

Abstract
Many new viruses have emerged in the last five decades. These newer genetically active agents have a major impact on the public health systems worldwide. Most emerging infections appear to be caused by pathogens already present in the environment, brought out of obscurity or given a selective advantage by changing conditions and afforded an opportunity to infect new host populations. Also on rare occasions, a new variant may evolve and cause a new disease. Altered virus transmission because of deforestation and environment change, ecological changes and agricultural development, commerce, technology, microbial adaptation and change, breakdown of public health measures, and deficiencies in public health infrastructure are the reasons for emergence and reemergence of infectious diseases in general and viral infections in particular.
Specific factors precipitating disease emergence can be identified in virtually all cases. Moreover, these factors are increasing in prevalence. This increase, together with the ongoing evolution of viral and microbial variants and selection for drug resistance, suggests that infections will continue to emerge and probably increase and emphasize the urgent need for effective surveillance and control. These viruses are rich source of emerging diseases due to the introduction of infections from other species in the zoonotic pool. In the near future, there is an urgent need to monitor collaboration between human–animal interface so that public health risks can be understood. A number of activities increase microbial traffic from animals to humans or disseminate microbes from isolated groups into new populations and as a result promote emergence and epidemics. In some cases, including many of the most novel infections, the agents are zoonotic crossing from their natural hosts into the human population because of many similarities. Vector-borne diseases also have a natural advantage of dissemination.
Keywords: Emerging infections, novel viruses, viral outbreaks, virus evolution, public health, risk factors, surveillance
Introduction
Following on from the discovery of Tobacco Mosaic Virus (TMV) in 1892 and Foot-and-Mouth disease virus in 1898, the first “filterable agent” to be discovered in humans was Yellow Fever (YF) virus in 1901 (Woolhouse et al., 2001). Landmark discoveries later were Rinderpest, Vaccinia and Rabies (1902–06), Avian leukosis and Poliomyelitis (1908–11), Bacteriophages (1915–17), and Spanish flu (1918–22; due to which 50 million people died). More than 65 viruses were described in the years 1920–27 (Li et al., 2009). Thereafter cultures of human Herpes Simplex Virus (HSV) and then YF virus were done, and first attenuated vaccine for YF was prepared (1929–31). First chorioallantoic membrane inoculation was done with fowlpox virus in 1931. Subsequently, nucleoprotein nature of viruses (1934–36), crystallization of TMV (1935–37), culture of polio viruses (1949), and DNA as genetic material of phages (1950–63) by Hershey Chase experiment were achieved and killed and live polio vaccines were prepared (Chua et al., 2007). Reoviruses were discovered (1953–64), single stranded Ribonucleic acid (ssRNA) of TMV was translated into viral proteins (1962), and in vitro synthesis of both single stranded deoxyribo nucleicacid (ssDNA) and ssRNA bacteriophage genome was successful (1967). Later in 1979–80, discovery and characterization of infectious naked RNA viroids were shown to be circular ssRNA, complete genomic sequence of hepatitis, cauliflower mosaic virus (CMV) (1982) and complete genomic sequence of TMV (1986–89) was achieved. Historically, major developments in virological technology mark filtration in 1890, complement fixation test in 1929, tissue culture in 1948, monoclonal antibodies in the 1970s, polymerase chain reaction (PCR) in 1985 and high-throughput sequencing in the 2000s (Antia et al., 2003).
New species of human viruses are still being identified, at a rate of three or four per year. Viruses make up about two-thirds of all new human pathogens. These new viruses differ widely in their importance, ranging from the rare and mild illness due to Menangle virus to the devastating public health impact of Human immunodeficiency virus 1 (HIV-1). On the contrary, a highly significant overrepresentation has been given by most human pathogen species of bacteria, fungi, or helminthes. Actually, it was the discovery of the HIV in the early 1980s that initiated a worldwide awareness and research interest in emerging novel viral pathogens (Anderson and Tong, 2010). Over the past several decades, new outbreaks of infections have led to the discovery of a diverse array of highly pathogenic viruses mainly those belonging to the Filoviridae, Arenaviridae, Bunyaviridae, Paramyxoviridae, Coronaviridae, Flaviviridae, Togaviridae, and Hepeviridae families. Some examples include BK virus, JC virus, Merkel cell polyomavirus (MCV or MCPyV), severe fever with thrombocytopenia syndrome virus (SFTSV), hantavirus (HTNV), and Sin Nombre virus (Bebber et al., 2007). Following the fatal cases of Lujo virus in southern Africa in 2008 (Briese et al., 2009), another arena virus, the Lassa virus, was first reported in Nigeria in 1969, reemerged in Guinea, Liberia, and Mali in 2009, Ghana in 2011, and Benin in 2014. Human metapneumovirus was first identified in the Netherlands in 2001 and was subsequently linked to an acute lower respiratory tract infection in children, similar to respiratory syncytial virus (RSV). Recently, in 2013, a novel avian influenza A strain (H7N9) of “bird flu” in China (Wolfe et al., 2004) and the Middle East respiratory syndrome related Coronavirus (MERS)-CoV have been identified (Corman et al., 2012). Of note, while 2015 was threatened by the reemergence of the Ebola virus, 2015/2016 has been challenged with the resurgence of the Zika virus (ZIKV). Despite substantial advancements in the understanding of the biology of pathogens, the breakthrough in prevention, their effects on public health and the global economy, and the emergence of novel pandemic viruses remains an enduring puzzle. The intricate “host–pathogen–environment” relationship remains the key to understanding the emergence/reemergence of pathogenic versus ecological approach to study the diversity of human viruses (defined as viruses for which there is evidence of natural infection of humans) (Jones et al., 2008). First, the temporal, geographical, and taxonomic patterns in the discovery of human viruses need to be described and co related. Second, the processes by which new human viruses emerge need to be understood. There are a number of definitions of “emergence.” All the stages of the process by which a virus shifts from not infecting humans at all to becoming a major human pathogen need to be understood. As experiences with HIV-1 and new variants of influenza A (and also with novel animal pathogens such as canine parvovirus) show, this shift can occur rapidly, over time scales of decades, years, or even months. Not all newly identified human virus species are “new” in the sense that they have only recently started to infect humans; many of them have been present in humans for a considerable time but have only recently been recognized (Cleaveland et al., 2007). Moreover, the “species” itself is an imprecise designation, especially in influenza-like viruses where different serotypes can have different epidemiologies and health impacts. Indeed, the demarcation between genus, species complex, species, and serotype (or other designations of subspecific variation) can be somewhat arbitrary (Cleaveland et al., 2001). Nonetheless, a study of currently recognized “species” is a natural starting point for attempts to characterize and interpret patterns of virus diversity. A comprehensive knowledge of important emerging/reemerging viral infections worldwide with their possible origins, evolution, natural reservoirs, human adaptations, and risk factors needs to be developed for individual novel agents for a better insight of the biologic and evolutionary mechanisms (Keeling and Rohani, 2008) (Fig. 4.1 ).
Figure 4.1.
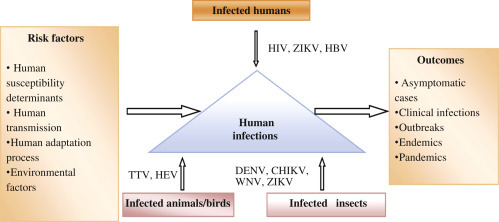
A depiction of important emerging/reemerging viral infections and their possible origins, evolutionary drivers, natural reservoirs, and risk factors. CHIKV, Chikungunya virus; DENV, dengue virus; HBV, Hepatitis B virus; HEV, hepatitis E virus; HIV, human immunodeficiency virus; TTV, Torque teno virus; WNV, West Nile virus; ZIKV, Zika virus.
Origin
Of the known viruses that infect humans, about 80% perpetuate naturally in nonhuman “reservoirs,” largely farm mammals and poultry and, to a lesser extent, in wild animals and arthropods. It is estimated that zoonotic infectious agents constitute about 60% of the known human pathogens and up to 75% of “emerging” human pathogens. The knowledge of such zoonosis and the diversity of these viruses in their known reservoirs are limited. Also the data on some of the domestic mammals harboring dozens of virus species is limited, and there is insufficient knowledge about the wild animals that are estimated to host thousands of virus species (Keusch et al., 2009). Emerging human viruses, such as novel influenza strains, human (h)CoV, Hendra virus, Nipah virus, and several others, are all linked to zoonosis. Recently, the deadly outbreak of MERS-CoV was linked to its zoonotic origin because of its close genetic homology to bat CoV, but not to any other known hCoV (Kilpatrick and Randolph, 2012). Current data shows that bats harbor the greatest diversity of CoVs, which vary from species to species and region to region. The Lyssaviruses of Rhabdoviridae are zoonotic human pathogens that cause fatal encephalitic disease. In addition, European bat lyssavirus type-1/-2, Bokeloh bat lyssavirus (Wolfe et al., 2007), and Australian bat lyssaviruses have been implicated in human fatalities. Rabies virus, the Archetypal lyssavirus, is historically one of the most dreaded viruses, with zoonotic potential in dogs, cats, and ferrets, and includes other domestic and wild mammals. Globally, several mammalian species, including the deer mouse, rice rat, and cotton rat, are recognized as potential reservoir of HTNV. In China, the HTNV and Seoul virus are zoonotically linked to the striped field mouse and the Norway rat, respectively. Also in China, the Fugong virus, a novel HTNV, has recently been identified in small oriental voles. HTNVs have also been detected in shrews and bats, but their link to human illness remains to be established. Moreover, in addition to humans and pigs, there is a growing chain of mammalian hosts for the hepatitis E virus (HEV) that includes deer, boar, mongoose, rabbit, rat, goat, camel, bat, ferret, moose, etc. Likewise, the highly prevalent Torque teno virus, of the newly established virus family, Anelloviridae, also infects pigs, cows, sheep, cats, dogs, and chickens. Arboviruses, such as dengue virus, chikungunya virus (CHIKV), ZIKV, and West Nile virus (WNV), are the arthropod-borne viruses that have reemerged in many tropical and subtropical regions in the last two decades (Taylor et al., 2001). Notably, ZIKV was known as a neglected tropical disease confined to Africa and Asia until an outbreak was reported in 2007 on Yap Island and in 2013/2014 on islands in the Pacific, thus expanding its geographical territory. The WNV remains the most important mosquito-borne encephalitis pathogen in North America, involving Culex sp. and the American Robin in its transmission cycle. Since its emergence in the West in 1999, it has undergone adaptive genetic changes as it spreads throughout North America (Woolhouse and Antia 2008). Furthermore, Crimean–Congo hemorrhagic fever virus (CCHFV) is considered to be one of the major emerging diseases spreading to and within the European nations, following the expanding distribution of anthropophilic ticks. Every year, >1000 cases of CCHFV, due to human-to-human transmission, are reported in southeastern Europe, including Turkey. SFTSV, a previously unidentified tick-borne bunyavirus, has recently emerged in China with fatality rates as high as 30% (Anderson and Tong, 2010) (Table 4.1 ).
Table 4.1.
Examples of Putative New Human Virus Species Reported From 2005 to 2009
| Virus | Family |
|---|---|
| Human bocavirus | Parvoviridae |
| Parvovirus 4 | Parvoviridae |
| KI polyomavirus | Polyomaviridae |
| Melaka virus | Reoviridae |
| WU polyomavirus | Polyomaviridae |
| Astrovirus MLB1 | Astroviridae |
| Bundibugyo ebolavirus | Filoviridae |
| Human bocavirus 2 | Parvoviridae |
| Human cosaviruses A–D | Picornaviridae |
| Human cosavirus E1 | Picornaviridae |
| Astrovirus VA1 | Astroviridae |
| Human papilloma virus 116 | Papillomaviridae |
| Klassevirus | Picornaviridae |
| Lujo virus | Arenaviridae |
Evolution and Adaptation to the Human Host
While DNA viruses are believed to have been evolving and diversifying for millions of years, most RNA viruses are likely to have a much more recent evolution and “human-adaptation” for only thousands of years. Owing to their error-prone polymerase/reverse-transcriptase (approx. 10−4/site/replication cycle) that seems to operate at optimal fidelity, RNA viruses exist as more genetically diversified populations than DNA viruses. Nevertheless, of the hitherto recognized 158 species of human RNA viruses consisting of 47 genera and 17 families, only a minority has adapted to humans (Simmonds, 2001). In contrast, of the known 91 DNA virus species with 22 genera and 8 families, nearly 87% are adapted to human hosts. In the human-adaptation process, the viral genetic mutations, reassortment, or virus–host genetic recombination might lead to the establishment of stable virus lineages in human populations. So, there is the possibility that such human-adapted viruses could circulate in the body asymptomatically and remain undiagnosed till their new clinical manifestations are reported. To understand this clearly, a currently isolated HEV genotype 3 from a chronic hepatitis E patient containing a recombinant virus–host RNA genome was shown to infect cultured human, pig and deer hepatocytes (Wolfe et al., 2004, Wolfe et al., 2007). It is assumed that there is a pool of human virus species still to be discovered. The composition of such a viral pool is dynamic, changing over time, that is, while some virus species tend to become extinct and others continue to evolve in their natural hosts. More commonly, new species arise as a result of jumps from one host to another, thus crossing the species barrier. Humans are, therefore, no more than “incidental” or “spillover” hosts for pathogens. However, only a minority of such viruses are capable of persisting in certain human populations (endemics) or spreading across populations (epidemics) in the absence of a reservoir. Differential host factors, such as age, health, physiology, nutritional status, exposure history, simultaneous infection with >1 pathogen, immunocompetence and genetics, are determinants to human susceptibility to an infection. The field of phylodynamics, combining a modeling framework for host, epidemiological, and molecular data, especially for RNA viruses, shows particular promise for understanding the patterns of viral evolution during epidemics (Gaynor et al., 2007). Moreover, the underappreciated aspect of growing human populations, global changes in land usage and the introduction of anthropophilic vectors create selective pressure on hosts and reservoirs. For example, both WNV and CHIKV evolved rapidly after being introduced to new locations and encountering new vectors. When Aedes albopictus rather than A. aegypti became the main vector of CHIKV in the Indian subcontinent after the 2004–09 epidemic, the same viral strain spread rapidly, and the subsequent mutants seemed to circulate and persist more efficiently. A unique molecular signature of the CHIKV epidemic was a single amino acid substitution (A226V) in the envelope protein (E1). This mutation was identified in 90% of the strains reported during the later phase and was associated with a high epidemic potential for CHIKV (Kilpatrick and Randolph, 2012). ZIKV, initially known to be transmitted by mosquitoes, was recently reported as being transmitted, albeit more rarely, via sexual contact, saliva, breast milk, blood transfusions and from mother to child (Cleaveland et al., 2001).
Evolutionary Drivers
The key to understanding the emergence/reemergence of novel viruses is to know the intricate “host–pathogen–environment” relationship in the evolution of pathogens. This emergence of infectious diseases in naive regions is caused due to the movement of pathogens by travel and trade, while local emergence is driven by a combination of environmental and social and environmental change. It is noted that virus transmission rates are higher in dense than in sparse populations and the spread is often enhanced by air travel or migration. Pathogens introduced into novel regions often cause explosive epidemics, followed by a declining incidence, whereas those that emerge locally due to land usage or social changes usually show consistent increases (Antia et al., 2003). A recent example is the emergence of ZIKV in Brazil in 2015. Phylogenetic studies suggested that ZIKV from the Pacific islands outbreak in 2013/2014 was probably introduced into Brazil during the FIFA World Cup or the 2014 FIA World Endurance Championship auto racing series. The dispersal of the virus in Brazil resulted in an explosive epidemic of Zika fever, and the infection spread to other countries due to frequent travel. While most human infections are known to have zoonotic origins, it is certain that alterations in the environment, due to industrialization and urbanization, are an important but completely neglected factor. Though the origins of most of the human viruses are not known so far, the great majority can be categorized as “crowd diseases” that require a relatively high host density to persist (Keusch et al., 2009).
Notably, the recent outbreaks of H1N1, hCoV, Hendra virus, Nipah virus, and MERS-CoV suggest that the Asia-Pacific region is the global hot spot for the emergence of novel RNA viruses. In this case an order-of-magnitude estimation of 1 such event per 100 years is broadly consistent with human demographic history. Notably, RNA viruses are known to incorporate drastic mutations in their genome, an example being the large duplication events in the G protein gene of Respiratory Syncytial Virus (RSV). Two such remarkable events were the 60-bp duplication in group B RSV in Argentina in 1999 and the 72-bp duplication in group A RSV in Canada in 2011. These new genotypes of RSV with their duplications, known as BA and ON1, respectively, spread to different geographical regions across the globe due to immunologically naive travelers. The mathematics of these spreading events is well known today and a sophisticated array of computational and mathematical models can be used to accurately back-predict such events. An example of this is the first case clusters of the severe acute respiratory syndrome (SARS) outbreak and the subsequent global spread, including the country-by-country distribution of human cases (Woolhouse et al., 2001). Recent investigations have evaluated the transmission dynamics of ZIKV infection using mathematical models. We may not ignore that wild animals constitute an important but poorly understood reservoir for known and undiscovered human pathogens, including viruses. Furthermore, the relative importance of an animal species as a source of human infection is a function of the prevalence of zoonotic agents in that species and the probability of close contact (direct or indirect) with susceptible humans. Clearly, these factors vary geographically, and changes in the patterns of human and animal disease will continue to result from socioeconomic and ecological changes at the human-to-animal interface.
Emergence as a Biological Process
Nonhuman Reservoirs
More than two-thirds of human virus species are zoonotic, that is, they are capable of infecting vertebrate hosts other than Homo sapiens. By far the most important nonhuman host taxa are other mammals, with rodents and ungulates most commonly identified as alternative hosts, followed by primates, carnivores, and bats. A minority of the zoonotic viruses (less than 20%) is also known to infect birds; very few have been reported from vertebrates other than mammals or birds (Antia et al., 2003).
The remaining viruses only naturally infect humans (these are sometimes referred to as “specialist” human pathogens). Some of these (e.g., hepatitis B) may have coevolved with humans over very long time periods; others (e.g., HIV-1) have much more recent origins. Some of both kinds are believed to have originated in other mammal or bird species, including HIV-1 (derived from a simian immunodeficiency virus found in chimpanzees); HIV-2 (sooty mangabeys); SARS virus (horseshoe bats); hepatitis B, human T-lymphotropic virus-1 and -2, dengue, and yellow fever (all primates); human coronavirus OC43, measles, mumps, and smallpox (all livestock); and influenza A (wildfowl). However, the origins of the majority of specialist human viruses is still not known and a gap in knowledge has prompted calls for an “origins initiative” (Simmonds, 2001).
Pathogen Pyramid
A useful conceptual framework for thinking about the emergence of novel viruses is the pathogen pyramid. The pyramid has four levels.
Each level represents a different degree of interaction between pathogens and humans, ranging from exposure through to epidemic spread. Some pathogens are able to progress from one level to the next:
Level 1 represents the exposure of humans to a novel pathogen, such as a virus. The source of viruses of interest is most likely to be other mammals or birds and “exposure” implies any route by which a particular viral infection might be acquired, whether by contact with blood, saliva, or feces; contamination of food and water; or via an arthropod vector. The rate of such exposure is determined by a combination of the distribution and ecology of the nonhuman host and human activities. It is likely that exposure to nonhuman viruses occurs commonly: a process referred to as “chatter.”
Level 2 represents the subset of viruses that are capable of infecting humans—that is, overcoming the “species barrier.” This is likely to reflect both the molecular biology of the virus (for example, is it capable of entering and replicating in human cells?) and the physiology of the exposed human (especially immunocompetence).
Level 3 represents the subset of viruses that cannot only infect humans but can also be transmitted from one human to another (by whatever route, including via arthropod vectors). Again, this will mainly reflect the host–pathogen interaction, especially whether it is possible for the virus to access tissues from which it can exit the host, such as the upper respiratory tract, lower gut, urogenital tract, skin, or blood (for some transmission routes).
Level 4 represents the subset of viruses that are sufficiently transmissible between humans to cause major outbreaks and/or become endemic in human populations without the requirement of a nonhuman reservoir. This equates with the epidemiological condition R O>1, that is, a single primary case generates, on average, more than one secondary case. This is a function of both the transmissibility of the virus (how infectious an infected host is and for how long) and properties of the human population (how human demography and behavior affect opportunities for transmission). From previous reviews of the literature, it is possible to put approximate numbers of virus species at each level of the pyramid. There are greater than 200 viruses at least at level 2. It has been reported that of the virus species known to infect domestic animals (livestock and companion animals)—to which humans are presumably routinely exposed—roughly one-third are also capable of infecting humans. The species barrier exists: but it is clearly very leaky. Based on data from roughly 50% of the viruses that can infect humans can also be transmitted by humans (level 3) and roughly 50% of those are sufficiently transmissible that R O may exceed one (level 4). That a significant minority of viruses (mammalian or avian) should be capable of extensive spread within human populations [or of rapidly becoming so is consistent with experience: there are several examples within the past hundred years alone (HIV-1, SARS, plus variants of influenza A) and many more in the past few millennia (e.g., measles, mumps, rubella, smallpox)]. It is noteworthy that the “shape” of the pathogen pyramid for viruses is very different to that for other kinds of pathogen (bacteria, fungi, protozoa, or helminths), of which much smaller fractions are capable of extensive spread in human populations (Towner et al., 2008). The most straightforward explanation for this is the much more rapid evolution of viruses (especially RNA viruses), allowing them to adapt to a new (human) host much more quickly than other kinds of pathogen.
Drivers of Emergence
“Drivers” of the emergence of novel viruses or other pathogens have been studied recently. These constitute a diverse set of environmental and biological factors, many of which—such as “urbanization” or “land use”—seem intuitively reasonable but are too broad to relate to mechanistic causes of emergence. Moreover, identification of drivers is usually a subjective exercise: there are very few formal tests of the idea that a specific driver is associated with the emergence of a specific pathogen or set of pathogens. In many cases, this would be a challenging exercise: many drivers have only indirect effects on emergence (e.g., climate change, which is often linked with changing distributions of disease vectors); and often an emergence event has multiple causes (good examples would be the emergence of Nipah virus or SARS coronavirus). Other ideas about the drivers of emergence are even harder to test formally. One such is that we are currently living through a “perfect storm” in which many potential drivers of emergence events (such as population growth, urbanization, global travel and trade, and intensification of livestock production) are acting in concert. Upward trends in many drivers can be quantified, but it is not entirely clear that the frequency of emergence events is increasing: one recent study suggested that it increased during the first decade of the HIV/AIDS pandemic but has been decreased thereafter. A slightly different way of thinking about the drivers of emergence is to draw an analogy between emerging pathogens and weeds. The idea here is that there is a sufficient diversity of pathogens available—each with their own biology and epidemiology—that any change in the human environment (but especially in the way that humans interact with other animals, domestic or wild) is likely to favor one pathogen or another, which responds by invading the newly accessible habitat. This implies that emerging pathogens possess different life-history characteristic to established, long-term endemic pathogens (Kilpatrick and Randolph, 2012).
Species Jumps
For viruses, one of the key steps in the emergence process is the jump between one host species and humans. (For other kinds of pathogens, there may be other sources of human exposure, notably environmental sources or the normally commensal skin or gut flora.) Various factors have been examined in terms of their relationship with a pathogen’s ability to jump into a new host species; these include taxonomic relatedness of the hosts, geographical overlap, and host range. The roles of both host relatedness and geographical proximity have been clearly established. Experimentally associations between the degree of cross-species transmission of bat lyssaviruses and both the geographical overlap between bat populations across the United States and the phylogenetically relatedness of the bat species involved have been established. Primate species tended to share more parasitic species if they were both more closely related and had sympatric distributions. A broad host range is also associated with the likelihood of a pathogen emerging or reemerging in human populations. An illustrative case study is bovine spongiform encephalopathy (BSE). After BSE’s emergence in the 1980s, well before it was found to infect humans as Variant Creutzfeldt-Jakob disease (vCJD), it rapidly became apparent that it could infect a wide range of hosts, including carnivores. This was in marked contrast to a much more familiar prion disease, scrapie, which was naturally restricted to sheep and goats. With hindsight, this observation might have led to public health concerns about BSE being raised earlier than they were. Host range is a highly variable trait among viruses: some, such as rabies, can infect a very wide range of mammals; others, such as mumps, specialize on a single species (humans). Moreover, for pathogens generally, host range seems to be phylogenetically labile, with even closely related species having very different host ranges. Hence, the biological basis of host range is relevant to understanding pathogen emergence (Jones et al., 2008).
Cell Receptor Usage and Host Range
One likely biological determinant of the ability of a virus to jump between species is whether or not they use a cell receptor that is highly conserved across different (mammalian) hosts. The viruses that use conserved receptors ought to be more likely to have a broad host range. A comprehensive review identified that, for 88 human virus species, only one cell receptor was identified. Although this was only 40% of the species of interest, 21 (out of 23) families were illustrated; this set constitutes a good cross section of corresponding taxonomic diversity. Of these 88 species, 22 use nonprotein receptors (e.g., heparin sulfate) and, of the remainder, 2 of the proteins were not entered in the UniProt database (making it impossible to determine whether the protein was “conserved” or not), leaving 64 species from 16 families. On the basis of a published studies of virus host ranges, these viruses have been accorded either a “narrow” host range (if the only nonhuman hosts they were known to infect were other primates) or a “broad” host range (if they were known to infect also other kinds of mammals or birds). Using the UniProt database it can be determined whether the cell receptor protein was “conserved” by quantifying the amino acid sequence homology between humans and mice. (For the subset of proteins where amino acid sequences data were also available for cows, pigs, or dogs, very similar patterns were found) The most prominent characteristic of the plot is that there are no instances of human viruses with wide host ranges that do not use highly conserved cell receptors (i.e., more than 90% amino acid sequence homology). Statistical analysis needs correction for phylogenetic correlation; viruses of the common family are both more likely to use the same cell receptor. This can be crudely (but conservatively) allowed by testing for an association between host range and receptor homology at the family; not species, level. This gives a statistically significant result (P=.015). Notably, the use of a conserved receptor is a necessary but not sufficient condition for a virus to have a broad host range encompassing different mammalian orders. It follows that a useful piece of knowledge about a novel mammalian virus, helping to predict whether or not it poses a risk to humans, would be to identify the cell receptor it uses. However, this may not always be practicable: at present, the cell receptor used by over half the viruses that infect humans is not known, and this fraction is considerably smaller for those that infect other mammals. The emergence of new human viruses is a long-standing and ongoing biological process. The first line of defense against emerging viruses is effective surveillance. There are three important key points in surveillance (Greninger et al., 2009). First, emerging viruses are everyone’s problem: the ease with which viruses can disperse, potentially worldwide within days, coupled with the very wide geographical distribution of emergence events, means that a coordinated, global surveillance network is essential if we are to ensure rapid detection of novel viruses. This immediately highlights the enormous national and regional differences in detection capacity, with the vast majority of suitable facilities located in Europe or North America. Second, reporting of unusual disease events is patchy, even once detected, reflecting both governance issues and lack of incentives. Third, we need to consider extending the surveillance effort to other mammal populations as well as humans, because these are the most likely source of new human viruses. Improving the situation will require both political will and considerable investment in infrastructure, human capacity and new tools. However, the benefits are potentially enormous (Gaynor et al., 2007). It is possible to forestall an emerging disease event, as experience with SARS has shown. However, our ability to achieve this is closely linked to our ability to detect such an event, and deliver effective interventions, as rapidly as possible. A better understanding of the emergence of new human viruses as a biological and ecological process will allow us to refine our currently very crude notions of the kinds of pathogens, or the kinds of circumstances, we should be most concerned about, and so direct our efforts at detection and prevention more efficiently.
Conclusions and Future Prospects
The frequent emergence or reemergence of deadly viral infections has significantly affected human health despite extraordinary progress in biomedical knowledge. High population density, rampant constructions, poor sanitation, changing climate, and the introduction of anthropophilic vectors create selective pressure on hosts and pathogen reservoirs. Although we know that a substantial fraction of well-identified mammalian viruses is responsible for human etiology, there are large numbers of evolving viruses waiting to infect and adapt. The unpredictable nature of novel infections, the rare occasions of outbreaks, the small numbers of confirmed cases, the asymptomatic occurrence, and occurrence in remote areas severely hamper the assessment of control and preventive modalities. Surveillance of the emerging human viruses must include livestock, wild animals, and potential arthropod vectors as well as their environments at an international level of coordination. Furthermore, individuals, such as livestock herders, zookeepers, hunters, rangers, and veterinarians working with reservoir or high-risk animals, must take hygienic and general universal containment measures.
Efficient vaccines are already available against important human viruses, such as HBV and human papillomavirus. Even though drug resistance in HBV and HIV has accelerated alarmingly, new generations of broad-spectrum antiviral agents as combination regimens hold promise. Moreover, specific geographical regions or interfaces between humans, livestock, wildlife, and the environment should be the targets for intense surveillance. In this way, further extensive research will surely enhance our understanding and capacity for predicting new pandemics and preparing control measures in advance. Furthermore, viral pathogen discovery is of critical importance to clinical microbiology, infectious diseases, and public health. Genomic approaches for pathogen discovery, including consensus PCR, microarrays, and unbiased next-generation sequencing (NGS), have the capacity to comprehensively identify novel microbes present in clinical samples. Although numerous challenges remain to be addressed, including the bioinformatics analysis and interpretation of large datasets, these technologies have been successful in rapidly identifying emerging outbreak threats, screening vaccines and other biological products for microbial contamination, and discovering novel viruses associated with both acute and chronic illnesses. Downstream studies such as genome assembly, epidemiologic screening, and cultures system or animal model of infection are necessary to establish an association of a candidate pathogen with disease. Although sometimes derided as a “fishing expedition” or a needle in haystack approach, pathogen discovery is, in actuality, a highly worthwhile scientific endeavor. Without a cause identified for many presumed infectious diseases, it is not possible to conduct downstream investigations in pathogenesis and host–microbial interactions nor is it possible to design effective vaccines or antimicrobial drugs to combat the associated illness. Potential applications of pathogen discovery range from outbreak investigation of emerging pathogens, to screening of blood products, vaccines, and other biologics for viral contaminants, to clinical diagnosis of unknown acute or chronic infectious diseases. The current availability of state-of-the-art genomic technologies such as pan-microbial microarrays and NGS provides an unprecedented opportunity to “cast a wide net” and surveys the full breadth of as-yet undiscovered pathogens in nature that pose significant threats to human health.
Acknowledgments
We acknowledge the total support provided by the Resource Centre National Institute of Virology, Pune, India, under the Indian Council of Medical Research, Directorate of Health Research and VIral Research and Diagnostic Laboratory (VRDL) India, in establishing VRDL at Department of Microbiology, Govt. Medical College, Jammu in Jammu and Kashmir, India.
References
- Anderson L.J., Tong S. Update on SARS research and other possibly zoonotic coronaviruses. Int. J. Antimicrob. Agents. 2010;36:S21–S25. doi: 10.1016/j.ijantimicag.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antia R., Regoes R.R., Koella J.C., Bergstrom C.T. The role of evolution in the emergence of infectious diseases. Nature. 2003;426:658–661. doi: 10.1038/nature02104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebber D.P., Marriot F.H.C., Gaston K.J., Harris S.A., Scotland R.W. Predicting unknown species numbers using discovery curves. Proc. R. Soc. B. 2007;274:1651–1658. doi: 10.1098/rspb.2007.0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briese T. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from Southern Africa. PLoS Pathog. 2009;5:e1000455. doi: 10.1371/journal.ppat.1000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua K.B. A previously unknown reovirus of bat origin is associated with an acute respiratory disease in humans. Proc. Natl. Acad. Sci. U.S.A. 2007;104:11 424–11 429. doi: 10.1073/pnas.0701372104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaveland S., Laurenson M.K., Taylor L.H. Diseases of humans and their domestic mammals: pathogen characteristics, host range and the risk of emergence. Philos. Trans. R. Soc. Lond. B. 2001;356:991–999. doi: 10.1098/rstb.2001.0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaveland S., Haydon D.T., Taylor L. Overviews of pathogen emergence: which pathogens emerge, when and why? Curr. Top. Microbiol. Immunol. 2007;315:85–111. doi: 10.1007/978-3-540-70962-6_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman V.M., Eckerle I., Bleicker T. Detection of a novel human coronavirus by real-time reverse-transcription polymerase chain reaction. Euro Surveill. 2012;17(39) doi: 10.2807/ese.17.39.20285-en. pii: 20285. [DOI] [PubMed] [Google Scholar]
- Gaynor A.M. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog. 2007;3:e64. doi: 10.1371/journal.ppat.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greninger A.I., Runckel C., Chiu C.Y., Haggerty T., Parsonnet J., Ganem D. The complete genome of klassevirus—a novel picornavirus in pediatric stool. Virol. J. 2009;6:82. doi: 10.1186/1743-422X-6-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K.E., Patel N.G., Levy M.A., Storeygard A., Balk D., Gittleman J.L. Global trends in emerging infectious diseases. Nature. 2008;451:990–994. doi: 10.1038/nature06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling M.J., Rohani P. Modelling Infectious Diseases in Humans and Animals. Princeton University Press; Princeton, NJ: 2008. [Google Scholar]
- Keusch G.T., Pappaioanou M., Gonzalez M.C., Scott K.A., Tsai P., editors. Sustaining Global Surveillance and Response to Emerging Zoonotic Diseases. The National Academies Press; Washington, DC: 2009. [PubMed] [Google Scholar]
- Kilpatrick A.M., Randolph S.E. Drivers, dynamics, and control of emerging vectorborne zoonotic diseases. Lancet. 2012;380:1946–1955. doi: 10.1016/S0140-6736(12)61151-9. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Barry P., Yeh E., Glaser C., Schnurr D., Delwart E. Identification of a novel human gammapapillomavirus species. J. Gen. Virol. 2009;90:2413–2417. doi: 10.1099/vir.0.012344-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P. Reconstructing the origins of human hepatitis viruses. Philos. Trans. R. Soc. Lond. B. 2001;356:1013–1026. doi: 10.1098/rstb.2001.0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor L.H., Latham S.M., Woolhouse M.E.J. Risk factors for human disease emergence. Philos. Trans. R. Soc. Lond. B. 2001;356:983–989. doi: 10.1098/rstb.2001.0888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towner J.S. Newly discovered Ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog. 2008;4:e1000212. doi: 10.1371/journal.ppat.1000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe N.D. Naturally acquired simian retrovirus infections in central African hunters. Lancet. 2004;363:932–937. doi: 10.1016/S0140-6736(04)15787-5. [DOI] [PubMed] [Google Scholar]
- Wolfe N.D., Dunavan C.P., Diamond J. Origins of major human infectious diseases. Nature. 2007;447:279–283. doi: 10.1038/nature05775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse M., Antia R. Emergence of new infectious diseases. In: Stearns S.C., Koella J.K., editors. Evolution in Health and Disease. second ed. Oxford University Press; Oxford, UK: 2008. pp. 215–228. [Google Scholar]
- Woolhouse M.E.J., Taylor L.H., Haydon D.T. Population biology of multi-host pathogens. Science. 2001;292:1109–1112. doi: 10.1126/science.1059026. [DOI] [PubMed] [Google Scholar]