

Abstract
Viruses are major pathogenic agents causing a variety of serious diseases in humans, other animals, and plants.
Drugs that combat viral infections are called antiviral drugs. There are no effective antiviral drugs for many viral infections. However, there are several drugs for influenza, a couple of drugs for herpesviruses, and some new antiviral drugs for treatment of HIV and hepatitis C infections.
The arsenal of antivirals is complex. As of March 2014, it consists of approximately 50 drugs approved by the FDA, approximately half of which are directed against HIV. Antiviral drug creation strategies are focused on two different approaches: targeting the viruses themselves or targeting host cell factors.
Direct virus-targeting antiviral drugs include attachment inhibitors, entry inhibitors, uncoating inhibitors, protease inhibitors, polymerase inhibitors, nucleoside and nucleotide reverse transcriptase inhibitors, nonnucleoside reverse-transcriptase inhibitors, and integrase inhibitors.
Protease inhibitors (darunavir, atazanavir, and ritonavir), viral DNA polymerase inhibitors (acyclovir, valacyclovir, valganciclovir, and tenofovir), and an integrase inhibitor (raltegravir) are included in the list of Top 200 Drugs by sales for the 2010s.
Keywords: Attachment inhibitors, Entry inhibitors, Integrase inhibitors, Nonnucleoside reverse-transcriptase inhibitors, Nucleoside and nucleotide reverse-transcriptase inhibitors, Polymerase inhibitors, Protease inhibitors, Uncoating inhibitors
Viruses are major pathogenic agents causing a variety of serious diseases in humans, other animals, and plants.
Viruses are one of the most widespread of all organisms and are capable of infecting every species of animal from mammals down to insects, plants, and even bacteria. It seems there are more species of viruses in the world than of all other creatures put together.
The most common viral infections are respiratory (infections of the nose, throat, upper airways, and lungs); gastrointestinal (gastroenteritis); liver (hepatitis); and skin (warts or other blemishes, rashes).
Viral diseases include influenza (causing fever, severe aching, and catarrh, often occurring in epidemics); severe acute respiratory syndrome (a form of pneumonia); chickenpox (disease caused by the herpes zoster virus, which manifests in a mild fever and a rash of itchy inflamed blisters); herpes (herpes simplex or herpes zoster, causing the eruption of small blister-like vesicles on the skin or mucous membranes); hepatitis (a disease characterized by inflammation of the liver); cold sores (diseases affecting mouth or genitals); measles (disease causing fever and a red rash on the skin, typically occurring in childhood); shingles (painful inflammation of the nerve ganglia, with a skin eruption often forming a girdle around the middle of the body); poliomyelitis (disease that affects the central nervous system that can cause temporary or permanent paralysis); acquired immunodeficiency syndrome (AIDS) caused by human immunodeficiency virus (HIV) (disease includes dry cough or shortness of breath, difficult or painful swallowing, diarrhea, white spots or unusual blemishes in and around the mouth, pneumonia-like symptoms, fever, vision loss, nausea, abdominal cramps, and vomiting); smallpox (disease started from fever, overall discomfort, headache, severe fatigue, severe back pain, vomiting. A few days later, flat, red spots appear on whole trunk, which become lesions. Occur first in the mouth and spread to the face, then to entire body); rabies (a contagious and fatal viral disease that causes madness and convulsions); dengue (jungle fever) causing sudden fever and acute pains in the joints); Ebola (fatal disease marked by fever and severe internal bleeding); and Lassa (fever with headaches, mouth ulcers, muscle aches, hemorrhages under the skin, heart and kidney failure, and a high mortality rate).
Viruses are transmitted in various ways. Some are swallowed, some are inhaled, some are spread sexually, and some are spread by the bites of insects, such as mosquitoes, certain biting flies, ticks or animals, or during transfusion with contaminated blood.
The deadliest, most horrifying diseases the world has ever seen have been initiated by viruses. A smallpox epidemic disaster is described in ancient Sanskrit medical texts, dating to about 1500 bc. In Europe, smallpox likely appeared by about 300 ad.
Rabies has a history dating back to 2300 bc, as described by Babylonians, who went mad and died after being bitten by dogs.
The bubonic plague arrived in Europe in 1347 when 12 Genoese trading ships docked at the Sicilian port. The ships carried goods that brought bubonic plague, which caused the Black Death, killing more than 20 million people in Europe—almost one-third of the continent’s population.
It is very believable that plague virus is very similar to the Ebola virus, the recent deadly virus named after the Ebola River in the Democratic Republic of the Congo, which is classified as one of the most dangerous pathogens on the planet.
There are still more than 100 million cases of dengue fever each year. Marburg and Ebola hemorrhagic fever have 90% fatality rates.
In 1847-1848, influenza swept through the Mediterranean to Western Europe.
In 1878, a disease causing high mortality in poultry became known as the “fowl plague.” Fowl plague is now called highly pathogenic avian influenza A.
In 1918-1919, the most famous disaster generated by viruses occurred: Spanish flu (influenza), which infected 20 to 40% of the world’s population and killed from 20 million to 100 million people. This pandemic happened in 1918 and is considered to be one of the worst in human history. Probably, no virus can claim credit for more worldwide pandemics and scares than the constantly mutating thousands of strains of influenza.
In 1957-1958, the “Asian flu” caused the second pandemic of the 20th century. It began in China and killed one million people worldwide.
In 1968-1969, the “Hong Kong flu” caused the last flu pandemic.
In 2009, a swine flu outbreak hit, originating in Mexico City and quickly spreading to more than 10 nations.
The first case of HIV infection in a human was identified in 1959. The first cases of HIV in the United States date back to 1981.
In 1984, the HIV, which causes HIV infection and AIDS, was discovered. The HIV infection is distinct from AIDS, the full-blown syndrome that, along with the consequences of a damaged immune system, is most often fatal. More than 78 million people have been infected with HIV since the start of the epidemic in the 1980s. AIDS-related illness are the sixth leading cause of death.
Viruses are small particles, much smaller than fungi or bacteria. These strange substances are something straddling between living and nonliving particles. Viruses are basically a pack of genetic material—either DNA or RNA—carried in a shell made up of protein. Some viruses have an additional layer called an envelope. Viruses vary in size and complexity, but common features for species of viruses are: nucleic acid, protein coat, lipid membrane (envelope). (Viruses can be divided into two major categories: enveloped and nonenveloped.) A virus particle, consisting of an outer protein shell called a capsid and an inner core of nucleic acid (either ribonucleic or deoxyribonucleic), is called a virion. Viruses are classified as DNA viruses or RNA viruses, depending on whether they use DNA or RNA to replicate. Further classification can be based on the nature of the genetic material that is packaged by the virus (single-stranded and double-stranded RNA viruses, analogue DNA viruses, and others). Viruses can’t even be considered cells. They can’t metabolize nutrients, produce and excrete wastes, move around on their own, or even reproduce unless they are inside another organism’s cells.
Coming into contact with host cells, they fuse themselves to the host cell and insert their genetic material into that cell, taking over its machinery to replicate themselves. Consequently, there is controversy regarding whether viruses should really be considered as living organisms.
The virus has to enter the cell to start it’s activity. As a first step, they must attach to a receptor on the cell surface. Each virus has its specific receptor and it is a vital component of the cell surface. The selectivity of the viruses determines the cell preference.
For instance, rhinoviruses have a preference for airways cells; HIV infects mainly T lymphocytes and macrophages with the clusters such as CD4, CCR5, and CXCR4 on the surface of immune cells; Epstein–Barr virus and rabies virus infect B lymphocytes carrying complement receptor type CR2. It is believed that influenza viruses infect cells in a multi-step process. The main targets of the influenza virus are the columnar epithelial cells of the trachea, bronchi and bronchioles. On the first step viruses are internalized via receptor-mediated endocytosis and then are trafficked along the endocytic pathway to endosomes. After that HA protein – hemagglutinin catalyzed fusion between the viral and endosomal membranes takes place, releasing viral ribonucleoproteins which are imported into the nucleus for viral gene expression and replication.
When a virus enters the body, it triggers the body’s immune defenses, such as lymphocytes and monocytes, which destroy the virus or the cells it has infected. If the body survives the virus attack, it becomes able to respond to a subsequent infection by the same virus (immunity, which can also be produced by vaccination). Vaccination is possible to prevent infections with some viruses, such as hepatitis B, varicella-zoster, influenzas A and B, Yellow fever, and poliovirus viruses; but not infections caused by HIV, hepatitis C, herpes simplex, cytomegalovirus, and most hemorrhagic fever viruses (except for Yellow fever virus).
One classification of viruses is based on pathogenic property and mode of transmission.
Respiratory viruses—influenza, rhinovirus, adenovirus—are usually acquired by inhalation of droplets and replicate in the respiratory tract. The polioviruses, rotaviruses, reoviruses, and some adenoviruses are enteric viruses. These are viruses that replicate in the gut and cause gastric infections. The most serious complications include meningitis, encephalitis, poliomyelitis, and myocarditis. Arboviruses infect insects that ingest vertebrate blood, replicate in tissue of the insect, and then are transmitted to the vertebrate host. Such viruses include flaviviruses, bunyaviruses, and some rhabdoviruses. Sexually transmitted viruses include HIV, herpes simplex, and papilloma viruses. Hepatitis viruses cause disease of the liver.
More than 30,000 different viruses have been isolated and studied to date. Historically, their classification was disease related. Now they are grouped at different hierarchical levels of order, family, subfamily, genus, and species on the basis of their properties.
Viral infections that cause persistent infections are viruses that belong to the Retroviridae family (HIV, which causes AIDS, and human T-cell lymphotrophic virus [HTLV], which causes leukemia); to the Flaviviridae family (hepatitis C [HCV] and to the Hepadnaviridae family (hepatitis B [HBV]), which cause chronic hepatitis and hepatocellular carcinoma; and to the Herpesviridae family (herpes simplex viruses 1 and 2 [HSV-1 and HSV-2]), which cause recurrent mucocutaneous infections and encephalitis. In addition, varicella zoster virus (VZV) causes recurrent neurological lesions; cytomegalovirus (CMV) causes retinitis, pneumonia, encephalitis; Epstein-Barr virus (EBV) causes lymphoproliferative disorders; and viruses that belong to the Papovaviridae family cause cervical carcinoma and warts.
Human viral infections associated with loss of work hours include rhinoviruses (influenza A virus causing influenza) and Caliciviruses, Norwalk viruses, and Astra diarrhea-causing viruses.
Viruses are also classified on the basis of morphology, chemical composition, and mode of replication, which reflects only a small part of the spectrum of the multitude of different viruses.
Viral diseases are not treatable with antibiotics, which can only cure bacterial diseases and infections.
Drugs that combat viral infections are called antiviral drugs. There are no effective antiviral drugs for many viral infections. However, there are several drugs for influenza, a couple of drugs for herpesviruses, and some new antiviral drugs for treatment of HIV and hepatitis C infections.
The arsenal of antivirals is complex. As of March 2014, it consists of approximately 50 drugs approved by the FDA, approximately half of which are directed against HIV. Antiviral drug creation strategies is focused on two different approaches: targeting the viruses themselves or targeting host cell factors. These approaches are widely reviewed 1., 2., 3., 4., 5., 6., 7., 8., 9., 10., 11., 12., 13., 14., 15., 16., 17., 18., 19., 20., 21., 22., 23., 24., 25., 26., 27., 28., 29., 30., 31., 32., 33., 34., 35., 36., 37., 38., 39., 40., 41., 42., 43., 44., 45., 46., 47., 48., 49., 50., 51., 52., 53., 54., 55., 56., 57., 58., 59., 60..
34.1. Direct Virus-Targeting Antiviral Drugs
Attachment Inhibitors
The first event in viral infection of the host cell is binding of the virus to the cell surface, which involves numerous interactions between the virion surface and the receptor. Some viruses have specific attachment sites widely distributed all over the host cell membrane that recognize molecules on the surface of virus particle. Many viruses use as an attachment site heparan sulphate proteoglycans found at the cell surface.
To block initial aspecific binding of virus to cells, polyanionic compounds (polysulfates, i.e., dextran sulfate, polysulfonates, polycarboxylates, polyoxometalates, and negatively charged albumins) have been suggested. A series of them were potent and selective inhibitors of respiratory viruses under experimental conditions.
Some pyrrolopyridine (6-azaindole) compounds, such as BMS-488043 (34.1.1) 61., 62. and BMS-663068 (34.1.2) [63], which block interactions between a virus and its receptor, thereby significantly reducing HIV-1 proliferation, have been proposed as anti-HIV therapeutics (Fig. 34.1 .).
Fig. 34.1.
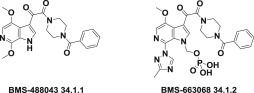
Compounds that block the interactions between a virus and its receptor.
Another example of compounds blocking initial binding of virus to cells are hydrolytic enzyme neuraminidase inhibitors.
The influenza neuraminidase is a surface glycoprotein that cleaves the cell-receptor sialic acid residues, thereby allowing the release of the virus to infect new cells [64]. Neuraminidase inhibitors are commonly used in both the prevention and the treatment of influenza.
Several neuraminidase inhibitors have been developed, including oseltamivir (Tamiflu) (34.1.3) and zanamivir (34.1.4), which were first used clinically as antiflu therapies [65]. Laninamivir (34.1.5) and peramivir (34.1.6) were also approved in northeast Asia (China, Japan and South Korea) recently. These agents are proven to be safe and effective alone or in combination for the treatment of uncomplicated influenzas [66] (Fig. 34.2 .).
Fig. 34.2.
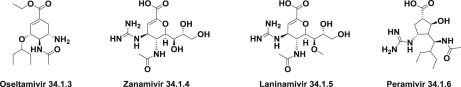
Neuraminidase inhibitors.
Another way of preventing binding of the virus to the host cell is by vaccination immunization using antibodies (specific immunoglobulins) against the infecting agent.
Entry Inhibitors
After attaching to host cells, the second step in the viral replication cycle is penetration. Enveloped viruses penetrate by fusion of the viral membrane with the cell membrane, while “naked” viruses penetrate the cell by phagocytosis of the virion and releasing viral genome into the host cytoplasm. For this reason entry inhibitors became one of the major concepts in the development of new antiviral drugs and play a very big role for antiviral therapies.
Fusion inhibitors are designed to block the conformational changes that are required for membrane fusion. Enfuvirtide (34.1.7), is the first and the only clinically approved fusion inhibitor that can inhibit a broad range of enveloped virus, particularly HIV strains. Enfuvirtide (Ac-Tyr-Thr-Ser-Leu-Ile-His-Ser-Leu-Ile-Glu-Glu-Ser-Gln-Asn-Gln-Gln-Glu-Lys-Asn-Glu-Gln-Glu-Leu-Leu-Glu-Leu-Asp-Lys-Trp-Ala-Ser-Leu-Trp-Asn-Trp-Phe-NH2) is the first of a novel class of peptide fusion inhibitors considered to be active against HIV-1 by disrupting the HIV-1 molecular machinery at the final stage of fusion with the target cell [67]. A series of modified peptides, including peptide fusion inhibitors possessing potent anti-HIV activity, namely, CP32M [68], sifuvirtide [69], and T2635 [70], have been synthesized and modified by pegylation, glycosylation, and other processes.
A plethora of small molecule fusion inhibitors have been synthesized, but because of the difficulty of their production and delivery, none has been approved for clinical use. Pleconaril (34.1.8), an entry inhibitor for nonenveloped viruses, prevents the virus from attaching itself to the host cell. It is the prime example of a human rhinovirus inhibitor 71., 72. to have been proposed. Other capsid-targeting molecules, namely, BTA-798 (34.1.9) 73., 74. and V-073 (34.1.10) 74., 75., are in clinical development.
The entry inhibitors primarily target the HIV-1 envelope glycoproteins or the cellular receptors, CD4, and the chemokine receptors involved in a number of biological processes. HIV-1 enters CD4-expressing cells via one or both of the chemokine receptors CCR5 and CXCR4. The CC-chemokine receptor 5 (CCR5) antagonist maraviroc (34.1.11) is the only approved drug of the novel class of “antiretroviral agents” that prevents the entry of HIV-1 into host cells by blocking the CCR5 coreceptor 74., 75. (Fig. 34.3 .).
Fig. 34.3.
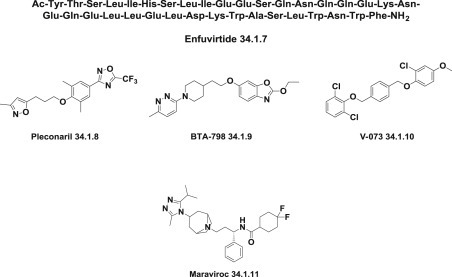
Entry inhibitors.
Uncoating Inhibitors
Another antiviral drug target is the uncoating step during viral infection, which is the process of capsid disintegration, retaining the virus in the encapsulated state, and not allowing the virus to release its genomic material into the host cell to interrupt the virus replicative cycle before it proceeds to the reverse transcriptase step. It is believed that such drugs work by blocking ion channels in the virus. Amantadine (34.1.12) 76., 77., 78., 79. and rimantadine (34.1.13) 78., 79., 80. are representatives of uncoating inhibitors specifically prevent release of influenza A virus in the cells, but to date no compounds have been shown to act at the uncoating of HIV or retroviruses (Fig. 34.4 .).
Fig. 34.4.

Uncoating inhibitors.
Protease Inhibitors
Proteases are essential enzymes that regulate number of processes such as infection, fertilization, allergic reactions, inflammation, blood clotting, cell growth and death, and bone remodeling. They have become a remarkable target for promising therapeutic agents.
Protease inhibitors are enzymes that are critical for diverse biological processes involved in the life cycle of viruses and their replication processes. Protease inhibitors have became very important constituents of the antiretroviral drugs armada. In particular, they prevent viral replication by blocking proteolytic cleavage of protein precursors that are necessary for the production of infectious particles. It should be noted that most viruses also encode proteases, which protect viral proteins by modulating host cell. Protease inhibitors were a major therapeutic breakthrough in the mid-1990s for the treatment of HIV infection, ushering in the era of highly active antiretroviral therapy. Among the anti-HIV drugs developed over the past two decades, inhibitors of HIV-1 protease and reverse transcriptase have found a very prominent clinical use; creation of a series of protease inhibitors is one of the great successes of antiviral drug design. Early research involved in a quest among suitable peptides. But finally it was concluded to decrease peptide-like features, minimize the size of molecules.
Currently approved protease inhibitors, which have been mainly created on principles of structure- and fragment-based drug design, include: nelfinavir (34.1.14), amprenavir (34.1.15), fosamprenavir (34.1.16), darunavir (34.1.17), saquinavir (34.1.18), atazanavir (34.1.19), indinavir (34.1.20), lopinavir (34.1.21), ritonavir (34.1.22), tipranavir (34.1.23), simeprevir (34.1.24). They share relative similarity in chemical structures and are recognized by suffix–navir and have become the most potent types of antiviral drugs (Fig. 34.5 .).
Fig. 34.5.
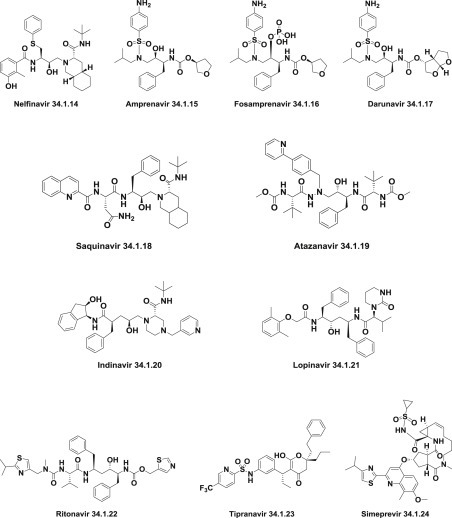
Protease inhibitors that act as antivirals.
Darunavir (34.1.17), atazanavir (34.1.19), and ritonavir (34.1.22) are drugs included in the list of Top 200 Drugs by sales for the 2010s.
Darunavir–Prezista
The synthesis of darunavir (34.1.17) 81., 82. was carried out via coupling mixed carbonate (34.1.27) with a benzene sulfonamide derivative of 1,3-diamino-4-phenylbutan-2-ol (34.1.34).
For this purpose, optically active bis-THF-ol (34.1.25) [83] was converted to mixed carbonate (34.1.27) by reaction with N,N′-disuccinimidyl carbonate (34.1.25) in the presence of triethylamine in methylene chloride.
For the synthesis of sulfonamide (34.1.34), commercially available (S)-1-((S)-oxiran-2-yl)-2-phenylethan-1-amine (34.1.29) was reacted with isobutylamine (34.1.28) in refluxing iso-propanol to give amino alcohol (34.1.30). Reaction of the resulting amino alcohol with p-nitrobenzenesulfonyl chloride (34.1.31) in the presence of aqueous NaHCO3 furnished the sulfonamide derivative (34.1.32). Catalytic hydrogenation of obtained product over 10% Pd-C in ethyl acetate effected reduction of the nitro group to the corresponding amine (34.1.33). The BOC group of the amine (34.1.33) was removed with the use of trifluoroacetic acid to produce diamine (34.1.34). Reaction of the diamine with the mixed carbonate (34.1.27) in the presence of triethylamine provided darunavir (34.1.17) in high yield (Scheme 34.1 .). Slight modifications of this Scheme have been proposed 84., 85., 86..
Scheme 34.1.
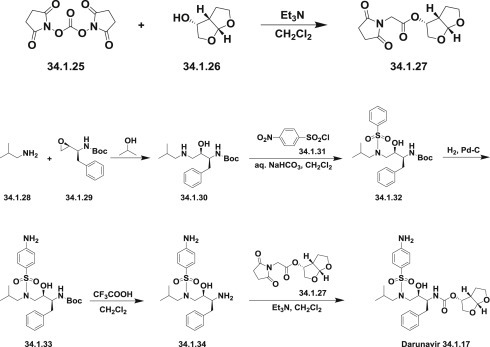
The synthesis of darunavir.
A series of darunavir analogues have displayed impressive enzymatic and antiviral properties and represent promising lead compounds for further optimization [87].
Darunavir is a unique and the most recent extremely potent protease inhibitor drug used to treat HIV infection. Darunavir usually is used in combination with ritonavir and other medicines. It maintains antiretroviral activity against a variety of multidrug-resistant HIV strains 88., 89., 90., 91., 92., 93., 94., 95., 96., 97..
Darunavir can cause serious, life-threatening side effects, including liver problems and severe skin reactions or rash.
Atazanavir–Reyataz
The original synthesis of atazanavir (34.1.19) 98., 99. was carried out through the reaction of the Boc-(pyridin-2-yl)benzyl)hydrazine (34.1.39) with the epoxide (34.1.40) prepared according to known procedures 100., 101. followed after deprotection by coupling with N-methoxycarbonyl-L-tert-leucine (34.1.42) to produce the desired atazanavir (34.1.19) (Scheme 34.2.).
Scheme 34.2.
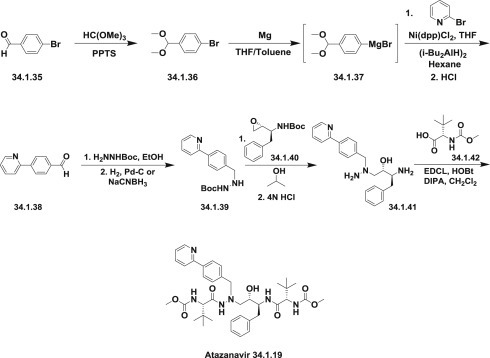
The original synthesis of atazanavir.
The required benzylhydrazine (34.1.39) was prepared by the coupling of 4-bromobenzaldehyde dimethyl acetal (34.1.36) to 2-bromopyridine catalyzed by the presence of 1,3-bis(diphenylphosphino)propane nickel (II) chloride and diisobutylaluminium hydride (current understanding of the coupling mechanism is limited) followed by acidic hydrolysis.
Obtained 4-(pyridin-2-yl)benzaldehyde (34.1.38) easily produced hydrazone with Boc-hydrazine in ethanol, which on hydrogenation on Pd-C catalyst or reduction with sodium cyanoborohydride provided the hydrazine building block (34.1.39).
Opening of the N-Boc–protected epoxide (34.1.40) with the Boc-protected benzylhydrazine (34.1.38) led to the symmetrically N-Boc–protected aza-dipeptide mimetic. Both of the Boc-protected groups were simultaneously cleaved off by acidic treatment (HCl in water/THF) and the obtained product (34.1.41) was coupled with N-methoxycarbonyl-L-tert-leucine (34.1.42) according to standard peptide synthesis procedures (carboxylic acid activator-carbodiimide-EDCl, racemization suppressor hydroxybenzotriazole [HOBt] base-triethylamine) to produce the target compound, atazanavir (34.1.19) (Scheme 34.2 .) The improved approach of the same strategy was published [102].
Another efficient and practical synthesis of atazanavir was developed by employing the diastereoselective reduction of ketomethylene aza-dipeptide (34.1.47) as the key and final step [103]. It was assembled from two key intermediates: hydrazide (34.1.44) and 1-amino-3-chloropropan-2-one derivative (34.1.46).
Hydrazide (34.1.44) was synthesized from the already known 4-(pyridin-2-yl)benzaldehyde (34.1.38), which in this special case was prepared by Kumada coupling of the Grignard reagent to 2-bromopyridine. The condensation of the aldehyde (34.1.38) with N-(methoxycarbonyl)-L-tert-leucinyl hydrazine (34.1.43) was carried out in ethanol. The resulting mixture was subjected to the next reaction without further purification. The quantitative hydrogenation with the use of Pd(OH)2/C in ethanol proceeded in high yield and high purity to produce the desired hydrazide (34.1.44).
The second key reagent, chloromethyl ketone (34.1.46), is prepared by reaction of (S)-3-amino-1-chloro-4-phenylbutan-2-one (34.1.45) with mixed anhydride, which in turn, was prepared from N-(methoxycarbonyl)-L-tert-leucine and isobutyl chloroformate. The coupling of the obtained chloromethyl ketone (34.1.46) with hydrazide (34.1.44) smoothly takes place in acetonitrile in presence of NaI and NaHCO3 to produce the ketone (34.1.47).
For the key and final step—reduction of obtained amino ketone (34.1.47)—a new reagent–solvent combination LiAlH(O-tBu)3 in Et2O was employed, which allowed synthesis of the desired syn-1,2-amino alcohol atazanavir (34.1.19) exclusively (Scheme 34.3 .) Other minor modifications of the presented two approaches have been proposed 104., 105., 106..
Scheme 34.3.
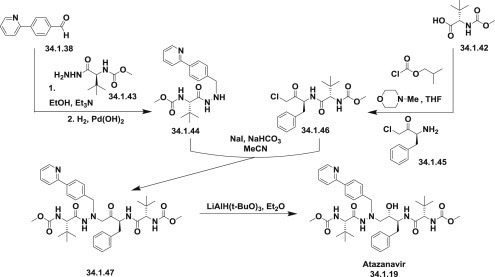
The synthesis of atazanavir.
Atazanavir is a novel, potent, safe, and generally well-tolerated inhibitor of the HIV protease. It was the first, and to date the only, protease inhibitor designed to be applied once daily. Atazanavir is expected to overcome the problems of earlier agents of this class of drugs, such as unfavorable adverse events like hyperlipidemia, diarrhea, and lipodystrophy. In combination with nucleoside reverse transcriptase inhibitors and boosted with ritonavir, it has been established as the preferred initial regimen in published guidelines 107., 108., 109., 110., 111., 112., 113., 114., 115., 116., 117., 118., 119..
Ritonavir–Norvir
The first publications 120., 121. describing the synthesis of ritonavir (34.1.22) started from methyl N-(benzyloxycarbonyl)-L-phenylalanine methyl ester (34.1.48), which on reduction with LiAlH4 was transformed to N-(benzyloxycarbonyl)-L-phenylalaninol (34.1.49). Oxidation of the N-(benzyloxycarbonyl)-L-phenylalaninol (34.1.49) with oxalyl chloride in DMSO at −60°C produced the corresponding aldehyde (34.1.50). The obtained aldehyde was dimerized with Zn dust in dichloromethane, in presence of vanadium trichloride, which catalyzes the pinacol coupling reaction, yielding (2S,3R,4R,5S)-2,5-bis(benzyloxy-carbonylamino)-1,6-diphenylhexane-3,4-diol (34.1.51) [along with the (2S,3S,4S,5S)-isomer]. The reaction of the diol (34.1.51) with α-acetoxyisobutyryl bromide (34.1.52) in hexane/dichloromethane produces (2S,3R,4R,5S)-2,5-bis(benzyloxycarbonylamino)-4-bromo-1,6-diphenylhexan-3-ol acetate ester (34.1.53), which was converted into the corresponding epoxide (34.1.54) with NaOH in dioxane/water. The reduction of the epoxide (34.1.54) with NaBH4 in THF followed by deprotection with CF3COOH produces (2S,3S,5S)-2,5-diamino-1,6-diphenylhexan-3-ol (34.1.55).
The obtained product was condensed with (5-thiazolylmethyl)(4-nitrophenyl)carbonate (34.1.56) in THF to produce (2S,3S,5S)-5-amino-1,6-diphenyl-2-(5-thiazolylmethoxycarbonylamino) hexan-3-ol (34.1.57). Finally, this compound was condensed with N-[N-(2-isopropylthiazol-4-ylmethyl)-N-methylaminocarbonyl]-L-valine (34.1.58) (EDCl, HOBt) in THF to produce the desired ritonavir (34.1.22) (Scheme 34.4 .).
Scheme 34.4.
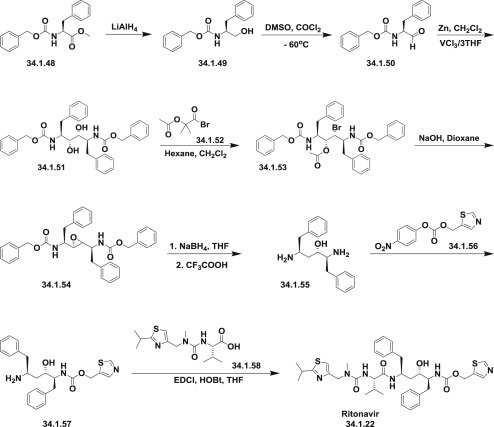
The synthesis of ritonavir.
(5-Thiazolylmethyl)(4-nitrophenyl)carbonate (34.1.56), used in Scheme 34.4., was synthesized from 5-thiazolylmethanol (34.1.65), which, in turn, was prepared via cyclization on heating of thioformamide (34.1.60) with 2-chloro-3-oxopropionic acid ethyl ester (34.1.61). The synthesis of thioformamide (34.1.60) was accomplished with P2S5 in ethyl ether from formamide (34.1.59). The starting β-aldehydoester (34.1.63) was obtained by condensation of ethyl chloroacetate (34.1.61) with ethyl formate (34.1.62) in the presence of t-BuOK in THF. The reduction of (34.1.64) with LiAlH4 in THF affords the 5-thiazolylmethanol (34.1.65), which was then esterified with 4-nitrophenyl chloroformate (34.1.66) in dichloromethane in the presence of 4-methylmorpholine to produce the desired product (34.1.56) (Scheme 34.5 .).
Scheme 34.5.
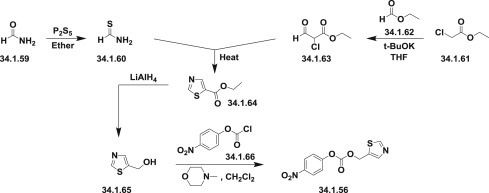
The synthesis of ritonavir (5-thiazolylmethyl)(4-nitrophenyl)carbonate.
The second thiazoly moiety-N-[N-(2-isopropylthiazol-4-ylmethyl)-N-methylaminocarbonyl]-L-valine (34.1.58) was synthesized by an analogues scheme.
The reaction of isobutyramide (34.1.67) with P2S5 in ethyl ether produced the corresponding thioamide (34.1.68), which was cyclized with 1,3-dichloroacetone (34.1.69) by means of MgSO4 in refluxing acetone, yielding 4-(chloromethyl)-2-isopropylthiazole (34.1.70). The reaction of the 4-(chloromethyl)-2-isopropylthiazole (34.1.70) with methylamine in water produced N-(2-isopropylthiazol-4-ylmethyl)-N-methylamine (34.1.71), which was condensed with N-(4-nitrophenoxycarbonyl)-L-valine methyl ester (34.1.73), which was synthesized by reaction of chloroformate (34.1.66) with L-valine methyl ester (34.1.72) in the presence of 4-methylmorpholine in dichloromethane.
Finally, this compound was converted into the corresponding free acid (34.1.58) with LiOH in dioxane/water mixture (Scheme 34.6 .).
Scheme 34.6.
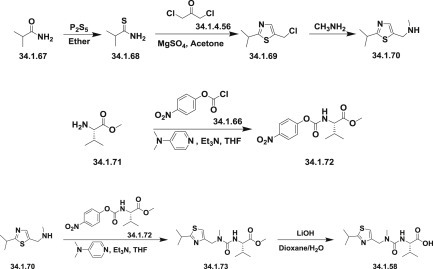
The synthesis of N-[N-(2-isopropylthiazol-4-ylmethyl)-N-methylaminocarbonyl]-L-valine.
A slightly different approach was demonstrated in patents 122., 123., which describe a new method for the preparation of a key diaminoalcohol intermediate (34.1.55) in the synthesis of ritonavir, has been described: The reaction of L-phenylalanine (34.1.74) with benzyl chloride using K2CO3 and KOH in hot water produced L-phenylalanine benzyl ester (34.1.75), which was condensed first with acetonitrile in presence of NaNH2 and then reacted with benzylmagnesium chloride, in methyl tert-butyl ether, yielding 5-amino-2-(dibenzylamino)-1,6-diphenylhex-4-en-3-one (34.1.76). The reduction of the obtained product with NaBH4 resulted in an enhanced enantioselectivity toward the desired (S,S,S)-enantiomer of 5-amino-2-(dibenzylamino)-1,6-diphenyl-3-hexanol (34.1.77). Finally, this compound was debenzylated by hydrogenation with ammonium formate over Pd/C in methanol/water to provide the target chiral diaminoalcohol (34.1.55) transformed to ritonavir (34.1.22) (Scheme 34.7 .).
Scheme 34.7.
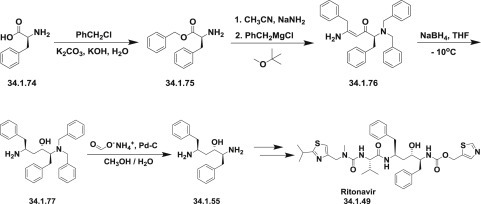
The synthesis of ritonavir.
Different variations of these approaches have been published 124., 125., 126., 127..
Ritonavir is a potent inhibitor of the protease encoded by the HIV-1 and is clinically applied to suppress HIV-1 replication in AIDS patients. Ritonavir does not cure HIV or AIDS, but it may slow the progress of the disease. It reduces the amount of virus in the body, has good oral bioavailability, and may increase the bioavailability of other protease inhibitors, including saquinavir, nelfinavir, and indinavir 128., 129., 130., 131., 132., 133.. Ritonavir, can cause clinically significant increases in serum levels of other protease inhibitors such as darunavir, fosamprenavir, lopinavir, and saquinavir, and is often used in combination with them.
Lopinavir–Kaletra
The first report of a new protease inhibitor candidate lopinavir (34.1.21) seems to be Abbott’s patent [134].
The core of lopinavir is identical to that of ritonavir. The 5-thiazolyl end group in ritonavir was replaced by the phenoxyacetyl group, and the 2-isopropylthiazolyl group in ritonavir was replaced by a modified valine in which the amino terminus had a six-membered cyclic urea attached.
Synthetic strategy employed for the synthesis of multikilogram quantities of lopinavir is very similar to that implemented for ritonavir (34.1.22), but using the protected version (34.1.79) of the “core” diamino alcohol (34.1.55), which was sequentially acylated with the acid chlorides of (S)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanoic acid (34.1.89) and 2-(2,6-dimethylphenoxy)acetic acid (34.1.94) 135., 136..
The bulk synthesis of protected diamino alcohol (34.1.79) was proposed 137., 138. by a method closely related to the method 122., 123. for ritonavir. For that purpose L-phenylalanine (34.1.74) was sequentially trialkylated with benzyl chloride using a K2CO3/water system at reflux to produce a tribenzylated product (34.1.75). A solution with generated acetonitrile anion in THF was added to the benzylated product (34.1.75) at less than −40°C to yield the cyanomethylketone (34.1.76), which was exposed to Grignard reagent–benzyl magnesium chloride to produce an enaminone (34.1.77). No racemization was observed in these two steps. The addition of the obtained enaminone (34.1.77) in THF/PrOH to a solution of NaBH4 and MsOH in THF at 5°C produced an intermediate aminoketone (34.1.78). No further reduction of the keto group occurs under these conditions. Reduction of the keto group could proceed by addition of a preformed solution of sodium tris(trifluoroacetoxy)borohydride in tetrahydrofuran [NaBH3(OTFA)], which produces a mixture of amino alcohols composed of 93% of the desired (2S)-5-amino-2-(dibenzylamino)-1,6-diphenylhexan-3-ol (34.1.79) along with 7% of the three undesired diastereomers. The crude mixture was debenzylated (Pd-C, HCONH4), and the product was purified by precipitation from iPrOH/HCl (aq) to produce (34.1.55) in greater than 99% purity and in high yield (Scheme 34.8 .).
Scheme 34.8.
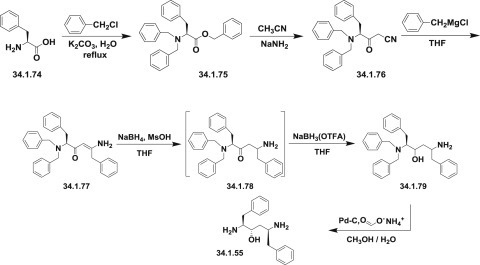
The synthesis of (2S)-5-amino-2-(dibenzylamino)-1,6-diphenylhexan-3-ol.
An efficient synthesis of each of the side chain moieties and their coupling with the “core” diamino alcohol derivatives was developed as follows: (S)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanoic acid (34.1.85) was prepared starting from L-valine (34.1.80), which was first converted to N-phenoxycarbonyl-L-valine (34.1.82) with phenylchloroformate (34.1.81). Accurate pH monitoring (pH 9.5 to 10.2) was necessary and LiOH was found to be a superior base. Control of pH was essential as the valine dimer and its derivatives were formed as reaction byproducts outside of this pH margin. LiCl was added to provide a lower freezing point to the aqueous solution and neutral Al2O3 was added to prevent gumming and emulsion formation during the course of the reaction.
Treatment of N-phenoxycarbonyl-L-valine (34.1.82) with 3-chloropropylamine hydrochloride (34.1.3) and solid NaOH in THF produced the unisolated salt of chloropropylurea (34.1.84), which was then treated with t-BuOK, effecting cyclization to produce the desired acid (34.1.85) in 75 to 85% yield and in greater than 99% enantiomeric excess. Acylation of (34.1.79) with synthesized acid (34.1.85) was initially achieved by well-known peptide coupling methods. Optimization of this transformation allowed the discovery of a more cost-effective method for implementing acyl chloride (34.1.86), which was easily prepared using thionyl chloride in THF at room temperature.
The reaction of dibenzylamino alcohols (34.1.79) with acyl chloride (34.1.86) in the presence of 3.0 equivalents of imidazole in EtOAc and DMF produced acylated intermediate (34.1.87) as a mixture of diastereomers, which, without any further purification, was subjected to debenzylation with Pd/C and HCO2NH4 in MeOH at 50°C, which proceeded without significant complications to produce (34.1.88).
Exposure of crude (34.1.88) to L-pyroglutamic acid (34.1.89) in dioxane at 50°C followed by cooling, allowed for the isolation of (S)-N-((2S,4S,5S)-5-amino-4-hydroxy-1,6-diphenylhexan-2-yl)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanamide (34.1.90) pyroglutamic salt as virtually a single diastereomer in high yield (Scheme 34.9 .).
Scheme 34.9.
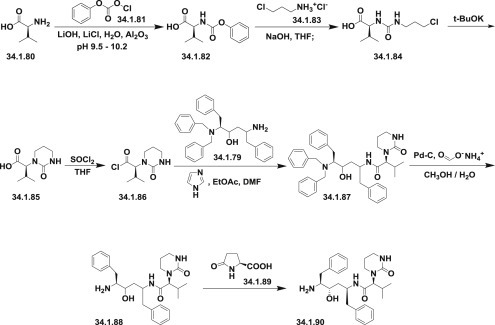
The synthesis of (S)-N-((2S,4S,5S)-5-amino-4-hydroxy-1,6-diphenylhexan-2-yl)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanamide.
Acyl chloride (34.1.92) was prepared by the reaction of 2-(2,6-dimethylphenoxy)acetic acid (34.1.91) with thionyl chloride in EtOAc, at room temperature adding a single drop of DMF, and warming the slurry to 50°C, which produced a clear solution of (34.1.92) that was used in the subsequent acylation of amine (34.1.90). Reaction of pyroglutamate salt (34.1.90) with acyl chloride (34.1.95) in ethyl acetate under Schotten-Baumann reaction conditions (use of a two-phase solvent system) in the presence of a water solution of NaHCO3 for liberation of free amine, produced the desired lopinavir (34.1.21) in high yield and purity (Scheme 34.10 .).
Scheme 34.10.
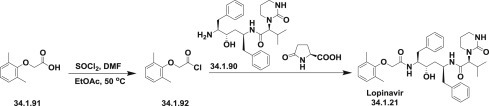
The synthesis of lopinavir.
Lopinavir is a novel and strong protease inhibitor developed from ritonavir with high specificity for HIV-1 protease 139., 140., 141.. It is indicated, in combination with other antiretroviral agents, for the treatment of HIV-1 infection. Numerous clinical trials have shown that lopinavir/ritonavir (Kaletra) is highly effective as a component of highly active antiretroviral therapy 142., 143..
Polymerase Inhibitors
The polymerases are enzymes essentially required for the replication of viruses. Viral DNA and RNA polymerases are responsible for copying the genetic materials of viruses, their transcription and replication, and therefore are central components in the life cycles of viruses.
Polymerase inhibitors block enzymatic function, thus preventing virus from multiplying. This group of antiviral medications consist of two classes: nucleoside inhibitors and nonnucleoside inhibitors. In contrast to the nucleoside inhibitors that bind to the active site of the polymerase, the nonnucleoside inhibitors bind to allosteric binding sites within the polymerase, thus blocking its action.
Along with nucleoside and nonnucleoside polymerase inhibitors there exist a class of nucleotide and nonnucleotide polymerase inhibitors.
Polymerase inhibitors are classified also as DNA or RNA polymerase inhibitors.
Nucleoside viral DNA polymerases are the specific target of a number of antiviral drugs currently used to inhibit viral replication. Most antiviral approved drugs which inhibit a DNA polymerase are nucleoside analogues. They represent the most productive source of antiviral agents. These agents need to be phosphorylated to their active form. Active forms inhibit polymerases by competing with natural substrates incorporation into the growing DNA chain, and in this way terminating viral DNA elongation.
The DNA polymerase inhibitors are acyclovir (34.1.93), valacyclovir (34.1.94), penciclovir (34.1.95), famciclovir (34.1.96), ganciclovir (34.1.97), and valganciclovir (34.1.98) (Fig. 34.6 .).
Fig. 34.6.
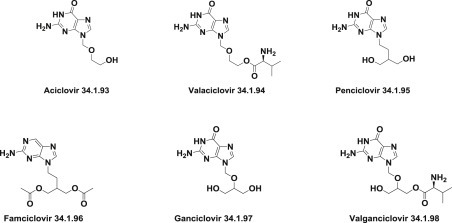
Structure of DNA polymerase inhibitors.
A special place among viral DNA polymerases inhibitors is occupied by foscarnet (Foscavir) (34.1.99), which selectively inhibits the pyrophosphate binding site on viral DNA polymerases, blocking the release of pyrophosphate from the terminal nucleoside, and does not affect human DNA polymerases (Fig. 34.7 .).
Fig. 34.7.
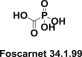
Structure of Foscavir.
Nucleoside and nucleotide DNA polymerase inhibitors are drugs identified by the suffix -ovir.
Viral RNA polymerase inhibitors are represented by the single drug ribavirin (34.1.100) (Fig. 34.8 .).
Fig. 34.8.
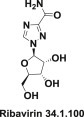
Structure of ribavirin.
Ribavirin (34.1.100) inhibits guanosine triphosphate formation, prevents capping of viral mRNA, and blocks derivative that resembles viral RNA-dependent RNA polymerase activity.
In recent years, antiviral drug discovery platforms utilizing high-throughput screening technology (HTS) have enabled discovery of many initial lead series of nonnucleoside viral RNA polymerase inhibitors.
The viral DNA polymerase inhibitors acyclovir, valacyclovir, valganciclovir, and tenofovir are included in the list of Top 200 Drugs by sales for the 2010s.
Acyclovir–Zovirax
The synthesis of the forerunner to polymerase inhibitors—acyclovir (34.1.93), as an antiherpes drug—was first described [144] and then disclosed in detail later [145].
The synthesis started with benzonitrile (34.1.101), which was heated at reflux in ethylene glycol for 72 hours to produce ethylene glycol monobenzoate (34.1.102). A cold mixture of the obtained ethylene glycol monobenzoate and paraformaldehyde in dry dichloroethane was saturated with dry HCl, producing 1-benzoyloxy-2-chloromethoxyethane (34.1.103). The last was added to a solution of 2,6-chloropurine (34.1.104) and triethylamine in dimethylformamide, and after the exothermic reaction the product—2,6-chloro-9-(2-benzoyloxyethoxymethyl)purine (34.1.105)—was separated. A solution of the 2,6-chloro-9-(2-benzoyloxyethoxymethyl)purine (34.1.105) in ammonia methanol solution was heated in autoclave at 95°C. As a result of the differences in the chemical reactivity in the 2- and 6- positions in the pyrimidine ring, selective substitution of the 6-chloro group takes place with simultaneous deprotection of the side chain to produce 2-chloro-9-(hydroxyethoxymethyl)adenine (34.1.106). Treatment of the last with nitrous acid, followed by reaction of deaminated intermediate with methanolic ammonia to displace the 2-chloro group, produces a moderate yield of acyclovir (34.1.93) (Scheme 34.11 .).
Scheme 34.11.
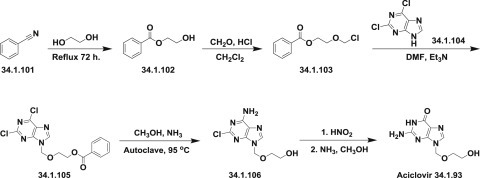
The synthesis of acyclovir.
A more efficient method to prepare acyclovir, which consists of alkylation of 2-chloro-6-iodopurine (34.1.109) with iodomethyl[(trimethylsilyl)oxy]ethyl ether (34.1.108), has been performed. The synthesis of (34.1.108) involved the reaction of 1,3-dioxolane (34.1.107) with trimethylsilyl iodide in cyclohexene to produce the desired side-chain moiety (34.1.108). Treatment of the anion of 2-chloro-6-iodo-purine (34.1.109) generated with NaH in dry DMF with prepared (34.1.108) at −63°C followed by hydrolysis and ammonolysis reactions, yielded the desired acyclovir (34.1.93). Hydrolysis was accomplished by adding aqueous solution of K2CO3 at room temperature to the solution of synthesized 2-chloro-9-[(2-hydroxyethoxy)methyl]-6-iodopurine (34.1.110) in dioxane. Ammonolysis occurred by heating of product of hydrolysis with NH3 in methanol in a sealed tube that was heated to 110°C to produce acyclovir (34.1.93) 146., 147. (Scheme 34.12 .).
Scheme 34.12.
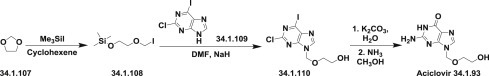
The synthesis of acyclovir.
A more convenient and economical synthesis of acyclovir has been reported [148]. The synthesis started from the easily available mixture of N,N′-diacetylguanines (34.1.112), which was prepared by acylation of guanine (34.1.111) with acetic anhydride/acetic acid mixture [149]. The obtained commixture of compounds was condensed with 2-oxabutane-1,4-diol diacetate (34.1.113) in the presence of p-toluenesulfonic acid or without solvent [149] or in DMSO [148], which produced a satisfactory yield of the desired separable crystalline intermediate (34.1.114), without the necessity for column purification. Deprotection of the compound (34.1.114) with 40% aqueous MeNH2 for 20 minutes at 100°C produced the acyclovir (34.1.96) for an overall yield of 33%. Effective hydrolysis of both acetyl protective groups is possible also by using ammonia in MeOH, 5% aqueous NaOH at room temperature, pyrrolidine at room temperature, via refluxing in glycol, whatever etc. Variations of this method have been reported 148., 149., 150. and reviewed 151., 152. (Scheme 34.13 .).
Scheme 34.13.
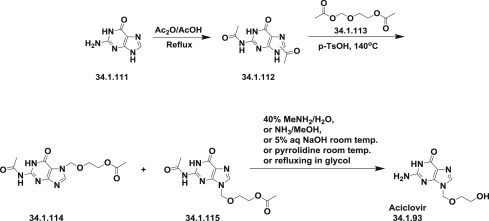
The synthesis of acyclovir.
Another approach [153] for the synthesis of acyclovir (34.1.93) was demonstrated; it started from the available 7-formamidofurazanopyrimidine (34.1.115) [154], which was alkylated with 2-(benzoyloxy)ethoxymethyl chloride (34.1.103) in dimethylformamide in the presence of triethylamine to produce a mixture of the desired compound (34.1.116) and some deformylated product. The mixture was reformylated with acetic-formic anhydride to produce crude (34.1.116). Reductive cleavage of the furazan ring in the obtained product with zinc dust in acetic acid followed by heating facilitated cyclization to produce 2-(methy1thio)adenine (34.1.117), the protective benzoyl group of which was cleaved with aqueous methylamine by heating on a steam bath to produce (34.1.118). The 2-methylthio group of (34.1.118) failed to react in liquid ammonia at 90°C. For this reason, the 6-amino group of the mentioned compound (34.1.118) was transformed to a hydroxyl group with sodium nitrite in acetic acid, which yielded (34.1.119). Subsequent amination with ammonia saturated ethanol at 140°C in an autoclave produced the desired acyclovir (34.1.93) (Scheme 34.14 .).
Scheme 34.14.
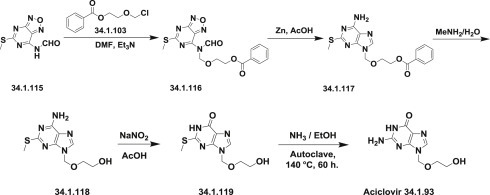
The synthesis of acyclovir.
Other approaches, which differ in details from described above, have been reviewed 151., 152..
Acyclovir is closely related to the natural component of DNA, guanine deoxyriboside, and acts to prevent the DNA replication of a DNA virus at concentrations far below those that affect cellular DNA.
The discovery of acyclovir, a nucleoside analogue, more than 30 years ago, represents a milestone. Acyclovir is the drug of choice for the prophylactic and curative treatment of herpes simplex (genital herpes) virus and varicella-zoster (shingles) virus infection, which remain a challenge in the 21st century. Acyclovir does not cure herpes, but may prevent a breakout of herpes sores or blisters. The viruses continue to live in the body even between outbreaks. But acyclovir decreases the severity and length of these outbreaks. It helps the sores heal faster, keeps new sores from forming, and decreases pain and itching. Acyclovir formulations include injection, oral and topical forms 153., 154., 155., 156..
Valacyclovir–Valtrex
Valacyclovir is a prodrug derived by esterifying acyclovir with L-valine. It is quickly absorbed and well tolerated. Upon administration, valacyclovir is rapidly and completely converted to acyclovir by enzymatic hydrolysis, which increases the oral bioavailability three- to fivefold.
In the first synthesis of valacyclovir (34.1.94) 157., 158., acyclovir (34.1.93) was condensed with N-carbobenzyloxy-L-valine (34.1.120) in DMF and in the presence of dicyclohexylcarbodiimide, providing N-carbobenzyloxy–protected valacyclovir (34.1.121), which was subjected to palladium catalyzed deprotection (Pd/Al2O3 in DMF) to furnish valacyclovir (34.1.94). An efficient and scaleable process according to this Scheme 34.15 was developed later [159].
Scheme 34.15.
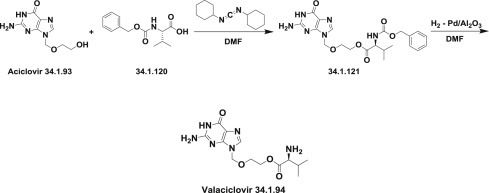
The synthesis of valacyclovir.
The same approach was implemented with another protecting group on an amino acid moiety and, valacyclovir was prepared by reaction of N-(Boc)-L-valine with acyclovir using 1-(3-dimethyl-aminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) as coupling agent and HCl in the deprotection step [160].
Valacyclovir is used for the treatment of herpes, varicella zoster, and cytomegaloviruses. A discussion on the clinical pharmacology, antiviral activity, clinical efficacy, and other therapeutic issues is presented in reviews 161., 162., 163., 164., 165., 166..
Valganciclovir–Valcyte
Valganciclovir (34.1.98) is a mono-L-valyl ester prodrug of the antiviral compound ganciclovir. Most of the known literature on the synthesis of valganciclovir involves the coupling of the protected form of ganciclovir (34.1.97) with N-protected L-valine derivatives followed by deprotection.
The synthesis of ganciclovir for the synthesis of valganciclovir started from epichlorohydrin (34.1.122), which on reaction with benzyl alcohol in presence of 50% aqueous NaOH (at room temperature) produced 1,3-di-O-benzylglycerol (34.1.123) in good yield. Chloromethylation of (34.1.123) with HCl and paraformaldehyde in methylene chloride gave the chloromethyl ether (34.1.124), reaction of which with potassium acetate in acetone yielded 2-O-(acetoxymethyl)-1,3-di-O-benzylglycerol (34.1.125). Condensation of the obtained product with a mixture of N,N′-diacetylguanines (34.1.112) in the presence of a catalytic amount of p-TsOH in sulfolane or DMF, produced a 3:2 mixture of N2-acetyl-9-[[1,3-bis(benzyloxy)-2-propoxy]methyl]guanine (34.1.126) and its corresponding N7 isomer. The desired isomer (34.1.126) was separated by crystallization from toluene. Debenzylation of the obtained product (34.1.126) using palladium hydroxide on carbon (cyclohexene, ethanol) produced compound (34.1.127), which was deacetylated with a concentrated NH4OH/methanol solution to produce ganciclovir (34.1.97) 167., 168. (Scheme 34.16 .).
Scheme 34.16.
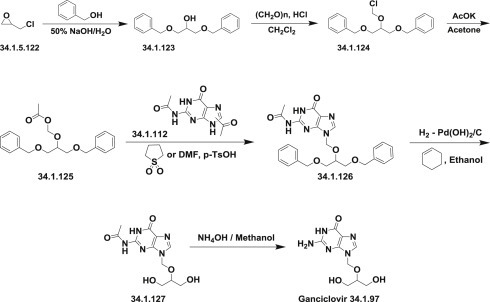
The synthesis of ganciclovir.
Other synthetic approaches of ganciclovir (34.1.100) have been reported 169., 170., 171., 172..
Valganciclovir was synthesized by method presented on the Scheme 34.17, which is closely related to presented above for ganciclovir.
Scheme 34.17.
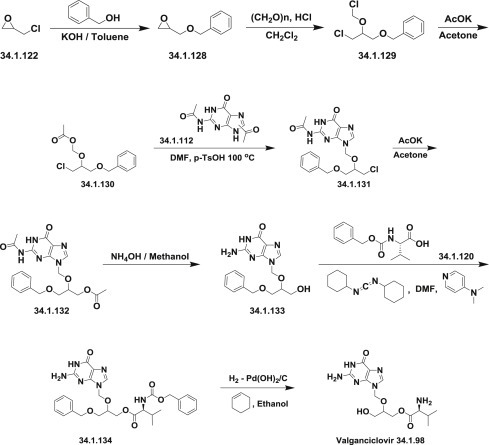
The synthesis of valganciclovir.
For this purpose, epichlorohydrin (34.1.122) was reacted with benzyl alcohol in the presence of powdered KOH in toluene at room temperature to produce benzyloxymethyloxirane (34.1.128). Gaseous HCl was bubbled into a stirred mixture of the obtained oxirane (34.1.132) and paraformaldehyde in dichloromethane to produce (1-chloro-2-chloromethoxy-3-benzyloxy)propane (34.1.129). This chloromethyl ether was reacted with potassium acetate in acetone to produce (1-chloro-2-acetoxymethoxy-3-benzyloxy)propane (34.1.130). A solution of diacetylguanine (34.1.112), (1-chloro-2-chloromethoxy-3-benzyloxy)propane (34.1.130), and p-TsOH in DMF was heated at 100°C for 6 hours to produce, after flash chromatography purification, a chloro derivative (34.1.131). The product from the previous step, and a large excess of potassium acetate in DMF, were heated to reflux for 5 hours to produce 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)methoxy-1-acetoxy-3-benzyloxy-propane (34.1.132). Stirring of the last in 30% ammonia/methanol at ambient temperature produced the deprotected compound (34.1.133). N-Benzyloxycarbonyl-L-valine (34.1.120)/dicyclohexylcarbodiimide complex prepared in dichloromethane was added to the suspension of (34.1.133) in DMF followed by 4-dimethylaminopyridine. The workup of the mixture after 18 hours produced valinate (34.1.134). The obtained product was deprotected by hydrogenation over palladium hydroxide in refluxing ethanol to produce the desired valganciclovir (34.1.98) 173., 174. (Scheme 34.17 .).
Valganciclovir (34.1. 98) has traditionally been synthesized by employing protection–deprotection strategies of either of the two hydroxy groups of optionally amino-protected ganciclovir and treating the monohydroxy-protected ganciclovir with protected L-valine (34.1.120). Most of the syntheses involve the use of key starting material ganciclovir (34.1.97).
In one approach, ganciclovir reacted with trimethyl orthoformate (or any other orthoformate) to produce cyclic orthoester (34.1.135), which was treated with formic acid in DMF/H2O to yield ganciclovir O-monoformate (34.1.136). The condensation of the ganciclovir O-monoformate (34.1.136) with N-benzyloxycarbonyl-L-valine- (34.1.120) produced the monovalinate (34.1.137), which upon deformylation with HCl in dichloromethane/methanol mixture of solvents gave rise to compound (34.1.138), which was deprotected by hydrogenation with Pd/C catalyst [175] to produce the desired valganciclovir (34.1.98) (Scheme 34.18 .).
Scheme 34.18.
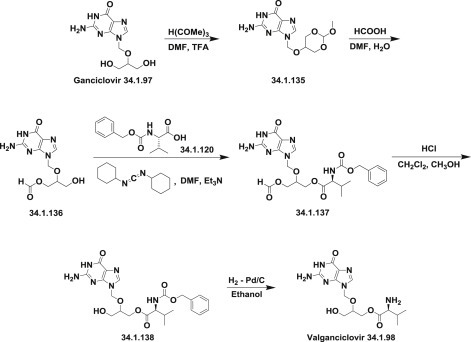
The synthesis of valganciclovir.
In another strategy, ganciclovir (34.1.97) was protected with tritylchloride or its derivatives in DMF in the presence of triethylamine and DMAP to produce the corresponding N,O-bis(trityl)–protected compound (34.1.139), which was condensed with N-(benzyloxycarbonyl)-L-valine (34.1.120) or N-(tert-butoxycarbonyl)-L-valine to yield the corresponding valine ester (34.1.140). Thereafter, the trityl-protecting group was removed using hot aqueous hydrochloric, acetic acid or trifluoroacetic acid, producing (34.1.141) followed by benzyloxycarbonyl removal by hydrogenation with Pd/C to obtain valganciclovir (34.1.98) 173., 176., 177., 178. (Scheme 34.19 .).
Scheme 34.19.
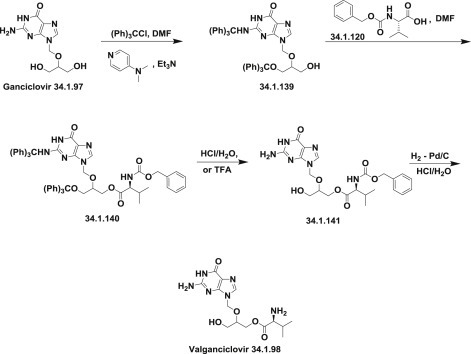
The synthesis of valganciclovir.
Many other closely related approaches also have been published 179., 180., 181., 182., 183., 184..
Valganciclovir is effective for the treatment of AIDS-related CMV retinitis, and for the prophylaxis of cytomegalovirus infection and disease in high-risk solid organ transplant recipients. The drug is generally well tolerated and has a similar tolerability profile. It is devoid of adverse events related to intravenous or indwelling catheter access associated with the use of intravenous ganciclovir 185., 186., 187., 188., 189., 190., 191., 192..
Nucleoside and Nucleotide Reverse Transcriptase Inhibitors
This group includes antiviral agents that are mainly recognized for the treatment of HIV, usually in combination with other retroviral drugs.
By 1980, HIV had spread to all continents. Identification of HIV as the causative agent of AIDS started a novel era in medicinal chemistry. As a result, the first antiretroviral agent, azidothymidine (Zidovudine, AZT) (34.1.146), was developed, which targeted the very nature of this class of viruses, reverse transcriptase, an enzyme that controls the replication of the genetic material of HIV. This enzyme is essential for HIV, but is not present in eukaryotic cells. These discoveries established the first class of antiretroviral agents: nucleoside and nucleotide reverse transcriptase inhibitors.
Nucleoside and nucleotide reverse transcriptase inhibitors are analogues of endogenous nucleosides and nucleotides. From the chemical point they can be represented as cytidine, guanosine, thymidine, and adenosine derivatives. They all are inactive in their parent forms and require phosphorylation steps by host cell kinases and phosphotransferases to form triphosphate derivatives capable of viral inhibition. In triphosphate forms, they compete with their corresponding endogenous deoxynucleoside triphosphates for incorporation by HIV reverse transcriptase. Once incorporated, they serve as chain-terminators of viral reverse transcripts, thus, acting on the viral replication cycle by inhibiting a critical step of proviral DNA synthesis prior to integration into the host cell genome 193., 194., 195., 196., 197., 198., 199..
The use of nucleoside and nucleotide reverse transcriptase inhibitors has revolutionized the treatment of infection by HIV and hepatitis-B virus. Nucleoside and nucleotide reverse transcriptase inhibitors became essential components in first-line therapy for HIV infection.
A variety of nucleoside reverse transcriptase inhibitors was synthesized, starting with azidothymidine (34.1.142), zalcitabine (34.1.143), stavudine (34.1.144), lamivudine (34.1.145), emtricitabine (34.1.146), didanosine (34.1.147), and abacavir (34.1.148), and, later, some nucleotide reverse transcriptase inhibitors, namely, cidofovir (34.1.149), adefovir (34.1.150), and tenofovir (34.1.151) appeared on the pharmaceutical market. Tenofovir (34.1.151) is used as tenofovir disoproxil fumarate, a prodrug for oral delivery, and is included in the list of Top 200 Drugs by sales for the 2010s (Fig. 34.9 .).
Fig. 34.9.
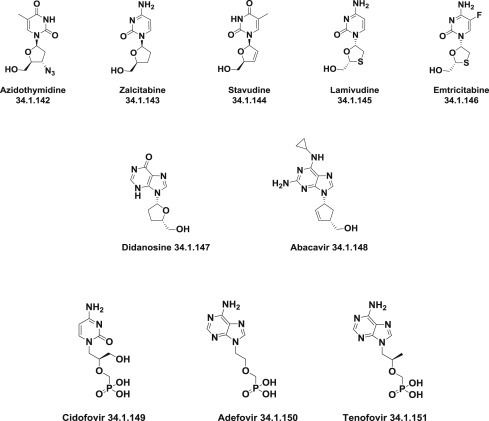
Nucleoside reverse transcriptase inhibitors.
Tenofovir–Viread
Two approaches for the synthesis of tenofovir (34.1.151) have been reported, both of which employ readily available reagents.
One is based on alkylation of adenine (34.1.152) in DMF in the presence of CsCO3 with chiral p-toluenesulfonyloxymethanephosphonate (34.1.153), which, in turn, was prepared in seven steps from D-(+)-isobutyl lactate [200]. Deprotection of the side chain of the obtained (34.1.154) using a standard deprotection/cleavage procedure with trimethylsilyl bromide to reflux acetonitrile produces tenofovir (34.1.151) [201] (Scheme 34.20 .).
Scheme 34.20.
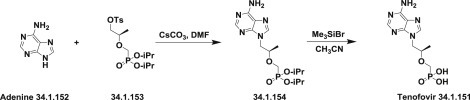
The synthesis of tenofovir.
In the second approach [202], adenine (34.1.152) was transformed to (R)-9-(2-hydroxypropyl)-adenine (34.1.156) by reaction with (R)-2-O-tetrahydropyranyl-1-O-p-toluenesulfonylpropane-1,2-diol (34.1.155) followed by deprotection with 0.25M sulfuric acid to produce the desired compound (34.1.156). After selective protection of N6 in (34.1.156) with benzoyl group, which was achieved by selective silylation with chlorotrimethylsilane in pyridine, followed by reaction with benzoyl chloride, the obtained 9-(R)-(2-hydroxypropyl)-N6-benzoyladenine (34.1.157) was alkylated with diisopropyl p-toluenesulfonyloxymethanephosphonate in DMF in the presence of sodium hydride to produce the compound (34.1.158). Debenzoylation of the N6 amino group was achieved using an ammonium hydroxide solution that produced (34.1.158), whose side chain was deprotected using trimethylsilyl bromide in reflux acetonitrile, which produced tenofovir (34.1.151) (Scheme 34.21 .)
Scheme 34.21.
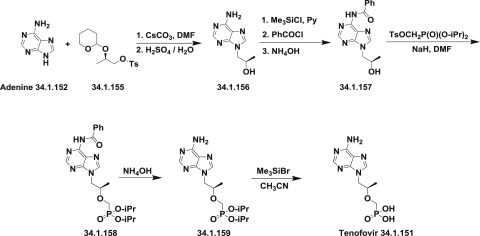
The synthesis of tenofovir.
The third approach was based on condensation of adenine (34.1.152) with propylene carbonate (34.1.160) to produce the (R)-9-(2-hydroxypropyl)-adenine (34.1.161). Coupling of the prepared adenine (34.1.161) with tosylated hydroxymethylphosphonate diester (34.1.162) using lithium tert-butoxide in THF, and after quenching obtained product with acetic acid and in dichloromethane/water mixture, the diethylphosphonate ester (34.1.163), which was subjected to excess bromotrimethylsilane in refluxing acetonitrile produced the desired tenofovir (34.1.151) [203] (Scheme 34.22 .).
Scheme 34.22.
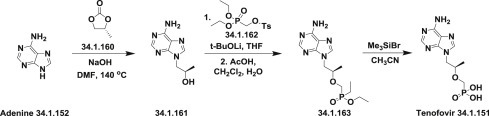
The synthesis of tenofovir.
This method was improved by converting it to a manufacturing process for the large-scale synthesis of tenofovir disoproxil fumarate 204., 205..
Tenofovir became available in 2001 and today it is an effective and widely used treatment for both HIV and hepatitis B virus infection.
Tenofovir is a component of the preferred first-line combination antiretroviral therapy. The efficacy, tolerability, prolonged half-life allowing for once-daily administration, and availability as a component of several fixed-dose formulations make tenofovir an attractive choice for treatment-naïve and treatment-experienced HIV-infected patients. It can be used in combination with other anti-HIV drugs 206., 207., 208., 209., 210., 211., 212., 213., 214., 215., 216., 217., 218., 219.. Since its approval in 2001, tenofovir has become one of the most frequently prescribed agents against HIV infection 206., 207., 208., 209., 210., 211., 212., 213., 214., 215., 216., 217., 218., 219..
Nonnucleoside Reverse-Transcriptase Inhibitors
Nonnucleoside reverse-transcriptase inhibitors represent one of the most significant classes of drugs for the treatment of AIDS/HIV infection and are a crucial component of current antiretroviral therapy 220., 221., 222., 223., 224..
These drugs are not competitive with nucleoside reverse transcriptase inhibitors. They are not incorporated into the viral DNA and work at a different site of the enzyme, preventing the enzyme’s action.
After the initial discovery of 1-(2-hydroxyethoxymethyl)-6-(phenylthio)thymine (HEPT) (34.1.164) followed by the synthesis of emivirine (34.1.165) and compounds of the IQP-0410 (34.1.166) series [225], several other chemical classes of compounds with nonnucleoside reverse transcriptase inhibitors properties were discovered. These include derivatives of α-anilinophenyl-acetamides (α-APA) such as loviride (34.1.167) and iminothioureas such as ITP (34.1.168) with activity against a wide variety of HIV-1 mutant strains [226], diaryltriazines such as R-106168 (34.1.169) [227], tetrahydroimidazo[4,5,1-jkj] 1., 4. benzodiazepin-2(1H)-one and -thione derivatives (TIBO) such as tivirapine (34.1.170) [228] and various other compounds. The hallmark of nonnucleoside reverse-transcriptase inhibitors has been their ability to interact with a specific site (“pocket”) of HIV-1 reverse-transcriptase (Fig. 34.10 .).
Fig. 34.10.
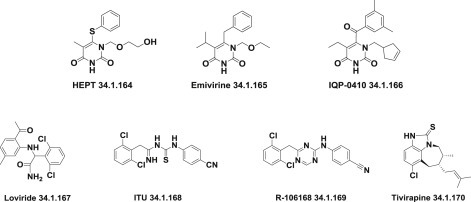
Nonnucleoside reverse-transcriptase inhibitors.
These findings followed with the discovery of the first drugs of this series, known as the first generation of nonnucleoside reverse-transcriptase inhibitors, which include efavirenz (34.1.171), the first nonnucleoside reverse transcriptase inhibitor (approved in 1998) 229., 230., 231., 232., delavirdine (34.1.172) 233., 234., 235., which is rarely used nowadays, and nevirapine (34.1.173) 236., 237., 238, 239.; all were commercialized as anti-HIV drugs (Fig. 34.11 .).
Fig. 34.11.
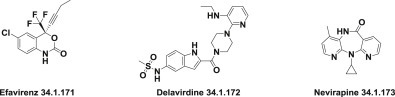
Commercialized nonnucleoside reverse-transcriptase inhibitors.
Sustained efforts in this area that were based on molecular modeling studies led to the identification of many promising hits, leads, and candidates, yielding the second-generation of nonnucleoside reverse-transcriptase inhibitors: etravirine (34.1.174), which was approved in 2008 240., 241., and rilpivirine (34.1.175) 242., 243., 244., 245., 246., 247., 248., 249., which was approved in 2011 (Fig. 34.12 .).
Fig. 34.12.
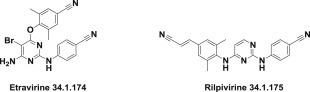
Second generation of nonnucleoside reverse-transcriptase inhibitors.
The nonnucleoside reverse-transcriptase inhibitors in clinical use today are efavirenz (34.1.171), delavirdine (34.1.172), nevirapine (34.1.173), etravirine (34.1.174), and rilpivirine (34.1.175).
Lersivirine (34.1.176) is a novel second-generation nonnucleoside reverse transcriptase inhibitor. Its development was recently stopped in Phase IIb clinical trials [250].
Capravirine (34.1.177) [251], is another second-generation nonnucleoside reverse-transcriptase inhibitor. Studies showed that it had no specific advantages over currently used drugs and, consequently, clinical trials were discontinued after Phase IIb trials (Fig. 34.13 .).
Fig. 34.13.
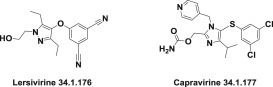
Novel second-generation nonnucleoside reverse-transcriptase inhibitors.
Human immunodeficiency virus infections are typically treated with drug combinations consisting of at least three different antiretroviral drugs; nonnucleoside reverse-transcriptase inhibitors are an essential component of antiretroviral therapy [252].
Integrase Inhibitors
Integrase inhibitors are a promising group of novel antiretroviral drugs that suppress the integrase-enzyme that facilitates the incorporation of HIV’s proviral DNA into the host cell genome and catalyzes a function vital to viral replication, via inhibiting the “integration” of the viral DNA into the hosts’ DNA genome 253., 254., 255., 256., 257..
Inhibitors of this enzyme represent the newest class of antiretroviral drugs in our armamentarium to treat viral infection.
Early integrase inhibitors included polyhydroxylated aromatic compounds, peptides, nucleotides, and DNA complexes, none of which were able to be developed into an effective drug.
The first major breakthrough was the discovery of the pyrrolo-diketo acids [258] as integrase inhibitors flowed by indole-diketo acids [259], naphthyridines 260., 261., 262., and dihydroxypyrimidine carboxamides [263], which finally led to discovery of raltegravir (34.1.178) 264., 265..
Raltegravir (34.1.178) was the first integrase inhibitor (it was approved in 2007) for the treatment of HIV infections 266., 267., 268., 269., 270., 271., 272., 273., 274., 275., 276., 277., 278., 279., 280., 281.. Raltegravir is included in the list of Top 200 Drugs by sales for the 2010s.
Elvitegravir (34.1.179), approved in late 2012, is another potent inhibitor of viral integrase 282., 283., 284.. Both raltegravir and elvitegravir are considered first-generation integrase inhibitors and are highly efficacious as first-line antiretroviral therapy.
Dolutegravir (34.1.180) is a second-generation integrase inhibitor that is currently under review by the FDA for marketing approval 285., 286., 287., 288., 289. (Fig. 34.14 .).
Fig. 34.14.
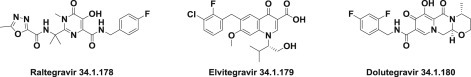
Integrase inhibitors.
Raltegravir–Isentress
The general synthetic route starts from the transformation of acetone cyanohydrin (34.1.181) to aminonitrile (34.1.182) under Strecker reaction conditions. Obtained aminonitrile (34.1.182) was converted to the N-Cbz–protected intermediate (34.1.183) using benzyl chloroformate in sodium carbonate water solution. For the preparation of amidoxime (34.1.184), hydroxylamine hydrochloride was added to a solution of (34.1.183) and potassium hydroxide in methanol. The amidoxime (34.1.184) was treated with dimethylacetylenedicarboxylate (34.1.185) in chloroform to produce (34.1.186), which was taken into xylene and heated at 145°C for 48 hours to be cyclized to pyrimidine-4-carboxylate (34.1.187). The obtained compound was benzoylated with benzoic acid chloride in pyridine to produce (34.1.188), which was purified by flash column chromatography and then N-methylated with dimethyl sulfate in dioxane using lithium hydride as a base, which provided the compound (34.1.189). The prepared (34.1.189) was hydrogenated in the presence of 10% Pd/C to produce the N-deprotected product (34.1.190), which was acylated with freshly prepared 5-methyl-1,3,4- oxadiazole-2-carbonyl chloride (34.1.191) in the presence of triethylamine in dichloromethane. Finally, refluxing prepared (34.1.192) overnight with p-fluorobenzylamine in methanol produced the desired raltegravir (34.1.178) 265., 267., 290. (Scheme 34.23 .). Further developments for efficient manufacturing of raltegravir are published 291., 292., 293., 294., 295..
Scheme 34.23.
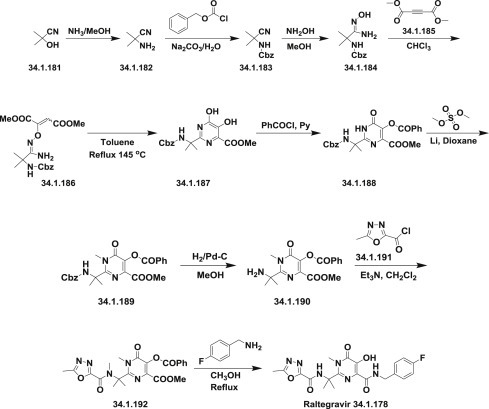
The synthesis of raltegravir.
Raltegravir is used along with other medications to treat HIV infection. It can cause serious, life-threatening side effects such as allergic reactions, skin reactions, and liver problems.
A number of other classes of compounds as potential antiviral drugs have attracted researcher’s attention in the last few years. Among them are methyltransferase inhibitors [296] (methyltransferase catalyzes the transfer of a methyl group from S-adenosyl-methionine to viral RNA, and is essential for the life cycle of many significant human pathogen viruses); helicase inhibitors 297., 298., 299. (helicases catalytically unwind duplex DNA or RNA and are required to displace the single-stranded genome after replication); neuraminidase inhibitors 300., 301. (neuraminidase inhibitors interfere with the release of virus from infected host cells); replication and transcription complex blockers 302., 303. (reagents that can efficiently block assembling of viral replication and transcription complex responsible for the production of the viral genome); and ribonucleoprotein complex inhibitors [304] (these inhibitors are thought to act as “molecule staples” that stabilize interactions between viral nucleoprotein monomers, promoting the formation of nonfunctional aggregates).
34.2. Indirect virus-targeting antivirals
While the antivirals currently in use exclusively target viral factors, several approaches now focus on cellular factors or pathways that indirectly interact with virus replication 305., 306., 307..
Among them are inhibitors of intracellular signaling cascades that are essential for virus replication.
Replication and transcription complex blockers block the formation of the viral replication and transcription complex, responsible for the production of the viral genome or other nucleic acids and ribonucleoprotein complex inhibitors which triggers the aggregation of and inhibits the nuclear accumulation of virally encoded nucleoprotein thereby inhibiting the replication of virus.
Among them a new compound – nucleozin (34.2.1) which inhibits the replication of various influenza A virus strains [308] (Fig. 34.15 .).
Fig. 34.15.
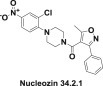
Structure of nucleozin.
References
- 1.De Clercq E. Antiviral drugs in current clinical use. J. Clin. Virol. 2004;30(2):115–133. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 2.De Clercq E. Antiviral drug discovery and development: where chemistry meets with biomedicine. Antiviral Res. 2005;67(2):56–75. doi: 10.1016/j.antiviral.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Balfour H.H., Jr. Antiviral drugs. N. Engl. J. Med. 1999;340(16):1255–1268. doi: 10.1056/NEJM199904223401608. [DOI] [PubMed] [Google Scholar]
- 4.Bonhoeffer S., May R.M., Shaw G.M., Nowak M.A. Virus dynamics and drug therapy. Proc. Natl. Acad. Sci. U. S. A. 1997;94(13):6971–6976. doi: 10.1073/pnas.94.13.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Clercq E. Antiviral agents active against influenza A viruses. Nat. Rev. Drug Discovery. 2006;5(12):1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boltz D.A., Aldridge J.R., Jr., Webster R.G., Govorkova E.A. Drugs in development for influenza. Drugs. 2010;70(11):1349–1362. doi: 10.2165/11537960-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biron K.K. Antiviral drugs for cytomegalovirus diseases. Antiviral Res. 2006;71(2-3):154–163. doi: 10.1016/j.antiviral.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Coen D.M., Schaffer P.A. Antiherpesvirus drugs: a promising spectrum of new drugs and drug targets. Nat. Rev. Drug Discovery. 2003;2(4):278–288. doi: 10.1038/nrd1065. [DOI] [PubMed] [Google Scholar]
- 9.Arora A., Mendoza N., Tyring S.K. Antiviral market overview. In: Gad S.C., editor. Development of Therapeutic Agents Handbook. Wiley; 2012. pp. 127–143. [Google Scholar]
- 10.De Clercq E. Antivirals: past, present and future. Biochem. Pharmacol. (Amsterdam, Neth.) 2013;85(6):727–744. doi: 10.1016/j.bcp.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 11.De Clercq E. Antiviral drugs. In: Krogsgaard-Larsen P., Stroemgaard K., Madsen U., editors. Textbook of Drug Design and Discovery. 4th ed. CRC Press; 2010. pp. 393–417. [Google Scholar]
- 12.De Clercq E. Highlights in the discovery of antiviral drugs: a personal retrospective. J. Med. Chem. 2010;53(4):1438–1450. doi: 10.1021/jm900932g. [DOI] [PubMed] [Google Scholar]
- 13.De Clercq E. The discovery of antiviral agents: ten different compounds, ten different stories. Med. Res. Rev. 2008;28(6):929–953. doi: 10.1002/med.20128. [DOI] [PubMed] [Google Scholar]
- 14.De Clercq E. Emerging antiviral drugs. Expert Opin. Emerg. Drugs. 2008;13(3):393–416. doi: 10.1517/14728214.13.3.393. [DOI] [PubMed] [Google Scholar]
- 15.De Clercq E. Antivirals: current state of the art. Future Virol. 2008;3(4):393–405. [Google Scholar]
- 16.De Clercq E. Status presens of antiviral drugs and strategies: part I: RNA viruses and retroviruses. Adv. Antiviral Drug Des. 2007;5:1–58. doi: 10.1016/S1075-8593(06)05001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Clercq E. Status presens of antiviral drugs and strategies: part II: RNA viruses (except retroviruses) Adv. Antiviral Drug Des. 2007;5:59–112. doi: 10.1016/S1075-8593(06)05002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Clercq E., Holy A. Case history: acyclic nucleoside phosphonates: a key class of antiviral drugs. Nat. Rev. Drug Discovery. 2005;4(11):928–940. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- 19.De Clercq E. Recent highlights in the development of new antiviral drugs. Curr. Opin. Microbiol. 2005;8(5):552–560. doi: 10.1016/j.mib.2005.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Clercq E. Strategies in the design of antiviral drugs. Nat. Rev. Drug Discovery. 2002;1(1):13–25. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- 21.De Clercq E. Antiviral drugs: current state of the art. J. Clin. Virol. 2001;22(1):73–89. doi: 10.1016/s1386-6532(01)00167-6. [DOI] [PubMed] [Google Scholar]
- 22.De Clercq E. Dancing with chemical formulae of antivirals: a personal account. Biochem. Pharmacol. (Amsterdam, Neth.) 2013;86(6):711–725. doi: 10.1016/j.bcp.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 23.De Clercq E. Dancing with chemical formulae of antivirals: a panoramic view (Part 2) Biochem. Pharmacol. (Amsterdam, Neth.) 2013;86(10):1397–1410. doi: 10.1016/j.bcp.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 24.De Clercq E. The design of drugs for HIV and HCV. Nat. Rev. Drug Discovery. 2007;6(12):1001–1018. doi: 10.1038/nrd2424. [DOI] [PubMed] [Google Scholar]
- 25.Burke J.D., Fish E.N. Antiviral strategies: the present and beyond. Curr. Mol. Pharmacol. 2009;2(1):32–39. doi: 10.2174/1874467210902010032. [DOI] [PubMed] [Google Scholar]
- 26.Meanwell N.A., Kadow J.F., Scola P.M. Antiviral agents. Annu. Rep. Med. Chem. 2002;37:133–147. [Google Scholar]
- 27.Eigen M. Error catastrophe and antiviral strategy. Proc. Natl. Acad. Sci. U. S. A. 2002;99(21):13374–13376. doi: 10.1073/pnas.212514799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z., Wang H., Du L.-Y., Chen K.-B., Xiao S.-L., Yu F., Zhang L.-H., Zhou D.-M. Discovery and development of antiviral drugs. J. Chin. Pharm. Sci. 2010;19(6):409–422. [Google Scholar]
- 29.Lou Z., Sun Y., Rao Z. Current progress in antiviral strategies. Trends Pharmacol. Sci. 2014;35(2):86–102. doi: 10.1016/j.tips.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones P.S. Strategies for antiviral drug discovery. Antiviral Chem. Chemother. 1998;9(4):283–302. [PubMed] [Google Scholar]
- 31.Tyring S.K. General (non-antiretroviral) antiviral drugs. Infect. Dis. Ther. 2005;37:123–289. [Google Scholar]
- 32.He H. Vaccines and antiviral agents. In: Romanowski V., editor. Current Issues in Molecular Virology: Viral Genetics and Biotechnological Applications. InTech; 2013. pp. 239–250. [Google Scholar]
- 33.Arbuthnot P., editor. Antiviral Drugs: Aspects of Clinical Use and Recent Advances. InTech; 2012. [Google Scholar]
- 34.Antonelli G., Turriziani O. Antiviral therapy: old and current issues. Int. J. Antimicrob. Agents. 2012;40(2):95–102. doi: 10.1016/j.ijantimicag.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Kazmierski W.M., editor. Antiviral Drugs: From Basic Discovery Through Clinical Trials. Wiley; 2011. [Google Scholar]
- 36.Chen T.-C., Weng K.-F., Chang S.-C., Lin J.-Y., Huang P.-N., Shih S.-R. Development of antiviral agents for enteroviruses. J. Antimicrob. Chemother. 2008;62(6):1169–1173. doi: 10.1093/jac/dkn424. [DOI] [PubMed] [Google Scholar]
- 37.Magri A., Bocchetta S., Burlone M.E., Minisini R., Pirisi M. Recent advances in HCV entry. Future Virol. 2014;9(2):189–205. [Google Scholar]
- 38.Hazuda D.J., Burroughs M., Howe A.Y.M., Wahl J., Venkatraman S. Development of boceprevir: a first-in-class direct antiviral treatment for chronic hepatitis C infection. Ann. N. Y. Acad. Sci. 2013;1291:69–76. doi: 10.1111/nyas.12218. [DOI] [PubMed] [Google Scholar]
- 39.Masgala A., Nikolopoulos G., Tsiodras S., Bonovas S., Sitaras N.M. Antiviral drugs in the prophylaxis of HBV infection. Curr. Med. Chem. 2012;19(35):5940–5946. [PubMed] [Google Scholar]
- 40.Mlynarczyk-Bonikowska B., Majewska A., Malejczyk M., Mlynarczyk G., Majewski S. Antiviral medication in sexually transmitted diseases. Part I: HSV, HPV. Mini-Rev. Med. Chem. 2013;13(13):1837–1845. doi: 10.2174/13895575113136660088. [DOI] [PubMed] [Google Scholar]
- 41.Wegzyn C.M., Wyles D.L. Antiviral drug advances in the treatment of human immunodeficiency virus (HIV) and chronic hepatitis C virus (HCV) Curr. Opin. Pharmacol. 2012;12(5):556–561. doi: 10.1016/j.coph.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 42.Gallay P.A. Cyclophilin inhibitors: a novel class of promising host-targeting ant-HCV agents. Immunol. Res. 2012;52(3):200–210. doi: 10.1007/s12026-011-8263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buti M., Esteban R. Drugs in development for hepatitis B. Drugs. 2005;65(11):1451–1460. doi: 10.2165/00003495-200565110-00001. [DOI] [PubMed] [Google Scholar]
- 44.Jordan R. Discovery and development of antiviral drugs for treatment of pathogenic human orthopoxvirus infections. RSC Drug Discovery Ser. 2013;32:81–110. [Google Scholar]
- 45.Arts E.J., Hazuda D.J. HIV-1 Targeting the host or the virus: current and novel concepts for antiviral approaches against influenza virus infection drug therapy. Perspect. Med. 2012;2(4) a007161/1-a007161/23. [Google Scholar]
- 46.Stellbrink H.-J. Antiviral drugs in the treatment of AIDS: what is in the pipeline? Eur. J. Med. Res. 2007;12(9):483–495. [PubMed] [Google Scholar]
- 47.Karmon S.L., Markowitz M. Next-generation integrase inhibitors. Drugs. 2013;73(3):213–228. doi: 10.1007/s40265-013-0015-5. [DOI] [PubMed] [Google Scholar]
- 48.Laver G. Antiviral drugs for influenza: Tamiflu past, present and future. Future Virol. 2006;1(5):577–586. [Google Scholar]
- 49.Hayden F.G. Antivirals for influenza: historical perspectives and lessons learned. Antiviral Res. 2006;71(2-3):372–378. doi: 10.1016/j.antiviral.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 50.Wee T., Jenssen H. Influenza drugs—current standards and novel alternatives. J. Antivirals Antiretrovirals. 2009;1(1):001–010. [Google Scholar]
- 51.Lee S.M.-Y., Yen H.-L. Targeting the host or the virus: current and novel concepts for antiviral approaches against influenza virus infection. Antiviral Res. 2012;96(3):391–404. doi: 10.1016/j.antiviral.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saravolac E.G., Wong J.P. Recent patents on development of nucleic acid-based antiviral drugs against seasonal and pandemic influenza virus infections. Recent Pat. Anti-Infect. Drug Discovery. 2007;2(2):140–147. doi: 10.2174/157489107780832622. [DOI] [PubMed] [Google Scholar]
- 53.Saravolac E.G., Wong J.P. Recent patents on development of nucleic acid-based antiviral drugs against seasonal and pandemic influenza virus infections. Front. Anti-Infect. Drug Discovery. 2010;1:409–425. doi: 10.2174/157489107780832622. [DOI] [PubMed] [Google Scholar]
- 54.Loregian A., Mercorelli B., Nannetti G., Compagnin C., Palu G. Antiviral strategies against influenza virus: towards new therapeutic approaches. Cell. Mol. Life Sci. 2014;71(19):3659–3683. doi: 10.1007/s00018-014-1615-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Park S., Kim Jin I., Park M.-S. Antiviral agents against influenza viruses. J. Bacteriol. Virol. 2012;42(4):284–293. [Google Scholar]
- 56.Driscoll J.S., editor. Antiviral drugs. (Wiley); 2002. [Google Scholar]
- 57.Gilbert C., Boivin G. Human cytomegalovirus resistance to antiviral drugs. Antimicrob. Agents Chemother. 2005;49(3):873–883. doi: 10.1128/AAC.49.3.873-883.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schang L.M. Herpes simplex viruses in antiviral drug discovery. Curr. Pharm. Des. 2006;12(11):1357–1370. doi: 10.2174/138161206776361174. [DOI] [PubMed] [Google Scholar]
- 59.Schafer J.J., Squires K.E. Integrase inhibitors: a novel class of antiretroviral agents. Ann. Pharmacother. 2010;44(1):145–156. doi: 10.1345/aph.1M309. [DOI] [PubMed] [Google Scholar]
- 60.Van Westreenen M., Boucher C.A.B. Classes of antiviral drugs. In: Boucher C.A.B., Galasso G.A., Katzenstein D.A., Cooper D.A., editors. Practical Guidelines in Antiviral Therapy. Elsevier; 2002. pp. 1–12. [Google Scholar]
- 61.Da L.-T., Quan J.-M., Wu Y.-D. Understanding the binding mode and function of BMS-488043 against HIV-1 viral entry. Proteins: Struct., Funct., Genet. 2011;79(6):1810–1819. doi: 10.1002/prot.23005. [DOI] [PubMed] [Google Scholar]
- 62.Yang Z., Zadjura L.M., Marino A.M., D’Arienzo C.J., Malinowski J., Gesenberg C., Lin P.-F., Colonno R.J., Wang T., Kadow J.F., Meanwell N.A., Hansel S.B. Utilization of in vitro Caco-2 permeability and liver microsomal half-life screens in discovering BMS-48(8043), a novel HIV-1 attachment inhibitor with improved pharmacokinetic properties. J. Pharm. Sci. 2010;99(4):2135–2152. doi: 10.1002/jps.21948. [DOI] [PubMed] [Google Scholar]
- 63.Chen K., Risatti C., Bultman M., Soumeillant M., Simpson J., Zheng B., Fanfair D., Mahoney M., Mudryk B., Fox R.J., Hsaio Y., Murugesan S., Conlon D.A., Buono F.G., Eastgate M.D. Synthesis of the 6-azaindole containing HIV-1 attachment inhibitor pro-drug, BMS-66(3068) J. Org. Chem. 2014;79(18):8757–8767. doi: 10.1021/jo5016008. [DOI] [PubMed] [Google Scholar]
- 64.Colman P.M., Varghese J.N., Laver W.G. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature (London, U. K.) 1983;303(5912):41–44. doi: 10.1038/303041a0. [DOI] [PubMed] [Google Scholar]
- 65.Ison M.G. Antivirals and resistance: influenza virus. Curr. Opin. Virol. 2011;1(6):563–573. doi: 10.1016/j.coviro.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 66.Ison M.G. Clinical use of approved influenza antivirals: therapy and prophylaxis. Influenza Other Respir. Viruses. 2013;7(Suppl. 1):7–13. doi: 10.1111/irv.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Joly V., Jidar K., Tatay M., Yeni P. Enfuvirtide: from basic investigations to current clinical use. Expert Opin. Pharmacother. 2010;11(16):2701–2713. doi: 10.1517/14656566.2010.522178. [DOI] [PubMed] [Google Scholar]
- 68.Yao X., Chong H., Zhang C., Qiu Z., Qin B., Han R., Waltersperger S., Wang M., He Y., Cui S. Structural basis of potent and broad HIV-1 fusion inhibitor CP32M. J. Biol. Chem. 2012;287(32):26618–26629. doi: 10.1074/jbc.M112.381079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang X., Wu H., Wang F. Sifuvirtide, a novel HIV-1 fusion inhibitor. In: Castanho M., Santos N.C., editors. Peptide Drug Discovery and Development: Translational Research in Academia and Industry. Wiley-VCH; 2011. pp. 231–243. [Google Scholar]
- 70.Eggink D., Langedijk J.P.M., Bonvin A.M.J.J., Deng Y., Lu M., Berkhout B., Sanders R.W. Detailed mechanistic insights into HIV-1 sensitivity to three generations of fusion inhibitors. J. Biol. Chem. 2009;284(39):26941–26950. doi: 10.1074/jbc.M109.004416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xiao C., McKinlay M.A., Rossmann M.G. Design of capsid-binding antiviral agents against human rhinoviruses. RSC Biomol. Sci. 2011;21(Structural Virology):319–337. [Google Scholar]
- 72.Zhang G., Zhou F., Gu B., Ding C., Feng D., Xie F., Wang J., Zhang C., Cao Q., Deng Y., Hu W., Yao K. In vitro and in vivo evaluation of ribavirin and pleconaril antiviral activity against enterovirus 71 infection. Arch. Virol. 2012;157(4):669–679. doi: 10.1007/s00705-011-1222-6. [DOI] [PubMed] [Google Scholar]
- 73.Thibaut H.J., De Palma A.M., Neyts J. Combating enterovirus replication: state-of-the-art on antiviral research. Biochem. Pharmacol. (Amsterdam, Neth.) 2012;83(2):185–192. doi: 10.1016/j.bcp.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 74.Dorr P., Stammen B., van der Ryst E. Discovery and development of maraviroc, a CCR5 antagonist for the treatment of HIV infection. In: Huang X., Aslanian R.G., editors. Case Studies in Modern Drug Discovery and Development. Wiley; 2012. pp. 196–226. [Google Scholar]
- 75.Lieberman-Blum S.S., Fung H.B., Bandres J.C. Maraviroc: a CCR5-receptor antagonist for the treatment of HIV-1 infection. Clin. Ther. 2008;30(7):1228–1250. doi: 10.1016/s0149-2918(08)80048-3. [DOI] [PubMed] [Google Scholar]
- 76.Hubsher G., Haider M., Okun M.S. Amantadine: the journey from fighting flu to treating Parkinson disease. Neurology. 2012;78(14):1096–1099. doi: 10.1212/WNL.0b013e31824e8f0d. [DOI] [PubMed] [Google Scholar]
- 77.Saito R., Li D., Sato M., Suzuki H. Amantadine. Virus Rep. 2006;3(1):40–47. [Google Scholar]
- 78.Fleming D.M. Managing influenza: amantadine, rimantadine and beyond. Int. J. Clin. Pract. 2001;55(3):189–195. [PubMed] [Google Scholar]
- 79.Hayden F.G. Amantadine and rimantadine—clinical aspects. In: Richman D.D., editor. Antiviral Drug Resistance. Wiley; 1996. pp. 59–77. [Google Scholar]
- 80.Hay A.J. Amantadine and rimantadine-mechanisms. In: Richman D.D., editor. Antiviral Drug Resistance. (Wiley); 1996. pp. 43–58. [Google Scholar]
- 81.Ghosh A.K., Leshchenko S., Noetzel M. Stereoselective photochemical 1,3-dioxolane addition to 5-alkoxymethyl-2(5H)-furanone: synthesis of bis-tetrahydrofuranyl ligand for HIV protease inhibitor UIC-94017 (TMC-114) J. Org. Chem. 2004;69(23):7822–7829. doi: 10.1021/jo049156y. [DOI] [PubMed] [Google Scholar]
- 82.Ghosh A.K., Kincaid J.F., Cho W., Walters D.E., Krishnan K., Hussain K.A., Koo Y., Cho H., Rudall C., Holland L., Buthod J. Potent HIV protease inhibitors incorporating high-affinity P2-ligands and (R)-[(hydroxyethyl)amino]sulfonamide isostere. Bioorg. Med. Chem. Lett. 1998;8(6):687–690. doi: 10.1016/s0960-894x(98)00098-5. [DOI] [PubMed] [Google Scholar]
- 83.Ghosh, A. K.; Leshchenko, S.; Noetzel, M. W. Method of preparing (3R,3aS,6aR)-3-hydroxyhexahydrofuro[2,3-b]furan and related compounds, WO 2004033462 (2004).
- 84.Erickson, J. W.; Gulnik, S. V., Fitness assay and associated methods, and applications to drug resistance and HIV protease inhibitors and other drugs with reduced resistance, WO 9967417 (1999).
- 85.Ghosh A.K., Sridhar P.R., Kumaragurubaran N., Koh Y., Weber I.T., Mitsuya H. Bis-tetrahydrofuran: a privileged ligand for darunavir and a new generation of HIV protease inhibitors that combat drug resistance. ChemMedChem. 2006;1(9):939–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 86.Ghosh A.K. Capturing the essence of organic synthesis: from bioactive natural products to designed molecules in today’s medicine. J. Org. Chem. 2010;75(23):7967–7989. doi: 10.1021/jo101606g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ghosh A.K., Gemma S., Simoni E., Baldridge A., Walters D.E., Ide K., Tojo Y., Koh Y., Amano M., Mitsuya H. Synthesis and biological evaluation of novel allophenylnorstatine-based HIV-1 protease inhibitors incorporating high affinity P2-ligands. Bioorg. Med. Chem. Lett. 2010;20(3):1241–1246. doi: 10.1016/j.bmcl.2009.11.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ruela Correa J.C., D’Arcy D.M., dos Reis Serra C.H., Nunes Salgado H.R. Darunavir: a critical review of its properties, use and drug interactions. Pharmacology. 2012;90(1-2):102–109. doi: 10.1159/000339862. [DOI] [PubMed] [Google Scholar]
- 89.de Bethune M.-P., Peeters A., Wigerinck P. From saquinavir to darunavir: the impact of 10 years of medicinal chemistry on a lethal disease. Methods Princ. Med. Chem. 2011;50:73–90. [Google Scholar]
- 90.Deeks E.D. Darunavir: a review of its use in the management of HIV-1 infection. Drugs. 2014;74(1):99–125. doi: 10.1007/s40265-013-0159-3. [DOI] [PubMed] [Google Scholar]
- 91.de Bethune M.-P., Sekar V., Spinosa-Guzman S., Vanstockem M., De Meyer S., Wigerinck P., Lefebvre E. Darunavir (prezista, TMC114): from bench to clinic, improving treatment options for HIV-infected patients. In: Kazmierski W.M., editor. Antiviral Drugs: From Basic Discovery Through Clinical Trials. Wiley; 2011. pp. 31–45. [Google Scholar]
- 92.Kogawa A.C., Salgado H.R.N. Characteristics, complexation and analytical methods of Darunavir. Br. J. Pharm. Res. 2014;4(11):1276–1286. [Google Scholar]
- 93.Phung B.-C., Yeni P. Darunavir: an effective protease inhibitor for HIV-infected patients. Expert Rev. Anti-Infect. Ther. 2011;9(6):631–643. doi: 10.1586/eri.11.48. [DOI] [PubMed] [Google Scholar]
- 94.El-Atrouni W.I., Temesgen Z. Darunavir. Drugs Today. 2007;43(10):671–679. doi: 10.1358/dot.2007.43.10.1131764. [DOI] [PubMed] [Google Scholar]
- 95.Molina J.-M., Hill A. Darunavir (TMC114): a new HIV-1 protease inhibitor. Expert Opin. Pharmacother. 2007;8(12):1951–1964. doi: 10.1517/14656566.8.12.1951. [DOI] [PubMed] [Google Scholar]
- 96.Sorbera L.A., Castaner J., Bayes M. Darunavir. Drugs Future. 2005;30(5):441–449. [Google Scholar]
- 97.McKeage K., Perry C.M., Keam S.J. Darunavir: a review of its use in the management of HIV infection in adults. Drugs. 2009;69(4):477–503. doi: 10.2165/00003495-200969040-00007. [DOI] [PubMed] [Google Scholar]
- 98.Bold G., Faessler A., Capraro H.-G., Cozens R., Klimkait T., Lazdins J., Mestan J., Poncioni B., Roesel J., Stover D., Tintelnot-Blomley M., Acemoglu F., Beck W., Boss E., Eschbach M., Huerlimann T., Masso E., Roussel S., Ucci-Stoll K., Wyss D., Lang M. New aza-dipeptide analogs as potent and orally absorbed HIV-1 protease inhibitors: candidates for clinical development. J. Med. Chem. 1998;41(18):3387–3401. doi: 10.1021/jm970873c. [DOI] [PubMed] [Google Scholar]
- 99.Fassler, A.; Bold, G.; Capraro, H-G.; Steiner, H. Process for the preparation of hydrazine derivatives useful as intermediates for the preparation of peptide analogs, PCT Int. Appl. (1997), WO 9746514 A1 19971211.
- 100.Thompson W.J., Fitzgerald P.M.D., Holloway M.K., Emini E.A., Darke P.L., McKeever B.M., Schleif W.A., Quintero J.C., Zugay J.A., Tucker T.J., Schwering J.E., Homnick C.F., Nunberg J., Springer J.P., Huff J.R. Synthesis and antiviral activity of a series of HIV-1 protease inhibitors with functionality tethered to the P1 or P1’ phenyl substituents: x-ray crystal structure assisted design. J. Med. Chem. 1992;35:1685–1701. doi: 10.1021/jm00088a003. [DOI] [PubMed] [Google Scholar]
- 101.Luly J.R., Dellaria J.F., Plattner J.J., Soderquist J.L., Yi N.A. Synthesis of protected aminoalkyl epoxides from R-amino acids. J. Org. Chem. 1987;52:1487–1492. [Google Scholar]
- 102.Xu Z., Singh J., Schwinden M.D., Zheng B., Kissick T.P., Patel B., Humora M.J., Quiroz F., Dong L., Hsieh D.-M., Heikes J.E., Pudipeddi M., Lindrud M.D., Srivastava S.K., Kronenthal D.R., Mueller R.H. Process research and development for an efficient synthesis of the HIV protease inhibitor BMS-23(2632) Org. Process Res. Dev. 2002;6(3):323–328. [Google Scholar]
- 103.Fan X., Song Y.-L., Long Y.-Q. An efficient and practical synthesis of the HIV protease inhibitor atazanavir via a highly diastereoselective reduction approach. Org. Process Res. Dev. 2008;12(1):69–75. [Google Scholar]
- 104.Giordano, C.; Pozzoli, C.; Benedetti, F., Process for the preparation of aryl-pyridinyl compounds, WO 2001027083 (2001).
- 105.Chen, W. Process for synthesizing atazanavir, WO 2009130534 (2009).
- 106.Simhadri, S.; Mohammad, Y.; Indukuri, V. S. K.; Gorantla, S. R., Preparation of atazanavir bisulfate, WO 2014030173 (2014).
- 107.Farajallah A., Bunch R.T., Meanwell N.A. Discovery and development of atazanavir. In: Kazmierski W.M., editor. Antiviral Drugs: From Basic Discovery Through Clinical Trials. Wiley; 2011. pp. 3–17. [Google Scholar]
- 108.Goldsmith D.R., Perry C.M. Atazanavir. Drugs. 2003;63(16):1679–1693. doi: 10.2165/00003495-200363160-00003. [DOI] [PubMed] [Google Scholar]
- 109.Busti A.J., Hall R.G., II., Margolis D.M. Atazanavir for the treatment of human immunodeficiency virus infection. Pharmacotherapy. 2004;24(12):1732–1747. doi: 10.1592/phco.24.17.1732.52347. [DOI] [PubMed] [Google Scholar]
- 110.Orrick J.J., Steinhart C.R. Atazanavir. Ann. Pharmacother. 2004;38(10):1664–1674. doi: 10.1345/aph.1D394. [DOI] [PubMed] [Google Scholar]
- 111.Havlir D.V., O’Marro S.D. Atazanavir: new option for treatment of HIV infection. Clin. Infect. Dis. 2004;38(11):1599–1604. doi: 10.1086/420932. [DOI] [PubMed] [Google Scholar]
- 112.Bentue-Ferrer D., Arvieux C., Tribut O., Ruffault A., Bellissant E. Clinical pharmacology, efficacy and safety of atazanavir: a review. Expert Opin. Drug Metab. Toxicol. 2009;5(11):1455–1468. doi: 10.1517/17425250903321514. [DOI] [PubMed] [Google Scholar]
- 113.Croom K.F., Dhillon S., Keam S.J. Atazanavir: a review of its use in the management of HIV-1 infection. Drugs. 2009;69(8):1107–1140. doi: 10.2165/00003495-200969080-00009. [DOI] [PubMed] [Google Scholar]
- 114.Wood R. Atazanavir: its role in HIV treatment. Expert Rev. Anti-Infect. Ther. 2008;6(6):785–796. doi: 10.1586/14787210.6.6.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.von Hentig N. Atazanavir/ritonavir: a review of its use in HIV therapy. Drugs Today. 2008;44(2):103–132. doi: 10.1358/dot.2008.44.2.1137107. [DOI] [PubMed] [Google Scholar]
- 116.Gianotti N., Soria A., Lazzarin A. Antiviral activity and clinical efficacy of atazanavir in HIV-1-infected patients: a review. New Microbiol. 2007;30(2):79–88. [PubMed] [Google Scholar]
- 117.Harrison T.S., Scott L.J. Atazanavir: a review of its use in the management of HIV infection. Drugs. 2005;65(16):2309–2336. doi: 10.2165/00003495-200565160-00010. [DOI] [PubMed] [Google Scholar]
- 118.Piliero P.J. Atazanavir: A novel once-daily protease inhibitor. Drugs Today. 2004;40(11):901–912. doi: 10.1358/dot.2004.40.11.872579. [DOI] [PubMed] [Google Scholar]
- 119.Piliero P.J. Atazanavir: a novel HIV-1 protease inhibitor. Expert Opin. Invest. Drugs. 2002;11(9):1295–1301. doi: 10.1517/13543784.11.9.1295. [DOI] [PubMed] [Google Scholar]
- 120.Kempf, D. J.; Norbeck, D. W.; Sham, H. L.; Zhao, C.; Sowin, T. J.; Reno, D. S.; Haight, A. R.; Cooper, A. J. Preparation of peptide analogs as retroviral protease inhibitors, WO 9414436 (1994).
- 121.Kempf D.J., Sham H.L., Marsh K.C., Flentge C.A., Betebenner D., Green B.E., McDonald E., Vasavanonda S., Saldivar A., Wideburg N.E., Kati W.M., Ruiz L., Zhao C., Fino L.M., Patterson J., Molla A., Plattner J.J., Norbeck D.W. Discovery of ritonavir, a potent inhibitor of HIV protease with high oral bioavailability and clinical efficacy. J. Med. Chem. 1998;41(4):602–617. doi: 10.1021/jm970636+. [DOI] [PubMed] [Google Scholar]
- 122.Stuk, T. L.; Allen, M. S.; Haight, A. R.; Kerdesky, F. A.; Langridge, D. C.; Leanna, M. R.; Lijewski, L. M.; Melcher, L.; Morton, H. E.; Robbins, T. A.; Sowin, T. J. Process for the stereoselective preparation of a substituted 2,5-diamino-3-hydroxyhexane as an intermediate for HIV protease inhibitors, US 5491253 (1996).
- 123.Haight, A. R.; Goodmonson, O. J.; Parekh, S. I.; Robbins, T. A.; Seif, L. S. Process for the preparation of a phenyl-disubstituted 2,5-diamino-3-hydroxyhexane, WO 9604232 (1996).
- 124.Tien, J J.; Menzia, J. A.; Cooper, A. J. Process for the preparation of HIV protease inhibiting peptide analogs, US 5567823 (1996).
- 125.Adamo I., Benedetti F., Berti F., Campaner P. Stereoselective hydroazidation of amino enones: synthesis of the ritonavir/lopinavir core. Org. Lett. 2006;8(1):51–54. doi: 10.1021/ol0524104. [DOI] [PubMed] [Google Scholar]
- 126.Cheng, Y.; Tanaka, H.; Baba, M. Preparation of 2′,3′-dideoxy and 2′,3′-didehydro nucleoside analogs as prodrugs for treating viral infections, most notably HIV, US 20040167096 (2004).
- 127.Bellani, P.; Frigerio, M.; Castoldi, P. A process for the synthesis of ritonavir, WO 2001021603 (2001).
- 128.Kempf D.J., Marsh K.C., Denissen J.F., McDonald E., Vasavanonda S., Flentge C.A., Green B.E., Fino L., Park C.H., Kong X.P. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc. Natl. Acad. Sci. U. S. A. 1995;92(7):2484–2488. doi: 10.1073/pnas.92.7.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lea A.P., Faulds D. Ritonavir. Drugs. 1996;52(4):541–546. doi: 10.2165/00003495-199652040-00007. discussion 547–548. [DOI] [PubMed] [Google Scholar]
- 130.Hoetelmans R.M.W., Meenhorst P.L., Mulder J.W., Burger D.M., Koks C.H.W., Beijnen J.H. Clinical pharmacology of HIV protease inhibitors: focus on saquinavir, indinavir, and ritonavir. Pharm. World Sci. 1997;19(4):159–175. doi: 10.1023/a:1008629608556. [DOI] [PubMed] [Google Scholar]
- 131.Hull M.W., Montaner J.S.G. Ritonavir-boosted protease inhibitors in HIV therapy. Ann. Med. 2011;43(5):375–388. doi: 10.3109/07853890.2011.572905. [DOI] [PubMed] [Google Scholar]
- 132.Sahali S., Chaix M.-L., Delfraissy J.-F., Ghosn J. Ritonavir-boosted protease inhibitor monotherapy for the treatment of HIV-1 infection. AIDS Rev. 2008;10(1):4–14. [PubMed] [Google Scholar]
- 133.Cooper C.L., van Heeswijk R.P.G., Gallicano K., Cameron D.W. A review of low-dose ritonavir in protease inhibitor combination therapy. Clin. Infect. Dis. 2003;36(12):1585–1592. doi: 10.1086/375233. [DOI] [PubMed] [Google Scholar]
- 134.Sham, H. L.; Norbeck, D. W.; Chen, X.; Betebenner, D. A. Preparation of peptide analogs as retroviral protease inhibitors, US 5914332 (1999).
- 135.Stoner E.J., Cooper A.J., Dickman D.A., Kolaczkowski L., Lallaman J.E., Liu J.-H., Oliver-Shaffer P.A., Patel K.M., Paterson J.B., Jr., Plata D.J., Riley D.A., Sham H.L., Stengel P.J., Tien J.-H.J. Synthesis of HIV protease inhibitor ABT-378 (Lopinavir) Org. Process Res. Dev. 2000;4(4):264–269. [Google Scholar]
- 136.Stoner E.J., Stengel P.J., Cooper A.J. Synthesis of ABT-378, an HIV protease inhibitor candidate: avoiding the use of carbodiimides in a difficult peptide coupling. Org. Process Res. Dev. 1999;3(2):145–148. [Google Scholar]
- 137.Stuk T.L., Haight A.R., Scarpetti D., Allen M.S., Menzia J.A., Robbins T.A., Parekh S.I., Langridge D.C., Tien J.-H.J., Pariza R.J., Kerdesky F.A. An efficient stereocontrolled strategy for the synthesis of hydroxyethylene dipeptide isosteres. J. Org. Chem. 1994;59(15):4040–4041. [Google Scholar]
- 138.Haight A.R., Stuk T.L., Menzia J.A., Robbins T.A. A convenient synthesis of enaminones using tandem acetonitrile condensation. Grignard addition, Tetrahedron Lett. 1997;38(24):4191–4194. [Google Scholar]
- 139.Nunes E.P., Santini de Oliveira M., Grinsztejn B. Lopinavir: the old champion. Future Virol. 2011;6(5):561–570. [Google Scholar]
- 140.Hurst M., Faulds D. Lopinavir. Drugs. 2000;60(6):1371–1379. doi: 10.2165/00003495-200060060-00009. [DOI] [PubMed] [Google Scholar]
- 141.Cvetkovic R.S., Goa K.L. Lopinavir/ritonavir: a review of its use in the management of HIV infection. Drugs. 2003;63(8):769–802. doi: 10.2165/00003495-200363080-00004. [DOI] [PubMed] [Google Scholar]
- 142.Chandwani A., Shuter J. Lopinavir/ritonavir in the treatment of HIV-I infection: a review. Ther. Clin. Risk Manage. 2008;4(5):1023–1033. doi: 10.2147/tcrm.s3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Oldfield V., Plosker G.L. Lopinavir/ritonavir: a review of its use in the management of HIV infection. Drugs. 2006;66(9):1275–1299. doi: 10.2165/00003495-200666090-00012. [DOI] [PubMed] [Google Scholar]
- 144.Schaeffer H.J., Beauchamp L., De Miranda P., Elion G.B., Bauer D.J., Collins P. 9-(2-Hydroxyethoxymethyl)guanine activity against viruses of the herpes group. Nature (London, U. K.) 1978;272(5654):583–585. doi: 10.1038/272583a0. [DOI] [PubMed] [Google Scholar]
- 145.Schaeffer, H. J. Compositions for treating viral infections and guanine acyclic nucleosides, US 4199574 (1980).
- 146.Keyser G.E., Bryant J.D., Barrio J.R. Iodomethyl ethers from 1,3-dioxolane and 1,3-oxathiolane: preparation of acyclic nucleoside analogs. Tetrahedron Lett. 1979;35:3263–3264. [Google Scholar]
- 147.Barrio J.R., Bryant J.D., Keyser G.E. A direct method for the preparation of 2-hydroxyethoxymethyl derivatives of guanine, adenine, and cytosine. J. Med. Chem. 1980;23(5):572–574. doi: 10.1021/jm00179a020. [DOI] [PubMed] [Google Scholar]
- 148.Matsumoto H., Kaneko C., Yamada K., Takeuchi T., Mori T., Mizuno Y. A convenient synthesis of 9-[(2-hydroxyethoxy)methyl]guanine (acyclovir) and related compounds. Chem. Pharm. Bull. 1988;36(3):1153–1155. doi: 10.1248/cpb.36.1153. [DOI] [PubMed] [Google Scholar]
- 149.Beauchamp L.M., Dolmatch B.L., Schaeffer H.J., Collins P., Bauer D.J., Keller P.M., Fyfe J.A. Modifications on the heterocyclic base of acyclovir: syntheses and antiviral properties. J. Med. Chem. 1985;28(8):982–987. doi: 10.1021/jm00146a002. [DOI] [PubMed] [Google Scholar]
- 150.Stimac A., Kobe J. A new synthesis of acyclovir prodrugs. N2-acetylacyclovir and 6-deoxyacyclovir. Synthesis. 1990;6:461–464. [Google Scholar]
- 151.Gao H., Mitra A.K. Synthesis of acyclovir ganciclovir and their prodrugs: a review. Synthesis. 2000;3:329–351. [Google Scholar]
- 152.Chu C.K., Cutler S.J. Chemistry and antiviral activities of acyclonucleosides. J. Het. Chem. 1986;23(2):289–319. [Google Scholar]
- 153.Kelley J.L., Schaeffer H.J. Purine acyclic nucleosides. Unambiguous synthesis of acyclovir via a furazano[3,4-d]pyrimidine. J. Heterocycl. Chem. 1986;23(1):271–273. [Google Scholar]
- 154.Taylor E.C., Beardsley G.P., Maki Y. New, general synthesis of 2-, 8-, and 9-substituted adenines. J. Org. Chem. 1971;36(21):3211–3217. doi: 10.1021/jo00820a029. [DOI] [PubMed] [Google Scholar]
- 155.Laskin O.L. Acyclovir. Pharmacology and clinical experience. Arch. Intern. Med. 1984;144(6):1241–1246. doi: 10.1001/archinte.144.6.1241. [DOI] [PubMed] [Google Scholar]
- 156.Richards D.M., Carmine A.A., Brogden R.N., Heel R.C., Speight T.M., Avery G.S. Acyclovir. A review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1983;26(5):378–438. doi: 10.2165/00003495-198326050-00002. [DOI] [PubMed] [Google Scholar]
- 157.Krenitsky, T. A.; Beauchamp, L. M. Acycloxic esters with valine and isoleucine as herpes virucides, EP 308065 (1989).
- 158.Beauchamp L.M., Krenitsky T.A. Acyclovir prodrugs: the road to valaciclovir. Drugs Future. 1993;18:619–628. [Google Scholar]
- 159.Prasada Raju V.V.N.K.V., Vedantham R., Khunt M.D., Mathad V.T., Dubey P.K., Chakravarthy A.K. An efficient and large scale process for synthesis of valacyclovir. Asian J. Chem. 2010;22(5):4092–4098. [Google Scholar]
- 160.Etinger, M. Y.; Yudovich, L. M.; Yuzefovich, M.; Nisnevich, G. A.; Dolitzki, B. Z.; Pertsikov, B.; Tishin, B.; Blasberger, D. Synthesis and purification of valaciclovir, WO 2003041647 (2003).
- 161.Acosta E.P., Fletcher C.V. Valaciclovir. Ann. Pharmacother. 1997;31(2):185–191. doi: 10.1177/106002809703100211. [DOI] [PubMed] [Google Scholar]
- 162.Beutner K.R. Valaciclovir: a review of its antiviral activity, pharmacokinetic properties, and clinical efficacy. Antiviral Res. 1995;28(4):281–290. doi: 10.1016/0166-3542(95)00066-6. [DOI] [PubMed] [Google Scholar]
- 163.Smiley M.L., Murray A., De Miranda P. Valaciclovir HCl (Valtrex): an acyclovir prodrug with improved pharmacokinetics and better efficacy for treatment of zoster. Adv. Exp. Med. Biol. 1996;394(Antiviral Chemotherapy 4):33–39. doi: 10.1007/978-1-4757-9209-6_4. [DOI] [PubMed] [Google Scholar]
- 164.Antman M.D., Gudmundsson O.S. Valaciclovir: a prodrug of acyclovir. Biotechnol.: Pharm. Aspects. 2007;5(Pt. 2, Prodrugs: Challenges and Rewards):669–676. [Google Scholar]
- 165.Tyring S.K., Baker D., Snowden W. Valaciclovir for herpes simplex virus infection: long-term safety and sustained efficacy after 20 years’ experience with acyclovir. J. Infect. Dis. 2002;186(Suppl. 1):S40–S46. doi: 10.1086/342966. [DOI] [PubMed] [Google Scholar]
- 166.Perry C.M., Faulds D. Valaciclovir: a review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in herpesvirus infections. Drugs. 1996;52(5):754–772. doi: 10.2165/00003495-199652050-00009. [DOI] [PubMed] [Google Scholar]
- 167.Verheyden, J. P. H.; Martin, J. C. 9-(1,3-Dihydroxy-2-propoxymethyl)guanine as antiviral agent, US 4423050 (1983). [DOI] [PMC free article] [PubMed]
- 168.Martin J.C., Dvorak C.A., Smee D.F., Matthews T.R., Verheyden J.P.H. 9-(1,3-Dihydroxy-2-propoxymethyl)guanine: a new potent and selective antiherpes agent. J. Med. Chem. 1983;26(5):759–761. doi: 10.1021/jm00359a023. [DOI] [PubMed] [Google Scholar]
- 169.Ogilvie K.K., Cheriyan U.O., Radatus B.K., Smith K.O., Galloway K.S., Kennell W.L. Biologically active acyclonucleoside analogs. II. The synthesis of 9-[[2-hydroxy-1-(hydroxymethyl)ethoxy]methyl]guanine (BIOLF-62) Can. J. Chem. 1982;60(24):3005–3010. [Google Scholar]
- 170.Field A.K., Davies M.E., DeWitt C., Perry H.C., Liou R., Germershausen J., Karkas J.D., Ashton W.T., Johnston D.B.R., Tolman R.L. 9-{[2-Hydroxy-1-(hydroxymethyl)ethoxy]methyl}guanine: a selective inhibitor of herpes group virus replication. Proc. Natl. Acad. Sci. U. S. A. 1983;80(13):4139–4143. doi: 10.1073/pnas.80.13.4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ashton W.T., Karkas J.D., Field A.K., Tolman R.L. Activation by thymidine kinase and potent antiherpetic activity of 2′-nor-2′-deoxyguanosine (2′NDG) Biochem. Biophys. Res. Commun. 1982;108(4):1716–1721. doi: 10.1016/s0006-291x(82)80109-5. [DOI] [PubMed] [Google Scholar]
- 172.McGee D.P.C., Martin J.C., Verheyden J.P.H. Synthesis of 9-[(1,3-dihydroxy-2-propoxy)methyl]guanine (DHPG) via condensation of N2,9-diacetylguanine with a sulfinylmethyl ether. Synth. Commun. 1988;18(14):1651–1660. [Google Scholar]
- 173.Nestor, J. J.; Womble, S. W.; Maag, H. 2-(2-Amino-1,6-dihydro-6-oxo-purin-9-yl)methoxy-1,3-propanediol L-monovaline ester [ganciclovir valine ester] as an antiviral with improved oral absorption, EP 694547 (1996).
- 174.Ramchandra, R. D.; Narayanrao, K. R.; Purushottam, P. V. Preparation of valganciclovir, EP 1837336 (2007).
- 175.Arzeno, H. B.; Humphreys, E. R.; Wong, J.; Roberts, C. R. Process for preparing 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)methoxy-1,3-propanediol (ganciclovir) mono-L-valinate ester, US 5840890 (1988).
- 176.Arzeno, H. B.; Humphreys, E. R. Preparation of L-monovaline ester of purine acyclic nucleosides as virucides, WO 9727196 (1997).
- 177.Dvorak, C. A.; Wren, D. L.; Fisher, L. E.; Axt, S. D.; Humphreys, E. R.; A., Humberto B.; Beard, C. C.; Nguyen, S. L.; Han, Y-K.; Roberts, C. R.; Lund, J. P.; Fatheree, P. R. Process for preparing 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)methoxy-1,3-propanediol (ganciclovir) mono-L-valinate ester, US 6040446 (2000).
- 178.Sharma, M. K.; Raina, S.; Panda, A. K.; Kumar, Y.; Khanduri, C. H. Preparation of ganciclovir mono-N-benzyloxycarbonyl-L-valinate ester, WO 2005092891 (2005).
- 179.Sorbera L.A., Castaner R., Castaner J. Valganciclovir hydrochloride. Drugs Future. 2000;25(5):474–480. [Google Scholar]
- 180.Arzeno, H. B.; Beard, C. C.; Fisher, L. E.; Prince, A. Preparation of L-monovaline ester of ganciclovir purine acyclic nucleosides as virucides, WO 9727194 (1970).
- 181.Arzeno, H. B. Preparation of L-monovaline ester of ganciclovir purine acyclic nucleosides as virucides, WO 9727195 (1997).
- 182.Arzeno, H. B.; Humphreys, E. R.; Wong, J-W.; Roberts, C. R. Preparation of L-monovaline ester of ganciclovir purine acyclic nucleosides as virucides, WO 9727197 (1997).
- 183.Arzeno, H. B.; Axt, S. D.; Beard, C. C.; Dvorak, C. A.; Fatheree, P. R.; Fisher, L. E.; Han, Y-K.; Humphreys, E. R.; Lund, J. P.; Nguyen, S. L.; Wren, D. L. Preparation of L-monovaline ester of purine acyclic nucleosides as virucides, WO 9727198 (1997).
- 184.Martin J.C., Tippe M.A., McGee D.P.C., Verheyden J.P.H. Synthesis and antiviral activity of various esters of 9-[(1,3-dihydroxy-2-propoxy)methyl]guanine. J. Pharm. Sci. 1987;76(2):180–184. doi: 10.1002/jps.2600760221. [DOI] [PubMed] [Google Scholar]
- 185.Curran M., Noble S. Valganciclovir. Drugs. 2001;61(8):1145–1150. doi: 10.2165/00003495-200161080-00013. [DOI] [PubMed] [Google Scholar]
- 186.Cvetkovic R.S., Wellington K. Valganciclovir: a review of its use in the management of CMV infection and disease in immunocompromised patients. Drugs. 2005;65(6):859–878. doi: 10.2165/00003495-200565060-00012. [DOI] [PubMed] [Google Scholar]
- 187.Razonable R.R., Paya C.V. Valganciclovir for the prevention and treatment of cytomegalovirus disease in immunocompromised hosts. Expert Rev. Anti-Infect. Ther. 2004;2(1):27–42. doi: 10.1586/14787210.2.1.27. [DOI] [PubMed] [Google Scholar]
- 188.Cocohoba J.M., McNicholl I.R. Valganciclovir: an advance in cytomegalovirus therapeutics. Ann. Pharmacother. 2002;36(6):1075–1079. doi: 10.1345/aph.1A393. [DOI] [PubMed] [Google Scholar]
- 189.Reusser P. Oral valganciclovir: a new option for treatment of cytomegalovirus infection and disease in immunocompromised hosts. Expert Opin. Invest. Drugs. 2001;10(9):1745–1753. doi: 10.1517/13543784.10.9.1745. [DOI] [PubMed] [Google Scholar]
- 190.Pescovitz M.D. Valganciclovir: recent progress. Am. J. Transplant. 2010;10(6):1359–1364. doi: 10.1111/j.1600-6143.2010.03112.x. [DOI] [PubMed] [Google Scholar]
- 191.Perrottet N., Decosterd L.A., Meylan P., Pascual M., Biollaz J., Buclin T. Valganciclovir in adult solid organ transplant recipients. Clin. Pharmacokinet. 2009;48(6):399–418. doi: 10.2165/00003088-200948060-00006. [DOI] [PubMed] [Google Scholar]
- 192.Maag H. Valganciclovir: a prodrug of ganciclovir. Biotechnol.: Pharm. Aspects. 2007;5(Pt. 2, Prodrugs: Challenges and Rewards, Part 2):677–686. [Google Scholar]
- 193.Balzarini J., De Clercq E. Nucleoside and nucleotide reverse transcriptase inhibitors. In: De Clercq E.D.A., editor. Antiretroviral Therapy. American Society Microbiolgy; 2001. pp. 31–62. [Google Scholar]
- 194.De Clercq E. New developments in anti-HIV chemotherapy. Biochim. Biophys. Acta, Mol. Basis Dis. 2002;1587(2-3):258–275. doi: 10.1016/s0925-4439(02)00089-3. [DOI] [PubMed] [Google Scholar]
- 195.Cihlar T., Ray A.S. Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine. Antiviral Res. 2010;85(1):39–58. doi: 10.1016/j.antiviral.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 196.Waters L., Boffito M. Pharmacology of current and investigational human immunodeficiency virus (HIV) Nucleoside/nucleotide reverse transcriptase inhibitors in adults. Anti-Infect. Agents Med. Chem. 2007;6(3):213–221. [Google Scholar]
- 197.Back D.J., Burger D.M., Flexner C.W., Gerber J.G. The pharmacology of antiretroviral nucleoside and nucleotide reverse transcriptase inhibitors: implications for once-daily dosing. JAIDS, J. Acquired Immune Defic. Syndr. 2005;39(Suppl. 1):S1–S23. doi: 10.1097/01.qai.0000168882.67942.3f. [DOI] [PubMed] [Google Scholar]
- 198.Das K., Arnold E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr. Opin. Virol. 2013;3(2):111–118. doi: 10.1016/j.coviro.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Das K., Arnold E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 2. Curr. Opin. Virol. 2013;3(2):119–128. doi: 10.1016/j.coviro.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Holy A., Rosenberg I. Preparation of 5′-O-phosphonylmethyl analogs of nucleoside-5′-phosphates, 5′-diphosphates and 5′-triphosphates. Coll. Czech. Chem. Comm. 1982;47(12):3447–3463. [Google Scholar]
- 201.Holy A., Dvorakova H., Masojidkova M. Synthesis of enantiomeric N-(2-phosphonomethoxy-propyl) derivatives of purine and pyrimidine bases. II. The synthon approach. Coll. Czech. Chem. Comm. 1995;60(8):1390–1409. [Google Scholar]
- 202.Holy A., Masojidkova M. Synthesis of enantiomeric N-(2-phosphonomethoxypropyl) derivatives of purine and pyrimidine bases. I. The stepwise approach. Coll. Czech. Chem. Comm. 1995;60(7):1196–1212. [Google Scholar]
- 203.Schultze L.M., Chapman H.H., Dubree N.J.P., Jones R.J., Kent K.M., Lee T.T., Louie M.S., Postich M.J., Prisbe E.J., Rohloff J.C., Yu R.H. Practical synthesis of the anti-HIV drug. PMPA, Tetrahedron Lett. 1998;39(14):1853–1856. [Google Scholar]
- 204.Barral K., Priet S., Sire J., Neyts J., Balzarini J., Canard B., Alvarez K. Synthesis, in vitro antiviral evaluation, and stability studies of novel α-borano-nucleotide analogues of 9-[2-(phosphonomethoxy)ethyl]adenine and (r)-9-[2-(phosphonomethoxy)propyl]adenine. J. Med. Chem. 2006;49(26):7799–7806. doi: 10.1021/jm060030y. [DOI] [PubMed] [Google Scholar]
- 205.Ripin D.H.B., Teager D.S., Fortunak J., Basha S.M., Bivins N., Boddy C.N., Byrn S., Catlin K.K., Houghton S.R., Jagadeesh S.T., Kumar K.A., Melton J., Muneer S., Rao L.N., Rao R.V., Ray P.C., Reddy N.G., Reddy R.M., Shekar K.C., Silverton T., Smith D.T., Stringham R.W., Subbaraju G.V., Talley F., Williams A. Process improvements for the manufacture of tenofovir disoproxil fumarate at commercial scale. Org. Process Res. Dev. 2010;14(5):1194–1201. [Google Scholar]
- 206.De Clercq E. Discovery and development of tenofovir disoproxil fumarate. In: Kazmierski W.M., editor. Antiviral Drugs: From Basic Discovery Through Clinical Trials. Wiley; 2011. pp. 85–101. [Google Scholar]
- 207.Kearney B.P., Flaherty J.F., Shah J. Tenofovir disoproxil fumarate: clinical pharmacology and pharmacokinetics. Clin. Pharmacokinet. 2004;43(9):595–612. doi: 10.2165/00003088-200443090-00003. [DOI] [PubMed] [Google Scholar]
- 208.Gallant J.E., Deresinski S. Tenofovir disoproxil fumarate. Clin. Infect. Dis. 2003;37(7):944–950. doi: 10.1086/378068. [DOI] [PubMed] [Google Scholar]
- 209.Chapman T.M., McGavin J.K., Noble S. Tenofovir disoproxil fumarate. Drugs. 2003;63(15):1597–1608. doi: 10.2165/00003495-200363150-00006. [DOI] [PubMed] [Google Scholar]
- 210.Grim S.A., Romanelli F. Tenofovir disoproxil fumarate. Ann. Pharmacother. 2003;37(6):849–859. doi: 10.1345/aph.1C388. [DOI] [PubMed] [Google Scholar]
- 211.Fung H.B., Stone E.A., Piacenti F.J. Tenofovir disoproxil fumarate: a nucleotide reverse transcriptase inhibitor for the treatment of HIV infection. Clin. Ther. 2002;24(10):1515–1548. doi: 10.1016/s0149-2918(02)80058-3. [DOI] [PubMed] [Google Scholar]
- 212.De Clercq E. Tenofovir: quo vadis anno 2012 (where is it going in the year 2012)? Med. Res. Rev. 2012;32(4):765–785. doi: 10.1002/med.21267. [DOI] [PubMed] [Google Scholar]
- 213.Celum C., Baeten J.M. Tenofovir-based pre-exposure prophylaxis for HIV prevention: evolving evidence. Curr. Opin. Infect. Dis. 2012;25(1):51–57. doi: 10.1097/QCO.0b013e32834ef5ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Perry C.M., Simpson D. Tenofovir disoproxil fumarate: in chronic hepatitis B. Drugs. 2009;69(16):2245–2256. doi: 10.2165/10482940-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 215.Buti M., Homs M. Tenofovir disoproxil fumarate in the treatment of chronic hepatitis B. Expert Rev. Gastroenterol. Hepatol. 2012;6(4):413–421. doi: 10.1586/egh.12.19. [DOI] [PubMed] [Google Scholar]
- 216.Jenh A.M., Thio C.L., Pham P.A. Tenofovir for the treatment of hepatitis B virus. Pharmacotherapy. 2009;29(10):1212–1227. doi: 10.1592/phco.29.10.1212. [DOI] [PubMed] [Google Scholar]
- 217.Pham P.A., Gallant J.E. Tenofovir disoproxil fumarate for the treatment of HIV infection. Expert Opin. Drug Metab. Toxicol. 2006;2(3):459–469. doi: 10.1517/17425255.2.3.459. [DOI] [PubMed] [Google Scholar]
- 218.Lyseng-Williamson K.A., Reynolds N.A., Plosker G.L. Tenofovir disoproxil fumarate: a review of its use in the management of HIV infection. Drugs. 2005;65(3):413–432. doi: 10.2165/00003495-200565030-00006. [DOI] [PubMed] [Google Scholar]
- 219.Foggia M., Nappa S., Bonadies G., Cotugno M., Di Filippo G., Borrelli F., Orlando R., Borgia G. Tenofovir disoproxil fumarate in the clinical practice: an overview. Anti-Infect. Agents Med. Chem. 2008;7(4):285–295. [Google Scholar]
- 220.James C., Preininger L., Sweet M. Rilpivirine: a second-generation nonnucleoside reverse transcriptase inhibitor. Am. J. Health-Syst. Pharm. 2012;69(10):857–861. doi: 10.2146/ajhp110395. [DOI] [PubMed] [Google Scholar]
- 221.Sahlberg C., Zhou X.-X. Development of non-nucleoside reverse transcriptase inhibitor inhibitors for anti-HIV therapy. Anti-Infect. Agents Med. Chem. 2008;7(2):101–117. [Google Scholar]
- 222.Pedersen O.S., Pedersen E.B. Non-nucleoside reverse transcriptase inhibitors: the NNRTI boom. Antiviral Chem. Chemother. 1999;10(6):285–314. doi: 10.1177/095632029901000601. [DOI] [PubMed] [Google Scholar]
- 223.Zhan P., Liu X. Novel HIV-1 non-nucleoside reverse transcriptase inhibitors: a patent review (2005 - 2010) Expert Opin. Ther. Pat. 2011;21(5):717–796. doi: 10.1517/13543776.2011.568481. [DOI] [PubMed] [Google Scholar]
- 224.Pauwels R., Andries K., Debyser Z., Van Daele P., Schols D., Stoffels P., De Vreese K., Woestenborghs R., Vandamme A.M., Janssen C.G.M., Cauwenbergh J.A.G., Desmyter J., Heykants J., Janssen M.A.C., De Clercq E., Janssen P.A.J. Potent and highly selective human immunodeficiency virus type 1 (HIV-1) inhibition by a series of α-anilinophenylacetamide derivatives targeted at HIV-1 reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 1993;90(5):1711–1715. doi: 10.1073/pnas.90.5.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Buckheit K.W., Yang L., Buckheit R.W., Jr. Development of dual-acting pyrimidinediones as novel and highly potent topical anti-HIV microbicides. Antimicrob. Agents Chemother. 2011;55(11):5243–5254. doi: 10.1128/AAC.05237-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Ludovici D.W., Kukla M.J., Grous P.G., Krishnan S., Andries K., de Bethune M.-P., Azijn H., Pauwels R., De Clercq E., Arnold E., Janssen P.A.J. Evolution of anti-HIV drug candidates. Part 1: From α-Anilinophenylacetamide (α-APA) to imidoyl thiourea (ITU) Bioorg. Med. Chem. Lett. 2001;11(17):2225–2228. doi: 10.1016/s0960-894x(01)00410-3. [DOI] [PubMed] [Google Scholar]
- 227.Ludovici D.W., Kavash R.W., Kukla M.J., Ho C.Y., Ye H., De Corte B.L., Andries K., de Bethune M.-P., Azijn H., Pauwels R., Moereels H.E.L., Heeres J., Koymans L.M.H., de Jonge M.R., Van Aken K.J.A., Daeyaert F.F.D., Lewi P.J., Das K., Arnold E., Janssen P.A.J. Evolution of anti-HIV drug candidates. Part 2: Diaryltriazine (DATA) analogues. Bioorg. Med. Chem. Lett. 2001;11(17):2229–2234. doi: 10.1016/s0960-894x(01)00411-5. [DOI] [PubMed] [Google Scholar]
- 228.De Clercq E. Perspectives of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Farmaco. 1999;54(1-2):26–45. doi: 10.1016/s0014-827x(98)00103-7. [DOI] [PubMed] [Google Scholar]
- 229.Yasuda N., Tan L. Efavirenz, a non-nucleoside reverse transcriptase inhibitor (NNRTI), and a previous structurally related development candidate. In: Yasuda N., editor. The Art of Process Chemistry. 2011. pp. 1–43. [Google Scholar]
- 230.Rakhmanina N.Y., van den Anker J.N. Efavirenz in the therapy of HIV infection. Expert Opin. Drug Metab. Toxicol. 2010;6(1):95–103. doi: 10.1517/17425250903483207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Best B.M., Goicoechea M. Efavirenz—still first-line king? Expert Opin. Drug Metab. Toxicol. 2008;4(7):965–972. doi: 10.1517/17425255.4.7.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Adkins J.C., Noble S. Efavirenz. Drugs. 1998;56(6):1055–1064. doi: 10.2165/00003495-199856060-00014. [DOI] [PubMed] [Google Scholar]
- 233.Adams W.J., Aristoff P.A., Jensen R.K., Morozowich W., Romero D.L., Schinzer W.C., Tarpley W.G., Thomas R.C. Discovery and development of the BHAP nonnucleoside reverse transcriptase inhibitor delavirdine mesylate. Pharm. Biotechnol. 1998;11:285–312. doi: 10.1007/0-306-47384-4_13. [DOI] [PubMed] [Google Scholar]
- 234.Freimuth W.W. Delavirdine mesylate, a potent non-nucleoside HIV-1 reverse transcriptase inhibitor. Adv. Exp. Med. Biol. 1996;394(4):279–289. doi: 10.1007/978-1-4757-9209-6_25. [DOI] [PubMed] [Google Scholar]
- 235.Scott L.J., Perry C.M. Delavirdine: a review of its use in HIV infection. Drugs. 2000;60(6):1411–1444. doi: 10.2165/00003495-200060060-00013. [DOI] [PubMed] [Google Scholar]
- 236.Murphy R.L., Montaner J. Nevirapine: a review of its development, pharmacological profile and potential for clinical use. Expert Opin. Invest. Drugs. 1996;5(9):1183–1199. [Google Scholar]
- 237.Grozinger K., Proudfoot J., Hargrave K. Discovery and development of nevirapine. In: Chorghade M.S., editor. Vol. 1. Wiley-Interscience; 2006. pp. 353–363. (Drug Discovery and Development). [Google Scholar]
- 238.Milinkovic A., Martinez E. Nevirapine in the treatment of HIV. Expert Rev. Anti-Infect. Ther. 2004;2(3):367–373. doi: 10.1586/14787210.2.3.367. [DOI] [PubMed] [Google Scholar]
- 239.Podzamczer D., Fumero E. The role of nevirapine in the treatment of HIV-1 disease. Expert Opin. Pharmacother. 2001;2(12):2065–2078. doi: 10.1517/14656566.2.12.2065. [DOI] [PubMed] [Google Scholar]
- 240.Schiller D.S., Youssef-Bessler M. Etravirine: a second-generation nonnucleoside reverse transcriptase inhibitor (NNRTI) active against NNRTI-resistant strains of HIV. Clin. Ther. 2009;31(4):692–704. doi: 10.1016/j.clinthera.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 241.Kakuda T.N., Scholler-Gyure M., Hoetelmans R.M.W. Clinical perspective on antiretroviral drug-drug interactions with the non-nucleoside reverse transcriptase inhibitor etravirine. Antiviral Ther. 2010;15(6):817–829. doi: 10.3851/IMP1652. [DOI] [PubMed] [Google Scholar]
- 242.Janssen P.A.J., Lewi P.J., Arnold E., Daeyaert F., de Jonge M., Heeres J., Koymans L., Vinkers M., Guillemont J., Pasquier E., Kukla M., Ludovici D., Andries K., de Bethune M.-P., Pauwels R., Das K., Clark A.D., Jr., Frenkel Y.V., Hughes S.H., Medaer B., De Knaep F., Bohets H., De Clerck F., Lampo A., Williams P., Stoffels P. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1e)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2- pyrimidinyl]amino]benzonitrile (R27(8474), rilpivirine) J. Med. Chem. 2005;48(6):1901–1909. doi: 10.1021/jm040840e. [DOI] [PubMed] [Google Scholar]
- 243.Sanford M. Rilpivirine. Drugs. 2012;72(4):525–541. doi: 10.2165/11208590-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 244.Fernandez-Montero J.V., Vispo E., Anta L., de Mendoza C., Soriano V. Rilpivirine: a next-generation non-nucleoside analogue for the treatment of HIV infection. Expert Opin. Pharmacother. 2012;13(7):1007–1014. doi: 10.1517/14656566.2012.667802. [DOI] [PubMed] [Google Scholar]
- 245.Garvey L., Winston A. Rilpivirine: a novel non-nucleoside reverse transcriptase inhibitor. Expert Opin. Invest. Drugs. 2009;18(7):1035–1041. doi: 10.1517/13543780903055056. [DOI] [PubMed] [Google Scholar]
- 246.Ripamonti D., Bombana E., Rizzi M. Rilpivirine: drug profile of a second-generation non-nucleoside reverse transcriptase HIV-inhibitor. Expert Rev. Anti-Infect. Ther. 2014;12(1):13–29. doi: 10.1586/14787210.2014.863708. [DOI] [PubMed] [Google Scholar]
- 247.Zaharatos G.J., Wainberg M.A. Update on rilpivirine: A new potent non-nucleoside reverse transcriptase inhibitor (NNRTI) of HIV replication. Ann. Med. 2013;45(3):236–241. doi: 10.3109/07853890.2012.732704. [DOI] [PubMed] [Google Scholar]
- 248.Schafer J.J., Short W.R. Rilpivirine, a novel non-nucleoside reverse transcriptase inhibitor for the management of HIV-1 infection: a systematic review. Antiviral Ther. 2012;17(8):1495–1502. doi: 10.3851/IMP2254. [DOI] [PubMed] [Google Scholar]
- 249.James C., Preininger L., Sweet M. Rilpivirine: a second-generation nonnucleoside reverse transcriptase inhibitor. Am. J. Health-Syst. Pharm. 2012;69(10):857–861. doi: 10.2146/ajhp110395. [DOI] [PubMed] [Google Scholar]
- 250.Platten M., Faetkenheuer G. Lersivirine-a new drug for HIV infection therapy. Expert Opin. Invest. Drugs. 2013;22(12):1687–1694. doi: 10.1517/13543784.2013.846325. [DOI] [PubMed] [Google Scholar]
- 251.Sorbera L.A., Castaner J., Bayes M. Capravirine. Drugs Future. 2003;28(12):1149–1158. [Google Scholar]
- 252.Deeks E.D. Emtricitabine/rilpivirine/tenofovir disoproxil fumarate single-tablet regimen: a review of its use in HIV infection. Drugs. 2014;74(17):2079–2095. doi: 10.1007/s40265-014-0318-1. [DOI] [PubMed] [Google Scholar]
- 253.Hazuda D., Iwamoto M., Wenning L. Emerging pharmacology: inhibitors of human immunodeficiency virus integration. Annu. Rev. Pharmacol. Toxicol. 2009;49:377–394. doi: 10.1146/annurev.pharmtox.011008.145553. [DOI] [PubMed] [Google Scholar]
- 254.Di Santo R. Inhibiting the HIV integration process: past, present, and the future. J. Med. Chem. 2014;57(3):539–566. doi: 10.1021/jm400674a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Johns B.A., Kawasuji T., Velthuisen E.J. HIV integrase inhibitors. RSC Drug Discovery Ser. 2013;32:149–188. [Google Scholar]
- 256.Metifiot M., Marchand C., Pommier Y. HIV integrase inhibitors: 20-year landmark and challenges. Adv. Pharmacol. (San Diego, CA, U. S.) 2013;67(Antiviral Agents):75–105. doi: 10.1016/B978-0-12-405880-4.00003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Dayam R., Gundla R., Al-Mawsawi L.Q., Neamati N. HIV-1 integrase inhibitors: 2005-2006 update. Med. Res. Rev. 2008;28(1):118–154. doi: 10.1002/med.20116. [DOI] [PubMed] [Google Scholar]
- 258.Hazuda D.J., Felock P., Witmer M., Wolfe A., Stillmock K., Grobler J.A., Espeseth A., Gabryelski L., Schleif W., Blau C., Miller M.D. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science (Washington, DC, U. S.) 2000;287:646–650. doi: 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- 259.Fujishita, T.; Yoshinaga, T.; Sato, A., Aromatic heterocycle compounds having HIV integrase inhibiting activities, WO2000039086 (2000).
- 260.Zhuang L., Wai J.S., Embrey M.W., Fisher T.E., Egbertson M.S., Payne L.S., Guare J.P., Jr., Vacca J.P., Hazuda D.J., Felock P.J., Wolfe A.L., Stillmock K.A., Witmer M.V., Moyer G., Schleif W.A., Gabryelski L.J., Leonard Y.M., Lynch J.J., Jr., Michelson S.R., Young S.D. Design and synthesis of 8-hydroxy-[1,6]-naphthyridines as novel inhibitors of HIV-1 integrase in vitro and in infected cells. J. Med. Chem. 2003;46:453–456. doi: 10.1021/jm025553u. [DOI] [PubMed] [Google Scholar]
- 261.Guare J.P., Wai J.S., Gomez R.P., Anthony N.J., Jolly S.M., Cortes A.R., Vacca J.P., Felock P.J., Stillmock K.A., Schleif W.A., Moyer G., Gabryelski L.J., Jin L., Chen I.W., Hazuda D.J., Young S.D. A series of 5-aminosubstituted 4-fluorobenzyl-8-hydroxy-[1,6]naphthyridine-7-carboxamide HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett. 2006;16:2900–2904. doi: 10.1016/j.bmcl.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 262.Hazuda D.J., Young S.D., Guare J.P., Anthony N.J., Gomez R.P., Wai J.S., Vacca J.P., Handt L., Motzel S.L., Klein H.G., Dornadula G., Danovich R.M., Witmer M.V., Wilson K.A., Tussey L., Schleif W.A., Gabryelski L.S., Jin L., Miller M.D., Casimiro D.R., Emini E.A., Shiver J.W. Integrase inhibitors and cellular immunity suppress retroviral replication in rhesus macaques. Science (Washington, DC, U. S.) 2004;305:528–532. doi: 10.1126/science.1098632. [DOI] [PubMed] [Google Scholar]
- 263.Summa V., Petrocchi A., Matassa V.G., Gardelli C., Muraglia E., Rowley M., Paz O.G., Laufer R., Monteagudo E., Pace P. 4,5-Dihydroxypyrimidine carboxamides and N-alkyl-5-hydroxypyrimidinone carboxamides are potent, selective HIV integrase inhibitors with good pharmacokinetic profiles in preclinical species. J. Med. Chem. 2006;49:6646–6649. doi: 10.1021/jm060854f. [DOI] [PubMed] [Google Scholar]
- 264.Anker M., Corales R.B. Raltegravir (MK-0518): a novel integrase inhibitor for the treatment of HIV infection. Expert Opin. Invest. Drugs. 2008;17:97–103. doi: 10.1517/13543784.17.1.97. [DOI] [PubMed] [Google Scholar]
- 265.Summa V., Petrocchi A., Bonelli F., Crescenzi B., Donghi M., Ferrara M., Fiore F., Gardelli C., Paz O.G., Hazuda D.J., Jones P., Kinzel O., Laufer R., Monteagudo E., Muraglia E., Nizi E., Orvieto F., Pace P., Pescatore G., Scarpelli R., Stillmock K., Witmer M.V., Rowley M. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 2008;51:5843–5855. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 266.Beare K.D., Coster M.J., Rutledge P.J. Diketoacid inhibitors of HIV-1 integrase: from L-708,906 to raltegravir and beyond. Curr. Med. Chem. 2012;19(8):1177–1192. doi: 10.2174/092986712799320565. [DOI] [PubMed] [Google Scholar]
- 267.Summa V., Pace P. Discovery and development of HIV integrase inhibitor raltegravir. In: Kazmierski W.M., editor. Antiviral Drugs: From Basic Discovery Through Clinical Trials. Wiley; 2011. pp. 181–195. [Google Scholar]
- 268.Rowley M. The discovery of raltegravir, an integrase inhibitor for the treatment of HIV infection. Prog. Med. Chem. 2008;46:1–28. doi: 10.1016/S0079-6468(07)00001-X. [DOI] [PubMed] [Google Scholar]
- 269.Evering T.H., Markowitz M. Raltegravir: an integrase inhibitor for HIV-1. Expert Opin. Invest. Drugs. 2008;17(3):413–422. doi: 10.1517/13543784.17.3.413. [DOI] [PubMed] [Google Scholar]
- 270.Hicks C., Gulick R.M. Raltegravir: the first HIV type 1 integrase inhibitor. Clin. Infect. Dis. 2009;48(7):931–939. doi: 10.1086/597290. [DOI] [PubMed] [Google Scholar]
- 271.Cocohoba J., Dong B.J. Raltegravir: the first HIV integrase inhibitor. Clin. Ther. 2008;30(10) doi: 10.1016/j.clinthera.2008.10.012. 2747–1765. [DOI] [PubMed] [Google Scholar]
- 272.Sayana S., Khanlou H. Raltegravir: the first in a new class of integrase inhibitors for the treatment of HIV. Expert Rev. Anti-Infect. Ther. 2008;6(4):419–426. doi: 10.1586/14787210.6.4.419. [DOI] [PubMed] [Google Scholar]
- 273.Temesgen Z., Siraj D.S. Raltegravir: first in class HIV integrase inhibitor. Ther. Clin. Risk Manage. 2008;4(2):493–500. doi: 10.2147/tcrm.s2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Croxtall J.D., Lyseng-Williamson K.A., Perry C.M. Raltegravir. Drugs. 2008;68(1):131–138. doi: 10.2165/00003495-200868010-00009. [DOI] [PubMed] [Google Scholar]
- 275.Calin R., Katlama C. The place of raltegravir in the clinical management of HIV-1 infection. Clin. Pract. (London, U. K.) 2013;10(4):427–438. [Google Scholar]
- 276.Rokas K.E.E., Brandon B.P., Shamroe C.L., Scott S.S., Millisor V.E., Bryant J.E., Weissman S.B. Role of raltegravir in HIV-1 management. Ann. Pharmacother. 2012;46(4):578–589. doi: 10.1345/aph.1Q616. [DOI] [PubMed] [Google Scholar]
- 277.Brainard D.M., Wenning L.A., Stone J.A., Wagner J.A., Iwamoto M. Clinical pharmacology profile of raltegravir, an HIV-1 integrase strand transfer inhibitor. J. Clin. Pharmacol. 2011;51(10):1375–1402. doi: 10.1177/0091270010387428. [DOI] [PubMed] [Google Scholar]
- 278.Okeke N.L., Hicks C. Role of raltegravir in the management of HIV-1 infection. HIV/AIDS. 2011;3:81–92. doi: 10.2147/HIV.S13985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Nunes E.P., Santini de Oliveira M., Grinsztejn B. Clinical use of raltegravir: a review. HIV Ther. 2010;4(5):531–542. [Google Scholar]
- 280.Burger D.M. Raltegravir: a review of its pharmacokinetics, pharmacology and clinical studies. Expert Opin. Drug Metab. Toxicol. 2010;6(9):1151–1160. doi: 10.1517/17425255.2010.513383. [DOI] [PubMed] [Google Scholar]
- 281.Evering T.H., Markowitz M. Raltegravir (MK-0518): an integrase inhibitor for the treatment of HIV-1. Drugs Today. 2007;43(12):865–877. doi: 10.1358/dot.2007.43.12.1146063. [DOI] [PubMed] [Google Scholar]
- 282.Shimura K., Kodama E.N. Elvitegravir: a new HIV integrase inhibitor. Antiviral Chem. Chemother. 2009;20(2):79–85. doi: 10.3851/IMP1397. [DOI] [PubMed] [Google Scholar]
- 283.Deeks E.D. Elvitegravir: a review of its use in adults with HIV-1 infection. Drugs. 2014;74(6):687–697. doi: 10.1007/s40265-014-0206-8. [DOI] [PubMed] [Google Scholar]
- 284.Ramanathan S., Mathias A.A., German P., Kearney B.P. Clinical pharmacokinetic and pharmacodynamic profile of the HIV integrase inhibitor elvitegravir. Clin. Pharmacokinet. 2011;50(4):229–244. doi: 10.2165/11584570-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 285.Karmon S.L., Markowitz M. Next-generation integrase inhibitors. Drugs. 2013;73(3):213–228. doi: 10.1007/s40265-013-0015-5. [DOI] [PubMed] [Google Scholar]
- 286.Wainberg M.A., Mesplede T., Quashie P.K. The development of novel HIV integrase inhibitors and the problem of drug resistance. Curr. Opin. Virol. 2012;2(5):656–662. doi: 10.1016/j.coviro.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 287.Shah B.M., Schafer J.J., De Simone J.A., Jr. Dolutegravir: a new integrase strand transfer inhibitor for the treatment of HIV. Pharmacotherapy. 2014;34(5):506–520. doi: 10.1002/phar.1386. [DOI] [PubMed] [Google Scholar]
- 288.Max B., Vibhakar S. Dolutegravir: a new HIV integrase inhibitor for the treatment of HIV infection. Future Virol. 2014;9(11):967–978. [Google Scholar]
- 289.McCormack P.L. Dolutegravir: a review of its use in the management of HIV-1 infection in adolescents and adults. Drugs. 2014;74(11):1241–1252. doi: 10.1007/s40265-014-0256-y. [DOI] [PubMed] [Google Scholar]
- 290.Belyk, K., M.; Morrison, H., G.; Jones, P.; Summa, V., Preparation of N-(4-fluorobenzyl)-5-hydroxy-1-methyl-2-(1-methyl-1-{[(5-methyl-1,3,4-oxadiazol-2-yl)carbonyl]amino}ethyl)-6-oxo-1,6-dihydropyrimidine-4-carboxamide potassium salts as HIV integrase inhibitors, WO 2006060712 (2006).
- 291.Humphrey G.R., Pye P.J., Zhong Y.-L., Angelaud R., Askin D., Belyk K.M., Maligres P.E., Mancheno D.E., Miller R.A., Reamer R.A., Weissman S.A. Development of a second-generation, highly efficient manufacturing route for the HIV integrase inhibitor raltegravir potassium. Org. Process Res. Dev. 2011;15(1):73–83. [Google Scholar]
- 292.Hunt J.A. Raltegravir (Isentress): the first-in-class HIV-1 integrase inhibitor. In: Li J.J., Johnson D.S., editors. Modern Drug Synthesis. Wiley; 2010. pp. 3–15. [Google Scholar]
- 293.Humphrey G.R., Zhong Y.-L. HIV integrase inhibitor: raltegravir. In: Yasuda N., editor. The Art of Process Chemistry. Wiley-VCH; 2011. pp. 165–190. [Google Scholar]
- 294.Patil G.D., Kshirsagar S.W., Shinde S.B., Patil P.S., Deshpande M.S., Chaudhari A.T., Sonawane S.P., Maikap G.C., Gurjar M.K. Identification, synthesis, and strategy for minimization of potential impurities observed in raltegravir potassium drug substance. Org. Process Res. Dev. 2012;16(8):1422–1429. [Google Scholar]
- 295.Gurjar, M. K.; Sonawane, S. P.; Maikap, G. S.; Patil, G. D.; Shinde, S. B.; Patil, P. S.; Mehta, S. S., Process for the preparation of raltegravir, WO 2013098854 (2013).
- 296.Sippl W., Jung M. DNA methyltransferase inhibitors. Methods Princ. Med. Chem. 2009;42(Epigenetic Targets in Drug Discovery):163–183. [Google Scholar]
- 297.Frick D.N., Lam A.M.I. Understanding helicases as a means of virus control. Curr. Pharm. Des. 2006;12(11):1315–1338. doi: 10.2174/138161206776361147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Xi X.G. Helicases as antiviral and anticancer drug targets. Curr. Med. Chem. 2007;14(8):883–915. doi: 10.2174/092986707780362998. [DOI] [PubMed] [Google Scholar]
- 299.Belon C.A., Frick D.N. Helicase inhibitors as specifically targeted antiviral therapy for hepatitis C. Future Virol. 2009;4(3):277–293. doi: 10.2217/fvl.09.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Amorim M.J., Kao R.Y., Digard P. Nucleozin targets cytoplasmic trafficking of viral ribonucleoprotein-Rab11 complexes in influenza A virus infection. J. Virol. 2013;87(8):4694–4703. doi: 10.1128/JVI.03123-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Gerritz S.W., Cianci C., Kim S., Pearce B.C., Deminie C., Discotto L., McAuliffe B., Minassian B.F., Shi S., Zhu S., Zhai W., Pendri A., Li G., Poss M.A., Edavettal S., McDonnell P.A., Lewis H.A., Maskos K., Mortl M., Kiefersauer R., Steinbacher S., Baldwin E.T., Metzler W., Bryson J., Healy M.D., Philip T., Zoeckler M., Schartman R., Sinz M., Leyva-Grado V.H., Hoffmann H.-H., Langley D.R., Meanwell N.A., Krystal M. Inhibition of influenza virus replication via small molecules that induce the formation of higher-order nucleoprotein oligomers. Proc. Natl. Acad. Sci. U. S. A. 2011;108(37):15366–15371. doi: 10.1073/pnas.1107906108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Belema M., Lopez O.D., Bender J.A., Romine J.L., St. Laurent D.R., Langley D.R., Lemm J.A., O’Boyle D.R., II., Sun J.-H., Wang C., Fridell R.A., Meanwell N.A. Discovery and development of hepatitis C virus NS5A replication complex inhibitors. J. Med. Chem. 2014;57(5):1643–1672. doi: 10.1021/jm401793m. [DOI] [PubMed] [Google Scholar]
- 303.Gentile I., Borgia F., Coppola N., Buonomo A.R., Castaldo G., Borgia G. Daclatasvir: the first of a new class of drugs targeted against hepatitis C virus NS5A. Curr. Med. Chem. 2014;21(12):1391–1404. doi: 10.2174/0929867321666131228222215. [DOI] [PubMed] [Google Scholar]
- 304.Muratore G., Goracci L., Mercorelli B., Foeglein A., Digard P., Cruciani G., Palu G., Loregian A. Small molecule inhibitors of influenza A and B viruses that act by disrupting subunit interactions of the viral polymerase. Proc. Natl. Acad. Sci. U. S. A. 2012;109(16):6247–6252. doi: 10.1073/pnas.1119817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Lou Z., Sun Y., Rao Z. Current progress in antiviral strategies. Trends Pharmacol. Sci. 2014;35(2):86–102. doi: 10.1016/j.tips.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Ludwig S. Targeting cell signaling pathways to fight the flu: towards a paradigm change in anti-influenza therapy. J. Antimicrob. Chemother. 2009;64(1):1–4. doi: 10.1093/jac/dkp161. [DOI] [PubMed] [Google Scholar]
- 307.Mercorelli B., Sinigalia E., Loregian A., Palu G. Human cytomegalovirus DNA replication: antiviral targets and drugs. Rev. Med. Virol. 2008;18(3):177–210. doi: 10.1002/rmv.558. [DOI] [PubMed] [Google Scholar]
- 308.Cheng H., Wan J., Lin M.-I., Liu Y., Lu X., Liu J., Xu Y., Chen J., Tu Z., Cheng Y.-S.E., Ding K. Design, synthesis, and in vitro biological evaluation of 1H-1,2,3-Triazole-4-carboxamide derivatives as new anti-influenza A agents targeting virus nucleoprotein. J. Med. Chem. 2012;55(5):2144–2153. doi: 10.1021/jm2013503. [DOI] [PubMed] [Google Scholar]