

Abstract
Virus infections continue to pose a substantial threat to human health. Unravelling the intricacies of immune defenses against viruses should lead to improved control of infections through the design of new vaccines and therapies. Our understanding of the fundamental cellular and molecular mechanisms involved in the immune systems response to virus infection has improved substantially in recent years. This wealth of new information and the promise of new insight from systems biology approaches continue to drive research in this field. Such knowledge has revealed why viruses sometimes induce immune dysfunction or trigger disastrous pathology and has paved the way for new therapies being tested against chronic and emerging infections. In this chapter, we briefly summarize the general concepts in immunity to virus infections and highlight some of the key challenges remaining for the future. Virus infections continue to pose a substantial threat to human health.
Keywords: Antiviral immunity, Innate immune responses to viruses, T cell and B cell responses to viruses, Memory T cells, Tissue-resident memory T cells, Immune exhaustion, Regulatory T cells, Immune pathology during virus infection
Viruses as obligate intracellular parasites require their host to replicate them and to facilitate their spread to others. In humans, most clinically relevant infections were derived from other animals, and this process continues. Recent examples include human immunodeficiency virus (HIV), Ebola virus, severe acute respiratory syndrome (SARS) virus, and Zika virus. Viral infections are rarely lethal, even if they are highly cytolytic to individual cells. Mortality commonly occurs when viruses jump species, when the virus undergoes a major antigenic change (i.e., influenza viruses), or when host immunity is compromised. HIV (Chapter 39) represents one of the most dramatic human examples of an exotic virus that kills its host. However, HIV kills slowly, providing ample time to spread to new hosts and an effective strategy for persistence in the species. Death or dire consequences following virus infection in mammals with inadequate immunity are well illustrated by observations that fetuses or neonates, especially if deprived of passive immunity, succumb to many agents well tolerated by healthy adults. The science of viral immunology seeks to understand mechanisms of virus–host interactions with a view to applying this knowledge to the design of effective vaccines and immunomodulators that control virus infections. These objectives are facilitated by an increasing wealth of immunological techniques, an expanding array of genetically manipulated animal models, and an abundance of high throughput technologies, which generate data that can be subjected to complex computational analysis. Such analyses can yield signatures indicative of optimal immunogenicity and vaccine efficacy or failure and can explain the variable outcome of infections in individual hosts. In most situations, defense against viruses involves multiple immune components, and the impact of a single mechanism varies greatly according to the method by which individual viruses enter, replicate, and spread within the host. In this chapter, we highlight the principal means by which the host achieves immunity after infection by viruses. Table 25.1 presents an overview.
TABLE 25.1.
Viral Infections and Immunity
| Viral Event | Obstacles | Time Course |
|---|---|---|
| Transmission | Mechanical and chemical barriers | 0 |
| Infection and replication | Innate immunity | 0 → |
| Infection stopped or spreads | Viral antigens transported to lymphoid tissues | Within 24 hours |
| Infection controlled | Specific antibodies and cell-mediated immunity | 4–10 days |
| Sterile immunity | Immune memory | 14 days to years |
| Viral persistence if infection not controlled | Immune disruption or evasion | Weeks to years |
Viral Entry and Infection
Access to target tissues presents numerous obstacles for entry and infection by most human viruses. Most effective of these are the mechanical barriers provided by skin and the mucosal surfaces, as well as the chemically hostile environment of the gut (Fig. 25.1 ). A number of common human viral pathogens enter through the gastrointestinal tract, including rotavirus, enteric adenoviruses, and hepatitis A virus (HAV). These are usually spread via person-to-person contact or contaminated food and water. Respiratory infections caused by influenza viruses, rhinoviruses, coronaviruses, measles virus, varicella-zoster virus (VZV), and respiratory syncytial virus (RSV) are often spread by aerosol transmission, as well as person-to-person contact. Many of the herpes viruses target the skin or the mucosae, such as herpes simplex virus (HSV) and VZV. HSV, in particular, can infect the oral and genital mucosae, the eye, and skin through small cuts and abrasions. Other herpes viruses, such as Epstein-Barr virus (EBV) and cytomegalovirus (CMV), target mucosae. CMV can also spread vertically from mother to baby or rarely via blood transfusions. Human papillomavirus (HPV) targets skin and mucosae and causes warts and may transform cells, inducing cancers, such as cervical cancer. Some viruses, such as West Nile virus, Dengue virus, Semliki forest virus, and Zika virus, can enter through skin via insect vectors. HIV and hepatitis B virus (HBV) are commonly spread via sexual contact. HIV, HBV, and hepatitis C virus (HCV) can also infect humans by direct entry into the bloodstream via transfusions or contaminated needles.
FIG 25.1.
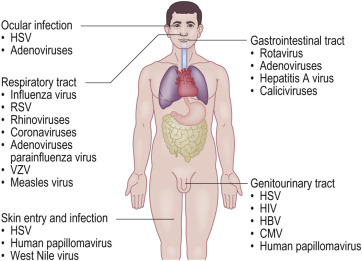
Common Routes of Entry and Infection for Human Viral Pathogens.
CMV, cytomegalovirus; HBV, hepatitis B virus; HIV, human immunodeficiency virus; HSV, herpes simplex virus; RSV, respiratory syncytial virus; VZV, varicella-zoster virus.
Most human viruses replicate only in certain target tissues, this being mainly the consequence of viral receptor distribution. Many viruses use two receptors, such as the use of the CD4 coreceptor and the chemokine receptor CCR5 on T cells by HIV. After attachment to a cellular receptor, viruses may fuse with the cell membrane or be endocytosed and then gain entry into the cytoplasm or nucleus by fusing with the vesicular membrane (enveloped viruses, such as HSV and HIV), or translocate across the cell membrane or induce lysis of the endocytic vesicle once in the cytoplasm (nonenveloped viruses, such as Norwalk virus and poliovirus).1 Viruses then utilize host cell machinery and specialized virally encoded proteins to replicate rapidly within the cell. Once they have multiplied within the cell, many viruses induce cytolysis to facilitate release of new infectious virions (e.g., poxviruses, poliovirus, and herpes viruses). Other viruses are released from infected cells by budding through the cell membrane in the absence of cell death (e.g., HIV and influenza virus). Having entered the body, however, viruses encounter numerous innate defenses and activate the components of adaptive immunity. The latter usually assures that clinical disease, if not infection, will not become evident. Successful exploitation of these defenses through the use of vaccines (Chapter 90) remains a central challenge for many human viruses, particularly those that cause chronic infections, such as HIV and HCV.2
Innate Immunity to Viruses
Viral infection induces an extensive array of defense mechanisms in the host. Innate defenses come into play to block or inhibit initial infection, to protect cells from infection, or to eliminate virus-infected cells. Innate mechanisms occur well before the effectors of adaptive immunity become active, but they are critical for the initiation of adaptive immunity via the elicitation of inflammation that promotes immune cell activation. The innate immune defenses are initiated via pattern recognition receptors (PRRs), which recognize pathogen-associated molecular patterns (PAMPs)3 (Chapter 3). These include transmembrane receptors of the Toll-like receptor (TLR) family, two families of intracellular receptors including the NOD-like receptors (NLRs) and the RIG-I–like helicases (RLHs), as well as the sensor molecule absent in melanoma-2 (AIM2). Additionally, the molecules cyclic guanosine monophosphate–adenosine monophosphate (GMP-AMP) synthase (cGAS), DDX41, IFI16, and Z-DNA–binding protein 1 (ZBP1) can sense cytosolic DNA (Table 25.2 ). These cellular sensors promote the expression of interleukin-1 (IL-1) and IL-18, type I (α/β) interferon (IFN-I), and a variety of IFN-stimulated genes and inflammatory cytokines, and chemokines. TLRs are cell surface or endosomal membrane–bound proteins expressed by numerous cells, including dendritic cells (DCs), macrophages, lymphocytes, and parenchymal cells. Expression of TLRs is largely inducible in most cell types, although some (TLR7/8/9) are constitutively expressed at high levels by specialized plasmacytoid DCs for rapid IFN production. Different TLR molecules recognize specific viral products, such as single- and double-stranded RNA (TLR 3 and TLR7/8, respectively) or double-stranded DNA (TLR9).
TABLE 25.2.
Sensors of Viral Infection
| Toll-Like Receptors (TLRs) | |
| TLR3 | dsRNA, MCMV, VSV, LCMV, HSV, EBV |
| TLR7 and TLR8 | ssRNA, Influenza virus, HIV, VSV |
| TLR9 | dsDNA, HSV, MCMV |
| TLR2 | MV hemagglutinin protein, HSV, HCMV |
| TLR4 | MMTV envelope protein, RSV |
| RIG-I-Like Helicases (RLHs) | |
| RIG-I | Influenza virus, VSV, HCV, JEV, MV, RSV, Sendai virus, EBV |
| MDA-5 | Poly(I:C), MV, Sendai virus, VSV, MCMV, Picornaviruses |
| NOD-Like Receptors (NLRs) | |
| NLRP3 | Influenza virus, Sendai virus, Adenovirus, Vaccinia virus |
| NOD2 | Influenza virus, VSV, RSV |
| Other sensors | |
| AIM2 | Vaccinia virus, MCMV |
| ZBP1 (DAI) | Cytosolic dsDNA, HSV |
| IFI16 | Cytosolic dsDNA, HSV |
| cGAS | Cytosolic dsDNA, HSV |
AIM2, absent in melanoma-2; IFI16, Gamma-interferon-inducible protein Ifi-16; cGAS, cyclic GMP-AMP Synthase; ZBP1, Z-DNA-binding protein 1; DAI, DNA-dependent activator of IFN; dsRNA, double-strand RNA; EBV, Epstein-Barr virus; HCMV, human cytomegalovirus; HCV, hepatitis C virus; HIV, human immunodeficiency virus; HSV, herpes simplex virus 1/2; JEV, Japanese encephalitis virus; LCMV, lymphocytic choriomeningitis virus; MCMV, murine cytomegalovirus; MDA-5, melanoma differentiation-associated gene; MMTV, mouse mammary tumor virus; MV, measles virus; NLR, NOD-like receptor; RLH, RIG-I-like helicase; RSV, respiratory syncytial virus; ssRNA, single-strand RNA; TLR, Toll-like receptor; VSV, vesicular stomatitis virus.
The RLHs retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene (MDA-5) mediate cytoplasmic recognition of viral nucleic acids. These activate mitochondrial antiviral signaling (MAVS) proteins to stimulate IFN-I production and activate inflammasomes, which are molecular complexes that facilitate the activation of caspases and induce the production of proinflammatory IL-1β and IL-18.4 NLRs are a second class of cytosolic sensors of PAMPs that activate inflammasomes via the adapter protein ASC. These include the NLRP (or NALP), NOD, and IPAF/NAIP receptors. Three major inflammasomes have been shown to be involved in antiviral immunity: the NLRP3 inflammasome, the RIG-I inflammasome, and the AIM2 inflammasome.3
 Key Concepts: Major Antiviral Innate Defense Mechanisms.
Key Concepts: Major Antiviral Innate Defense Mechanisms.
- Acting to block infection:
- Natural antibodies
- Complement components
- Some cytokines and chemokines
- Acting to protect cells from infection:
- Interferon-α/β
- Interferon-γ (IL-γ)
- IL-1, IL-18
- Acting to destroy or inhibit virus-infected cells:
- Natural killer (NK) cells
- Natural killer T cells (NKT cells)
- Macrophages
- Neutrophils
- γδ T cells
- Nitric oxide
- Involved in regulating antiviral inflammatory response:
- ILs-1, 6, 10, 12, 18, 23, 33
- Transforming growth factor (TGF)-β
- Chemokines (CCL2, 3, 4, 5)
Alt-text: Unlabelled box
The innate defense system consists of multiple cellular components and many specialized proteins. The longest known and best-studied antiviral proteins are the α/β IFNs, which act by binding to the type I IFN receptor and result in the transcription of more than 100 IFN-stimulated genes. One consequence of this “antiviral state” is the inhibition of cell protein synthesis and the prevention of viral replication.5 Multiple leukocyte subsets are involved in innate defense, including macrophages, DCs, neutrophils, natural killer (NK) cells, natural killer T cells (NKT cells), and γδT cells. Furthermore, tissue cells, including fibroblasts, epithelial cells, and endothelial cells, express PRRs and respond to viral infection via the production of innate cytokines, including IFN-I and IL-1. IFN-I is a critical link between the innate and adaptive immune system, via activation of DCs and T cells, as well as protecting T cells from NK cell-mediated attack.6 IFN-Is can also activate NK cells and induce other cytokines that promote NK responses, such as IFN-γ and IL-12. NK cells produce proinflammatory cytokines; they can kill infected cells and interact with DCs, and are an important component of innate defense against viruses. NK cells can protect against some herpes viruses, which downregulate major histocompatibility complex (MHC) expression in the cells they infect. NK cells are also important in resistance to mouse and human CMV and possibly to HIV, influenza virus, and Ebola virus.7 NK cells have also recently been shown to possess traits of adaptive immunity and, like T and B cells, can form populations of memory cells.8 NK cells are regulated by an array of activating and inhibitory receptors, whose expression and function are just beginning to be understood. Uninfected cells are usually protected from NK cell cytolysis as they deliver negative signals, such as high expression of MHC molecules. In contrast, virus-infected cells are killed either because they deliver positive signals or because they lack adequate MHC-negative signals. NK cells may also control excessive immune responses to viruses by killing CD4+ T cells and indirectly regulating cytotoxic T lymphocyte (CTL) responses. NKT cells may provide some antigen-specific innate immune protection against certain viruses, such as influenza virus.9
Several classes of innate host proteins function in antiviral defense. These include natural antibodies, which may play a role in defense against some viral infections, as well as pentraxins and complement proteins.10 Some viruses may be directly inactivated by complement activation or be destroyed by phagocytic cells that bind and ingest complement-bound virions. Several proinflammatory cytokines and chemokines induced by virus infection also play key roles in defense. Foremost among these is IL-1 and other members of the IL-1 family, including IL-18 and IL-33.11 These cytokines influence both innate and adaptive immune cells and play critical roles in antiviral defense. Other antiviral cytokines are produced early following infection, such as TNF-α, IFN-γ, IL-12, IL-6, and chemokines, such as MIP-1α. In particular, IL-12 is a potent inducer of IFN-γ from NK cells. Inflammatory chemokines may also play an important role in innate antiviral defense by orchestrating macrophage, neutrophil, DC, and NK cell responses at the site of infection. Not only are these components of innate immunity involved in mediating initial protection against viruses; several components (e.g., the PRRs; the cytokines IFN-I, IL-I, IL-33, and IL-12; and phagocytes, including macrophages, monocytes, and DCs) serve to shape the nature and effectiveness of the subsequent adaptive response to viral pathogens. For instance, DCs require innate signals, such as IFN-I and IL-12, for maturation and optimal T-cell activation. Furthermore, CD8+ T cells responding to viruses need IFN-I and IL-33 signals for expansion and memory formation. Thus both the magnitude and the type of innate response induced by virus infection have a marked influence on the generation of adaptive immune responses.
Adaptive Immunity to Viruses
Innate immunity generally only slows, rather than stops, viral infection, allowing time for the adaptive immune response to begin. The two major divisions of adaptive immunity, antibody-mediated and T-cell–mediated, are mainly directed at different targets. Antibodies usually function by binding to free viral particles and, in so doing, block infection of the host cell (Chapter 15). In contrast, T cells act principally by recognizing and destroying virus-infected cells or by orchestrating an inflammatory response that includes several antiviral components (Chapters 16, 17). As all viruses replicate within cells and many can spread directly between cells without reentering the extracellular environment, resolution of infection is reliant more on T-cell function than on antibody function. However, broadly neutralizing antiviral antibodies have the potential to be effective therapies against many different human infections, including HIV, influenza viruses, and Ebola virus. Recent advances have allowed researchers to isolate and identify human monoclonal antibodies (mAbs) against these and other pathogens,12 offering promise of new therapies as well as significant insight for vaccine design. Antiviral antibodies are also very important as an immunoprotective barrier against reinfection. It is the presence of antibodies at portals of entry—most often mucosal surfaces—that is of particular relevance to influenza, HSV, and HIV infections.13 Yet, how to generate vaccines that induce optimal antibody responses, including broadly neutralizing antibodies, remains an important unsolved problem.
Initiation of adaptive immunity is closely dependent on early innate mechanisms that activate antigen-presenting cells (APCs), principally subsets of DCs. APCs and lymphocytes are drawn into lymphoid tissues by chemokine and cytokine signals and are retained there for a few days to facilitate effective intercellular interactions. The architecture of the secondary lymphoid tissues supports the coordinated interactions among the cells of the adaptive immune system14 through a network of supportive stromal cells and local chemokine gradients15(Chapter 2). The induction events occur in lymph nodes draining an infection site or in the spleen if virus enters the bloodstream. The passage of viral antigens to lymph nodes usually occurs in DCs. Some viruses are able to compromise the function of APCs, such as HSV and measles virus, which can inhibit DC maturation.
B-cell activation occurs following antigen encounter in the B-cell follicles, and possibly the T-cell zones, in the spleen or lymph nodes. Some activated B cells become short-lived plasma cells, whereas others move to the edges of the B-cell follicles and interact with antigen-specific helper CD4+ T cells via presentation of antigenic peptides on B-cell MHC class II molecules. These Bcl6-dependent CD4 T follicular helper (Tfh) cells are specialized for providing help for B-cell responses and are needed to promote and regulate B-cell responses.16 Activated B cells initiate germinal center (GC) reactions with the help of CD4 Tfh cells, ensuring somatic hypermutation and affinity maturation for the selection of high-affinity, antibody-producing, long-lived plasma cells, as well as memory B cells.17 At the molecular level, upregulation of the transcription factors Blimp-1, XBP-1, and IRF-4 dictates plasma cell formation, whereas Pax-5 expression delineates B cells destined for GC reactions and the memory B-cell lineage.
Antibody binding to epitopes expressed by native proteins at the surface of free virions usually blocks viral attachment or penetration of target cells. Sometimes the consequence is viral lysis (with complement proteins also involved), opsonization, or sensitization for destruction by Fc receptor–bearing cells that mediate antibody-dependent cellular cytotoxicity (ADCC). Occasionally, however, Fc receptor binding of antibody-bound virus may facilitate infection and result in more severe tissue damage. This occurs in Dengue fever and may happen in some instances in HIV infection. The antibody involved in the protection of mucosal surfaces in humans is predominantly secretory immunoglobulin A (IgA), but serum-derived IgG may also be protective, particularly in such sites as the vaginal mucosa.13 Both antibody isotypes act mainly to block infection of epithelial cells, although in some instances, the antibody may transport antigen from within the body across epithelial cells to the outside. Mucosal antibody persists for a much shorter period compared with serum antibody, which explains, in part, why immunity to mucosal pathogens is usually of much shorter duration compared with immunity to systemic viral infections.
Key Concepts: Antiviral T- and B-Cell Immunity.
| Effector Systems | Recognized Molecules | Control Mechanisms |
|---|---|---|
| Antibody | Surface proteins or virions | Neutralization of virus, opsonization, or destruction of infected cells by ADCC |
| Antibody + complement | Surface proteins expressed on infected cells | Infected cell destruction by ADCC or complement-mediated lysis |
| Mucosal antibody (IgA) | Surface proteins or virions | Viral neutralization, opsonization, and transcytosis |
| CD4+ T cells | Viral peptides (10–20 mers) presented on MHC class II surface, internal or nonstructural proteins presented by APCs | Antiviral cytokine and chemokine production; help for CD8+ T-cell and B-cell responses; killing infected cells; regulatory functions to reduce immunopathology |
| CD8+ T cells | Viral peptides (8–10 mers) presented on MHC class I surface, internal, or nonstructural proteins presented on infected cells or by cross-presentation | Killing infected cells or purging virus without cell death; antiviral cytokine and chemokine production |
ADCC, antibody-dependent cellular cytotoxicity; APC, antigen-presenting cell; IgA, immunoglobulin A; MHC, major histocompatibility complex.
Alt-text: Unlabelled box
Like B-cell responses, T-cell responses to viral infections also begin within lymphoid tissues. Specific CD8+ CTL precursors recognize antigen in the context of MHC class I–peptide antigen complexes on DCs. The CD8+ T cells become activated, proliferate, and differentiate into effectors. Expansion of these naïve antigen-specific precursors is considerable, often exceeding 10 000-fold, and results in an effector population that can account for 40% or more of a host's total CD8+ T-cell population (Fig. 25.2 ). Various factors, including antigen and APCs, costimulatory molecules (e.g., CD28 and 4–1BB), and inflammatory cytokines (e.g., IFN-I and IL-12) are required to program the development of functional effector lymphocytes. In some infections, CD4+ T-cell help is also important to prime robust CTL responses via signals, including CD40 that are delivered to DCs.18 Activated CTL effectors then exit lymphoid organs and access almost all body locations via the bloodstream. However, effectors do not stay activated for long once the virus is cleared, and approximately 95% die by a process termed activation-induced cell death. Following this contraction phase, the remaining cells differentiate into memory cells, which remain as a more or less stable population in the host for many years. They represent an expanded pool of CTL precursors that can be activated upon secondary encounter with antigen and provide enhanced protection upon reinfection with the same virus (see next section). Although much of our knowledge of T-cell responses to viruses has been obtained from murine studies, it is increasingly clear that the fundamental principles are the same or similar in humans.19
FIG 25.2.
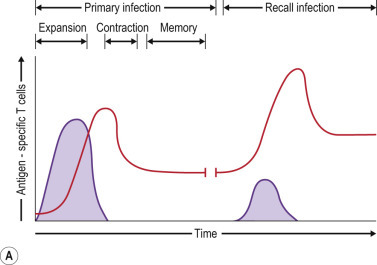
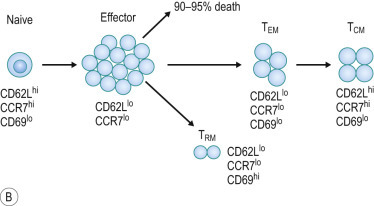
Expansion/Contraction/Memory Phases of Adaptive Immunity and Memory Cell Subsets.
(A) Dynamics of primary and secondary (recall) T-cell responses to viral infection. Both primary and recall T-cell responses undergo expansion and contraction phases, followed by stable immune memory. Recall responses induce a larger effector pool and reduced contraction further boosting the memory pool. (B) Effector and memory T-cell differentiation. Antigen stimulation expands effector cells, most of which die during the contraction phase. Effector memory T (TEM) cells that are formed gradually convert to central memory T (TCM) cells over time, with corresponding changes in surface marker expression. Some effector T cells develop into resident memory T (TRM) cells that persist in the tissues and do not reenter the circulation.
T-cell immunity against a particular virus involves both CD4+ and CD8+ T-cell subsets that recognize peptides derived from viral antigens bound to surface MHC proteins (class II and class I, respectively) (Chapters 5, 6). Complexes of viral peptides bound to MHC class II proteins are generated by APCs from scavenged and processed virus-infected cells or viral particles. Antigen–MHC class I complexes are expressed on the surface of infected cells, and antigen can also be transferred to APCs from infected cells by a process known as cross-presentation. Recent experiments in mice have also demonstrated a role for transfer of antigen between DCs as they migrate from infected tissues to the lymphoid tissues. Multiple subsets of DCs exist and specialize somewhat in antigen presentation on MHC-I or MHC-II.20 During the process of activation, T cells can receive signals from multiple DC types in a temporally controlled sequence that coordinates CD4+ and CD8+ T-cell interactions.18 Use of MHC class I and class II tetramers to directly visualize antigen-specific CD8+ and CD4+ T-cell responses, respectively, has demonstrated the significant size of T-cell responses to viruses, such that the majority of the activated T cells seen at the peak of the response are virus-specific.
CTLs function by recognizing virus-infected cells and killing them; this often involves perforins and cytotoxic granules containing granzymes. Effector CTLs can also induce death in target cells following engagement of the Fas ligand on the CTL with Fas on target cells. Both pathways lead to apoptosis of the target cell, involving the degradation of nucleic acids, including those of the virus. Alternatively, CD8+ T cells also mediate defense through the release of various cytokines after antigen recognition. Some of the cytokines and chemokines most highly produced by CTLs include IFN-γ, TNF-α, lymphotoxin-α, and RANTES (CCL5) (Chapters 9, 10). These cytokines can have multiple antiviral effects on infected cells and on the cells around them, including purging of virus from infected cells without killing the cells. This is particularly important for such viruses as HSV, which infects nonrejuvenating cells, such as nerve cells.
CD4+ T cells are involved in antiviral defense as well as being modulators of inflammatory reactions to viruses. Multiple functional subsets of CD4+ T cells are recognized based largely on the types of cytokines produced when they recognize antigen. CD4+ T cells are more broadly reactive than CD8+ T cells; they recognize larger peptides processed from viral proteins and are restricted by MHC class II. These CD4+ T cells participate in antiviral immunity in several ways. They can act as helper cells for the development of high-affinity antibody responses and for more functional CD8+ T-cell responses.16, 21 Additionally, CD4+ T cells act as effectors and orchestrate inflammatory reactions, which either serve a protective function or, in some cases, become prolonged causing chronic tissue damage (Chapter 16). The latter can happen in HCV-mediated hepatitis and HSV-mediated stromal keratitis. Occasionally, CD4+ T cells can mediate direct cytotoxicity, but they are less effective than CD8+ T cells. The principal subsets of CD4+ T cells involved in inflammatory reactions are T helper-1 (Th1) cells (producing mainly IFN-γ, tumor necrosis factor [TNF]-α, IL-2) and Th17-producing cells (IL-17a and IL-22). A third effector subset, Th2 cells producing (IL-4, IL-5, and IL-13), also participates in inflammatory reactions, although in the case of viruses, these are usually more tissue damaging than protective. This situation can occur in responses to RSV infection. Regulatory T cells (Tregs) are a further subset of CD4+ T cells of particular importance, since these cells largely act to regulate the function of effector subsets and, in so doing, influence the severity and duration of inflammatory reactions22 (Chapter 18). Tregs produce antiinflammatory cytokines, such as IL-10 and TGF-β, and can be distinguished from other CD4+ subsets by their expression of a unique transcription factor FoxP3. The balance of CD4+ T-cell subset representation in response to a virus infection is critical. In situations where responses become overtly tissue damaging and chronic, the balance favors effector subsets. In such situations, changing the balance to favor Tregs can result in diminished lesions.
Immunological Memory
Immunological memory is a cardinal feature of adaptive immunity. The goal of vaccinology is to induce long-lived immunological memory to protect against reinfection (Chapter 90). Following infection with certain viruses, memory can be exceptionally long-lived, potentially for the life of the host (e.g., yellow fever and smallpox viruses).19, 23 Memory is defined by the persistence of specific lymphocytes and antibody-producing plasma cells rather than that of antigen to induce continuous lymphocyte activation. Humoral memory to viruses involves long-lived plasma cells in bone marrow, which provide a continuous low-level source of serum antibody. This maintenance of humoral immunity also involves a population of homeostatically maintained memory B cells, which may be required to maintain stable numbers of long-lived plasma cells over time. The pool of memory T cells is regulated by low-level homeostatic division controlled by the cytokines IL-7 and IL-15. For memory CD8+ T cells, IL-7 is primarily important for survival, whereas IL-15 is crucial for low-level proliferation to maintain the size of the memory T-cell pool.
Key Concepts: Principles of Antiviral Immunity.
Many human viral infections are successfully controlled by the immune system.
Certain emerging viruses may overwhelm the immune system and cause severe morbidity and mortality.
Other viruses have developed mechanisms to overwhelm or evade the immune system and persist.
Individuals with defects in innate or adaptive immunity demonstrate more severe viral infections.
T-cell immunity is more important for control than are antibodies in many viral infections.
Antibodies are important to minimize reinfection, particularly at mucosal sites.
Immune memory is often sufficient to prevent secondary disease, although not in all viral infections.
Tissue-specific immune memory may be important to rapidly protect against reinfection at peripheral sites (e.g., skin and mucosae).
Alt-text: Unlabelled box
Immunological memory is defined by a pool of antigen-specific cells whose increased frequency enables rapid control of viral reinfection (see Fig. 25.2). IL-7Rα-expressing effector T cells are the precursors of this memory pool. This population of cells, which constitutes about 5–10% of the effector pool, preferentially survives the contraction phase and gradually differentiates into a stable memory population.24 Upon reinfection, these memory cells can be rapidly activated and, by virtue of their increased frequency, mediate more rapid clearance of the viral pathogen. Moreover, repeated stimulation of memory cells via multiple infections with the same virus, or prime-boost vaccine regimes, further increases the size of the antigen-specific memory T-cell pool.25 Restimulation also affects the activation status and tissue distribution of memory T cells, which may enhance protection from viral infection in mucosal and other tissues.
Experiments in humans and mice have demonstrated that memory T cells are heterogeneous. Memory T cells were divided into effector memory (TEM) and central memory (TCM) subsets, defined by expression of two surface molecules involved in T-cell migration: CD62L and CCR7.24 The CD62LloCCR7lo TEM subset is found primarily in nonlymphoid tissues and the spleen, whereas the CD62LhiCCR7hi TCM subset is largely present in lymph nodes and the spleen. The current model predicts that effector T cells form the TEM subset and that these cells gradually convert to a TCM phenotype over time (Fig. 25.2B). Although the conditions that control the rate of this conversion are unknown, it is likely that the amounts of antigen and inflammatory signals received during the effector phase greatly influence this. It has also been shown that CD4+ T-cell help is required for the generation of long-lived memory CD8+ T cells, via interactions with DCs.21
Studies suggest that TCM are capable of mounting stronger proliferative responses following reinfection. Tissue-specific homing of TEM cells permits them to enter sites of potential viral infection, such as skin and mucosae. However, we now know that many memory T cells found at sites of previous viral infections take up long-term residence in tissues.26 This includes skin, intestines, lungs, the liver, and the brain. These resident memory T cells (TRM cells) are sequestered from the circulation and provide rapid protection against viruses, such as HSV, in skin, where they localize with a unique dendritic morphology and undergo slow surveillance of the tissue (Fig. 25.3 ). Notably, activation of TRM cells can trigger enhanced early inflammation to drive local immunity. This is in contrast to TEM cells, which continue to migrate through nonlymphoid tissues, rather than being sequestered in peripheral tissues, and also differs from the CD8+ and CD4+ TCM, which migrate largely through lymphoid organs (spleen and lymph nodes). These differences may define the physiological raison d'être for these memory T-cell subsets, highlighting that measurement of memory T cells in human peripheral blood is a poor representation of the total-body memory T cell pool.
FIG 25.3.
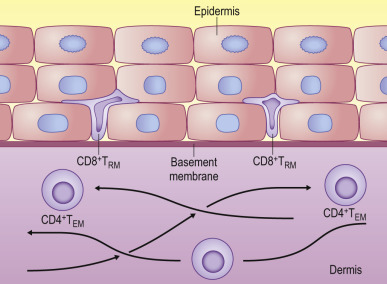
Unique Subsets of Memory CD8+ and CD4+ T Cells Reside Within Peripheral Tissues, at Sites of Previous Viral Infection, and Provide Rapid Protection Against Reinfection.
Resident memory CD8+ T cells (TRM) remain localized in the epidermis in skin after herpes simplex virus (HSV) infection. Resident memory CD4+ T (TEM) cells continue to migrate through the dermal layers of skin, with access to blood and lymphoid tissues.
TRM cells can be detected in tissues by using markers, such as CD69 and CD103, although these are imperfect identifiers, including in human tissues. TRM cells in different anatomical locations share a common genetic signature and require common transcription factors for their formation. Yet, these cells also adopt unique gene expression that is imprinted by the tissue environment, and presumably imparts specialized functions on TRM cells in each location. However, memory in certain peripheral tissues, such as lungs, appears to wane over time, suggesting that memory T cells may not persist in sufficient numbers in this site. This rationalizes a need for vaccines that induce optimal numbers of memory T cells in tissues as well as blood.
Immune Evasion and Immunity to Chronic Viral Infections
Many, if not all, viruses employ immune blunting or delay tactics to circumvent aspects of the immune system, allowing them time to replicate further or escape detection (Table 25.3 ).27 One such mechanism may involve killing or infecting APCs. Viruses may also delay or prevent apoptosis induced by CTLs within infected cells. Other viral evasion measures aimed at the CD8+ T cell–mediated antiviral defense system inhibit antigen processing, thereby minimizing effector CTL induction. To escape CTL killing, many viruses also downregulate the MHC molecules on the surface of infected cells. In addition, viruses may produce various mimics or modulators/inhibitors of cytokines, chemokines, or other components of the immune system or their receptors. Viruses also resort to antigenic hypervariability to escape antibody or T-cell recognition. This can occur during transmission from host to host (e.g., influenza virus), or within hosts during chronic infection through the generation of viral escape mutants. The latter is particularly important for HIV and HCV infections.
TABLE 25.3.
Mechanisms and Examples of Viral Immune Evasion
| Mechanism | Example |
|---|---|
| Interference with viral antigen processing and presentation | HSV (ICP47), EBV (EBNA-1), HIV (Nef, Tat), HPV (E5), CMV (UL6) |
| Evasion of NK cell function | HIV (Nef), EBV (EBNA-1), CMV (UL40, UL18) |
| Inhibition of cell apoptosis | Adenovirus (RID complex and E1B), HIV (Nef), EBV (BHRF-1) |
| Destruction of T cells | HIV |
| Interference with antiviral cytokines and chemokines | EBV (IL-10 homologue), CMV(US28 chemokine receptor homologue), vaccinia virus (IL-18-binding protein), HIV (Tat chemokine activity) |
| Inhibition of complement action | HSV, pox viruses |
| Inhibition of DC maturation | HSV, vaccinia virus |
| Frequent antigenic variation | Influenza virus, HIV |
| Infection of immune privileged site | Measles virus, VZV and HSV (neurons) |
| Immune exhaustion | HIV, HCV, HBV |
CMV, cytomegalovirus; DC, dendritic cell; EBV, Epstein–Barr virus; HBV, hepatitis B virus; HCV, hepatitis C virus; HIV, human immunodeficiency virus; HPV, human papillomavirus; HSV, herpes simplex virus; IL-18, interleukin-18; NK, natural killer; RID, receptor internalization and degradation; VZV, varicella-zoster virus.
The success of many viral pathogens rests in their ability to subvert the host immune response. The most successful human viruses can escape the immune system and persist for the life of the host.28 Two well-studied examples of this are CMV and EBV. T-cell responses to these viruses are prominent and readily detectable in humans, and yet the immune system is unable to clear either pathogen completely. However, these viruses generally remain undetectable in immunocompetent individuals. Other viral infections, such as those caused by the herpes viruses HSV and VZV, are marked by periods of latency when no virus can be detected. Yet, periods of viral reactivation, often triggered by stress, can lead to episodes of disease. These are controlled by the immune response, which plays a central role in controlling herpes virus latency.29
Many of the most medically important human viruses are associated with persistent viremia. These include those causing chronic infections, such as HIV, HCV, HBV, and human T-lymphotropic virus (HTLV), among others. Such chronic viral infections are marked by high levels of persisting antigen and can result in skewed T-cell immunodominance hierarchies, altered tissue localization of immune cells, and severely impaired T-cell function.30 This altered T-cell function is hierarchical and results in functional T-cell defects ranging from reduced cytokine production and altered proliferative capacity (exhaustion) to death (deletion) of the responding T cells (Fig. 25.4 ).
FIG 25.4.
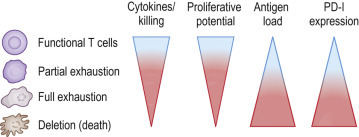
Hierarchical Model of T-Cell Exhaustion During Persistent Viral Infection.
T-cell function (cytokine production, killing, and proliferative potential) is negatively influenced by increasing levels of antigen. Low levels of persistent antigen may lead to partial loss of function and intermediate levels of programmed death (PD)-1 expression. High, sustained levels of antigen over time can lead to full loss of function, high levels of PD-1, and eventually cell death (deletion).
Sustained viral antigen levels and inflammation are responsible for this immune dysfunction. This is in stark contrast to normal memory T-cell development, which occurs in the absence of persisting antigen (see previous section). Studies have demonstrated that signaling through multiple inhibitory receptors expressed on the cell surface contributes to exhaustion during chronic infections.30 This includes the receptor programmed death (PD)-1, expression of which may be essential for preventing excessive immunopathology by effector T cells and yet appears to contribute directly to failed immunity to HIV infection and other chronic human viral infections. Although the molecular mechanisms of exhaustion remain unclear, differential involvement of transcription factors and altered gene expression define exhausted T cells. These studies implicated multiple inhibitory receptors as a potential therapeutic targets, and although combinations of these checkpoint inhibitor blockade therapies are proving highly beneficial to the treatment of certain cancers,31 similarly efficacious responses have yet to be demonstrated during chronic virus infection.
Outcomes of Virus Infection: Immunity or Immunopathology
Typically, individual humans respond to a virus infection in different ways. When the common cold or even pandemic influenza infection occurs, only a small percentage of exposed persons may develop overt clinical disease. In the prevaccine days, poliomyelitis was a much-feared consequence of poliovirus infection, but only a very small percentage of infected persons developed the paralyzing complications. Similarly, only an unfortunate few develop life-threatening meningoencephalitis following infection with the insect-transmitted West Nile virus. It is particularly characteristic of chronic viral infections that clinical expression is highly variable. With HCV, for example, in 70–80% of patients, some form of chronic liver disease develops, and the virus is not cleared. However, in up to 30%, the infection is controlled, the virus is cleared, and immunity to reinfection develops. The latter group of individuals make a type of immune response that includes protective antibodies along with an appropriate pattern of T-cell responsiveness.32
We do not fully understand the reasons for the varying outcomes of virus infections in different persons, and almost certainly multiple factors are involved. Many of these factors impact the response pattern made by the innate immune system, which, in turn, affects the magnitude and type of adaptive immune response that occurs. Some of the circumstances that do influence the outcome of infection include genetic susceptibility of the host, the age of the host when infected, the dose and route of infection, the variable induction in the host of antiinflammatory cells and proteins, and the presence of concurrent infections and past exposure to cross-reactive antigens.32
Immunopathology and Autoimmunity
Immune responses against virus-infected cells often result in tissue damage, especially if cell killing is involved or if there is extensive recruitment and activation of inflammatory cell types, such as macrophages and sometimes neutrophils. If the response is brief and is quickly repaired, it is usually deemed an immunoprotective event. A prolonged tissue-damaging effect resulting from an immune reaction against viruses is considered immunopathology. Such situations most commonly involve persistent viruses, which are themselves often mildly cytodestructive in the absence of an immune reaction. Chronic tissue damage initiated by viruses can also result in development of an autoreactive and an occasionally oncogenic response. For example, some autoimmune diseases may be initiated or exacerbated by viral infections, but no named virus has been regularly incriminated as a cause of human autoimmune disease.33 Circumstantial evidence exists for a virus link in multiple sclerosis (MS), insulin-dependent diabetes, and possibly systemic lupus erythematosus (SLE). In MS, many viruses have been isolated from patients, although no specific one has been tied to the disease etiology. The current hypothesis is that viral infections set up an inflammatory environment that may exacerbate or tip the balance toward disease in genetically susceptible individuals.
Immunopathological reactions involving viruses have several mechanisms, but T cells are usually involved as orchestrators of inflammatory events (Table 25.4 ). The clearest example of immunopathology involving a virus is lymphocytic choriomeningitis virus (LCMV) in the mouse. This model has dominated ideas and has set several paradigms in viral immunology in general. The first virus-induced immunopathological lesions recognized were glomerulonephritis and arteritis, noted in mice persistently infected with LCMV. The lesions were assumed to represent inflammatory reactions to tissue-entrapped immune complexes that activate complement. Similar immune complex–mediated lesions occur in other infections, including lung lesions found in severe influenza, respiratory syncytial virus infection, viral hepatitis, and arthritis. However, only rarely have viral antigens been shown to contribute to the antigen component of the complex. An example where the inclusion of viral antigen in immune complexes has been demonstrated is chronic HBV infection of humans. Autoimmune diseases, such as SLE, also result from immune complex–mediated tissue damage. However, evidence linking viruses to the etiology or pathogenesis of SLE is scarce, since the immune complexes in SLE do not appear to include viral antigens at any stage.
TABLE 25.4.
Lesions Resulting From Immunopathology
| Primarily involving CD8+ T cells acting as cytotoxic T lymphocytes or sources of proinflammatory cytokines | Murine lymphocytic choriomeningitis virus; |
| Hepatitis B virus (HBV)–induced chronic hepatitis | |
| Coxsackie B virus–induced diabetes | |
| Coxsackie B virus–induced myocarditis | |
| Demyelination caused by some strains of mouse coronavirus and Theiler virus | |
| Primarily involving CD4+ T cells that produce Th1 cytokines | Demyelination caused by some strains of mouse coronavirus and Theiler's virus; |
| Herpes simplex virus (HSV)–induced stromal keratitis | |
| Involvement of CD4+ T cells that produce Th2 cytokines | Respiratory syncytial virus (RSV)–induced pulmonary lesions |
| Involvement of antibody | Glomerulonephritis in chronic hepatitis B |
| Dengue hemorrhagic fever |
Thanks largely to the LCMV model, it is clear that CD8+ T-cell recognition of viral antigens can result in tissue damage. In LCMV infection, damage occurs in the leptomeninges of immunocompetent mice infected intracerebrally. Hepatitis can also occur in mice infected intravenously. Neither lesion becomes evident if the CD8+ T-cell response is suppressed. CD8+ T cell–mediated immunopathology can be a causative mechanism of chronic hepatitis associated with HCV and HBV infection, although the tissue damage also involves inflammatory CD4+ T cells. Additional viral immunopathology models where lesions result primarily from CD8+ T-cell involvement include myocarditis and insulin-dependent diabetes associated with coxsackie B virus infection. In both instances, CD8+ T cells mainly orchestrate events, but tissue damage may result from the bystander effects of cytokines and other molecules, such as lipid mediators, metalloproteinases, and components of the oxygen burst. Although coxsackie virus can be a cause of diabetes in the mouse, attempts to relate viral infection directly to the etiology of human diabetes have so far failed.
 Clinical Relevance: Hypothesized Role of Viruses in Autoimmunity.
Clinical Relevance: Hypothesized Role of Viruses in Autoimmunity.
Molecular mimicry: similar epitopes shared by virus and host
Bystander activation: chronic release of cytokines and host antigens activates local autoreactive lymphocytes
Viral persistence: chronic viral antigen presentation on host cells leads to prolonged immunopathology
Alt-text: Unlabelled box
Immunopathological reactions against viruses can also involve subsets of CD4+ T cells, which can be either Th1 or Th17 or both. One well-studied example involves persistent infection with Theiler virus in mice.34 This infection causes a demyelinating syndrome that resembles the autoimmune disease experimental allergic encephalomyelitis. In both situations, CD4+ T cells that produce Th1 cytokines appear to serve as pathological mediators. Furthermore, in both models an increase in the involvement of myelin-derived autoantigens occurs as the disease progresses. Once again, such observations indicate the possible role of a virus in an autoimmune disease. With the Theiler virus model, the virus persists in the nervous system and chronically stimulates CD4+ T cells to secrete an array of cytokines. The demyelinating events appear to result from cytokine action on oligodendrocytes. Myelin components, such as myelin basic protein, proteolipid protein, and myelin oligodendroglial glycoprotein, may be released and can participate as additional antigen in immunoinflammatory events. This scenario is referred to as epitope spreading.
Another model of virus-induced immunopathology that mainly involves the Th1 subset of CD4+ T cells is stromal keratitis caused by HSV infection (Fig. 25.5 ).35 The pathogenesis of this immunopathological lesion is unusual in that it occurs and progresses when viral antigens can no longer be demonstrated. The chronic immunoinflammatory lesions are mainly orchestrated by CD4+ T cells, but multiple early events induce the subsequent pathology. Viral replication, the production of certain cytokines and chemokines (IL-1, IL-6, IL-12, and CXCL8), recruitment of inflammatory cells (e.g., neutrophils), and neovascularization of the avascular cornea all precede immunopathology. Recently, it has become evident that Th17 T cells participate in stromal keratitis lesions. The role of Th17 T cells as orchestrators of inflammatory reactions has been a major research focus, especially in lesions of autoimmune diseases.36 When Th17 T cells are the principal mediators of tissue damage, abundant neutrophils are recruited to inflammatory sites, with such cells being mainly responsible for tissue damage.
FIG 25.5.
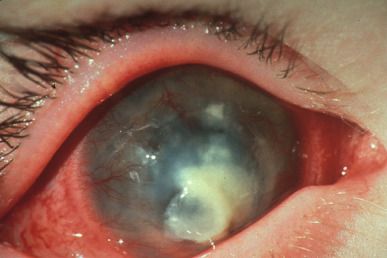
Example of Herpetic Stromal Keratitis (Hsk) in the Human Eye After Herpes Simplex Virus-1 (HSV-1) Infection.
Inflammation of the eye and eyelid can be observed, as well as neovascularization and substantial necrosis, ulceration, and opacity of the cornea.
A further mechanism of viral-induced immunopathology and autoimmunity is molecular mimicry.33 Molecular mimicry represents shared antigenic epitopes, either B- or T-cell antigen, between the host and virus. This concept originated with streptococci and their association with rheumatic fever. With human autoimmune disease, there is little direct support for viral molecular mimicry; however, some animal models have been used to prove the theoretical case, where a viral antigen is expressed as a self-protein in the islet cells of the pancreas. In this model, subsequent infection with the virus induces diabetes. However, this is not true mimicry and may be more closely related to viral antigen persistence in a model such as Theiler disease.
As discussed previously, the outcome of a T-cell response to a virus may be critically dependent on the balance of the T-cell-type response thus, tissue damage is likely to be more severe and prolonged if CD8+ or Th1 and Th17 CD4+ T cells are predominant. Lesions become milder and may resolve when the balance favors Tregs. Accordingly, therapeutic approaches that can shift the balance of T cells are under trial.
Key Concepts: Phases of Immunity Affected by Regulatory T Cells (Tregs).
Interference with antigen presentation by dendritic cells
Inhibition of T-cell proliferation
Inhibition of molecules involved in tissue-specific migration of effector cells
Inhibition of T-cell effector functions in lymphoid and nonlymphoid tissues
Alt-text: Unlabelled box
Translational Research Opportunities
Reversing T-cell exhaustion in patients suffering from chronic infections or cancer will be a key clinical target in the near future. The discovery of multiple inhibitory receptors on exhausted T cells (e.g., PD-1, LAG-3, 2B4, TIM-3) has provided the opportunity to selectively improve T-cell function through blockade of these inhibitory receptors. This may be combined with blockade of immunosuppressive cytokines (e.g., IL-10) or enhancement of signals stimulatory to the response (e.g., IL-7 therapy), as well as with more traditional antiviral therapies and vaccination. The challenge that lies ahead will be in determining which combination of inhibitory and stimulatory signals will need to be manipulated in different diseases and in different groups of patients.
The design of a new generation of vaccines to target diseases, such as HIV and influenza, may require tailor-made solutions for patients who respond poorly to vaccination or respond improperly, as with adverse effects, such as autoimmune reactions. High-throughput approaches now allow for the generation of a molecular signature of vaccination or infection.37 Such systems' biology approaches are expected to result in novel screening for immune protection parameters after vaccination. In the near future, this should also assist in the formulation of new vaccines containing key immune activators, such as those that stimulate certain subsets of T cells or induce appropriate homing molecule expression on these cells to direct them to tissues where they are required to mediate protection (e.g., mucosal sites, or skin).
 On the Horizon: Pressing Issues in Need of Solutions.
On the Horizon: Pressing Issues in Need of Solutions.
Design of new vaccines that induce broadly neutralizing antibodies
Design of new vaccines that induce tissue-resident and circulating memory T-cell subsets
Overcoming immune dysfunction during chronic viral infections for successful viral clearance
Improving the efficacy of vaccines to viruses using systems biology approaches
Therapies for reducing immunopathology during viral infections
Alt-text: Unlabelled box
In some individuals, viral infections cause mild, or sometimes debilitating, tissue damage. Factors that influence whether a viral infection results in immunopathology varies from individual to individual. These factors include age, the route of infection, preexisting immunity, host genetics, and the host's viral burden or virome. Our knowledge of the influence of these factors on the outcome of viral infection is expected to improve rapidly in the coming decades. Recent advances have shed considerable light on the various proinflammatory and antiinflammatory mediators produced during viral infections. These represent key targets for novel therapies in the near future via the use of small-molecule inhibitors.
Conclusions
Humans are infected by many pathogenic viruses. In most cases, these infections are controlled by the immune system with limited damage to the host. However, certain viruses, particularly in cases where the host's immune system is impaired, can cause significant damage to the host's tissues. As our understanding of the mechanisms underlying innate immune defenses, antigen presentation, T- and B-cell responses, and Tregs continues to improve, so too does the ability to design better vaccines and therapies to boost the immune control of viral infections. Although this remains a challenging goal, particularly for many human viruses, such as HIV, HCV, and HSV, these rapid advances continue to provide many avenues for further investigation.
Acknowledgments
Barry T. Rouse is supported by grants from the National Institutes of Health and Scott N. Mueller by the Australian Research Council and the Australian National Health and Medical Research Council.
 Please check your eBook at https://expertconsult.inkling.com/ for self-assessment questions. See inside cover for registration details.
Please check your eBook at https://expertconsult.inkling.com/ for self-assessment questions. See inside cover for registration details.
References
- 1.Marsh M, Helenius A. Virus entry: open sesame. Cell. 2006;124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bachmann MF, Jennings GT. Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol. 2010;10:787–796. doi: 10.1038/nri2868. [DOI] [PubMed] [Google Scholar]
- 3.Iwasaki A. A virological view of innate immune recognition. Annu Rev Microbiol. 2012;66:177–196. doi: 10.1146/annurev-micro-092611-150203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanneganti TD. Central roles of NLRs and inflammasomes in viral infection. Nat Rev Immunol. 2010;10:688–698. doi: 10.1038/nri2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 6.Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol. 2015;15:231–242. doi: 10.1038/nri3806. [DOI] [PubMed] [Google Scholar]
- 7.Jost S, Altfeld M. Control of human viral infections by natural killer cells. Annu Rev Immunol. 2013;31:163–194. doi: 10.1146/annurev-immunol-032712-100001. [DOI] [PubMed] [Google Scholar]
- 8.O'Sullivan TE, Sun JC, Lanier LL. Natural killer cell memory. Immunity. 2015;43:634–645. doi: 10.1016/j.immuni.2015.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Godfrey DI, Uldrich AP, McCluskey J. The burgeoning family of unconventional T cells. Nat Immunol. 2015;16:1114–1123. doi: 10.1038/ni.3298. [DOI] [PubMed] [Google Scholar]
- 10.Bottazzi B, Doni A, Garlanda C. An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu Rev Immunol. 2010;28:157–183. doi: 10.1146/annurev-immunol-030409-101305. [DOI] [PubMed] [Google Scholar]
- 11.Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corti D, Lanzavecchia A. Broadly neutralizing antiviral antibodies. Annu Rev Immunol. 2013;31:705–742. doi: 10.1146/annurev-immunol-032712-095916. [DOI] [PubMed] [Google Scholar]
- 13.Iwasaki A. Antiviral immune responses in the genital tract: clues for vaccines. Nat Rev Immunol. 2010;10:699–711. doi: 10.1038/nri2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qi H, Kastenmuller W, Germain RN. Spatiotemporal basis of innate and adaptive immunity in secondary lymphoid tissue. Annu Rev Cell Dev Biol. 2014;30:141–167. doi: 10.1146/annurev-cellbio-100913-013254. [DOI] [PubMed] [Google Scholar]
- 15.Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol. 2009;9:618–629. doi: 10.1038/nri2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- 17.Corcoran LM, Tarlinton DM. Regulation of germinal center responses, memory B cells and plasma cell formation-an update. Curr Opin Immunol. 2016;39:59–67. doi: 10.1016/j.coi.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 18.Bedoui S, Heath WR, Mueller SN. CD4+ T cell help amplifies innate signals for primary CD8+ T cell immunity. Immunol Rev. 2016;272:52–64. doi: 10.1111/imr.12426. [DOI] [PubMed] [Google Scholar]
- 19.Ahmed R, Akondy RS. Insights into human CD8+ T-cell memory using the yellow fever and smallpox vaccines. Immunol Cell Biol. 2011;89:340–345. doi: 10.1038/icb.2010.155. [DOI] [PubMed] [Google Scholar]
- 20.Merad M, Sathe P, Helft J. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4+ T cells in CD8+ T cell memory. Nat Rev Immunol. 2016;16:102–111. doi: 10.1038/nri.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Veiga-Parga T, Sehrawat S, Rouse BT. Role of regulatory T cells during virus infection. Immunol Rev. 2013;255:182–196. doi: 10.1111/imr.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amanna IJ, Slifka MK, Crotty S. Immunity and immunological memory following smallpox vaccination. Immunol Rev. 2006;211:320–337. doi: 10.1111/j.0105-2896.2006.00392.x. [DOI] [PubMed] [Google Scholar]
- 24.Mueller SN, Gebhardt T, Carbone FR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. 2013;31:137–161. doi: 10.1146/annurev-immunol-032712-095954. [DOI] [PubMed] [Google Scholar]
- 25.Jameson SC, Masopust D. Diversity in T cell memory: an embarrassment of riches. Immunity. 2009;31:859–871. doi: 10.1016/j.immuni.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol. 2016;16:79–89. doi: 10.1038/nri.2015.3. [DOI] [PubMed] [Google Scholar]
- 27.Finlay BB, McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124:767–782. doi: 10.1016/j.cell.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 28.Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 29.Rouse BT, Kaistha SD. A tale of 2 alpha-herpesviruses: lessons for vaccinologists. Clin Infect Dis. 2006;42:810–817. doi: 10.1086/500141. [DOI] [PubMed] [Google Scholar]
- 30.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baumeister SH, Freeman GJ, Dranoff G. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol. 2016;34:539–573. doi: 10.1146/annurev-immunol-032414-112049. [DOI] [PubMed] [Google Scholar]
- 32.Rouse BT, Sehrawat S. Immunity and immunopathology to viruses: what decides the outcome? Nat Rev Immunol. 2010;10:514–526. doi: 10.1038/nri2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujinami RS, von Herrath MG, Christen U. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev. 2006;19:80–94. doi: 10.1128/CMR.19.1.80-94.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olson JK, Ercolini AM, Miller SD. A virus-induced molecular mimicry model of multiple sclerosis. Curr Top Microbiol Immunol. 2005;296:39–53. doi: 10.1007/3-540-30791-5_3. [DOI] [PubMed] [Google Scholar]
- 35.Biswas PS, Rouse BT. Early events in HSV keratitis—setting the stage for a blinding disease. Microbes Infect. 2005;7:799–810. doi: 10.1016/j.micinf.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 36.Korn T, Bettelli E, Oukka M. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 37.Pulendran B. Systems vaccinology: probing humanity's diverse immune systems with vaccines. Proc Natl Acad Sci USA. 2014;111:12300–12306. doi: 10.1073/pnas.1400476111. [DOI] [PMC free article] [PubMed] [Google Scholar]