An official website of the United States government
Here's how you know
Official websites use .gov
A
.gov website belongs to an official
government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you've safely
connected to the .gov website. Share sensitive
information only on official, secure websites.
As a library, NLM provides access to scientific literature. Inclusion in an NLM database does not imply endorsement of, or agreement with,
the contents by NLM or the National Institutes of Health.
Learn more:
PMC Disclaimer
|
PMC Copyright Notice
Since January 2020 Elsevier has created a COVID-19 resource centre with free information in English and Mandarin on the novel coronavirus COVID-19. The COVID-19 resource centre is hosted on Elsevier Connect, the company's public news and information website. Elsevier hereby grants permission to make all its COVID-19-related research that is available on the COVID-19 resource centre - including this research content - immediately available in PubMed Central and other publicly funded repositories, such as the WHO COVID database with rights for unrestricted research re-use and analyses in any form or by any means with acknowledgement of the original source. These permissions are granted for free by Elsevier for as long as the COVID-19 resource centre remains active.
Humans have been fighting disease since the dawn of civilization and will probably do so till its end. Every century has had its medical revolution. But the last century was special: we started to uncover the most fundamental biological mechanisms designed to keeps us healthy every second of our lives. The discoveries of these mechanisms, collectively referred to as the immune system, have had an unprecedented positive impact on human health. Understanding the nature of infectious diseases, the discovery of antibiotics and vaccination campaigns have already saved hundreds of millions of lives. And this is just the beginning.
Viruses fail to fulfill the classic criteria for a living organism, i.e., they have no metabolism of their own and they use the reproductive mechanism of their host animals, plants, or bacteria for their own reproduction. This inability to reproduce outside host cells makes it impossible for them to survive independently. If they are not living organisms, what are they?
They are basically programs that are added into the genome of their hosts, hijacking their reproductive mechanisms to produce more viruses—very much like computer viruses.
Because viruses use the hosts’ metabolic functions, inhibitors such as antibiotics have no effect on them—there is no metabolism to be disrupted. The only point of possible intervention is viral interaction with the host (Fig. 5.5 and Fig. 5.6).
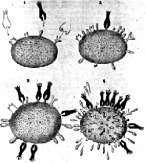
How viruses attack cells. Left: bacteriophages attack Escherichia coli.
Right: A retrovirus attacks a human cell.
There are two kinds of viruses—enveloped viruses and naked viruses. The genome of naked viruses is protected by a capsid (Engl. core), whereas enveloped viruses form their envelope by pinching off a bud from the host’s cell membrane, into which they insert virus-coded proteins. This lipid–protein viral envelope contains a capsid, similar to that of naked viruses (Fig. 5.8
).
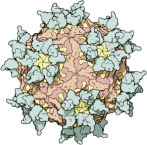
Above: subunits of a virus combine to form a capsid. Right: The polio virus, which was fully synthesized in the test tube for the first time in 2002, consists of a long RNA strand in a hollow protein capsid.
Unlike microorganisms, viruses only require their nucleic acid and a host cell for their reproduction. Some viruses, however, also contain the enzymes they need for replication. This applies to retroviruses that contain reverse transcriptase in their capsid (see Chapter: The Wonders of Gene Technology).
Viruses are completely dependent on their host cells as far as the reproduction of their nucleic acid and the synthesis of their proteins is concerned.
Some viruses reproduce using the lytic cycle (Greek lysis, dissolution—a word that also gave the name to Fleming’s lysozyme, Chapter: Enzymes: Molecular Supercatalysts for Use at Home and in Industry), during which the virus is released and destroys the host cell.
Other viruses prefer a nonlytic cycle in which they form buds that are pinched off from the cell membrane (this is how it works in enveloped viruses such as influenza viruses or HIV).
All viruses contain a single type of nucleic acid: RNA or DNA. They are classified according to their nucleic acid, their protein covers, and their host specificity.
Examples of RNA viruses include the AIDS virus HIV, the influenza viruses, the measles virus, the rabies virus, and a plant virus known as tobacco mosaic virus (TMV, see Chapter: The Wonders of Gene Technology). The latter two are rod-shaped. Other viruses in this category include the picornavirus group, e.g., poliovirus and rhinovirus, which is responsible for colds (Fig. 5.5
), and the Ebola virus that has caused so many deaths in Africa.
The SARS (Severe Acute Respiratory Syndrome) virus is a major cause for concern, especially in Hong Kong, China and Canada (Figure 5.1, Figure 5.3
). This is another RNA virus, called a coronavirus, because its surface resembles a crown (corona, Latin for crown).
In spite of the SARS (Severe Acute Respiratory Syndrome) outbreak, my biotechnology lectures in Hong Kong continued. The topic, of course, was “detection of viruses”…
The SARS virus is a coronavirus. It works in a different way from, say, the AIDS virus. It introduces its single-stranded RNA into the host cell and uses RNA-dependent RNA polymerase to produce mirror image copies.
DNA viruses include, e.g., papovaviruses, which mostly cause warts, but some species may cause tumors. Others are Variola(smallpox) and Vaccinia(cowpox) viruses, herpes viruses, adenoviruses (causing infections of the mucous membrane), bacteriophages that attack bacteria (Greek phagein, to eat), and baculoviruses that exclusively attack insects.
A new global threat could come from combined bird and human influenza viruses.
5.2. How Viruses Attack Cells
Viruses always bind first to the surfaces of cells (Fig. 5.6
). DNA viruses such as bacteriophages inject their genetic material (double-stranded DNA) into the bacterial cell (Fig. 5.6, left). With the help of the bacterial cell, they produce enzymes (DNA and RNA polymerase) that are used to synthesize DNA and mRNA. The viral mRNA is synthesized by the bacterial RNA polymerase and read by the bacterial ribosome. Thus, the bacterial cell produces the viral protein envelope as well as its DNA from bacterial building blocks. Eventually, the parts that make up a bacteriophage assemble join to form a complete bacteriophage that lyses the host cell.
In some cases, however, viral DNA is inserted into bacterial DNA without lyzing the cell. Such DNA is called dormant viral DNA, which will only be released and reproduced in later bacterial generations. In animal cells (Fig. 5.6, right), viruses bind to receptors on the cell surface, and the protein envelope merges with the cell membrane to let the virus enter.
In RNA viruses of the retrovirus group (e.g., HIV), single-stranded RNA enters the cell and is converted into double-stranded DNA with the help of an enzyme carried by the virus (reverse transcriptase, Chapter: The Wonders of Gene Technology).
The transcribed viral DNA is inserted into chromosomal DNA in the nucleus. The transcription mechanism of the host cell (RNA polymerase) then transcribes it into mRNA, which becomes a blueprint for the synthesis of viral proteins in the ribosomes. These include nonstructural proteins which are the cause of the pathogenicity of many viruses. The newly produced viral RNA and the viral proteins assemble to form new viruses which exit the cell.
Only very few types of viruses integrate their genome into that of the host. These include herpes viruses and retroviruses. Genome integration enables a virus to remain stable in the genome of the host cell over many generations of cell division. The infection is dormant and the infected organism may display absolutely no sign of any disease or malfunction.
In other cases, viruses such as the hepatitis B virus or some papilloma viruses cause DNA breakages when integrating host cell genomes. Such “abortive integration” is partly responsible for the development of tumors. For example, nearly 80% of all cervical cancers are caused by specific variants of HPV (human papilloma virus).
The search for strategies against the attack of viruses is a major undertaking (Box 5.1
). It is conceivable that specific antibodies (see later in the Chapter) could trap and neutralize viruses before they dock onto the host cell and invade it. Antibodies could also prevent invasion by masking the relevant binding sites on the target cells so that the viruses would fail to recognize them (Fig. 5.9
). Antibodies can also tag viruses so that they can be recognized and destroyed by immune system cells, such as macrophages and granulocytes.
Box 5.1. Antiviral Drugs.
There are many strategies to try to stop the spread of HIV. Attempts are being made to interfere with every single stage of the viral reproduction cycle.
1.
Docking on to cells that have not yet been infected: The viral envelope protein gp120 binds to the CD4 receptor on the surface of helper T lymphocytes.
Antibodies to CD4 could saturate the docking sites on the cell. An alternative would be to synthesize CD4 molecules, which could be injected into the bloodstream and bind to gp120 in the viral envelope, thus preventing infection. Both methods work in the lab, but could lead to immunological complications in vivo, as CD4 or anti-CD4 would interfere with the interaction of natural CD4 and its ligands.
2.
The inhibition of reverse transcriptase is an effective method. As HIV is a retrovirus, its RNA must first be transcribed into DNA. Compounds such as azidothymidine (AZT, also known as zidovudine), lamivudine, and dideoxyinosine (ddi) are structurally analogous to nucleotides and can be used “by mistake” by reverse transcriptase to produce a defective vital genome.
The first potent active agent against herpes, i.e., acyclovir, is another drug that works as an inhibitor through structural analogy to nucleotides, blocking reverse transcriptase (revertase). It is very effective as an ointment to treat herpes simplex and herpes zoster (shingles). Other inhibitors block the active site of HIV reverse transcriptase (e.g., nevirapine and delavirdine).
3.
Antisense RNA is an RNA copy that is precisely complementary to the HIV genome. Antisense RNA does not code for proteins and has thus no function in the cell. As the viral genome is single-stranded RNA, which is released during infection, it could immediately bind to the “waiting” antisense RNA to form a stable, “useless” RNA/RNA hybrid which would not be able to produce a provirus. Such a therapeutic approach would involve gene therapy or stem cells (see Chapter: Analytical Biotechnology and the Human Genome).
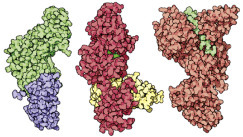
Computer-aided drug design: Drugs can be designed and tested on computers. Automated docking methods are used to find the best docking sites on a biomolecule. If the predicted bond is strong enough, the molecule can be synthesized and its activity tested. The best site for saquinavir is shown in red.
HIV protease (top) and AIDS drugs indinavir, saquinavir, ritonavir, and nelfinavir (top left to bottom right).
The antisense drug fomivirsen has been successfully used for the treatment of a viral eye infection in AIDS patients and has saved their eyesight.
4.
Inhibition of HIV protease. Drugs that inhibit protease are a triumph of modern medicine and molecular design. The protease cleaves off the long polypeptide chains produced by the virus and cuts them into small fragments precisely when they are needed to pack the new viruses. The drug that firmly binds to the protease and blocks its action prevents the virus from maturing to its infectious stage.
How antibodies neutralize a virus: Only the binding arms of the antibodies are shown here (in light blue), not their “feet.” The diagram clearly shows how viral spikes are covered by the antibodies and thus neutralized.
Retroviruses can be fought with reverse transcriptase inhibitors (see Chapter: The Wonders of Gene Technology) that prevent the transformation of RNA into DNA. Another approach is to use antisense RNA—an exact mirror image (see Chapter: Analytical Biotechnology and the Human Genome) of the viral RNA—which it can bind and inactivate (Box 5.1).
A fairly recent strategy consists of employing short double-stranded RNA sections (RNAi—the i standing for interference. More details in Chapter: Myocardial Infarction, Cancer, and Stem Cells: Biotechnology Is a Life Saver). Artificially created RNAi, between 21 and 23 nucleotides long, was used by German scientist Tom Tuschl to silence mammalian genes without triggering a disruptive interferon response (which would have led to the degradation of all RNA present). Since then, it has been possible to silence specific genes—e.g., for HIV, the nef, rev, gag, and pol genes. There are also initial successes in the fight against influenza and hepatitis C viruses.
Many HIV therapeutics are based on inhibitors of virus-coded protease, which plays an important part in the maturation process of viral proteins. All
these different routes are followed in contemporary AIDS research (Box 5.1). As the HIV virus mainly attacks T-helper cells that control the human immune response, it should be possible to strengthen the immune response by providing genetically engineered cytokines: single molecules such as interleukin-2 that modulate immune responses. In a first step, the virus could be put out of action with “chemical weapons,” and then immune cells could be stimulated by interleukin-2.
Some viruses cause infected cells to produce another type of cytokines: interferons. The secreted or artificially introduced interferon binds to specific receptor molecules on the surface of other, uninfected cells, making them resistant to the virus.
Just like the lymphokine interleukin-2 (IL-2), interferons were first enthusiastically hailed as the wonder drugs of the future that would be able to cure a host of diseases from the common cold to cancer. However, they did not fulfill these unrealistic expectations. Their effectiveness as a cure is limited, whereas the side effects are often considerable. However, like IL-2, they have their place in the treatment of human disease, usually in combination with other medications (see Chapter: Myocardial Infarction, Cancer, and Stem Cells: Biotechnology Is a Life Saver).
5.3. How the Body Defends Itself Against Infections—Humoral Immune Response Through Antibodies
When Europeans conquered the Americas, they were assisted by biological weapons of which they were not aware at the time—bacteria and viruses. These killed a large proportion of the native inhabitants. Between the 16th and the 19th century, the influx of conquerors and settlers into the Americas and Oceania brought measles, smallpox, influenza,
typhoid, diphtheria, malaria, mumps, whooping cough, the plague, tuberculosis, and yellow fever, whereas the natives had just one fatal pathogen “on their side”—syphilis. It seems that epidemics were something unknown on the American continent.
Box 5.2. The Expert’s View: Testing for HIV Infection.
There are many reasons for wanting to establish an individual’s HIV status, i.e., to test whether or not he or she is infected with HIV. For the individual, knowing one’s status is key to benefiting from the enormous medical advances that have occurred over the past 30 years: with modern antiretroviral therapy, most infected people can enjoy a high quality of life while living with HIV rather than dying from AIDS. In addition, large numbers of HIV tests are done to ensure the safety of blood donated for transfusion and to screen pregnant women, so that measures can be taken to reduce the risk of mother-to-child transmission.
Before any HIV test is done, however, the individual’s informed consent should be obtained.
The term “AIDS test” should be avoided: AIDS is a clinically defined condition that develops in most HIV-infected individuals after a number of years of infection; testing is done to identify the presence of HIV.
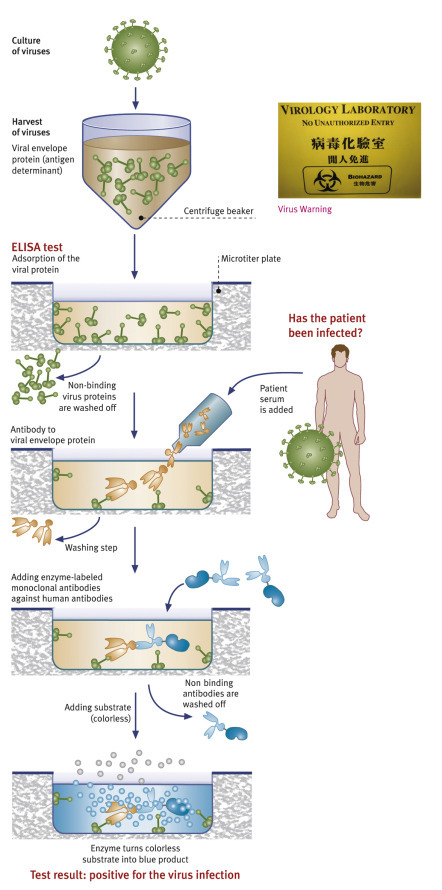
HIV infection is normally diagnosed indirectly, through detecting virus-specific antibodies. Virtually all HIV-infected individuals will form such antibodies, but unfortunately these do not confer immunity, in contrast to most other viral infections.
There are various tests to detect antibodies. Most commonly, so-called enzyme-linked immunosorbent assays (ELISA) are used for screening purposes. Such screening tests have a very high sensitivity, i.e., they are able to identify positive samples. Typically, far less than 1 in 1000 positive samples will yield a false-negative test result. This is achieved through the use of appropriate antigens (to which patients’ antibodies react in the test) and careful optimization of the whole assay (designed to make the antigen–antibody reaction visible), in order to minimize the chance of obtaining a false-negative result and thus missing the diagnosis of an infection.
On the other hand, the specificity, i.e., the ability to correctly identify negative samples, of screening tests is usually not as high; this means that occasionally a sample will have a positive (or, rather, reactive) result although it does not actually contain antibodies against HIV. This nonspecific reactivity can be caused by numerous factors, most of which are not associated with any pathological condition. A reactive (“positive”) screening test on its own does not necessarily mean that the person tested is infected with HIV!
The Human immunodeficiency virus (HIV) is the cause of Acquired Immunodeficiency Syndrome (AIDS). It is a retrovirus—an enveloped virus that has an RNA genome.
To replicate, the RNA genome undergoes reverse transcription into DNA with the help of reverse transcriptase. Another enzyme, known as integrase, helps to integrate the viral DNA into the host genome. In AIDS, the immune system begins to fail, leading to life-threatening opportunistic infections. Infection with HIV can be transferred via blood, semen, vaginal fluid, preejaculate, or breast milk.
These bodily fluids may contain free virus particles or viruses within infected immune cells. The three major transmission routes are unprotected sexual intercourse, contaminated needles, and transmission from an infected mother to her baby at birth or through breast milk. HIV primarily infects vital cells in the human immune system such as T-helper cells (specifically T-cells), macrophages, and dendritic cells. HIV infection leads to low levels of T-cells through different mechanisms. When T cell numbers fall below a critical level, cell-mediated immunity is lost, and the body becomes increasingly susceptible to opportunistic infections.
If untreated, eventually most HIV-infected individuals develop AIDS and die, while about one in ten remain healthy for many years, with no noticeable symptoms.
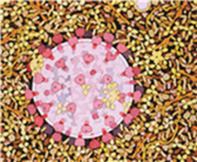
AIDS virus in the blood, surrounded by Y-shaped antibodies.
For this reason, each reactive screening test result must be confirmed by at least one confirmatory assay. This can be the so-called Western blot (obligatory in the United States and Germany) or a series of different tests applied in a defined sequence (algorithm). Only if this confirmatory testing confirms the sample’s reactivity can HIV infection be diagnosed and the patient be told that he or she is HIV-positive. A second blood sample should then also be sent for testing to confirm the specimen’s identity.
While the sensitivity and specificity of a specific HIV assay are usually known, another performance parameter is more relevant in practice. We do not actually know the “true” HIV status of the patient tested but we have to deduce it from the test results.
The positive predictive value (PPV) is the probability with which a positive test result indicates a truly infected patient; vice versa, the negative predictive value (NPV) is the probability with which a patient’s negative test result reflects that he/she is truly uninfected. These predictive values depend not only on the sensitivity and specificity of the test used but also on the HIV prevalence of the population tested. Unfortunately, this statistical phenomenon is often misused to “prove” the alleged uselessness of HIV testing. In groups with a very low HIV prevalence (e.g., carefully selected blood donors), the majority of those with a reactive screening test result are indeed not infected. But this is exactly why all reactive screening tests must be confirmed before a diagnosis is made; it is not a reason to decry HIV tests as useless! In high prevalence populations, the vast majority of reactive test results sadly reflect true positivity; this is why the World Health Organization’s guidelines stipulate simpler confirmatory algorithms in such settings.
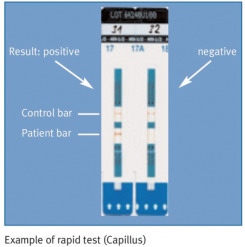
In some settings, so-called rapid/simple test devices (also called point-of-care tests) are preferable to laboratory-based testing. These can often be done on capillary blood (obtained from the tip of a finger), are easy to perform, require minimal equipment, and typically produce results that are available within half an hour or less.
Rapid tests are valuable if the result is needed quickly: in emergency rooms, after needle stick injuries, etc. They can also help to reduce the rate of “unclaimed” test results (i.e., patients not returning to get their test results) and are the only feasible option in many resource-constrained settings. An algorithm consisting of different rapid tests may even be used for confirmatory purposes, obviating the need to send samples to a laboratory at all. However, quality control of rapid testing is a formidable but extremely important challenge.
All tests based on the detection of HIV-specific antibodies have one problem in common: They do not recognize individuals in the very early stages of infection while the body is still mounting an immune response. The length of time until antibodies become detectable is known as the “window period.” Approaches are available to significantly shorten the “window”: direct diagnosis by isolating an infectious virus or by detecting viral antigens or viral genomic material (nucleic acid). Virus isolation requires cell culturing in a specialized laboratory and is therefore impractical and expensive.
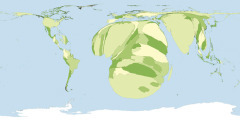
Prof. Mark Newman (University of Michigan) and his team have created new types of maps: On top, there is normal map of the world, showing the sizes of the countries of the world in proportion to their actual size on the surface of the planet and retaining their actual shapes. It is possible, however, to redraw the map with the sizes of countries made bigger or smaller in order to represent something of interest. Such maps are called cartograms and can be an effective and natural way of portraying geographic or social data. Below, the cartogram shows the number of inhabitants with HIV/AIDS (Maps courtesy of Mark Newman, UMICH).
HIV infection in humans is now a pandemic. As of 2013 the Joint United Nations Programme on HIV/AIDS (UNAIDS) and the World Health Organization (WHO) estimate that AIDS has killed more than 39 million people since it was first recognized in December 1981. AIDS is one of the most destructive pandemics in recorded history. In 2015, AIDS claimed an estimated 364 million lives. It is estimated that about 0.6% of the world’s living population is infected with HIV.
A third of these deaths are occurring in sub-Saharan Africa, retarding economic growth and increasing poverty. According to current estimates, HIV is set to infect 90 million people in Africa, resulting in a minimum estimate of 18 million orphans. Antiretroviral treatment reduces both the mortality and the morbidity of HIV infection, but routine access to antiretroviral medication is not available in all countries yet.
Testing for viral p24 antigen has become an integral component of testing, using fourth-generation screening assays that detect viral antigen alongside specific antibodies, and this method thus offers a much reduced “window period.” Testing for viral genome (nucleic acid testing=NAT), e.g., using polymerase chain reaction (PCR), is done under certain circumstances: to exclude infectivity in blood donors and to diagnose HIV infection in patients with suspected primary infection and in babies born to HIV-infected mothers.
In such babies, passively (transplacentally) acquired maternal HIV antibodies are normally detectable up to around 12–15 months of age.
Therefore, children of HIV-positive mothers will initially have positive HIV antibody test results, until they have eliminated maternal antibodies. Fortunately, the majority of these babies will not themselves be infected; good prevention of mother-to-child transmission (PMTCT) programs can reduce the rate of vertical transmission to below 1%. NAT allows the early identification of infected babies and thus the initiation of appropriate clinical management.
When antibody tests are impractical, one can test for proviral cDNA in leukocytes using a qualitative (“yes or no?”) assay to diagnose infection. The quantification of HIV RNA in blood plasma (“how much?”), the so-called viral load, may serve as a prognostic marker, to monitor the success of therapy and to estimate infectiousness. It has become a very important tool in the context of antiretroviral therapy. However, viral load tests are not intended to diagnose infection and may occasionally give false low-positive results in noninfected individuals.
In the right hands and done by skilled professionals, tests for HIV infection are nowadays extremely reliable and can give a definitive answer in almost all cases. If done in time, HIV testing allows avoidance of serious disease or even death, through antiretroviral treatment and reduction of the risk of transmission to others through preventative measures.
Born in Frankfurt am Main, Germany, Wolfgang Preiser studied medicine and later specialized in medical virology at J.W. Goethe University in his hometown and at University College London, UK. During the outbreak of severe acute respiratory syndrome (SARS) in 2003, he was involved in the identification of the etiologic agent and was sent to China as a temporary advisor to the World Health Organization (WHO). Since 2005, he has been Professor and Head of the Division of Medical Virology at Stellenbosch University in South Africa. His research focuses on developing and evaluating novel forms of laboratory diagnosis of viral infections and on tropical and emerging viruses.
Stephen Korsman is a medical virologist at the Walter Sisulu University/Mthatha National Health Laboratory Service Pathology Department in South Africa. He earned his MD in 1999 and became a Fellow of the College of Pathologists of South Africa as a clinical virologist in 2005. He was awarded a MMed in clinical virology in 2006. His fields of interest are mother-to-child transmission of HIV, emerging infectious diseases, molecular diagnostics, and medical education.
US Food and Drug Administration (FDA), Center for Biologics Evaluation and Research (CBER): Licensed/Approved HIV, HTLV and Hepatitis Tests: http://www.fda.gov/cber/products/testkits.htm
This is the 14th edition of a regularly updated medical textbook that provides a comprehensive and up-to-date overview of the treatment of HIV Infection (825 pages, ISBN: 3-924774-50-1—ISBN13: 978-3-924774-50-9). Full text available free online at http://hivmedicine.com/
According to Jared Diamond, the inequality of weapons between Red Indians or Indios and Europeans was caused by the presence of the large cattle herds of the settled Eurasian farmers. These herds became the breeding ground for acute and endemic diseases that later spread to similarly crowded human settlements (Box 5.4
).
Box 5.4. Biotech History: Jared Diamond: Lethal microbes.
The importance of lethal microbes in human history. In his 1998 Pulitzer Prize–winning book “Guns, Germs, and Steel—A Short History of Everybody for the Last 13,000 Years,” Professor Jared Diamond (b. 1937) starts an inquiry into the reasons why Europe and the Middle East became the cradles of modern societies. He kindly permitted the reprint of a drastically shortened extract about the role of microbes in human history:
The importance of lethal microbes in human history is well illustrated by European’s conquest and depopulation of the New World.
Far more Native Americans died in bed from Eurasian germs than on the battlefield from European guns and swords.
Those germs undermined Indian resistance by killing most Indians and their leaders and by sapping the survivors’ morale. For instance, in 1519 Cortés landed on the coast of Mexico with 600 Spaniards, to conquer the fiercely militaristic Aztec Empire with a population of many millions. That Cortés reached the Aztec capital of Tenochtitlán, escaped with the loss of “only” two-thirds of his force, and managed to fight his way back to the coast demonstrates both Spanish military advantages and the initial naïveté of the Aztecs. But when Cortés’s next onslaught came, the Aztecs were no longer naive and fought street by street with the utmost tenacity.
What gave the Spaniards a decisive advantage was smallpox, which reached Mexico in 1520 with one infected slave arriving from Spanish Cuba. The resulting epidemic proceeded to kill nearly half of the Aztecs, including Emperor Cuitláhuac. Aztec survivors were demoralized by the mysterious illness that killed Indians and spared Spaniards, as if advertising the Spaniard’s invincibility. By 1618, Mexico’s initial population of about 20 million had plummeted to about 1.6 million.
A woodcut of the Four Horsemen of the Apocalypse (1497–1498) by Albrecht Dürer (1471–1528, one of the greatest German artists).
Traditionally, the four horsemen stand for Pestilence, War, Famine, and Death.
From the King James Version of the Bible, Revelation Chapter 6, Environmental Biotechnology: From One-Way Streets to Traffic Circles, verses 2–8:
“And I saw, and behold a white horse: and he that sat on him had a bow; and a crown was given unto him: and he went forth conquering, and to conquer. And when he had opened the second seal, I heard the second beast say, Come and see. And there went out another horse that was red: and power was given to him that sat thereon to take peace from the earth, and that they should kill one another: and there was given unto him a great sword. And when he had opened the third seal, I heard the third beast say, Come and see. And I beheld, and lo a black horse; and he that sat on him had a pair of balances in his hand. And I heard a voice in the midst of the four beasts say, A measure of wheat for a penny, and three measures of barley for a penny; and see thou hurt not the oil and the wine. And when he had opened the fourth seal, I heard the voice of the fourth beast say, Come and see. And I looked, and behold a pale horse: and his name that sat on him was Death, and Hell followed with him. And power was given unto them over the fourth part of the earth, to kill with sword, and with hunger, and with death, and with the beasts of the earth.”
Pizarro had similarly grim luck when he landed on the coast of Peru in 1531 with 168 men to conquer the Inca Empire of millions. Fortunately for Pizarro and unfortunately for the Incas, smallpox had arrived overland around 1526, killing much of the Inca population, including both the emperor Huayna Capac and his designated successor. The result of the throne’s being left vacant was that two other sons of Huayna Capac, Atahualpa and Huascar, became embroiled in a civil war that Pizarro exploited to conquer the divided Incas.
When we in the United States think of the most populous New World societies existing in 1492, only those of the Aztecs and the Incas tend to come to our minds. We forget that North America also supported populous
Francisco Pizarro asked the Inca leader Atahualpa to meet him and his body guards unarmed. He knew that if he had the Emperor he would have the entire Inca Empire, and all the gold which it held. With a blast of their cannons, Pizarro’s men slaughtered all the Incas in the square of Cajamarca.
Indian societies in the most logical place, the Mississippi Valley, which contains some of our best farmland today. In that case, however, conquistadores contributed nothing directly to the societies’ destruction; Eurasian germs, spreading in advance, did everything.
The 11th and last Aztec emperor, Cuauhtémoc, surrenders in August 1521, and is presented to Cortés. The words in the legend “Ycpolinh q mexuca” are translated as “Now the Mexica [Aztecs] were finished.”
When Hernando de Soto became the first European conquistador to march through the southeastern United States, in 1540, he came across Indian town sites abandoned 2 years earlier because the inhabitants had died in epidemics. These epidemics had been transmitted from coastal Indians infected by Spaniards visiting the coast. The Spaniard’s microbes spread to the interior in advance of the Spaniards themselves.
De Soto was still able to see some of the densely populated Indian towns lining the lower Mississippi. After the end of his expedition, it was a long time before Europeans again reached the Mississippi Valley, but Eurasian microbes were now established in North America and kept spreading.
By the time of the next appearance of Europeans on the lower Mississippi, that of French settlers in the late 1600s, almost all of those big Indian towns had vanished. Their relics are the great mound sites of the Mississippi Valley. Only recently have we come to realize that many of the mound-building societies were still largely intact when Columbus reached the New World, and that they collapsed (probably as a result of disease) between 1492 and the systematic European exploration of the Mississippi.
When I was young, American schoolchildren were taught that North America had originally been occupied by only about one million Indians. That low number was useful in justifying the white conquest of what could be viewed as an almost empty continent. However, archaeological excavations, and scrutiny of descriptions left by the very first European explorers on our coasts now suggest an initial number of around 20 million Indians. For the New World as a whole, the Indian population decline in the century or two following Columbus’s arrival is estimated to have been as large as 95%.
The main killers were Old World germs to which Indians had never be exposed, and against which they therefore had neither immune nor genetic resistance. Smallpox, measles, influenza, and typhus competed for top rank among the killers. As if these had not been enough, diphtheria, malaria, mumps, pertussis, plague, tuberculosis, and yellow fever came up close behind. In countless cases, whites were actually there to witness the destruction occurring when the germs arrived.
For example, in 1837 the Mandan Indian tribe, with one of the most elaborate cultures in our Great Plains, contracted smallpox from a steamboat travelling up the Missouri River from St. Louis. The population of one Mandan village plummeted from 2000 to fewer than 40 within a few weeks.
An Aztec drawing representing patients affected by smallpox at different stages.
While over a dozen major infectious diseases of Old World origins became established in the New World, perhaps not a single major killer reached Europe from the Americas. The sole possible exception is syphilis, whose area of origin remains controversial.
The one-sidedness of that exchange of germs becomes even more striking when we recall that large, dense human populations are a prerequisite for the evolution of crowd diseases. If recent reappraisals of the pre-Columbian New World population are correct, it was not far below the contemporary population of Eurasia. Some New World cities, like Tenochtitlán, were among the world’s most populous cities at the time. Yet Tenochtitlán didn’t have awful germs waiting in store for the Spaniards. Why not?
Smallpox (also known by the Latin name Variola) is a highly contagious disease unique to humans.
The main reason becomes clear, however, if we ask a simple question: From what microbes could any crowd diseases of the Americas have evolved? We’ve seen that Eurasian crowd diseases evolved from diseases of domesticated herd animals. Significantly, there were many such animals in Eurasia. But there were only five animals that became domesticated in the Americas.
In turn, this extreme paucity of domestic animals in the New World reflects the paucity of wild starting material. About 80% of the big wild mammals of the Americas became extinct at the end of the last Ice Age, around 13,000 years ago. The few domesticates that remained to Native Americans were not likely sources of crowd diseases, compared with cows and pigs. Muscovy ducks and turkeys don’t live in enormous flocks, and they’re not cuddly species (like young lambs) with which we have much physical contact.
The historical importance of animal-derived diseases extends far beyond the collision of the Old and the New Worlds. Eurasian germs played a key role in decimating native peoples in many other parts of the world, including Pacific islanders, Aboriginal Australians, and the Khoisan peoples (Hottentots and Bushmen) of southern Africa. Cumulative mortalities of these previously unexposed peoples from Eurasian germs ranged from 50% to 100%. For instance, the Indian population of Hispaniola declined from around 8 million, when Columbus arrived in AD 1492, to zero by 1535. Measles reached Fiji with a Fijian chief returning from a visit to Australia in 1875, and proceeded to kill about one-quarter of all Fijians then alive (after most Fijians had already been killed by epidemics beginning with the first European visit, in 1791).
Syphilis, gonorrhea, tuberculosis, and influenza arriving with Captain Cook in 1779, followed by a big typhoid epidemic in 1804 and numerous “minor” epidemics, reduced Hawaii’s population from around half a million in 1779–84,000 in 1853, the year when smallpox finally reached Hawaii and killed around 10,000 of the survivors. The examples could be multiplied almost indefinitely.
However, germs did not act solely to Europeans’ advantage. While the New World and Australia did not harbor native epidemic diseases awaiting Europeans, tropical Asia, Africa, Indonesia, and New Guinea certainly did. Malaria throughout the tropical Old World, cholera in tropical Southeast Asia, and yellow fever in tropical Africa were (all still are) the most notorious of the tropical killers. They posed the most serious obstacle to European colonization of the tropics, and they explain why the European colonial partitioning of New Guinea and most of Africa was not accomplished until nearly 400 years after European ship traffic, they emerged as the major impediment to colonization of the New World tropics as well. A familiar example is the role of those two diseases in aborting the French effort, and nearly aborting the ultimately successful American effort, to construct the Panama Canal.
There is no doubt that Europeans developed a big advantage in weaponry, technology, and political organization over most of the non-European peoples that they conquered.
But that advantage alone doesn’t fully explain how initially so few European immigrants came to supplant so much of the native population of the Americas and some other parts of the world. That might not have happened without Europe’s sinister gift to other continentsthe germs evolving from Eurasians’ long intimacy with domestic animals.
For Jared Diamond’s bio, see p. 26 in Chapter 1, Beer, Bread, and Cheese: The Tasty Side of Biotechnology.
Cited Literature:
Diamond J (1998) Guns, germs, and steel. A short history of everybody for the last 13,000 years. Vintage, Random House, London, pp. 210–213 and 214
This explains why outbreaks of infectious diseases as we know them are a fairly recent phenomenon. Smallpox appeared for the first time in 1600 BC, mumps and the plague at 400 BC, and cholera and louse-borne epidemic typhoid only in the 16th century.
Over several hundred years, the Eurasian population had been able to develop immunity to the diseases and reached a stalemate. The inhabitants of the New World, by contrast, did not have time to arm their totally unprepared immune systems and were almost completely wiped out by the pathogens (Box 5.4).
How does the immune system protect us? Let us just resort to a simplified answer at the moment, as the immune system is so complex that a full explanation would go beyond an introduction to biotechnology.
The immune system distinguishes between “self” and “nonself” and has the ability to produce a hundred million (108) different antibody specificities and over a trillion (1012) different T-cell receptors. It consists of two closely interconnected systems with parallel actions—the humoral and the cellular immune response.
The humoral immune response (Latin: humor=liquid) uses soluble proteins, antibodies also known as immune-globulins (Box 5.3
), as recognition elements. There are also humoral defense factors, including lysozyme (see Chapter: Enzymes: Molecular Supercatalysts for Use at Home and in Industry), interferons, and other cytokines. Antibodies bind to foreign molecules or cells, thus labeling them as intruders and encouraging phagocytosis by macrophages. Antibodies are produced by plasma cells, which, in turn, are derived from B-cells.
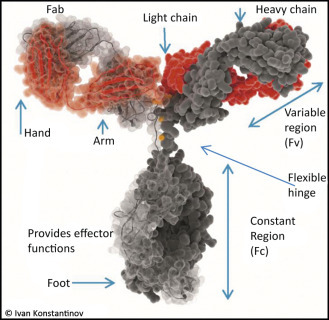
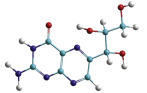
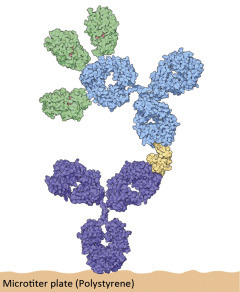
The two light chains (yellow) and the two heavy chains (red) combine to form a variable fragment that binds to the antigen and holds it in two “hands,” The variable region contains up to 20 hypervariable amino acids that allow billions of combinations for the recognition of antigens.
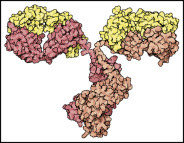
Antibodies are components of the immune systems of vertebrates that protect against intruders. Their task is to specifically recognize pathogens, bind to them and thereby label them for the immune system. They also neutralize the toxins released by many pathogens.
Three antibody fragments (Fab) bind simultaneously to the epitopes of an antigen (green). The enzyme shown is the familiar lysozyme (see Chapter: Enzymes: Molecular Supercatalysts for Use at Home and in Industry). The contours of the second Fab and the “foot” of the antibody (Fc section) have been added.
Antibody molecules consist of four protein chains—two light chains or L-chains (“light” in this context means molecular weights of 25 kD) and two heavy chains or H-chains with molecular weights of 55 kD. They are held together by disulfide bonds. The “foot” is the same for all antibodies and is called the constant region (Fc, fragment constant). The Fc part holds the antibody on the surface of the cell or provides the signals to activate other elements of the immune system against its bound target.
How is it possible for an organism to produce a specific antibody to virtually any antigen? How can it recognize the incredibly large number of possible antigens? As it would not be economical to have a specific gene for each of the approximately 100,000,000 different antibodies, the immune system resorts to an ingenious trick.
Immunotechnologists Frank Breitling and Stefan Dübel explain it as follows: Just as it is possible to build any number of different houses with a few types of standardized bricks, cells use standardized polypeptide elements to build any modular antibody. Thus, only a few hundred of these polypeptide elements need to be coded for in the genome. Some large building blocks code for the constant regions in the antibody.
The antigen-binding specificity of an antibody, however, is mediated by a small proportion of the whole protein. These are the variable regions. They, too, consist of three to four different modules which are combined individually in each cell during the differentiation of B-lymphocytes.
B-cells or B-Lymphocytes derived their name from Bursa fabricii—a lymphatic organ that is unique to birds which lies in the end section of the cloaca (Fig. 5.13
). Lymphocytes develop into B-lymphocytes in that bursa. If the bursa were removed from a chicken, it would become very susceptible to bacterial infections and would be unable to produce antibodies.
B-cells (B-lymphocytes) were named after the lymphatic organ Bursa fabricii that is found exclusively in birds, in the last section of the cloaca.
A foreign macromolecule (or a cell or virus) that elicits an immune response is called an antigen. Antibodies do not target the whole antigen, but only a portion of it, which is known as epitope or antigenic determinant.
An infection mobilizes several cooperating immune cell populations. B-lymphocytes carry antibodies as recognition molecules on their surfaces (membrane-bound immunoglobulins). However, they are usually not activated by circulating antigens. These are taken up by antigen-presenting cells—either macrophages or dendritic cells. They process the antigen, move antigen fragments to the surface and present it to T-helper cells. The fragments on these antigen-presenting cells act as a stimulus on the T-cells to produce interleukin-2, which, in turn, activates the B-cells that have also been in contact with the antigen. These begin to proliferate and form a cell clone (clonal selection). Some of the resulting daughter cells become memory cells, which ensure a rapid immune response in the case of reinfection, while others develop into antibody-producing plasma cells.
The freely circulating antibodies bind to the antigen and cells that may carry it, thus labeling the enemy for destruction by other components of the immune system.
These mechanisms are supported by the body’s own complement system, a cascade involving about 30 proteins, and by ADCC (antibody-dependent cell-mediated cytotoxicity), the destruction of foreign or abnormal cells by specific immune system killer cells.
Whether dissolved in the blood plasma or cell-bound, both the complement and ADCC systems form a defense line against microorganisms (e.g., bacteria, fungi, or parasites). Because of their powerful cell-destroying properties, they can cause tissue damage when not properly regulated, which may occur due to various diseases (heart attack, systemic Lupus erythematodes or rheumatoid arthritis).
5.4. Cellular Immune Response: Killer T-Cells
When the thymus gland is removed from young animals, they become susceptible to infection, similar to chickens after removal of the bursa (Fig. 5.13). The number of lymphocytes (white blood cells) drops dramatically after a thymectomy. Because they originate in the thymus gland, these lymphocytes were called T-lymphocytes or T-cells.
Soluble antibodies (Section 5.3), although very effective against pathogens outside cells, provide almost no protection against viruses and mycobacteria (such
as leprosy and tuberculosis). These are shielded from the antibodies by the host cell membrane. Fortunately for us, evolution has found a cunning defense strategy—cell mediated immune response (Fig. 5.10
).
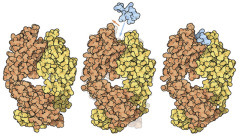
How antibodies bind to antigens: A deep cavity binds to a small fullerene molecule (top, Buckminster fullerene, shown in red), while the large flat surface of an antigen-binding site binds to a protein, lysozyme (bottom, also shown in red).
Cytotoxic T-lymphocytes or killer T-cells are always on the lookout for foreign components on the surface of all cells they encounter, and will destroy these cells where they find them (Figure 5.11, Figure 5.12
). This is not an easy task, especially because some intruders take great care not to display any antigens, but host cells have an ingenious “cut-and-display” mechanism (through proteasomes).
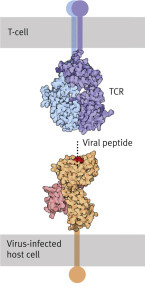
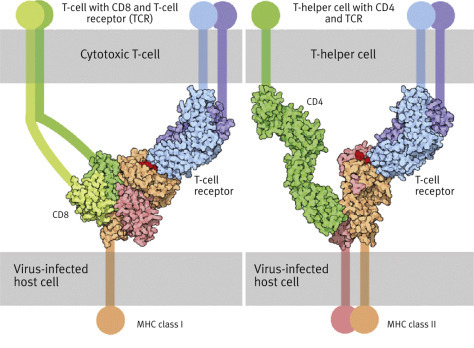
above: T-killer cell (small, in the foreground) attacking a virus-infected cell. Below left: T-cells with CD8 (green) and T-cell receptor (TCR, blue). Below right: T-helper cell with CD4 (green) and T-cell receptor (TCR, blue). Both dock onto the virus-infected cell presenting a viral peptide (red).
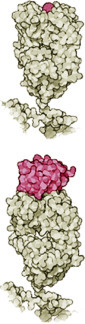
They cut out a sample of small peptides found in the cytosol, which originate from the breakdown of the intruder’s proteins. These peptides are transported to the surface of the cell membrane where they are presented by cell membrane proteins (Fig. 5.11) that have been coded by the major histocompatibility complex (MHC).
MHC proteins belong either to class I or class II (Fig. 5.12). MHC proteins of class I in an infected cell hold on very tightly to the peptides to be presented in order to enable specific receptors of a T-killer cell to bond to them. When foreign peptides are found, they act as a killer signal is emitted by the T-lymphocyte, which triggers a programmed cell death mechanism or apoptosis, i.e., “cell suicide in the interest of the whole organism.”
Cytotoxic cells carry an additional protein called CD8 (CD stands for cluster of differentiation) that recognizes complexes of MHC class I protein and the presented alien peptide. When such a complex is recognized, the T-cell secretes the protein perforin which opens pores 10 nm in diameter in the membrane of the target cell. Proteases (granzymes) are secreted into the now permeable target cell. When they die, the target cells degrade all of their constituents: their own and that of the pathogen virus or bacterium. The T-cell detaches itself from the target cell and proliferates, having proved to be an effective tool against the intruder.
Not all T-cells are cytotoxic or killer cells. T-helper cells stimulate the multiplication of B-lymphocytes and cytotoxic T-cells (Fig. 5.12). They are thus indispensable in the fight against extracellular as well as intracellular pathogens.
T-helper cells are also activated through the recognition of antigens on the surface of antigen-presenting cells—usually dendritic cells or macrophages. The antigen is presented to the T-helper cells as a peptide fragment that has been derived from the foreign protein by the antigen-presenting cell (what has been described as being “processed” earlier on). The recognition of the antigen by the T-helper cells depends on the MHC proteins class II on the antigen presenting cell. When MHC proteins class II present a peptide, it is a call for help: “This cell has been in contact with a pathogen,” whereas the message of class I is: “This cell has succumbed to the attacker. Trigger self-destruction.”
T-helper cells use a T-cell receptor and a protein (CD4) on their surfaces that carries an extracellular immunoglobulin-like domain (structured like an antibody, Fig. 5.12). The recognition of the complex triggers events that do not lead to the death of the cell, but stimulates the T-helper cells to secrete lymphokines, including interleukin-2 and interferon-γ. IL-2 stimulates the proliferation of those B-cells that have also been in contact with the antigen. These turn into antibody-producing plasma cells in the blood.
The essential role of helper T-lymphocytes is illustrated by HIV, which preferentially infects this type of immune system cell. By doing so, it interferes with the proliferation of B-cells and the production of antibodies—any antibodies. The result is immunodeficiency, the hallmark of untreated AIDS.
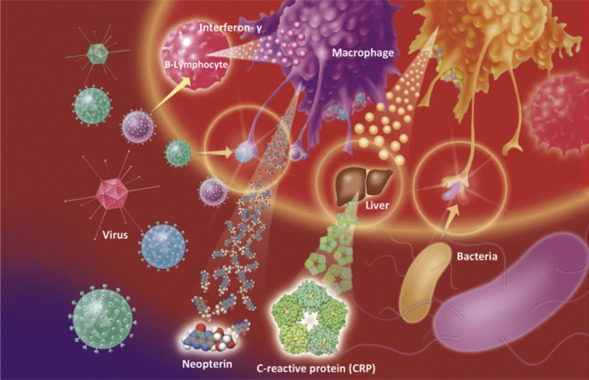
Neopterin is a newly discovered signaling substance that is activated in viral infections. This low-molecular-weight compound is secreted by macrophages when stimulated by interferon-γ. As Dietmar Fuchs and his research group (Fig. 5.25
) in Innsbruck (Austria), showed, this can be used to provide a rapid test for viral infections. A high concentration of neopterin indicates that a viral attack is taking place, even if the culprit virus has not been identified yet. This can be useful in early diagnosis, and it is an invaluable indicator for blood banks to check donors for viral infections. A neopterin test is routinely carried out at least by all Austrian blood banks.
Top: Dietmar Fuchs (University of Innsbruck, Austria), the world’s leading expert on neopterin, which is a signaling molecule produced by macrophages after a virus attack. Neopterin can be traced shortly after any kind of viral attack. Bottom: neopterin molecule.
5.5. The First Vaccination: Cowpox Against Smallpox
If the English country doctor Edward Jenner (1749–1823) were to repeat his famous experiment of 1796 (Fig. 5.15
) today, he would soon find himself in prison. He injected 8-year-old James Phipps with a sample from a cowpox pustule of dairy girl Sarah Nelmes (Fig. 5.15). Then, 2 months later, he injected the boy with a potentially lethal dose of smallpox. This would nowadays be a violation of even the most lax safety guidelines, only exceeded by the experiments of Louis Pasteur (Box 5.5
). However, the boy survived, and Jenner helped to revolutionize medicine. The first vaccine
(Latin vacca, cow) in history had been found, although it has been said that there were already active smallpox vaccinations as early as 1000 BC.
Edward Jenner vaccinating Joseph Meister (top). Jenner’s drawing of the cowpox infected arm of dairy maid Sarah Nelmes (below).
Box 5.5. Biotech History: Inoculation.
“A nonmedically qualified individual uses material of unknown composition and toxicity to treat patients, including a child, who may be suffering from a potentially fatal illness. The individual does not even try to obtain informed consent, but publishes patients’ names and addresses to help publicize some astounding claims. Moreover, like fraudulent quacks the world over, the individual keeps details of the ‘treatment’ secret, so that its validity cannot be independently validated. Perhaps worst of all, this reckless person injects human beings with an extremely virulent microbe before conducting tests in animals. Some patients die, and a close collaborator who is a medical doctor dissociates himself from his colleague’s work.
The person who took these risks, yet emerged with thunderous acclaim for his astonishing triumph in defeating rabies, was Louis Pasteur.”
This is a quote from Bernard Dixon, Power unseen. How microbes rule the world. Pasteur himself said that he was extremely lucky: “chance favors the prepared mind.” This also applies to his other pioneering achievements. Just like his predecessor Edward Jenner (1749–1823), who applied the first vaccines, he flouted several ethical principles. He postulated that the pathogen would be found in the spinal cord, although it was a completely unknown microbe then. The pathogen could be attenuated by removing the spinal cord from rabbits that had been inoculated with spinal cord material from a rabid dog and letting it age.
On July 6, 1885, he injected little Joseph Meister with some of the aged rabbit spinal cord, although he could not be sure it contained the virus. Nowadays, the torpedo-shaped rabies viruses, members of the Rhabdovirus family, can be made visible under the electron microscope (Fig. 5.5). In Pasteur’s day, they remained invisible. In contrast to Jenner’s vaccine, Pasteur’s had been produced in the lab.
Jenner, however, was on comparatively safe ground, having noticed that cowpox is innocuous. In the 11th century AD, Chinese doctors had observed that people who had survived a smallpox infection were resistant to recurring infection. In ancient China, toddlers were therefore infected with smallpox on purpose. Given the high general infant mortality, the risks associated with the vaccination appeared acceptable. In China, there was a smallpox goddess called Chuan Hsing Hua Chieh, and the equivalent Hindu goddess was Shitala mata. Vaccine reactions were less pronounced when the vaccine came from patients with a mild form of smallpox. The use of such smallpox vaccines also spread to Europe and became quite popular in the second half of the 18th century under the name of “variolation.”
In 1721, Lady Mary Wortley Montagu (1689–1762) had her daughter “variolated”—the first person in England to have been “officially” vaccinated. Experiments had been carried out on prisoners and orphans, which gave British doctors the confidence to inoculate members of the Royal family.
However, it was Edward Jenner who demonstrated that there was a generally low-risk method to achieve the same purpose, and his vaccination method was one of the reasons for the decline in smallpox, without which the Industrial Revolution could not have taken place. The true significance of Jenner’s discovery, however, was highlighted much later through Louis Pasteur’s research. Pasteur had been experimenting with Pasteurella multocidia, a poultry pathogen, and one of his cultures had been forgotten in the lab for several weeks before Pasteur used it on his chickens. He found that not only did the chickens survive the infection, but also became immune to further infections with the pathogen.
Earlier on, in 1881, Pasteur had been able to demonstrate in public that sheep could be vaccinated to protect them from anthrax. He had also been the savior of silkworm breeding, as well as having expounded the scientific principles of wine fermentation. Pasteur was definitely successful, and his successful fight against rabies became the foundation of the Institut Pasteur in Paris, as Pasteur was able to draw conclusions about immunological reactions that led to the development of a whole range of vaccination methods.
Robert Koch (1843–1910) was the founder of medical bacteriology. In a controversy with the argumentative Louis Pasteur, he was the first to show that cholera, anthrax, tuberculosis, and the plague are caused by specific bacteria. His research on tuberculosis earned Koch the Nobel Prize for Medicine or Physiology in 1905.
Numerous research trips to India, Japan, and African countries yielded valuable results in tropical medicine and parasitology, such as information about the plague, malaria, sleeping sickness (African trypanosomiasis), and cholera pathogens.
Emil von Behring (1854–1917) introduced a new vaccination procedure that became known as passive vaccination, which involves injecting antiserum (serum therapy). From 1880 to 1889, he was a medical officer (Stabsarzt) in the Prussian army and started teaching at the Institute of Hygiene and Infectious Diseases in Berlin where he became an assistant of Robert Koch. This is where he began his collaboration with the Japanese doctor and microbiologist Shibasaburo Kitasato (1856–1931).
The pharmacy of the future, as imagined in Behring’s day.
In 1890, Behring had his first successes in treating diphtheria in animals. Diphtheria was one of the most dreaded infections at the time, dubbed the “children’s destroying angel.” In cooperation with Paul Ehrlich (1854–1915), Behring succeeded in creating an antiserum to it in 1893 and saved the lives of many children.
In 1895, Emil Behring was nominated director of the Institute of Hygiene at the University of Marburg, Germany. In 1901—4 years before his much-revered teacher Robert Koch—he was awarded the Nobel Prize for Medicine and Physiology for his discovery of antibodies and the production of vaccines. He was given the noble title “von.”
In 1904, he founded a factory called BehringWerke in Marburg, where sera against diphtheria and tetanus were produced in large quantities.
Nowadays, biotechnology is revolutionizing vaccination—making it possible to develop new vaccines in a very short time while dramatically reducing the risk involved. Jenner had correctly observed that overcoming cowpox conveyed lifelong immunity in humans, not only to cowpox, but also to smallpox. What Jenner did not know was that the cowpox virus is closely related to the smallpox virus. As explained above, in case of infection by either virus, lymphocytes in the blood can trigger an alarm at the intrusion of antigens, causing on a command to other cells to produce antibodies. Antibodies, in turn, tag the pathogens for macrophages to destroy virus-infected cells and viral particles (Fig. 5.7
).
Body defenses against infections. An extensive description is given in Chapter 9, Myocardial Infarction, Cancer, and Stem Cells: Biotechnology Is a Life Saver. This picture shows macrophages, antibodies, and T-cells.
As mentioned above, B-cells proliferate after antigen contact and activation by T-helper cells.
While a proportion of their offspring produce large quantities of antibodies to tag the intruders, some become memory cells that enable a fast immune reaction, should the organism be reinfected in the future.
Memory B-cells can remain in the system for life—conveying lifelong immunity to the relevant antigen.
Jenner was in luck because the similarity in the structure of cowpox and smallpox ensured that immunity was acquired not only for the innocuous cowpox, but also for the far more dangerous smallpox. It is thus possible to prepare the immune system for the attack of life-threatening pathogens by exposing it to innocuous ones.
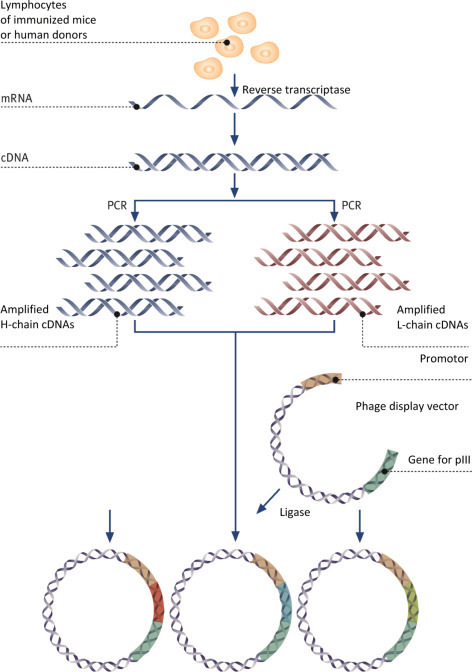
Designer Antibodies!
Following success in creating a range of therapeutic antibodies, antibody phage display is now beginning to revolutionize the discovery of antibodies for research.
The advantages are numerous: not only can animal trials be dispensed with and many animal lives saved, but the method has also large potential for miniaturization, which saves costs and increases throughput. Above all, however, it gives researchers the opportunity to exercise full and detailed control over the biochemical properties of the selected antibody.
During the in vitro selection process, the biochemical conditions can be adjusted at will—for instance, the antibody can be selected to bind to just one individual allosteric conformation of a protein, or to a particular epitope.
It is also possible to add soluble competitors during the selection of the antibody in order to counter-select for unwanted cross-reactions.
Through further miniaturization and parallelization of selection and production, a complete set of monoclonal antibodies to all proteins of the human genome could now be created at reasonable cost.
Antibodies can also be custom-made for various typical assays. For instance, Escherichia coli can biotinylate the recombinant antibodies while they are being produced in vivo, or couple them with an enzyme (alkaline phosphatase) during the actual production process.
All these options offered by antibody phage display go far beyond what could be obtained from an immunized animal.
Prof. Stefan Dübel, Technische Universität Braunschweig.
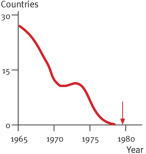
The last smallpox patient in the world, the Somali Ali Maow Maalin, was discharged from hospital on October 26, 1977 (Fig. 5.16
). Over the following 2 years, the world population was scrutinized for smallpox and finally declared smallpox-free.
The last patient in the world infected with smallpox, 23-year-old Somali Maow Maalin, in 1977. 200–300 million dollars have been spent on the eradication of smallpox in the world. Without antibiotics, Maow Maalin would not have survived the disease.
Only two laboratories in the world still store stocks of smallpox viruses (one hopes!)—the international WHO reference labs in Atlanta (United States) and near Novosibirsk (Russia). There has been a debate about the wisdom of keeping the remaining stocks. In view of possible bioterrorist attacks, however, all industrial nations keep a stock of smallpox vaccines. Who would be confident enough to say that smallpox viruses will not get into the wrong hands? It may already have happened (Fig. 5.17
).
At the beginning of the 19th century, half a million people per year contracted smallpox in Germany alone. One in ten died, and faces scarred by smallpox were a common sight. But this sad fact belongs to the past. For the first time in history, a disease has been eradicated due to vaccination.
Unfortunately, there are not many pathogens that have such close innocuous relations, and only with Louis Pasteur, born the year before Jenner died, did the systematic search for vaccines begin (Box 5.3).
5.6. Contemporary Vaccination
Nowadays, vaccination relies on toxoids for vaccination, killed or weakened live pathogens, and antigens produced by recombinant DNA technology. Thanks to the success of genetic engineering, research is also being done on modified live vaccines and peptide vaccines (Figure 5.18, Figure 5.19, Figure 5.20
).
Top: Antibiotics are ineffective in viral infections, and the only useful medication is often an antiinflammatory painkiller. (French poster text: The flu virus is everywhere… Have you got aspirin?)
Bottom: The first polio vaccine was the inactivated polio vaccine. It was developed by Jonas Salk and came into use in 1955.
The oral polio vaccine was developed by Albert Sabin (shown on stamp) and came into commercial use in 1961.
Toxoids are extracts from toxins released by pathogens. They are neutralized (sometimes using formalin) but can still stimulate the body’s immune system, when injected. Vaccines against tetanus and diphtheria belong to this class (Fig. 5.21
).
After smallpox, rinderpest (cattle plague) was the next disease that was eradicated globally. Measles will be targeted next.
The tetanus pathogenClostridium tetani (the one who dwells in the ground), e.g., can infect an open wound and inject a neurotoxic protein into the bloodstream. This results in spastic paralysis—it used to be a shocking sight among soldiers wounded in battle.
The tetanus vaccine consists of an inactivated neurotoxin, requiring a booster every 10 years to maintain a sufficient number of antibodies circulating in the system.
Cholera, polio, and typhoid vaccines consist of chemically killed bacteria or viruses. In other words, these vaccines contain pathogens that cannot cause the disease, but that retain all the antigens.
Cholera vaccine, e.g., cannot cause an outbreak of the disease, even though it contains the cholera bacterium toxin (it has been rendered ineffective). Cholera vaccine is simply swallowed: it is an active oral vaccine. It is said to be active because the organism produces its own antibodies against the killed bacteria and the toxin.
Cholera vaccination provides strong protection against the disease: approximately 90%.
Adults and children from 6 years on are given two vaccinations, between 1 and 6 weeks apart. Protection begins 8 days after the vaccination and lasts about 2 years.
Rubella and measles vaccinations rely on attenuated (weakened) pathogens. Unfortunately, there have been a number of incidents in which the pathogens had not been sufficiently attenuated.
Various genetically engineered vaccines for humans and animals have been in use since 1985, e.g., against foot-and-mouth disease in cattle. A recombinant vaccine has been recently developed against strains of HPVs that cause the majority (80%) of all cervical cancers. How effective these vaccines truly are against preventing cancer is not yet known.
The first genetically engineered vaccine designed for humans was approved in 1986 in the United States. It protects against hepatitis B, a chronic disease caused by a DNA virus (HBV) that affects some 240 million people. 686,000 people die every year due to hepatitis B. It causes one of the most frequent infectious diseases worldwide, alongside tuberculosis and HIV. Up to 25% of people affected die from HBV sequels that include cirrhosis or carcinoma of the liver. The virus is endemic in South East Asia and in sub-Saharan Africa. Thanks to vaccination programs, its presence in the Western European and US populations has been reduced to 0.1% chronic virus carriers.
The conventional production of hepatitis B vaccine was facing huge problems. In contrast to most other microorganisms, hepatitis B viruses cannot be bred in conventional nutrient media or animal embryos (such as fertilized chicken eggs). Infected blood had to be used instead. The vaccine manufacturing process consisted of isolating viruses from the blood of infected carriers, detaching viral envelope proteins with detergents (which mostly destroy the viruses), and purifying them. These envelope proteins provoked immune reactions and thus made the vaccines.
Infected blood is, of course, dangerous to work with. All members of the lab had to be immunized, i.e., vaccinated, and the work was carried out in isolated secure labs. The matter was further complicated because each batch had to be tested on chimpanzees (of which, for ethical reasons, there were only a limited number available) in order to make sure no live virus remained.
A full year was needed to produce a batch of hepatitis B vaccine in this way. Unsurprisingly, only a very limited amount of natural vaccine was available and only high-risk groups could be vaccinated.
The new genetically engineered vaccine against hepatitis is produced by genetically modified eukaryotic cells in culture: yeasts or mammalian cells. Both types of cells produce a viral surface protein. Because no virus is present at any stage, the new vaccine can never cause hepatitis.
Vaccine production in E. coli has not proved to be very effective because bacteria are not able to imitate the protein modifications carried out by pathogens, such as glycosylations in particular (see end of Chapter: The Wonders of Gene Technology).
DNA vaccines constitute a possible alternative to vaccines against proteins. The idea here is to introduce an individual gene that encodes an antigen and express it inside the organism.
Making a DNA vaccine would be very simple. Unfortunately, this approach is limited because of unwanted immune reactions, i.e., allergies.
Above: Blind monks examining an elephant, a work of Japanese artist Hanabusa Itcho (1652–1724). This is as an allegory to the immune system.
Below: How hybridoma cells for monoclonal antibodies are made: Lymphocytes are isolated from the spleen of a mouse and fused (either using a chemical such as polyethylene glycol or electrofusion) with myeloma cells, i.e., immune cells that have turned malignant.
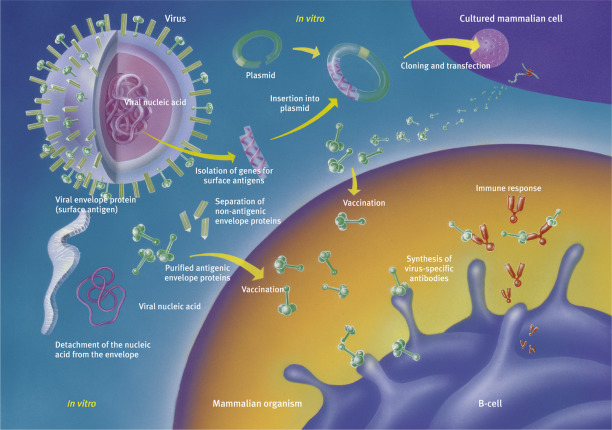
Recombinant vaccines represent a major breakthrough in the field. They can be manufactured whenever a surface protein of a pathogen can be shown to cause an immune response.
The gene responsible for this protein is isolated from the pathogen’s genome and inserted it into that of harmless microorganisms such as bakers’ yeast or mammalian organisms, which could then produce large amounts of the protein. There is an added bonus to the method: viral contamination of the vaccine is impossible.
The most comprehensive and disastrous vaccination program ever carried out in Germany started in late October 2009, after the outbreak of the swine flu virus. Little information was available about the vaccine’s benefits and risks and the public opinion about mass vaccination was hesitant and skeptical.
In late 2011, the Pandemrix vaccine reached its expiration date. Some 16 million doses, worth 130 million Euros, were burned at a temperature of 1000°C, contributing got one of the greatest flops in the history of German health services.
5.7. Live Vaccines
Rabies has been an almost worldwide scourge for most of history, with the exception of North Western Europe, Japan, Australia, and a few Pacific islands.
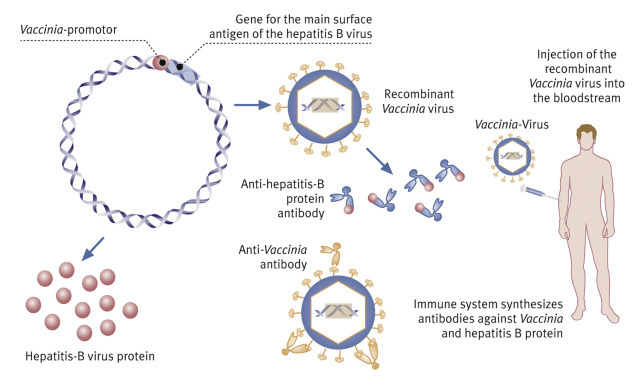
But nowadays, foxes in Europe’s woodlands are biotechnologically protected. Baits are laced with live vaccines. In live vaccines, innocuous viruses such as the cowpox virus (Vaccinia) are used as vectors to transport foreign genes.
Vaccinia double-stranded viral DNA comprises 180,000 base pairs. In 1982, it was shown that at least two larger sections of the DNA are not needed for reproduction. These can therefore be replaced with foreign DNA. Genes that code for envelope proteins with an antigenic effect can thus be stably inserted into the viral genome in a way that does not affect the ability of the vector virus to infect mammalian cells.
Up to 20 foreign genes can be simultaneously introduced in this way, preceded by Vaccinia DNA promoter sequences that switch on their expression, yielding antigenic proteins (Fig. 5.14
). The method has been successful in animal experiments, resulting in the production of surface antigens of hepatitis B, rabies, herpes simplex, and influenza viruses.
A queue of donors at the reception desk of the Guangzhou bloodbank in China. Rapid “doorkeeper” tests for syphilis and hepatitis detect infected individuals upfront who will then be barred from giving blood. It would be great to have a rapid neopterin test here!
Right: One-step production of modern vaccines against both smallpox and hepatitis.
Developing an effective vaccine against HIV has proved a formidable challenge, ever since the first trials back in 1987. There are numerous challenges to be overcome: HIV inactivates the body’s capacity to produce antibodies, the very system that makes vaccination possible. HIV antigens can rapidly change through mutation, and killed HIV is poorly antigenic. Nevertheless, at least one trial, started in Thailand in 2003, is showing promise by reducing infection rates in tested volunteers by 26%. The recombinant vaccine, dubbed RV144, consist of an innocuous form of canarypox, a bird virus harmless for humans, containing genetically engineered versions of three HIV genes. The HIV proteins that are expressed pose no risk whatsoever, but elicit an immune reaction in the organism.
Edible vaccines are highly controversial. In Chapter 7, Green Biotechnology, we will look at the creation of transgenic plants. Specifically marked (perhaps through blue pigment, see Fig. 7.52) bananas or potatoes could be used to produce such oral vaccines and be ingested as food, the antigen traversing the gastrointestinal tract and conferring immunization.
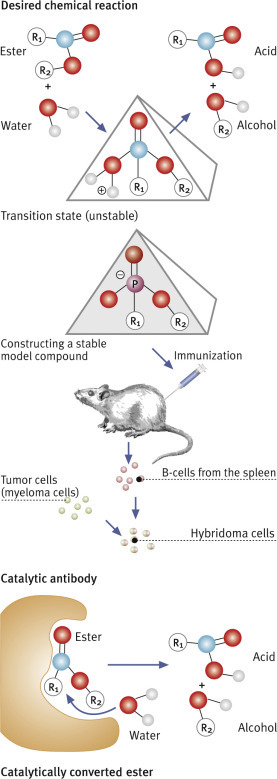
5.8. Monoclonal Antibodies
1975 saw a ground-breaking publication. Cesar Milstein (1927–2002, Box 5.8
), an Argentinian who had fled the military dictatorship in his native country and worked in Cambridge, and the German Georges Köhler (1946–1995), described a method to produce monoclonal antibodies. These are antibodies with identical molecular structure and specificity. Monoclonal antibodies ushered in a new era in biochemical analysis and medical diagnostics and therapeutics. The unique ability of the immune system to recognize certain structures at a molecular level could now be harnessed by humans. Unsurprisingly, the two scientists were awarded the Nobel Prize in 1984 (Box 5.6
).
Box 5.8. Biotech History: Monoclonal Antibodies.
Georges Köhler (1946–1995) who had studied in Freiburg, Germany and written his PhD thesis in Basel, Switzerland, went to Cambridge in 1974 to work in Cesar Milstein’s (1927–2002) lab for 2 years.
Milstein had found a way around the problem that it is not possible to grow normal lymphocytes in cell colonies to provide a supply of antibodies. He cultured myeloma cells instead, which he had obtained from mice. These are cells descending from lymphocytes which have turned malignant. They have retained the ability of lymphocytes to produce antibodies, while also having the cancer cell characteristic of being immortal. If it were possible to interbreed them somehow, i.e., fuse them with normal lymphocytes of known specificity, would the daughter cells produce those specific antibodies alongside the myeloma antibodies? In their experiment, they used erythrocytes (red blood cells) from sheep as an antigen and injected them into mice. Once the immune reaction had fully developed, they took out the spleen where lymphocytes form in large quantities. They mashed the spleen tissue and mixed it with myeloma cell cultures, adding polyethylene glycol, a chemical compound that facilitates cell fusion. They hoped that this would result in hybrids with the desired properties.
And indeed, this cellular mix and match fulfilled expectations and produced spectacular results. Using the sheep erythrocytes as test antigen, Köhler and Milstein identified a considerable number of hybrid cells that recognized the erythrocytes as foreign and produced antibodies to them. Single hybrids could be grown in cultures, having inherited the immortality trait from their myeloma cell parents. They also produced the antibody of known specificity which they had inherited from their lymphocyte parent. These cells were called hybridoma cells.
After being awarded the Nobel Prize in 1984, Georges Köhler was asked in an interview why he and Milstein had not patented their method. After all, they could have earned millions, given the billions of dollars in sales of monoclonal antibodies.
Köhler replied: “Prof. Milstein told the relevant people on the Medical Research Council that we had found something that could be patented, but we did not get any reply. So we were not bothered and publicized our method. We are scientists, not businessmen.
I don’t think scientists should patent anything. At the time, we did not think long and hard about it, but decided spontaneously, from the heart. It would have meant that I would have had to learn how to deal with money, how to negotiate licenses. It would have changed my whole personality, and I don’t think it would have done me any good.”
Box 5.6. The Expert’s View: Why is there still no vaccine for HIV?
Looking at the limitations of antiretroviral medication that is currently available, it seems that an effective vaccine would be the only way to defeat AIDS. Why, you may well ask, has such a vaccine not yet been developed? After all, AIDS has been around for over 40 years and in the past decade alone, around one billion dollars were invested in HIV vaccine research.
The answer is rather trivial—the HI virus poses an unprecedented challenge for research. Not only are there two main types (HIV 1 and 2)—including many subtypes of HIV 1 with nine strains of HIV 1 M alone, which further divide into many recombinant types specific to geographical regions. In one defined strain, the amino sequence of a single antigen such as the envelope protein Env can vary by 20% from one isolate to the other. Such enormous antigen diversity is very unusual indeed in a viral pathogen.
Apparently, HIV has an unmatched ability to change its surface characteristics and constantly frustrate the immune system. Not surprisingly, conventional vaccines targeting a permanent characteristic for an antibody to bind do not have a chance.
Experiments that were started in the 1990s in the hope of generating virus-neutralizing antibodies through vaccination did not fulfill their promise. They all failed. One of the more recent disappointments was a vaccine from a company called VaxGen, targeting the gp120 envelope protein. The vaccine did not convey any immune protection in phase II clinical studies. Since then, the idea of finding a highly specific antibody has been abandoned.
What compounds matters further is that the components of defense mechanisms in the blood prohibiting the propagation of the virus are very different from those in the first line of defense in the genital tract, where infection must be prevented in the first place.
So far, researchers have not even been able to find an immunological cellular tag that would show the effectiveness of a vaccine in a simple test.
This, in turn, makes it difficult to predict the success of vaccine studies in humans. A good immune response to a vaccine in preliminary tests does not necessarily mean that the vaccine will prevent infection in real life.
Researchers had long been hoping that studying what is called long-term nonprogressors (LTNP)—individuals infected with HIV, but not developing AIDS—would lead them to an ideal immunological tag.
At first, cytotoxic T lymphocytes looked like good candidates, as this cell type was often very active in LTNPs, whereas they remained unremarkable in patients dying of AIDS. However, recent research disproved this assumption, but hinted at a correlation between a low number of virus copies per microliter of blood (viral load) and multifunctional CD4+ and CD8+ T-cells (lymphocytes that express both interleukin-2 and interferon-γ and have a receptor for interleukin-7). These T-lymphocytes are considered to be memory cells that release a range of cytokines upon contact with a viral antigen, thus slowing down multiplication of the virus.
However, the Amsterdam cohort study of 2006 showed that the presence of multifunctional memory cells did nothing to predict the viral load, nor was it a negatively correlated with the progression of symptomless infection to AIDS.
Not an all-purpose vaccine
It had been observed that individuals infected with a “crippled” HIV virus will not develop AIDS for a longer period of time (more than 17 years in one case). This had raised hopes that it would be possible to use attenuated live vaccines, and indeed, virus variants lacking the Nef gene (or other gene sections required for virus multiplication) protected a large proportion of monkeys from HIV-1 infection.
Meanwhile, it became clear that even variants lacking three essential gene sections could prove to be a biological time bomb if used as a vaccine. Ruth M. Ruprecht and colleagues at the Dana Faber Cancer Institute in Boston showed that artificially modified HIV variants can make laboratory animals ill and eventually kill them. So much for the idea of using attenuated live vaccines.
In recent years, all hope has been resting on genetically engineered vaccines. A new strategy relies on the use of plasmid DNA vaccines administered together with recombinant vectors. These vectors are tuned to express several HIV-1 antigens simultaneously. Such DNA vaccines, however, must be administered several times to elicit a measurable immune response, which will only develop if specific adjuvants are added to the vaccines. Here, much research still needs to be done.
Top: The author in a rapid test at a blood bank in Guangzhou (China). Negative, fortunately! Bottom: All blood donations must be tested for HIV and various types of hepatitis so that the blood products are safe.
Attenuated adenoviruses or orthopox viruses seem to be obvious candidates as recombinant vectors.
So far, several DNA vaccine candidates have been identified in proof-of-concept studies. Recently, a DNA vaccine containing an adenoviral vector expressing three different HIV-1 B-strain antigens (gag, pol, and nef) were selected for a make-or-break test, a randomized controlled field study involving 3000 subjects.
Above: An AIDS virus in the blood, surrounded by Y-shaped antibodies. Below: HIV is tested in a lab South Africa.
However, an intermediate analysis of what became known as the STEP study was so unsatisfactory that the study was abandoned prematurely. It neither protected vaccinated individuals from HIV infection, nor was the virus load in the blood of infected vaccinated individuals lower than in the control group.
Even worse—HIV infection in individuals receiving a synthetic vaccine was even more widespread than in control individuals who had not been vaccinated.
Researchers, who were shocked by the findings, assume that the adenoviral vector itself could have activated the immune system and thus disabled the intended protective effect of the three HIV antigens. All in all, two decades of research into DNA vaccines have led to widespread resignation among scientists.
Over the past decade, AIDS researchers have therefore lowered their ambitions as to what an effective vaccine should achieve. A protection rate of just 60% is now considered to be a success.
By comparison—the vaccine for hepatitis B, a virus also transmitted through blood products and sexual intercourse, has a success rate of nearly 100%.
An all-purpose HIV vaccine does not seem to be on the horizon at least for the next 20 years.
What is more conceivable is the development of a range of vaccines that will meet the specific needs of different at-risk groups, such as noninfected gay people, adolescents in developing countries, or HIV carriers. In the latter case, this would be an antidisease vaccine that does not prevent infection, but strengthens the immune system so that it can fight the pathogen and prevent the development of full-blown AIDS.
The actual effectiveness of a vaccine candidate and possible serious, but rare side effects can only be adequately evaluated and quantified once several tens of thousands of individuals have been vaccinated in different endemic areas. This would involve the planning of many multicentered vaccination studies, which would then have to be approved, carried out, and evaluated. It is therefore thought that it will probably take another two decades until a truly reliable HIV vaccine with no side effects will be available.
Even if problems on the biomedical side could be dealt with more quickly than expected, there are hurdles that cannot be overcome with ever-new tweaks developed by high-tech laboratories.
In order to keep the duration of a study as short as possible and for statistical as well as cost reasons, the vaccination studies must be carried out where the infection rate is highest in a given timespan, and that is sub-Saharan Africa.
In the wake of the STEP study fiasco, health authorities in developing countries demand unequivocal evidence that a new vaccine at least does no harm. Whether it can help, is another matter.
Prof. Hermann Feldmeier teaches Tropical Medicine and Infectious Disease Epidemiology at the Charité, Humboldt University, Berlin, Germany.
The method involves immunizing an animal, retrieving spleen tissue from it, growing a mixed culture of myeloma and spleen cells, fusing them, and then selecting and breeding hybridoma cells (Box 5.7
). What makes the procedure so revolutionary is the easy manufacturing of large amounts of specific antibodies.
Box 5.7. How Antibodies are Obtained.
It has been known since the research by Behring and Kitasato around 1890 that specifically binding molecules can be isolated from blood.
The classical method is the immunization of laboratory animals by injecting them with an antigen. The immunization process must be repeated several times successfully before antibodies can be isolated from the animals’ sera. The Shanghai goat shown in Fig. 5.27
was inoculated with highly purified protein from a human heart muscle (h-FABP, which stands for heart Fatty-Acid-Binding Protein), and from its blood, h-FABP antibodies were isolated. The antibodies are a mixture of molecules that bind to various sites (epitopes) on the antigen. The strength of these bonds varies. Each of these specific antibodies is produced by its own B-lymphocyte clone in the blood, and the immune response relies on the reproduction of several different clones. These are called polyclonal antibodies.
Polyclonal antibodies are obtained from rabbits and—in larger quantities—from goats. Pictured here is “our” goat in Shanghai that was injected with antigens (in our case the heart attack marker FABP) to stimulate the production of antibodies.
Center: The method developed by Köhler and Milstein uses the hybridoma technique. In a first step, the lab animal (usually a mouse) is inoculated, and the antibodies to the antigen are first produced in the spleen, then circulate in the blood and lymph systems. These are a range of antibodies, derived from various cell clones, i.e., polyclonal antibodies. However, the aim was to produce large quantities of homogenous antibodies that only target a single epitope.
The antibodies are not taken from the blood this time, but from the spleen of the inoculated mouse. The large number of B-lymphocytes can be easily isolated. B-lymphocytes originate from bone marrow stem cells and reproduce in the spleen and lymph nodes, producing antigen-specific clones or differentiating into plasma cells or memory cells. A B-lymphocyte can only produce its very specific antibody.
The B-lymphocytes undergo in vitro fusion with myeloma cells, which are tumor cells that are easily cultured. The resulting hybridoma cells give rise to clones that produce uniform antibodies. These are called monoclonal antibodies.
The selected clones have the immortality of cancer cells combined with the antibody production of lymphocytes, and are therefore ready to be produced in unlimited quantities. However, the screening process is very labor- and material-intensive, as thousands of clones must be separately cultured and tested.
A third alternative is recombinant antibodies that are not produced in animals (in vivo), but in bacterial or cell cultures (in vitro).
Opera buffs are familiar with the plot of Carl Maria von Weber’s “Freischütz” where young Max tries to get hold of magic bullets that always find their target (Fig. 5.30
). Such magic bullets were also the dream of Paul Ehrlich, and monoclonal antibodies are just that in modern medicine, where they are widely used as diagnostic and therapeutic molecules (Fig. 5.22
).
If Samiel, the devil in the opera “Der Freischütz,” had been around nowadays, he would have given away monoclonal antibodies rather than magic bullets.
Left: How antibodies form complexes with antigens: The cartoon shows how Y-shaped antibodies (molecules shaped like kangaroos) bind antigens (e.g., viruses, kangaroo babies in the cartoon) to their two arms and form large complexes that render the solution cloudy. This is how the first immune reactions were technically measured. Where there was no antigen, no complexes causing turbidity could form.
Consider a viral infection, e.g., with the body producing antibodies as a response to a viral attack. Monoclonal antibodies against this particular virus can be used to identify the virus in the body fluids of the patient (see Chapter: Analytical Biotechnology and the Human Genome, ELISA test for viruses). Such tests have already become routine for HIV and hepatitis (Figure 5.23, Figure 5.24
).
An ELISA (enzyme-linked immunosorbent assay) can detect whether a patient has produced antibodies against the virus in question, i.e., if he/she has been infected with it.
A polystyrol ELISA plate contains 8 ×12 =96 wells for immune reactions to take place.
It is filled using a multichannel pipette. The eight pipette tips are exchangeable.
Often, however, it is not the virus itself that is identified. This would require very sensitive tests. Instead, monoclonal antibodies detect the antibodies produced by the patient in response to the infection. Because it can take weeks until these are produced in significant quantities, a recent HIV infection can go undetected.
When the SARS epidemic emerged in Hong Kong in 2003, it proved tricky to detect viruses quickly and reliably, even using high-tech diagnostics such as the polymerase chain reaction (PCR, see Chapter: Analytical Biotechnology and the Human Genome). The problem was compounded by the fact that the sensitivity of PCR is such that even contamination from the air in the lab would yield positive results. As a result, healthy patients were misdiagnosed as having SARS (false-positives) and became infected with SARS while in hospital.
What is true for viruses applies to other kinds of pathogens as well, and even for pathological surface structures of body cells. In certain kinds of cancer, e.g., tumor cells display specific membrane proteins. These tumor markers can help the immune system to recognize them as tumor cells and are called tumor markers. It is possible to produce monoclonal antibodies against such tumor markers, which can then be used in diagnostics to determine the size and position of the tumor in the body. Based on these results, a better decision can be made with regards to the treatment of the tumor, be it surgery or radiotherapy.
This is how the tumor is tracked down: A monoclonal antibody is radioactively labeled in a chemical in vitro process and then injected into the patient’s bloodstream.
The antibodies circulate throughout the body, but are immobilized if they encounter a tumor marker. Thus, after a while, a cluster of radioactivity emerges where the tumor is. In order to keep radiation to a minimum, only low-level radioactivity is used in the process, with measuring tools that are sensitive enough to detect it.
Monoclonal antibodies also hold the key for improved anticancer therapies. The main problem with traditional, small molecule chemotherapies is that even the best cancer-killing drugs cannot specifically target cancer cells. They are as potent as they are toxic to healthy cells! Monoclonal antibodies, coupled with the cytotoxic drugs, could be the magic bullets that deliver them to their targets. Such antibody-drug conjugates, like ado-trastuzumab emtansine (Kadcyla), which targets a particularly aggressive type of breast cancer (see below), have opened a brand new era in anticancer therapeutics.
But even without attached toxic molecules, monoclonal antibodies can offer effective therapies with little off-target toxicity. The first major success of what has become known as targeted therapy is rituximab, a drug used to treat non-Hodgkin lymphoma (see below),
A century has passed since Paul Ehrlich spoke of his vision of antibodies as magic bullets (Fig. 5.37
), and it has indeed come true.
Paul Ehrlich (left). Below, Erlich with his Japanese disciple Sachahiro Hata.
Bottom: Ehrlich’s side chain theory. Amazing visionary!
Further exciting developments are in store. Bispecific antibodies can now be produced in which each of the two arms binds to a different antigen, acting in effect as two drugs together. Antibodies can also be glycoengineered to enhance their interactions with the other components of the immune system. We have entered the golden age of therapeutic antibodies!
Antibodies with enzymatic activities open up further possibilities—catalytic antibodies, or abzymes, combine the endless selection variability of antibodies with the amazing catalytic power of enzymes (Fig. 5.29
).
The production of catalytic antibodies, in this case catalytic antibodies for ester hydrolysis (diagram left). Below: Despite its crucial significance in synthetic chemistry, the Diels-Alder reaction cannot be catalyzed by natural enzymes. The two starting compounds form a short-lived instable intermediate complex (shown in red) that decomposes into sulfur dioxide and the end product. Enzymes act by stabilizing the intermediate state. In order to turn an antibody into an enzyme, the intermediate state of the product must be chemically stabilized or mimicked.
The green compound is such a stable imitation. It is then injected into mice and provokes an antibody reaction. The antibodies have catalytic action, i.e., they convert the starting products, although their effectiveness is not as dramatic as that of true enzymes. The diagram shows a top view of the one “hand” of the antibody.
5.9. Catalytic Antibodies
Can antibodies act as enzymes? Given that both antibodies and enzymes bind to molecules in a similar way, i.e., through induced fit, Richard Lerner and his team at the Scripps Clinic in California had the idea of building antibodies with catalytic properties.
Enzymes drastically reduce the activation energy required for the reaction to occur by binding not to the substrate itself, but to a transition state of the substrate (see Chapter: Enzymes: Molecular Supercatalysts for Use at Home and in Industry). Thus, the transition state is stabilized, while economizing on energy. The time needed to establish a reaction equilibrium can often be divided by a factor of 108–1012.
Lerner’s idea was the following: If it were possible to develop an antibody against transition states of compounds, it should be possible for the antibody to catalyze the relevant reactions. In order to produce antibodies, antigens were required that would elicit an immune response in a lab animal. However, the transition state of a substrate is so unstable that it does not exist as a free molecule, and it is therefore impossible to make a lab animal produce antibodies. The problem was solved by chemically building models that perfectly resembled the chemical and spatial structure of the actual transition state of a substrate, but were stable (Fig 5.29).
Here is an example: in ester hydrolysis (cleavage by inserting water into an ester), the reaction of the planar ester molecule (atoms lying in the same plane) involves a transition state in which the atoms are arranged as a tetrahedron (atoms sitting in the corners of the tetrahedron) before reaching the final planar shape (acid and alcohol). The ester is electrically neutral, while the transition state is polarized through positive and negative charges.
A stable model compound was found where phosphorus substitutes the central carbon atom of the ester (Fig. 5.29). The molecule imitates the geometry and charge distribution of the unstable transition state. The model compound was bound to a carrier molecule which successfully elicited an immune reaction in mice. Later, hybridoma cells were created with the ability to bind to the model compound.
Ester was added to various monoclonal antibodies obtained in this way. Some of them showed no effect. These were probably specific to a part of the molecule that is not relevant to the transition state.
Other antibodies, however, sped up the reaction by a 1000-fold. Not bad for the beginning!
5.10. Recombinant Antibodies
Before the emergence of gene technology, the traditional way of obtaining antibodies was by immunization (polyclonal antibodies isolated directly from animal serum) or by hybridoma technology (monoclonal antibodies) (Box 5.7). These techniques are labor-intensive and imply animal research.
Nowadays, it is possible to produce antibodies without animals, using only cell cultures and viruses. The technique yields the antigen-binding fragments (Fab fragments) of antibodies, the very regions responsible for the endless variety of binding specificities. Traditionally, Fab fragments (Box 5.3) could only be obtained from complete antibodies by cleavage with proteases. The procedure has now been replaced by a smart recombinant DNA method.
Recombinant antibodies are also monoclonal (derived from a single clone) and highly specific, binding precisely to their epitope.
The crucial sites for antibody binding are the variable regions (Fv). They lack the stabilizing disulfide bridges of constant chains and need extra stabilization. This is usually done by chaining peptides that turn them into single protein strands. They are also elongated by a tag that modifies theirbiochemical or surface-binding properties. This results in single-chain Fv fragments (scFv fragments).
Why produce recombinant antibodies, since the immune system can do so on its own?
For one thing, the absence of animal involvement should please animal lovers. But it also offers better safety, since animals can transmit viruses that are potentially dangerous to humans.
The production of antibodies using E.coli cells is also vastly cheaper and faster than production in hybridoma cells. Finally, recombinant antibodies rely on the entire range of molecular biology techniques developed for E.coli, offering unsurpassed ease and biotech power! (Box 5.9
)
Box 5.9. The Expert’s View: Frances S. Ligler: Sensing with Antibodies to Detect Something Lethal.
In September and October of 2001, several cases of anthrax broke out in the United States. It motivated efforts to define biodefense and biosecurity, where more limited definitions of biosafety had focused on unintentional or accidental impacts of agricultural and medical technologies. The Center for Disease Control (CDC) has defined and categorized bioterrorism agents according to priority. Category A agents are biological agents with both a high potential for adverse public health impact and that also have a serious potential for large-scale dissemination. The Category A agents are anthrax (by Bacillus anthracis bacterium), smallpox (by Vaccinia virus), plague (by Yersinia pestis bacterium), botulism (by Clostridium botulinum), tularemia (Rabbit Fever by Francisella tularensis bacterium), and viral hemorrhagic fevers (Ebola and Marburg viruses).
The human body has developed elegant methods for the recognition of hazardous molecules and pathogens. The most well-known and best understood of these protective mechanisms involve antibodies. One of the most alluring characteristics of antibodies is that they can be generated to recognize most molecules with a unique shape and perform that recognition function in a way that is highly specific. However, in contrast to enzymes, antibodies do not convert the bound molecule (antigen) into a product.
How to Generate a Signal?
One needs a label to get a signal out of an antibody-antigen reaction, and one needs a sensor to detect and quantify the signal.
ELISAs and immunodipsticks are already on the market: pregnancy tests are using colloidal gold to label a detector antibody to detect human choriogonadotropine (hcG). These tests are onetime tests, carried out only when needed.
But how can one monitor continuously the environment, e.g., to protect us from bioterrorist attacks? Early antibody-based sensor devices in the years 1975–1985 relied on two big leaps:
The first was that antibodies could be integrated with optical and electronic hardware to provide a direct readout of antigen binding that was more sensitive than the human eye.
Recently, anthrax has gained the potential to be a major threat through bioterrorism. It is an effective weapon because it forms sturdy spores that may be stored for years, and which rapidly lead to lethal infections when inhaled.
Anthrax is caused by an unusually large bacterium, Bacillus anthracis. Once its spores lodge in the skin or in the lungs, it rapidly begins growth and produces a deadly three-part toxin.
These toxins are frighteningly effective, designed for maximum lethality.
They combine two functions: part is a delivery mechanism that seeks out cells, and part is a toxic enzyme that rapidly kills the cell. In anthrax toxins, there is one delivery molecule, termed “protective antigen” because of its use in anthrax vaccines (shown on the left). It delivers the other two parts, edema factor and lethal factor (center and right), which are the toxic components that attack cells.
The second concept was that antibodies could bind their target antigens after being immobilized on a sensor surface instead of binding the antigen in solution to generate a detection event.
The development of diode lasers, light emitting diodes (LEDs), photodiodes, CCD and CMOS cameras, and other small, inexpensive optical and electronic components sparked advances in sensor hardware development. However, solving the problem of maintaining antibody functionality after immobilization had to be solved first in order to justify device development. This is where my lab made its first major contribution to the field in the late 1980s.
How to keep antibodies functional?
Intact IgG antibodies are Y-shaped molecules, with binding sites at the end of each arm of the Y. The prevalent scientific thinking was that the best approach to immobilization would be one that bound the feet of the Y to the sensing surface, leaving the arms to wave free into the sample.
The two primary methods of doing this relied on using either the carbohydrate side chains in the Fc (or “legs region”) of the Y or on fragmenting the Y (by enzymes like papain, see Chapter: The Wonders of Gene Technology) in order to generate a free thiol (–SH) group for attachment. While these approaches were moderately successful, they did not address the two most critical problems in preserving the antigenbinding capability of the immobilized antibodies: the “fried egg” effect and the “Gulliver” effect.
Antibodies, like many other proteins, like to adsorb to surfaces. The initial interaction of the hydrophilic outside of the protein with the more hydrophobic surface is weak; however, over time, the hydrophobic inside of the protein begins to interact with and bind tightly to the surface.
Visualize a raw egg inside the shell with the hydrophobic “yolk” safely surrounded by the more hydrophilic egg white. When the shell is broken and the egg plopped onto a hot surface, the white spreads out, leaving the yolk in close proximity to the frying pan. An antibody undergoing such a conformational change is definitely not in the optimum configuration for antigenbinding. In order to avoid the fried egg effect, the surface must be modified to be as hydrophilic as possible, and direct interactions between the surface and the antibody are discouraged.
In Jonathan Swift’sGulliver’s Travels, Gulliver awakens on the shores of Lilliput to find himself tied down by the Little People using lots of tiny ropes. He cannot move his arms or legs. Initial methods for attaching antibodies to surfaces used crosslinking molecules that attached the antibodies to the sensor surface in lots of places, limiting the ability of the binding arms of Y to function away from the surface. An ideal crosslinker would bind the antibody to the sensor surface in only a few places, and preferably in the legs of the Y.
In the late 1980s, my lab first used a special class of crosslinkers to attach antibodies to a sensor surface modified with a hydrophilic film. These “heterobifunctional” crosslinkers had one end that bound only to the antibody and an opposite end that bound only to the modified surface so that antibodies could not be inadvertently attached to each other.
The use of the silane films and heterobifunctional crosslinkers was rapidly accepted and is now widely employed for antibody immobilization, avoiding the “fried egg effect.”
The Gulliver effect
Our second breakthrough in antibody immobilization solved the Gulliver problem. It is well known that the B-vitamin biotin binds very tightly to the egg white protein avidin. Though not the first investigators to attach antibodies through a biotin bridge to avidin on a surface, we demonstrated that antibody function could be optimized by attaching only two or three biotins to each antibody. The resulting method of attaching the antibody to the surface had several advantages: The avidin provided a nice, hydrophilic cushion between the antibody and the sensor surface to prevent the “fried egg effect.” The limitation on the number of crosslinks prevented the Gulliver effect. And finally, the limitation on the number of modifications to the antibody minimized the chance of directly damaging the active site of the antibody.
Biosensors using immobilized antibodies
The devices sensing the binding of antigens to antibodies have become incredibly sophisticated, but with that sophistication, they have become much simpler to operate and much more reliable in performance. For example, the first fielded fiber-optic biosensor weighed 150 pounds, fluids were manually manipulated, and samples were tested for only one target at a time. Now you can purchase a fiber-optic biosensor that is fully automated, tests for eight targets simultaneously, and fits in a backpack along with an air sampler. Another version of this system has been flown on a very small unmanned plane as part of a 10-pound payload and was able to identify bacteria while flying through the air.
Most of these immunosensors use the following (simplified) principle (see big Figure!): The capture antibodies are bound (via avidin and biotin) onto a glass surface. This can be a fiber-optic or a simple glass slide. A laser beam is directed through the glass to the surface that carries the antibodies. If the glass is covered with a fluid, two different refraction indices apply. If the beam hits the surface at less than the critical angle, total internal reflection (TIR) of the light beam occurs, generating an evanescent wave on the glass surface which penetrates 100 nm into the liquid solution—precisely the operating region of detector antibodies! In fact, two detector antibodies are used. When the first or capture antibody binds to the antigen, a second antibody carrying a fluorescent tracer binds to the caught antigen to form a sandwich.
The evanescent wave excites the fluorescent tracers: they start to emit light, indicating that the antigen has been bound and detected. Unbound detector antibodies are not excited, as they are not within the reach of the evanescent wave. The fluorescent signal is detected, filtered, and amplified.
These immunosensors are sensitive to parts per billion or better—e.g., a tablespoon of drugs or explosives in an Olympic-sized swimming pool.
Swallow: Unmanned Airborne Vehicle equipped with air sampler (tube extending from nose) and antibody-based biosensor for remote identification of biothreat agents.
Immunosensor using the evanescent wave effect for exciting labels bound to detector antibodies (for details, see running text).
Array biosensors have been developed in order to simultaneously monitor for large numbers of targets. These systems rely on an array of antibodies in discrete spots on a flat surface.
Sample and fluorescent tracer molecules cause fluorescent complexes to form on some of the antibody spots but not on others. The identity of the target can be determined by the location of the spots that light up. The intensity of the signal provides quantitative information on the amount of the target present in the sample.
Users of antibody-based biosensors have become less concerned with how they work than with how easily and cheaply they can be used. To make them easier to use, small plumbing systems (microfluidics) are becoming increasingly used.
Advances in optics offer new opportunities for increased sensitivity and reductions in size and cost. Silicon technology is producing better and better integrated optical waveguides for highly multiplexed analyses. Arrays of single photon detectors may offer the opportunity to detect one target if the antibody binding is sufficiently long-lived and if the background can be sufficiently reduced.
The production of devices based on organic polymers, such as organic LEDs, transistors, and photodiodes, is also very exciting because they should be relatively simple to integrate with biological recognition elements and polymerbased microfluidics to form monolithic, inexpensive, or disposable sensors.
Enzyme-linked Immunosorbent Assay (ELISA): a capture antibody is immobilized to a surface and binds the antigen. The detector antibody is labelled with an enzyme and forms a “sandwich” structure. After washing steps and adding colorless substrates, a colored product is formed (proportional to antigen concentration).
Novel antibody-based biosensors are highly sensitive and have proved their worth in the detection and monitoring of pesticides in agriculture, toxins and pathogens in homogenized foods, disease markers in clinical fluids, and biothreat agents in air and water.
Thus, analytical biotechnology helps to make our lives safer.
The author, Dr Frances Ligler, competing in the hardest 100-mile horse race in the United States, the Tevis Cup Race. This race is held in the Sierra Nevada mountains. She does like climbing mountains, in both scientific and equestrian endeavors.
Frances S. Ligler is currently the Navy’s Senior Scientist for Biosensors and Biomaterials. She is a Member of the Bioengineering Section of the National Academy of Engineering. She earned a BS from Furman University and both a DPhil and a DSc from Oxford University.
Currently she is working in the fields of biosensors and microfluidics. She has published over 290 full-length articles in scientific journals, which have been cited over 4000 times, and has 24 issued patents.
She is the winner of the Navy Superior Civilian Service Medal and of awards like the National Drug Control Policy Technology Transfer Award, the Chemical Society Hillebrand Award, Navy Merit Award, NRL Technology Transfer Award, and others. In 2003, she was awarded the Homeland Security Award by the Christopher Columbus Foundation and the Presidential Rank of Distinguished Career Professional by President Bush.
Cited Literature:
Ligler FS, Taitt CR (2002) Optical Biosensors: Present and Future. Elsevier Science, NY
5.11. Recombinant Antibody Libraries
Even a very successful fusion of myeloma (cancer) and spleen cells into hybridoma cells can only yield a few dozen different antibodies—a hundred at the most. This is not terribly impressive, keeping in mind that our own immune system is capable of producing 100,000 antibodies with different specificities. How could we tap into this vast potential?
Recombinant antibody generation techniques bypass the ineffective hybridoma-generating fusion step. After immunization of an animal with an antigen, spleen cells are collected, and their mature mRNA is isolated (see Chapter: The Wonders of Gene Technology).
Using reverse transcriptase, the single-stranded mRNA is copied into double-stranded copy DNA (cDNA). PCR (see Chapter: Analytical Biotechnology and the Human Genome) is then used to produce millions of copies of cDNA encoding the light and heavy antibody chains (Fig. 5.31
).
Preparing a recombinant antibody gene library for phage display.
The cDNA copies are cut with restriction endonucleases, resulting in fragments with sticky ends, which are then cloned into bacteriophage λ- derived vectors. Two different libraries are created: one with DNA encoding the heavy chains (H-chains) and the other with DNA encoding the light chains (L-chains). The two phage libraries are then combined with a third helper phage, and the mixture is used to infect a lawn of E.coli cells growing in a Petri dish.
Infection with all three phages creates recombinant phage particles that lyze E. coli cells, yielding plaques (“puddles”) containing billions of phages. If recombination brings matching DNA sequences for H and L chains in the same phage, the plaque will also contain Fab fragments. To find such phages, an imprint of the plaques is made on a nitrocellulose filter. Like all proteins, Fab fragments bind firmly to the filter. After adding a radioactively labeled antigen and washing out unbound antigen, the filter is put on X-ray film. Black spots of the film will show which plaques contain binding Fab fragments and the phages that made them!
Once the plaques are identified, Fab-encoding DNA is isolated so that it can be inserted into bacterial or mammalian vectors.
This increases the availability of possible antibodies at least by a 1000-fold, compared to standard hybridoma technology.
5.12. Piggyback or Phage Display
The B-lymphocytes needed by the immune system are selected thanks to a membrane-bound antibody on their surface (see above). An antigen (e.g., of a virus) that binds to it and IL-2, produced by T-helper cells, stimulate the B-lymphocytes to divide (clonal selection). The antibody indicates the “street number” of the relevant gene that produces it in its parent lymphocyte. In other words, the antibody carries its gene piggyback, as if in a huge backpack (Fig. 5.32
).
Another way of producing a gene library of human antibodies outside the human immune system is through transgenic mice.
In these mice, their own antibody genes have been removed and replaced by a comprehensive set of human genes.
After immunization, hybridoma technology (Box 5.7, Box 5.8) is used to obtain cell lines from the spleen, which secrete human antibodies.
This method has become another widely used source for therapeutic human antibodies alongside phage display.
It is easy to identify the B-lymphocytes with the desired antibody because it makes a clone. But could we do the same in bacteria by reading the “street number” in a simpler way, to fish out the right gene? That would be a genetic engineer’s dream!
This dream was turned into reality by George P. Smith (b. 1941, Fig. 5.34
) at the University of Missouri in Columbia, Missouri, United States, in 1985. He solved the problem not using bacteria, but bacteriophage M13. Unlike phage λ (see Chapter: The Wonders of Gene Technology), it is a filamentous phage, and with a much smaller genome. It consists of circular single-stranded DNA, containing only a few genes for capsid proteins and for a simple infection cycle (Box 5.10
).
Left: George P. Smith used bacteriophage M13 for his phage display method.
Right: Escherichia coli being infected with phages.
Box 5.10. The Expert’s View: The many aspects of a Superorganism—a Bee Hive and its Immune Defenses.
Honey bees (Apis mellifera) form colonies whose complexity and functionality never cease to amaze. Superorganisms pose their very own challenges, risks, and opportunities—for their members as well as for researchers.
In the summer months, a bee colony would comprise up to 50,000 worker bees, several hundred drones, and just one queen. All worker bees descend from the same mother, but they are not necessarily full sisters because several drones mated with the queen on her nuptial flight, thus contributing to several genetically distinct patrilines.
Cooperation between members of a bee colony yields synergetic effects that open up completely new perspectives for the biology of the insects, and new properties are emerging that could not be found in individual bees or small groups of bees.
The super-physiology that characterizes a superorganism, however, has many things in common with the physiology of individual organisms. Its homeostasis is maintained by multiple feedback loops. Honey bees create the world in which they spend the major part of their lives and control their living conditions to an extent unheard of in other organisms. Having synthesized their own building material, they build wax honeycombs of almost crystalline regularity in a process of self-organization. Their body heat keeps the wax malleable at the right temperature and ensures that it will flow into the most energy-efficient—hexagonal—structure. The ambient temperature generated by the bees in the process also affects the development of bees in the brood nest. The bee colony can thus influence characteristics of their members beyond direct genetics—it determines their epigenetics as well.
The temperature in the brood nest is a major factor that shapes the characteristics and abilities in the pupal stage of development. Larvae live in open cells, where they are fed by nurse bees. Once they reach the pupal stage, their cells are closed with a wax cap and they undergo metamorphosis from pupa to adult bee. State-of-the-art thermal imaging revealed that bees squeeze their thorax tightly to the capped cells, vibrating their flight muscles and raising their body temperature to 43°C.
Newly built honeycomb cell. The cells are still round and will only be transformed into the more energetically efficient hexagonal shape after being warmed up by the honey bees.
The heat thus generated keeps the brood nest at a temperature of 35°C. Some heater bees also use empty cells that are optimally distributed for even heat distribution over the (mostly capped) brood area so that their heat can reach a larger number of cells. We know that minute differences in temperature during the pupal stage will affect lifespan, cognitive abilities, and immune response in adult bees. During the preceding larval stage, temperature is not controlled as rigorously and the larvae are fed royal jelly produced in the hypopharyngal glands of nurse bees, which is functionally equivalent to mother’s milk in mammals. It contains immunopeptides such as defensin, but also low-molecular-weight compounds (e.g., 10-hydroxyl-2-trans-decenoic acid and acetylcholine), which are bacteriocidal and fungicidal and compensate for the rather weak larval immune system.
The bee colony’s brood nest with well-developed larvae in open cells, as well as capped cells where pupae are undergoing metamorphosis into adult bees.
RFID (Radio Frequency Identification) chips attached to the bees‘ backs record behavior patterns over their lifetime—a method that could be applied to a virtually unlimited number of individual bees.
House bees removing a diseased pupa from a capped cell.
The immunity system of an entire community is also referred to as social immunity and comprises a complex of interdependencies between the different developmental stages, which can only be understood using high-tech methods.
These include the use of RFID (Radio Frequency Identification) chips that are attached to numerous individual bees as soon as they hatch. These allow researchers to follow and record aspects of behavioral patterns in individual honey bees throughout their lives.
RFID recordings of flight patterns of honey bees infected with mites revealed that the orientation system of such bees was severely impaired when returning to the hive from foraging trips.
Densely crowded beehives with temperatures around 35°C in the brood area, combined with high air humidity, provide ideal conditions for the spread of all kinds of infections. It is all the more surprising that honey bees have so far been able to withstand this infection pressure. The only explanation is that during 30 million years of evolution, bee colonies have developed many defense lines at different levels.
False-color thermography showing temperature distribution in the bodies of honey bees. Heater bees heating their thorax up to over 43°C are clearly visible.
An exogenic defense line is propolis, the resin used to seal cracks in the beehive. It originates exclusively from plants—secretions of tree buds and bark. Propolis is rich in antiseptic and antibacterial components and is therefore a disinfectant as well as repair material. The cells of honeycombs are lined with it. The endogenic defenses of a colony are summarized in the term social immunity. They include hygienic procedures, such as the constant cleaning of honeycomb cells and the personal hygiene regimens of colony members. The spread of disease is prevented by worker bees pushing diseased adult bees out of the hive and removing dead larvae and pupae from the brood cells. How the bees can detect dead pupae inside capped brood cells is still a mystery. Like all invertebrates, honey bees do not have an adaptive immune system. They cannot produce pathogen-specific antibodies. This is compensated for by a further sophistication of the innate immune system, which is more ancient in evolutionary terms. It gives effective protection against microbial pathogens. The underlying principle, the humoral immune response, involves transient de novo synthesis of a wide range of antimicrobial peptides (AMPs). These AMPs are synthesized in the bees‘ fatbody (liver-like organ) and secreted into their hemolymph (blood equivalent). They usually eliminate microbial pathogens by attacking their membranes and cell wall constituents.
Resistance to AMPs has so far not been observed, which makes them attractive candidates for the development of new antibiotics in human medicine, e.g., for the treatment of wounds.
In order to study the immune response of bees, larvae had to be raised in vitro, through the entire developmental cycle of larva, pupa, and adult bee. Newly hatched larvae were collected from a brood area and transferred to tissue-culturing plates, the wells of which were filled with a suitable feed solution. Once the larvae have reached the prepupal stage, they are again transferred into wells lined with paper tissue. They are no longer fed, to reproduce the situation in a capped brood cell.
Raising bees in vitro ensures that the studies are carried out in a stable and sterile environment. When bacteria are injected into inflicted wounds using fine glass capillaries, the artificially raised larvae show a strong humoral immune response. This becomes evident when analyzing hemolymph samples 24 hours and 48 hours after injection on a denaturing polyacrylamide gel. At least three low-molecular-weight AMPs (hymenoptaecin, defensing, and abaecin) are found, and to discover whether the samples show actual antimicrobial activity, a growth inhibition test is carried out. Samples 3 and 6 show large areas of inhibited growth.
Gel electrophoretic analysis of induced immunopeptides in the hemolymph of bee larvae that had wounds inflicted and infected with E. coli bacteria. A growth inhibition test demonstrates antimicrobial activity in the relevant samples.
The decoding of the bee genome in 2006 enables us to study systematically the molecular processes involved in fending off pathogens in individual bees and the entire superorganism.
After studying biology, geography, and physics, Jürgen Tautz (b. 1949) was awarded a PhD in sensory ecology at the University of Konstanz. He continued to work on the bioacoustics of insects, fish, and frogs before founding the BEEgroup at the University of Würzburg in 1994. The group carried out basic research on the biology of honey bees. Alongside his scientific work (over 140 publications, including 30 title stories, some of which appeared in Science and Nature), Jürgen Tautz has become a well-known science communicator, sharing his enthusiasm for Life Sciences with the wider public.
The European Molecular Biology Organization (EMBO) recognized him as one of the outstanding science communicators in 2005, 2007, and 2008. In 2012, he received the Science Communicator Award from the German Research Foundation DFG.
Hildburg Beier (b.1943), Professor of Biochemistry at the University of Würzburg, joined the BEEgroup after retiring in September 2008. Her areas of expertise include splicing of tRNA-precursors, RNA ligases, gene expression in RNA viruses, and cellular and humoral immune response in insects.
The cunning virus enters E. coli via its sex pili, introducing its single-stranded DNA through an F-pilus. The DNA acts as a template to form double-stranded DNA in the cell, which is not inserted into the bacterial genome, but replicated between 100 and 200 times. When the bacterium divides, each daughter cell receives multiple copies of the phage DNA.
M13 is considerate enough not to kill its host and only to slow down its growth. After single-stranded DNA copies have been made and packed into filamentous protein envelopes, the phage particles are released. A tubule is formed by 2700 type pVIII proteins. Most remarkable, however, are the five proteins of type pVII and pIX at one end and pIII and pIV at the other end of the phage (Fig. 5.33
).
Left: Frank Breitling, who thought of this method on a visit to the Heidelberg outdoor swimming pool with Stefan Dübel. Message: Even scientists should sometimes relax to get new ideas!
This remarkable feature is that pIII remains functional even after integration of foreign sequences. Smith hypothesized that if a foreign gene were inserted into the pIII gene of the phage, it should show up as a foreign protein on the phage envelope, on its surface. And it worked!
This method, called phage display was first used to find the gene for a strongly binding growth hormone.
Recombinant antibodies that have been licensed by the FDA.
Four hundred clinical studies on antibody therapies are currently under way, 42 of which have already reached phase III and II/III and 174 of which are in phase I or I/II (according to Stefan Dübel, May 2005)
5.13. Phage Display for High Affinity Growth Hormone
The search focused on variants of the human growth hormone (hGH) with enhanced binding to the hGH receptor. The fragments in the peptide sequence that were responsible for receptor bonding were already known. “Degenerate” oligonucleotides were created that coded for all possible amino acids in these positions. These hGH gene variants were inserted into a M13 vector next to the pIII gene, to ensure that they would form pIII-hGH fusion proteins. This library was then used to infect E. coli.
This infection led to the emergence of M13 phages carrying the normal pIII protein in their envelope, with an hGH variant attached to it. Each phage displayed one single hGH variant. How could the most suitable one be identified?
A suspension of phages was passed through a separation column filled with polymer beads cross-linked to the hGH receptor (an isolated protein, Fig 5.35
) and was firmly bonded (more about the method in Chapter: The Wonders of Gene Technology: purification of insulin using bonded antibodies). Phages that did not carry hGH on the surface passed through the column straightaway, not bonding anywhere (Fig. 5.33). Phages with only weak bonds also travelled through the column, but the “good” phages bound to the receptors and could be detached later, using a weak acid.
Human growth hormone (hGH, red) and the binding of hGH to its receptor (green and blue).
These binding phages were reintroduced to E. coli and selected again in the column. The procedure was repeated six times, yielding phages with the highest hGH receptor affinities from the available pool. Some of these super phages were cloned and their DNA sequences identified. Thus, an improved version of growth hormone could be produced—an example of protein design.
Can this procedure also be used for antibodies? Very much so! Genes for the antibody fragment scFv were obtained from an antibody library and packed into M13 phages. They reproduced in E. coli and carried the relevant fragment on the surface like a street number!
The selection process is called panning, in analogy with the sorting process in gold pans. A new gold rush has begun, and the targets are cancer cells. Sales of antibodies made by Phage Display have increased linearly from ZERO in 2004 to $4 billion in 2008 and to staggering $11 billion in 2013.
5.14. Ongoing Hope for Cancer Patients—Antibody Targeted Therapies
“Imagine a new cancer treatment, based on the cruise missiles principle. A submicroscopic rocket with an automatic search head is launched in the body through injection. It searches out cancer cells and destroys them without attacking normal healthy tissue. Such a wonder weapon does not exist yet, but there are indications that it could be available in the near future.” These words were published in 1981, in a newspaper that is definitely not prone to hype, namely the Wall Street Journal.
The author of this prophetic article did not exaggerate: the advent of monoclonal antibodies has revolutionized the treatment of many cancers. Today, more than a dozen approved anticancer MAbs are benefitting millions of patients worldwide, and several hundred are at various stages of development.
All anticancer monoclonal antibodies abide one fundamental principle, called targeted therapy. As in tumor diagnostics, therapeutic antibodies recognize cell-surface antigens that differentiate cancer cells from their healthy counterparts.
But they do much more than simply tag cancer cells: they inhibit their growth and may even promote their death via multiple mechanisms.
Thus, by binding well-chosen membrane proteins, monoclonal antibodies can target key mechanisms that make the cells cancerous (hence the term targeted therapy).
Trastuzumab, one of the oldest anticancer antibodies, approved in 1998, targets HER2 (human epidermal growth factor receptor 2), a membrane protein important in controlling cell growth in a variety of tissues. As it turns out, some very aggressive forms of breast cancer cells overexpress the HER2 gene and depend strongly on this overexpression for their growth. The binding of trastuzumab to HER2 inhibits the corresponding growth-signaling pathway and prevents cancer cells from dividing.
HER2 is an example of an oncogene (a mutated gene that promotes cancer). Strong dependence on oncogenes exhibited by some cancers is called oncogene addiction. And just like with a drug addict, blocking the addiction has drastic consequences!
In addition to inhibiting the HER2 signaling pathway necessary for cancer progression, trastuzumab, like other monoclonal antibodies, is capable of activating the direct killing of cancer cells by the immune system. One such mechanism is ADCC (Antibody-Dependent Cell Cytotoxicity). In ADCC, the constant part (Fc) of a monoclonal antibody tagging a cancer cells binds the Fc receptor of a cytotoxic T lymphocyte that can literally drill holes in the neighbouring cancer cell. Another mechanism, called CDC (Complement-Dependent Cytotoxicity), relies directly on blood proteins that can also cause the death of cells tagged by an antibody.
For a cancer antibody therapy to work, one must first identify a target that differentiates a particular cancer cell from a normal one. Such a target may unveil a mechanism that drives not only the corresponding cancer, but other diseases as well. For example, rituximab, the oldest anticancer antibody, approved in 1997, targets CD20, a membrane protein that is present on most non-Hodgkin lymphoma B-lymphocytes, but not on their healthy counterparts.
But rituximab has also been proven effective in other diseases where CD20-expressing cells play some role: rheumatoid arthritis, multiple sclerosis, and a range of other autoimmune diseases.
Understanding a mechanism important in cancer may provide precious therapeutic clues for other, seemingly unrelated diseases!
The first generation of therapeutic antibodies are chimeric in nature, with two-thirds made up of human protein sequences (the constant region) and one third made of mouse sequences (the Fab region). The presence of animal sequences is a potential concern for unwanted side effects; subsequent antibodies were engineered to contain less and less nonhuman components. Second generation therapeutic antibodies are humanized: they contain less than 10% of nonhuman sequences (in the antigen binding region). Third generation therapeutic antibodies are said to be fully human because only human genes have been employed to build them (either in humanized mice or through phage display technologies). Chimeric antibodies are usually well-tolerated; humanized antibodies have fewer side effects, and human antibodies should be the safest of all.
Not only has the safety of monoclonal antibodies improved over the years, but so has their potency. For instance, antibody glycosylation (the addition of complex sugars to the protein backbone) can strongly affect the process of antibody-dependent cell killing. This process has proven very difficult to standardize in cell culture, resulting in significant batch to batch variation in potency. This problem is now effectively addressed by chemists that have learned how to fine-tune glycosylation in vitro, using a process called glycoengineering.
The killing potential of an anticancer antibody can be strongly enhanced by making it carry a toxic payload. Trastuzumab, for instance, can be further “weaponized” by crosslinking the antibody to emtansine, a very potent cytotoxic molecule. Emtansine on its own is too toxic to administer to cancer patients, but the ado-trastuzumab emtansine antibody-drug conjugate delivers the deadly payload only where it is needed, inside the cancer cell.
There are many other exciting developments in the field of anticancer antibody therapeutics. Bispecific antibodies can bind to two different targets on the same cell, making it more difficult for cancer cells to develop resistances. And artificially connecting parts of antibodies to other molecular components of the immune system can yield completely new therapeutic approaches. One such amazingly spectacular approach is proving a life-saver, if not a cure, for more and more patients suffering from very aggressive forms of leukemia and lymphoma. It consists of genetically engineering a cytotoxic T-lymphocyte that carries an Fc receptor directly fused to a specific antigen binding region of an antibody. The resulting super killer cell, called a CART-cell (Chimeric Antigen Receptor T-cell), is capable of continuously killing cancer cells as they arise, keeping the patient in good health! (Fig. 5.36
)
Paul Ehrlich’s name for an antibody was the magic bullet, a name emphasizing its selective toxicity for certain pathogens. The first magic bullet was a chemotherapeutic drug called salvarsan and is known in the English-speaking world as arsphenamine. It was developed in 1909 by Paul Ehrlich and Sahachiro Hata (1873–1838, Fig. 5.37). The German name is derived from Latin salvare—to save, sanus, healthy—and arsenic and could be interpreted as “healing arsenic.” It is a milestone in drug research, as for the first time, a targeted antimicrobial drug was available to tackle a dangerous infectious disease. The effectiveness of arsphenamine was such that some infections like syphilis could be cured with a single injection (Figure 5.38, Figure 5.39
).
Side-chain theory (German, Seitenkettentheorie) is a theory proposed by Paul Ehrlich to explain the immune response in living cells. Ehrlich theorized from very early in his career that chemical structure could be used to explain why the immune response occurred in reaction to infection. His drawings are amazingly similar to the later discovered antibodies.
But this was only the first of the incredible advances made in the field of immunotherapy in the last 100 years. Has Ehrlich’s dream of magic bullets come true? It has indeed (Fig. 5.40
).
He that will not apply new remedies must tolerate old evils.
Movie star Edward C Robinson (1893–1973), an anti-Nazi-activist, played the Jewish scientist Paul Ehrlich in an expensive Hollywood production in 1940.
Cited and Recommended Literature
●
Cann J (2015) Principles of Molecular Virology (6th edn.) Academic Press New York. In my opinion, the best textbook on this subject. Cann explains that there is more biological diversity within viruses than in all the rest of the bacterial, plant, and animal kingdoms put together.
●
McKane L, Kandell J (2000) Microbiology (3rd edn.) McGraw-Hill, New York. Colorful and easy-to-read textbook, highly recommended.
●
Goldsby RA, Kindt TK, Osborne BA and Kuby J (2006) Immunology, 6th edn., W.H. Freeman, New York. Reliable introduction in immunology.
●
Sompayrac LM (2012) How Pathogenic Viruses Think, Jones and Bartlett.
●
Sompayrac LM (2002) How the Immune System Works (2nd edn.) Blackwell Cambridge. From same author. Another good read!
●
Dübel S (2006) Recombinant therapeutic antibodies (Mini-review) Appl Microbiol Biotechnol. DOI 10.1007/s00253-006-0810-y. Short and very informative review, comprehensible even for newcomers.
●
Dübel S, Reichert JM (2014) Handbook of Therapeutic Antibodies. Wiley-Blackwell. Most comprehensive reference!
●
Dixon B (1994) Power Unseen: How Microbes Rule the World. WH Freeman/Spektrum Oxford NY Heidelberg. Very amusing and interesting reading!



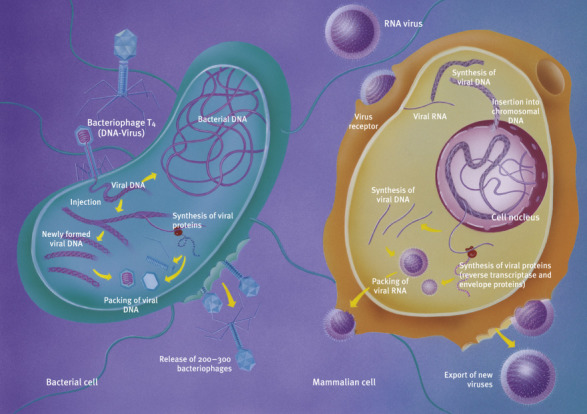
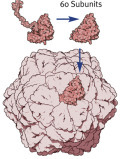
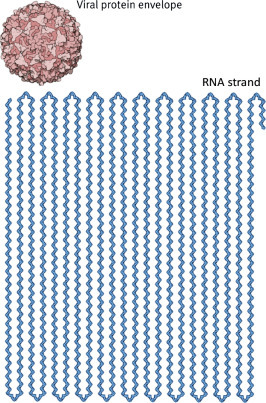



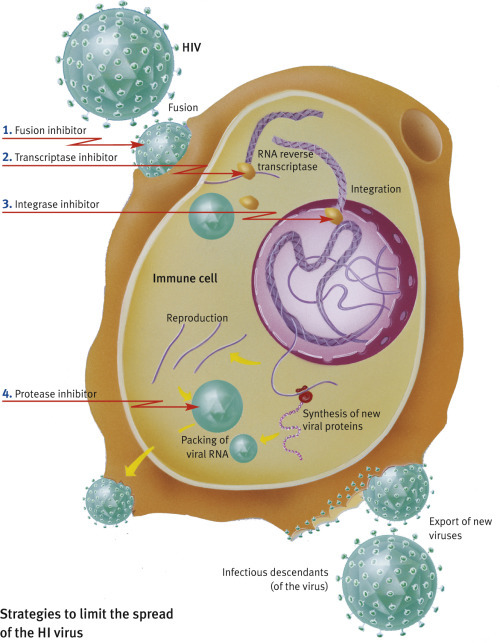
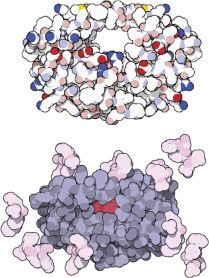 Computer-aided drug design: Drugs can be designed and tested on computers. Automated docking methods are used to find the best docking sites on a biomolecule. If the predicted bond is strong enough, the molecule can be synthesized and its activity tested. The best site for saquinavir is shown in red.
Computer-aided drug design: Drugs can be designed and tested on computers. Automated docking methods are used to find the best docking sites on a biomolecule. If the predicted bond is strong enough, the molecule can be synthesized and its activity tested. The best site for saquinavir is shown in red.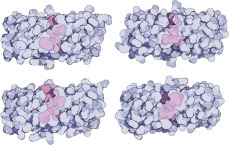 HIV protease (top) and AIDS drugs indinavir, saquinavir, ritonavir, and nelfinavir (top left to bottom right).
HIV protease (top) and AIDS drugs indinavir, saquinavir, ritonavir, and nelfinavir (top left to bottom right).