

Abstract
Many viruses replicate and assemble in subcellular microenvironments called virus factories or ‘viroplasm.’ Virus factories increase the efficiency of replication and at the same time protect viruses from antiviral defenses. We describe how viruses reorganize cellular membrane compartments and cytoskeleton to generate these ‘mini-organelles’ and how these rearrangements parallel cellular responses to stress such as protein aggregation and DNA damage.
Keywords: Aggresome, Autophagosome, Autophagy, Double membrane vesicle, PML-nb, Replicase, Spherule, Viroplasm, Virus factory, Virus replication
Introduction
Viruses are relatively simple life forms which use a protein capsid to carry short stretches of genomic material from one cell to another. All viruses are obligate intracellular parasites and need to enter cells to replicate. Nucleic acid genomes delivered to the cytoplasm provide a template to generate new genomes which in turn initiate the synthesis of mRNA that encode viral proteins. These processes require the assembly of complex replication machines that ensure that genome replication and packaging are carefully coordinated. For many viruses this is achieved by concentrating virus genomes, replicase proteins, and the host proteins required for replication in a ‘mini-organelle’ called a virus factory or ‘viroplasm’ (Netherton et al., 2007, Fernandez de Castro et al., 2012).
All viruses synthesize mRNA and use host ribosomes to make viral proteins. Viruses are grouped into the ‘RNA’ or ‘DNA’ viruses according to the nature of their genome. The (+) strand genomes make an mRNA that is immediately translatable, and the complimentary RNA or DNA sequence is called negative (−) strand. This means that the genomes of (+) strand RNA viruses can function as mRNA while the (−) strand RNA viruses need to be delivered into the cell with an RNA-dependent RNA polymerase (RdRp) that uses the negative template to make the (+) strand. In both cases virus replication occurs in association with cytoplasmic organelles and (+) RNA is translated by ribosomes into protein. The genomes of most DNA viruses are made from double-stranded DNA and most DNA viruses replicate in the nucleus because they need to recruit host proteins for DNA replication. The DNA viruses also show temporal gene expression with genes being expressed at early, intermediate, and late times during infection. Early genes are expressed before the onset of replication while late genes are normally used for replication and assembly. The small DNA viruses, for example, polyoma and papillomaviruses have limited coding capacity and do not make their own DNA-dependent DNA polymerase (DdDp). The large DNA viruses, for example herpes viruses and poxviruses encode their own DdDp, but the herpes viruses still rely on nuclear factors for genome replication. Interestingly, the poxviruses encode all the enzymes needed for DNA and mRNA synthesis, and replicate in the cytoplasm.
Evolution has not been blind to viral infection and cells make several proteins that recognize viral genomes as danger signals because they are different from host DNA or RNA. The RNA viruses are a good example because replication of RNA from an RNA (rather than DNA) template generates a double-stranded RNA (dsRNA) intermediate that is not generated by the host cell. Recognition of dsRNA in plants, insects, and other primitive animals results in gene silencing while in vertebrates this leads to activation of interferon (Randall and Goodbourn, 2008). Cells also contain sensors for viral DNA that activate proinflammatory responses (Rathinam and Fitzgerald, 2011). The concentration of replicase proteins within virus factories would be expected to provide a potent signal for activation of these defenses. Viruses have overcome this problem by using virus factories to shield replication intermediates from recognition by nucleic acid sensors, but at the same time the factories retain connections to the cytoplasm to allow export of nucleic acid to sites of genome packaging.
RNA Viruses
(+) Strand RNA Viruses Generate Mini-Organelles from Membranes Derived from the Secretory Pathway
The genomes of the (+) strand RNA viruses encode a single polyprotein synthesized by cellular ribosomes. The capsid proteins and the nonstructural proteins required for replication and are generated from the polyprotein by viral, and sometimes host, proteases. In each case nonstructural proteins with hydrophobic domains act as a scaffold to target the RdRp to the cytoplasmic face of membrane-bound organelles where assembly of the replicase complex induces membrane curvature (reviewed Cottam et al., 2009, den Boon et al., 2010, den Boon and Ahlquist, 2010). These remodeled membranes vary greatly between different families of (+) strand RNA viruses but are thought to be formed by similar mechanisms where their dimensions are determined by the assembly of the replicase proteins during invagination. This process is analogous to the way that virus capsids vary in size and shape depending on the assembly of capsid subunits (Ahlquist, 2006).
Spherules produced by the Togaviridae and nodaviruses
The mammalian alphaviruses, for example, Semliki Forest virus (SFV), Sindbis virus (SINV), and the rubivirus rubella virus are members of the ‘Togavividae’ and encode a single polyprotein called nsp1234 that contains the helicase, methytransferase, and RdRp. Proteolytic processing releases the RdRp (P4) which binds to P1,2,3 and is targeted to membranes by a hydrophobic domain in P1 (Figure 1(a) ). The structural proteins are generated as a second polyprotein encoded by a sub-genomic message which is formed when the RdRp transcribes RNA from an internal initiation site. A signal sequence exposed after removal of the capsid protein directs synthesis of envelope proteins by ribosomes attached to the endoplasmic reticulum (ER). The ‘Togaviridae’ generate large ‘cytopathic vacuoles’ with 50 nm diameter invaginations called spherules aligned along the inside face of the vacuole. The vacuoles contain replicase proteins and are derived from endosomes and lysosomes. Spherules can be generated from many different membrane-bound organelles. Flock house virus, an insect virus, generates spherules in the outer membrane of mitochondria (Figures 1(b) and 1(c); Miller et al., 2001) while Brome mosaic virus, a plant virus, generates spherules in the ER. In each case the spherules are of similar size and contain approximately 100 copies of the replicase protein packed along the inner membrane surface (Kopek et al., 2007). The interior of each spherule is connected to the cytosol by a thin membrane neck that surrounds a channel wide enough to allow passage of (+) RNA into the cytosol allowing assembly with capsid proteins. Spherule formation involves the assembly of replicase proteins on the cytoplasmic face of membrane-bound organelles where they induce negative curvature forcing membrane to invaginate into the organelle. Multivesicular body formation is topologically similar where ESCRT (endosomal sorting complex required for transport) proteins assemble on the cytoplasmic face of endosomes generating vesicles that bud away from the cytoplasm into the endosome. Recent work has shown that proteins of the ESCRT-III complex are required for proper formation of spherules where they may drive spherule closure and at the same time maintain the channel that allow genomes to pass into the cytosol (Barajas et al., 2014, Diaz et al., 2015).
Figure 1.

Formation of spherules during replication of alphaviruses. (a) The alphaviruses encode a single polyprotein called nsp1234 that contains the helicase, methytransferase, and RdRp. Proteolytic processing releases the RdRp (P4) which is targeted to membranes by a hydrophobic domain in P1 (i) The structural proteins (ii) synthesized as a second polyprotein (C,E2,6k,E1) are directed by ribosomes to the endoplasmic reticulum. Assembly of replicase proteins induces membrane curvature and invagination forming a spherule (iii). The interior of each spherule is connected to the cytosol by a thin membrane neck that surrounds a channel wide enough to allow passage of (+) RNA into the cytosol allowing assembly with capsid proteins (iv). In the case of Bromovirus and tombusvirus, ESCRT proteins (green) recruited to the spherule by replicase proteins stabilze the membrane neck and prevent closure of the spherule. (b) Spherules generated in mitochondria by flock house virus. Electron micrographs (i) generated from a tilt series were processed digitally to generate the tomogram in (ii). (c) A 3D reconstruction of spherules shows the likely organization of replicase proteins of flock house virus modeled as 7 nm diameter spheres (green) lining the inner surface of the spherule.
Reproduced from Kopek, B.G., Perkins, G., Miller, D.J., Ellisman, M.H., Ahlquist, P., 2007. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biology 5, e220.
Convoluted membranes and membrane webs generated by coronaviruses and flaviviruses
The flaviviruses, for example, yellow fever virus, West Nile virus, and hepatitis C (HCV) also generate a single polyprotein that, in common with picornaviruses, is processed to generate structural and nonstructural proteins (Figure 2(a) ). The flaviviruses are enveloped viruses and the initial processing of the polyprotein exposes signal sequences that direct synthesis and processing of the polyprotein by the ER. The NS5B protein at the C-terminus of the polyprotein is targeted to the cytoplasmic face of the ER by hydrophobic domains in NS4A, NS5A, and NS5B. The flaviviruses generate 80–100 nm diameter invaginations into an ER membrane connected to spherical vesicles and convoluted membranes (Figure 2(b)). These invaginations are less well organized than spherules, even so the spherical vesicles contain the viral polymerase and dsRNA and each has a pore opening to the cytosol that could allow exit of viral RNA for packaging with capsid proteins and envelopment (Welsch et al., 2009, Romero-Brey et al., 2012). The genome of the coronaviruses (CoV) is similar in organization to the flaviviruses, but these viruses are the most complex of the (+) strand RNA viruses and encode as many as 15 nonstructural proteins. Severe acute respiratory syndrome coronavirus (SARS-CoV) induces double membrane vesicles (DMVs) that are between 150 and 300 nm diameter connected to the ER (Figure 2(c); Knoops et al., 2008). The infected cells also contain convoluted tubular and reticular membranes, and later during infection ‘vesicular packets’ appear where vesicles are surrounded by a common outer ER membrane (Knoops et al., 2008). It is thought that replication occurs in the convoluted membranes and that genomes are transported to vesicular packets for envelopment and budding.
Figure 2.
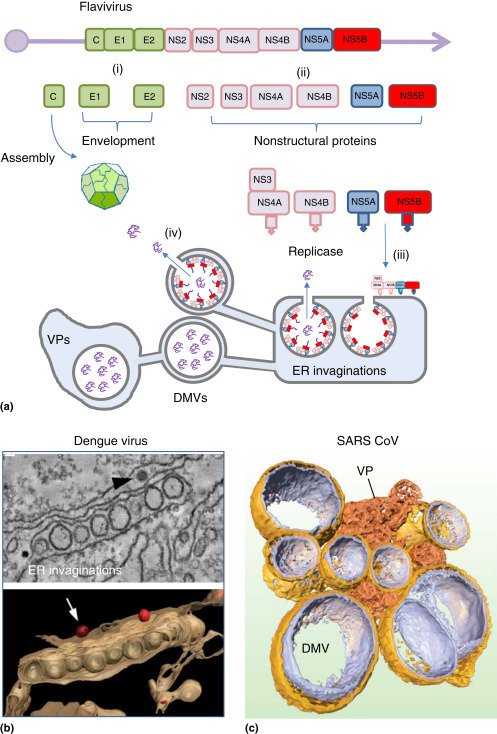
Formation of double-membraned vesicles by flaviviruses and coronaviruses. (a) The flaviviruses polyprotein encodes the capsid protein (C), envelope proteins (E1, E2) and the nonstructural proteins (NS2–NS5) required for replication. (i) Removal of the capsid protein by the NS2 protease exposes a signal peptide that directs synthesis of envelope proteins by the ER. (ii) Hydrophobic domains in NS4A, NS4B, NS5A and NS5B direct posttranslational insertion into the cytoplasmic face of the ER. (iii) NS5B is the RdRp. Assembly of replicase proteins induces ER invaginations and double membrane vesicles (DMV) and membraneous webs, also known as vesicular packets (VP). (b) Endoplasmic reticulum (ER) invaginations generated during dengue virus infection. Electron micrographs generated from a tilt series (top) were processed digitally to generate the tomogram (bottom). The 3D reconstruction shows the likely organization of ER invaginations. The arrow indicates viruses that may receive genomes from ER invaginations during assembly and envelopment. Reproduced from Welsch, S., Miller, S., Romero-Brey, I., et al., 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host & Microbe 5, 365–375. (c) The coronavirus polyprotein has the same overall organization as the flaviviruses but encodes as many as 15 nonstructural proteins. The figure shows double membrane vesicles and vesicular packets generated from the ER during SARS coronavirus infection. Electron micrographs generated from a tilt series were processed digitally to generate the tomogram. The 3D reconstruction shows the likely organization of interconnected DMVs (gold and silver) and vesicular packets (orange).
Reproduced from Knoops, K., Kikkert, M., Worm, S.H., et al., 2008. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biology 6, e226.
Membrane tubes and DMVs generated by picornaviruses
DMVs formed by membrane rearrangement
Picornaviruses, for example poliovirus, coxsackievirus, foot and mouth disease virus (FMDV), and the rhinoviruses that cause the common cold generate a single polyprotein that encodes the structural and nonstructural proteins (Figure 3(a) ). This is cleaved by the viral protease 3Cpro to generate capsid proteins from VP1-4 and replicase proteins from 2ABC and 3ABCD. The RdRp is encoded by 3Dpol which is targeted to membranes by hydrophobic domains in 3A, 2B, and 2C. Picornaviruses generate large numbers of membrane vesicles which vary between 200 nm and 400 nm diameter and some have double membranes (DMVs) while others have single membranes. These DMVs were observed in the first electron micrographs generated from cells infected with viruses in the 1960s (Dales et al., 1965) and their precise origins are still in dispute. The sizes and relative numbers of vesicles vary greatly between picornaviruses families and it is not clear whether single or DMVs house the RNA polymerase. For enteroviruses, such as poliovirus and coxsakievirus, replication may take place on tubular membranes generated during the log phase of virus production, and these may been seen as single membrane vesicles in cross section in electron micrographs (Limpens et al., 2011, Belov et al., 2012). Single membrane tubes may gain curvature following progressive assembly of replicase proteins or through recruitment of reticulon proteins (see below) leading to formation of DMVs (Figure 3(a)). The DMVs generated during picornavirus replication lack an obvious opening to the cytosol making it possible that they seal and are a by-product of replicase assembly and used to store viral RNA.
Figure 3.

Formation of membrane tubes and double membrane vesicles by picornaviruses. (a) The picornaviruses polyprotein (i) encodes structural proteins (VP1–VP4) that generate the capsid and nonstructural proteins (2ABC and 3ABCD), required for replication. (ii) Processing of the polyprotein by 3Cpro generates the replicase from 2B, 2C, 2BC, 3A, and 3Dpol that are targeted to the cytoplasmic face of the endoplasmic reticulum by hydrophobic domains in 2B, 2C and 3A (iii). Replicase assembly leads to the generation of membrane tubes, the origins of the tubes are unknown but are likely to be derived from ER or cis Golgi. Later during infection tubes gain curvature, possibly powered by reticulon proteins (iv) and form double membrane vesicles (DMVs). (b) Electron micrograph of vesicles and tubes generated during coxsackievirus infection. The image is one of several tomographic slices used to generate rendered tomograms in panel C. Reproduced from Limpens, R.W., van der Scharr, H.M., Kumar, D., et al., 2011. The transformation of enterovirus replication structures: A three dimensional study of single and double membraned compartments. mBio 2, e00166-11. (c) Side and top views of surfaced rendered models of membrane vesicles showing single membrane tubes (green), ER (blue) and open (orange) and closed (yellow) DMVs.
Reproduced from Knoops, K., Kikkert, M., Worm, S.H., et al., 2008. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biology 6, e226.
Double membrane autophagosomes
The DMVs seen in cells infected with enteroviruses may be formed from autophagosomes. Autophagosomes are generated by a membrane trafficking pathway called autophagy that delivers portions of the cytosol to lysosomes for degradation. Autophagy is activated during many viral infections and capture by autophagosomes can lead to destruction of viruses in lysosomes. It was therefore a surprise when it was discovered that poliovirus benefits from autophagy, with virus yields increasing some 10-fold when autophagy is activated during infection (Jackson et al., 2005, Wileman, 2006). The poliovirus replicase proteins associate with autophagosomes during infection and co-expression of 2BC and 3A in cells generates autophagosomes and DMVs (Schlegel et al., 1996, Suhy et al., 2000). It is currently believed that poliovirus 2BC and 3A activate autophagy during infection allowing replication to take place on the surface of autophagosomes. The enteroviruses infect via the gastrointestinal tract and are resistant to acid pH and proteases. It is likely that they can survive in lysosomes and there is evidence that low pH promotes maturation of the poliovirus capsid before release from cells by exocytosis (Richards and Jackson, 2012). Autophagosomes are used as sites of assembly for the replicases of other picornaviruses such as coxsackieviruses, human enterovirus 71, and encephalomyocarditis, but the role played by lysosomes during virus release is less clear. Unlike the enteroviruses, FMDV is very susceptible to low pH and this facilitates disassembly of the virus during endocytosis and cell entry. This sensitivity to low pH makes it unlikely that the FMDV would survive in autophagosomes or lysosomes. Autophagy is, however, activated during FMDV infection but this takes place early during infection before the nonstructural proteins are synthesized, and can be activated by empty capsids and UV inactivated viruses that cannot replicate (Berryman et al., 2012). It is thought that FMDV capsids activate autophagy during cell entry and this is consistent with the co-localization of capsid proteins with autophagosomes.
Host cell contribution to membrane rearrangements
The assembly of replicase proteins during spherule formation induces negative curvature resulting in a membrane invagination that moves away from the cytosol into the organelle. The formation of DMVs may require additional proteins because the generation of the outer membrane requires induction of positive curvature, allowing the curved face of the DMV to extend into the cytoplasm. The positive curvature of ER tubes is maintained by a family of proteins called reticulons which have hydrophobic domains that insert into the outer leaflet of the phospholipid bilayer (Diaz and Ahlquist, 2012, Tang et al., 2007). The picornavirus 2C proteins that provide a scaffold for the 3Dpol polymerase and target the replicase complex to membranes bind reticulon proteins and replication is reduced in the absence of reticulons. Most DMVs are derived, at least in part, from the ER, making it possible that reticulons play a key role in the formation of DMVs.
The lipid composition of membranes is also important for the replication of many different (+) strand RNA viruses and they are inhibited by drugs that block fatty acid and sterol synthesis (Castorena et al., 2010, Kapadia and Chisari, 2005, Perez et al., 1991, Rassmann et al., 2007). Lipids can also provide sites for assembly of replicase complexes. The RdRp (3Dpol) of poliovirus and coxsackievirus, for example, binds phophatidylinositol-4-phosphate (PI4-P). This means that recruitment of the 3Dpol polymerase can be increased if membranes contain increased levels of PI4-P. For some viruses this is achieved by manipulation of host proteins that control membrane traffic through the secretory pathway. The 3A protein of poliovirus and coxsackievirus increases the activity of the Arf1 GTPase and its guanine nucleotide exchange factor GBF1. Under normal circumstances activation of Arf1 by GBF1 results in formation of the COP1-coated vesicles that carry proteins from the Golgi to the ER. Activation of GBF1 by the viral 3A protein, however, results in recruitment of an alternative Arf1 effector protein, phosphatidylinositol-4-kinase-IIIβ (PI4-IIIβ) to membranes (Hsu et al., 2010). The enzyme phosphorylates PI4 to generate PIP-4 providing membranes enriched in PIP-4 which recruit 3Dpol. A similar mechanism may operate for flaviviruses (Reiss et al., 2011, Berger et al., 2009) and coronaviruses (Verheije et al., 2008) where replication and membrane remodeling are reduced following inhibition of the Arf1 GTPases or phosphatidylinositol-4-kinase-III.
(−) Strand RNA Viruses Replicate in Factories Assembled in the Golgi
The ‘Bunyaviridae’ are good examples of (−) strand RNA viruses and include Hantavirus, Rift Valley fever virus, and Crimean Congo hemorrhagic fever virus which cause severe diseases in man. The Bunyamwera virus serves as a good model for this family of viruses. The genome is segmented into three (−) strand RNA strands named according to their size. The long (L) segment encodes the RdRp, the medium segment encodes a polyprotein that is processed by the ER to make the envelope proteins Gn and Gc which travel to the Golgi apparatus, while the small segment encodes the nuclear protein (NP). One copy of each segment is packaged within the virus and each segment binds one copy of the RdRp and the rest of the RNA strand is protected by several NP proteins. On entering the cell the RdRp copies each (−) strand to make (+) strands that can be read as mRNA. Infection of cells with Bunyamwera virus results in the formation of tubules within the Golgi apparatus which form the virus factory (Fontana et al., 2008). These tubules contain the RdRp, NP, and viral RNA. It is likely that they function as specialized spherules for generating new genome segments which are assembled with the RdRp and NP. The formation of the factory in the Golgi apparatus would facilitate delivery of RNA segments budding into late Golgi compartments during virus assembly and envelopment.
DNA Viruses
The Nucleo-Cytoplasmic Large DNA Viruses Use Aggresomes to Generate Pericentriolar Factories
The nucleo-cytoplasmic large DNA viruses (NCLDV) are a family of large DNA viruses with genomes ranging from 150 kb to 1.2 MB. The family includes the poxviruses iridoviruses, African swine fever virus (ASFV), phycodnaviruses, and mimiviruses that share several conserved core genes. The NCLDV infect a wide range of hosts including mammals, arthropods, fish, and marine plankton, but all replicate and assemble in large factories located close to the nucleus at the microtubule organizing center (MTOC) (Figure 4 ). The factories contain viral DNA and structural proteins but lack cellular membranes and are very similar to inclusions called aggresomes that form during protein misfolding diseases (Heath et al., 2001, Wileman, 2007). Protein misfolding leads to the formation of small protein aggregates in the cytosol which are transported along microtubules by dynein motors to the MTOC where they encounter the major cellular pools of chaperones and proteasomes. If aggregates fail to fold, or exceed the proteolytic capacity of the proteasomes, they accumulate within an aggresome surrounded by mitochondria and a cage made from the vimentin filaments (Figure 4(a)). The inclusions also contain ubiquitin and a linker protein called p62/SQSTM1 that binds ubiquitin and LC3. LC3 is the major membrane protein of the autophagosome and p62 is used to remove ubiqutinated proteins from aggresomes into autophagosomes which deliver them to lysosomes for degradation. Virus factories and aggresomes share many features in common including recruitment of chaperones, proteasomes, and mitochondria and confinement within a vimentin cage. Many viruses are recognized by dynein after entering cells and delivered to the MTOC. It is possible that incoming virus capsids stimulate an aggresome response because they appear foreign or misfolded to cells (Wileman, 2007). For the NCLDV this may be beneficial and provide transport to a site for replication, for other viruses it may represent an innate immune response leading to confinement at the MTOC and degradation.
Figure 4.

Aggresomes and perinuclear sites generated by nucelocytoplasmic large DNA viruses. (a) (i) Misfolded proteins aggregate in the cytoplasm and bind to cytoplasmic dynein. (ii) Dynein motors move aggregated proteins by retrograde transport to the microtubule organizing center (MTOC). (iii) An aggresome forms and induces vimentin rearrangement and recruits of mitochondria and chaperones. (iv) Poxvirus cores enter the cytoplasm and bind dynein and microtubules and begin RNA replication. (v) Poxvirus cores are disassembled at the MTOC by proteasomes dynein and microtubules and begin DNA replication. (vi) DNA replication and virus assembly take place in factories located at the MTOC. (b) Virus factory generated by African swine fever virus. The image shows a vimentin cage (red) surrounding a virus factory containing newly assembled viruses (green). The nucleus is blue. (c) Electron micrograph African swine fever virus factory showing viruses at different stages of assembly. Fully assembled viruses are seen as hexagons in cross section. Those with dark centers have packaged DNA genomes.
The factories formed by poxviruses and ASFV are the best characterized. During cell entry the poxviruses release a viral core into the cytoplasm which binds dynein motor proteins (Figure 4(a)). The core immediately starts the synthesis of at least 100 early genes using a viral DNA-dependent RNA polymerase. The mRNA transcribed from early genes by the polymerase are delivered into the cytosol as the core travels to the MTOC. The early proteins encoded by the mRNA are used to break open the core to release viral DNA through a process requiring proteasome-dependent degradation of ubiquitinated capsid proteins. Poxviruses are unusual DNA viruses because they encode all the enzymes needed for DNA and mRNA synthesis and do not need to travel to the nucleus. The viral genomes remain at the MTOC where they create a factory by providing a template for DNA replication and generation of viral mRNA encoding intermediate and late genes. Interestingly, for poxviruses it appears that intermediate and late proteins are synthesized within the factory rather than in the cytoplasm. This means that viral mRNA is retained within the factory and that the factory recruits cellular factors needed for translation. Detailed analysis of poxvirus factories shows that cavities within the viral DNA recruit viral mRNA, intermediate and late proteins, and host cell translation initiation factors such as eIF4g and eIF4e. The results suggest that translation of viral mRNA takes place on ribosomes recruited into the factory, and that sites of translation are segregated from sites of genome replication (Katsafanas and Moss, 2007). Viral structural proteins are retained in the factory until they are assembled into new viruses which are carried into the cytosol along microtubules.
Similar to poxviruses, infection of cells by ASFV also results in delivery of a nucleoprotein core particle into the cytosol. The cores deliver the genome to the nucleus where it provides a template for the formation of short precursor fragments that are exported to the cytosol for replication. The formation of the ASFV factory begins with the removal of cellular material from the MTOC where it is replaced by a high concentration of vimentin filaments arranged into an ‘aster’ aligned along microtubules. This site also recruits chaperones and mitochondria. Following the onset of DNA replication the vimentin filaments are rearranged into a cage surrounding virus DNA and delineate the edges of the factory (Figure 4(b)). The factory is kept at the MTOC by microtubules and becomes dispersed through the cytosol if microtubules are depolymerized. The rearrangement of vimentin into a cage requires phosphorylation of the N-terminal domain of vimentin that is important for filament assembly by host cell calcium calmodulin-dependent kinase II (CamKinase II). This phosphorylation is required for replication because inhibition of CamKinase II blocks late gene expression (Stefanovic et al., 2005). In common with poxviruses, ASFV structural proteins do not leave the factory until they are assembled into new viruses (Figure 4(c)). At late stages in the lifetime of the factory the MTOC is lost and microtubules form bundles in the cytosol. It is thought that this may weaken the factory to facilitate release of viruses into the cytosol.
The DNA Viruses Generate Replication Compartments in the Nucleus
Most DNA viruses replicate in the nucleus because they need nuclear factors to generate new genomes (reviewed in Schmid et al., 2014, Jiang and Imperiale, 2012). DNA viruses entering the cell are transported by dynein motors along microtubules to the MTOC where they are transferred to the nuclear pore complex for disassembly and release of the DNA genome into the nucleus (Figure 5 ). The viral genome must recruit host factors required for replication and establish sites for DNA replication and transcription in the nucleus, and at the same time evade cellular defenses against infection. The delivery of viral DNA to the nucleus initiates large-scale rearrangement of nuclear structures and the formation of replication compartments that can expand to fill the nucleus late during infection. In many cases the RCs form next to nuclear substructures called promyelocytic leukemia nuclear bodies (PML-NB), ND10s or PODs which store proteins involved in apoptosis, chromatin remodeling, and DNA repair (reviewed in Everett, 2013, Boutell and Everett, 2013). Many of the components of the PML-NBs are upregulated by interferon suggesting they play a role in defense against infection. The RCs initially form close to the nuclear envelope, possibly where the viral DNA first enters the nucleus. A key question has centered on determining whether sites of DNA virus replication are indeed PML-NBs, and understanding how a virus would survive in a compartment designed to inhibit replication. The interaction of DNA viruses with PML-NBs is best studied for herpes simplex virus-1 (HSV-1), but is likely to be similar during the replication of other DNA viruses.
Figure 5.

Replication compartments generated by DNA viruses in the nucleus. (a) Herpes virus replication sites in the nucleus. DNA viruses are transferred from the MTOC to the nuclear pore complex for disassembly and release of the DNA genome into the nucleus. In many cases the RCs form close to the nuclear envelope next to a nuclear substructures called promyelocytic leukemia nuclear bodies (PML-NB) which vary between 100 and 1000 µm in diameter and contain PML Daxx, Sp100, and ATRX (i). PML-NBs have also been called nuclear aggresomes (ii) because they accumulate misfolded proteins, cellular chaperones, proteasome subunits, and ubiquitinated proteins. Viral DNA entering the nucleus lacks chromatin and exposes a damage signal that recruits PML-NB proteins to add chromatin to genomes and repress transcription (iii). For HSV-1 this cellular defense is countered by the regulatory protein ICP0 which targets PML for degradation (iv). The residue of the PML body (v) may form the ‘assemblon’ that is seen late during infection which contains tegument proteins and aggregated capsids. The assemblon may serve as a site of virus assembly early during infection and become the VICE domain/nuclear aggresome at later times when structural proteins are made in excess. Fully assembled capsids (vi) pass across the nuclear envelope and enter the cytoplasm and envelop in the trans-Golgi network before release from the cell. (b) Recruitment of PML-NB proteins to sites of HSV-1 replication. Cells were infected with a strain of virus (ΔICP0) lacking ICP0 to slow dispersal of PML-NB proteins. The fluorescence micrograph shows the virus genome located through detection of transcriptional activator ICP4 (green), PML protein (red) and DNA damage response protein γH2AX (blue). The PML protein has redistributed to sites containing viral DNA.
Reproduced from Everett, R.D., 2013. The spatial organisation of DNA virus genomes in the nucleus. PLoS Pathogens 9, e1003386.
Use of promeyelocytic leukemia nuclear bodies (PML-NB) by herpes virus
PML-NBs vary between 100 µm and 1000 µm in diameter and contain PML, the major structural protein, and Daxx, Sp100, and ATRX which regulate chromatin assembly and inhibit transcription. The PML-NBs do not move through the nucleus but PML proteins exchange rapidly with the surrounding nucleoplasm. Recruitment into the PML-NB is dependent on addition of ubiquitin-like SUMO proteins to PML, and the presence of SUMO interaction motifs in Daxx and Sp100. Viral DNA entering the nucleus lacks chromatin and this may expose a damage signal that recruits PML-NB proteins which would add chromatin to genomes and repress transcription (Figure 5). For HSV-1 this cellular defense is countered by the regulatory protein ICP0 which is expressed very early during infection. The ubiquitin E3 ligase activity of ICP0 ubiquitinates PML leading to degradation of PML by proteasomes and loss of PML-NBs from the nucleus of infected cells. Evidence that PML-NBs suppress virus replication before they are dispersed by ICP0 comes from the observation that HSV-1 strains expressing defective ICP0s fail to degrade PML, have reduced replication and can fail to proceed to lytic infection. Herpes viruses also use structural proteins to combat PML-NBs. The herpes virus nucleoprotein core is surrounded by a layer of tegument proteins (i.e., viral matrix) that lie beneath the envelope. The human cytomegalovirus tegument phosphoprotein pp71 locates to PML-NBs soon after cell entry and induces degradation of PML, similarly the major tegument protein of Epstein-Barr virus BNRF1 disassembles the Daxx–ATRX complex. Daax–ATRX complexes are also displaced from PML-NBs by specific capsid proteins of adenoviruses and papillomaviruses that travel to the nucleus following cell entry.
PML-NBs have also been called nuclear aggresomes because they are closely associated with nuclear inclusions that accumulate misfolded proteins, cellular chaperones, proteasome subunits, and ubiquitinated proteins (Fu et al., 2005). Aggregated proteins, for example, the polyQ repeat proteins associated with Huntington’s disease or spinocerebellar ataxis accumulate close to, or within PML-NBs which fuse together to form larger structures. Nuclear aggresomes also appear during herpes virus infection, and in some reports they are called virus-induced-chaperone-enriched (VICE) domains. These domains appear adjacent to sites of replication soon after the dispersal of PML-NBs by ICP0. Proteomic analysis has shown that 20 or more host proteins may be recruited to replication sites. These include proteins involved in DNA replication and transcription, chromatin remodeling and DNA repair, and RNA polymerase II which is used to transcribe the viral genome. As seen for the poxviruses the sub-compartmentalization of the virus factory through the formation of a VICE domain may facilitate replication by sequestering structural proteins away from sites of genome replication and transcription. VICE domains may also store viral proteins, such as the UL6 viral portal protein, and pp65 tegument protein, that are prone to aggregation when they are made in excess over levels needed for the assembly of new viruses. Evidence that aggregation of viral proteins is detrimental to replication comes from studies of HCMV strains expressing a defective tegument protein called UL97. UL97 is a kinase that can reduce protein aggregation (Prichard et al., 2005). Drugs that inhibit the kinase activity of UL97, or mutations that abolish kinase activity, reduce replication and induce the formation of aggresomes containing tegument proteins.
Successful subversion of the cellular defenses provided by the PML-NB allows the virus to initiate DNA replication and transcription. Precisely where this occurs in the nucleus is not known because the PML-NB markers are dispersed by ICP0. The genomes of HSV-1 expressing a defective ICP0 accumulate in large PML-NBs making it likely that replication takes place within the residue of the PML-NB or VICE domain. Once genomes have been generated in the factory they have to be assembled with capsids. Capsid proteins are synthesized on ribosomes in the cytosol and imported into the nucleus where they assemble with scaffold proteins and a portal protein to generate empty immature capsids. Import of the genome through the portal results in proteolysis and loss of the scaffold and formation of the mature capsid. These events take place in the nucleus and early studies identified an ‘assemblon’ as a site of virus assembly, seen late during infection which contains tegument proteins and aggregated capsids. The assemblon may serve as a site of virus assembly early during infection and becomes the VICE domain/nuclear aggresome at later times when structural proteins are made in excess over levels that can be packaged into viruses. Fully assembled capsids pass across the nuclear envelope and enter the cytoplasm where they are enveloped in the trans-Golgi network before release from the cell.
See also
INTRACELLULAR INFECTIOLOGY: INFECTIOUS AGENTS | Cell Biology of Virus Infection
Biographies

Prof. Tom Wileman trained in cell biology and immunology at Washington University and Harvard Medical Schools in the USA between 1982 and 1994. His research focused on the identification of the macrophage mannose endocytosis receptor, the cloning of genes for T-cell antigen receptors and understanding mechanisms of ER-related protein degradation. He was appointed Assistant Professor at Harvard Medical School in 1991. In 1994 he joined the Pirbright Institute in the UK as Head of Immunology where he applied his understanding of cell biology to study how viruses use cellular organelles to facilitate replication. In 2005 he moved to the Norwich Medical School at the University of East Anglia as Professor of Molecular Virology where he has studied the role played by autophagy in controlling virus replication, particularly how viral proteins activate autophagy, and the consequences this has for the survival of viruses within cells. His current work is developing mice with conditional tissue-specific expression of autophagy genes to study the impact of autophagy on infection ‘in vivo.’

Dr. Chris Netherton trained in Biochemistry at the Universities of Nottingham and Sussex between 1994 and 2001. His PhD focused on the characterization of a family of secretory proteins encoded by African swine fever virus. Since then his research has encompassed various aspects of the intracellular response to viral infection including secretion, endoplasmic reticulum stress, and the relationship between viral assembly sites, and protein misfolding pathways. He also studies the immune response to viral infection and was the first to show that Mx protein could inhibit the replication of a large DNA virus. More recently he has been studying the interplay between the autophagy pathway and viral infection.

Dr. Penny Powell trained in molecular cell biology at St. Louis Medical School and Harvard Medical School in the USA between 1984 and 1994. Her work centered on cloning genes for lysosomal enzymes to follow how defects in intracellular trafficking cause lysosomal storage diseases, and also on cloning vascular growth factors involved in the growth of new blood vessels. She was recruited back to the UK as a Senior Scientist to the Pirbright Institute to study viral proteins which inhibit inflammatory and innate immune responses during viral infection. Currently, a Senior Lecturer at Norwich Medical School, University of East Anglia, she studies innate immune responses to viruses, including interferon signaling, RNA interference, stress, and autophagy. Her current work is developing the use of intestinal organoids or 'mini-guts' derived from stem cells isolated from small intestinal crypts to study innate defenses against infections of the gut.
References
- Ahlquist P. Parallels among positive-strand RNA viruses, reverse transcribing viruses and double stranded RNA viruses. Nature Reviews Microbiology. 2006;4:371–382. doi: 10.1038/nrmicro1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barajas D., Fernandez de Castro Martin I., Pogany J., Risco C., Nagy P.D. Noncononical role for the host Vps4 AAA+ ATPase ESCRT protein in the formation of Tomato Bushy Stunt Virus replicase. PLoS Pathogens. 2014;10:e1004087. doi: 10.1371/journal.ppat.1004087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov G.A., Nair V., Hansen B.T. Complex dynamic development of poliovirus membraneous replication complexes. Journal of Virology. 2012;86:302–312. doi: 10.1128/JVI.05937-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger K.L., Cooper J.D., Heaton N.S. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:7577–7582. doi: 10.1073/pnas.0902693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryman S., Brooks E., Burman A. Foot-and-mouth disease virus induces autophagosomes during cell entry via a Class III Phosphatidylinositol 3-Kinase-Independent pathway. Journal of Virology. 2012;86:12940–12953. doi: 10.1128/JVI.00846-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boon J.A., Ahlquist P. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annual Review of Microbiology. 2010;64:241–256. doi: 10.1146/annurev.micro.112408.134012. [DOI] [PubMed] [Google Scholar]
- den Boon J.A., Diaz A., Ahlquist P. Cytoplasmic viral replication complexes. Cell Host & Microbe. 2010;8:77–85. doi: 10.1016/j.chom.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutell C., Everett R.D. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. Journal of General Virology. 2013;94:465–481. doi: 10.1099/vir.0.048900-0. [DOI] [PubMed] [Google Scholar]
- Castorena K.M., Stapleford K.A., Miller D.J. Complementary transcriptomic, lipidomic and targeted functional genetic analyses in cultured Drosophila cells highlight the role of glycerophospholipid metabolism in Flock House virus RNA replication. BMC Genomics. 2010;11:183. doi: 10.1186/1471-2164-11-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottam E., Pierini R., Roberts R., Wileman T. Origins of membrane vesicles generated during replication of positive-strand RNA viruses. Future Virology. 2009;4:473–485. [Google Scholar]
- Dales S., Eggers H.J., Tamm I., Palade G.E. Electron microscopic study of the formation of poliovirus. Virology. 1965;26:379–389. doi: 10.1016/0042-6822(65)90001-2. [DOI] [PubMed] [Google Scholar]
- Diaz A., Ahlquist P. Role of host reticulon proteins in rearranging membranes for positive strand RNA virus replication. Current Opinion in Microbiology. 2012;15:519–524. doi: 10.1016/j.mib.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz A., Zhang J., Ollwerther A., Wang X., Ahlquist P. Host ESCRT proteins are required for bromovirus RNA replication compartment assembly and function. PLoS Pathogens. 2015;11(3):e1004742. doi: 10.1371/journal.ppat.1004742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R.D. The spatial organisation of DNA virus genomes in the nucleus. PLoS Pathogens. 2013;9:e1003386. doi: 10.1371/journal.ppat.1003386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez de Castro I., Volonte L., Risco C. Virus factories: Biogenesis and structural design. Cellular Microbiology. 2012;15:24–34. doi: 10.1111/cmi.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana J., Lopez-Montero N., Elliott R.M., Fernandez J.J., Risco C. The unique architecture of Bunyamwera virus factories around the Golgi complex. Cellular Microbiology. 2008;10:2012–2028. doi: 10.1111/j.1462-5822.2008.01184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L., Gao Y.-S., Tousson A. Nuclear aggresomes form by fusion of PML-associated aggregates. Molecular Biology of the Cell. 2005;16:4905–4917. doi: 10.1091/mbc.E05-01-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath C.M., Windsor M., Wileman T. Aggresomes resemble sites specialized for virus assembly. Journal of Cell Biology. 2001;153:449–456. doi: 10.1083/jcb.153.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu N.Y., Ilnytska O., Belov G. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson W.T., Giddings T.H., Jr., Taylor M.P. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biology. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M., Imperiale M.J. Design stars: How small DNA viruses remodel the host nucleus. Future Virology. 2012;7:445–459. doi: 10.2217/FVL.12.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapadia S.B., Chisari F.V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proceedings of the National Aademy of Sciences of the United States of America. 2005;102:2561–2566. doi: 10.1073/pnas.0409834102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsafanas G.C., Moss B. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host & Microbe. 2007;2:221–228. doi: 10.1016/j.chom.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoops K., Kikkert M., Worm S.H. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biology. 2008;6:e226. doi: 10.1371/journal.pbio.0060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopek B.G., Perkins G., Miller D.J., Ellisman M.H., Ahlquist P. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biology. 2007;5:e220. doi: 10.1371/journal.pbio.0050220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limpens R.W., van der Scharr H.M., Kumar D. The transformation of enterovirus replication structures: A three dimensional study of single and double membraned compartments. mBio. 2011;2:e00166-11. doi: 10.1128/mBio.00166-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D.J., Schwartz M.D., Ahlquist P. Flock house virus RNA replicates on outer mitochondrial membranes in Drosophila cells. Journal of Virology. 2001;75:11664–11676. doi: 10.1128/JVI.75.23.11664-11676.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netherton C., Moffat K., Brooks E., Wileman T. A guide to viral inclusions, membrane rearrangements, factories, and viroplasm produced during virus replication. Advances in Virus Research. 2007;70:101–182. doi: 10.1016/S0065-3527(07)70004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez L., Guinea R., Carrasco L. Synthesis of Semliki Forest virus RNA requires continuous lipid synthesis. Virology. 1991;183:74–82. doi: 10.1016/0042-6822(91)90119-v. [DOI] [PubMed] [Google Scholar]
- Prichard M.N., Britt W.J., Daily S.L., Hartline C.B., Kern E.R. Human cytomegalovirus UL97 kinase is required for the normal intranuclear distribution of pp65 and virion morphogenesis. Journal of Virology. 2005;79:15494–15502. doi: 10.1128/JVI.79.24.15494-15502.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall R.E., Goodbourn S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. Journal of General Virology. 2008;98:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- Rassmann A., Henke A., Jarasch N. The human fatty acid synthase: A new therapeutic target for coxsackievirus B3-induced diseases? Antiviral Research. 2007;76:150–158. doi: 10.1016/j.antiviral.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Rathinam V.A.K., Fitzgerald K.A. Innate immune sensign of DNA viruses. Virology. 2011;411:153–162. doi: 10.1016/j.virol.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss S., Rebhan I., Backes P. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host & Microbe. 2011;9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards A.L., Jackson W.T. Intracellular vesicle acidification promotes maturation of infectious poliovirus particles. PLoS Pathogens. 2012;8:e10003046. doi: 10.1371/journal.ppat.1003046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Brey I., Merz A., Chiramel A. Three-Dimensional Architecture and Biogenesis of Membrane Structures Associated with Hepatitis C Virus Replication. PLoS Pathogens. 2012;8(12) doi: 10.1371/journal.ppat.1003056. e1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel A., Giddings T.H., Jr., Ladinsky M.S., Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. Journal of Virology. 1996;70:6576–6588. doi: 10.1128/jvi.70.10.6576-6588.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M., Speiseder T., Dobner T., Gonzalez R.A. DNA virus replication compartments. Journal of Virology. 2014;88:1404–1420. doi: 10.1128/JVI.02046-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic S., Windsor M., Nagata K.-I., Inagaki M., Wileman T. Vimentin rearrangement during African swine fever virus infection involves retrograde transport along microtubules and phosphorylation of vimentin by calcium calmodulin kinase II. Journal of Virology. 2005;79:11766–11775. doi: 10.1128/JVI.79.18.11766-11775.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhy D.A., Giddings T.H., Jr., Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. Journal of Virology. 2000;74:8953–8965. doi: 10.1128/jvi.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.F., Yang S.Y., Wu B.W. Reticulon 3 binds the 2C protein of enterovirus 71 and is required for replication. Journal of Biological Chemistry. 2007;282:5888–5898. doi: 10.1074/jbc.M611145200. [DOI] [PubMed] [Google Scholar]
- Verheije M.H., Raaben M., Mari M. Mouse hepatitis coronavirus RNA replication depends on GBF1-mediated ARF1 activation. PLoS Pathogens. 2008;4:e1000088. doi: 10.1371/journal.ppat.1000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsch S., Miller S., Romero-Brey I. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host & Microbe. 2009;5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wileman T. Aggresomes and autophagy generate sites for viral infection. Science. 2006;312:875–878. doi: 10.1126/science.1126766. [DOI] [PubMed] [Google Scholar]
- Wileman T. Aggresomes and pericentriolar sites of virus assembly: Cellular defense or viral design? Annual Review of Microbiology. 2007;61:149–167. doi: 10.1146/annurev.micro.57.030502.090836. [DOI] [PubMed] [Google Scholar]