

Publisher Summary
Specific diagnoses of a viral infection are described in this chapter that are generally of five types: those that demonstrate the presence of infectious virus; those that detect viral antigens; those that detect viral nucleic acids; those that demonstrate the presence of an agent-specific antibody response; and those that directly visualize the virus. Most available routine tests are agent dependent—that is, they are designed to detect a specific virus and will give a negative test result even if other viruses are present in the sample. For this reason, agent-independent tests such as virus isolation and electron microscopy are still used to identify the unexpected or unknown agent in a clinical sample. Traditional methods such as virus isolation are still widely used; however, many are too slow to have any direct influence on the clinical management of an index case. For some economically important viruses, standardized diagnostic tests and reagents of good quality are available commercially; assays have been miniaturized to conserve reagents and decrease costs; instruments have been developed to automate tests, again often decreasing costs; and computerized analyses aid in making the interpretation of results as objective as possible in addition to facilitating reporting, record keeping, and billing. Respiratory diseases, diarrheal diseases of neonates, and some mucocutaneous diseases may be caused by a variety of different infectious agents, including viruses. Rapid and accurate identification of the causative agent can be the basis for establishing a management plan that prevents additional losses in the stable, kennel, flock, or herd.
Tests to support or establish a specific diagnosis of a viral infection are of five general types: (1) those that demonstrate the presence of infectious virus; (2) those that detect viral antigens; (3) those that detect viral nucleic acids; (4) those that demonstrate the presence of an agent-specific antibody response; (5) those that directly visualize (“see”) the virus. Most available routine tests are agent dependent—that is, they are designed to detect a specific virus and will give a negative test result even if other viruses are present in the sample. For this reason, agent-independent tests such as virus isolation and electron microscopy are still used to identify the unexpected or unknown agent in a clinical sample. Traditional methods such as virus isolation are still widely used; however, many are too slow to have any direct influence on clinical management of an index case. A major thrust of the developments in diagnostic sciences continues to be toward rapid methods that provide a definitive answer in less than 24 hours or, optimally, even during the course of the initial examination of the animal. A second major area of interest and focused effort is the development of multiplexed tests that can screen simultaneously for several pathogens from a single sample. The best of these methods fulfill five prerequisites: speed, simplicity, diagnostic sensitivity, diagnostic specificity, and low cost. For some economically important viruses: (1) standardized diagnostic tests and reagents of good quality are available commercially; (2) assays have been miniaturized to conserve reagents and decrease costs; (3) instruments have been developed to automate tests, again often decreasing costs; (4) computerized analyses aid in making the interpretation of results as objective as possible in addition to facilitating reporting, record keeping, and billing.
Although less impressive in veterinary medicine in comparison with human medicine (for reasons of economic return on investment and range of tests required across each species), there has been recent expansion in the number of commercially available rapid diagnostic kits. These tests detect viral antigens, allowing a diagnosis from a single specimen taken directly from the animal during the acute phase of the illness, or they test for the presence of virus-specific antibody. Solid-phase enzyme immunoassays (EIAs) or enzyme-linked immunosorbent assays (ELISAs), in particular, have revolutionized diagnostic virology for both antigen and antibody detection, and are now methods of choice in many situations. For laboratory-based diagnosis, polymerase chain reaction (PCR) technology is now widely used to detect viral nucleic acids in clinical specimens, offering a very rapid alternative to other methods of virus detection. Quantitative PCR assays, in particular, facilitate the very rapid, sensitive, and specific identification of many known pathogenic viruses, and automation of these assays allows the processing of large numbers of samples in short periods of time (high sample-throughput). Another major advantage of quantitative PCR assays is that they provide an objective estimate of viral load in a clinical sample. Research efforts in PCR continue, to move testing from the laboratory to the field, particularly for high-consequence agents with which rapidity of diagnosis is critically important.
The provision, by a single laboratory, of a comprehensive service for the diagnosis of viral infections of domesticated animals is a formidable undertaking. Viruses in more than 130 different genera and belonging to 35 families cause infections of veterinary significance. Add to these numbers the rapidly expanding array of viruses that occur in wildlife and fish, and it is not surprising that no single laboratory can have the necessary specific reagents available or the skills and experience for the detection and identification of all viruses of all animal species. For this reason, veterinary diagnostic laboratories tend to specialize [e.g., in diseases of food animals, companion animals, poultry, fish, or laboratory species, or in diseases caused by exotic viruses (foreign animal diseases)]. Contacting the laboratory to determine its specific capabilities should be a first step in submitting specimens for testing. Table 5.1 provides a general guide to diagnostic tests currently used in veterinary medicine. These will be defined in more detail later in this chapter.
Table 5.1.
Principles and Objectives of Diagnostic Methods
| Principle | Method | Specimens/Findings | Characteristics |
|---|---|---|---|
| Visual Information Leading to a Presumptive Diagnosis | |||
| Review of the disease history, clinical examination, chemistry, hematology, etc. | Subject animal and its body fluids/Abnormal values | Essence of differential and rule out diagnoses; presumptive diagnosis determines the specimens and methods for further testing | |
| Pathology, histopathology, ultrastructural pathology | Animals, organs, tissues, cells/Characteristic lesions, inclusion bodies | Although slow and expensive, still important in veterinary diagnostics | |
| Detection of viruses by electron microscopy | Tissues, cells, secretions, excretions, vesicular contents/Particles of uniform, characteristic morphology | Rapid; sensitive enough with many diseases, especially diarrheas; expensive; technically demanding, expertise unavailable in many settings | |
| Detection and Identification of Viral Antigens | |||
| Enzyme immunoassay methods (e.g., antigen-capture enzyme immunoassay) | Tissues, cells, secretions, excretions/Reaction of viral antigen with antibody of known specificity | Rapid, sensitive and specific. Most common methods in use today | |
| Immunochromatography, immunogold-binding assays (the equivalent of the home pregnancy test) | Blood, secretions, excretions/Viral antigen identified by reaction with antibody of known specificity | Rapid, sensitive, specific, suitable for testing of individual specimens in the clinical setting | |
| Immunofluorescence | Tissues and cells/Viral antigen identified in situ by reaction with antibody of known specificity | Rapid, sensitive and specific. Localization of antigen in specific cells adds to confidence in diagnosis; technically demanding | |
| Immunohistochemistry (immunoperoxidase staining) | Tissues and cells/Viral antigen identified in situ by reaction with antibody of known specificity | Slow, but sensitive and specific. Localization of antigen in specific cells adds to confidence in diagnosis; technical expertise involved is more like an extension of histopathology | |
| Immunoelectron microscopy | Tissues, cells, secretions, excretions/Character and aggregation of virus by specific antibody of known specificity | Extension of diagnostic electron microscopy. Rapid, sensitive and specific. Expensive and technically demanding; expertise unavailable in many settings | |
| Direct Detection and Identification of Viral Nucleic Acids | |||
| Hybridization methods, including in-situ hybridization, Southern blot hybridization and dot-blot filter hybridization methods | Extracts from tissues, cells, secretions, excretions/Viral nucleic acid identified by reaction with specific DNA probe | Dot-blot methods are rapid, simple to carry out, very sensitive, and with suitable reagents very specific. Largely being replaced with polymerase chain reaction (PCR) procedures | |
| PCR, reverse transcriptase-PCR, real-time PCR, and amplification by isothermal amplification | Extracts from tissues, cells, secretions, excretions/Viral nucleic acid specifically amplified using primer sets and then identified by various methods such as fragment size analysis, labeled DNA probes, probe hydrolysis, and partial sequencing | Some methods can be subject to contamination, causing false-positive results. Nevertheless, because of incredible sensitivity and specificity, becoming used very widely in circumstances where the “state of the art” is required. Automation and new methods for identifying amplified products are leading to quicker, more reliable, and less expensive tests | |
| Viral genomic sequencing and partial sequencing | Extracts from tissues, cells, secretions, excretions/Viral nucleic acid specifically amplified, usually via PCR and then subjected to automated sequencing, usually of only 100–300 bases in selected genomic regions | When combined with automated genome amplification methods and computer-based analyses of results, this becomes the new “gold standard” in identifying a virus | |
| Oligonucleotide fingerprinting and restriction endonuclease mapping | Extracts from tissues, cells, secretions, excretions/Viral nucleic acid amplified, usually via PCR or growing the virus in cell culture, then restriction enzyme digestion and gel electrophoresis to determine characteristic banding patterns (“viral bar-coding”) | Very slow, expensive, difficult to automate, and complex to analyze. Methods largely being replaced with PCR and sequencing | |
| Virus Isolation and Identification | |||
| Virus isolation in cultured cells | Tissues, cells, secretions, excretions/Specimens inoculated into suitable cell cultures and presence of virus detected by various methods, usually immunological methods | Relatively slow, expensive, and technically demanding. However, this is the only method that provides a virus isolate for further testing (e.g., strain typing) and is therefore widely used in reference centers | |
| Virus isolation in animals | Tissues, cells, secretions, excretions/Specimens inoculated into animals, usually newborn or 3-week-old mice, usually by the intracerebral or intraperitoneal routes, with sickness or death as indication of viral growth. Identification of virus by various methods, usually immunological methods | Even slower, more expensive, and technically demanding than virus isolation in cell culture. However, for viruses that do not grow well in cell culture, this is the only method that provides a virus isolate for further testing (e.g., strain typing) and is therefore still used in reference centers in special circumstances | |
| Detection and Quantitation of Antiviral Antibodies (Serologic Diagnosis) | |||
| Enzyme immunoassay (EIA)–enzyme-linked immunosorbent assay (ELISA) | Serum/Specimens tested for presence of specific antibodies indicating recent or past infection | Rapid, sensitive, and specific; the pillar of retrospective diagnosis for many clinical and epidemiological purposes. In many cases, paired sera are needed to confirm infection or recent vaccination | |
| IgM class-specific antibody EIA–ELISA | Serum/Specimens tested for presence of specific IgM antibodies indicating recent infection | Rapid, sensitive, and specific; becoming the pillar of serologic diagnosis of recent infection in human medicine, with limited development in veterinary medicine. In many cases a single serum suffices | |
| Serum (virus) neutralization assay | Serum/Specimens tested for presence of specific antibodies indicating recent or past infection | Cell culture-based method; slow, expensive, and technically demanding. However, this is the “gold standard” of serology, as neutralizing antibodies correlate best with immune protection | |
| Immunoblotting (Western blotting) | Serum/Specimens tested for presence of specific antibodies indicating recent or past infection | Slow, expensive, and technically demanding, mostly used as confirmatory test | |
| Indirect immunofluorescence assay | Serum/Specimens tested for presence of specific antibodies indicating recent or past infection | Rapid, sensitive, but subject to uncontrollable, non-specific reactions | |
| Hemagglutination-inhibition assay | Serum/Specimens tested for presence of specific antibodies indicating recent or past infection | Rapid, sensitive, and specific; widely used for retrospective diagnosis for epidemiological and regulatory purposes. Still a pillar in avian virus diagnostics and for many mammalian virus diseases | |
| Immunodiffusion | Serum/Specimens tested for presence of specific antibodies indicating recent or past infection | Rapid, but can lack sensitivity and subject to specificity problems. There are very good tests available for some diseases | |
Rationale for Specific Diagnosis
Why bother to establish a definitive laboratory diagnosis of a viral infection? In earlier times when laboratory diagnostic testing was in its infancy, diagnosis of diseases related to viral infections was achieved mainly on the basis of clinical history and signs, and/or gross pathology and histopathology; laboratory test results were viewed as confirmatory data. This is no longer the case, for several reasons: (1) the recent development of rapid test formats for specific and sensitive identification of individual viral infections; (2) many clinical cases occur as disease complexes that cannot be diagnosed on the basis of clinical signs or pathology alone—for example, the canine and bovine respiratory disease complexes; (3) diagnostic medicine, especially that pertaining to companion animals, increasingly demands reliable and specific antemortem diagnoses; (4) legal/regulatory actions for diseases of production animals and zoonoses can require identification of the specific agents involved, avian influenza being a relevant contemporary example. Other areas in which laboratory testing data is essential are considered below.
At the Individual Animal or Individual Herd Level
Diseases in which the management of the animal or its prognosis is influenced by the diagnosis. Respiratory diseases (e.g., in a broiler facility, acute respiratory disease in a boarding kennel, shipping fever in a cattle feedlot), diarrheal diseases of neonates, and some mucocutaneous diseases may be caused by a variety of different infectious agents, including viruses. Rapid and accurate identification of the causative agent can be the basis for establishing a management plan (biosecurity, vaccination, antimicrobial treatment) that prevents additional losses in the stable, kennel, flock, or herd.
Certification of freedom from specific infections. For diseases in which there is life-long infection—such as bovine and feline leukemia virus infection, persistent bovine viral diarrhea virus infection, equine infectious anemia, and certain herpesvirus infections—a negative test certificate or history of appropriate vaccination is often required as a condition of sale, for exhibition at a state fair or show, or for competitions and/or international movement.
Artificial insemination, embryo transfer, and blood transfusion. Males used for semen collection and females used in embryo transfer programs, especially in cattle, and blood donors of all species are usually screened for a range of viruses to minimize the risk of viral transmission to recipient animals.
Zoonoses. Viruses such as rabies, Rift Valley fever, Hendra, influenza, eastern, western, and Venezuelan equine encephalitis are all zoonotic, and are of sufficient public health significance as to require relevant veterinary diagnostic laboratories to establish the capability for accurate detection of these agents. Early warning of a potential influenza virus epidemic through diagnosis of infection and/or disease in an individual poultry flock or in affected swine allows the implementation of control programs to eradicate the infection and/or restrict movement of exposed animals. As an example, laboratory identification of rabies virus in a dog, skunk, or bat that has bitten a child provides the basis for treatment decisions.
At the State, Country, and International Level
Epidemiologic and economic awareness. Provision of a sound veterinary service in any state or country depends on knowledge of prevailing diseases, hence epidemiologic studies to determine the prevalence and distribution of particular viral infections are frequently undertaken. Such programs are also directed against specific zoonotic, food-borne, water-borne, rodent-borne, and arthropod-borne viruses. Internationally, the presence of specific livestock diseases in a country or region requires notification to the Office Internationale des Epizooties (the OIE, syn. the World Organization for Animal Health), which records the occurrence of these notifiable diseases in the approximately 175 member countries of the organization.
Test and removal programs. For infections caused by viruses such as equine infectious anemia virus, Marek's disease virus, bovine herpesvirus 1, pseudorabies virus, and bovine viral diarrhea virus, it is possible to reduce substantially the incidence of disease or eliminate the causative virus from herds or flocks by test and removal programs. The elimination of pseudorabies virus from commercial swine facilities in the United States is an example of where differential laboratory tests [the so-called differentiation/discrimination of infected from vaccinated animals (DIVA) test, which discriminates between naturally infected and vaccinated animals] were essential to the eradication effort.
Surveillance programs in support of enzootic disease research and control activities. Surveillance of viral infections based on laboratory diagnostics is central to all epidemiologic research, whether to determine the significance of a particular virus in a new setting, to unravel the natural history and ecology of a virus in a particular host animal population, to establish priorities and means of control, or to monitor and evaluate control programs.
Surveillance programs in support of exotic disease research and control activities. The countries of Europe, North America, Australia, New Zealand, and Japan are usually free of many devastating diseases of livestock such as foot-and-mouth disease, classical swine fever, African swine fever, and fowl plague that are still enzootic in other parts of the world. However, periodic incursions of these feared exotic diseases into previously free areas occur with alarming regularity and very substantial adverse economic impact. Thus it is of the utmost importance that the clinical diagnosis of a suspected high-consequence viral infection be confirmed quickly and accurately. Many countries maintain or share the use of specialized biocontainment laboratories devoted to rapid and accurate diagnosis and research on high-consequence viruses that cause economically devastating “foreign animal diseases.”
Prevention of new, emerging, and re-emerging viral diseases of animals. Continuous surveillance of animal populations for evidence of new viruses, new diseases, and new epizootics is essential if new threats are to be dealt with rapidly and comprehensively. New viruses and new virus–disease associations continue to be discovered, virtually every year. Vigilance by astute veterinary clinicians as well as by diagnosticians and epidemiologists is essential for early recognition of such occurrences.
Collection, Packaging, and Transport of Specimens
The chance of detecting a virus depends critically on the attention given by the attending veterinarian to the collection of specimens. Clearly, such specimens must be taken from the right site, from the most appropriate animal, and at the right time. The right time for virus detection is as soon as possible after the animal first develops clinical signs, because maximal amounts (titers) of virus are usually present at the onset of signs and often then decrease rapidly during the ensuing days. Specimens for virus detection taken as a last resort when days or weeks of empirical therapy have failed are almost invariably a useless endeavor and a waste of consumer and laboratory resources. Similarly, the incorrect collection and storage of specimens, and the submission of inappropriate specimens, will diminish the likelihood of accurate diagnostic laboratory success.
The site from which the specimen is collected will be influenced by the clinical signs and knowledge of the pathogenesis of the suspected agent(s) (Table 5.2 ). In viral respiratory infection in cattle, for example, the most important diagnostic specimens that should be collected include nasal or throat swabs or transtracheal wash fluid from live animals, and lung tissue and lymph nodes from dead animals; whole-blood samples from this type of case are often useless because the causative viral agents (bovine respiratory syncytial virus, bovine herpesvirus 1, bovine coronavirus, etc.) may not produce detectable concentrations of virus in blood samples (viremia). Likewise, for routine enteric cases (diarrhea), feces would be the primary sample in calves with rotavirus, coronavirus, or torovirus infections, with whole-blood being useful only if bovine virus diarrhea virus was a likely cause. Timing of sample collection is also critical, particularly with enteric cases, as detection of rotavirus may not be possible more than 48 hours after the onset of clinical signs. PCR tests do extend the sampling period because of their high analytical sensitivity and their ability to detect viral nucleic acids even if the causative virus is already complexed with neutralizing antibodies, but this longer detection period does not eliminate the need to be attentive to timing. Furthermore, the extended detection of viral nucleic acid by PCR assays increases the likelihood of false-positive results, wherein a virus detected by PCR is not the actual cause of the affected animal's disease.
Table 5.2.
Specimens Appropriate for Laboratory Diagnosis of Various Clinical Syndromes in the Live Animal
| Syndrome | Specimen |
|---|---|
| Respiratory | Nasal or throat swab; nasopharyngeal aspirate, tracheal wash fluid |
| Enteric | Feces |
| Genital | Genital swab |
| Eye | Conjunctival swab |
| Skin | Vesicle swab or scraping; biopsy of solid lesion |
| Central nervous system | Cerebrospinal fluid |
| Generalized | Nasal swaba, fecesa, blood leukocytesa, serum, urine |
| Biopsy | Relevant organ |
| Any disease | Blood for serologyb |
Depending on presumed pathogenesis.
Blood allowed to clot, serum kept for assay of antibody.
Tissue specimens should always be taken from any part of the body where lesions are observed, either by surgical biopsy or at necropsy of dead animals, as it is critical that laboratory findings be reconciled with lesions that are manifest in the affected animal. Thus separate samples should be split between material that will be fixed (formalin or other fixative) and material that will remain unfixed for virus detection assays such as immunohistochemical staining, PCR testing, or virus isolation.
Because of the lability of many viruses, specimens intended for virus isolation must always be kept cold and moist, which requires preparation ahead of time. In collection of specimens such as swabs, the discussion immediately turns to viral transport media. The various transport media consist of a buffered salt solution to which has been added protein (e.g., gelatin, albumin, or fetal bovine serum) to protect the virus against inactivation and antimicrobials to prevent the multiplication of bacteria and fungi. A transport medium designed for bacteria or mycoplasma should not be used for virus sampling unless it has been proven not to be inhibitory for the intended test. Separate samples should be collected for bacterial testing. In general, specimens correctly collected and maintained for virus isolation will be acceptable for antigen and nucleic acid detection testing. An example of a kit containing materials suitable for the collection and transportation of specimens is shown in Figure 5.1 .
Figure 5.1.

Kit for the correct collection and transport of specimens to maximize the chances of obtaining a valid laboratory diagnosis of a clinical case. The diagnostic laboratories can provide such kits, which contain materials both for collecting specimens and for proper shipping of the specimens that meet transportation standards. Items that may be included are: kit shipping box; insulated pouch, freezer packs, 95-kPa-rated specimen pouch; 95-kPa-rated formalin jars (1 small, 1 large); sealable plastic bags (small- and medium-sized); absorbent material sufficient to absorb all fluid in the shipment; serum blood collection tubes: ethylenediamine tetra-acetic acid (EDTA) blood collection tubes; blood collection needles; syringes; scalpels; buffered saline; alcohol swabs; history form; mailing label; formalin shipping label; shipping declaration forms as defined by the country of origin.
(Courtesy of B. Thompson, Animal Health Diagnostic Center, College of Veterinary Medicine, Cornell University.)
Specimens should be forwarded to the testing laboratory as soon as possible. With courier services increasingly available throughout the world, overnight delivery services have greatly decreased the time interval required for agent detection, and also greatly increased the rate of diagnostic success (pathogen detection rate). Specimens should not be frozen but should be kept cold (refrigeration temperature), if delivery to the laboratory will be within several days. While viability is not necessary for PCR assays and direct antigen detection, maintaining the specimens under optimum condition for virus isolation will also enhance detection by these other techniques. Specimens should never be sent to the diagnostic laboratory without a detailed clinical history of the animal and/or herd from which the specimens are derived. Clinical histories assist diagnosticians in selecting the most appropriate tests for the specimens received and permit a dialogue with the clinician over additional specimens if needed. Similarly, a detailed and accurate description of the nature and distribution of the lesions in affected animals is critical if samples are to be submitted for histopathological evaluation, regardless of whether the tissue specimens were obtained at necropsy or at surgical biopsy.
Packaging and specimen labeling and identification may be a mundane topic, but attention to these details maximizes the likelihood of safe arrival of the specimens at the laboratory and prevents legal sanctions over incorrectly shipped hazardous materials. The submitter should have an understanding of local transport regulations, which in most instances mirror international air transport regulations, and pack diagnostic specimens accordingly. Although specimens may have been dispatched locally by land transport, often shipments will be partially transported by air over even short to moderate distances, without the knowledge of the shipper. The specimens should be protected from breaking in transit and should be sent refrigerated (but not frozen), with “cold packs.” Wherever possible, sampling should include specimens that allow the use of several diagnostic tests, as no single test will provide an unambiguous diagnosis in all cases.
Diagnosis of Viral Infections by Gross Evaluation and Histopathology
The gross and histological evaluation of tissues from animals with presumptive viral diseases is still a useful and important diagnostic method. If biopsy/necropsy samples are collected for possible histopathological diagnosis of viral infections, then the appropriate tissue specimens in the appropriate fixative—routinely formalin—are required. If special procedures are to be requested, such as electron microscopy or frozen sections for immunohistochemical staining, the receiving laboratory should be consulted for procedural and material details. It is critical that a thorough, accurate history and description of the lesions in affected animals accompany the submitted specimens.
The great benefit of pathology is that it can provide unambiguous confirmation of specific viral diseases, especially when done in conjunction with appropriate laboratory virological testing. In contrast, the mere demonstration of a particular virus, or seroconversion of an animal to that virus, is not necessarily proof of disease causality. Thus laboratory demonstration of a specific virus combined with compatible clinical signs and lesions in the affected animal strongly reinforces confidence in a specific diagnosis. Similarly, the identification of characteristic lesions in an animal without associated detection of the relevant virus should stimulate additional laboratory efforts to confirm or refute the tentative diagnosis.
Methods of Detection of Viruses
Detection of Viruses by Electron Microscopy
Perhaps the most obvious method of virus detection/identification is direct visualization of the virus itself (Figure 5.2 ). The morphology of most viruses is sufficiently characteristic to identify the image as a virus and to assign an unknown virus to the correct family. In the context of the particular case (e.g., detection of parapoxvirus in a scraping from a pock-like lesion on a cow's teat), the method may provide an immediate definitive diagnosis. Non-cultivable viruses may also be detectable by electron microscopy. Beginning in the late 1960s, electron microscopy was the means to the discovery of several new families of previously non-cultivable viruses, notably rotaviruses, noroviruses, astroviruses, and toroviruses, and unknown members of recognized families such as adenoviruses and coronaviruses. Even today, non-cultivable viruses such as those in the genus Anellovirus (torque teno viruses) have been identified by electron microscopy in samples from humans and a variety of animals.
Figure 5.2.

Diagnostic electron microscopy. The morphology of most viruses is sufficiently characteristic to assign an unknown virus to the correct family. In this case, direct negative staining of vesicular fluid revealed large numbers of herpesvirus particles, allowing a presumptive diagnosis of infectious bovine rhinotracheitis. Magnification: ×10,000.
Two general procedures can be applied to virus detection by electron microscopy: negative-stain electron microscopy and thin-section electron microscopy. For the negative stain procedure, virus particles in a fluid matrix are applied directly to a solid support designed for the procedure. Contrast stains are applied and the virus particles are directly visualized by electron microscopy. Thin-section electron microscopy can be used directly on fixed tissue samples, usually containing “viral” inclusions from the affected animal or on cell cultures growing an unidentified virus. Low sensitivity is the biggest limitation of electron microscopy as a diagnostic tool, followed by the need for expensive equipment and a highly skilled microscopist. To detect virus particles by negative-stain electron microscopy, the fluid matrix must contain approximately 106 virions per ml. Such concentrations are often surpassed in clinical material such as feces and vesicle fluid, or in virus-infected cell cultures, but not in respiratory mucus, for instance. Aggregation of virus particles by specific antiserum (immunoelectron microscopy) can enhance sensitivity and provide provisional identity of the agent. For thin-section electron microscopy, most of the cells in the tissue sample must contain virus if virions are likely to be visualized. Routine electron microscopy procedures have been largely replaced with more sensitive and less expensive procedures such as antigen-capture tests or immunostaining techniques, but because electron microscopy is an agent-independent test, it still has use in specialized cases and in facilities with the necessary equipment and expertise.
Detection of Viruses by Isolation
Despite the explosion of new techniques for “same-day diagnosis” of viral disease by demonstration of viral antigen or viral nucleic acid in specimens, virus isolation in cell culture remains an important procedure. Theoretically at least, a single viable virion present in a specimen can be grown in cultured cells, thus expanding it to produce enough material to permit further detailed characterization. Virus isolation remains the “gold standard” against which newer methods must be compared, but nucleic acid detection tests, particularly quantitative PCR assays, are challenging that paradigm.
There are several reasons why virus isolation remains as a standard technique in many non-commercial laboratories. Until recently it was the only technique that could detect the unexpected—that is, identify a totally unanticipated virus, or even discover an entirely new agent. Accordingly, even those laboratories well equipped for rapid diagnosis may also inoculate cell cultures in an attempt to isolate a virus. Metagenomic and “deep sequencing” techniques can detect unknown agents (so-called pathogen mining), but few laboratories outside subsidized research programs have the resources to routinely apply this technology. Culture is the easiest method of producing a supply of live virus for further examination by molecular methods (genome sequencing, antigenic variation, etc.). Research and reference laboratories, in particular, are always on the lookout for new viruses within the context of emerging diseases; such viruses require comprehensive characterization, as recently shown by the quickly evolving strains of influenza virus. Moreover, large quantities of virus must be grown in cultured cells to produce diagnostic antigens and reagents such as monoclonal antibodies. Until recently, vaccine development has also been reliant on the availability of viruses grown in culture, although this may quickly change in the future with the increasing sophistication of recombinant DNA technology.
The choice of cell culture strategy for the primary isolation of an unknown virus from clinical specimens is largely empirical. Primary cells derived from fetal tissues of the same species usually provide the most sensitive cell culture substrates for virus isolation. Continuous cell lines derived from the homologous species are, in many cases, an acceptable alternative. As interest in wildlife diseases increases, most laboratories are challenged to have the necessary cell cultures to “match” with the affected species. Testing strategies for challenging cases tend to reflect the creativity and bias of the diagnostic virologist and the particular laboratory, although the clinical signs exhibited by the affected animals will often suggest which virus might be present. Most laboratories also select a cell line that is known to grow many types of viruses, in case an unanticipated agent is present. Arthropod cell cultures are used frequently as a parallel system for isolating “arboviruses.” Even with the best cell culture systems available, some viruses such as papillomaviruses will not grow in traditional cell culture conditions. Special culture systems such as organ cultures and tissue explants can be of value, but contact should be made with the testing laboratory to determine their capabilities before requesting such specialized and sophisticated diagnostic expertise.
Historically, when standard methods had failed to diagnose what appeared to be an infectious disease, inoculation of the putative natural host animal was used to define the infectious nature of the problem and to aid in the eventual isolation of the agent. This practice has largely been abandoned, as a result of costs and animal welfare concerns. Some specialized laboratories still have the capability to inoculate suckling mice, a system that has been valuable for isolating arboviruses that resist cultivation in cell cultures. Embryonated hens' eggs are still used for the isolation of influenza A viruses, even though cell cultures [Madin–Darby canine kidney (MDCK) cells] are now more commonly used. Many avian viruses also replicate much better in eggs than in cell cultures derived from chick embryo tissues, and there is a lack of widely available avian cell lines for routine virus isolation procedures. According to the virus of interest, the diagnostic specimen is inoculated into the amniotic cavity, or the allantoic cavity, the yolk sac, onto the chorioallantoic membrane or, in rare instances, intravenously into the vessels of the shell membrane and embryo. Evidence of viral growth may be seen on the chorioallantoic membrane (e.g., characteristic pocks caused by poxviruses), but otherwise other means are used to detect viral growth (e.g., death of the embryo, hemagglutination, immunofluorescence or immunohistochemical staining of viral antigens, or antigen-capture ELISA).
Attempts to isolate viruses require stringent attention by the clinician to the details of sample collection and transport, because success depends on the laboratory receiving a specimen containing viable virus. Contact with the testing laboratory before specimen collection is strongly advised in order to clarify the sampling strategy, assess shipping requirements, and alert the laboratory to the number and type of specimens being shipped. Having cell cultures available on the day of arrival of a specimen can enhance the success of isolation. There is no such thing as an emergency (“stat”) virus isolation; each virus has its own biological clock and no amount of concern will speed up the replication cycle. For viruses such as the alphaherpesviruses, a successful isolation can be evident as cytopathic effect in the inoculated cell cultures within 2–3 days, whereas others are considerably slower and require repeated serial passage. In general, the time for detection will depend on the laboratory's procedures for identifying virus in the culture system. For instance, non-cytopathic bovine viral diarrhea virus can be detected by virus isolation as early as 3 days postinoculation or as late as 3 weeks, depending on laboratory procedures. Procedures for routinely detecting and identifying virus in inoculated cell cultures include immunofluorescence or immunohistochemical staining of the infected monolayer, antigen-capture ELISA, nucleic acid detection tests such as PCR, hemadsorption, or even negative-stain electron microscopy for unknown isolates.
Detection of Viral Antigens
The direct detection of viral antigens in a clinical sample can be achieved in as little as 15 minutes with some immunoassays, or the procedure can take several days if extensive sample preparation and staining is involved. Viable virus is generally not required in the specimen for a positive antigen detection test result, but the timing of sample collection is as important with these assays as it is for virus isolation. Analytical sensitivity varies across the various test modalities, ranging from detection of a single infected cell to assays that require as much as 105 antigen units. The advance that revolutionized this type of testing was the development of monoclonal antibodies. These reagents are highly specific in their binding to antigen and, once developed, provide a virtually inexhaustible supply of the same material for test consistency.
The downside to antigen detection tests is that many antigens are altered or masked by tissue fixation. Furthermore, they are agent specific, thus a test for canine parvovirus cannot detect the presence of canine coronavirus in the specimen, which would require a separate and additional agent-specific test.
Immunofluorescence Staining
Immunofluorescence or fluorescent antibody staining is an antigen-detection test that is used primarily on frozen tissue sections, cell “smears,” or cultured cells; formalin-fixed tissue samples are generally not useful with this procedure. Antigen is detected through the binding to the sample matrix of specially modified, agent-specific antibodies. The modification is the “tagging” of the antibody with a fluorochrome that absorbs ultraviolet light of a defined wavelength, but emits light at a higher wavelength. The emitted light is detected optically with a special microscope equipped with filters specific for the emission wavelength of the fluorochrome. The fluorochrome can be bound directly to the agent-specific antibody (direct immunofluorescence) or it can be attached to an anti-immunoglobulin molecule that recognizes the agent-specific antibody (indirect immunofluorescence) (Figure 5.3 A). The indirect method enhances the sensitivity of the test, but may also increase background.
Figure 5.3.

(A) Immunofluorescence. Left: Direct method. Right: Indirect method. (B) Immunohistochemistry. Left: Direct method. Right: Indirect method.
Immunofluorescence staining does require specialized equipment, including a cryostat for sectioning frozen tissue along with a fluorescent microscope for detecting the bound antibody. Immunofluorescence has proven to be of great value in the identification of viral antigens in infected cells taken from animals or in cultured cells inoculated with specimens from infected animals. For certain viral diseases, specimens that include virus-infected cells can easily be collected from the mucous membrane of the upper respiratory tract, genital tract, eye, or skin, simply by swabbing or scraping the infected area with reasonable firmness. Cells are also present in mucus aspirated from the nasopharynx or in fluids from other sites, including tracheal and bronchial lavages, or pleural, abdominal, or cerebrospinal fluids. Respiratory infections with parainfluenzaviruses, orthomyxoviruses, adenoviruses, and herpesviruses are particularly amenable to rapid diagnosis (less than 3 hours test time) by immunofluorescence staining. The method can also be applied to tissue—for example, biopsies for the diagnosis of herpesvirus diseases, or at necropsy on brain tissue from a raccoon showing neurological signs as a result of infection with canine distemper virus or rabies virus (Figure 5.4 ).
Figure 5.4.
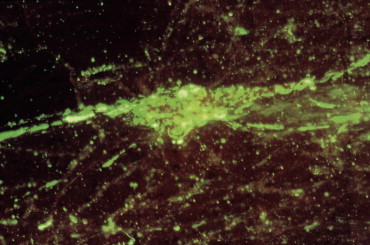
Direct fluorescent antibody stain of brain tissue for rabies virus. Frozen tissue sections from the brain of a bovine showing abnormal neurological signs were fixed in cold acetone and stained with a commercial reagent containing three monoclonal antibodies specific for the nucleocapsid of rabies virus. Antibodies were labeled with fluorescein. Positive staining is noted in a Perkinje cell.
(Courtesy of J. Galligan, New York State Department of Health.)
Immunohistochemical (Immunoperoxidase) Staining
In principle, immunohistochemical staining is very similar to immunofluorescence staining of viral antigens, but with several key differences (Figure 5.3B). The “tag” used in immunohistochemical staining is an enzyme, generally horseradish peroxidase. The enzyme reacts with a substrate to produce a colored product that can be visualized in the infected cells with a standard light microscope. The tissue sample will often be formalin fixed, which permits testing of the specimen days to weeks after sampling, without the need for low-temperature storage. Another major advantage for the immunohistochemical staining technique is that it involves an amplification process wherein the product of the reaction increases with increasing incubation, whereas immunofluorescence staining generates a real-time signal that does not get stronger with a longer incubation period. Furthermore, immunohistochemically stained slides can be kept for extended periods of time for several observations, whereas the immunofluorescence slides deteriorate more rapidly. Immunofluorescence does have the advantage of speed; immunohistochemical staining on formalin-fixed tissues requires more than 24 hours to give results. Perhaps the greatest benefit of immunohistochemical staining is that it readily facilitates comparison of virus distribution and cellular localization in tissue sections to determine whether or not viral antigen distribution coincides with that of any lesions that are present (Figure 5.5 ).
Figure 5.5.

Immunohistochemical staining of bovine viral diarrhea virus (BVDV)-infected tissue. A formalin-fixed kidney specimen from an acutely ill calf was reacted with monoclonal antibody 15.c.5. Binding was detected using goat-antimouse serum tagged with horseradish peroxidase. Substrate for the enzyme was 3-amino-9-ethyl carbazole. Dark staining cells are positive for BVDV antigen.
Enzyme Immunoassay—Enzyme-Linked Immunosorbent Assay
Enzyme immunoassays (EIAs)—often referred to as enzyme-linked immunosorbent assays (ELISAs)—have revolutionized diagnostic testing procedures. Assays can be designed to detect antigens or antibodies. Although EIAs have high relative sensitivity, samples may still require more than 105 virus particles/ml for positive reactions with many tests. This level of sensitivity still makes these tests highly valuable, particularly in group settings, where any positive animal defines the herd status. Assays may be conducted on a single sample in the veterinarian's clinic or on many hundred samples at the same time, using automated systems in centralized laboratories. Some commonly used antigen detection test kits include those specific for feline leukemia virus, canine parvovirus, bovine viral diarrhea virus, rotavirus, and influenza virus. There are many different types of EIA tests that differ in their geometricproperties, detector systems, amplification systems and sensitivity. Not all possible tests will be discussed, as the basic test principles apply to all.
Most EIAs are solid-phase enzyme immunoassays; the “capture” antibody is attached to a solid substrate, typically the wells of polystyrene or polyvinyl microtiter plates. The simplest format is a direct EIA (Figure 5.6 ). Virus and/or soluble viral antigens from the specimen are allowed to bind to the capture antibody. After unbound components are washed away, an enzyme-labeled antiviral antibody (the “detector” antibody) is added; various enzymes can be linked to the antibody, but horseradish peroxidase and alkaline phosphatase are the most commonly used. After a washing step, an appropriate organic substrate for the particular enzyme is added and readout is based on the color change that follows. The colored product of the reaction of the enzyme on the substrate can be detected visually or read by a spectrophotometer to measure the amount of enzyme-conjugated antibody bound to the captured antigen. The product of the enzyme reactions can be modified to produce a fluorescent or chemiluminescent signal to enhance sensitivity. With all such assays, extensive validation testing must be carried out to determine the cut-off values of the test, which define the diagnostic sensitivity and diagnostic specificity of the test.
Figure 5.6.
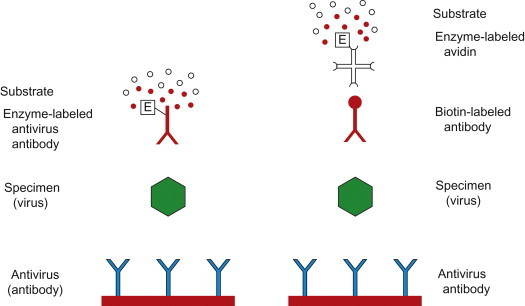
Enzyme immunoassay (EIA, also called enzyme-linked immunosorbent assay—ELISA) for the detection of virus and/or viral antigen. Left: Direct method. Right: Indirect method using biotinylated antibody, enzyme (e.g., peroxidase)-labeled avidin, and an enzyme substrate and chromogen for color reaction.
Indirect EIAs are widely used because of their greater analytical sensitivity, but the increase in sensitivity is usually accompanied by a loss of specificity. In this test format, the detector antibody is unlabeled and a second labeled (species-specific) anti-immunoglobulin is added as the “indicator” antibody (Figure 5.6). Alternatively, labeled staphylococcal protein A, which binds to the Fc moiety of IgG of many mammalian species, can be used as the indicator in indirect immunoassays. Monoclonal antibodies have especially facilitated the development of EIA tests, because they provide a consistent supply of highly sensitive and specific reagents for commercial tests. However, any variation (antigenic variation of the virus target) in the specific epitopes recognized by specific monoclonal antibodies can lead to loss of binding and loss of test sensitivity because of false-negative results.
EIAs have been adapted to formats for use in veterinary clinics on single animal specimens (Figure 5.7 ).
Figure 5.7.
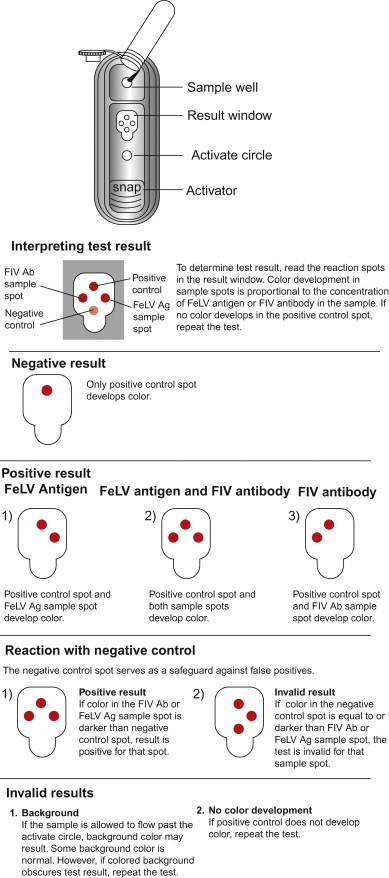
Commercial enzyme immunoassay device for clinical use on a single animal. This kit is for the simultaneous detection of feline leukemia virus (FeLV) antigen (Ag) and feline immunodeficiency virus (FIV) antibody (Ab) in feline serum, plasma, or whole blood. The detection of FeLV group-specific antigen is diagnostic of FeLV infection, and the detection of specific antibody to FIV is indicative of infection. The test utilizes a monoclonal antibody to FeLV p27, inactivated FIV antigen, and positive and negative controls. A conjugate mixture contains enzyme-conjugated antibody to p27 and enzyme-conjugated antigen. When the conjugate and the test sample are mixed, conjugated monoclonal antibody will bind to p27 antigen (if present). The sample–conjugate mixture is then added to the “Snap” device and flows across the spotted matrix. The matrix-bound p27 antibody (FeLV spot) will capture the p27-conjugated antibody complex, whereas the matrix-bound FIV antigen (FIV spot) will capture the FIV antibody-conjugated antigen complex. The device is then activated (snapped), releasing wash, and substrate reagents stored within the device. Color development in the FeLV antigen sample spot indicates the presence of FeLV antigen, whereas color development in the FIV antibody sample spot indicates the presence of FIV antibody.
(Courtesy of Idexx Laboratories, Inc.)
Immunochromatography
Immunochromatography simply refers to the migration of antigen or antigen–antibody complexes through a filter matrix or in a lateral flow format—for example, using nitrocellulose strips. In most formats, a labeled antibody binds to the antigen of interest. The antigen–antibody complexes are then immobilized in the support matrix by an unlabeled antibody bound to the matrix. All controls are included in the membrane as well, and results are seen as colored spots or bands, as one of the test reagents is conjugated to colloidal gold or a chromogenic substance. This test format is especially convenient for point-of-care testing, as the test process is simple and each test unit contains both positive and negative controls to assess test validity.
Detection of Viral Nucleic Acids
Developments in the area of nucleic acid technology in the past few years have relegated some (earlier) techniques to the annals of history with respect to their use in diagnostic testing. For example, classic hybridization techniques are not typically amenable to use for routine testing, especially with the requirement for rigorous quality-control standards. The most dramatic changes in nucleic acid detection technology have been in the evolution of polymerase chain reaction (PCR) testing, and the equally important standardization of nucleic acid extraction procedures. In addition, the rapid advances in nucleotide sequencing technology, oligonucleotide synthesis, and development of genetic databases permit inexpensive sequence analysis that has replaced less rigorous procedures for comparing genetic changes in virus strains and isolates. Current technology permits PCR amplification of virus “populations” with direct sequencing of the amplified products from the clinical specimen without the potential introduction of cell culture selection bias. More recent developments permit the detection and characterization of unknown agents (viral metagenomics). With the developments in nanotechnology, one could anticipate the future advent of inexpensive nucleic acid detection units that could reliably detect infectious agents when used in the clinician's office or in the field, without the need for highly trained personnel.
Nucleic acid detection methods are invaluable when dealing with: (1) viruses that cannot be cultured readily; (2) specimens that contain inactivated virus as a result of prolonged storage, fixation of tissue, or transport; (3) latent infections in which the viral genome lies dormant and infectious virus is absent; (4) virus complexed with antibody as would be found in the later stages of an acute infection or during some persistent viral infections. However, the added sensitivity provided by amplification of viral nucleic acid can actually create new problems. Unlike the situation with bacterial pathogens, it has usually been the case that merely detecting a pathogenic virus in a lesion, or from a clinically ill animal, has been considered evidence of its etiologic role (causal relationship). As detection methods have become increasingly sensitive and testing includes more agents, questions of viral “passengers” become more pertinent. Indeed, with viruses such as bluetongue virus, viral nucleic acid can be detected in the blood of previously infected ruminants several months after infectious virus has been cleared. Furthermore, with bovine herpesvirus 1 as an example, detection of viral nucleic acid does not address whether it is present as a consequence of acute infection, reactivation of a latent infection, or vaccination.
Polymerase Chain Reaction
The PCR assay is an in-vitro method for the enzymatic synthesis of specific DNA sequences using two oligonucleotide primers, usually of about 20 residues (20-mers), that hybridize to opposite strands and flank the region of interest in the target DNA; the primer pairs are sometimes referred to as forward and reverse primers (Figure 5.8 ). Primers are necessary to provide the DNA polymerase with a substrate upon which to add new nucleotides, and to direct the reaction to the specific region of the DNA for amplification. Primers can also be designed to provide “tags” on the amplified products for purposes of detection. Computer programs are used for the design of optimum primer sets and to predict the parameters (time/temperature) for the reactions. Where there are either known mismatched bases or anticipated mismatches between the primer and target sequences, the primers can be made to be degenerate—sets of primers with different bases at a given location. This can increase the diagnostic sensitivity of the test, as more genetic variants can be detected. For PCR, reactions are carried out in a thermocycler under carefully controlled conditions of ionic strength, temperature, primer concentration, and nucleotide concentration. Repetitive cycles involving template denaturation by heating, primer annealing, and extension of the annealed primers by DNA polymerase result in the exponential accumulation of a specific DNA fragment the termini of which are defined by the 5′ ends of the primers. The primer extension products synthesized in one cycle serve as templates in the next, hence the number of target DNA copies approximately doubles every cycle; 20 cycles yields about a millionfold amplification.
Figure 5.8.
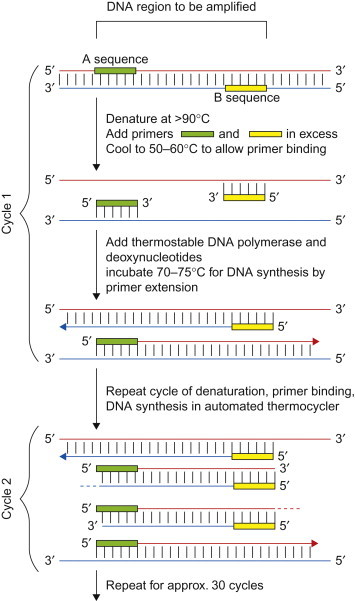
Amplification of part of a DNA sequence by the polymerase chain reaction. Oligonucleotide primers must first be made according to the sequences of either end of the portion of DNA to be amplified. After the DNA has been denatured by heating, the primers can hybridize to the complementary sequences on the opposite strand. In the presence of heat-resistant DNA polymerase and deoxynucleotide triphosphates, two new copies of the desired region are produced. The cycles of melting, annealing, and extension are repeated rapidly; each time, the amount of target DNA sequence doubles. After the first few cycles, virtually all the templates consist of just the short region chosen for amplification. After 30 cycles, taking about 3 hours, this region bounded by the chosen primers has been amplified many millionfold.
(Courtesy of I. H. Holmes and R. Strugnell.)
Since the introduction of the concept of PCR in 1983, there have been numerous changes to virtually every facet of the process. Incorporation of a thermostable DNA polymerase permitted high temperature denaturation and strand separation of the synthesized products, which eliminated the need to replenish the polymerase at each cycle. The use of a thermostable polymerase also increased the specificity of the reaction, as cycling could be done under more stringent annealing conditions; specifically, higher annealing temperatures reduce mismatch base pairing which can lead to false-positive results. In order to increase the sensitivity of the test, a “nested” PCR procedure was developed. In this procedure, one set of primers was used to do an initial amplification of a target area and the product of the first reaction became the template for a second PCR test in which new primers targeted a region internal to the first set of primers. This amplification of amplified product greatly increased the sensitivity of the test, but greatly increased the chances for false-positive results through contamination of test materials by the initial amplified product. Further developments in quantitative PCR technology have markedly reduced the use of nested procedures.
The development of reverse transcriptase polymerase chain reaction (RT-PCR) methods to detect RNA sequences was a major advance in cell biology and viral diagnostics. For RT-PCR, the RNA is first transcribed into cDNA using a DNA polymerase capable of using RNA as a template, such as retrovirus reverse transcriptase. Newer reverse transcriptase enzymes have been developed that permit synthesis of the cDNA strand at higher temperatures, which increases the analytical sensitivity and specificity of the reaction. In single-tube RT-PCR tests, all components for both reactions are placed in the reaction tube at the onset of the testing. The cDNA synthesis step is followed immediately by the PCR reaction. In this test format, there is no opportunity for products of one reaction to cross contaminate another, because the reaction tube is never opened until the end of the testing protocol. Advances such as the single-tube test greatly increased the reliability of PCR test results by virtually eliminating laboratory contamination problems.
Methods for Detection of Amplified Products
In the initial era of PCR testing, the amplified products were detected by analyzing the reaction products by gel separation. Amplified products of a defined sized were visualized by using fluorescent dyes that bound to the oligonucleotides separated in agarose gels. A “band” at the appropriate size was taken as a positive test for the presence of an agent in a sample. Methods were developed to increase the sensitivity of detecting bands in the gels, but even with enhanced sensitivity, this detection procedure had one major flaw—the reaction tube had to be opened in order to assess the status of the sample. Many laboratory areas became contaminated with the amplified reaction products, with false-positive results frequently obtained from subsequent samples run in the facility. Heroic efforts were made to avoid the false-positive problem, but suspicion of positive test results became prevalent and still linger. Fortunately, technology provided an answer that has come to dominate PCR testing: real-time PCR testing (Figure 5.9 ). This major technology advance was facilitated by the development of a thermocycler with a fluorimeter that could accurately measure (quantify) the accumulation of PCR product (amplicons) in the reaction tube as it was being made—that is, in real-time. Product is measured by increases in fluorescence intensity generated by several different fluorescent reporter molecules, including non-specific DNA binding dyes (SYBR Green I), TaqMan® probes (Figure 5.9A), and molecular beacons as examples. Once reactants are added to the reaction tubes, the tubes need never to be opened again, thus preventing any opportunity for laboratory contamination. The real-time detection systems are also more sensitive than standard gel systems, and added assay specificity is achieved through the use of reaction detection probes, because signal is generated only if the probe sequence is also able to bind to the target sequence.
Figure 5.9.
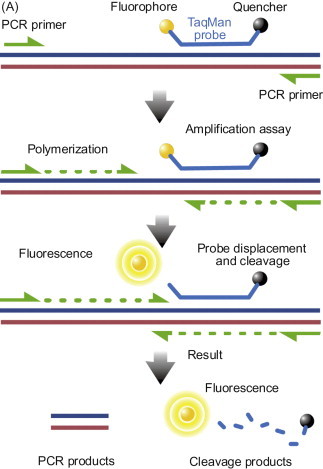

(A) TaqMan® probe chemistry mechanism. These probes rely on the 5′-3′ nuclease activity of Taq DNA polymerase to cleave a dual-labeled probe during hybridization to the complementary target sequence. (B) Real-time quantitative PCR data. Reaction curves for a test run to assess assay conditions using dilutions of an RNA transcript (copy number control) of a cloned segment of canine pneumovirus. The vertical lines represent the Ct value, which is the number of PCR cycles required for the fluorescent signal to cross the threshold value. TaqMan© probe was labeled with FAM (6-carboxy fluorescein; the reporter dye) at the 5′ end and BHQ (Black Hole Quencher; the quencher) at the 3′ end.
Another advantage of the real-time system is that the process can be quantitative. Under optimum conditions, the amount of the amplicon increases by a factor of 10 with each 3.3 amplification cycle (Figure 5.9B). With real-time systems, the generation of product is recorded at each cycle. The amount of product generated in a test reaction can be compared with a copy number control and, with proper extraction controls in the system, a direct measure of the amount of starting sequence can be determined. In humans, for example, this feature has particular value in monitoring responses over time to drug treatments for infections with hepatitis C and human immunodeficiency viruses.
A further variation in PCR testing that is becoming more commonly used is multiplex PCR. In this method, two or more primer pairs specific for different target sequences are included in the same amplification reaction. In this manner, testing can be done for several agents at the same time and in the same assay tube, thereby saving time and costs. With real-time, multiplex PCR assays, several probes with different fluorescent molecules can be detected simultaneously. This type of application is useful in evaluation of samples from disease complexes, such as acute respiratory disease in dogs. Issues of test sensitivity must be addressed in this format, because several reactions must compete for common reagents in the reaction, thus an agent in high copy number might mask the presence of one at low copy number.
Advantages and Limitations of the Polymerase Chain Reaction Technology
Given the explosion in use and availability of PCR assays in virological testing, consideration should be given to the potential benefits and limitations of these assays. The PCR assay is especially useful in the detection of viruses that are difficult to grow in culture, such as certain enteric adenoviruses, papillomaviruses, astroviruses, coronaviruses, noroviruses, and rotaviruses. PCR can be used on any sample that is appropriate for virus isolation; the decision to do PCR as opposed to other virus detection tests is based on speed, cost, and laboratory capability. PCR tests also may be preferred for the initial identification of zoonotic viruses such as rabies virus, certain poxviruses, or influenza viruses, to minimize the risk of exposure for laboratory personnel as amplification of infectious virus is not necessary for detection.
A limitation of PCR or any nucleic acid amplification technique can be the matrix in which the target sample is embedded. Material in the sample matrix can inhibit the enzymes on which the assay is based, which has been a constant source of concern when dealing with fecal samples and, to some extent, milk samples. Extraction controls need to be included in these types of sample in order to detect problems with the amplification process itself (rather than lack of specific template). Furthermore, PCR and simple nucleic acid amplification tests are agent specific, thus no signal will be generated if the primers do not match the sequence of any virus contained in the sample. With earlier direct PCR assays, and especially with nested PCR assays, false-positive test results were a very significant concern as a result of the ease of laboratory contamination with amplified product. With the availability of single-tube real-time PCR testing formats and real-time PCR tests, this problem has largely been eliminated, although correct performance of PCR assays remains a technically challenging process. Performance of real-time PCR assays is being continually improved with standardized reagent kits, robust instrumentation, standardized extraction protocols, and defined laboratory operating procedures, and this nucleic acid detection test format has become the mainstay of testing laboratories. However, test interpretation still requires evaluation of whether or not a particular test result (either positive or negative) is biologically relevant, which in turn requires a global assessment of history, clinical signs, and lesions in the particular animal from which the sample was obtained.
Microarray (Microchip) Techniques
Another technological advance that is impacting the field of diagnostics is the advent of microarrays or microchips. The microchip for nucleic acid detection is a solid support matrix onto which have been “printed” spots, each containing one of several hundred to several thousand oligonucleotides. Increasingly, these oligonucleotides can represent conserved sequences from virtually all viruses represented in the various genetic databases, or can be customized to represent only viruses from a given species involved in a specific disease syndrome, such as acute respiratory disease in cattle. The basis of the test is the capture by these oligonucleotides of randomly amplified labeled nucleic acid sequences from clinical specimens. The binding of a labeled sequence is detected by laser scanning of the chip and software programs assess the strength of the binding. From the map position of the reacting oligonucleotides, the software identifies the species of virus in the clinical sample. This type of test was used to determine that the virus responsible for severe acute respiratory syndrome (SARS) was a coronavirus. With knowledge of the oligonucleotide sequences that bound the unknown agent, primers can be made to eventually determine the entire nucleotide sequence of a new species of virus. The low cost of oligonucleotides synthesis, development of laser scanning devices, nucleic acid amplification techniques, and software development have made this technology available in specialized laboratories. A variation of this technique is the re-sequencing microarray. These arrays consist of a set of overlapping probes, which may differ from each other at a single base. The strength of binding to the individual probes in the family provides information on the genetic sequence of the amplified product. With current technology, the microarray approach is only available in specialized reference laboratories and it would not be used for routine diagnostic testing. In the standard format, this technique would probably not detect a new virus family not represented in a current database, because oligonucleotides for the new agent would not be included on the microchip.
Gene Amplification by Isothermal Amplification
For nucleic acid amplification, it is necessary to continually displace the newly synthesized product so that another copy of the sequence can be made. With PCR, the strand displacement is achieved with temperature: the 95°C temperature maximum melts (separates) the DNA strands, permitting binding of new primers. Isothermal amplification is a technique that does not require the temperature cycling and accompanying equipment used in PCR. Two techniques using different polymerases to achieve sequence amplification have been developed for isothermal amplification: nucleic acid sequence-based amplification (NASBA) and loop-mediated isothermal amplification (LAMP). If these techniques show significant advantages over PCR, one would expect the availability of an expanding array of available tests.
Nucleic Acid (Viral Genomic) Sequencing
Perhaps no area in molecular biology has advanced so rapidly as nucleic acid sequencing. With speed and capacity has come low cost, so that direct sequencing of complete viral genomes is now commonplace. Older techniques such as restriction mapping and oligonucleotide fingerprinting that were used to detect genetic differences among virus isolates have been displaced by sequencing methodology. In the area of diagnostics, new viruses are being discovered by techniques that take advantage of random nucleic acid amplification and low-cost sequencing. There are several basic techniques with numerous modifications that are too detailed to discuss individually. In general, the process involves random amplification of enriched nucleic acid samples or total nucleic acid samples, followed by sequencing of all the amplified products. The process works best if host-cell nucleic acids can be eliminated from the samples by nuclease treatment or subtraction of host sequences by hybridization to normal cell sequences immobilized on solid supports. No prior knowledge of the viral sequence is needed, and there is no need for any virus-specific reagents. Computer analysis of the sequenced material can identify sequences that are closely or distantly related to those of specific virus families. The method used to construct the entire genome, if this is desired, is somewhat dependent upon the number and size of sequences identified, but methods are available to “walk” down the entire genome from a single viral sequence. With these types of nucleic acid detection protocols, unknown viruses can be discovered and characterized without the requirement that they first be propagated in cell culture.
Detection and Quantitation of Virus-Specific Antibodies (Serologic Diagnosis)
The detection of an immune response to an infectious agent has, for the most part, relied on determining the antibody response of the host to the agent of interest. This approach measures only one limb of the adaptive immune response (humoral immunity); techniques for reliably measuring the cell-mediated responses have not been routinely available or cost effective. For many situations, measurement of antibody responses remains a valuable technique for defining the infection status of animals. Serological tests can be used to: (1) define whether an animal has ever been infected by a particular virus; (2) determine if a specific virus (or other pathogen) is linked to a clinical event; (3) determine if an animal has responded to a vaccination. For the serologic diagnosis of an acute viral disease in an individual animal, the classic approach has been to test paired sera—that is, an acute and a convalescent serum from the same animal, for a change in titer (fourfold or greater) of virus-specific antibody. The acute-phase serum sample is taken as early as possible in the illness; the convalescent-phase sample usually at least 2 weeks later. Given this time line, diagnosis based on this approach is said to be “retrospective.” In recent years this approach has been complemented by serologic methods for detecting virus-specific IgM antibodies—in many viral diseases a presumptive diagnosis may be made on the basis of detecting IgM antibody in a single acute-phase serum specimen—for example, West Nile virus infection of horses.
To assess whether an animal has ever been infected with certain viruses, serological testing can be more reliable than trying to detect the virus itself. For example, serological testing is used to screen horses for exposure to equine infectious anemia virus, cattle for bovine leukemia virus, and goats for caprine arthritis encephalitis virus. In these instances, the number of infected cells in chronically infected animals may be too low for even PCR detection, but infection generally stimulates an antibody response that is readily detected by various tests. Serological testing is also widely used both during virus eradication programs and in the certification of animals for movement and trade.
Use of serological tests to assess vaccine efficacy can be an important aspect of an infectious disease management program. In many countries, purchase of vaccine can be done by the animal owner. Antibody testing of selected animals can provide the practitioner with valuable insight as to whether the immunization program of the producer is being performed correctly. As eradication programs expand for diseases of production animals, marker vaccines are more frequently being used and so-called DIVA serological assays can distinguish whether a given antibody response is caused by vaccine or natural infection. For herpesvirus infections such as bovine herpesvirus 1, it is essential to determine whether an antibody response is the result of infection, because infection invariably leads to latency. Movement of a latently infected animal into a negative herd can result in an outbreak of disease, thus gene deletion “marker” vaccines were developed to facilitate differentiation of vaccinated and naturally infected cattle.
Serum Specimens for Serologic Assays
For most serological tests, serum is the sample of choice. However, some tests have been validated using plasma as well as serum. Communication with the testing laboratory is necessary when fluids other than serum are being collected, in order to avoid having to re-sample the animal when serum is the only acceptable test material. Antibodies in serum are very stable to moderate environmental conditions. Standard protocols call for serum to be kept cold, but freezing of the sample is not necessary unless several weeks will elapse between collection and testing. Antibodies can even be detected from blood samples dried onto filter paper and stored for months before testing.
As with other aspects of diagnostic testing, technological advances continue to modify how antibodies to specific viruses are detected. In most cases, the newer technologies are applied to those tests that have some commercial potential. In veterinary medicine, there are many tests for agents that may be of minor importance but useful in certain situations. Tests available for these agents may be the first ones developed with older testing technology. As viruses of wildlife species assume greater importance through public awareness, it will be necessary to develop additional serological tests, because species-specific tests for domestic species cannot be used. All serological test types will not be discussed in detail (below), but readers should be aware that other test formats may become available and continuing communication with their testing laboratory is the most efficient way to learn about the tests available for each species and for each virus.
Enzyme Immunoassay—Enzyme-Linked Immunosorbent Assay
Enzyme immunoassays (EIAs, ELISA) are the serologic assays of choice for the qualitative (positive or negative) or quantitative determination of viral antibodies because they are rapid, relatively cost effective, and may not require the production of infectious virus for antigen if recombinant antigens are used. In the EIA test format for antibody detection, viral antigen is bound to a solid matrix. Serum is added and, if antibodies to the antigen are present in the sample, they bind to it. In direct EIA tests, the bound antibody is detected by an anti-species antibody tagged with an enzyme. With addition of the enzyme substrate, a color reaction develops that can be assessed either visually or with a spectrophotometer. Controls run with the sample define whether the test is acceptable and which samples in the test are positive. Kinetics-based EIAs offer the advantage that quantitative assays can be based on a single dilution of serum. The product of the enzyme reaction is determined several times over a short interval. Software programs convert the rate of product development to the amount of antibody bound to the antigen.
A disadvantage of direct EIA tests is that they are species specific. A test developed for canine distemper virus antibodies in a dog cannot be used to determine the presence or absence of antibodies to the same virus in a lion. To obviate this problem, competitive or blocking EIA tests have been developed. In this test format, an antibody that binds to the antigen of interest (usually a monoclonal antibody) is tagged with the enzyme. Unlabeled antibody that can bind to the same site as the monoclonal antibody will compete with the labeled monoclonal antibody for that site. A reduction in the binding of the labeled monoclonal antibody indicates that the sample did contain antibody (Figure 5.10 ). In this test format, the species of the unlabeled antibody is not a factor. The diagnostic sensitivity and specificity of EIA tests, whether direct or indirect, have been greatly enhanced by the development of monoclonal antibodies and the production of recombinant antigens.
Figure 5.10.

Competitive enzyme-linked immunosorbent assay (cELISA) for caprine arthritis encephalitis viral (CAEV) antibodies. Undiluted serum samples in duplicate are added to antigen-coated wells of a commercial cELISA test for CAEV antibodies. After removal of the test sera, an antibody specific for CAEV antigen and coupled to horseradish peroxidase is added. The detector antibody is removed after incubation, and a substrate is added to detect the presence of bound detector antibody. If there are antibodies specific for CAEV in the test sera, these antibodies bound to antigen will prevent the detector antibody from binding. A positive sample will therefore show less enzyme product (color) than the negative controls. Cut-off values are determined by reading the intensity of the reaction with a spectrophotometer, although visual inspection can usually detect positive samples. Wells A1–2 and G11–12: positive controls; wells B1–2 and H11–12: negative controls; wells D1–2, H1–2, B3–4, F3–4, C5–6, D5–6, A7–8, E7–8, G7–8, and B9–10: samples positive for antibodies to CAEV.
In a widely used format for test kits that can be run in a practitioner's office, the test serum flows through a membrane filter that has three circular areas impregnated with antigen, two of which have already interacted with a positive and a negative serum, respectively (Figure 5.7). After the test serum flows through the membrane and a washing step is completed, a second anti-species antibody with an enzyme linked to it is added and the membrane is again rinsed before the addition of the enzyme substrate. The result is read as a color change in the test sample circle, which is compared against the color change in the positive control and no change in the negative control. Such single-patient tests are relatively expensive compared with the economies of testing hundreds of sera in a single run in a fully automated laboratory. The great savings in time and effort to send samples to the laboratory, in addition to the fact that decisions can be made while both client and patient are still in the consulting room, make single tests attractive and useful in the immediate clinical management of critically ill animals.
Serum (Virus) Neutralization Assay
As virus isolation is considered the gold standard for the detection of virus against which other assays must be compared, the serum (virus) neutralization test has historically been the gold standard, when available, for the detection and quantitation of virus-specific antibodies. Neutralizing antibody also attracts great interest because it is considered a direct correlate of protective antibody in vivo. For the assay of neutralizing antibody, two general procedures are available: the constant-serum–variable-virus method and the constant-virus–variable-serum method. Although the constant-serum–variable-virus method may be a more sensitive assay, it is rarely used because it utilizes relatively large amounts of serum, which may not be readily available. The basis of the neutralization assay is the binding of antibody to infectious virus, thus preventing the virus from initiating an infection in a susceptible cell. The growth of the virus is detected by its ability to kill the cell (cytopathic effect) or by its ability to produce antigen in the infected cells that is detected by immunofluorescence or immunohistochemistry. The amount of antibody in a sample is determined by serial dilution of the sample and “challenging” each of these dilutions with a standard amount of virus (constant-virus–variable-serum method). The last dilution that shows neutralization of the virus is defined as the endpoint and the titer of the serum is the reciprocal of the endpoint dilution; for example, an endpoint of 1:160 equates to a titer of 160. The disadvantages of serum neutralization tests are that they are relatively slow to generate a result, require production of infectious virus for the test, and have a constant high overhead cost in maintaining cell culture facilities for the test. These assays have the benefit of being species independent and, as such, are very useful in wildlife studies. With new agents, a serum neutralization test can be operational with several weeks of isolating the virus, whereas EIA test development may take months or even years to validate.
Immunoblotting (Western Blotting)
Western blotting tests simultaneously but independently measure antibodies against several proteins of the agent of interest. There are four key steps to western blotting. First, concentrated virus is solubilized and the constituent proteins are separated into discrete bands according to their molecular mass (M r), by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Secondly, the separated proteins are transferred electrophoretically (“blotted”) onto nitrocellulose to immobilize them. Thirdly, the test serum is allowed to bind to the viral proteins on the membrane. Fourthly, their presence is demonstrated using a radio-labeled or, most commonly, an enzyme-labeled anti-species antibody. Thus immunoblotting permits demonstration of antibodies to some or all of the proteins of any given virus, and can be used to monitor the presence of antibodies to different antigens at different stages of infection. Although this procedure is not routinely used in a diagnostic setting with viruses, western blots were central to the identification of immunogenic proteins in a variety of viruses. Similarly, the assay is used in the analysis of samples for the presence of prion proteins in ruminant tissues. Western blots are more of a qualitative test than a quantitative one, and are not easily standardized from laboratory to laboratory. For this reason, ELISAs and bead-based assays are preferred test formats.
Indirect Immunofluorescence Assay
Indirect immunofluorescence assays are used for the detection and quantitation of antibody; specifically, these are tests that use virus-infected cells (usually on glass microscope slides) as a matrix to capture antibodies specific for that virus. Serial dilutions of test serum are applied to individual wells of the cell substrate and usually an anti-species antibody with a fluorescent tag is then added as the detector of antibody binding. Slides are read with a fluorescent microscope and scored as positive if the infected cell shows a fluorescent pattern consistent with the antigen distribution of the virus used. This test is rapid (less than 2 hours) and can be used to determine the isotype of the reacting antibody if one uses an anti-isotype-specific serum such as an anti-canine IgM. Non-specific fluorescence can be an issue, particularly with animals that have been heavily vaccinated as they may contain anti-cell antibodies that will bind to uninfected cells and mask specific anti-virus fluorescence. Test slides for some agents can be purchased, so that laboratories offering this test need not have infectious virus or a cell culture facility.
Hemagglutination-Inhibition Assay
For those viruses that hemagglutinate red blood cells of one or another species, such as many of the arthropod-borne viruses, influenza viruses, and parainfluenza viruses, hemagglutination-inhibition assays have been widely used. For detecting and quantitating antibodies in the serum of animals, the methods are sensitive, specific, simple, reliable, and quite inexpensive. In spite of all of the technological advances, hemagglutination inhibition assays remain the mainstay for determining antibody responses to specific influenza A viruses. The principle of the assay is simple—virus binds to red blood cells through receptors on their surface. Antiviral antibodies bind to these receptors and block hemagglutination. Serum is diluted serially in the wells of the microtiter plate, usually in twofold steps, and to each well a constant amount of virus, usually four or eight hemagglutinating units, is added. The reciprocal of the highest dilution of serum that inhibits the agglutination of the red blood cells by the standardized amount of virus represents the hemagglutination-inhibition titer of the serum (Figure 5.11 ). Care should be taken in interpreting many prior sero-surveys based on results of hemagglutination inhibition tests, particularly for paramyxoviruses, as non-specific inhibitors of agglutination produced many false-positive test results in some of those studies.
Figure 5.11.

Hemagglutination inhibition (HI) test for detecting antibodies specific for canine influenza virus (H3N8). Sera treated to remove non-specific agglutinins and non-specific inhibitors of agglutination are diluted (twofold) in buffered saline in the wells of a 96-well microtiter plate. Following the dilution operation, an equal volume of canine influenza virus (≈4 hemagglutinin units) is added to each well and the plate incubated for 30 minutes. An equal volume of turkey red blood cells (0.5% suspension) is then added to each well. The HI reactions are determined when the control-cell wells show complete settling (button) of the red blood cells. Rows A, B: titration of viral suspension used in test, showing correct amount of hemagglutinin added to test wells. Row C: red blood cell control. Rows D–H: test canine sera. Wells D–H1: test serum control showing no non-specific agglutination of red blood cells at lowest dilution tested. HI titers are the reciprocal of the last dilution showing inhibition of agglutination by test virus. Row D: HI titer=64; row E: HI titer <4; row F:HI titer=8; row G: HI titer=2048; row H: HI titer=256.
Immunodiffusion
Historically, agar gel immunodiffusion (AGID) assays were used for the specific diagnosis of a number of viral infections and diseases, including bluetongue, hog cholera, influenza, equine infectious anemia (Coggins test), and bovine leukemia. These assays are very simple to perform, they utilize inexpensive materials, and they do not require production of infectious material by the testing laboratory. Often crude cell extracts or even tissue extracts from infected animals can be used as the test antigen. AGID tests are relatively fast, easily controlled, but lacked sensitivity as compared with later developed EIA tests. Furthermore, they are strictly qualitative (providing a simple yes/no answer) and cannot be automated (Figure 5.12 ). Thus most of these tests have been replaced with EIA tests.
Figure 5.12.

Agar gel immunodiffusion test for antibody detection for viral agents such as BTV, EIA, EHD, and influenza A viruses. Spatially defined wells are produced in a semi-solid matrix such as agar or agarose. Into the central well is placed the test antigen (AG). Serum containing antibodies (AS) to the virus of interest is placed in alternating wells around the central antigen well. Test sera (1,2,3) are placed in the remaining wells. Plates are incubated for 24–48 hrs to allow the development of visible precipitin lines between the antigen and the test sera. Well 1 = weak positive; well 2 = negative; well 3 = strong positive.
IgM Class-Specific Antibody Assay
A rapid antibody-based diagnosis of a viral infection or disease can be made on the basis of a single acute-phase serum by demonstrating virus-specific antibody of the IgM class. Because IgM antibodies appear early after infection but drop to low levels within 1–2 months and generally disappear altogether within 3 months, they are usually indicative of recent (or chronic) infection.
The most common method used is the IgM antibody capture assay, in which the viral antigen is bound on a solid-phase substrate such as a microtiter well. The test serum is allowed to react with this substrate and the IgM antibodies “captured” by the antigen are then detected with labeled anti-IgM antibody matched to the species from which the specimen was obtained.
New Generation Technologies
As with nucleic acid technologies, technological developments for analyte detection are rapidly evolving, and a substantial number of potentially novel platforms for serological assays have been developed that have not yet been fully validated for routine diagnostic use. It is beyond the scope of this text to provide an exhaustive catalog of these technologies, many of which will never find their way into routine diagnostic use. However, one technology that has demonstrated particular promise in both the clinical and research arena is xMAP®, developed by Luminex. The success of this testing platform probably reflects the maturity of existing technologies that were combined to provide a versatile analyte detection system. xMAP® combines a flow cytometry platform, uniquely labeled microspheres, digital signal processing, and standard chemical coupling reactions to provide a system that can be used to detect either proteins or nucleic acids (Figure 5.13 ). The microspheres carry unique dyes (up to 100 different ones) that emit fluorescent signals that identify the individual beads coupled with a specific ligand. For antibody detection tests, the antigen of interest is coupled to a specific bead. The beads are exposed to the test serum and the bound antibody is detected with an anti-species antibody tagged with a reported dye. The microspheres are analyzed in a flow cytometer in which lasers excite both the bead dyes and the reporter dyes. Multiple beads for each antigen are analyzed in each test, providing independent readings of the reaction.
Figure 5.13.

Multiplex assay for the detection of cytokines. The feasibility of this type of multiplex assay resides in the ability to make microspheres with unique fluorescent signatures and with surface-reactive groups that can be used to bind various ligands. For the cytokine assay, sets of microspheres are individually coated with antibodies specific for cytokines in the screening panel. The microspheres are mixed together and reacted with the test specimen. The binding is detected by anti-cytokine antibodies with a fluorescent tag. The fluorescent signature of the individual microspheres and the fluorescent signal from the bound antibodies are read in a cell-sorting device using lasers for excitation of the dyes. The unique signatures of the microspheres permit a quantitative analysis of up to 100 different reactants in a single specimen.
(Courtesy of B. Wagner, Cornell University.)
One distinct advantage of this system is its multiplex capability. Theoretically, 100 or more different antigens can be assessed for antibody reactivity in a single assay. For maximum sensitivity and specificity, recombinant antigens are needed to eliminate extraneous proteins that would reduce specific antigen density on the beads and increase non-specific background reactivity that can confuse test interpretation. Advantages of this bead-based system are: (1) it utilizes small sample volumes; (2) it can be multiplexed; (3) it has been reported to be more sensitive than standard ELISA tests; (4) it can be less expensive than many serology tests; (5) it can be more rapid than ELISA tests, particularly when testing for antibodies to several antigens. As an example, this test platform is ideal for the antibody screening tests that are necessary for maintaining research rodent colonies in which antibody responses to several agents are monitored and for which sample volumes are often limiting. This platform can also provide DIVA testing as would be applied for control of important regulatory disease such as foot-and-mouth disease. As an example, recombinant antigens representing the capsid proteins present in inactivated foot-and-mouth disease virus vaccines along with non-structural viral protein can be coupled to different beads to analyze the antibody profile of a suspect animal. In a single assay, the test can provide evidence of vaccination—response to capsid antigen only—or of a natural infection—response to both types of proteins. One could envision this type of bead-based assay as a quantitative western blot, in that reactivity to several antigens can be assessed. As eradication programs progress for viral diseases of production animals, it is very likely that the requirement for this type of DIVA testing will only increase. The disadvantage for antibody detection is the need for recombinant antigens to achieve acceptable sensitivity, and high validation costs associated with multiplex reactions.
Interpretation of Laboratory Findings
As with any laboratory data, the significance of specific results obtained from the virology laboratory must be interpreted in light of the clinical history of the animal from which the sample was collected. To some extent, the significance of any result is also influenced by the type of virus that was detected. A fluorescent-antibody positive test for rabies virus on a bat found in a child's bedroom will elicit a public health response in the absence of clinical data, whereas a positive serological test for bovine leukemia virus from the dam of an aborted fetus is likely to be an irrelevant finding if the animal is from an enzootic region. With multiplex PCR testing, it may be possible to detect several different viruses, bacteria, and mycoplasma species in a single dog with acute respiratory disease, raising the obvious question, “what is significant?” Are the virus signals due to a recent vaccination, reactivation of a herpesvirus, or “footprints” of the etiological agent? Clearly, several sources of data must be integrated by the clinician to arrive at a coherent treatment strategy. However, it is also clear that the speed, the number, and the reliability of virus detection tests have changed the way in which clinicians use laboratory test results, and these results are having greater impact on treatment and management decisions. When attempting to interpret the significance of the detection of a specific virus in a clinical specimen, one may be guided by the following considerations.
The site from which the virus was isolated. For example, one would be quite confident about the etiological significance of equine herpesvirus 1 detected in the tissues of a 9-month-old aborted equine fetus with typical gross and microscopic lesions. However, recovery of an entero-virus from the feces of a young pig may not necessarily be significant, because such viruses are often associated with inapparent infections.
The epidemiologic circumstances under which the virus was isolated. Interpretation of the significance of a virus isolation result is much more meaningful if the same virus is isolated from several cases of the same illness in the same place and time.
The pathogenetic character of the virus detected. Knowledge that the virus detected is nearly always etiologically associated with frank disease—that is, rarely is found as a “passenger”—engenders confidence that the finding is significant.
The identity of the specific virus. The detection of foot-and-mouth disease virus in any ruminant in a virus-free country would, in and of itself, be the cause for great alarm. Similarly, the identification of mouse hepatitis virus in a free colony, or koi herpesvirus amongst highly valuable ornamental fish, would trigger a substantial response.
Interpretation of Serologic Laboratory Findings
A significant (conventionally, fourfold or greater) increase in antibody titer between acute and convalescent samples is the basis, albeit in retrospect, for linking a specific virus with a clinical case of a particular disease. However, one must always be aware of the vaccination status of the animal, as sero-responses to vaccines, especially live-attenuated virus vaccines, may be indistinguishable from those that occur after natural infections. The demonstration of antibody in a single serum sample can be diagnostic of current infection in an unvaccinated animal (e.g., with retroviruses and herpesviruses), because these viruses establish life-long infections. However, in such circumstances there is no assurance that the persistent virus was responsible for the disease under consideration. Assays designed to detect IgM antibody provide evidence of recent or current infection. A summary of the major strengths and limitations of the several alternative approaches to the serological diagnosis of viral infections is given in Table 5.1.
Detection of antiviral antibody in pre-suckle newborn cord or venous blood provides a basis for specific diagnosis of in-utero infections. This approach was used, for example, to show that Akabane virus was the cause of arthrogryposis-hydranencephaly in calves. Because transplacental transfer of immunoglobulins does not occur in most domestic animals, the presence of either IgG or IgM antibodies in pre-suckle blood is indicative of infection of the fetus.
Sensitivity and Specificity
The interpretation and value of a particular serologic test is critically dependent on an understanding of two key parameters: diagnostic sensitivity and diagnostic specificity. The diagnostic sensitivity of a given test is expressed as a percentage and is the number of animals with the disease (or infection) in question that are identified as positive by that test, divided by the total number of the animals that have the disease (or infection) (Table 5.3 ). For example, a particular EIA used to screen a population of cattle for antibody to bovine leukemia virus may have a diagnostic sensitivity of 98%—that is, of every 100 infected cattle tested, 98 will be diagnosed correctly and 2 will be missed (the false-negative rate=2%). In contrast, the diagnostic specificity of a test is a measure of the percentage of those without the disease (or infection) who yield a negative result. For example, the same EIA for bovine leukemia virus antibody may have a diagnostic specificity of 97%—that is, of every 100 uninfected cattle, 97 will be diagnosed correctly as negative, but 3 will be diagnosed incorrectly as infected (the false-positive rate=3%). Whereas diagnostic sensitivity and diagnostic specificity are fixed percentages intrinsic to the particular diagnostic assay and the population of animals used to validate the test, the predictive value of an assay is affected greatly by the prevalence of the disease (or infection) in the test population. Thus, if the same EIA is used to screen a high-risk population with a known bovine leukemia prevalence of 50%, the predictive value of the assay will be high, but if it is used to screen a population with a known prevalence of 0.1%, the great majority of the 3.1% of animals that test positive will in fact be false-positives and will require follow-up with a confirmatory test of much higher specificity. This striking illustration draws attention to the importance of selecting diagnostic assays with a particular objective in mind. An assay with high diagnostic sensitivity is required when the aim is to screen for a serious infection or when eradication of the disease is the aim, in which case positive cases must not be missed. An assay (usually based on an independent technology) with very high diagnostic specificity is required for confirmation that the diagnosis is correct.
Table 5.3.
Calculations of Accuracy of Serologic Testing
| Test Table | |||
|---|---|---|---|
|
Reference Test Results |
|||
| + | − | ||
| New Test Results | + | TP | FP |
| − | FN | TN | |
| Sensitivity | The sensitivity of a test is the probability that it will produce a true positive result when used on an infected population (as compared to a reference or “gold standard”). After inserting the test results into a table set up like the Test Table above, the sensitivity of a test can be determined by calculating: |
| Specificity | The specificity of a test is the probability that a test will produce a true negative result when used on a noninfected population (as determined by a reference or “gold standard”). After inserting the test results into a table set up like the Test Table above, the specificity of a test can be determined by calculating: |
| Positive Predictive Value | The positive predictive value of a test is the probability that a person is infected when a positive test result is observed. In practice, predictive values should only be calculated from cohort studies or studies that legitimately reflect the number of people in that population who are infected with the disease of interest at that time. This is because predictive values are inherently dependent upon the prevalence of infection. After inserting results into a table set up like the Test Table above, the positive predictive value of a test can be determined by calculating: |
| Negative Predictive Value | The negative predictive value of a test is the probability that a person is not infected when a negative test result is observed. This measure of accuracy should only be used if prevalence is available from the data. (See note in positive predictive value definition.) After inserting test results into a table set up like the Test Table above, the negative predictive value of a test can be determined by calculating: |
FP, number of false positive specimens; FN, number of false negative specimens; TP, number of true positive specimens; TN, number of true negative specimens.
The analytic sensitivity of a given immunoassay is a measure of its ability to detect small amounts of antibody (or antigen). For instance, EIAs and serum neutralization assays generally display substantially higher analytical sensitivity than AGID tests. Improvements in analytical sensitivity may be obtained by the use of purified reagents and sensitiveinstrumentation. However, the analytical specificity of an immunoassay is a measure of its capacity to discriminate the presence of antibody directed against one virus versus another. This quality is influenced mainly by the purity of the key reagents, especially the antigen when testing for antibody and the antibody when testing for antigen.