

Abstract
Alzheimer’s disease (AD) is a major problem of health and disability, with a relevant economic impact on society (e.g., €177 billion in Europe). Despite important advances in pathogenesis, diagnosis, and treatment, The primary causes of AD remain elusive, accurate biomarkers are not well characterized, and available pharmacological treatments are not cost-effective. As a complex disorder, AD is polygenic and multifactorial: hundreds of defective genes distributed across the human genome may contribute to its pathogenesis (with the participation of diverse environmental factors, cerebrovascular dysfunction, and epigenetic phenomena) and lead to amyloid deposition, neurofibrillary tangle formation, and premature neuronal death. Future perspectives for the global management of AD predict that structural and functional genomics and proteomics may help in the search for reliable biomarkers, and that pharmacogenomics may be an option in optimizing drug development and therapeutics.
Keywords: Alzheimer’s disease, APOE, Biomarkers, CYPs, Genetics, Genomics, Pathogenesis, Pharmacogenomics, Treatment
27.1. Overview
Since the identification of its pathogenic features by Alois Alzheimer in 1906, more than 90,000 papers have been published on Alzheimer’s disease (AD) to date (2.5 million references on cancer since 1818; 1.6 million on cardiovascular disorders since 1927; and 1.01 million on central nervous system disorders since 1893) [1]. The number of people affected by dementia is becoming a public and socioeconomic concern in many countries all over the world, independent of economic conditions. The growth of the elderly population is a common phenomenon in both developed and developing countries, bringing about future challenges in terms of health policy and disability rates.
In the United States, rates for the leading causes of death are heart disease (200.2 per 100,000), cancer (180.7 per 100,000), and stroke (43.6 per 100,000). AD is the fifth leading cause of death in people older than 65 years of age, representing 71,600 deaths per year. AD affects approximately 5.4 million individuals in the United States and is estimated to affect up to 16 million by 2050 [2]. Disability caused by senility and dementia affects 9.2 per 1000 in the population aged 65–74 years, 33.5 per 1000 in those within the 75–84 range, and 83.4 per 1000 in the population over 85 years [3], [4]. In low- to middle-income countries, dementia makes the largest contribution to disability, with a median population-attributable prevalence fraction of 25.1%, followed by stroke (11.4%), limb impairment (10.5%), arthritis (9.9%), depression (8.3%), eyesight problems (6.8%), and gastrointestinal impairments (6.5%) [5].
In Western countries, AD is the most prevalent form of dementia (45–60%), followed by vascular dementia (30–40%), and mixed dementia (10–20%), which in people older than 85 years of age may account for more than 80% of cases.
The different forms of dementia pose several challenges to society and to the scientific community: (1) they represent an epidemiological problem and a socioeconomic, psychological, and family burden; (2) most of them have an obscure/complex pathogenesis; (3) their diagnosis is not easy and lacks specific biomarkers; and (4) their treatment is difficult and inefficient.
In terms of economic burden, approximately 10–20% of direct costs are associated with pharmacological treatment, with a gradual increase that parallels the severity of the disease. A Canadian study [6] shows that the mean total cost to treat patients with very mild AD is $367 per month, compared with $4063 per month for patients with severe or very severe AD. Only 20–30% of patients with dementia respond appropriately to conventional drugs, and the onset of adverse drug reactions imposes the need for other drugs to neutralize side effects, thus multiplying the initial cost of the pharmacological treatment and the health risk for the patients [7]. Wimo et al. [8] studied the economic impact of dementia in Europe in the EU-funded Eurocode project and found that the total cost of dementia in EU27 countries in 2008 was estimated to be €160 billion (€22,000 per dementia patient per year), of which 56% were costs of informal care. The corresponding costs for the whole of Europe were €177 billion. Informal caregiver costs were the largest cost component, accounting for about half to just over 60% of total societal costs, depending on the country and AD severity [9].
In addition (and related) to the problem of direct and indirect costs for the management of dementia, there is an alarming abuse of inappropriate psychotropic drug consumption worldwide. Antipsychotic medications are taken by more than 30% of elderly patients with dementia [10], and conventional antipsychotics are associated with a higher risk of all-cause mortality among nursing home residents [11].
Abuse, misuse, self-prescription, and uncontrolled medical prescription of CNS drugs are becoming major problems with unpredictable consequences for brain health. The pharmacological management of dementia is an issue of special concern because of the polymedication required to modulate its symptomatic complexity where cognitive decline, behavioral changes, and psychomotor deterioration coexist. In parallel, a growing body of fresh knowledge is emerging on the pathogenesis of dementia, together with data on the neurogenomics and pharmacogenomics of CNS disorders. The incorporation of this new armamentarium of molecular pathology and genomic medicine into daily medical practice, together with educational programs for the correct use of drugs, must help researchers and clinicians to (1) understand AD pathogenesis; (2) establish an early diagnosis; and (3) optimize therapeutics either as a preventive strategy or as formal symptomatic treatment [7], [12].
27.2. Toward a Personalized Medicine for Dementia and Neurodegenerative Disorders
Common features of neurodegenerative disorders include the following:
-
•
Polygenic/complex disorders in which genetic, epigenetic, and environmental factors are involved
-
•
Deterioration of higher activities of the CNS
-
•
Multifactorial dysfunction in several brain circuits
-
•
Accumulation of toxic proteins in the nervous tissue
For instance, the neuropathological hallmarks of AD (amyloid deposition in senile plaques, neurofibrillary tangle formation, and neuronal loss) are merely the phenotypic expression of a pathogenic process in which different gene clusters and their products are potentially involved [7], [12].
A large number of the genes that form the structural architecture of the human genome are expressed in the brain in a time-dependent manner along the lifespan. The cellular complexity of the CNS (103 different cell types) and synapses (each of the 1011 neurons in the brain having around 103–104 synapses with a complex multiprotein structure integrated by 103 different proteins) requires very powerful technology for gene expression profiling, which is still in its very early stages and is not devoid of technical obstacles and limitations [13]. Transcripts of 16,896 genes have been measured in different CNS regions. Each region possesses its own unique transcriptome fingerprint that is independent of age, gender, and energy intake. Fewer than 10% of genes are affected by age, diet, or gender, with most of these changes occurring between middle and old age. Gender and energy restriction have robust influences on the hippocampal transcriptome of middle-aged animals. Prominent functional groups of age- and energy-sensitive genes are those encoding proteins involved in DNA damage responses, mitochondrial and proteasome functions, cell fate determination, and synaptic vesicle trafficking [14].
The introduction of novel procedures in an integral genomic medicine protocol for CNS disorders and dementia is imperative in drug development and in clinical practice in order to improve diagnostic accuracy and to optimize therapeutics. Personalized strategies, adapted to the complexity of each case, are essential to depict a clinical profile based on specific biomarkers correlating with individual genomic profiles [7], [15].
Our understanding of the pathophysiology of CNS disorders and dementia has advanced dramatically during the last 30 years, especially in terms of their molecular pathogenesis and genetics. The drug treatment of CNS disorders has also made remarkable strides with the introduction of many new drugs for the treatment of schizophrenia, depression, anxiety, epilepsy, Parkinson’s disease, and AD, among many other quantitatively and qualitatively important neuropsychiatric disorders.
Improvement in terms of clinical outcome, however, has fallen short of expectations, with up to one-third of patients continuing to experience clinical relapse or unacceptable medication-related side effects in spite of efforts to identify optimal treatment regimes with one or more drugs. Potential reasons for this historical setback might be: (1) that the molecular pathology of most CNS disorders is still poorly understood; (2) that drug targets are inappropriate, not fitting into the real etiology of the disease; (3) that most treatments are symptomatic but not antipathogenic; (4) that the genetic component of most CNS disorders is poorly defined; and (5) that the understanding of genome–drug interactions is very limited [7], [12].
The optimization of CNS therapeutics requires the establishment of new postulates regarding (1) the costs of medicines, (2) the assessment of protocols for multifactorial treatment in chronic disorders, (3) the implementation of novel therapeutics addressing causative factors, and (4) the establishment of pharmacogenomic strategies for drug development [12]. Personalized therapeutics based on individual genomic profiles implies the characterization of five types of gene clusters:
-
•
Genes associated with disease pathogenesis
-
•
Genes associated with the mechanism of action of drugs
-
•
Genes associated with drug metabolism (phase I and II reactions)
-
•
Genes associated with drug transporters
-
•
Pleiotropic genes involved in multifaceted cascades and metabolic reactions
27.3. Genomics of Alzheimer’s Disease
More than 3000 genes distributed across the human genome have been screened for association with AD during the past 30 years [16]. In the Alzgene database [17] there are 695 genes potentially associated with AD, of which the top ten are (in decreasing order of importance): APOE (19q13.2), BIN1 (2q14), CLU (8p21–p12), ABCA7 (19p13.3), CR1 (1q32), PICALM (11q14), MS4A6A (11q12.1), CD33 (19q13.3), MS4A4E (11q12.2), and CD2AP (6p12). Potentially defective genes associated with AD represent about 1.39% (35,252.69 Kb) of the human genome, which is integrated by 36,505 genes (3,095,677.41 Kb). The highest number of AD-related defective genes concentrate on chromosomes 10 (5.41%; 7337.83 Kb), 21 (4.76%; 2289.15 Kb), 7 (1.62%; 2584.26 Kb), 2 (1.56%; 3799.67 Kb), 19 (1.45%; 854.54 Kb), 9 (1.42%; 2010.62 Kb), 15 (1.23%; 1264.4 Kb), 17 (1.19%; 970.16 Kb), 12 (1.17%; 1559.9 Kb), and 6 (1.15%; 1968.22 Kb), with the highest proportion (related to the total number of genes mapped on a single chromosome) located on chromosome 10 and the lowest on chromosome Y [18] (Figure 27.1 ).
Figure 27.1.

Distribution of AD-related genes in the human genome.
The genetic and epigenetic defects identified in AD can be classified into four major categories: Mendelian mutations; susceptibility SNP; mtDNA mutations; and epigenetic changes. Mendelian mutations affect genes directly linked to AD, including 32 mutations in the amyloid beta precursor protein (APP) gene (21q21)(AD1), 165 mutations in the presenilin 1 (PSEN1) gene (14q24.3)(AD3), and 12 mutations in the presenilin 2 (PSEN2) gene (1q31–q42) (AD4) [16], [17], [18], [19], [20]. PSEN1 and PSEN2 are important determinants of γ-secretase activity responsible for proteolytic cleavage of APP and NOTCH receptor proteins. Mendelian mutations are very rare in AD (1:1000). Mutations in exons 16 and 17 of the APP gene appear with a frequency of 0.30% and 0.78%, respectively, in AD patients. Likewise, PSEN1, PSEN2, and microtubule-associated protein Tau (MAPT)(17q21.1) mutations are present in less than 2% of cases. Mutations in these genes confer specific phenotypic profiles to patients with dementia: amyloidogeneic pathology associated with APP, PSEN1, and PSEN2 mutations and tauopathy associated with MAPT mutations represent the two major pathogenic hypotheses for AD [16], [17], [18], [19], [20], [21].
Multiple polymorphic risk variants can increase neuronal vulnerability to premature death (see Appendix A). Among these susceptibility genes, the apolipoprotein E (APOE) gene (19q13.2)(AD2) is the most prevalent as a risk factor for AD, especially in those subjects harboring the APOE-4 allele (Figure 27.2 ), whereas carriers of the APOE-2 allele might be protected against dementia. APOE-related pathogenic mechanisms are also associated with brain aging and with the neuropathological hallmarks of AD [16].
Figure 27.2.
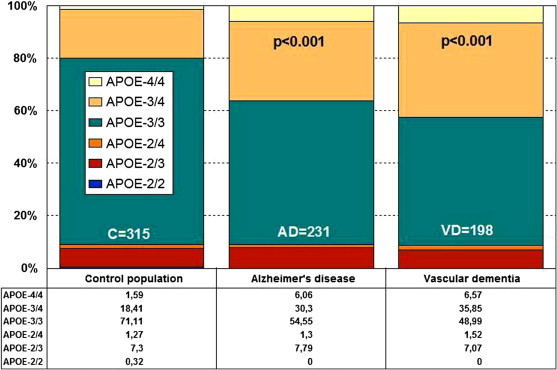
Distribution and frequency of APOE genotypes in AD and vascular dementia.
Source: Adapted from Cacabelos[18].
27.4. Pathogenic Events
The dual amyloidogenic-tauopathic theory of AD has dominated the pathogenic universe of AD-related neurodegeneration (and divided the research community) for the past 50 years, nourished by the presence of APP, PSEN1, PSEN2, and MAPT mutations in a very small number of cases with early-onset AD. Nevertheless, this theory does not explain AD pathogenesis in full, and consequently novel (or complementary) theories have been emerging recently and during the past decades. A summary of the pathogenic events in AD is given in the following sections.
27.4.1. Genomic Defects
As a complex polygenic/multifactorial disorder, in which hundreds of polymorphic variants of risk might be involved (Appendix A, Figure 27.1), AD fulfils the “golden rule” of complex disorders, according to which the larger the number of genetic defects distributed in the human genome, the earlier the onset of the disease and the poorer its therapeutic response to conventional treatments; conversely, the smaller the number of pathogenic SNPs, the later the onset of the disease and the better its therapeutic response to different pharmacological interventions [12], [16], [22], [23], [24], [25], [26], [27], [28]. Genetic variation associated with different diseases interferes with microRNA-mediated regulation by creating, destroying, or modifying microRNA (miRNA) binding sites. miRNA-target variability is a ubiquitous phenomenon in the adult human brain which may influence gene expression in physiological and pathological conditions. AD-related SNPs interfere with miRNA gene regulation and affect AD susceptibility. Significant interactions include target SNPs present in seven genes related to AD prognosis with the miRNAs miR-214, -23a and -23b, -486-3p, -30e*, -143, -128, -27a and -27b, -324-5p, and -422a. The dysregulated miRNA network contributes to aberrant gene expression in AD [29], [30], [31].
27.4.2. Epigenetic Phenomena
Epigenetic factors have emerged as important mediators of development and aging, gene–gene and gene–environmental interactions, and the pathophysiology of complex disorders. Major epigenetic mechanisms (DNA methylation, histone modifications and chromatin remodeling, and noncoding RNA regulation) may contribute to AD pathology [30], [31].
27.4.3. Cerebrovascular Dysfunction
Vascular and metabolic dysfunctions are key components in AD pathology throughout the course of disease. Although common denominators between vascular and metabolic dysfunction are oxidative stress and Aβ [32], genetic factors and cardiovascular risk factors may also account for the cerebrovascular damage present in AD [33]. Inherited polymorphisms of the vascular susceptibility gene Ninjurin2 (NINJ2) are associated with AD risk [34]. Endothelial dysfunction has been implicated as a crucial event in the development of AD.
Breakdown of the blood–brain barrier (BBB) as a result of disruption of tight junctions and transporters leads to increased leukocyte transmigration and is an early event in the pathology of many CNS disorders. BBB breakdown leads to neuroinflammation and oxidative stress, with mitochondrial dysfunction. The high concentration of mitochondria in cerebrovascular endothelial cells might account for the sensitivity of the BBB to oxidant stressors [35], [36].
Chronic brain hypoperfusion may be sufficient to induce premature neuronal death and dementia in vulnerable subjects [16], [23], [24], [25], [37], [38], [39]. APOE-related changes in cortical oxygenation and hemoglobin consumption are evident, as revealed by brain optical topography analysis, and reflect that APOE-4 carriers exhibit deficient brain hemodynamics and a poorer panneocortical oxygenation than do APOE-3 or APOE-2 carriers [18]. Hypoperfusion in frontal, parietal, and temporal regions is a common finding in AD. White matter hyperintensities (WMH) correlate with age and with disease severity [40].
Cerebral amyloid angiopathy (CAA) accounts for the majority of primary lobal intracerebral hemorrhages (ICH) among the elderly, and represents the cause of 20% of spontaneous ICHs in patients over 70 years of age. The basis for this disease process is the deposition and formation of eventually destructive amyloid plaques in the walls of brain vessels, predominantly arterial but not excluding venules and capillaries. CAA and CAA-associated microhemorrhages may also participate in the pathogenesis of AD [41]. Aβ deposition in asymptomatic elderly individuals is associated with lobar MH (LMH).
LMH is present in 30.8% of AD, 35.7% of MCI, and 19.1% of controls [42]. Neurovascular dysfunction in AD leads to reduced clearance across the BBB and accumulation of neurotoxic Aβ peptides in the brain. The ABC transport protein P-glycoprotein (P-gp, ABCB1) is involved in the export of Aβ from the brain into the blood. P-gp, LRP1, and RAGE mRNA expression is reduced in mice treated with Aβ1–42. In addition to the age-related decrease in P-gp expression, Aβ1–42 itself downregulates the expression of P-gp and other Aβ transporters, which could exacerbate the intracerebral accumulation of Aβ and thereby accelerate neurodegeneration in AD and cerebral β-amyloid angiopathy [43].
27.4.4. Phenotypic Expression of Amyloid Deposits and Neurofibrillary Tangles
β-Amyloid deposits in senile and neuritic plaques and hyperphosphorylated tau proteins in neurofibrillary tangles (NFT) are extracellular and intracellular expressions, respectively, of the AD neuropathological phenotype, together with selective neuronal loss in hippocampal and neocortical regions. Aβ plaque in the brain is the primary (postmortem) diagnostic criterion of AD. The main component of senile plaques is Aβ, a 39–43 amino acid peptide, generated by the proteolytic cleavage of amyloid precursor protein (APP) by the action of beta- and gamma-secretases. Aβ is neurotoxic, and this neurotoxicity is related to its aggregation state [16], [17], [18], [19], [20], [21].
27.4.5. Neuronal Apoptosis
Neuronal loss is a pathognomonic finding in AD and the final common path of multiple pathogenic mechanisms leading to neurodegeneration in dementia. Atrophy of the medial temporal lobe, especially the hippocampus and the parahippocampal gyrus, is considered to be AD’s most predictive structural brain biomarker. The medial and posterior parts of the parietal lobe seem to be preferentially affected, compared to the other parietal lobe parts [18].
27.4.6. Neurotransmitter Deficits
An imbalance of different neurotransmitters (glutamate, acetylcholine, noradrenaline, dopamine, serotonin, and some neuropeptides) has been proposed as the neurobiological basis of behavioral symptoms in AD. Altered reuptake of neurotransmitters by vesicular glutamate transporters (VGLUTs), excitatory amino acid transporters (EAATs), the vesicular acetylcholine transporter (VAChT), the serotonin reuptake transporter (SERT), or the dopamine reuptake transporter (DAT) are involved in the neurotransmission imbalance in AD. Protein and mRNA levels of VGLUTs, EAAT1-3, VAChT, and SERT are reduced in the disease [44].
27.4.7. Oxidative Stress
Oxidative damage is a classic pathogenic mechanism of neurodegeneration [36], [45]. It is greater in brain tissue from patients with AD than age-matched controls. Tayler et al. [46] studied the timing of this damage in relation to other pathogenic AD processes. Antioxidant capacity is elevated in AD and directly related to disease severity as indicated by the Braak tangle stage and the amount of insoluble Aβ. Accumulation of Aβ has been shown in brain mitochondria of AD patients and in AD transgenic mouse models. The presence of Aβ in mitochondria leads to free radical generation and neuronal stress.
A novel mitochondrial Aβ-degrading enzyme, presequence protease (Pre), has been identified in the mitochondrial matrix. hPreP activity is decreased in AD human brains and in the mitochondrial matrix of AD transgenic mouse brains (TgmAβPP and TgmAβPP/ABAD). Mitochondrial fractions isolated from AD brains and TgmAβPP mice have higher levels of 4-hydroxynonenal, an oxidative product. Cytochrome c oxidase activity is significantly reduced in the AD mitochondria. Decreased PreP proteolytic activity, possibly due to enhanced ROS production, may contribute to Aβ accumulation in mitochondria, leading to mitochondrial toxicity and neuronal death in AD [47].
27.4.8. Cholesterol and Lipid Metabolism Dysfunction
Cholesterol seems to be intimately linked with the generation of amyloid plaques, which are central to AD pathogenesis. APOE variants are determinants in cholesterol metabolism and diverse forms of dyslipoproteinemia [12], [48]. Cholesterol protects the Aβ-induced neuronal membrane disruption and inhibits beta-sheet formation of Aβ on the lipid bilayer [49]. Jones et al. [50] found a significant over-representation of association signals in pathways related to cholesterol metabolism and the immune response in both of the two largest genome-wide association studies for LOAD.
27.4.9. Neuroinflammation and Immunopathology
Several genes associated with immune regulation and inflammation show polymorphic variants of risk in AD, and abnormal levels of diverse cytokins have been reported in the brain, CSF, and plasma of AD patients [16], [23]. The activation of inflammatory cascades has been consistently demonstrated in AD pathophysiology, in which reactive microglia are associated with Aβ deposits and clearance. Resident microglia fail to trigger an effective phagocytic response to clear Aβ deposits, although they mainly exist in an “activated” state. Oligomeric Aβ (oAβ) can induce more potent neurotoxicity when compared with fibrillar Aβ (fAβ). Aβ(1–42) fibrils, not Aβ(1–42) oligomers, increase microglial phagocytosis [51]. Among several putative neuroinflammatory mechanisms, the TNF-α signaling system has a central role in this process. In AD, TNF-α levels are altered in serum and CSF. The abnormal production of inflammatory factors may accompany the progression from mild cognitive impairment (MCI) to dementia. Abnormal activation of the TNF-α signaling system, represented by increased expression of sTNFR1, is associated with a higher risk of progression from MCI to AD [52].
27.4.10. Neurotoxic Factors
Old and new theories suggest that different toxic agents, from metals (e.g., aluminium, copper, zinc, iron) to biotoxins and pesticides, might contribute to neurodegeneration. Dysfunctional homeostasis of transition metals is believed to play a role in AD pathogenesis [18].
27.4.11. Other Players
Many novel pathogenic mechanisms potentially involved in AD neurodegeneration have been proposed in recent times. Moreover, there has been a revival of some old hypotheses. Examples of pathogenic players in AD, other than those just discussed, include the Ca2+ hypothesis, insulin resistance, NGF imbalance, glycogen synthase kinase-3 (GSK-3), advanced glycation end products (AGEs) and their receptors (RAGE), the efflux transporter P-glycoprotein (P-gp), c-Abl tyrosine kinase, post-transcriptional protein alterations that compromise the proteasome system and the chaperone machinery (HSPB8–BAG3), autophagy as a novel Aβ-generating pathway, hypocretin (orexin), cathepsin B, Nogo receptor proteins, adipocytokines and CD34+ progenitor cells, CD147, impairment of synaptic plasticity (PSD-95), anomalies in neuronal cell division and apoptosis, stem cell factor (SCF), telomere shortening, deficiency in repair of nuclear and mitochondrial DNA damage, and microDNAs [18].
27.5. Biomarkers and Comorbidity
AD’s phenotypic features represent the biomarkers to be used as diagnostic predictors and the expression of pathogenic events to be modified with an effective therapeutic intervention. Important differences have been found in the AD population (as compared with healthy subjects) in different biological parameters, including blood pressure, glucose, cholesterol and triglyceride levels, transaminase activity, hematological parameters, metabolic factors, thyroid function, brain hemodynamic parameters, and brain mapping activity [7], [23], [24], [25], [53], [54], [55], [56], [57], [58], [59].
These clinical differences are clear signs of comorbidity rather than typical features of AD. Blood pressure values, glucose levels, and cholesterol levels are higher in AD than in healthy elderly subjects. Approximately 20% of AD patients are hypertensive, 25% are diabetics, 50% are hypercholesterolemic, and 23% are hypertriglyceridemic. More than 25% of patients exhibit high GGT activity, 5–10% show anemic conditions, 30–50% show an abnormal cerebrovascular function characterized by poor brain perfusion, and more than 60% have an abnormal electroencephalographic pattern, especially in frontal, temporal, and parietal regions, as revealed by quantitative EEG (qEEG) or computerized mapping [7], [12], [23], [54]. Significant differences are currently seen between females and males, indicating the effect of gender on the phenotypic expression of the disease. In fact, the prevalence of dementia is 10–15% higher in females than in males from 65–85 years of age. All of these parameters are highly relevant when treating AD patients, because some of them reflect a concomitant pathology that also needs therapeutic consideration.
AD biomarkers can be differentiated into several categories: (1) neuropathological markers; (2) structural and functional neuroimaging markers; (3) neurophysiological markers (EEG, qEEG, brain mapping); (4) biochemical markers in body fluids (e.g., blood, urine, saliva, CSF); and (5) genomic markers (structural and functional genomics, proteomics, metabolomics).
27.5.1. Neuropathology
Plaques and tangles in the hippocampus and cortex are still considered the seminal findings in AD neuropathology and are the conventional means of establishing the boundary between amyloidopathies and tauopathies; however, both phenotypic markers are also present in normal brains, in more than 60% of cases with traumatic brain injury, and in many other brain disorders [60].
27.5.2. Structural and Functional Neuroimaging
Structural and functional neuroimaging techniques (MRI, fMRI, PET, SPECT) are essential tools in the diagnosis of dementia, although the specificity of visual observations in degenerative forms of dementia is of doubtful value. Nevertheless, these procedures are irreplaceable for a differential diagnosis. There is a characteristic regional impairment in AD that involves mainly the temporo–parietal association cortices, the mesial temporal structures, and, to a more variable degree, the frontal association cortex. This pattern of functional impairment can provide a biomarker for diagnosis of AD and other neurodegenerative dementias at the clinical stage of mild cognitive impairment, and for monitoring its progression. Healthy young APOE ɛ4 carriers have smaller hippocampal volumes than APOE ɛ2 carriers.
The difference in hippocampal morphology is cognitively/clinically silent in young adulthood, but can render APOE ɛ4 carriers more prone to the later development of AD, possibly because of lower reserve cognitive capacity [61]. LOAD patients exhibit a selective parahippocampal white matter (WM) loss, while EOAD patients experience a more widespread pattern of posterior WM atrophy. The distinct regional distribution of WM atrophy reflects the topography of gray matter (GM) loss. ApoE ɛ4 status is associated with a greater parahippocampal WM loss in AD. The greater WM atrophy in EOAD than in LOAD fits with the evidence that EOAD is a more aggressive form of the disease [62]. FDG-PET is quantitatively more accurate than perfusion SPECT.
Regional metabolic and blood flow changes are closely related to clinical symptoms, and most areas involved in these changes also develop significant cortical atrophy. FDG-PET is complementary to amyloid PET, which targets a molecular marker that does not have a close relation to current symptoms. FDG-PET is expected to play an increasing role in diagnosing patients at an early stage of AD and in clinical trials of drugs aimed at preventing or delaying the onset of dementia [63]. Functional neuroimaging biomarkers are becoming popular, with the introduction of novel tracers for brain amyloid deposits. Amyloid deposition causes severe damage to neurons many years before onset of dementia via a cascade of several downstream effects.
Positron emission tomography (PET) tracers for amyloid plaque are desirable for early diagnosis of AD, particularly to enable preventative treatment once effective therapeutics is available. The amyloid imaging tracers flutemetamol, florbetapir, and florbetaben labeled with 18F have been developed for PET. These tracers are currently undergoing formal clinical trials to establish whether they can be used to accurately image fibrillary amyloid, and to distinguish patients with AD from normal controls and those with other diseases that cause dementia [63].
27.5.3. Neurophysiology
There is a renewed interest in the use of computerized brain mapping as a diagnostic aid and as a monitoring tool in AD [64]. Electroencephalography (EEG) studies in AD show an attenuation of average power within the alpha band (7.5–13 Hz) and an increase in power in the theta band (4–7 Hz) [65]. APOE genotypes influence brain bioelectrical activity in AD. In general, APOE-4 carriers tend to exhibit a slower EEG pattern from early stages [16], [18], [66].
27.5.4. Biochemistry of Body Fluids
Other biomarkers of potential interest include cerebrospinal fluid (CSF) and peripheral levels of Aβ42, protein tau, histamine, interleukins, and some other novel candidate markers such as chitinase 3-like 1 (CHI3L1) protein [7], [16], [25], [67], [68], [69]. The concentration of the 42-amino-acid form of Aβ (Aβ1–42) is reduced in the CSF of AD patients, which is believed to reflect the AD pathology, with plaques in the brain acting as sinks. Novel C-truncated forms of Aβ (Aβ1–14, Aβ1–15, and Aβ1–16) were identified in human CSF. The presence of these small peptides is consistent with a catabolic amyloid precursor protein cleavage pathway by β- followed by α-secretase. Aβ1–14, Aβ1–15, and Aβ1–16 increase dose-dependently in response to γ-secretase inhibitor treatment, while Aβ1–42 levels are unchanged [70].
Kester et al. [71] investigated change over time in CSF levels of amyloid-beta 40 and 42 (Aβ40 and Aβ42), total tau (tau), tau phosphorylated at threonine 181 (ptau-181), isoprostane, neurofilaments heavy (NfH) and neurofilaments light (NfL). Aβ42, tau, and tau phosphorylated at threonine 181 differentiated between diagnosis groups, whereas isoprostane, NfH, and NfL did not. In contrast, effects of follow-up time were found only for nonspecific CSF biomarkers: levels of NfL decreased, and levels of isoprostane, Aβ40, and tau increased over time. An increase in isoprostane was associated with progression of mild cognitive impairment in AD and with cognitive decline. Contrary to AD-specific markers, nonspecific CSF biomarkers show change over time, which potentially can be used to monitor disease progression in AD.
27.5.5. Genomics and Proteomics
Structural markers are represented by SNPs in genes associated with AD, polygenic cluster analysis, and genome-wide studies (GWSs). Functional markers attempt to correlate genetic defects with specific phenotypes (genotype–phenotype correlations). In proteomic studies, several candidate CSF protein biomarkers have been assessed in neuropathologically confirmed AD, nondemented (ND) elderly controls, and non-AD dementias (NADD). Markers selected included apolipoprotein A-1 (ApoA1), hemopexin (HPX), transthyretin (TTR), pigment epithelium-derived factor (PEDF), Aβ1–40, Aβ1–42, total tau, phosphorylated tau, α-1 acid glycoprotein (A1GP), haptoglobin, zinc α-2 glycoprotein (Z2GP), and apolipoprotein E (ApoE). Concentrations of Aβ1–42, ApoA1, A1GP, ApoE, HPX, and Z2GP differed significantly among AD, ND, and NADD subjects. The CSF concentrations of these three markers distinguished AD from ND subjects with 84% sensitivity and 72% specificity, with 78% of subjects correctly classified.
By comparison, Aβ1–42 alone gave 79% sensitivity and 61% specificity, with 68% of subjects correctly classified. For the diagnostic discrimination of AD from NADD, only the concentration of Aβ1–42 was significantly related to diagnosis, with a sensitivity of 58% and a specificity of 86% [72]. Carrying the APOE-ɛ4 allele was associated with a significant decrease in CSF Aβ1–42 concentrations in middle-aged and older subjects. In AD, Aβ1–42 levels are significantly lower in APOEɛ4 carriers compared to noncarriers. These findings demonstrate significant age effects on CSF Aβ1–42 and pTau181 across the lifespan, and also suggest that a decrease in Aβ1–42, but an increase in pTau181 CSF levels, is accelerated by the APOEɛ4 genotype in middle-aged and older adults with normal cognition [73].
Han et al. [74] carried out a GWAS to better define the genetic backgrounds of normal cognition, mild cognitive impairment (MCI), and AD in terms of changes in CSF levels of Aβ1–42, T-tau, and P-tau181P. CSF Aβ1–42 levels decreased with APOE gene dose for each subject group. T-tau levels tended to be higher among AD cases than among normal subjects. CYP19A1 “aromatase” (rs2899472), NCAM2, and multiple SNPs located on chromosome 10 near the ARL5B gene demonstrated the strongest associations with Aβ1–42 in normal subjects.
Two genes found to be near the top SNPs, CYP19A1 (rs2899472) and NCAM2 (rs1022442), have been reported as genetic factors related to the progression of AD. In AD subjects, APOE ɛ2/ɛ3 and ɛ2/ɛ4 genotypes were associated with elevated T-tau levels, and the ɛ4/ɛ4 genotype was associated with elevated T-tau and P-tau181P levels. Blood-based markers reflecting core pathological features of AD in presymptomatic individuals are likely to accelerate the development of disease-modifying treatments.
Thambisetty et al. [75] performed a proteomic analysis to discover plasma proteins associated with brain Aβ burden in nondemented older individuals. A panel of 18 2DGE plasma protein spots effectively discriminated between individuals with high and low brain Aβ. Mass spectrometry identified these proteins, many of which have established roles in Aβ clearance, including a strong signal from ApoE. A strong association was observed between plasma ApoE concentration, and Aβ burden in the medial temporal lobe. Targeted voxel-based analysis localized this association to the hippocampus and entorhinal cortex. APOE ɛ4 carriers also showed greater Aβ levels in several brain regions relative to ɛ4 noncarriers. Both peripheral concentration of the ApoE protein and the APOE genotype may be related to early neuropathological changes in brain regions vulnerable to AD pathology even in the nondemented elderly.
27.6. Therapeutic Strategies
Modern therapeutic strategies in AD are aimed at interfering with the main pathogenic mechanisms potentially involved in AD [7], [12], [16], [18], [23], [24], [28], [53], [54], [55], [56], [57], [58], [59] (Box 27.1). Starting in the early 1990s, the neuropharmacology of AD was dominated by acetylcholinesterase inhibitors, represented by tacrine, donepezil, rivastigmine, and galantamine [76], [77], [78]. Memantine, a partial NMDA antagonist, was introduced in the 2000s for the treatment of severe dementia [79]; and the first clinical trials with immunotherapy, to reduce amyloid burden in senile plaques, were withdrawn due to severe ADRs [80], [81]. After the initial promise of β- and γ-secretase inhibitors [82], [83] and novel vaccines [84], [85] devoid of severe side effects, during the past few years no relevant drug candidates have dazzled the scientific community with their capacity to halt disease progression; however, a large number of novel therapeutic strategies for the pharmacological treatment of AD have been postulated, with some apparent effects in preclinical studies (see Box 27.1).
Box 27.1. Experimental Strategies for the Pharmacological Treatment of Alzheimer’s Disease.
New cholinesterase inhibitors
Cholinergic receptor agonists
Monoamine regulators
Diverse natural compounds derived from vegetal sources:
Alkaloids from the calabar bean (Physostigma venenosum)
Huperzine A from Huperzia serrata
Galantamine from the snowdrop Galanthus woronowii
Cannabinoids (cannabidiol) from Cannabis sativa
Saffron (Crocus sativus)
Ginseng (Panax species)
Sage (Salvia species)
Lemon balm (Melissa officinalis)
Polygala tenuifolia
Nicotine from Nicotiana species
Grape seed polyphenolic extracts
Fuzhisan, a Chinese herbal medicine
Resveratrol
Xanthoceraside
Garlic (Allium sativum)
Linarin from Mentha arvensis and Buddleja davidii
Carotenoids (e.g., retinoic acid, all-trans retinoic acid, lycopene and β-carotene)
Curcumin from the rhizome of Curcuma longa
Decursinol from the roots of Angelica gigas
Bacopa monniera LINN (Syn. Brahmi)
Olive oil
Phytoestrogens
Walnut extract
Erigeron annuus leaf extracts
Epigallocatechin-3-gallate
Luteolin
The brown algae (Ecklonia cava)
Gami-Chunghyuldan (standardized multiherbal medicinal formula)
Punica granatum extracts
Plants of different origin:
Yizhi Jiannao
Drumstick tree (Moringa oleifera)
Ginkgo/Maidenhair tree (Ginkgo biloba)
Sicklepod (Cassia obtisufolia)
Sal Leaved Desmodium (Desmodium gangeticum)
Lemon Balm (Melissa officinalis)
Garden sage, common sage (Salvia officinalis)
Immunotherapy and treatment options for tauopathies:
Tau kinase inhibitors
2-Aminothiazoles
Phosphoprotein phosphatase 2A (PP2A) inhibitors
c-Jun N-terminal kinase (JNKs) inhibitors
p38 MAP kinase inhibitors (CNI-1493)
Harmine (β-carboline alkaloid)
Immunotherapy and Aβ breakers for AD-related amyloidopathy:
Active and passive immunization
Secretase inhibitors (β- and γ-)
Neostatins
Neurosteroids
Phosphodiesterase inhibitors
Protein phosphatase methylesterase-1 inhibitors
Histone deacetylase inhibitors
mTOR inhibitors
Peroxisome proliferator-activated receptor agonists
P-glycoprotein regulators
Nuclear receptor agonists
Glycogen synthase kinase-3β (GSK-3β) regulators
Histamine H3 receptor inverse agonists
Estrogens
Kynurenine 3-monooxygenase inhibitors
Chaperones (small heat shock proteins (sHSPs); Hsp90 inhibitors and HSP inducers)
microRNAs (miRNAs) and gene silencing (RNA interference)(RNAi)
Miscellaneous strategies:
Sodium fullerenolate
Glucagon-like peptide -1 (GLP-1)
Chemokines
Macrophage inflammatory protein-2 (MIP-2)
Stromal cell-derived factor-1α (SDF-1α)
Cyclooxygenase-1 and cyclooxygenase-2 inhibitors
Bone morphogenetic protein 9 (BMP-9)
Granulocyte colony stimulating factor (G-CSF)/AMD3100 (CXCR4 antagonist)
Vitamins (A, B, C, D)
ω-3 Polyunsaturated fatty acids (n-3 PUFAs)
Docosahexaenoic acid (DHA, C22:6 n-3)
Sphingosylphosphorylcholine
Citidine-5-diphosphocholine (CDP-choline)
Cathepsin B inhibitors
Pituitary adenylate cyclase–activating polypeptide
NAP (Davunetide)
Transcription factor specificity protein 1 (Sp1) inhibitors (tolfenamic acid)
- TNF inhibitors:
- 2-(2,6-Dioxopiperidin-3-yl)phthalimidine EM-12 dithiocarbamates
- N-substituted 3-(Phthalimidinp-2-yl)-2,6-dioxopiperidines
- 3-substituted 2,6-Dioxopiperidines
Pyrrolo[3,2-e][1,2,4]triazolo[1,5-a]pyrimidine (SEN1176)
Latrepirdine
Leucettines
Dihydropyridines (inhibitors of L-type calcium channels)
Brain-penetrating angiotensin-converting enzyme (ACE) inhibitors
NADPH oxidase inhibitors (Apocynin)
Heterocyclic indazole derivatives (inhibitors of serum- and glucocorticoid-inducible-kinase 1 [SGK1])
IgG-single-chain Fv fusion proteins
27.6.1. Immunotherapy
There are two main modalities of immunotherapy for AD: (1) passive immunotherapy, with the administration of monoclonal Aβ-specific antibodies [86]; and (2) active immunization with the Aβ42 antigen [87], [88] or Aβ-conjugated synthetic fragments bound to a carrier protein, thus avoiding potential problems associated with mounting a T-cell response directly against Aβ [89]. A new approach—delivering Aβ42 in a novel immunogen-adjuvant manner consisting of sphingosine-1-phosphate (S1P)-containing liposomes, administered to APP/PS1 transgenic mice before and after the detection of AD-like pathology in the brain—has recently been developed [85].
The results from this novel vaccine (EB101) indicate that active immunization significantly prevents and reverses the progression of AD-like pathology and also clears prototypical neuropathological hallmarks in transgenic mice. This new approach strongly induces T-cell, B-cell, and microglial immune response activation, avoiding the Th1 inflammatory reaction [90].
The rationale for amyloid immunotherapy in AD [91] is based on the following assumptions:
-
•
β-amyloid plaques and their aggregated, proto-fibrillar, and oligomeric precursors contain immunologic neo-epitopes that are absent from the full-length amyloid precursor protein (APP), as well as from its soluble proteolytic derivatives restricted to brain tissue; consequently, β-amyloid-based immunotherapies designed to selectively target pathologic neo-epitopes present on Aβ oligomers, protofibrils, or fibrils should not cause autoimmune disease in unaffected tissues throughout the organism
-
•
β-amyloid buildup precedes neurodegeneration and functional loss, and either the prevention of its formation or its removal can be expected to result in the slowing or the prevention of neurodegeneration
-
•
β-amyloid can cause the formation of neurofibrillary tangles in vivo and in vitro. The removal of β-amyloid, or the prevention of its buildup, has the potential not only to correct β-amyloid-related toxicity, but also to prevent the formation of neurofibrillary tangles
-
•
Conformational changes of endogenously occurring proteins and the formation of insoluble aggregates are commonly associated with neurodegeneration and brain disease, so the removal or prevention of these pathologic protein aggregates is also a therapeutic goal in the principle of immunotherapy
-
•
Immunotherapy works in experimental animals and in initial clinical trials: both active immunization and passive antibody transfer consistently reduce brain β-amyloid load, improve β-amyloid-related memory impairments, and protect neurons against degeneration in many independent experiments using different mouse models and primates [90]
Since Aβ immunotherapy has a limited clearance effect of tau aggregates in dystrophic neurites, the development of an alternative therapy that directly targets pathological tau has become crucial. Increased levels of tau oligomers have been observed in the early stage of AD, prior to the detection of neurofibrillary tangles (NFT) formed by aggregation and accumulation of the microtubule-associated protein tau [92]. Several approaches have been taken to treat AD by targeting tau, such as the following:
-
1.
The inhibition of tau hyperphosphorylation, by a kinase inhibitor of soluble aggregated tau formation, which also prevents related motor deficits [93].
-
2.
Activation of the proteolytic pathway, by the degrading action of calpain [94] and puromycin-sensitive aminopeptidase [95].
-
3.
The stabilization of microtubules, treating tauopathies by functionally binding and stabilizing microtubules with mt-binding protein tau [96] and paclitaxel, a drug proven effective in restoring affected axonal transport and motor impairments [97].
-
4.
Tau clearance by immunotherapy in this case, the tau active vaccination uses phosphorylated antigens of tau fragments associated with neurofibrillary tangles [98] that results in an efficient reduction of both soluble and insoluble tau active fragments, reducing phosphorylated NFTs in AD-like mouse brains.
Preclinical studies have shown clear evidence that Aβ immunization therapy provides protection and reverses the pathological effects of AD in transgenic mouse models [99]. This strategy seems to improve cognition performance [100] after Aβ42 immunization, in addition to causing an effective reduction in Aβ pathology. A recent immunization study has proven that a fragment of the Aβ peptide bound to polylysines activates the immune response that diminishes AD-like pathology in APP transgenic mice. This result reinforces the notion that the immune-conjugate approach is an effective means of Aβ immunotherapy, and also that the entire Aβ peptide is not necessary for its efficacy. It is in accordance with the hypothesis that specific antibodies directed against the amino-terminal and/or central region of the amyloid peptide provide beneficial protection against amyloid pathology. Passive immunization studies have also been conducted with promising experimental results, showing that a humoral response alone, without Aβ cellular response, is sufficient to reduce the β-amyloid burden and reverse memory deficits [101].
Among the drugs and vaccines currently under development to treat the pathological effects of AD, the most promising are bapineuzumab, solanezumab, CAD106, and EB101. Solanezumab is a monoclonal antibody raised against Aβ13–28 that recognizes an epitope in the core of the amyloid peptide, binding selectively to soluble Aβ and with low affinity for the fibrillar Aβ form [102]. Thus, it presents fewer adverse events than does bapineuzumab, which binds to Aβ amyloid plaques more strongly than soluble Aβ [103]. There are a few other monoclonal antibodies against Aβ that have properties different from those of bapineuzumab, such as PF-04360365, which specifically targets the free carboxy-terminus of Aβ1–40, MABT5102A, which binds with equally high affinity to Aβ monomers, oligomers, and fibrils, and GSK933776A, which targets the N-terminus of Aβ.
Specific anti-Aβ antibodies are present in pooled preparations of intravenous immunoglobulin (IVIg or IGIV), which has already been approved by the FDA for the treatment of a variety of neurological conditions. Current results from these studies have shown that IVIg treatment may also be an efficacious alternative approach in the treatment of AD neuropathologies [90], [104].
Avoiding both the strong Th1 effects of the QS-21 adjuvant and the T-cell epitopes at the C-terminus of Aβ, CAD106 consists of a short N-terminal fragment of Aβ attached to a virus-like particle, with no additional adjuvant [105]. This therapeutic agent is currently in phase II trials. Affiris is testing two short 6-amino-peptides (AD01, AD02), administered with aluminum hydroxide as adjuvant, that mimic the free N-terminus of Aβ and therefore cause cross-reactivity with the native peptide in phase I trials [106]. In terms of prevention and therapeutic treatment, the EB101 vaccine showed for the first time the effectiveness of combining a liposomal immunogen-adjuvant with an Aβ antigen to induce an effective immunological response combined with an anti-inflammatory effect in preclinical studies using APP/PS1 transgenic mice [85], [90].
The EB101 vaccine immunization process has shown a marked positive effect as a preventive and therapeutic treatment, reducing amyloidosis-induced inflammation as an effective Th2 immunomodulator. Moreover, this vaccine proved to stimulate innate immunity and enable effective phagocytosis to clear amyloid and neurofibrillary tangles, which are among the major hallmarks of AD-like neuropathology observed. A few other vaccines are currently under development, and recent studies have opened up new perspectives in the immunization approach to AD pathology; in particular, gene-gun-mediated genetic immunization with the Aβ42 gene [107] shows that self-tolerance can be broken in order to produce a humoral response to the Aβ42 peptide with minimal cellular response.
27.7. Pharmacogenomics
AD patients may take 6–12 different drugs per day for the treatment of dementia-related symptoms, including memory decline (conventional antidementia drugs, neuroprotectants), behavioral changes (antidepressants, neuroleptics, sedatives, hypnotics), and functional decline. Such drugs may also be taken for the treatment of concomitant pathologies (epilepsy, cardiovascular and cerebrovascular disorders, parkinsonism, hypertension, dyslipidemia, anemia, arthrosis, etc). The co-administration of several drugs may cause side effects and ADRs in more than 60% of AD patients, who in 2–10% of cases require hospitalization. In more than 20% of patients, behavioral deterioration and psychomotor function can be severely altered by polypharmacy. The principal causes of these iatrogenic effects are (1) the inappropriate combination of drugs, and (2) the genomic background of the patient, which is responsible for his/her pharmacogenomic outcome.
Pharmacogenomics account for 30–90% of the variability in pharmacokinetics and pharmacodynamics. The genes involved in the pharmacogenomic response to drugs in AD fall into five major categories:
-
•
Genes associated with AD pathogenesis and neurodegeneration (APP, PSEN1, PSEN2, MAPT, PRNP, APOE, and others)
-
•
Genes associated with the mechanism of action of drugs (enzymes, receptors, transmitters, messengers)
-
•
Genes associated with drug metabolism (phase I (CYPs) and phase II reactions (UGTs, NATs))
-
•
Genes associated with drug transporters (ABCs, SLCs)
-
•
Pleiotropic genes involved in multifaceted cascades and metabolic reactions (APOs, ILs, MTHFR, ACE, AGT, NOS, etc) [18] (Figure 27.1)
27.7.1. Pathogenic Genes
In more than 100 clinical trials for dementia, APOE has been used as the only gene of reference for the pharmacogenomics of AD [7], [12], [15], [16], [22], [23], [24], [25], [26], [27], [28], [53], [54], [55], [56], [57], [58], [59]. Several studies indicate that the presence of the APOE-4 allele differentially affects the quality and extent of drug responsiveness in AD patients treated with cholinergic enhancers (tacrine, donepezil, galantamine, rivastigmine), neuroprotective compounds (nootropics), endogenous nucleotides (CDP-choline), immunotrophins (anapsos), neurotrophic factors (cerebrolysin), rosiglitazone, or combination therapies [108], [109], [110]; however, controversial results are frequently found that are due to methodological problems, study design, and patient recruitment in clinical trials.
The major conclusion in most studies is that APOE-4 carriers are the worst responders to conventional treatments [7], [12], [15], [16], [22], [23], [24], [25], [26], [27], [28], [53], [54], [55], [56], [57], [58], [59]. When APOE and CYP2D6 genotypes are integrated in bigenic clusters and the APOE+CYP2D6-related therapeutic response to a combination therapy is analyzed in AD patients, it becomes clear that the presence of the APOE-4/4 genotype is able to convert pure CYP2D6*1/*1 extensive metabolizers (EMs) into full poor responders to conventional treatments, indicating the existence of a powerful influence of the APOE-4 homozygous genotype on the drug-metabolizing capacity of pure CYP2D6 EMs. In addition, a clear accumulation of APOE-4/4 genotypes is observed among CYP2D6 poor (PMs) and ultrarapid metabolizers (UMs) [12].
27.7.2. Genes Involved in the Mechanism of Action of CNS Drugs
Most genes associated with the mechanism of action of CNS drugs encode receptors, enzymes, and neurotransmitters on which psychotropic drugs act as ligands (agonists, antagonists), enzyme modulators (substrates, inhibitors, inducers), or neurotransmitter regulators (releasers, reuptake inhibitors) [111]. In the case of conventional antidementia drugs, tacrine, donepezil, rivastigmine and galantamine are cholinesterase inhibitors, and memantine is a partial NMDA antagonist (Table 27.1 ).
Table 27.1.
Pharmacogenomic Profile of Antidementia Drugs
| Donepezil | |
|---|---|
| Category | Antidementia agent/cholinesterase inhibitor |
| Mechanism | Centrally active, reversible acetylcholinesterase inhibitor; increases acetylcholine available for synaptic transmission in CNS |
| Genes | |
| Pathogenic | APOE, CHAT |
| Mechanistic | CHAT, ACHE, BCHE |
| Metabolism: substrate | CYP2D6 (major), CYP3A4 (major), UGTs, ACHE |
| Metabolism: inhibitor | ACHE, BCHE |
| Transporter | ABCB1 |
| Galantamine | |
| Category | Antidementia agent/cholinesterase inhibitor |
| Mechanism | Reversible and competitive acetylcholinesterase inhibition leading to increased concentration of acetylcholine at cholinergic synapses; modulates nicotinic acetylcholine receptor; may increase glutamate and serotonin levels |
| Genes | |
| Mechanistic | APOE, APP |
| Pathogenic | ACHE, BCHE, CHRNA4, CHRNA7, CHRNB2 |
| Metabolism: substrate | CYP2D6 (major), CYP3A4 (major), UGT1A1 |
| Metabolism: inhibitor | ACHE, BCHE |
| Memantine | |
| Category | Antidementia Drug; N-methyl-d-aspartate Receptor Antagonist |
| Mechanism | Binds preferentially to NMDA receptor-operated cation channels; may act by blocking glutamate actions, mediated in part by NMDA receptors. Antagonists: GRIN2A, GRIN2B, GRIN3A, HTR3A, CHRFAM7A |
| Genes | |
| Pathogenic | APOE, PSEN1, MAPT |
| Mechanistic | GRIN2A, GRIN2B, GRIN3A, HTR3A, CHRFAM7A |
| Metabolism: inhibitor | CYP1A2 (weak), CYP2A6 (weak), CYP2B6 (strong), CYP2C9 (weak), CYP2C19 (weak), CYP2D6 (strong), CYP2E1 (weak), CYP3A4 (weak) |
| Pleiotropic | APOE, MAPT, MT-TK, PSEN1 |
| Rivastigmine | |
| Category | Antidementia Agent/Cholinesterase Inhibitor |
| Mechanism | Increases acetylcholine in CNS through reversible inhibition of its hydrolysis by cholinesterase |
| Genes | |
| Pathogenic | APOE, APP, CHAT |
| Mechanistic | ACHE, BCHE, CHAT, CHRNA4, CHRNB2 |
| Metabolism: inhibitor | ACHE, BCHE |
| Pleiotropic | APOE, MAPT |
| Tacrine | |
| Category | Antidementia agent/cholinesterase inhibitor |
| Mechanism | Elevates acetylcholine in cerebral cortex by slowing degradation of acetylcholine |
| Genes | |
| Pathogenic | APOE |
| Mechanistic | ACHE, BCHE, CHRNA4, CHRNB2 |
| Metabolism: substrate | CYP1A2 (major), CYP2D6 (minor), CYP3A4 (major) |
| Metabolism: inhibitor | ACHE, BCHE, CYP1A2 (weak) |
| Transporter | SCN1A |
| Pleiotropic | APOE, MTHFR, CES1, LEPR, GSTM1, GSTT1 |
Source: Cacabelos [113].
27.7.3. Genes Involved in Drug Metabolism
Drug metabolism includes phase I reactions (i.e., oxidation, reduction, hydrolysis) and phase II conjugation reactions (i.e., acetylation, glucuronidation, sulphation, methylation) (Table 27.2 ). The principal enzymes with polymorphic variants involved in phase I reactions are the following: cytochrome P450 monooxygenases (CYP3A4/5/7, CYP2E1, CYP2D6, CYP2C19, CYP2C9, CYP2C8, CYP2B6, CYP2A6, CYP1B1, CYP1A1/2), epoxide hydrolase, esterases, NQO1 (NADPH-quinone oxidoreductase), DPD (dihydropyrimidine dehydrogenase), ADH (alcohol dehydrogenase), and ALDH (aldehyde dehydrogenase). The major enzymes involved in phase II reactions include UGTs (uridine 5′-triphosphate glucuronosyl transferases), TPMT (thiopurine methyltransferase), COMT (catechol-O-methyltransferase), HMT (histamine methyl-transferase), STs (sulfotransferases), GST-A (glutathione S-transferase A), GST-P, GST-T, GST-M, NAT1 (N-acetyl transferase 1), NAT2, and others (Table 27.2).
Table 27.2.
Drug Metabolism-Related Genes
| Phase I Enzymes | |
|---|---|
| Alcohol dehydrogenases | ADH1A, ADH1B, ADH1C, ADH4, ADH5, ADH6, ADH7, ADHFE1 |
| Aldehyde dehydrogenases | ALDH1A1, ALDH1A2, ALDH1A3, ALDH1B1, ALDH2, ALDH3A1, ALDH3A2, ALDH3B1, ALDH3B2, ALDH4A1, ALDH5A1, ALDH6A1, ALDH7A1, ALDH8A1, ALDH9A1, AOX1 |
| Aldo-keto reductases | AKR1A1, AKR1B1, AKR1C1, AKR1D1 |
| Amine oxidases | MAOA, MAOB, SMOX |
| Carbonyl reductases | CBR1, CBR3, CBR4 |
| Cytidine deaminase | CDA |
| Cytochrome P450 family | CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2A7, CYP2A13, CYP2B6, CYP2C18, CYP2C19, CYP2C8, CYP2C9, CYP2D6, CYP2D7P1, CYP2E1, CYP2F1, CYP2J2, CYP2R1, CYP2S1, CYP2W1, CYP3A4, CYP3A5, CYP3A7, CYP3A43, CYP4A11, CYP4A22, CYP4B1, CYP4F2, CYP4F3, CYP4F8, CYP4F11, CYP4F12, CYP4Z1, CYP7A1, CYP7B1, CYP8B1, CYP11A1, CYP11B1, CYP11B2, CYP17A1, CYP19A1, CYP20A1, CYP21A2, CYP24A1, CYP26A1, CYP26B1, CYP26C1, CYP27A1, CYP27B1, CYP39A1, CYP46A1, CYP51A1, POR, TBXAS1 |
| Cytochrome b5 reductase | CYB5R3 |
| Dihydropyrimidine dehydrogenase | DPYD |
| Esterases | AADAC, CEL, CES1, CES1P1, CES2, CES3, CES5A, ESD, GZMA, GZMB, PON1, PON2, PON3, UCHL1, UCHL3 |
| Epoxidases | EPHX1, EPHX2 |
| Flavin-containing monooxygenases | FMO1, FMO2, FMO3, FMO4, FMO5, FMO6P |
| Glutathione reductase/peroxidases | GSR, GPX1, GPX2, GPX3, GPX4, GPX5, GPX6, GPX7 |
| Peptidases | DPEP1, METAP1 |
| Prostaglandin-endoperoxide synthases | PTGS1, PTGS2 |
| Short-chain dehydrogenases/reductases | DHRS1, DHRS2, DHRS3, DHRS4, DHRS7, DHRS9, DHRS12, DHRS13, DHRSX, HSD11B1, HSD17B10, HSD17B11, HSD17B14 |
| Superoxide dismutase | SOD1, SOD2 |
| Xanthine dehydrogenase | XDH |
| Phase II Enzymes | |
| Amino acid transferases | AGXT, BAAT, CCBL1 |
| Dehydrogenases | NQO1, NQO2, XDH |
| Esterases | CES1, CES2, CES3, CES4, CES5A |
| Glucuronosyl transferases | DDOST, UGT1A1, UGT1A10, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT2A1, UGT2A3, UGT2B10, UGT2B11, UGT2B15, UGT2B17, UGT2B28, UGT2B4, UGT2B7, UGT3A1, UGT8 |
| Glutathione transferases | GSTA1, GSTA2, GSTA3, GSTA4, GSTA5, GSTCD, GSTK1, GSTM1, GSTM2, GSTM3, GSTM4, GSTM5, GSTO1, GSTO2, GSTP1, GSTT1, GSTT2, GSTZ1, MGST1, MGST2, MGST3, PTGES |
| Methyl transferases | AS3MT, ASMT, COMT, GAMT, GNMT, HNMT, INMT, NNMT, PNMT, TPMT |
| N-Acetyl transferases | AANAT, ACSL1, ACSL3, ACSL4, ACSM1, ACSM2B, ACSM3, GLYAT, NAT1, NAT2, NAA20, SAT1 |
| Thioltransferase | GLRX |
| Sulfotransferases | SULT1A1, SULT1A2, SULT1A3, SULT1B1, SULT1C1, SULT1C2, SULT1C3, SULT1C4, SULT1E1, SULT2A1, SULT2B1, SULT4A1, SULT6B1, TST, CHST1, CHST2, CHST3, CHST4, CHST5, CHST6, CHST7, CHST8, CHST9, CHST10, CHST11, CHST12, CHST13, GAL3ST1 |
Note: See Appendix B for long-form names of genes listed.
Among these enzymes, CYP2D6, CYP2C9, CYP2C19, and CYP3A4/5 are the most relevant in the pharmacogenetics of CNS drugs [15], [111] (Table 27.1). Approximately 18% of neuroleptics are major substrates of CYP1A2 enzymes, 40% of CYP2D6, and 23% of CYP3A4; 24% of antidepressants are major substrates of CYP1A2 enzymes, 5% of CYP2B6, 38% of CYP2C19, 85% of CYP2D6, and 38% of CYP3A4; 7% of benzodiazepines are major substrates of CYP2C19 enzymes, 20% of CYP2D6, and 95% of CYP3A4 [15], [111]. Most CYP enzymes exhibit ontogenic-, age-, sex-, circadian-, and ethnic-related differences [112].
In dementia, as in any other CNS disorder, CYP genomics is a very important issue, since in practice more than 90% of patients with dementia are daily consumers of psychotropics. Furthermore, some acetylcholinesterase inhibitors (the most prescribed antidementia drugs worldwide) are metabolized via CYP enzymes (Table 27.1). Most CYP enzymes display highly significant ethnic differences, indicating that the enzymatic capacity of these proteins varies depending upon the polymorphic variants present in their coding CYP genes.
The practical consequence of this genetic variation is that the same drug can be differentially metabolized according to the genetic profile of each subject, and that, if an individual’s pharmacogenomic profile is known, his/her pharmacodynamic response is potentially predictable. This is the cornerstone of pharmacogenetics. In this regard, the CYP2D6, CYP2C19, CYP2C9, and CYP3A4/5 genes and their respective protein products deserve special consideration.
27.7.3.1. CYP2D6
CYP2D6 is a 4.38 kb gene with 9 exons mapped on 22q13.2. Four RNA transcripts of 1190–1684 bp are expressed in the brain, liver, spleen, and reproductive system, where 4 major proteins of 48–55 kDa (439–494 aa) are identified. It is a transport enzyme of the cytochrome P450 subfamily IID or multigenic cytochrome P450 superfamily of mixed-function monooxygenases. The cytochrome P450 proteins are monooxygenases which catalyze many reactions involved in drug metabolism and synthesis of cholesterol, steroids, and other lipids. CYP2D6 localizes to the endoplasmic reticulum and is known to metabolize as many as 25% of commonly prescribed drugs, and more than 60% of current psychotropics. Its substrates include debrisoquine, an adrenergic-blocking drug; sparteine and propafenone, both antiarrhythmic drugs; and amitryptiline, an antidepressant. CYP2D6 is highly polymorphic in the population.
There are 141 CYP2D6 allelic variants, of which -100C > T, -1023C > T, -1659G > A, -1707delT, -1846G > A, -2549delA, -2613-2615delAGA, -2850C > T, -2988G > A, and -3183G > A represent the ten most important [113], [114], [115]. Different alleles result in the extensive, intermediate, poor, and ultrarapid metabolizer phenotypes, characterized by normal, intermediate, decreased, and multiplied ability to metabolize the enzyme’s substrates, respectively. The hepatic cytochrome P450 system is responsible for the first phase in the metabolism and elimination of numerous endogenous and exogenous molecules and ingested chemicals. P450 enzymes convert these substances into electrophilic intermediates, which are then conjugated by phase II enzymes (e.g., UDP glucuronosyltransferases, N-acetyltransferases) to hydrophilic derivatives that can be excreted. According to the database of the World Guide for Drug Use and Pharmacogenomics [113], 982 drugs are CYP2D6-related: 371 are substrates, more than 300 are inhibitors, and 18 are CYP2D6 inducers.
In healthy subjects, extensive metabolizers (EMs) account for 55.71% of the population; intermediate metabolizers (IMs) account for 34.7%; poor metabolizers (PMs), 2.28%; and ultrarapid metabolizers (UMs), 7.31%. Remarkable worldwide interethnic differences exist in the frequency of the PM and UM phenotypes [116], [117], [118]. On average, approximately 6.28% of the world’s population belongs to the PM category. Europeans (7.86%), Polynesians (7.27%), and Africans (6.73%) show the highest rate of PMs, whereas Orientals (0.94%) show the lowest [116]. The frequency of PMs among Middle Eastern populations, Asians, and Americans is in the range of 2–3%. CYP2D6 gene duplications are relatively infrequent among Northern Europeans, but in East Africa the frequency of alleles with duplication of CYP2D6 is as high as 29% [119]. In Europe, there is a North–South gradient in the frequency of PMs (6–12% of PMs in Southern European countries, and 2–3% of PMs in Northern latitudes) [111].
In AD, EMs, IMs, PMs, and UMs are 56.38%, 27.66%, 7.45%, and 8.51%, respectively, and in vascular dementia, they are, respectively, 52.81%, 34.83%, 6.74%, and 5.62% (Figure 27.3 ). There is an accumulation of AD-related risk genes in PMs and UMs. EMs and IMs are the best responders, and PMs and UMs are the worst responders to a combination therapy of cholinesterase inhibitors, neuroprotectants, and vasoactive substances. The pharmacogenetic response in AD appears to depend on the networking activity of genes involved in drug metabolism and genes involved in AD pathogenesis [7], [12], [15], [16], [22], [23], [24], [25], [26], [27], [28], [53], [54], [55], [56], [57], [58], [59].
Figure 27.3.

Distribution and frequency of CYP2D6 phenotypes in AD and vascular dementia.
EM—extensive metabolizer; IM—intermediate metabolizer; PM—poor metabolizer; UM—ultrarapid metabolizer.
Source: Adapted from Cacabelos[18].
27.7.3.2. CYP2C9
CYP2C9 is a gene (50.71 kb) with 9 exons mapped on 10q24. An RNA transcript of 1860 bp is mainly expressed in hepatocytes, where a protein of 55.63 kDa (490 aa) can be identified. More than 600 drugs are CYP2C9-related: 311 act as substrates (177 major, 134 minor); 375, as inhibitors (92 weak, 181 moderate, and 102 strong); and 41 as inducers of the CYP2C9 enzyme [113]. There are 481 CYP2C9 SNPs. By phenotype (Figure 27.4 ), in the control population, PMs represent 7.04%, IMs 32.39%, and EMs 60.56%. In AD, PMs, IMs, and EMs are 6.45%, 37.64%, and 55.91%, respectively, and in vascular dementia they are 3.61%, 28.92%, and 67.47%, respectively [18] (Figure 27.4).
Figure 27.4.
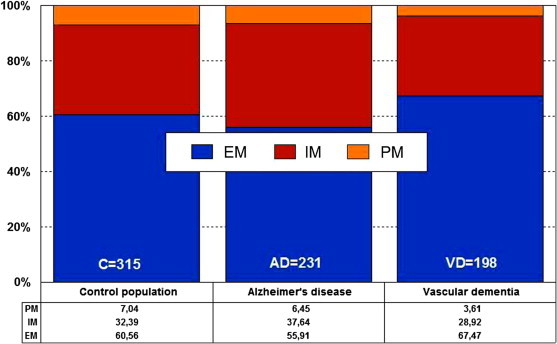
Distribution and frequency of CYP2C9 phenotypes in AD and vascular dementia.
EM—extensive metabolizer; IM—intermediate metabolizer; PM—poor metabolizer.
Source: Adapted from Cacabelos[18].
27.7.3.3. CYP2C19
CYP2C19 is a gene (90.21 kb) with 9 exons mapped on 10q24.1q24.3. RNA transcripts of 1901 bp, 2395 bp, and 1417 bp are expressed in liver cells, where a protein of 55.93 kDa (490 aa) has been identified. Nearly 500 drugs are CYP2C19-related, with 281 acting as substrates (151 major, 130 minor), 263 as inhibitors (72 weak, 127 moderate, and 64 strong), and 23 as inducers of the CYP2C19 enzyme [113]. About 541 SNPs have been detected in the CYP2C19 gene. The frequencies of the three major CYP2C19 geno-phenotypes in the control population are CYP2C19-*1/*1-EMs, 68.54%; CYP2C19-*1/*2-IMs, 30.05%; and CYP2C19-*2/*2-PMs, 1.41%. EMs, IMs, and PMs account for 69.89%, 30.11%, and 0%, respectively, in AD, and 66.27%, 30.12%, and 3.61%, respectively, in vascular dementia [18] (Figure 27.5 ).
Figure 27.5.
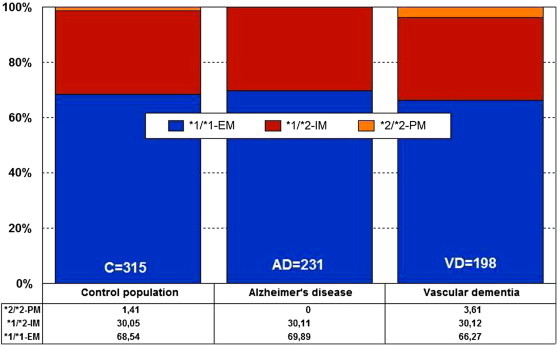
Distribution and frequency of CYP2C19 pheno- genotypes in AD and vascular dementia.
EM—extensive metabolizer; IM—intermediate metabolizer; PM—poor metabolizer.
Source: Adapted from Cacabelos[18].
27.7.3.4. CYP3A4/5
CYP3A4 is a gene (27.2 kb) with 13 exons mapped on 7q21.1. RNA transcripts of 2153 bp, 651 bp, 564 bp, 2318 bp, and 2519 bp are expressed in intestine, liver, prostate, and other tissues, where four protein variants of 57.34 kDa (503 aa), 17.29 kDa (153 aa), 40.39 kDa (353 aa), and 47.99 kDa (420 aa) have been identified. The human CYP3A locus contains the three CYP3A genes (CYP3A4, CYP3A5, and CYP3A7), three pseudogenes, and a novel CYP3A gene termed CYP3A43. The gene encodes a putative protein with 71.5–75.8% identity with the other CYP3A proteins. The predominant hepatic form is CYP3A4, but CYP3A5 contributes significantly to total liver CYP3A activity.
CYP3A4 metabolizes more than 1900 drugs: 1033 act as substrates (897 major, 136 minor); 696, as inhibitors (118 weak, 437 moderate, and 141 strong); and 241, as inducers of the CYP3A4 enzyme [113]. About 347 SNPs have been identified in the CYP3A4 gene (CYP3A4*1A: wild-type), 25 of which are of clinical relevance. Concerning CYP3A4/5 polymorphisms in AD, 82.75% of cases are EMs (CYP3A5*3/*3), 15.88% are IMs (CYP3A5*1/*3), and 1.37% are UMs (CYP3A5*1/*1). Unlike other human P450s (CYP2D6, CYP2C19), there is no evidence of a “null” allele for CYP3A4 [113].
27.7.3.5. CYP Clustering
The construction of a genetic map integrating the most prevalent CYP2D6+CYP2C19+CYP2C9 polymorphic variants in a trigenic cluster yields 82 different haplotype-like profiles. The most frequent trigenic genotypes in the AD population are *1*1-*1*1-*1*1 (25.70%), *1*1-*1*2-*1*2 (10.66%), *1*1-*1*1-*1*1 (10.45%), *1*4-*1*1-*1*1 (8.09%), *1*4-*1*2-*1*1 (4.91%), *1*4-*1*1-*1*2 (4.65%), and *1*1-*1*3-*1*3 (4.33%). These 82 trigenic genotypes represent 36 different pharmacogenetic phenotypes.
According to these trigenic clusters, only 26.51% of patients show a pure 3EM phenotype, 15.29% are 2EM1IM, 2.04% are pure 3IM, 0% are pure 3PM, and 0% are 1UM2PM (the worst possible phenotype). This implies that only one-quarter of the population normally process the drugs that are metabolized via CYP2D6, CYP2C9, and CYP2C19 (approximately 60% of the drugs in current use) [12]. Taking into consideration the data available, it might be inferred that at least 20–30% of the AD population may exhibit an abnormal metabolism of cholinesterase inhibitors and/or other drugs that undergo oxidation via CYP2D6-related enzymes.
Approximately 50% of this population cluster shows an ultrarapid metabolism, requiring higher doses of cholinesterase inhibitors in order to reach a therapeutic threshold. The other 50% of the cluster exhibit a poor metabolism, displaying potential adverse events at low doses. If we take into account that approximately 60–70% of therapeutic outcomes depend on pharmacogenomic criteria (e.g., pathogenic mechanisms associated with AD-related genes), it can be postulated that pharmacogenetic and pharmacogenomic factors are responsible for 75–85% of therapeutic response (efficacy) in AD patients treated with conventional drugs [12], [15], [16], [17], [18], [22], [23], [24], [25], [28], [53], [54], [55], [56], [57], [58], [59].
27.7.4. Drug Transporters
ABC genes—especially ABCB1 (ATP-binding cassette, subfamily B, member 1P-glycoprotein-1, P-gp1, Multidrug Resistance 1, MDR (17q21.12), ABCC1 (9q31.1), ABCG2 (White121q22.3), and other genes of this family—encode proteins that are essential for drug metabolism and transport. The multidrug efflux transporters P-gp, the multidrug resistance-associated protein 4 (MRP4), and the breast cancer resistance-protein (BCRP), located on endothelial cells lining the brain vasculature, play important roles in limiting the movement of substances into the brain and in enhancing their efflux from the brain.
Transporters also cooperate with phase I/phase II metabolism enzymes by eliminating drug metabolites. Their major features are their capacity to recognize drugs belonging to unrelated pharmacological classes and their redundancy, by which a single molecule can act as a substrate for different transporters. This ensures efficient neuroprotection against xenobiotic invasions. The pharmacological induction of ABC gene expression is a mechanism of drug interaction, which may affect substrates of the upregulated transporter; overexpression of MDR transporters confers resistance to anti-cancer agents and CNS drugs [120], [121].
Also of importance for CNS pharmacogenomics are transporters encoded by genes of the solute carrier superfamily (SLC) and solute carrier organic (SLCO) transporter family, which are responsible for the transport of multiple endogenous and exogenous compounds, including folate (SLC19A1), urea (SLC14A1, SLC14A2), monoamines (SLC29A4, SLC22A3), aminoacids (SLC1A5, SLC3A1, SLC7A3, SLC7A9, SLC38A1, SLC38A4, SLC38A5, SLC38A7, SLC43A2, SLC45A1), nucleotides (SLC29A2, SLC29A3], fatty acids (SLC27A1-6), neurotransmitters (SLC6A2[noradrenaline transporter]), SLC6A3[dopamine transporter], SLC6A4[serotonin transporter, SERT], SLC6A5, SLC6A6, SLC6A9, SLC6A11, SLC6A12, SLC6A14, SLC6A15, SLC6A16, SLC6A17, SLC6A18, SLC6A19), glutamate (SLC1A6, SLC1A7), and others [122].
Some organic anion transporters (OAT), which belong to the solute carrier (SLC) 22A family, are also expressed at the BBB, and regulate the excretion of endogenous and exogenous organic anions and cations [123]. The transport of amino acids and di- and tripeptides is mediated by a number of different transporter families, and the bulk of oligopeptide transport is attributable to the activity of members of the SLC15A superfamily (peptide transporters 1 and 2 (SLC15A1[PepT1]) and SLC15A2[PepT2], and peptide/histidine transporters 1 and 2 (SLC15A4[PHT1] and SLC15A3[PHT2]). ABC and SLC transporters expressed at the BBB may cooperate to regulate the passage of different molecules into the brain [124]. Polymorphic variants in ABC and SLC genes may also be associated with pathogenic events in CNS disorders and drug-related safety and efficacy complications [111], [122].
27.7.5. Pleiotropic Activity of APOE in Dementia
APOE is the prototypical paradigm of a pleiotropic gene with multifaceted activities in physiological and pathological conditions [16], [22]. ApoE is consistently associated with the amyloid plaque marker for AD. APOE-4 may influence AD pathology interacting with APP metabolism and Aβ accumulation, enhancing hyperphosphorylation of tau protein and NFT formation, reducing choline acetyltransferase activity, increasing oxidative processes, modifying inflammation-related neuroimmunotrophic activity and glial activation, altering lipid metabolism, lipid transport, and membrane biosynthesis in sprouting and synaptic remodeling, and inducing neuronal apoptosis [16], [23], [24], [25].
To address the complex misfolding and aggregation that initiates the toxic cascade resulting in AD, Petrlova et al. [26] developed a 2,2,6,6-tetramethylpiperidine-1-oxyl-4-amino-4-carboxylic acid spin-labeled amyloid-β (Aβ) peptide to observe its isoform-dependent interaction with the ApoE protein. Oligomer binding involves the C-terminal domain of ApoE, with ApoE3 reporting a much greater response through this conformational marker. ApoE3 displays a higher affinity and capacity for the toxic Aβ oligomer. ApoE polymorphism and AD risk can largely be attributed to the reduced ability of ApoE4 to function as a clearance vehicle for the toxic form of Aβ. MAPT and APOE are involved in the pathogenic mechanisms of AD, and both the MAPT H1/H1 genotype and the APOE ɛ4 allele lead to a more rapid progression to dementia among MCI subjects, probably mediating an increased rate of amyloid-β and tau brain deposition [27].
The distribution of APOE genotypes in the Iberian peninsula is as follows: APOE-2/2 0.32%; APOE-2/3 7.3%; APOE-2/4 1.27%; APOE-3/3 71.11%; APOE-3/4 18.41%; and APOE-4/4 1.59% [18] (Figure 27.2). These frequencies are very similar in Europe and in other Western societies. There is a clear accumulation of APOE-4 carriers among patients with AD (APOE-3/4 30.30%, APOE-4/4 6.06%) and vascular dementia (APOE-3/4 35.85%, APOE-4/4 6.57%) as compared to controls (Figure 27.2). Different APOE genotypes confer specific phenotypic profiles to AD patients [15], [16], [22]. Some of these profiles may add risk or benefit when patients are treated with conventional drugs, and in many instances the clinical phenotype demands the administration of additional drugs that increase the complexity of therapeutic protocols.
From studies designed to define APOE-related AD phenotypes [7], [12], [23], [24], [25], [28], [53], [54], [55], [56], [57], [58], [59], several conclusions can be drawn, which are shown in Box 27.2. These 20 major phenotypic features clearly illustrate the biological disadvantage of APOE-4 homozygotes and the potential consequences that these patients may experience when they receive pharmacological treatment for AD and/or concomitant pathologies [7], [12], [23], [24], [25], [28], [53], [54], [55], [56], [57], [58], [59].
Box 27.2. Key Conclusions Regarding APOE-Related AD Phenotypes.
-
1.
The age at onset is 5–10 years earlier in approximately 80% of AD cases harboring the APOE-4/4 genotype.
-
2.
The serum levels of ApoE are lowest in APOE-4/4, intermediate in APOE-3/3 and APOE-3/4, and highest in APOE-2/3 and APOE-2/4.
-
3.
Serum cholesterol levels are higher in APOE-4/4 than in other genotypes.
-
4.
HDL-cholesterol levels tend to be lower in APOE-3 homozygotes than in APOE-4 allele carriers.
-
5.
LDL-cholesterol levels are systematically higher in APOE-4/4 than in any other genotype.
-
6.
Triglyceride levels are significantly lower in APOE-4/4.
-
7.
Nitric oxide levels are slightly lower in APOE-4/4.
-
8.
Serum and cerebrospinal fluid Aβ levels tend to differ between APOE-4/4 and the other most frequent genotypes (APOE-3/3, APOE-3/4).
-
9.
Blood histamine levels are dramatically reduced in APOE-4/4 as compared to the other genotypes.
-
10.
Brain atrophy is markedly increased in APOE-4/4>APOE-3/4>APOE-3/3.
-
11.
Brain mapping activity shows a significant increase in slow wave activity in APOE-4/4 from the early stages of the disease.
-
12.
Brain hemodynamics, as reflected by reduced brain blood flow velocity and increased pulsatility and resistance indices, is significantly worse in APOE-4/4 (and in APOE-4 carriers in general, as compared with APOE-3 carriers); brain hypoperfusion and neocortical oxygenation is also more deficient in APOE-4 carriers.
-
13.
Lymphocyte apoptosis is markedly enhanced in APOE-4 carriers.
-
14.
Cognitive deterioration is faster in APOE-4/4 patients than in carriers of any other APOE genotype.
-
15.
In approximately 3–8% of AD cases, some dementia-related metabolic dysfunctions accumulate more in APOE-4 carriers than in APOE-3 carriers.
-
16.
Some behavioral disturbances, alterations in circadian rhythm patterns, and mood disorders are slightly more frequent in APOE-4 carriers.
-
17.
Aortic and systemic atherosclerosis is more frequent in APOE-4 carriers.
-
18.
Liver metabolism and transaminase activity differ in APOE-4/4 with respect to other genotypes.
-
19.
Hypertension and other cardiovascular risk factors accumulate in APOE-4 carriers.
-
20.
APOE-4/4 carriers are the poorest responders to conventional drugs.
27.7.6. Pharmacogenomics of Antidementia Drugs
The following list describes the pharmacogenomics of the most common antidementia drugs (Table 27.1).
Donepezil: is a centrally active, reversible acetylcholinesterase inhibitor that increases the acetylcholine available for synaptic transmission in the CNS. The therapeutic response of donepezil is influenced by pathogenic gene variants (APOE, CHAT), as well as mechanistic gene polymorphic variants (CHAT, ACHE, and BCHE). It is a major substrate of CYP2D6, CYP3A4, ACHE, and UGTs; it inhibits ACHE and BCHE; and it is transported by ABCB1 [113].
Galantamine: is a reversible and competitive acetylcholinesterase inhibitor leading to increased concentration of acetylcholine at cholinergic synapses. It also modulates nicotinic acetylcholine receptors and may increase glutamate and serotonin levels. APOE, APP, ACHE, BCHE, CHRNA4, CHRNA7, and CHRNB2 variants may potentially influence galantamine efficacy and safety. Galantamine is a major substrate of CYP2D6, CYP3A4, and UGT1A1, and an inhibitor of ACHE and BCHE [113].
Rivastigmine: is a cholinesterase inhibitor that increases acetylcholine in the CNS through reversible inhibition of its hydrolysis by cholinesterase. APOE, APP, CHAT, ACHE, BCHE, CHRNA4, CHRNB2, and MAPT variants may affect its pharmacokinetics and pharmacodynamics [113].
Tacrine: is the first FDA-approved antidementia drug. Its use was stopped due to hepatotoxicity. Tacrine is a cholinesterase inhibitor that elevates acetylcholine in the cerebral cortex by slowing degradation of acetylcholine. ACHE, BCHE, CHRNA4, CHRNB2, APOE, MTHFR, CES1, LEPR, GSTM1, and GSTT1 variants may affect its therapeutic and toxic effects. Tacrine is a major substrate of CYP1A2 and CYP3A4, a minor substrate of CYP2D6, and is transported via SCN1A. It is an inhibitor of ACHE, BCHE, and CYP1A2 [113].
Memantine: is an N-Methyl-D-Aspartate (NMDA) receptor antagonist that binds preferentially to NMDA receptor-operated cation channels. It may act by blocking the actions of glutamate, mediated in part by NMDA receptors, and it is also an antagonist of GRIN2A, GRIN2B, GRIN3A, HTR3A, and CHRFAM7A. Several pathogenic (APOE, PSEN1, MAPT) and mechanistic gene variants (GRIN2A, GRIN2B, GRIN3A, HTR3A, CHRFAM7A) may influence its therapeutic effects. Memantine is a strong inhibitor of CYP2B6 and CYP2D6, and a weak inhibitor of CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2E1, and CYP3A4 [113].
27.7.7. Multifactorial Therapy
Some studies using a multifactorial approach also have shown that diverse pharmacogenomic factors may influence efficacy and safety. In one of these studies [15], [58], patients with dementia received the following for three months: a multifactorial therapy integrated by CDP-choline (500 mg/day, p.o.), Nicergoline (5 mg/day, p.o.), Sardilipin (E-SAR-94010) (LipoEsar®)(250 mg, t.i.d.), and Animon Complex® (2 capsules/day)—a nutraceutical compound integrated by a purified extract of Chenopodium quinoa (250 mg), ferrous sulphate (38.1 mg equivalent to 14 mg of iron), folic acid (200 μg), and vitamin B12 (1 μg) per capsule (RGS: 26.06671/C).
Patients with chronic deficiencies of iron (<35 μg/mL), folic acid (<2.5 ng/mL), or vitamin B12 (<150 pg/mL) received an additional supplement of iron (80 mg/day), folic acid (5 mg/day), and B complex vitamins (B1, 15 mg/day; B2, 15 mg/day; B6, 10 mg/day; B12, 10 μg/day; nicotinamide, 50 mg/day), respectively, to maintain stable levels of serum iron (50–150 μg/mL), folic acid (5–20 ng/mL) and vitamin B12 levels (500–1000 pg/mL) in order to avoid the negative influence of all these metabolic factors on cognition. Patients with hypertension (>150/85 mmHg) received Enalapril (20 mg/day).
The frequency of APOE genotypes was APOE-2/3, 7.97%; APOE-2/4, 1.18%; APOE-3, 58.95%; APOE-3/4, 27.32%; and APOE-4/4, 4.58%. Cognitive function (as assessed by MMSE); 20.51 ± 6.51 vs. 21.45 ± 6.95, p < 0.0000000001; ADAS-Cog, 22.94 ± 13.87 vs. 21.23 ± 12.84, p < 0.0001; ADAS-Non-Cog, 5.26 ± 4.18 vs. 4.15 ± 3.63, p < 0.0000000001; ADAS-Total, 27.12 ± 16.93 vs. 24.28 ± 15.06, p < 0.00009) improved after treatment. Mood (HAM-A, 11.35 ± 5.44 vs. 9.79 ± 4.33, p < 0.0000000001; HAM-D, 10.14 ± 5.23 vs. 8.59 ± 4.30, p < 0.0000000001) also improved. Glucose levels did not change.
Total cholesterol levels (224.78 ± 45.53 vs. 203.64 ± 39.69 mg/dL, p < 0.0000000001), HDL-cholesterol levels (54.11 ± 14.54 vs. 52.54 ± 14.86 mg/dL, p < 0.0001), and LDL-cholesterol levels (148.15 ± 39.13 vs. 128.89 ± 34.83 mg/dL, p < 0.0000000001) were significantly reduced. Folate (7.07 ± 3.61 vs. 18.14 ± 4.23 ng/mL, p < 0.000000001) and vitamin B12 levels (459.65 ± 205.80 vs. 689.78 ± 338.82 pg/mL, p < 0.000000001) also increased, and both TSH and T4 levels remained unchanged after treatment. The response rate in terms of cognitive improvement was as follows: 59.74% responders (RRs), 24.44% nonresponders (NRs), and 15.82% stable responders (SRs) (no change in MMSE score after three months of treatment). The response rate in cholesterol levels was very similar: 57.78% RRs, 28.50% NRs, and 13.72% SRs [15].
27.7.7.1. APOE-Related Cognitive Function Changes
In this study, the basal MMSE score differed in APOE-2/3 carriers with respect to APOE-2/4 (p < 0.02), APOE-3/4 (p < 0.004), and APOE-4/4 (p < 0.0009), in APOE-3/3 vs. APOE-3/4 (p < 0.0005), and in APOE-3/3 vs. APOE-4/4 (p < 0.002). The best responders were APOE-3/3 (p < 0.0000000001) >APOE-3/4 (p < 0.00001) >APOE-4/4 carriers (p < 0.05). Patients harboring the APOE-2/3 and APOE-2/4 genotypes did not show any significant improvement. The response rate by genotype was the following: APOE-2/3: 44.26% RRs, 36.07% NRs, 19.67% SRs; APOE-2/4: 55.56% RRs, 44.44% NRs, 0.0% SRs; APOE-3/3: 63.42% RRs, 21.06% NRs, 15.52% SRs; APOE-3/4: 56.94% RRs, 27.75% NRs, 15.31% SRs; and APOE-4/4: 51.43% RRs, 28.57% NRs, 20.00% SRs [15] (Figure 27.6, Figure 27.7 ).
Figure 27.6.
APOE-related cognitive performance in response to multifactorial therapy in patients with dementia.
Tb—basal MMSE score prior to treatment; Tt—MMSE score after 3 months treatment in total sample. E2/3b—basal MMSE score in APOE-2/3 carriers; E2/3t—MMSE score after treatment in APOE-2/3 carriers; E2/4b—basal MMSE score in APOE-2/4 carriers; E2/4t—MMSE score after treatment in APOE-2/4 carriers; E3/3b—basal MMSE score in APOE-3/3 carriers; E3/3t—MMSE score after treatment in APOE-3/3 carriers; E3/4b basal MMSE score in APOE-3/4 carriers; E3/4t—MMSE score after treatment in APOE-3/4 carriers; E4/4b—basal MMSE score in APOE-4/4 carriers; E4/4—MMSE score after treatment in APOE-4/4 carriers.
Source: Adapted from Cacabelos et al.[15].
Figure 27.7.

APOE-related cognitive response rate in patients with dementia treated with multifactorial therapy.
27.7.7.2. APOE-Related Changes in Blood Pressure Values
Systolic blood pressure (SBP) was significantly reduced in patients with the APOE-3/3 (p < 0.00007) and APOE-3/4 genotypes (p < 0.01), and diastolic blood pressure exhibited a similar pattern (APOE-3/3, p < 0.005; APOE-3/4, p < 0.01), with no changes in either SBP or DBP in APOE-2/3, APOE-2/4, and APOE-4/4 carriers [15].
27.7.7.3. APOE-Related Blood Lipid Response to Sardilipin
Basal cholesterol levels were significantly different in patients with the APOE-2/3 genotype vs. APOE-3/3 (p < 0.007), vs. APOE-3/4 (p < 0.001), vs. APOE-4/4 (p < 0.00002); APOE-2/4 vs. APOE-4/4 (p < 0.01); APOE-3/3 vs. APOE-4/4 (p < 0.005); and APOE-3/4 vs. APOE-4/4 (p < 0.01).
The highest cholesterol levels were seen in APOE-4/4> APOE-3/4> APOE-3/3. All patients showed a clear reduction in cholesterol levels after treatment with Sardilipin. This was particularly significant in APOE-3/3 (p < 0.0000000001) > APOE-3/4 (p < 0.00000008) > APOE-4/4 (p < 0.002) > APOE-2/3 (p < 0.02) > APOE-2/4 carriers (p: 0.26). The response rate by genotype was as follows: APOE-2/3: 63.93% RRs, 29.51% NRs, 6.56% SRs; APOE-2/4: 44.44% RRs, 22.22% NRs, 33.34% SRs; APOE-3/3: 54.32% RRs, 28.16% NRs, 17.52% SRs; APOE-3/4: 53.59% RRs, 31.58% NRs, 14.83% SRs; APOE-4/4: 65.71% RRs, 20.00% NRs, 14.29% SRs [15].
HDL-cholesterol levels significantly decreased in APOE-3/3 (p < 0.001) > APOE-3/4 (p < 0.05), with no significant changes in patients with other genotypes. In contrast, LDL-cholesterol levels showed changes identical to those observed in total cholesterol, with similar differences among genotypes at baseline and almost identical decreased levels after treatment (APOE-3/3, p > 0.0000000001 >APOE-3/4, p < 0.00001 >APOE-2/3, p < 0.0004 >APOE-4/4, p < 0.001 >APOE-2/4, p:0.31) [15].
Sardilipin (E-SAR-94010, LipoEsar®, LipoSea®) is a natural product extracted from the marine species Sardina pilchardus by means of nondenaturing biotechnological procedures. The main chemical compounds of LipoEsar® are lipoproteins (60–80%), whose micelle structure probably mimics that of physiological lipoproteins involved in lipid metabolism. In preclinical studies, Sardilipinhas been shown to be effective in:
-
1.
Reducing blood cholesterol (CHO), triglyceride (TG), uric acid (UA), and glucose (Glu) levels, as well as liver alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activity.
-
2.
Enhancing immunological function by regulating both lymphocyte and microglia activity.
-
3.
Inducing antioxidant effects mediated by superoxide dismutase activity.
-
4.
Improving cognitive function [15].
According to these results, it appears that the therapeutic response of patients with dyslipidemia to Sardilipin is APOE-related. The best responders were patients with APOE-3/3 >APOE-3/4 >APOE-4/4. Patients with the other APOE genotypes (2/2, 2/3, 2/4) did not show any hypolipidemic response to this novel compound. In patients with dementia, the effects of Sardilipin were very similar to those observed in patients with chronic dyslipidemia, suggesting that the lipid-lowering properties of Sardilipin are APOE-dependent [15] (Figure 27.8 ).
Figure 27.8.

APOE-related total cholesterol levels in response to multifactorial therapy in patients with dementia.
Tb—basal cholesterol levels prior to treatment; Tt—cholesterol levels after 3 months treatment in total sample. E2/3b—basal cholesterol levels in APOE-2/3 carriers; E2/3t—total cholesterol levels after treatment in APOE-2/3 carriers; E2/4b—basal cholesterol levels in APOE-2/4 carriers; E2/4t—total cholesterol levels after treatment in APOE-2/4 carriers; E3/3b—basal cholesterol levels in APOE-3/3 carriers; E3/3t—total cholesterol levels after treatment in APOE-3/3 carriers; E3/4b—basal cholesterol levels in APOE-3/4 carriers; E3/4t—total cholesterol levels after treatment in APOE-3/4 carriers; E4/4b—basal cholesterol levels in APOE-4/4 carriers; E4/4—total cholesterol levels after treatment in APOE-4/4 carriers.
Source: Adapted from Cacabelos et al.[15].
27.8. Future Perspective
To make AD a global health priority in the coming years, conceptual and procedural changes are needed on several grounds, such as (1) political, administrative, economic, legal, ethical, industrial, regulatory and educational issues; (2) novel biomarkers (genomics, proteomics, molecular neuroimaging) as diagnostic aids; (3) innovative therapeutics; (4) pharmacogenomics in clinical practice to optimize therapeutics; and (5) selective preventive plans for the population at risk.
There is disharmony concerning the public and governmental interest in dementia and its social, medical, and economic implications. The diagnosis and management of dementia is dissimilar in Europe, North America, Latin America, Asia, Africa, and Oceania. The economic/cultural status of each country (developed versus developing), the particular epidemiology of aging and dementia in each latitude, national standards of education, health priorities (infectious diseases versus degenerative diseases), and the quality and efficiency of medical services are conditioning factors for investing (or not investing) national resources in dementia as a health priority.
Educational programs, international guidelines, and consensus protocols for the management of dementia are necessary for global harmonization, for professionals from different countries to speak the same conceptual language, and to improve cost-effectiveness ratios [125], [126], [127], [128]. There are many legal issues (e.g., informed consent, lawsuit, testament, tutorship) and ethical issues (e.g., clinical trials, use of genetic information, institutionalization) that deserve more attention in order to humanize the end of life in the very frail conditions under which demented patients survive.
The updating of regulatory issues is also a matter of deep concern. Regulatory aspects of drug development are not universal, with notable peculiarities in the European Union (EMA), the United States (FDA), and Japan (Koseisho). Because the costs of dementia cannot be fully assumed by more than 60% of the European population, European authorities must take into account this circumstance when health reform is implemented in the coming years [8], [18].
Genomics, transcriptomics, proteomics, and metabolomics will revolutionize medicine in the next decades. Genetic testing is gaining acceptance among physicians and patients in different countries [128], [129], [130], [131], although Americans, Europeans, and Japanese differ notably in their knowledge, beliefs, and attitudes regarding genetic testing for AD [128], [131], [132]. The validation of protocols for genomic screening will contribute to the implementation of structural genomics, functional genomics, and proteomics as diagnostic aids and therapeutic targets [133].
An accurate diagnosis of AD demands the use of reliable biomarkers in routine protocols at a reasonable price [68]. Levels of specific secreted cellular signaling proteins in cerebrospinal fluid or plasma correlate with pathological changes in the AD brain; therefore, proteomic analysis of these levels can be used to discover said biomarkers [134]. It is likely that the best biomarkers result from a combination of genomic, transcriptomic, and proteomic analyses of body fluids. The measurement of these biomarkers correlates with brain imaging markers and cognitive performance [73], [74], [75].
New initiatives for the prevention of dementia (global versus selective prevention) will also emerge [135], together with new insights into the role of nutrition and nutrigenomics in brain function and neurodegeneration [59], [136]. In terms of prevention, it must be taken into consideration that neuronal death and Aβ accumulation starts many years before the onset of the disease, and that preventive strategies should be selective to protect the population at risk. For this purpose, accurate biomarkers are essential, and surrogate markers are needed to facilitate primary prevention.
Without doubt, the highest priority for the coming decade will be an intense search for novel therapeutic options in the form of both symptomatic treatments and preventive strategies. Past failures must be studied by researchers and the pharmaceutical industry in order to avoid unnecessary expenses in redundant trials that lead nowhere. Combination treatments require further evaluation and more sophisticated strategies than dual combinations [137], [138]. The administration of psychotropic drugs to demented patients should be reduced and predicted with pharmacogenetic markers to minimize side effects, cerebrovascular risk, and cognitive deterioration.
Priority areas for pharmacogenetic research are the prediction of serious adverse drug reactions (ADRs) and the determination of efficacy variation [139]. Both are necessary in CNS disorders and dementia to cope with efficacy and safety issues associated with current psychotropics and antidementia drugs, as well as new CNS drugs. With regard to the future of pharmacogenomics as a practical discipline, several issues should be addressed:
-
•
The education of physicians in medical genomics and pharmacogenomics is fundamental (less than 2% of clinicians are familiar with genomic science)
-
•
Genomic screening of gene clusters involved in pharmacogenomic outcomes must become a clinical routine (without genetic testing, there is no pharmacogenetics)
-
•
Each patient must be a carrier of a pharmacogenetic card [140] indicating what kind of drugs he/she can take and which medications he/she should avoid
-
•
Regulatory agencies should request pharmacogenetic data from the pharmaceutical industry when applying for drug approval
-
•
Pharmacogenetic data must be incorporated into patient information leaflets and the pharmaceutical vade mecum
-
•
New guidelines for daily praxis, such as those given in the World Guide for Drug Use and Pharmacogenomics [113], will promote understanding of the relationship between drugs and genes to make drug prescription truly personalized
27.9. Conclusion
AD is a major health problem that comes with a high cost to society. As a clinical entity, AD is a polygenic/complex disorder in which many different gene clusters may be involved. Most genes screened to date belong to different proteomic and metabolomic pathways that potentially affect AD pathogenesis, represented by accumulation of Aβ deposits in senile plaques, intracellular NFTs with hyperphosphorylated tau, and neuronal loss.
The presence of the APOE-4 allele of the apolipoprotein E gene seems to be a major risk factor for both degenerative and vascular dementia, and APOE variants are directly involved in AD pathogenesis at multiple levels. Specific biomarkers (structural and functional genomic markers, proteomic markers in body fluids, neuroimaging markers) are needed for an accurate AD diagnosis. Current pharmacological treatment of AD with cholinesterase inhibitors (donepezil, rivastigmine, galantamine) and memantine is not cost-effective; moreover, the overuse of psychotropic drugs in patients with dementia contributes to deteriorating cognitive and psychomotor functions.
Old treatments addressed memory impairment. New treatments are oriented to halting disease progression by interfering with Aβ accumulation, NFT formation, oxidative stress, neuroinflammation, and cerebrovascular damage. Over the past few years diverse candidate drugs have been investigated in AD models, but not one has reached the market. Since only 25–30% of the population is an extensive metabolizer for drugs metabolized via CYP2D6, CYP2C9, and CYP2C19 enzymes, it seems reasonable to use pharmacogenomic procedures as a way to optimize AD therapeutics, thus reducing ADRs and unnecessary costs. The therapeutic response to conventional drugs in patients with AD is genotype-specific, with CYP2D6-PMs, CYP2D6-UMs, and APOE-4/4 carriers shown to be the worst responders. APOE and CYP2D6 may cooperate, as pleiotropic genes, in the metabolism of drugs and hepatic function.
If we know the pharmacogenomic profiles of patients who require treatment with antidementia drugs and/or psychotropic drugs currently in use, we may be able to achieve the following benefits:
-
•
Identifying candidate patients with the ideal genomic profile to receive a particular drug
-
•
Adapting the dose in more than 90% of cases according to the condition of EM, IM, PM, or UM, which will limit the occurrence of direct side effects in 30–50% of cases
-
•
Reducing drug interactions by 30–50% (avoiding the administration of inhibitors or inducers able to modify the normal enzymatic activity on a particular substrate)
-
•
Enhancing efficacy
-
•
Eliminating unnecessary costs (>30% of pharmaceutical direct costs) deriving from the consequences of inappropriate drug selection and overmedication to mitigate ADRs [18]
Appendix A
Selected Genes Potentially Associated with Alzheimer’s Disease
| Locus | Size (Kb) | Symbol | Title/Gene | OMIM | Other Related Diseases |
|---|---|---|---|---|---|
| 1p13.1 | 52.32 | NGF | Nerve growth factor (beta polypeptide) | 162030 | Hereditary sensory and autonomic neuropathy type V, allergic rhinitis |
| 1p13.3 | 5.95 | GSTM1 | Glutathione S-transferase mu 1 | 138350 | Cancer |
| 1p13.3 | 7.11 | GSTM3 | Glutathione S-transferase mu 3 (brain) | 138390 | Cancer |
| 1p13.3 | 20.38 | CSF1 | Colony-stimulating factor 1 (macrophage) | 120420 | |
| 1p13-p12 | 20.94 | HMGCS2 | 3-hydroxy-3-methylglutaryl-CoA synthase 2 (mitochondrial) | 600234 | HMG-CoA synthase-2 deficiency |
| 1p21 | 232.03 | COL11A1 | Collagen, type XI, alpha 1 | 120280 | Fibrochondrogenesis, Marshall syndrome, Stickler syndrome type II, lumbar disc herniation |
| 1p21.3-p13.1 | 88.38 | SORT1 | Sortilin 1 | 602458 | |
| 1p22.2 | 18.49 | GBP2 | Guanylate binding protein 2, interferon-inducible | 600412 | |
| 1p31.3 | 44.38 | TM2D1 | TM2-domain containing 1 | 610080 | |
| 1p32 | 134.20 | ERI3 | ERI1 exoribonuclease family member 3 | 609917 | |
| 1p32.3 | 37.62 | DHCR24 | 24-dehydrocholesterol reductase | 606418 | Desmosterolosis |
| 1p32.3 | 204.31 | ZFYVE9 | Zinc finger, FYVE-domain containing 9 | 603755 | |
| 1p34 | 85.79 | LRP8 | Low-density lipoprotein receptor-related protein 8, apolipoprotein e receptor | 602600 | Myocardial infarction, major depressive disorder |
| 1p34.3 | 34.93 | LCK | Lymphocyte-specific protein tyrosine kinase | 153390 | SCID due to LCK deficiency |
| 1p36.1 | 128.30 | ECE1 | Endothelin-converting enzyme 1 | 600423 | Hirschsprung disease, cardiac defects, autonomic dysfunction, essential hypertension |
| 1p36.13-q31.3 | 3.81 | APH1A | APH1A gamma secretase subunit provided | 607629 | |
| 1p36.1-p34 | 115.01 | HSPG2 | Heparan sulfate proteoglycan 2 | 142461 | Dyssegmental dysplasia Silverman-Handmaker type, Schwartz-Jampel syndrome type 1, tardive dyskinesia |
| 1p36.22 | 12.87 | TARDBP | TAR DNA binding protein | 605078 | Amyotrophic lateral sclerosis 10 with or without FTD, frontotemporal lobar degeneration TARDBP-related |
| 1p36.3 | 83.64 | TP73 | Tumor protein p73 | 601990 | Neuroblastoma |
| 1p36.3 | 20.37 | MTHFR | Methylenetetrahydrofolate reductase (NAD(P)H) | 607093 | Homocystinuria due to MTHFR deficiency, neural tube defects, schizophrenia, thromboembolism, occlusive vascular disease, colon cancer, acute leukemia |
| 1p36-p35 | 14.28 | HTR6 | 5-Hydroxytryptamine (serotonin) receptor 6, G protein-coupled | 601109 | |
| 1q21 | 35.76 | CTSS | Cathepsin S | 116845 | |
| 1q21 | N/A | AD13 | Alzheimer disease 13 | 611152 | |
| 1q21 | 3.64 | S100A1 | S100 calcium binding protein A1 | 176940 | Cardiomyopathies |
| 1q21.3 | 11.55 | FAM63A | Family with sequence similarity 63, member A | N/A | |
| 1q21.3 | 12.10 | CHRNB2 | Cholinergic receptor, nicotinic, beta 2 (neuronal) | 118507 | Nocturnal frontal lobe epilepsy 3 |
| 1q21-q22 | 66.10 | NTRK1 | Neurotrophic tyrosine kinase, receptor, type 1 | 191315 | Insensitivity to pain with anhidrosis, medullary thyroid carcinoma, self-mutilating behavior, mental retardation |
| 1q21-q23 | 1.05 | APCS | Amyloid P component, serum | 104770 | Secondary amyloidosis |
| 1q22 | 57.51 | LMNA | Lamin A/C | 150330 | Emery-Dreifuss muscular dystrophy 2, Emery-Dreifuss muscular dystrophy 3, familial partial lipodystrophy 2, muscular dystrophy, limb girdle muscular dystrophy type 1B, dilated cardiomyopathy 1A, Charcot-Marie-Tooth disease type 2B1, Hutchinson-Gilford progeria syndrome, heart-hand syndrome of Slovenian type, Malouf syndrome, mandibuloacral dysplasia, lethal restrictive dermopathy |
| 1q22 | 11.92 | FDPS | Farnesyl diphosphate synthase | 134629 | |
| 1q22-q23 | 15.68 | NCSTN | Nicastrin | 605254 | Acne inversa 1 |
| 1q22-q23 | 6.72 | USF1 | Upstream transcription factor 1 | 191523 | Hyperlipidemia |
| 1q23 | 3.95 | FCER1G | Fc fragment of IgE, high-affinity I, receptor forgamma polypeptide | 147139 | |
| 1q23.2 | 2.30 | CRP | C-reactive protein, pentraxin-related provided | 123260 | |
| 1q24.2 | 206.52 | POU2F1 | POU class 2 homeobox 1 | 164175 | |
| 1q25 | 64.97 | SOAT1 | Sterol O-acyltransferase 1 | 102642 | |
| 1q25 | N/A | AD14 | Alzheimer disease 14 | 611154 | |
| 1q25.2-q25.3 | 8.62 | PTGS2 | Prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase) | 600262 | |
| 1q31-q32 | 4.89 | IL10 | Interleukin 10 | 124092 | Rheumatoid arthritis |
| 1q31-q42 | 25.53 | PSEN2 | Presenilin 2 (Alzheimer disease 4) | 600759 | Dilated cardiomyopathy 1V |
| 1q32 | 95.63 | CFH | Complement factor H | 134370 | Hemolytic-uremic syndrome, chronic hypocomplementemic nephropathy, basal laminar drusen, complement factor H deficiency, macular degeneration 4 |
| 1q32 | 145.64 | CR1 | Complement component (3b/4b) receptor 1 (Knops blood group) | 120620 | CR1 deficiency, systemic lupus erythematosus |
| 1q32-q41 | 48.77 | HSD11B1 | Hydroxysteroid (11-beta) dehydrogenase 1 | 600713 | Cortisone reductase deficiency 2, obesity, insulin resistance |
| 1q41-q42 | 47.41 | PARP1 | Poly (ADP-ribose) polymerase 1 | 173870 | Xeroderma pigmentosum, Fanconi anemia, diabetes type I |
| 1q42.2 | 12.07 | AGT | Angiotensinogen (serpin peptidase inhibitor, clade A, member 8) | 106150 | Renal tubular dysgenesis, non-familial structural atrial fibrillation, inflammatory bowel disease, essential hypertension, preeclampsia |
| 1q43 | 108.70 | MTR | 5-methyltetrahydrofolate-homocysteine methyltransferase | 156570 | Methylcobalamin deficiency type cblG, neural tube defects |
| 2p12-p11.1 | 1140 | CTNNA2 | Catenin (cadherin-associated protein), alpha 2 | 114025 | |
| 2p16.3 | 78.41 | RTN4 | Reticulon 4 | 604475 | |
| 2p21 | 68.97 | LHCGR | Luteinizing hormone/choriogonadotropin receptor | 152790 | Leydig cell adenoma with precocious puberty, Leydig cell hypoplasia with hypergonadotropic hypogonadism, Leydig cell hypoplasia with pseudohermaphroditism, female luteinizing hormone resistance, male precocious puberty |
| 2p22-p21 | 51.91 | EIF2AK2 | Eukaryotic translation initiation factor 2-alpha kinase 2 | 176871 | |
| 2p25 | 66.53 | ADAM17 | ADAM metallopeptidase domain 17 | 603639 | Neonatal inflammatory skin and bowel disease |
| 2q14 | 11.48 | IL1A | Interleukin 1, alpha | 147760 | Rheumatoid arthritis |
| 2q14 | 7.02 | IL1B | Interleukin 1, beta | 147720 | |
| 2q14 | 59.31 | BIN1 | Bridging integrator 1 | 601248 | Centronuclear myopathy |
| 2q14.2 | 16.12 | IL1RN | Interleukin 1 receptor antagonist | 147679 | Interleukin 1 receptor antagonist deficiency, microvascular complications of diabetes 4 |
| 2q21.1 | 88.75 | KCNIP3 | Kv channel interacting protein 3, calsenilin | 604662 | |
| 2q21.2 | 1900 | LRP1B | Low-density lipoprotein receptor-related protein 1B | 608766 | |
| 2q24-q31 | 235.50 | LRP2 | Low-density lipoprotein receptor-related protein 2 | 600073 | Donnai-Barrow syndrome, facio-oculoacousticorenal syndrome |
| 2q34 | 75.67 | CREB1 | cAMP responsive element binding protein 1 | 123810 | Angiomatoid fibrous histiocytoma |
| 3p21.31 | 7.18 | CCR2 | Chemokine (C-C motif) receptor 2 | 601267 | |
| 3p21.31 | 6.07 | CCR5 | Chemokine (C-C motif) receptor 5 (gene/pseudogene) | 601373 | Insulin-dependent diabetes mellitus 22 |
| 3p25 | 146.51 | PPARG | Peroxisome proliferator-activated receptor gamma | 601487 | Carotid intimal medial thickness 1, insulin resistance, lipodystrophy 3, obesity, diabetes type 2, cancer |
| 3p26.2 | 16.73 | OGG1 | 8-oxoguanine DNA glycosylase | 601982 | Renal cell carcinoma |
| 3q13.3 | 272.47 | GSK3B | Glycogen synthase kinase 3 beta | 605004 | Parkinson disease |
| 3q21.3 | 88.66 | RAB7A | RAB7A, member RAS oncogene family | 602298 | Charcot-Marie-Tooth disease type 2B |
| 3q22.1 | 32.87 | TF | Transferrin | 190000 | Atransferrinemia |
| 3q22-q24 | N/A | AD15 | Alzheimer disease 15 | 611155 | |
| 3q25.2 | 104.08 | MME | Membrane metallo-endopeptidase | 120520 | Membranous glomerulonephritis, neutral endopeptidase deficiency |
| 3q26.1-q26.2 | 64.56 | BCHE | Butyrylcholinesterase | 177400 | |
| 3q26.2-qter | 15.50 | APOD | Apolipoprotein D | 107740 | |
| 3q27 | 8.26 | AHSG | Alpha-2-HS-glycoprotein | 138680 | |
| 3q28 | 1.51 | SST | Somatostatin | 182450 | |
| 4p13 | 404.59 | APBB2 | Amyloid beta (A4) precursor protein binding, family B, member 2 | 602710 | |
| 4p14 | 11.55 | UCHL1 | Ubiquitin carboxyl-terminal esterase L1 (ubiquitin thiolesterase) | 191342 | Parkinson disease 5 |
| 4p14-p13 | 78.93 | RFC1 | Replication factor C (activator 1) 1, 145kDa | 102579 | |
| 4p16.1 | 550.19 | SORCS2 | Sortilin-related VPS10 domain-containing receptor 2 | 606284 | |
| 4p16.3 | 28.90 | LRPAP1 | Low-density lipoprotein receptor-related protein-associated protein 1 | 104225 | |
| 4q13.3 | 17.16 | ALB | Albumin | 103600 | Analbuminemia, dysalbuminemic hyperthyroxinemia, dysalbuminemic hyperzincemia |
| 4q13-q21 | 3.21 | IL8 | Interleukin 8 | 146930 | Bronchiolitis |
| 4q21 | 114.20 | SNCA | Synuclein, alpha (non-A4 component of amyloid precursor) | 163890 | Lewy body dementia, Parkinson disease 1, Parkinson disease 4 |
| 4q25 | 491.92 | COL25A1 | Collagen, type XXV, alpha 1 | 610004 | |
| 4q25 | 14.85 | CASP6 | Caspase 6, apoptosis-related cysteine peptidase | 601532 | |
| 4q27 | 29.00 | ANXA5 | Annexin A5 | 131230 | Recurrent pregnancy loss |
| 4q32 | 21.80 | TLR2 | Toll-like receptor 2 | 603028 | Colorectal cancer |
| 4q32.1 | 9.11 | LRAT | Lecithin retinol acyltransferase (phosphatidylcholine-retinol O-acyltransferase) | 604863 | Leber congenital amaurosis 14, retinal dystrophy, retinitis pigmentosa |
| 5p15.3 | 52.64 | SLC6A3 | Solute carrier family 6 (neurotransmitter transporter, dopamine), member 3 | 126455 | Epilepsy, parkinsonism-dystonia, attention-deficit hyperactivity disorder, Parkinson disease |
| 5q13.1 | 86.07 | PIK3R1 | Phosphoinositide-3-kinase, regulatory subunit 1 (alpha) | 171833 | Agammaglobulinemia 7, insulin resistance |
| 5q13.3-q14 | 24.93 | HMGCR | 3-hydroxy-3-methylglutaryl-CoA reductase | 142910 | |
| 5q14.1 | 209.33 | ARSB | Arylsulfatase B | 611542 | Mucopolysaccharidosis type VI (Maroteaux-Lamy) |
| 5q15 | 112.65 | CAST | Calpastatin | 114090 | |
| 5q21 | 294.01 | EFNA5 | Ephrin-A5 | 601535 | |
| 5q31 | 105.89 | FGF1 | Fibroblast growth factor 1 (acidic) | 131220 | |
| 5q31 | 6.34 | APBB3 | Amyloid beta (A4) precursor protein-binding, family B, member 3 | 602711 | |
| 5q31.1 | 1.97 | CD14 | CD14 molecule | 158120 | |
| 5q31-q32 | 2.04 | ADRB2 | Adrenoceptor beta 2, surface | 109690 | Nocturnal asthma, obesity, diabetes type 2 |
| 5q32 | 491.97 | PPP2R2B | Protein phosphatase 2, regulatory subunit B, beta | 604325 | Spinocerebellar ataxia 12 |
| 5q34 | 180.24 | WWC1 | WW and C2 domain–containing 1 | 610533 | |
| 5q35.3 | 17.08 | DBN1 | Drebrin 1 | 126660 | |
| 6p12 | 16.28 | VEGFA | Vascular endothelial growth factor A | 192240 | Microvascular complications of diabetes 1 |
| 6p12 | 149.48 | CD2AP | CD2-associated protein | 604241 | Focal segmental glomerulosclerosis 3 |
| 6p21 | 38.96 | GLP1R | Glucagon-like peptide 1 receptor | 138032 | |
| 6p21.1 | 4.68 | TREM2 | Triggering receptor expressed on myeloid cells 2 | 605086 | Nasu-Hakola disease |
| 6p21.3 | 7.96 | HFE | Hemochromatosis | 613609 | Hemochromatosis, microvascular complications of diabetes 7, porphyria cutanea tarda, porphyria variegata |
| 6p21.3 | 4.31 | UBD | Ubiquitin D | 606050 | |
| 6p21.3 | 3.42 | HLA-A | Major histocompatibility complex, class I, A | 142800 | |
| 6p21.3 | 12.26 | DDX39B | DEAD (Asp-Glu-Ala-Asp) box polypeptide 39B | 142560 | Rheumatoid arthritis |
| 6p21.3 | 2.77 | TNF | Tumor necrosis factor | 191160 | Asthma, vascular dementia, migraine without aura, insulin resistance, cancer |
| 6p21.3 | 2.43 | HSPA1A | Heat shock 70kDa protein 1A | 140550 | |
| 6p21.3 | 16.91 | PPP1R10 | Protein phosphatase 1, regulatory subunit 10 | 603771 | |
| 6p21.3 | 3.36 | AGER | Advanced glycosylation end product-specific receptor | 600214 | Diabetes |
| 6p21.3 | 16.94 | TAP2 | Transporter 2, ATP-binding cassette, subfamily B (MDR/TAP) | 170261 | Bare lymphocyte syndrome type I due to TAP2 deficiency, Wegener-like granulomatosis, ankylosing spondylitis, insulin-dependent diabetes mellitus, celiac disease |
| 6p21.3 | 47.89 | C2 | Complement component 2 | 613927 | C2 deficiency |
| 6p21.3 | 20.62 | C4B | Complement component 4B (Chido blood group) | 120820 | C4B deficiency, systemic lupus erythematosus |
| 6p21.3 | 20.62 | C4A | Complement component 4A (Rodgers blood group) | 120810 | C4A deficiency, systemic lupus erythematosus, type I diabetes mellitus |
| 6p21.3 | 13.05 | MICB | MHC class I polypeptide-related sequence B | 602436 | |
| 6p21.3 | 7.11 | RXRB | Retinoid X receptor, beta | 180246 | |
| 6p21.33 | 11.72 | MICA | MHC class I polypeptide-related sequence A | 600169 | |
| 6p22.1 | 21.01 | PGBD1 | PiggyBac transposable element derived 1 | N/A | |
| 6p23 | 462.38 | ATXN1 | Ataxin 1 | 601556 | Spinocerebellar ataxia 1 |
| 6p25.3-p24.3 | 176.61 | F13A1 | Coagulation factor XIII, A1 polypeptide | 134570 | Factor XIIIA deficiency |
| 6p25-p24 | 199.05 | NEDD9 | Neural precursor cell expressed, developmentally downregulated 9 | 602265 | Cancer metastasis |
| 6q21 | 213.12 | FYN | FYN oncogene related to SRC, FGR, YES | 137025 | |
| 6q21 | 49.75 | SNX3 | Sorting nexin 3 | 605930 | |
| 6q25.1 | 412.78 | ESR1 | Estrogen receptor 1 | 133430 | Breast cancer, atherosclerosis, migraine, myocardial infarction, endometrial cancer, osteoporosis |
| 6q25.3 | 14.21 | SOD2 | Superoxide dismutase 2, mitochondrial | 147460 | Microvascular complications of diabetes 6, cardiomyopathy, premature aging, sporadic motor neuron disease, cancer |
| 6q27 | 18.54 | TBP | TATA box binding protein | 600075 | Spinocerebellar ataxia 17, Parkinson disease |
| 7p15.1 | 7.68 | NPY | Neuropeptide Y | 162640 | Elevated cholesterol levels, higher alcohol consumption, metabolic diseases, cardiovascular diseases |
| 7p21 | 4.86 | IL6 | Interleukin 6 (interferon, beta 2) | 147620 | Crohn disease-associated growth failure, diabetes, intracranial hemorrhage in brain cerebrovascular malformations, Kaposi sarcoma, rheumatoid arthritis |
| 7p22 | 8.92 | NUDT1 | Nudix- (nucleoside diphosphate-linked moiety X)-type motif 1 | 600312 | |
| 7q11.2 | 77.09 | CD36 | CD36 molecule (thrombospondin receptor) | 173510 | Platelet glycoprotein IV deficiency, macrothrombocytopenia, coronary heart disease |
| 7q21 | 1440 | MAGI2 | Membrane-associated guanylate kinase, WW and PDZ domain-containing 2 | 606382 | |
| 7q21.12 | 209.46 | ABCB1 | ATP-binding cassette, subfamily B (MDR/TAP), member 1 | 171050 | Inflammatory bowel disease 13 |
| 7q21.3 | 26.22 | PON1 | Paraoxonase 1 | 168820 | Coronary artery disease, coronary artery spasm 2, microvascular complications of diabetes 5 |
| 7q21.3 | 36.50 | PON3 | Paraoxonase 3 | 602720 | |
| 7q21.3 | 30.21 | PON2 | Paraoxonase 2 | 602447 | Coronary artery disease, diabetes |
| 7q22 | 517.73 | RELN | Reelin | 600514 | Lissencephaly 2 (Norman-Roberts type) |
| 7q22.1 | 12.18 | SERPINE1 | Serpin peptidase inhibitor, clade E (nexin, plasminogen activator inhibitor type 1), member 1 | 173360 | Plasminogen activator inhibitor-1 deficiency, thrombophilia |
| 7q31.1 | 42.20 | PPP1R3A | Protein phosphatase 1, regulatory subunit 3A | 600917 | Insulin resistance |
| 7q31.1 | 36.40 | CAV1 | Caveolin 1, caveolae protein, 22kDa | 601047 | Lipodystrophy type 3 |
| 7q31-q32 | 30.06 | DLD | Dihydrolipoamide dehydrogenase | 238331 | Dihydrolipoamide dehydrogenase deficiency, maple syrup urine disease |
| 7q34 | 17.78 | EPHA1 | EPH receptor A1 | 179610 | Cancer |
| 7q36 | 23.54 | NOS3 | Nitric oxide synthase 3 (endothelial cell) | 163729 | Coronary artery spasm 1, hypertension, ischemic stroke, placental abruption |
| 7q36 | 4.15 | CDK5 | Cyclin-dependent kinase 5 | 123831 | |
| 7q36 | 59.28 | PAXIP1 | PAX-interacting (with transcription, activation domain) protein 1 | 608254 | |
| 7q36 | N/A | AD10 | Alzheimer disease-10 | 609636 | |
| 8p11.2 | 8.38 | STAR | Steroidogenic acute regulatory protein | 600617 | Lipoid adrenal hyperplasia |
| 8p11.22 | 108.28 | ADAM9 | ADAM metallopeptidase domain 9 | 602713 | Cone-rod dystrophy 9 |
| 8p12 | 3.67 | ADRB3 | Adrenoceptor beta 3 | 109691 | Obesity |
| 8p12 | 32.96 | PLAT | Plasminogen activator, tissue | 173370 | Hyperfibrinolysis, thrombophilia |
| 8p12 | 29.86 | EIF4EBP1 | Eukaryotic translation initiation factor 4E binding protein 1 | 602223 | |
| 8p12-q22 | N/A | AD12 | Alzheimer disease 12 | 611073 | |
| 8p21-p12 | 17.90 | CLU | Clusterin | 185430 | Neoplasms |
| 8p22 | 9.97 | NAT2 | N-acetyltransferase 2 (arylamine N-acetyltransferase) | 612182 | Cancer |
| 8p22 | 28.19 | LPL | Lipoprotein lipase | 609708 | Hyperlipidemia, lipoprotein lipase deficiency |
| 8p22 | 25.61 | CTSB | Cathepsin B | 116810 | Esophageal adenocarcinoma, neoplasms |
| 8q13 | 2.24 | CRH | Corticotropin releasing hormone | 122560 | |
| 8q22 | 87.63 | DPYS | Dihydropyrimidinase | 613326 | Dihydropyrimidinuria |
| 8q24.1 | 81.79 | ENPP2 | Ectonucleotide pyrophosphatase/phosphodiesterase 2 | 601060 | |
| 9p13.3 | 3.05 | SIGMAR1 | Sigma nonopioid intracellular receptor 1 | 601978 | Amyotrophic lateral sclerosis 16 |
| 9p13.3 | 16.68 | VCP | Valosin-containing protein | 601023 | Amyotrophic lateral sclerosis 14 with or without frontotemporal dementia, inclusion body myopathy with early-onset Paget disease and frontotemporal dementia |
| 9p21 | 26.74 | CDKN2A | Cyclin-dependent kinase inhibitor 2A | 600160 | Melanoma and neural system tumor syndrome, orolaryngeal cancer, pancreatic cancer, cutaneous malignant melanoma 2 |
| 9p21.3 | 126.31 | CDKN2B-AS1 | CDKN2B antisense RNA 1 | 613149 | Cardiovascular diseases, cancer, intracranial aneurysm, type-2 diabetes, periodontitis, endometriosis, frailty in the elderly, glaucoma |
| 9p24 | 32.69 | VLDLR | Very low-density lipoprotein receptor | 192977 | Cerebellar hypoplasia and mental retardation with or without quadrupedal locomotion 1 |
| 9p24.1 | 42.20 | IL33 | Interleukin 33 | 608678 | |
| 9q13-q21.1 | 244.83 | APBA1 | Amyloid beta (A4) precursor protein-binding, family A, member 1 | 602414 | |
| 9q21.2 | 294.71 | PRUNE2 | Prune homolog 2 (Drosophila) | 610691 | |
| 9q21.2-q21.3 | 48.29 | UBQLN1 | Ubiquilin 1 | 605046 | Parkinson disease |
| 9q21.33 | 210.79 | DAPK1 | Death-associated protein kinase 1 | 600831 | |
| 9q21.33 | 74.06 | GOLM1 | Golgi membrane protein 1 | 606804 | |
| 9q22.1 | 355.04 | NTRK2 | Neurotrophic tyrosine kinase, receptor, type 2 | 600456 | Obesity, mood disorders |
| 9q22.1 | N/A | AD11 | Alzheimer disease 11 | 609790 | |
| 9q31.1 | 169.23 | GRIN3A | Glutamate receptor, ionotropic, N-methyl-D-aspartate 3A | 606650 | |
| 9q31.1 | 147.25 | ABCA1 | ATP-binding cassette, subfamily A (ABC1), member 1 | 600046 | HDL deficiency type 2, Tangier disease, coronary artery disease |
| 9q33.1 | 13.32 | TLR4 | Toll-like receptor 4 | 603030 | Colorectal cancer, macular degeneration |
| 9q33.3 | 6.54 | HSPA5 | Heat shock 70kDa protein 5 (glucose-regulated protein, 78kDa) | 138120 | |
| 9q34 | 22.98 | DBH | Dopamine beta-hydroxylase (dopamine beta-monooxygenase) | 609312 | Dopamine beta-hydroxylase deficiency |
| 9q34 | 40.10 | TRAF2 | TNF receptor-associated factor 2 | 601895 | |
| 9q34 | 21.69 | ABCA2 | ATP-binding cassette, subfamily A (ABC1), member 2 | 600047 | |
| 9q34.3 | 114.12 | RXRA | Retinoid X receptor, alpha | 180245 | |
| 10 | 51.74 | ENTPD7 | Ectonucleoside triphosphate diphosphohydrolase 7 | N/A | |
| 10p12 | 401.08 | CACNB2 | Calcium channel, voltage-dependent, beta 2 subunit | 600003 | Brugada syndrome 4 |
| 10p12.31 | 161.34 | C10orf112 | Chromosome 10 open reading frame 112 | N/A | |
| 10p13 | 38.20 | OPTN | Optineurin | 602432 | Amyotrophic lateral sclerosis 12, glaucoma |
| 10p13 | N/A | AD7 | Alzheimer disease 7 | 606187 | |
| 10p14-p13 | 27.42 | PTPLA | Protein tyrosine phosphatase-like (proline instead of catalytic arginine), member A | 610467 | |
| 10p15.2 | 35.12 | PITRM1 | Pitrilysin metallopeptidase 1 | N/A | |
| 10q | N/A | AD6 | Alzheimer disease 6 | 605526 | |
| 10q11.2 | 71.94 | ALOX5 | Arachidonate 5-lipoxygenase | 152390 | Atherosclerosis, cancer |
| 10q11.2 | 56.01 | CHAT | Choline O-acetyltransferase | 118490 | Myasthenic syndrome associated with episodic apnea |
| 10q11.2 | 3.38 | DKK1 | Dickkopf WNT signaling pathway inhibitor 1 | 605189 | |
| 10q21 | 14.09 | TFAM | Transcription factor A, mitochondrial | 600438 | |
| 10q21 | 707.23 | ANK3 | Ankyrin 3, node of Ranvier (ankyrin G) | 600465 | |
| 10q21 | 134.12 | TET1 | Tet methylcytosine dioxygenase 1 | 607790 | |
| 10q21 | 65.24 | SGPL1 | Sphingosine-1-phosphate lyase 1 | 603729 | |
| 10q21.1 | 16.52 | CDK1 | Cyclin-dependent kinase 1 | 116940 | |
| 10q21.3 | 175.08 | LRRTM3 | Leucine-rich repeat transmembrane neuronal 3 | 610869 | |
| 10q21.3 | 33.72 | SIRT1 | Sirtuin 1 | 604479 | |
| 10q22.2 | 1780 | CTNNA3 | Catenin (cadherin-associated protein), alpha 3 | 607667 | |
| 10q22.2 | 6.40 | PLAU | Plasminogen activator, urokinase | 191840 | Quebec platelet disorder |
| 10q22.3 | 768.22 | KCNMA1 | Potassium large conductance calcium-activated channel, subfamily M, alpha member 1 | 600150 | Generalized epilepsy and paroxysmal dyskinesia |
| 10q23 | 1.38 | CH25H | Cholesterol 25-hydroxylase | 604551 | |
| 10q23.2-q23.3 | 38.34 | LIPA | Lipase A, lysosomal acid, cholesterol esterase | 613497 | Cholesteryl ester storage disease, Wolman disease |
| 10q23.3 | 105.34 | PTEN | Phosphatase and tensin homolog | 601728 | Bannayan-Riley-Ruvalcaba syndrome, Cowden syndrome 1, endometrial carcinoma, Lhermitte-Duclos syndrome, macrocephaly/autism syndrome, malignant melanoma, PTEN hamartoma tumor syndrome, squamous cell carcinoma, thyroid carcinoma, VATER association with macrocephaly and ventriculomegaly, glioma, meningioma, prostate cancer |
| 10q23.32 | 104.42 | HECTD2 | HECT domain-containing E3 ubiquitin protein ligase 2 | N/A | |
| 10q23.33 | 5.73 | HHEX | Hematopoietically expressed homeobox | 604420 | |
| 10q23-q25 | 122.41 | IDE | Insulin-degrading enzyme | 146680 | Type 2 diabetes mellitus |
| 10q23-q25 | 624.13 | SORCS3 | Sortilin-related VPS10 domain-containing receptor 3 | 606285 | |
| 10q23-q25 | 591.05 | SORCS1 | Sortilin-related VPS10 domain-containing receptor 1 | 606283 | |
| 10q24 | 23.92 | COX15 | Cytochrome c oxidase assembly homolog 15 (yeast) | 603646 | Cardioencephalomyopathy due to cytochrome c oxidase deficiency 2, Leigh syndrome due to cytochrome c oxidase deficiency |
| 10q24 | 69.20 | ABCC2 | ATP-binding cassette, subfamily C (CFTR/MRP), member 2 | 601107 | Dubin-Johnson syndrome |
| 10q24.1 | 25.25 | FAS | Fas cell surface death receptor | 134637 | Autoimmune lymphoproliferative syndrome type IA, squamous cell carcinoma, autoimmune lymphoproliferative syndrome |
| 10q24.2 | 134.34 | DNMBP | Dynamin binding protein | 611282 | |
| 10q24.3 | 50.88 | ALDH18A1 | Aldehyde dehydrogenase 18 family, member A1 | 138250 | Cutis laxa type IIIA, hyperammonemia, hypoornithinemia, hypocitrullinemia, hypoargininemia, hypoprolinemia, cataracts, connective tissue diseases |
| 10q24.3 | 7.00 | CYP17A1 | Cytochrome P450, family 17, subfamily A, polypeptide 1 | 609300 | 17,20-lyase deficiency, 17-alpha-hydroxylase/17,20-lyase deficiency, pseudohermaphroditism, adrenal hyperplasia |
| 10q24.31 | 17.82 | SCD | Stearoyl-CoA desaturase (delta-9-desaturase) | 604031 | |
| 10q24.33 | 261.38 | SH3PXD2A | SH3 and PX domains 2A | N/A | |
| 10q24.33 | 5.50 | CALHM1 | Calcium homeostasis modulator 1 | 612234 | |
| 10q25.1 | 13.27 | GSTO1 | Glutathione S-transferase omega 1 | 605482 | |
| 10q25.1 | 30.55 | GSTO2 | Glutathione S-transferase omega 2 | 612314 | |
| 10q25.3 | 2.86 | ADRB1 | Adrenoceptor beta 1 | 109630 | Heart failure |
| 10q26.3 | 128.60 | EBF3 | Early B-cell factor 3 | 607407 | Glioblastoma multiforme, gastric carcinoma |
| 10q26.3 | 53.38 | HTRA1 | HtrA serine peptidase 1 | 602194 | CARASIL syndrome, macular degeneration |
| 10q26.3 | 374.23 | ADAM12 | ADAM metallopeptidase domain 12 | 602714 | |
| 11p11.2 | 20.97 | MAPK8IP1 | Mitogen-activated protein kinase 8 interacting protein 1 | 604641 | Noninsulin-dependent diabetes mellitus |
| 11p13 | 67.17 | BDNF | Brain-derived neurotrophic factor | 113505 | Central hypoventilation syndrome, anorexia nervosa, bulimia nervosa, memory impairment, obsessive-compulsive disorder |
| 11p15 | 24.29 | APBB1 | Amyloid beta (A4) precursor protein-binding, family B, member 1 (Fe65) | 602709 | |
| 11p15.1 | 3.72 | SAA1 | Serum amyloid A1 | 104750 | Atherosclerosis, rheumatoid arthritis, Crohn’s disease, neoplasms |
| 11p15.5 | 3.40 | DRD4 | Dopamine receptor D4 | 126452 | Autonomic nervous system dysfunction, novelty-seeking personality, attention-deficit hyperactivity disorder |
| 11p15.5 | 11.24 | CTSD | Cathepsin D | 116840 | Breast cancer, neuronal ceroid lipofuscinosis type 10 |
| 11p15.5 | 1.43 | INS | Insulin | 176730 | Insulin-dependent diabetes mellitus type 2, permanent neonatal diabetes mellitus, diabetes mellitus type 1, familial hyperproinsulinemia with or without diabetes |
| 11p15.5 | 1.59 | HBG2 | Hemoglobin, gamma G | 142250 | Transient neonatal cyanosis |
| 11q12.1 | 13.06 | MS4A6A | Membrane-spanning 4-domains, subfamily A, member 6A | 606548 | |
| 11q12.2 | 41.84 | MS4A4E | Membrane-spanning 4-domains, subfamily A, member 4E | 608401 | |
| 11q13 | 3.06 | GSTP1 | Glutathione S-transferase pi 1 | 134660 | Cancer |
| 11q13 | 14.31 | INPPL1 | Inositol polyphosphate phosphatase-like 1 | 600829 | Opsismodysplasia, breast cancer |
| 11q13.3 | 6.66 | GAL | Galanin/GMAP prepropeptide | 137035 | |
| 11q14 | 112.71 | PICALM | Phosphatidylinositol-binding clathrin assembly protein | 603025 | Acute myeloid leukemia, T-cell acute lymphoblastic leukemia, malignant lymphomas |
| 11q14.1 | 202.53 | GAB2 | GRB2-associated binding protein 2 | 606203 | |
| 11q22.2-q22.3 | 25.73 | CASP4 | Caspase 4, apoptosis-related cysteine peptidase | 602664 | |
| 11q22.2-q22.3 | 20.87 | IL18 | Interleukin 18 (interferon-gamma-inducing factor) | 600953 | |
| 11q22.3 | 8.33 | MMP1 | Matrix metallopeptidase 1 (interstitial collagenase) | 120353 | Epidermolysis bullosa dystrophica, arthritis, chronic obstructive pulmonary disease |
| 11q22.3 | 7.82 | MMP3 | Matrix metallopeptidase 3 (stromelysin 1, progelatinase) | 185250 | Coronary heart disease, arthritis |
| 11q23 | 3.05 | APOA5 | Apolipoprotein A-V | 606368 | Hyperchylomicronemia, hypertriglyceridemia, hyperlipoproteinemia type V |
| 11q23 | 2.59 | APOA4 | Apolipoprotein A-IV | 107690 | |
| 11q23.2-q23.3 | 30.57 | BACE1 | Beta-site APP-cleaving enzyme 1 | 604252 | |
| 11q23.2-q24.2 | 181.56 | SORL1 | Sortilin-related receptor, L(DLR class) A repeats containing | 602005 | |
| 11q23.3 | 3.16 | APOC3 | Apolipoprotein C-III | 107720 | Hyperalphalipoproteinemia 2, hypertriglyceridemia |
| 11q23-q24 | 1.87 | APOA1 | Apolipoprotein A-I | 107680 | Amyloidosis, combined ApoA-I and apoC-III deficiency, corneal clouding, hypoalphalipoproteinemia, Tangier disease, systemic non-neuropathic amyloidosis |
| 11q24 | 74.99 | APLP2 | Amyloid beta (A4) precursor-like protein 2 | 104776 | |
| 11q25 | 12.32 | ACAD8 | Acyl-CoA dehydrogenase family, member 8 | 604773 | Isobutyryl-CoA dehydrogenase deficiency |
| 12p11.23-q13.12 | N/A | AD5 | Alzheimer disease 5 | 602096 | |
| 12p12 | 418.61 | GRIN2B | Glutamate receptor, ionotropic, N-methyl D-aspartate 2B | 138252 | Mental retardation |
| 12p12.1 | 7.11 | IAPP | Islet amyloid polypeptide | 147940 | Diabetes type 2 |
| 12p13 | 4.55 | PKP2P1 | Plakophilin 2 pseudogene 1 | 602861 | Arrhythmogenic right ventricular dysplasia 9 |
| 12p13 | 3.95 | GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | 138400 | |
| 12p13 | 7.18 | GNB3 | Guanine nucleotide binding protein (G protein), beta polypeptide 3 | 139130 | Essential hypertension, obesity |
| 12p13.2 | 150.85 | LRP6 | Low-density lipoprotein receptor-related protein 6 | 603507 | Coronary artery disease |
| 12p13.2-p12.3 | 13.89 | OLR1 | Oxidized low density lipoprotein (lectin-like) receptor 1 | 602601 | Myocardial infarction, atherosclerosis |
| 12p13.3 | 37.84 | NCAPD2 | Non-SMC condensin I complex, subunit D2 | N/A | |
| 12p13.31 | 10.31 | TAPBPL | TAP binding protein-like | 607081 | |
| 12p13.31 | 48.26 | A2M | Alpha-2-macroglobulin | 103950 | Alpha-2-macroglobulin deficiency |
| 12q12 | 144 | LRRK2 | Leucine-rich repeat kinase 2 | 609007 | Parkinson disease 8 |
| 12q13 | 79.39 | TFCP2 | Transcription factor CP2 | 189889 | |
| 12q13 | 118.56 | ATF7 | Activating transcription factor 7 | 606371 | |
| 12q13.11 | 63.49 | VDR | Vitamin D (1,25-dihydroxyvitamin D3) receptor | 601769 | Involutional osteoporosis |
| 12q13.11 | 29.04 | KANSL2 | KAT8 regulatory NSL complex subunit 2 | N/A | |
| 12q13.11 | 28.54 | CCNT1 | Cyclin T1 | 143055 | Neoplasms |
| 12q13.3 | 84.86 | LRP1 | Low-density lipoprotein receptor-related protein 1 | 107770 | |
| 12q14 | 45.33 | CAND1 | Cullin-associated and neddylation-dissociated 1 | 607727 | |
| 12q23-q24.1 | 5.68 | PLA2G1B | Phospholipase A2, group IB (pancreas) | 172410 | |
| 12q24.11 | 61.42 | KIAA1033 | KIAA1033 | N/A | |
| 12q24.2 | 43.10 | ALDH2 | Aldehyde dehydrogenase 2 family (mitochondrial) | 100650 | Esophageal cancer alcohol-related |
| 12q24.2-q24.31 | 153.66 | NOS1 | Nitric oxide synthase 1 | 163731 | Stroke |
| 13q22.1 | 18.54 | KLF5 | Kruppel-like factor 5 (intestinal) | 602903 | |
| 13q34 | 25.17 | DAOA | D-amino acid oxidase activator | 607408 | Schizophrenia, bipolar affective disorder |
| 14q13.1 | 8.63 | PNP | Purine nucleoside phosphorylase | 164050 | Immunodeficiency due to purine nucleoside phosphorylase deficiency |
| 14q21 | 114.25 | SOS2 | Son of sevenless homolog 2 (Drosophila) | 601247 | |
| 14q22.1 | 241.61 | FRMD6 | FERM domain-containing 6 | 614555 | |
| 14q23.2 | 111.52 | ESR2 | Estrogen receptor 2 (ER beta) | 601663 | |
| 14q24 | 71.97 | MTHFD1 | Methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 1, methenyltetrahydrofolate cyclohydrolase, formyltetrahydrofolate synthetase | 172460 | Abruptio placentae, spina bifida folate-sensitive |
| 14q24.3 | 87.26 | PSEN1 | Presenilin 1 | 104311 | Familial acne inversa 3, dilated cardiomyopathy 1U, frontotemporal dementia, Pick disease |
| 14q24.3 | 13.44 | NPC2 | Niemann-Pick disease, type C2 | 601015 | Niemann-Pick disease type C2 |
| 14q24.3 | 21.86 | DLST | Dihydrolipoamide S-succinyltransferase (E2 component of 2-oxo-glutarate complex) | 126063 | |
| 14q24.3 | 3.46 | FOS | FBJ murine osteosarcoma viral oncogene homolog | 164810 | |
| 14q24.3 | 5.82 | NGB | Neuroglobin | 605304 | |
| 14q31 | 62.31 | SEL1L | Sel-1 suppressor of lin-12-like (C. elegans) | 602329 | |
| 14q32 | 76.35 | MOK | MOK protein kinase | 605762 | |
| 14q32.1 | 13.95 | SERPINA1 | Serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 1 | 107400 | Emphysema due to AAT deficiency, emphysema-cirrhosis due to AAT deficiency, hemorrhagic diathesis due to antithrombin Pittsburgh, chronic obstructive pulmonary disease |
| 14q32.1 | 11.68 | SERPINA3 | Serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 3 | 107280 | Alpha-1-antichymotrypsin deficiency, occlusive cerebrovascular disease |
| 14q32.1 | 42.88 | CYP46A1 | Cytochrome P450, family 46, subfamily A, polypeptide 1 | 604087 | |
| 14q32.3 | 72.36 | KLC1 | Kinesin light chain 1 | 600025 | |
| 14q32.32 | 26.40 | AKT1 | V-akt murine thymoma viral oncogene homolog 1 | 164730 | Breast cancer, colorectal cancer, Cowden syndrome 6, ovarian cancer, proteus syndrome, schizophrenia |
| 15q11-q12 | 196.68 | APBA2 | Amyloid beta (A4) precursor protein-binding, family A, member 2 | 602712 | |
| 15q13.1 | 32.42 | CHRFAM7A | CHRNA7 (cholinergic receptor, nicotinic, alpha 7, exons 5–10) and FAM7A (family with sequence similarity 7A, exons A–E) fusion | 609756 | |
| 15q14 | 139.70 | CHRNA7 | Cholinergic receptor, nicotinic, alpha 7 (neuronal) | 118511 | Schizophrenia, myoclonic epilepsy |
| 15q21.1 | 130.54 | CYP19A1 | Cytochrome P450, family 19, subfamily A, polypeptide 1 | 107910 | Aromatase deficiency, aromatase excess syndrome |
| 15q21.1 | 2.09 | EID1 | EP300 interacting inhibitor of differentiation 1 | 605894 | |
| 15q21-q23 | 136.90 | LIPC | Lipase, hepatic | 151670 | Hepatic lipase deficiency, noninsulin-dependent diabetes mellitus |
| 15q22 | 153.67 | ADAM10 | ADAM metallopeptidase domain 10 | 602192 | |
| 15q22.2 | 31.58 | APH1B | APH1B gamma secretase subunit | 607630 | |
| 15q22.31 | 48.35 | SNX1 | Sorting nexin 1 | 601272 | |
| 15q22.33 | 129.34 | SMAD3 | SMAD family member 3 | 603109 | |
| 15q24 | 28.24 | CHRNA3 | Cholinergic receptor, nicotinic, alpha 3 (neuronal) | 118503 | Lung cancer |
| 15q24.1 | 21.11 | CSK | C-src tyrosine kinase | 124095 | |
| 15q25.1 | 63.28 | IREB2 | Iron-responsive element binding protein 2 | 147582 | |
| 15q26 | 150.50 | MEF2A | Myocyte enhancer factor 2A | 600660 | Coronary artery disease 1 with myocardial infarction |
| 16p13.3 | 17.87 | UBE2I | Ubiquitin-conjugating enzyme E2I | 601661 | |
| 16p13.3 | 14.60 | MEFV | Mediterranean fever | 608107 | Familial Mediterranean fever |
| 16q12 | 29.56 | VPS35 | Vacuolar protein sorting 35 homolog (S. cerevisiae) | 601501 | Parkinson disease 17 |
| 16q21 | 21.92 | CETP | Cholesteryl ester transfer protein, plasma | 118470 | Hyperalphalipoproteinemia |
| 16q22 | 28.10 | NAE1 | NEDD8-activating enzyme E1 subunit 1 | 603385 | |
| 16q22.1 | 17.23 | NQO1 | NAD(P)H dehydrogenase, quinone 1 | 125860 | Tardive dyskinesia, cancer |
| 17p11.2 | 25.66 | SREBF1 | Sterol regulatory element-binding transcription factor 1 | 184756 | |
| 17p12 | 139.28 | COX10 | Cytochrome c oxidase assembly homolog 10 (yeast) | 602125 | Charcot-Marie-Tooth type 1A, hereditary neuropathy with liability to pressure palsies |
| 17p13 | 72.14 | MYH13 | Myosin, heavy-chain 13, skeletal muscle | 603487 | |
| 17p13.1 | 8.80 | TNK1 | Tyrosine kinase, nonreceptor, 1 | 608076 | |
| 17p13.1 | 19.15 | TP53 | Tumor protein p53 | 191170 | Adrenal cortical carcinoma, breast cancer, choroid plexus papilloma, colorectal cancer, hepatocellular carcinoma, Li-Fraumeni syndrome, nasopharyngeal carcinoma, osteosarcoma, pancreatic cancer, basal-cell carcinoma 7, glioma |
| 17p13.1 | 31.63 | MYH8 | Myosin, heavy-chain 8, skeletal muscle, perinatal | 160741 | Carney complex variant, trismus-pseudocamptodactyly syndrome |
| 17q | 2.22 | PNMT | Phenylethanolamine N-methyltransferase | 171190 | Essential hypertension |
| 17q11.2 | 4.17 | CDK5R1 | Cyclin-dependent kinase 5, regulatory subunit 1 (p35) | 603460 | |
| 17q11.2 | 43.97 | BLMH | Bleomycin hydrolase | 602403 | |
| 17q11.2 | 39.58 | SLC6A4 | Solute carrier family 6 (neurotransmitter transporter, serotonin), member 4 | 182138 | Sudden infant death syndrome, aggressive behavior, depression, obsessive-compulsive disorder |
| 17q11.2 | 31.68 | THRA | Thyroid hormone receptor, alpha | 190120 | Hypothyroidism nongoitrous 6 |
| 17q11.2 | 0,086 | MIR144 | MicroRNA 144 | 612070 | |
| 17q11.2-q12 | 43.76 | NOS2 | Nitric oxide synthase 2, inducible | 163730 | Hypertension |
| 17q11.2-q12 | 1.93 | CCL2 | Chemokine (C-C motif) ligand 2 | 158105 | Psoriasis, rheumatoid arthritis, atherosclerosis, spina bifida |
| 17q12 | 1.91 | CCL3 | Chemokine (C-C motif) ligand 3 | 182283 | Human immunodeficiency virus type 1 |
| 17q21.1 | 133.95 | MAPT | Microtubule-associated protein tau | 157140 | Pick disease, frontotemporal dementia, cortico-basal degeneration, progressive supranuclear palsy, Parkinson disease, tauopathy and respiratory failure |
| 17q21.1 | 0,445 | STH | Saitohin | 607067 | |
| 17q21.32 | 7.98 | GRN | Granulin | 138945 | Frontotemporal lobar degeneration with ubiquitin-positive inclusions, primary progressive aphasia, neuronal ceroid lipofuscinosis 11 |
| 17q21-q22 | N/A | GPSC | Gliosis, familial progressive subcortical | N/A | |
| 17q23.1 | 11.08 | MPO | Myeloperoxidase | 606989 | Myeloperoxidase deficiency |
| 17q23.2 | 83.06 | APPBP2 | Amyloid beta precursor protein (cytoplasmic tail) binding protein 2 | 605324 | |
| 17q23.3 | 21.32 | ACE | Angiotensin I converting enzyme (peptidyl-dipeptidase A) 1 | 106180 | Renal tubular dysgenesis, benign serum increase of angiotensin I-converting enzyme, myocardial infarction, stroke, severe acute respiratory syndrome, microvascular complications of diabetes 3 |
| 17q24.3 | 158.72 | BPTF | Bromodomain PHD finger transcription factor | 601819 | |
| 17q24-q25 | 87.63 | GRB2 | Growth factor receptor-bound protein 2 | 108355 | |
| 18p11.2 | 33.49 | MC2R | Melanocortin 2 receptor (adrenocorticotropic hormone) | 607397 | Glucocorticoid deficiency due to ACTH unresponsiveness |
| 18q12.1 | 33.62 | DSC1 | Desmocollin 1 | 125643 | |
| 18q12.1 | 7.26 | TTR | Transthyretin | 176300 | Amyloidotic polyneuropathy, euthyroid hyperthyroxinaemia, amyloidotic vitreous opacities, cardiomyopathy, oculoleptomeningeal amyloidosis, meningocerebrovascular amyloidosis, carpal tunnel syndrome |
| 19p13 | 14.48 | PIN1 | Peptidylprolyl cis/trans isomerase, NIMA-interacting 1 | 601052 | Cancer |
| 19p13.2 | 113.86 | DNM2 | Dynamin 2 | 602378 | Axonal Charcot-Marie-Tooth disease type 2M, Charcot-Marie-Tooth disease type B, centronuclear myopathy |
| 19p13.2 | 44.47 | LDLR | Low-density lipoprotein receptor | 606945 | Familial hypercholesterolemia |
| 19p13.2 | N/A | AD9 | Alzheimer disease 9 | 608907 | |
| 19p13.2-p13.1 | 41.35 | NOTCH3 | Notch 3 | 600276 | Cerebral arteriopathy with subcortical infarcts and leukoencephalopathy |
| 19p13.3-13.2 | 42.82 | C3 | Complement component 3 | 120700 | C3 deficiency, atypical hemolytic uremic syndrome, macular degeneration age-related 9 |
| 19p13.3 | 27.06 | GNA11 | Guanine nucleotide binding protein (G protein), alpha 11 (Gq class) | 139313 | |
| 19p13.3 | 25.47 | ABCA7 | ATP-binding cassette, subfamily A (ABC1), member 7 | 605414 | |
| 19p13.3 | 10.90 | APBA3 | Amyloid beta (A4) precursor protein-binding, family A, member 3 | 604262 | |
| 19p13.3 | 9.29 | GRIN3B | Glutamate receptor, ionotropic, N-methyl-D-aspartate 3B | 606651 | |
| 19p13.3-p13.2 | 181.75 | INSR | Insulin receptor | 147670 | Diabetes mellitus insulin-resistant with acanthosis nigricans, hyperinsulinemic hypoglycemia 5, leprechaunism, Rabson-Mendenhall syndrome |
| 19p13.3-p13.2 | 15.78 | ICAM1 | Intercellular adhesion molecule 1 | 147840 | |
| 19q13 | 12.47 | TOMM40 | Translocase of outer mitochondrial membrane 40 homolog (yeast) | 608061 | |
| 19q13.1 | 23.02 | TGFB1 | Transforming growth factor, beta 1 | 190180 | Camurati-Engelmann disease |
| 19q13.1 | 11.30 | APLP1 | Amyloid beta (A4) precursor-like protein 1 | 104775 | |
| 19q13.12 | 11.91 | GAPDHS | Glyceraldehyde-3-phosphate dehydrogenase, spermatogenic | 609169 | |
| 19q13.12 | 1.41 | PSENEN | Presenilin enhancer 2 homolog (C. elegans) | 607632 | Acne inversa 2 |
| 19q13.2 | 43.09 | PVRL2 | Poliovirus receptor-related 2 (herpesvirus entry mediator B) | 600798 | Multiple sclerosis |
| 19q13.2 | 3.61 | APOE | Apolipoprotein E | 107741 | Hyperlipoproteinemia type III, lipoprotein glomerulopathy, sea-blue histiocyte disease, macular degeneration, myocardial infarction |
| 19q13.2 | 4.69 | APOC1 | Apolipoprotein C-I | 107710 | |
| 19q13.2 | 3.26 | APOC4 | Apolipoprotein C-IV | 600745 | Coronary artery disease |
| 19q13.2 | 3.58 | APOC2 | Apolipoprotein C-II | 608083 | Hyperlipoproteinemia type Ib |
| 19q13.2 | 12.36 | BCAM | Basal cell adhesion molecule (Lutheran blood group) | 612773 | |
| 19q13.3 | 38.76 | CLPTM1 | Cleft lip and palate-associated trans-membrane protein 1 | 612585 | |
| 19q13.3 | 14.94 | CD33 | CD33 molecule | 159590 | |
| 19q13.3 | 6.61 | NR1H2 | Nuclear receptor subfamily 1, group H, member 2 | 600380 | |
| 19q13.3 | 54.03 | MARK4 | MAP/microtubule affinity-regulating kinase 4 | 606495 | |
| 19q13.32 | 21.59 | EXOC3L2 | Exocyst complex component 3-like 2 | N/A | |
| 19q13.32 | 3.06 | BLOC1S3 | Biogenesis of lysosomal organelles complex1, subunit 3 | 609762 | Hermansky-Pudlak syndrome 8 |
| 19q13.33 | 47.86 | CARD8 | Caspase recruitment domain family, member 8 | 609051 | Rheumatoid arthritis |
| 19q13.43 | 9.76 | GALP | Galanin-like peptide | 611178 | Neuroblastic tumor |
| 20p | N/A | AD8 | Alzheimer disease 8 | 607116 | |
| 20p11.21 | 4.28 | CST3 | Cystatin C | 604312 | Cerebral amyloid angiopathy, macular degeneration 11 |
| 20p13 | 15.44 | PRNP | Prion protein | 176640 | Creutzfeldt-Jakob disease, fatal familial insomnia, Gerstmann-Straussler disease, Huntington disease-like 1, kuru, prion disease |
| 20pter-p12 | 6.55 | PRND | Prion protein 2 (dublet) | 604263 | |
| 20q13.2-q13.3 | 18.09 | CHRNA4 | Cholinergic receptor, nicotinic, alpha 4 (neuronal) | 118504 | Nocturnal frontal lobe epilepsy type 1 |
| 20q13.31 | 5.38 | PCK1 | Phosphoenolpyruvate carboxykinase 1 (soluble) | 614168 | Cytosolic phosphoenolpyruvate carboxykinase deficiency |
| 21q11 | 98.17 | SAMSN1 | SAM domain, SH3 domain, and nuclear localization signals 1 | 607978 | |
| 21q21.1 | 134.54 | TMPRSS15 | Transmembrane protease, serine 15 | 606635 | Enterokinase deficiency |
| 21q21.1 | 541.88 | NCAM2 | Neural cell adhesion molecule 2 | 602040 | |
| 21q21.3 | 290.59 | APP | Amyloid beta (A4) precursor protein | 104760 | Cerebral amyloid angiopathy |
| 21q22.1 | 291.96 | KCNJ6 | Potassium inwardly rectifying channel, subfamily J, member 6 | 600877 | |
| 21q22.11 | 102.95 | EVA1C | Eva-1 homolog C (C. elegans) | N/A | |
| 21q22.11 | 3.79 | DNAJC28 | DnaJ (Hsp40) homolog, subfamily C, member 28 | N/A | |
| 21q22.13 | 147.82 | DYRK1A | Dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 1A | 600855 | Down syndrome, mental retardation 7 |
| 21q22.2 | 129.73 | DOPEY2 | Dopey family member 2 | 604803 | |
| 21q22.3 | 261.50 | RUNX1 | Runt-related transcription factor 1 | 151385 | Acute myeloid leukemia, platelet disorder with associated myeloid malignancy |
| 21q22.3 | 108.80 | BACE2 | Beta-site APP-cleaving enzyme 2 | 605668 | Down syndrome |
| 21q22.3 | 97.56 | ABCG1 | ATP-binding cassette, subfamily G (WHITE), member 1 | 603076 | |
| 21q22.3 | 23.17 | CBS | Cystathionine-beta-synthase | 613381 | Cystathionine beta-synthase deficiency, homocystinuria, hyperhomocysteinemic thrombosis |
| 21q22.3 | 50.19 | MCM3AP | Minichromosome maintenance complex component 3-associated protein | 603294 | |
| 21q22.3 | 6.50 | S100B | S100 calcium binding protein B | 176990 | Down syndrome, epilepsy, amyotrophic lateral sclerosis, melanoma, type I diabetes |
| 22q11.21 | 28.24 | COMT | Catechol-O-methyltransferase | 116790 | Panic disorder, schizophrenia |
| 22q11.21 | 26.88 | RTN4R | Reticulon 4 receptor | 605566 | Schizophrenia |
| 22q11.23 | 0.845 | MIF | Macrophage migration inhibitory factor (glycosylation-inhibiting factor) | 153620 | Rheumatoid arthritis |
| 22q11.23 | 8.15 | GSTT1 | Glutathione S-transferase theta 1 | 600436 | Carcinoma |
| 22q13.1 | 13.15 | HMOX1 | Heme oxygenase (decycling) 1 | 141250 | Heme oxygenase-1 deficiency, chronic obstructive pulmonary disease |
| 22q13.2 | 21.30 | SEPT3 | Septin 3 | 608314 | |
| 22q13.31 | 93.16 | PPARA | Peroxisome proliferator-activated receptor alpha | 170998 | Hyperapobetalipoproteinemia |
| Xp11.2 | 3.12 | HSD17B10 | Hydroxysteroid (17-beta) dehydrogenase 10 | 300256 | 17-beta-hydroxysteroid dehydrogenase X deficiency, 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency, mental retardation |
| Xp11.23 | 115.86 | MAOB | Monoamine oxidase B | 309860 | |
| Xp11.3 | 91.92 | MAOA | Monoamine oxidase A | 309850 | Brunner syndrome |
| Xp11.3-p11.23 | 4.50 | TIMP1 | TIMP metallopeptidase inhibitor 1 | 305370 | |
| Xp21.1 | 68.97 | OTC | Ornithine carbamoyltransferase | 300461 | Ornithine transcarbamylase deficiency, Duchenne muscular dystrophy |
| Xq12 | 186.59 | AR | Androgen receptor | 313700 | Spinal and bulbar muscular atrophy of Kennedy, prostate cancer, complete androgen insensitivity, hypospadia type 1 |
| Xq21.3 | 843.97 | PCDH11X | Protocadherin 11 X-linked | 300246 | |
| Xq21.3 | N/A | AD16 | Alzheimer disease 16 | 300756 |
Appendix B
Long-Form Names for Genes Listed in Table 27.2
ADH1A: Alcohol dehydrogenase 1A (class I), alpha polypeptide
AADAC: Arylacetamide deacetylase
AANAT: Aralkylamine N-acetyltransferase
ACSL1: Acyl-CoA synthetase long-chain family member 1
ACSL3: Acyl-CoA synthetase long-chain family member 3
ACSL4: Acyl-CoA synthetase long-chain family member 4
ACSM1: Acyl-CoA synthetase medium-chain family member 1
ACSM2B: Acyl-CoA synthetase medium-chain family member 2B
ACSM3: Acyl-CoA synthetase medium-chain family, member 3
ADH1B: Alcohol dehydrogenase 1B (class I), beta polypeptide
ADH1C: Alcohol dehydrogenase 1C (class I), gamma polypeptide
ADH4: Alcohol dehydrogenase 4 (class II), pi polypeptide
ADH5: Alcohol dehydrogenase 5 (class III), chi polypeptide
ADH6: Alcohol dehydrogenase 6 (class V)
ADH7: Alcohol dehydrogenase 7 (class IV), mu or sigma polypeptide
ADHFE1: Alcohol dehydrogenase, iron containing, 1
AGXT: Alanine-glyoxylate aminotransferase
AKR1A1: Aldo-keto reductase family 1, member A1 (aldehyde reductase)
AKR1B1: Aldo-keto reductase family 1, member B1 (aldose reductase)
AKR1C1: Aldo-keto reductase family 1, member C1
AKR1D1: Aldo-keto reductase family 1, member D1
ALDH1A1: Aldehyde dehydrogenase 1 family, member A1
ALDH1A2: Aldehyde dehydrogenase family 1, subfamily A2
ALDH1A3: Aldehyde dehydrogenase family 1, subfamily A3
ALDH1B1: Aldehyde dehydrogenase 1 family, member B1
ALDH2: Aldehyde dehydrogenase 2 family (mitochondrial)
ALDH3A1: Aldehyde dehydrogenase 3 family, member A1
ALDH3A2: Aldehyde dehydrogenase 3 family, member A2
ALDH3B1: Aldehyde dehydrogenase 3 family, member B1
ALDH3B2: Aldehyde dehydrogenase 3 family, member B2
ALDH4A1: Aldehyde dehydrogenase 4 family, member A1
ALDH5A1: Aldehyde dehydrogenase 5 family, member A1
ALDH6A1: Aldehyde dehydrogenase 6 family, member A1
ALDH7A1: Aldehyde dehydrogenase 7 family, member A1
ALDH8A1: Aldehyde dehydrogenase 8 family, member A1
ALDH9A1: Aldehyde dehydrogenase 9 family, member A1
AOX1: Aldehyde oxidase 1
AS3MT: Arsenic (+3 oxidation state) methyltransferase
ASMT: Acetylserotonin O-methyltransferase
BAAT: Bile acid CoA: amino acid N-acyltransferase (glycine N-choloyltransferase)
CBR1: Carbonyl reductase 1
CBR3: Carbonyl reductase 3
CBR4: Carbonyl reductase 4
CCBL1: Cysteine conjugate-beta lyase, cytoplasmic
CDA: Cytidine deaminase
CEL: Carboxyl ester lipase
CES1: Carboxylesterase 1
CES1P1: Carboxylesterase 1 pseudogene 1
CES2: Carboxylesterase 2
CES3: Carboxylesterase 3
CES5A: Carboxylesterase 5A
CHST1: Carbohydrate (keratan sulfate Gal-6) sulfotransferase 1
CHST2: Carbohydrate (N-acetylglucosamine-6-O) sulfotransferase 2
CHST3: Carbohydrate (chondroitin 6) sulfotransferase 3
CHST4: Carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 4
CHST5: Carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 5
CHST6: Carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 6
CHST7: Carbohydrate (N-acetylglucosamine 6-O) sulfotransferase 7
CHST8: Carbohydrate (N-acetylgalactosamine 4-0) sulfotransferase 8
CHST9: Carbohydrate (N-acetylgalactosamine 4-0) sulfotransferase 9
CHST10: Carbohydrate sulfotransferase 10
CHST11: Carbohydrate (chondroitin 4) sulfotransferase 11
CHST12: Carbohydrate (chondroitin 4) sulfotransferase 12
CHST13: Carbohydrate (chondroitin 4) sulfotransferase 13
COMT: Catechol-O-methyltransferase
CYB5R3: Cytochrome b5 reductase 3
CYP1A1: Cytochrome P450, family 1, subfamily A, polypeptide 1
CYP1A2: Cytochrome P450, family 1, subfamily A, polypeptide 2
CYP1B1: Cytochrome P450, family 1, subfamily B, polypeptide 1
CYP2A6: Cytochrome P450, family 2, subfamily A, polypeptide 6
CYP2A7: Cytochrome P450, family 2, subfamily A, polypeptide 7
CYP2A13: Cytochrome P450, family 2, subfamily A, polypeptide 13
CYP2B6: Cytochrome P450, family 2, subfamily B, polypeptide 6
CYP2C8: Cytochrome P450, family 2, subfamily C, polypeptide 8
CYP2C9: Cytochrome P450, family 2, subfamily C, polypeptide 9
CYP2C18: Cytochrome P450, family 2, subfamily C, polypeptide 18
CYP2C19: Cytochrome P450, family 2, subfamily C, polypeptide 19
CYP2D6: Cytochrome P450, family 2, subfamily D, polypeptide 6
CYP2D7P1: Cytochrome P450, family 2, subfamily D, polypeptide 7 pseudogene 1
CYP2E1: Cytochrome P450, family 2, subfamily E, polypeptide 1
CYP2F1: Cytochrome P450, family 2, subfamily F, polypeptide 1
CYP2J2: Cytochrome P450, family 2, subfamily J, polypeptide 2
CYP2R1: Cytochrome P450, family 2, subfamily R, polypeptide 1
CYP2S1: Cytochrome P450, family 2, subfamily S, polypeptide 1
CYP2W1: Cytochrome P450, family 2, subfamily W, polypeptide 1
CYP3A4: Cytochrome P450, family 3, subfamily A, polypeptide 4
CYP3A5: Cytochrome P450, family 3, subfamily A, polypeptide 5
CYP3A7: Cytochrome P450, family 3, subfamily A, polypeptide 7
CYP3A43: Cytochrome P450, family 3, subfamily A, polypeptide 43
CYP4A11: Cytochrome P450, family 4, subfamily A, polypeptide 11
CYP4A22: Cytochrome P450, family 4, subfamily A, polypeptide 22
CYP4B1: Cytochrome P450, family 4, subfamily B, polypeptide 1
CYP4F2: Cytochrome P450, family 4, subfamily F, polypeptide 2
CYP4F3: Cytochrome P450, family 4, subfamily F, polypeptide 3
CYP4F8: Cytochrome P450, family 4, subfamily F, polypeptide 8
CYP4F11: Cytochrome P450, family 4, subfamily F, polypeptide 11
CYP4F12: Cytochrome P450, family 4, subfamily F, polypeptide 12
CYP4Z1: Cytochrome P450, family 4, subfamily Z, polypeptide 1
CYP7A1: Cytochrome P450, family 7, subfamily A, polypeptide 1
CYP7B1: Cytochrome P450, family 7, subfamily B, polypeptide 1
CYP8B1: Cytochrome P450, family 8, subfamily B, polypeptide 1
CYP11A1: Cytochrome P450, family 11, subfamily A, polypeptide 1
CYP11B1: Cytochrome P450, family 11, subfamily B, polypeptide 1
CYP11B2: Cytochrome P450, family 11, subfamily B, polypeptide 2
CYP17A1: Cytochrome P450, family 17, subfamily A, polypeptide 1
CYP19A1: Cytochrome P450, family 19, subfamily A, polypeptide 1
CYP20A1: Cytochrome P450, family 20, subfamily A, polypeptide 1
CYP21A2: Cytochrome P450, family 21, subfamily A, polypeptide 2
CYP24A1: Cytochrome P450, family 24, subfamily A, polypeptide 1
CYP26A1: Cytochrome P450, family 26, subfamily A, polypeptide 1
CYP26B1: Cytochrome P450, family 26, subfamily B, polypeptide 1
CYP26C1: Cytochrome P450, family 26, subfamily C, polypeptide 1
CYP27A1: Cytochrome P450, family 27, subfamily A, polypeptide 1
CYP27B1: Cytochrome P450, family 27, subfamily B, polypeptide 1
CYP39A1: Cytochrome P450, family 39, subfamily A, polypeptide 1
CYP46A1: Cytochrome P450, family 46, subfamily A, polypeptide 1
CYP51A1: Cytochrome P450, family 51, subfamily A, polypeptide 1
DDOST: Dolichyl-diphosphooligosaccharide-protein glycosyltransferase subunit (non-catalytic)
DHRS1: Dehydrogenase/reductase (SDR family) member 1
DHRS2: Dehydrogenase/reductase (SDR family) member 2
DHRS3: Dehydrogenase/reductase (SDR family) member 3
DHRS4: Dehydrogenase/reductase (SDR family) member 4
DHRS7: Dehydrogenase/reductase (SDR family) member 7
DHRS9: Dehydrogenase/reductase (SDR family) member 9
DHRS12: Dehydrogenase/reductase (SDR family) member 12
DHRS13: Dehydrogenase/reductase (SDR family) member 13
DHRSX: Dehydrogenase/reductase (SDR family) X-linked
DPEP1: Dipeptidase 1 (renal)
DPYD: Dihydropyrimidine dehydrogenase
EPHX1: Epoxide hydrolase 1, microsomal (xenobiotic)
EPHX2: Epoxide hydrolase 2, microsomal (xenobiotic)
ESD: Esterase D
FMO1: Flavin containing monooxygenase 1
FMO2: Flavin containing monooxygenase 2
FMO3: Flavin containing monooxygenase 3
FMO4: Flavin containing monooxygenase 4
FMO5: Flavin containing monooxygenase 5
FMO6P: Flavin containing monooxygenase 6 pseudogene
GAL3ST1: Galactose-3-O-sulfotransferase 1
GAMT: Guanidinoacetate N-methyltransferase
GLRX: Glutaredoxin (thioltransferase)
GLYAT: Glycine-N-acyltransferase
GNMT: Glycine N-methyltransferase
GPX1: Glutathione peroxidase 1
GPX2: Glutathione peroxidase 2 (gastrointestinal)
GPX3: Glutathione peroxidase 3 (plasma)
GPX4: Glutathione peroxidase 4
GPX5: Glutathione peroxidase 5
GPX6: Glutathione peroxidase 6 (olfactory)
GPX7: Glutathione peroxidase 7
GSR: Glutathione reductase
GSTA1: Glutathione S-transferase alpha 1
GSTA2: Glutathione S-transferase alpha 2
GSTA3: Glutathione S-transferase alpha 3
GSTA4: Glutathione S-transferase alpha 4
GSTA5: Glutathione S-transferase alpha 5
GSTCD: Glutathione S-transferase, C-terminaldomain containing
GSTK1: Glutathione S-transferase kappa 1
GSTM1: Glutathione S-transferase mu 1
GSTM2: Glutathione S-transferase mu 2 (muscle)
GSTM3: Glutathione S-transferase mu 3 (brain)
GSTM4: Glutathione S-transferase mu 4
GSTM5: Glutathione S-transferase mu 5
GSTO1: Glutathione S-transferase omega 1
GSTO2: Glutathione S-transferase omega 2
GSTP1: Glutathione S-transferase pi 1
GSTT1: Glutathione S-transferase theta 1
GSTT2: Glutathione S-transferase theta 2
GSTZ1: Glutathione S-transferase zeta 1
GZMA: Granzyme A (granzyme 1, cytotoxic T-lymphocyte-associated serine esterase 3)
GZMB: Granzyme B (granzyme 2, cytotoxic T-lymphocyte-associated serine esterase 1)
HNMT: Histamine N-methyltransferase
HSD11B1: Hydroxysteroid (11-beta) dehydrogenase 1
HSD17B10: Hydroxysteroid (17-beta) dehydrogenase 10
HSD17B11: Hydroxysteroid (17-beta) dehydrogenase 11
HSD17B14: Hydroxysteroid (17-beta) dehydrogenase 14
INMT: Indolethylamine N-methyltransferase
MAOA: Monoamine oxidase A
MAOB: monoamine oxidase B
METAP1: Methionyl aminopeptidase 1
MGST1: Microsomal glutathione S-transferase 1
MGST2: Microsomal glutathione S-transferase 1
MGST3: Microsomal glutathione S-transferase 3
NAA20: N(alpha)-acetyltransferase 20, NatB catalytic subunit
NAT1: N-acetyltransferase 1 (arylamine N-acetyltransferase)
NAT2: N-acetyltransferase 2 (arylamine N-acetyltransferase)
NNMT: Nicotinamide N-methyltransferase
NQO1: NAD(P)H dehydrogenase, quinone 1
NQO2: NAD(P)H dehydrogenase, quinone 2
PNMT: Phenylethanolamine N-methyltransferase
PON1: Paraoxonase 1
PON2: Paraoxonase 2
PON3: Paraoxonase 3
POR: P450 (cytochrome) oxidoreductase
PTGES: Prostaglandin E synthase
PTGS1: Prostaglandin-endoperoxide synthase 1 (prostaglandin G/H synthase and cyclooxygenase)
PTGS2: Prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase)
SAT1: Spermidine/spermine N1-acetyltransferase 1
SMOX: Spermine oxidase
SOD1: Superoxide dismutase 1, soluble
SOD2: Superoxide dismutase 2, mitochondrial
SULT1A1: Sulfotransferase family, cytosolic, 1A, phenol-preferring, member 1
SULT1A2: Sulfotransferase family, cytosolic, 1A, phenol-preferring, member 2
SULT1A3: Sulfotransferase family, cytosolic, 1A, phenol-preferring, member 3
SULT1B1: Sulfotransferase family, cytosolic, 1B, member 1
SULT1C1: Sulfotransferase family, cytosolic, 1C, member 1
SULT1C2: Sulfotransferase family, cytosolic, 1C, member 2
SULT1C3: Sulfotransferase family, cytosolic, 1C, member 3
SULT1C4: Sulfotransferase family, cytosolic, 1C, member 4
SULT1E1: Sulfotransferase family 1E, estrogen-preferring, member 1
SULT2A1: Sulfotransferase family, cytosolic, 2A,dehydroepiandrosterone (DHEA)-preferring, member 1
SULT2B1: Sulfotransferase family, cytosolic, 2B, member 1
SULT4A1: Sulfotransferase family 4A, member 1
SULT6B1: sulfotransferase family, cytosolic, 6B, member 1
TBXAS1: Thromboxane A synthase 1 (platelet)
TPMT: Thiopurine S-methyltransferase
TST: Thiopurine S-methyltransferase
UCHL1: Ubiquitin carboxyl-terminal esterase L1 (ubiquitin thiolesterase)
UCHL3: Ubiquitin carboxyl-terminal esterase L3 (ubiquitin thiolesterase)
UGT1A1: UDP glucuronosyltransferase 1 family, polypeptide A1
UGT1A3: UDP glucuronosyltransferase 1 family, polypeptide A3
UGT1A4: UDP glucuronosyltransferase 1 family, polypeptide A4
UGT1A5: UDP glucuronosyltransferase 1 family, polypeptide A5
UGT1A6: UDP glucuronosyltransferase 1 family, polypeptide A6
UGT1A7: UDP glucuronosyltransferase 1 family, polypeptide A7
UGT1A8: UDP glucuronosyltransferase 1 family, polypeptide A8
UGT1A9: UDP glucuronosyltransferase 1 family, polypeptide A9
UGT1A10: UDP glucuronosyltransferase 1 family, polypeptide A10
UGT2A1: UDP glucuronosyltransferase 2 family, polypeptide A1, complex locus
UGT2A3: UDP glucuronosyltransferase 2 family, polypeptide A3
UGT2B10: UDP glucuronosyltransferase 2 family, polypeptide B10
UGT2B11: UDP glucuronosyltransferase 2 family, polypeptide B11
UGT2B15: UDP glucuronosyltransferase 2 family, polypeptide B15
UGT2B17: UDP glucuronosyltransferase 2 family, polypeptide B17
UGT2B28: UDP glucuronosyltransferase 2 family, polypeptide B28
UGT2B4: UDP glucuronosyltransferase 2 family, polypeptide B4
UGT2B7: UDP glucuronosyltransferase 2 family, polypeptide B7
UGT3A1: UDP glycosyltransferase 3 family, polypeptide A1
UGT8: UDP glycosyltransferase 8
XDH: Xanthine dehydrogenase
References
- 1.National Center for Biotechnology Information. The NCBI database. http://www.ncbi.nlm.nih.gov/pubmed/; 2013 [accessed 19.06.13].
- 2.Suehs BT, Davis CD, Alvir J, van Amerongen D, Pharmd NC, Joshi AV. The clinical and economic burden of newly diagnosed Alzheimer’s disease in a medicare advantage population. Am J Alzheimers Dis Other Demen. 2013;28(4):384–392. doi: 10.1177/1533317513488911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.National Center for Health Statistics. Health, United States, 2009: With special feature on medical technology. Hyattsville, MD. 2010. [PubMed]
- 4.Centers for Disease Control and Prevention. <http://www.cdc.gov/DataStatistics/>. Updated March 6 2012 [accessed 19.06.13].
- 5.Sousa RM, Ferri CP, Acosta D, Albanese E, Guerra M, Huang Y. Contribution of chronic diseases to disability in elderly people in countries with low and middle incomes: a 10/66 Dementia Research Group population-based survey. Lancet. 2009;374(9704):1821–1830. doi: 10.1016/S0140-6736(09)61829-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herrmann N, Tam DY, Balshaw R, Sambrook R, Lesnikova N, Lanctôt KL. The relation between disease severity and cost of caring for patients with Alzheimer disease in Canada. Can J Psychiatry. 2010;55(12):768–775. doi: 10.1177/070674371005501204. [DOI] [PubMed] [Google Scholar]
- 7.Cacabelos R. The path to personalized medicine in mental disorders. In: Ritsner MS, editor. Vol. 4. Springer; Netherlands: 2009. pp. 3–63. (The handbook of neuropsychiatric biomarkers, endophenotypes and genes). [Google Scholar]
- 8.Wimo A, Jönsson L, Gustavsson A, McDaid D, Ersek K, Georges J. The economic impact of dementia in Europe in 2008-cost estimates from the Eurocode project. Int J Geriatr Psychiatry. 2011;26(8):825–832. doi: 10.1002/gps.2610. [DOI] [PubMed] [Google Scholar]
- 9.Wimo A, Reed CC, Dodel R, Belger M, Jones RW, Happich M. The GERAS study: a prospective observational study of costs and resource use in community dwellers with Alzheimer’s disease in three European countries: study design and baseline findings. J Alzheimers Dis. 2013;36(2):385–399. doi: 10.3233/JAD-122392. [DOI] [PubMed] [Google Scholar]
- 10.Kamble P, Chen H, Sherer JT, Aparasu RR. Use of antipsychotics among elderly nursing home residents with dementia in the US: an analysis of National Survey Data. Drugs Aging. 2009;26(6):483–492. doi: 10.2165/00002512-200926060-00005. [DOI] [PubMed] [Google Scholar]
- 11.Liperoti R, Onder G, Landi F, Lapane KL, Mor V, Bernabei R. All-cause mortality associated with atypical and conventional antipsychotics among nursing home residents with dementia: a retrospective cohort study. J Clin Psychiatry. 2009;70(10):1340–1347. doi: 10.4088/JCP.08m04597yel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cacabelos R. Pharmacogenomics and therapeutic strategies for dementia. Expert Rev Mol Diagn. 2009;9(6):567–611. doi: 10.1586/erm.09.42. [DOI] [PubMed] [Google Scholar]
- 13.Anderson CNG, Grant SGN. High throughput protein expression screening in the nervous system-needs and limitations. J Physiol. 2006;575(Pt 2):367–372. doi: 10.1113/jphysiol.2006.113795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu X, Zhan M, Duan W, Prabhu V, Brenneman R, Wood W. Gene expression atlas of the mouse central nervous system: impact and interactions of age, energy intake and gender. Genome Biol. 2007;8(11):R234. doi: 10.1186/gb-2007-8-11-r234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cacabelos R, Fernández-Novoa L, Martínez-Bouza R, McKay A, Carril JC, Lombardi V. Future trends in the pharmacogenomics of brain disorders and dementia: influence of APOE and CYP2D6 variants. Pharmaceuticals. 2010;3(10):3040–3100. [Google Scholar]
- 16.Cacabelos R, Fernández-Novoa L, Lombardi V, Kubota Y, Takeda M. Molecular genetics of Alzheimer’s disease and aging. Methods Find Exp Clin Pharmacols. 2005;27(Suppl. A):1–573. [PubMed] [Google Scholar]
- 17.Alzheimer Research Forum. Alzforum. <http://www.alzgene.org/> Published 1996. Updated April 18 2011 [accessed 19.06.13].
- 18.Cacabelos R. Alzheimer’s disease 2011: where are we heading? Gen-T. 2011;8:54–86. [Google Scholar]
- 19.Selkoe DJ, Podlisny MB. Deciphering the genetic basis of Alzheimer’s disease. Annu Rev Genomics Hum Genet. 2002;3:67–99. doi: 10.1146/annurev.genom.3.022502.103022. [DOI] [PubMed] [Google Scholar]
- 20.Suh YH, Checler F. Amyloid precursor protein, presenilins, and α-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer’s disease. Pharmacol Rev. 2002;54(3):469–525. doi: 10.1124/pr.54.3.469. [DOI] [PubMed] [Google Scholar]
- 21.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 22.Takeda M, Martínez R, Kudo T, Tanaka T, Okochi M, Tagami S. Apolipoprotein E and central nervous system disorders: reviews of clinical findings. Psychiatry Clin Neurosci. 2010;64(6):592–607. doi: 10.1111/j.1440-1819.2010.02148.x. [DOI] [PubMed] [Google Scholar]
- 23.Cacabelos R. Pharmacogenomics in Alzheimer’s disease. Methods Mol Biol. 2008;448:213–357. doi: 10.1007/978-1-59745-205-2_10. [DOI] [PubMed] [Google Scholar]
- 24.Cacabelos R, Takeda M. Pharmacogenomics, nutrigenomics and future therapeutics in Alzheimer’s disease. Drugs Future. 2006;31(Suppl. B):5–146. [Google Scholar]
- 25.Cacabelos R. The application of functional genomics to Alzheimer’s disease. Pharmacogenomics. 2003;4(5):597–621. doi: 10.1517/phgs.4.5.597.23795. [DOI] [PubMed] [Google Scholar]
- 26.Petrlova J, Hong HS, Bricarello DA, Harishchandra G, Lorigan GA, Jin LW. A differential association of apolipoprotein E isoforms with the amyloid-β oligomer in solution. Proteins. 2011;79(2):402–416. doi: 10.1002/prot.22891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Samaranch L, Cervantes S, Barabash A, Alonso A, Cabranes JA, Lamet I. The effect of MAPT H1 and APOE ɛ4 on transition from mild cognitive impairment to dementia. J Alzheimers Dis. 2010;22(4):1065–1071. doi: 10.3233/JAD-2010-101011. [DOI] [PubMed] [Google Scholar]
- 28.Cacabelos R. Donepezil in Alzheimer’s disease: from conventional trials to pharmacogenetics. Neuropsychiatr Dis Treat. 2007;3(3):303–333. [PMC free article] [PubMed] [Google Scholar]
- 29.Mallick B, Ghosh Z. A complex crosstalk between polymorphic microRNA target sites and AD prognosis. RNA Biol. 2011;8(4):665–673. doi: 10.4161/rna.8.4.15584. [DOI] [PubMed] [Google Scholar]
- 30.Qureshi IA, Mehler MF. Advances in epigenetics and epigenomics for neurodegenerative diseases. Curr Neurol Neurosci Rep. 2011;11(5):464–473. doi: 10.1007/s11910-011-0210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Enciu AM, Popescu BO, Gheorghisan-Galateanu A. MicroRNAs in brain development and degeneration. Mol Biol Rep. 2012;39(3):2243–2252. doi: 10.1007/s11033-011-0973-1. [DOI] [PubMed] [Google Scholar]
- 32.Murray IV, Proza JF, Sohrabji F, Lawler JM. Vascular and metabolic dysfunction in Alzheimer’s disease: a review. Exp Biol Med (Maywood) 2011;236(7):772–782. doi: 10.1258/ebm.2011.010355. [DOI] [PubMed] [Google Scholar]
- 33.Ettorre E, Cerra E, Marigliano B, Vigliotta M, Vulcano A, Fossati C. Role of cardiovascular risk factors (CRF) in the patients with mild cognitive impairment (MCI) Arch Gerontol Geriatr. 2012;54(2):330–332. doi: 10.1016/j.archger.2011.04.025. [DOI] [PubMed] [Google Scholar]
- 34.Lin KP, Chen SY, Lai LC, Huang YL, Chen JH, Chen TF. Genetic polymorphisms of a novel vascular susceptibility gene, Ninjurin2 (NINJ2), are associated with a decreased risk of Alzheimer’s disease. PLoS One. 2011;6(6):e20573. doi: 10.1371/journal.pone.0020573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Romero A, Cacabelos R, Oset-Gasque MJ, Samadi A, Marco-Contelles J. Novel tacrine-related drugs as potential candidates for the treatment of Alzheimer’s disease. Bioorg Med Chem Lett. 2013;23(7):1916–1922. doi: 10.1016/j.bmcl.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 36.Aliev G, Palacios HH, Cacabelos P, Cacabelos R, Burzynski G, Burzynski SR. Mitochondria specific antioxidants and their derivatives in the context of the drug development for neurodegeneration and cancer. Drug Des. 2013;2:1. [Google Scholar]
- 37.Grammas P, Martínez J, Miller B. Cerebral microvascular endothelium and the pathogenesis of neurodegenerative diseases. Expert Rev Mol Med. 2011;13:e19. doi: 10.1017/S1462399411001918. [DOI] [PubMed] [Google Scholar]
- 38.Cacabelos R, Fernández-Novoa L, Lombardi V, Corzo L, Pichel V, Kubota Y. Cerebrovascular risk factors in Alzheimer’s disease: brain hemodynamics and pharmacogenomic implications. Neurol Res. 2003;25(6):567–580. doi: 10.1179/016164103101202002. [DOI] [PubMed] [Google Scholar]
- 39.Cacabelos R, Fernández-Novoa L, Corzo L, Amado L, Pichel V, Lombardi V. Phenotypic profiles and functional genomics in Alzheimer’s disease and in dementia with a vascular component. Neurol Res. 2004;26(5):459–480. doi: 10.1179/016164104225017677. [DOI] [PubMed] [Google Scholar]
- 40.Kim JH, Hwang KJ, Kim JH, Lee YH, Rhee HY, Park KC. Regional white matter hyperintensities in normal aging, single domain amnestic mild cognitive impairment, and mild Alzheimer’s disease. J Clin Neurosci. 2011;18(8):1101–1106. doi: 10.1016/j.jocn.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 41.Chen H, Zhang JH. Cerebral amyloid angiopathy-related microhemorrhages in Alzheimer’s disease: a review of investigative animal models. Acta Neurochir Suppl. 2011;111:15–17. doi: 10.1007/978-3-7091-0693-8_3. [DOI] [PubMed] [Google Scholar]
- 42.Yates PA, Sirisriro R, Villemagne VL, Farquharson S, Masters CL, Rowe CC, AIBL Research Group Cerebral microhemorrhage and brain β-amyloid in aging and Alzheimer disease. Neurology. 2011;77(1):48–54. doi: 10.1212/WNL.0b013e318221ad36. [DOI] [PubMed] [Google Scholar]
- 43.Brenn A, Grube M, Peters M, Fischer A, Jedlitschky G, Kroemer HK. Beta-amyloid downregulates MDR1-P-glycoprotein (Abcb1) expression at the blood-brain barrier in mice. Int J Alzheimers Dis. 2011;2011:690121. doi: 10.4061/2011/690121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen KH, Reese EA, Kim HW, Rapoport SI, Rao JS. Disturbed neurotransmitter transporter expression in Alzheimer’s disease brain. J Alzheimers Dis. 2011;26(4):755–766. doi: 10.3233/JAD-2011-110002. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Aliev G, Obrenovich ME, Tabrez S, Jabir NR, Reddy VP, Li Y. Link between cancer and Alzheimer disease via oxidative stress induced by nitric oxide-dependent mitochondrial DNA overproliferation and deletion. Oxid Med Cell Longev. 2013;2013:962984. doi: 10.1155/2013/962984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tayler H, Fraser T, Miners JS, Kehoe PG, Love S. Oxidative balance in Alzheimer’s disease: relationship to APOE, Braak tangle stage, and the concentrations of soluble and insoluble amyloid-β. J Alzheimers Dis. 2010;22(4):1363–1373. doi: 10.3233/JAD-2010-101368. [DOI] [PubMed] [Google Scholar]
- 47.Alikhani N, Guo L, Yan S, Du H, Pinho CM, Chen JX. Decreased proteolytic activity of the mitochondrial amyloid-β degrading enzyme, PreP peptidasome, in Alzheimer’s disease brain mitochondria. J Alzheimers Dis. 2011;27(1):75–87. doi: 10.3233/JAD-2011-101716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mathew A, Yoshida Y, Maekawa T, Sakthi Kumar DS. Alzheimer’s disease: cholesterol a menace? Brain Res Bull. 2011;86(1–2):1–12. doi: 10.1016/j.brainresbull.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 49.Qiu L, Buie C, Reay A, Vaughn MW, Cheng KH. Molecular dynamics simulations reveal the protective role of cholesterol in beta amyloid protein-induced membrane disruptions in neuronal membrane mimics. J Phys Chem B. 2011;115(32):9795–9812. doi: 10.1021/jp2012842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones L, Holmans PA, Hamshere ML, Harold D, Moskvina V, Ivanov D. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS One. 2010;5(11):e13950. doi: 10.1371/journal.pone.0013950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan XD, Zhu YG, Lin N, Zhang J, Ye QY, Huang HP. Microglial phagocytosis induced by fibrillar beta-amyloid is attenuated by oligomeric beta-amyloid: implications for Alzheimer’s disease. Mol Neurodegener. 2011;6:45. doi: 10.1186/1750-1326-6-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diniz BS, Teixeira AL, Ojopi EB, Talib LL, Mendonça VA, Gattaz WF. Higher serum sTNFR1 level predicts conversion from mild cognitive impairment to Alzheimer’s disease. J Alzheimers Dis. 2010;22(4):1305–1311. doi: 10.3233/JAD-2010-100921. [DOI] [PubMed] [Google Scholar]
- 53.Cacabelos R, Martínez-Bouza R. Genomics and pharmacogenomics of dementia. CNS Neurosci Ther. 2011;17(5):566–576. doi: 10.1111/j.1755-5949.2010.00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cacabelos R. Pharmacogenomics and therapeutic prospects in Alzheimer’s disease. Expert Opin Pharmacother. 2005;6(12):1967–1987. doi: 10.1517/14656566.6.12.1967. [DOI] [PubMed] [Google Scholar]
- 55.Cacabelos R. Pharmacogenomics and therapeutic prospects in dementia. Eur Arch Psychiatry Clin Neurosci. 2008;258(Suppl. 1):28–47. doi: 10.1007/s00406-007-1006-x. [DOI] [PubMed] [Google Scholar]
- 56.Cacabelos R. Pharmacogenetic basis for therapeutic optimization in Alzheimer’s disease. Mol Diagn Ther. 2007;11(6):385–405. doi: 10.1007/BF03256262. [DOI] [PubMed] [Google Scholar]
- 57.Cacabelos R, Llovo R, Fraile C, Fernández-Novoa L. Pharmacogenetic aspects of therapy with cholinesterase inhibitors: the role of CYP2D6 in Alzheimer’s disease pharmacogenetics. Curr Alzheimer Res. 2007;4(4):479–500. doi: 10.2174/156720507781788846. [DOI] [PubMed] [Google Scholar]
- 58.Cacabelos R. Molecular pathology and pharmacogenomics in Alzheimer’s disease: polygenic-related effects of multifactorial treatments on cognition, anxiety, and depression. Methods Find Exp Clin Pharmacol. 2007;29(Suppl. A):1–91. [PubMed] [Google Scholar]
- 59.Cacabelos R. Pharmacogenomics, nutrigenomics and therapeutic optimization in Alzheimer’s disease. Aging Health. 2005;1(2):303–348. [Google Scholar]
- 60.Braak H, del Tredici K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011;121(2):171–181. doi: 10.1007/s00401-010-0789-4. [DOI] [PubMed] [Google Scholar]
- 61.Lu PH, Thompson PM, Leow A, Lee GJ, Lee A, Yanovsky I. Apolipoprotein E genotype is associated with temporal and hippocampal atrophy rates in healthy elderly adults: a tensor-based morphometry study. J Alzheimers Dis. 2011;23(3):433–442. doi: 10.3233/JAD-2010-101398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Canu E, Frisoni GB, Agosta F, Pievani M, Bonetti M, Filippi M. Early and late onset Alzheimer’s disease patients have distinct patterns of white matter damage. Neurobiol Aging. 2012;33(6):1023–1033. doi: 10.1016/j.neurobiolaging.2010.09.021. [DOI] [PubMed] [Google Scholar]
- 63.Herholz K, Ebmeier K. Clinical amyloid imaging in Alzheimer’s disease. Lancet Neurol. 2011;10(7):667–670. doi: 10.1016/S1474-4422(11)70123-5. [DOI] [PubMed] [Google Scholar]
- 64.Vialatte FB, Dauwels J, Maurice M, Musha T, Cichocki A. Improving the specificity of EEG for diagnosing Alzheimer’s disease. Int J Alzheimers Dis. 2011;2011:259069. doi: 10.4061/2011/259069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roh JH, Park MH, Ko D, Park KW, Lee DH, Han C. Region and frequency specific changes of spectral power in Alzheimer’s disease and mild cognitive impairment. Clin Neurophysiol. 2011;122(11):2169–2176. doi: 10.1016/j.clinph.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 66.Canuet L, Tellado I, Couceiro V, Fraile C, Fernandez-Novoa L, Ishii R. Resting-state network disruption and APOE genotype in Alzheimer’s disease: a lagged functional connectivity study. PLoS One. 2012;7(9):e46289. doi: 10.1371/journal.pone.0046289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cacabelos R, Fernández-Novoa Corzo L, Pichel V, Lombardi V, Kubota Y. Genomics and phenotypic profiles in dementia: implications for pharmacological treatment. Methods Find Exp Clin Pharmacol. 2004;26(6):421–444. [PubMed] [Google Scholar]
- 68.Hampel H, Frank R, Broich K, Teipel SJ, Katz RG, Hardy J. Biomarkers for Alzheimer’s disease: academic, industry and regulatory perspectives. Nat Rev Drug Discov. 2010;9(7):560–574. doi: 10.1038/nrd3115. [DOI] [PubMed] [Google Scholar]
- 69.Cacabelos R, Corzo L, Fernández-Novoa L, Lombardi V. Histamine in Alzheimer’s disease pathogenesis: biochemistry and functional genomics. Methods Find Exp Clin Pharmacol. 2004;26(Suppl. 2):9–16. [Google Scholar]
- 70.Portelius E, Mattsson N, Andreasson U, Blennow K, Zetterberg H. Novel Aβ isoforms in Alzheimer’s disease – their role in diagnosis and treatment. Curr Pharm Des. 2011;17(25):2594–2602. doi: 10.2174/138161211797416039. [DOI] [PubMed] [Google Scholar]
- 71.Kester MI, Scheffer PG, Koel-Simmelink MJ, Twaalfhoven H, Verwey NA, Veerhuis R. Serial CSF sampling in Alzheimer’s disease: specific versus non-specific markers. Neurobiol Aging. 2012;33(8):1591–1598. doi: 10.1016/j.neurobiolaging.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 72.Roher AE, Maarouf CL, Sue LI, Hu Y, Wilson J, Beach TG. Proteomics-derived cerebrospinal fluid markers of autopsy-confirmed Alzheimer’s disease. Biomarkers. 2009;14(7):493–501. doi: 10.3109/13547500903108423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Popp J, Lewczuk P, Frommann I, Kölsch H, Kornhuber J, Maier W. Cerebrospinal fluid markers for Alzheimer’s disease over the lifespan: effects of age and the APOEɛ4 genotype. J Alzheimers Dis. 2010;22(2):459–468. doi: 10.3233/JAD-2010-100561. [DOI] [PubMed] [Google Scholar]
- 74.Han MR, Schellenberg GD, Wang LS. Alzheimer’s disease neuroimaging initiative. Genome-wide association reveals genetic effects on human Aβ42 and τ protein levels in cerebrospinal fluids: a case control study. BMC Neurol. 2010;10:90. doi: 10.1186/1471-2377-10-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thambisetty M, Tripaldi R, Riddoch-Contreras J, Hye A, An Y, Campbell J. Proteome-based plasma markers of brain amyloid-β deposition in non-demented older individuals. J. Alzheimers Dis. 2010;22(4):1099–1109. doi: 10.3233/JAD-2010-101350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loveman E, Green C, Kirby J, Takeda A, Picot J, Payne E. The clinical and cost-effectiveness of donepezil, rivastigmine, galantamine and memantine for Alzheimer’s disease. Health Technol Assess. 2006;10(1):1–160. doi: 10.3310/hta10010. [DOI] [PubMed] [Google Scholar]
- 77.Cacabelos R, Álvarez A, Lombardi V, Fernández-Novoa L, Corzo L, Pérez P. Pharmacological treatment of Alzheimer disease: from psychotropic drugs and cholinesterase inhibitors to pharmacogenomics. Drugs Today (Barc) 2000;36(7):415–499. [PubMed] [Google Scholar]
- 78.Giacobini E. The brain cholinergic system in health and disease. In: Giacobini E, Pepeu G, editors. Cholinesterases in human brain: the effect of cholinesterase inhibitors on Alzheimer’s disease and related disordersp. Informa Healthcare; Oxon, UK: 2006. pp. 235–264. [chapter 18] [Google Scholar]
- 79.Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Möbius HJ. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med. 2003;348(14):1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 80.Schenk DB, Seubert P, Grundman M, Black R. Aβ immunotherapy: lessons learned for potential treatment of Alzheimer’s disease. Neurodegener Dis. 2005;2(5):255–260. doi: 10.1159/000090365. [DOI] [PubMed] [Google Scholar]
- 81.Wisniewski T, Boutajangout A. Vaccination as a therapeutic approach to Alzheimer’s disease. Mt Sinai J Med. 2010;77(1):17–31. doi: 10.1002/msj.20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Strooper B, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol. 2010;6(2):99–107. doi: 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shelton CC, Zhu L, Chau D, Yang L, Wang R, Djaballah H. Modulation of gamma-secretase specificity using small molecule allosteric inhibitors. Proc Natl Acad Sci USA. 2009;106(48):20228–20233. doi: 10.1073/pnas.0910757106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lambracht-Washington D, Qu BX, Fu M, Eagar TN, Stüve O, Rosenberg RN. DNA beta-amyloid(1–42) trimer immunization for Alzheimer disease in a wild-type mouse mode. JAMA. 2009;302(16):1796–1802. doi: 10.1001/jama.2009.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carrera I, Etcheverría I, Fernández-Novoa L, Lombardi L, Vigo C, Cacabelos R. Development of the EuroEspes EB101 vaccine against Alzheimer’s disease. Gen-T. 2011;8:38–52. [Google Scholar]
- 86.Wisniewski T, Konietzko U. Amyloid-beta immunisation for Alzheimer’s disease. Lancet Neurol. 2008;7(9):805–811. doi: 10.1016/S1474-4422(08)70170-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Singh S, Kushwah AS, Singh R, Farswan M, Kaur R. Current therapeutic strategy in Alzheimer’s disease. Eur Rev Med Pharmacol Sci. 2012;16(12):1651–1664. [PubMed] [Google Scholar]
- 88.Grill JD, Cummings JL. Current therapeutic targets for the treatment of Alzheimer’s disease. Expert Rev Neurother. 2010;10(5):711–728. doi: 10.1586/ern.10.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tabira T. Immunization therapy for Alzheimer disease: a comprehensive review of active immunization strategies. Tohoku J Exp Med. 2010;220(2):95–106. doi: 10.1620/tjem.220.95. [DOI] [PubMed] [Google Scholar]
- 90.Carrera I, Cacabelos R. Novel immunotherapeutic procedures for prevention of Alzheimer’s disease. Drug Des. 2013;2:107. [Google Scholar]
- 91.Galimberti D, Ghezzi L, Scarpini E. Immunotherapy against amyloid pathology in Alzheimer’s disease. J Neurol Sci. 2013;333(1–2):50–54. doi: 10.1016/j.jns.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 92.Maeda S, Sahara N, Saito Y, Murayama S, Ikai A, Takashima A. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer’s disease. Neurosci Res. 2006;54(3):197–201. doi: 10.1016/j.neures.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 93.Boutajangout A, Sigurdsson EM, Krishnamurthy PK. Tau as a therapeutic target for Alzheimer’s disease. Curr Alzheimer Res. 2011;8(6):666–677. doi: 10.2174/156720511796717195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Johnson GV. Tau phosphorylation and proteolysis: insights and perspectives. J Alzheimers Dis. 2006;9(Suppl. 3):243–250. doi: 10.3233/jad-2006-9s326. [DOI] [PubMed] [Google Scholar]
- 95.Kudo LC, Parfenova L, Ren G, Vi N, Hui M, Ma Z. Puromycin-sensitive aminopeptidase (PSA/NPEPPS) impedes development of neuropathology in hPSA/TAU(P301L) double-transgenic mice. Hum Mol Genet. 2011;20(9):1820–1833. doi: 10.1093/hmg/ddr065. [DOI] [PubMed] [Google Scholar]
- 96.Trojanowski JQ, Smith AB, Huryn D, Lee VM. Microtubule-stabilising drugs for therapy of Alzheimer’s disease and other neurodegenerative disorders with axonal transport impairments. Expert Opin Pharmacother. 2005;6(5):683–686. doi: 10.1517/14656566.6.5.683. [DOI] [PubMed] [Google Scholar]
- 97.Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci USA. 2005;102(1):227–231. doi: 10.1073/pnas.0406361102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010;30(49):16559–16566. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 100.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Chishti MA. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408(6815):979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 101.Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7(3):369–372. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- 102.Imbimbo BP, Ottonello S, Frisardi V, Solfrizzi V, Greco A, Seripa D. Solanezumab for the treatment of mild-to-moderate Alzheimer’s disease. Expert Rev Clin Immunol. 2012;8(2):135–149. doi: 10.1586/eci.11.93. [DOI] [PubMed] [Google Scholar]
- 103.Panza F, Frisardi V, Imbimbo BP, D’Onofrio G, Pietrarossa G, Seripa D. Bapineuzumab: anti-β-amyloid monoclonal antibodies for the treatment of Alzheimer’s disease. Immunotherapy. 2010;2(6):767–782. doi: 10.2217/imt.10.80. [DOI] [PubMed] [Google Scholar]
- 104.Magga J, Puli L, Pihlaja R, Kanninen K, Neulamaa S, Malm T. Human intravenous immunoglobulin provides protection against Aβ toxicity by multiple mechanisms in a mouse model of Alzheimer’s disease. J Neuroinflammation. 2010;7:90. doi: 10.1186/1742-2094-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kingwell K. Alzheimer disease: amyloid-β immunotherapy CAD106 passes first safety test in patients with Alzheimer disease. Nat Rev Neurol. 2012;8(8):414. doi: 10.1038/nrneurol.2012.128. [DOI] [PubMed] [Google Scholar]
- 106.Delrieu J, Ousset PJ, Caillaud C, Vellas B. ‘Clinical trials in Alzheimer’s disease’: immunotherapy approaches. J Neurochem. 2012;120(Suppl. 1):186–193. doi: 10.1111/j.1471-4159.2011.07458.x. [DOI] [PubMed] [Google Scholar]
- 107.Qu BX, Xiang Q, Li L, Johnston SA, Hynan LS, Rosenberg RN. Abeta42 gene vaccine prevents Abeta42 deposition in brain of double transgenic mice. J Neurol Sci. 2007;260(1–2):204–213. doi: 10.1016/j.jns.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Roses AD. The medical and economic roles of pipeline pharmacogenetics: Alzheimer’s disease as a model of efficacy and HLA-B(*)5701 as a model of safety. Neuropsychopharmacology. 2009;34(1):6–17. doi: 10.1038/npp.2008.153. [DOI] [PubMed] [Google Scholar]
- 109.Roses AD, Saunders AM, Huang Y, Strum J, Weisgraber KH, Mahley RW. Complex disease-associated pharmacogenetics: drug efficacy, drug safety, and confirmation of a pathogenic hypothesis (Alzheimer’s disease) Pharmacogenomics J. 2007;7(1):10–28. doi: 10.1038/sj.tpj.6500397. [DOI] [PubMed] [Google Scholar]
- 110.Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 2006;6(4):246–254. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- 111.Cacabelos R. Pharmacogenomics of central nervous system (CNS) drugs. Drug Dev Res. 2012;73:461–476. [Google Scholar]
- 112.Xie HG, Kim RB, Wood AJ, Stein CM. Molecular basis of ethnic differences in drug disposition and response. Annu Rev Pharm Toxicol. 2001;41:815–850. doi: 10.1146/annurev.pharmtox.41.1.815. [DOI] [PubMed] [Google Scholar]
- 113.Cacabelos R, editor. World guide for drug use and pharmacogenomics. EuroEspes Publishing; Corunna, Spain: 2012. [Google Scholar]
- 114.Division of Pharmacology/Neurobiology, Biozentrum, University of Basel, Switzerland. The human cytochrome P450 (CYP) allele nomenclature database. <http://www.cypalleles.ki.se/>; 2013 [accessed 19.06.13].
- 115.Stanford University. PharmGK. <http:// www.pharmgkb.org> Published 2001 [accessed 19.06.13].
- 116.Isaza CA, Henao J, López AM, Cacabelos R. Isolation, sequence and genotyping of the drug metabolizer CYP2D6 gene in the Colombian population. Meth Find Exp Clin Pharmacol. 2000;22(9):695–705. doi: 10.1358/mf.2000.22.9.802286. [DOI] [PubMed] [Google Scholar]
- 117.Mizutani T. PM frequencies of major CYPs in Asians and Caucasians. Drug Metab Rev. 2003;35(2–3):99–106. doi: 10.1081/dmr-120023681. [DOI] [PubMed] [Google Scholar]
- 118.Ozawa S, Soyama A, Saeki M, Fukushima-Uesaka H, Itoda M, Koyano S. Ethnic differences in genetic polymorphisms of CYP2D6, CYP2C19, CYP3As and MDR1/ABCB1. Drug Metab Pharmacokin. 2004;19(2):83–95. doi: 10.2133/dmpk.19.83. [DOI] [PubMed] [Google Scholar]
- 119.Weinshilboum RM, Wang L. Pharmacogenetics and pharmacogenomics: development, science, and translation. Annu Rev Genomics Hum Genet. 2006;7:223–245. doi: 10.1146/annurev.genom.6.080604.162315. [DOI] [PubMed] [Google Scholar]
- 120.Marquez B, Van Bambeke F. ABC multidrug transporters: target for modulation of drug pharmacokinetics and drug-drug interactions. Curr Drug Targets. 2011;12(5):600–620. doi: 10.2174/138945011795378504. [DOI] [PubMed] [Google Scholar]
- 121.Haufroid V. Genetic polymorphisms of ATP-binding cassette transporters ABCB1 and ABCC2 and their impact on drug disposition. Curr Drug Targets. 2011;12(5):631–646. doi: 10.2174/138945011795378487. [DOI] [PubMed] [Google Scholar]
- 122.Cacabelos R. The metabolomics paradigm of pharmacogenomics in complex disorders. Metabolomics. 2012;2:5. [Google Scholar]
- 123.Hosoya K, Tachikawa M. Roles of organic anion/cation transporters at the blood-brain and blood-cerebrospinal fluid barriers involving uremic toxins. Clin Exp Nephrol. 2011;15(4):478–485. doi: 10.1007/s10157-011-0460-y. [DOI] [PubMed] [Google Scholar]
- 124.Carl SM, Lindley DJ, Couraud PO, Weksler BB, Romero I, Mowery SA. Knipp GT.ABC and SLC transporter expression and Pot substrate characterization across the human CMEC/D3 blood-brain barrier cell line. Mol Pharm. 2010;7(4):1057–1068. doi: 10.1021/mp900178j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr., Kawas CH. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Goldman JS, Hahn SE, Catania JW, LaRusse-Eckert S, Butson MB, Rumbaugh M. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical Genetics and the National Society of Genetic Counselors. Genet Med. 2011;13(6):597–605. doi: 10.1097/GIM.0b013e31821d69b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Schipper HM. Presymptomatic apolipoprotein E genotyping for Alzheimer’s disease risk assessment and prevention. Alzheimers Dement. 2011;7(4):e118–e123. doi: 10.1016/j.jalz.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 130.Kopits IM, Chen C, Roberts JS, Uhlmann W, Green RC. Willingness to pay for genetic testing for Alzheimer’s disease: a measure of personal utility. Genet Test Mol Biomarkers. 2011;15(12):871–875. doi: 10.1089/gtmb.2011.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ohara T, Ninomiya T, Kubo M, Hirakawa Y, Doi Y, Hata J. Apolipoprotein genotype for prediction of Alzheimer’s disease in older Japanese: the hisayama study. J Am Geriatr Soc. 2011;59(6):1074–1079. doi: 10.1111/j.1532-5415.2011.03405.x. [DOI] [PubMed] [Google Scholar]
- 132.Akinleye I, Roberts JS, Royal CD, Linnenbringer E, Obisesan TO, Fasaye GA. Differences between African American and white research volunteers in their attitudes, beliefs and knowledge regarding genetic testing for Alzheimer’s disease. J Genet Couns. 2011;20(6):650–659. doi: 10.1007/s10897-011-9377-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sowell RA, Owen JB, Butterfield DA. Proteomics in animal models of Alzheimer’s and Parkinson’s disease. Ageing Res Rev. 2009;8(1):1–17. doi: 10.1016/j.arr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Britschgi M, Rufibach K, Bauer Huang SL, Clark CM, Kaye JA, Li G. Modeling of pathological traits in Alzheimer’s disease based on systemic extracellular signaling proteome. Mol Cell Proteomics. 2011;10(10) doi: 10.1074/mcp.M111.008862. M111.008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Khachaturian ZS, Petersen RC, Gauthier S, Buckholtz N, Corey-Bloom JP, Evans B. A roadmap for the prevention of dementia: the inaugural Leon Thal Symposium. Alzheimers Dement. 2008;4(3):156–163. doi: 10.1016/j.jalz.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cacabelos R. Role of nutrition in the prevention of Alzheimer’s disease. Aging Health. 2005;1(3):359–362. [Google Scholar]
- 137.Patel L, Grossberg GT. Combination therapy for Alzheimer’s disease. Drugs Aging. 2011;28(7):539–546. doi: 10.2165/11591860-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 138.Álvarez XA, Cacabelos R, Sampedro C, Couceiro V, Aleixandre M, Vargas M. Combination treatment in Alzheimer’s disease: results of a randomized, controlled trial with cerebrolysin and donepezil. Curr Alzheimer Res. 2011;8(5):583–591. doi: 10.2174/156720511796391863. [DOI] [PubMed] [Google Scholar]
- 139.Need AC, Motulsky AG, Goldstein DB. Priorities and standards in pharmacogenetic research. Nat Genet. 2005;37(7):671–681. doi: 10.1038/ng1593. [DOI] [PubMed] [Google Scholar]
- 140.Carril JC, Martínez R, Fernández-Novoa L, Carrera I, Corzo L, Fraile C. The EuroEspes pharmacogenetic card. Personalization in pharmacological treatment. Gen-T. 2011;3:89–102. [Google Scholar]