of complete medium is prewarmed in a water bath. After removing the frozen vial from liquid nitrogen, it is immediately placed into a 37°C water bath and gently swirled until two-thirds of the content are completely thawed. The vial is wiped with 70% ethanol and placed in a biosafety cabinet where 1 mL of the prewarmed medium is added in a drop-wise fashion to the partially thawed vial to minimize the osmotic stress imposed upon the cells when DMSO is diluted. The contents of the now completely thawed vial are transferred also in a drop-wise fashion to the remaining 9 mL of complete medium and centrifuged at 300 × g for 3 minutes. After aspirating the supernatant, the cell pellet can be washed once in medium to remove residual cryopreservatives. Cells are then resuspended in complete medium and transferred to a cell culture vessel. Cell attachment should occur within 24 hours.
Note: The viability of cells after cryopreservation is impacted by their ability to cope with the stressors of freezing and thawing. It is therefore recommended to perform the thawing process as swiftly as possible. When handling cryovials that have been frozen with glycerol as the cryoprotectant, the thawing process can be simplified by diluting the cryopreserved cells ten times directly into complete, prewarmed medium, avoiding the centrifugation and washing step.
Applications
Model Systems in Health and Disease
Cell culture is one of the most important techniques in cellular and molecular biology since it provides a platform to investigate the biology, biochemistry, physiology (e.g., aging) and metabolism of wild-type cells and diseased cells. The interaction and route of infection between wild-type cells and pathogenic agents (e.g., bacteria and viruses) can also be studied in specific cocultures. Furthermore, immortalized cancer cell lines have given researchers insight into the biology of cancer and through the selective treatment of wild-type cells with UV radiation, viruses, and toxins, causative agents of tumorigenicity have been identified. Finally, human-induced pluripotent stem cells (hIPSCs) have been derived from individuals with inherited disorders and differentiated toward the affected cell type in which the disease manifests. These hIPSC-derived somatic cells are suitable platforms for the studying molecular mechanisms of a disease in a dish.
Drug Development and Drug Testing
Cell culture tools can also be applied to screen novel chemicals, cosmetics, and drug compounds for their efficacy and assess drug cytotoxicity in specific cell types. Detoxifying cell types such as hepatocytes and kidney cells are oftentimes of high interest for these purposes. When using cell cocultures or diseased cells obtained from individual patients, it is also possible to screen for drugs to selectively target specific cell types (e.g., in cancer treatment), at doses that are nontoxic and with minimized side-effects for the patient. Furthermore, large-scale cells cultures can serve for the generation of genetically engineered proteins, antibodies, hormones. and biopharmaceuticals that can be isolated and used therapeutically.
Virology and Vaccine Production
Cell culture with mammalian cells offers a host for viruses to replicate, allowing researchers to study their growth rates, development, and conditions required for their infectious cycle. Furthermore, the attenuated viruses used in vaccines against polio, measles, chicken pox, rabies, and hepatitis B are raised in animal cell cultures.
Tissue Regeneration and Transplantation
hIPSCs, embryonic stem cells, and adult stem cells have the capacity to regenerate and differentiate into specialized cell types that can be used as replacement tissues or organs. These cell cultures are oftentimes performed in a 3D protein matrix that allows cells to self-organize into functional cell clusters (organoids).
Genetic Engineering and Gene Therapy
The expression of specific genes and their impact on cells can be studied by the introduction of new genetic material (e.g., DNA, RNA) into the nucleus of cultured mammalian cells. Similarly, the importance of genes in regulating specific pathways can be observed through silencing them. Oftentimes, viral vectors or specialized enzymes are used to carry out these tasks. Altering the genome of cells can also aid in restoring dysfunctional genes in patients.
The overarching benefit of using cell culture techniques to address these basic scientific and translational research questions is the homogeneity and reproducibility of data that can be generated using clonal cell lines. Studying an isolated, simplified cellular system in a well-defined and controlled environment limits the exposure of confounding effects inherent to an in vivo system and therefore allows for the generation of simplified but robust data sets.
Scenario
Culturing hIPSCs to study inherited liver diseases: Small pieces of skin biopsies obtained from patients with an inherited liver disease can be cultured in a Petri dish with fibroblast growth medium. Primary fibroblast cultures will emerge from the skin tissue after 2–3 days. Upon reaching ~80% confluency, the outgrown primary fibroblasts can be isolated from the skin tissue using enzymatic digestion with collagenase and subcultured in new vessels. This generates a pure population of fibroblasts, which can be scaled-up and virally transduced to express pluripotent genes (e.g., OCT4, NANOG, TRA-1-60), thereby “reprogramming” somatic cells to become hIPSCs [8]. hIPSCs in culture will pack together tightly in flat colonies with sharp edges and are characterized by their high nucleo-cytoplasmic ratio, ability to self-renew, and capacity to form cells of all three germ layers. As such, hIPSCs can be differentiated toward definitive endoderm using Wnt pathway activation and Activin A. Cells at this stage of differentiation will migrate from their colonies into a monolayer, downregulate their pluripotency genes and express markers of their endoderm fate (e.g., SOX17, CXCR4, GSC). Further changes in the media composition and exogenous growth factors added to the definitive endoderm cells will direct them toward foregut endoderm and subsequent specification to hepatic endoderm. The final stage of the hepatic differentiation medium yields hepatocyte-like cells that secrete albumin and other serum proteins, take up low-density lipoprotein (LDL), store glycogen, and metabolize drugs. Importantly, these cells will also display the disease phenotypes observed in the patient from which the skin biopsy was initially obtained. While this process allows researchers to study and rescue disease mechanisms ex vivo [9], it also sheds light on liver development and the emergence of disease in utero.
Key Limitations
Discrepancies between Cellular Environments in vitro and in vivo
One of the pillars of cell culture research is the design of a defined cellular environment in which single variables can be manipulated in order to monitor cellular responses. To achieve this goal, the cellular environment in vitro is oftentimes oversimplified and relies, for example, on a single cell type cultured in a monolayer. However, data generated from such a cellular system does not truly phenocopy the intricate cellular interactions between different cell types and extracellular matrices of an in vivo environment. To address this drawback, there is currently significant research into the design of cell cocultures that allow paracrine signaling between cells that cohabitate space in vivo, as well as bioartifical matrices that facilitate cellular growth in their native 3D orientation. The goal is the design of cellular systems that mimic the complexity of the multicellular in vivo niche, yet also allow standardization for cell culture assays.
Discrepancies between Gene Expression in Primary Cells and Immortal Cell Lines
The oftentimes most relevant cell types for addressing translational research questions—primary cells—are in fact very difficult to isolate and culture in vitro due to their limited proliferation and functional capacity ex vivo. To delay senescence, viral transfection of primary cells can sequester tumor-suppressor proteins, thereby extending the number of possible passages and allowing the emergence of immortal cell lines. Although this facilitates their culture ex vivo, this technique also introduces the expression of carcinogenic genes. In addition, immortalized cell lines can acquire mutations during subculturing that can further interfere with the cellular phenotype and create a nonphysiological cell culture system.
Troubleshooting
| Problem | Possible Reason | Suggested Solution |
|---|---|---|
| Lack of viable cells upon thawing | Incorrect storage | Obtain new stock stored in liquid nitrogen that have not been thawed |
| Incorrect thawing | Thaw cells quickly but gradually, dilute frozen cells drop-wise with prewarmed medium, handle cells gently, and only centrifuge at low speeds | |
| Glycerol exposure to light | If the cryopreservative agent glycerol was used and exposed to light, its by-product acrolein may be toxic for the cells | |
| Lack of cell attachment to culture vessel after subculturing | Residual digestive enzyme activity | Thoroughly wash cells in prewarmed medium before replating |
| Excessive digestion of cells | Reduce digestion time and block enzymatic activity using inhibitor (FBS for Trypsin) | |
| Mycoplasma contamination | Perform routine Mycoplasma PCR tests on cultures | |
| Slow cell growth | Incorrect growth medium | Growth medium must fit the requirements of the culture cell line and (if applicable) contain serum that has been screened |
| Depletion or breakdown of essential cell culture components | Ensure presence of growth-promoting factors and substitute unstable components such as glutamine with GlutaMax | |
| Incorrect storage of medium and supplements | Follow manufacturer’s instructions closely | |
| Passage number is too high | The proliferation rate of cell cultures may cease with continuous subcultures—cells should be replaced with low-passage stocks | |
| Confluency is too low | Enhance concentration of initial plating density | |
| Confluency is too high | Cells should be passaged in their log-phase (at around ~80% confluency) | |
| Mycoplasma contamination | Perform routine Mycoplasma PCR tests on cultures | |
| Rapid shift in medium pH | Incorrect CO2 tension | Adjust CO2 concentration of incubator based on HCO3− concentrations of medium: 2.0 g/L requires CO2 levels of 5%, while 3.7 g/L rely on 10% supplementary CO2 |
| Lack of gas exchange | Loosen caps of tissue culture flasks | |
| Insufficient bicarbonate buffering in medium | Add HEPES (10–25 mM) | |
| Bacterial contamination | Investigate cultures under light microscope | |
| Cell death | Lack of CO2 or fluctuating temperature | Monitor CO2 and temperature levels of incubator and do not leave cells outside the incubator for extended periods of time |
| Accumulation of toxins | Regularly replace cell culture medium | |
| Incorrect osmotic pressure | Review osmolality of cell culture medium and potential effects of added drug compounds or HEPES |
Conclusion
This chapter has described the vast possibilities to employ cell culture techniques to address basic and translational research questions and has explained the necessary considerations for setting up a cell culture lab. It has also shown essential practices and techniques for successfully working with cell lines and explained the conditions required for creating a cellular environment that mimics their in vivo niche.
References
- 1.HSE/ACDP. Biological agents: managing the risks in laboratories and healthcare premises. 2005.
- 2.Drexler HG, Uphoff CC. Mycoplasma contamination of cell cultures: Incidence, sources, effects, detection, elimination, prevention. Cytotechnology. 2002;39:75–90. doi: 10.1023/A:1022913015916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Merten O. Virus contaminations of cell cultures – A biotechnological view. Cytotechnology. 2002;39:91–116. doi: 10.1023/A:1022969101804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan C, Kumar C, Bohl S, Klingmueller U, Mann M. Comparative Proteomic Phenotyping of Cell Lines and Primary Cells to Assess Preservation of Cell Type-specific Functions. Mol Cell Proteomics. 2009;8(3):443–450. doi: 10.1074/mcp.M800258-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartz MA, Both G, Lechene C. Effect of cell spreading on cytoplasmic pH in normal and transformed fibroblasts. Proc Natl Acad Sci USA. 1989;86:4525–4529. doi: 10.1073/pnas.86.12.4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strober W. Trypan blue exclusion test of cell viability. Current Protocols in Immunology, Appendix 3B. 2001 doi: 10.1002/0471142735.ima03bs21. [DOI] [PubMed] [Google Scholar]
- 7.Lovelock JE, Bishop MW. Prevention of freezing damage to living cells by diemthyl sulphoxide. Nature. 1959;16(183):1394–1395. doi: 10.1038/1831394a0. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 9.Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. Journal Clin Invest. 2010;120(9):3127–3136. doi: 10.1172/JCI43122. http://dx.doi.org/10.1172/JCI43122DS1. [DOI] [PMC free article] [PubMed] [Google Scholar]