

Abstract
Prominent in the current stage of drug development, antiviral compounds can be efficiently prepared through cycloaddition reactions. The chapter reports the use of classical Diels–Alder and their hetero version for the design and synthesis of compounds that were tested for their antiviral activities against a variety of viruses. Furthermore, 1,3-dipolar cycloaddition reactions of selected 1,3-dipoles, such as azides, nitrones, and nitrile oxides, are reviewed in the light of their application in the preparation of key intermediates for antiviral synthesis. A few examples of [2+2] cycloaddition reactions are also presented. The products obtained from these pericyclic reaction approaches were all tested for their activities in terms of blocking the virus replication, and the relevant biological data are highlighted.
Keywords: Cycloaddition reactions; antiviral compounds; Diels–Alder reactions; hetero Diels–Alder reactions; 1,3-dipolar cycloadditions; azides; nitrones; nitrile oxides; nitrile imines; azomethine ylides; [2+2] cycloadditions
Cycloaddition: general aspects
Cycloaddition reactions, whose applications are the main topic of the present book and which were classically classified within the group of pericyclic reactions, have been exhaustively studied by many research groups during the last 50 years. These investigations have been pursued both on the basis of experimental research and upon theoretical methodologies based on ab initio calculations.1 The evolution of theory and the use of increasingly powerful computers and software increased the level of theory applied to more complex molecular systems. The electron density analysis, as proposed by Domingo’s Molecular Electron Density theory,2 promises to go into deep of cycloaddition reaction mechanisms, and the methods seem to offer novel opportunities with respect to the studies based on simple molecular orbital (MO) interactions, as well as on the Frontier MO (FMO) theory, typically employed in the last century. New theoretical investigations, based on accurate experimental studies, built new, solid, reactivity models for these relevant types of organic reactions that allow a profound rationalization; the new findings imply strong and significant repercussions on cycloaddition reactions and applications in modern organic chemistry and synthesis, specifically. The matter is quite complex, and more and more discussions will appear in literature in the next years as the level of complexity of the theoretical investigation increases with the development of new computational clusters.
The complex matter regarding mechanisms and potential synthetic applications of cycloaddition reactions has been and is still constantly discussed in literature as the level of complexity of theoretical investigations and solution demands increases. The synthesis of molecules with high level of complexity and increasing importance, mainly in the medicinal chemistry field, places the topic of cycloaddition in front of the dichotomy: to be useful and participate in the pivotal step of a synthesis or to be relegated to a simple chemical curiosity, not applicable to solve tremendous needs in modern organic chemistry. Several books and reviews3 have been published from the late 1960s to nowadays dealing with general and more specific aspects of cycloaddition chemistry.
Cycloaddition reactions are a mature topic of organic chemistry but abundantly used in different areas where chemistry represents the pivotal step for reaching the desired target. Books dedicated to cycloaddition reactions deal typically with the synthesis of heterocycles, general organic synthesis applications, specific applications in natural product synthesis, or they describe the use of a class of organic compounds as partners in cycloaddition reactions.3 In 2016, a book appeared in the Topic in Current Chemistry Collections dealing with the bioorthogonal chemistry edited by Vrabel and Carrell. This represents a first example of a book dedicated to the view of cycloadditions in modern organic chemistry. However, other subjects still demand a general review since in recent years pericyclic reactions were extensively applied to different chemistry areas such as chemical biology, biological processes, catalyzed cycloaddition reactions, delivery of antiviral or anticancer drugs, targeting of biological active molecules, material chemistry with applications in medicine, photovoltaic processes, photochemistry, energy harvesting, and so on. None of these topics, where cycloaddition reactions spread out, has received a unifying overview in a single book that could offer a solid background for future development in these and other subjects. Moreover, the chronological structure of the references regarding a general topic allows the reader to recognize the relevance of specific subjects during the past decades and the evolution of the synthesis with respect to specific active compounds. This book aims to fill the gap, shedding a new light over pericyclic reactions and demonstrating how these valuable tools may elegantly solve synthetic and mechanistic problems in modern organic chemistry.
The Diels–Alder (DA) reactions are one of the general classes of cycloadditions, which include also hetero DA (HDA) reactions, [2+2] cycloadditions, and 1,3-dipolar cycloaddition reactions (Fig. 1.1 ).
Figure 1.1.

General schemes for [4+2], [2+2], and [3+2] cycloaddition reactions.
Within this frame, both the inter- and intramolecular versions of these reactions are widely used in organic synthesis. In many cases, these reactions occur at room temperature, but harsher reaction conditions are sometimes required to get the final compounds. Moreover, the use of catalysts has been increasingly developed not only in the way to accelerate the reactions but mainly to ensure enantioselective and diasteroselective processes.
Most DA reactions involve a diene bearing electron-donating substituents and an electron-poor dienophile. However, DA exists also in the inverse electron demand version where the substituents located on diene and dienophile display reversed electronic properties with respect to the classical DA reaction. The two cycloaddends must have complementary electronic character to ensure the usefulness of the DA cycloaddition reactions (Fig. 1.2 ).
Figure 1.2.
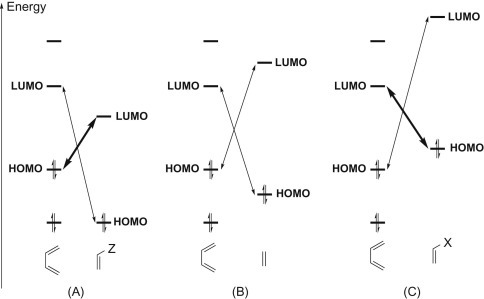
FMO relative energies. (A) Dienophile with low-energy LUMO; (B) diene+dienophile; (C) dienophile with high-energy HOMO. FMO, Frontier molecular orbital.
The monumental work of Rolf Huisgen et al. in the 1960s led to the general concept of 1,3-dipolar cycloaddition reactions. These extremely valuable synthetic tools permit the preparation of molecules of higher complexity than the reactants. Few reactions rival these processes in synthetic utility, mainly in the field of heterocyclic chemistry. Five-membered heterocycles are the synthetic target of these reactions and their ability to produce heterocycles extends the importance of the 1,3-dipolar cycloaddition to other areas of organic chemistry, not strictly related to the ring formation processes. Moreover, the introduction of new chiral catalysts allowed for the control of new stereocenters derived from the cycloaddition reactions with the valuable stereochemical outcome borne by the newly synthesized molecules. If these molecules are drugs, this is a key point for determining the success of a synthetic approach.
Mechanisms, reactivities, and selectivities of these reactions have been completely rationalized according to modern theoretical investigations. However, the traditional references to these concepts will still be used to illustrate basic ideas that are in the background of the subjects treated in this book. Figures and schemes will focus reader attention on the pivotal steps of the syntheses highlighting the role of cycloaddition reaction in achieving the best results both in terms of chemical yields and selectivities and in terms of biological activities.
Cycloaddition for antiviral compounds
The virus
The smallest and simplest biological structure, called a virus (from Latin: poison), lacks any type of cellular organization and is forced to live within cells, being intracellular parasites. Viral infections deeply marked human history, as the Spanish conquest of South American territories showed in a paradigmatic picture of death. The development of new and powerful diagnostic methodologies allowed for an increasing capacity to identify new viruses. From the 1960s forward, new frightening hemorrhagic African fevers were discovered and attributed to the filoviruses, while the AIDS appeared in 1981, although HIV was already present in the human population in the 1930s.
Another emergency is the reappearance of viral infections due to virus mutations and resistance to antiviral therapies. This is the case of the influenza viruses; the virus surface proteins have changed and are not recognizable by the human immune system any longer. Sometimes the virus genoma can be exchanged from humans to animals and back to humans, determining new pandemic situations and social alarms. Viral infections are quite detrimental since, contrary to bacteria, viruses cannot be defeated with antibiotics. Until very recently, a small number of antiviral compounds could be used in the pharmacological treatment of these diseases. Luckily, not only are viruses in progress, so are chemical research and synthetic approaches to new and powerful methods for the contrast to viral infections. The demand of antiviral compounds is constantly increasing and chemistry is constantly under examination to furnish novel strategies for a large-scale preparation of active molecules.4
Diels–Alder and hetero Diels–Alder reactions and the antivirals
To start discussing the application of DA cycloaddition to the synthesis of value-added molecules, it is worth citing the case of the influenza viruses and specifically the H1N1 virus and how neuraminidase (NA) inhibitors can be prepared with different synthetic approaches, having in common the pivotal step represented by the DA cycloaddition reaction. The research subject offers also a brilliant example of joint efforts from academia and pharmaceutical industries. The influenza infection is known to be a serious health concern, and the cause of morbidity and mortality in many countries. Current options for its treatment and prevention sometimes have severe limitations, underscoring the need for new, effective antiinfluenza agents. Influenza NA is one of the two major surface glycoproteins expressed by influenza viruses. NA promotes the cleavage of the terminal sialic acid residues attached to glycoproteins and glycolipids.5 Therefore, NA represents a potential target for developing antiinfluenza agents. In fact, in 1999 a merged research group by Gilead Sciences Inc. and Roche Discovery proposed the synthesis of a 1,4,5,6-tetrahydropyridazine derivative and its C-5 epimer, which possess side chains similar to compound GS4071. These compounds were synthesized via HDA reaction, and evaluated as influenza NA inhibitors. Both compounds exhibited a μM range of influenza NA inhibitory activity.6 The strategy followed is based on an HDA cycloaddition reaction between the hydrazone 3 and alkene 4 in the presence of sodium carbonate in acetonitrile to afford a 1:3 mixture of the desired cycloadduct 5 and its regioisomer in 87% yield (Scheme 1.1 ).
Scheme 1.1.

Synthetic strategy for (5S,6S)-6-acetamido-5-amino-1-(2-ethylbutanoyl)-1,4,5,6-tetrahydropyridazine-3-carboxylic acid 6.
After removal of the Boc group on 5 and saponification, the amino compound 6 could be easily separated from its regioisomer by simple chromatography. The influenza NA inhibitory activity of compound 6 is reported in Scheme 1.1. Compound 6, having a similar side chain and stereochemistry (trans) to GS4071, exhibited IC50 of 6 μM against influenza A and 62 μM against influenza B. Increased activity was resulted from converting the amino group of 6 into the guanidine functionality. The relatively weak NA inhibitory activity of 6 compared to that of GS4071 can be explained from conformational and binding studies. These investigations revealed that the NH2 and NHAc groups of 6 occupy the pseudo-axial positions, rather than the normally preferred pseudo-equatorial positions that are adopted on binding to influenza NA, indicating that an energy penalty is incurred upon the binding. In addition, the X-ray structure of 6 bound to NA reveals a different binding mode for the 3-pentyl side chain as compared with that of GS4071. In the case of GS4071, the two ethyl groups of the 3-pentyl side chain bind in two different pockets. However, presumably owing to the partly planar nature of the amide bond, the 3-pentyl side chain of 6 poorly fits and exposes the hydrophobic group to water.6
A fruitful collaboration between the University of Manchester and a research group of a big chemical company, GlaxoSmithKline, was established in 2001 for the synthesis of new inhibitors of influenza virus sialidases. Structurally related cyclohexene derivatives, Zanamivir 7 and GS4071 (administered in prodrug form as Oseltamivir), are known to interfere with viral replication by acting as potent inhibitors of virus sialidases. These enzymes, located on the viral surface, cleave the α-glycosidic bond of terminal sialic acid residues. Zanamivir 7 and GS4071 bind to the active site of a sialidase in an analogous manner. Whereas the trihydroxypropyl side chain binds to a hydrophilic pocket via hydrogen bonding interactions, the pentyloxy function is accommodated in a hydrophobic pocket. The importance of the substituent structures has been demonstrated in several binding studies and structural modification by replacement of selected substituents aimed to tune properly the inhibitory activity. For example, the replacement of the trihydroxypropyl side chain by a dialkylcarbamoyl entity leads to compounds that retain potency against influenza A sialidase but lose effectiveness against influenza B sialidase. Surprisingly, in this series, the guanidino function appears to play no role in binding to influenza A sialidase. Also the location of the C C double bond seems to be critical and the retrosynthetic analysis has to take into account the generation of regioisomers. The synthesis of two novel compounds, whose inhibition activity was evaluated in comparison with the GS4071, is based upon a DA tactic between the nitro acrylamide 8 and the diene 9 (Scheme 1.2 ).7
Scheme 1.2.

Synthetic strategy for Zanamivir-like compounds 14a,b.
The reactions afford a separable 2:1 mixture of the regioisomeric cycloadducts 10a,b in 40% overall yield. The synthesis proceeds by using just the regioisomer 10a that was reduced by Al(Hg) and acetylated to give 11, subsequently transformed in three steps into the α-hydroxy ester 12 in 53% yield as a mixture of diastereoisomers (ratio 3:1). Martin’s sulfurane dehydration reaction provided a 1:1.3 mixture of compounds 14a and 14b (68% yield after chromatography). Finally, hydrolysis of the ester 13a [LiOH, MeOH–H2O (9:1), 2 h] was uneventful, leading to the desired acid 14a in 70% yield. Under similar conditions, the ester 13b afforded a 4:2:2:1 mixture of products; the most prevalent component [isolated in a near-pure state as a syrup (24% yield) by addition of EtOAc to the mixture and decantation of the liquid] was identified as the required acid 14b.
The inhibitory activities of the acids 14a,b, compared with Zanamivir 7, against influenza A and B sialidases are shown in Scheme 1.2 in the inset. Both compounds are highly selective for influenza A sialidase by a factor of over 700 for 14a and over 1300 for 14b. Against influenza A sialidase, the acid 14b is about 3 times less active than Zanamivir 7 whereas the acid 14a is about 40 times less active. These findings indicate that the presence of a basic function is not a requirement for high potency. They also reveal that the position of the double bond contributes to, but is not critical for high activity against influenza A sialidase.7
The same authors further explored the substituent effects as well as the potentialities of the synthesized compounds in racemic forms by preparing the nitro-cyclohexene 17 by DA cycloaddition of the nitro-olefin 15a and the diene 16. The product was obtained in good yields (56%) and submitted to a sequence of well-established chemical conversions to get the final acids 18a,b in excellent yields (Scheme 1.3 ).8
Scheme 1.3.

[4+2] Cycloaddition reaction strategy for GS4071-like compounds.
The inhibitory activities of the acids 18a,b against influenza A and B sialidases in comparison with GS4071 are shown in Scheme 1.3. Only compound 18a showed any appreciable activity. Compared with GS4071, it was 70 times less active against influenza A sialidase and 280 times less active against influenza B sialidase. These results indicate that the amino group of GS4071 contributes to but is not obligatory for high potency, although it is important in binding to the sialidase. The poor activity of the acid 18b demonstrates the inadequacy of the syn-arrangement of the pentoxy and acetylamino groups, a relevant structure–activity relationship (SAR) information for future-planned syntheses of potential active analogs. From the synthetic point of view, the excellent endo-stereoselectivity realized in the DA reactions is attributable to the presence of the O-acetyl substituent in the oxybuta-1,3-diene. In conclusion, the sialidase inhibitory properties of the acids 18a,b provide new insights into SAR of antiinfluenza agents.
The relevance of the contrast to influenza pandemic is a worldwide concern. As told before, pandemic is attributed to a mutation of influenza virus proteins that allows the virus to evade the human immune system. As result of today’s extensive global transport, a local influenza epidemic cannot be restricted to a specific area. Because of the high mutation frequency of influenza virus, an effective antiinfluenza drug should target fundamental molecular processes that are essential and specific for the life cycle of the virus. This principle is based on the hypothesis that structures of fundamental proteins are conserved even in mutant viruses, and NA belongs to these virus proteins. An influenza virion budding from an infected cell binds to a terminal sialic acid residue on the host cell surface glycoprotein with hemagglutinin (HA: Fig. 1.3 ). NA hydrolytically cleaves the glycosidic bond of sialic acid to release the virus from the host cell surface. This process liberates the budding virion from the infected cell and is essential for spreading the infection. As expected, the active site of NA is highly conserved across the influenza A and B virus strains. Therefore, an NA inhibitor is a prime candidate for broad spectrum antiinfluenza drugs.
Figure 1.3.

Schematic representation of an influenza virion budding from a host cell.
From: Shibasaki, M.; Kanai, M. Eur. J. Org. Chem. 2008, 1839–1850.
Binding studies provided molecular-level structural information for the design of NA inhibitors. Several research groups, both from academia and pharmaceutical industries, have investigated the topic and proposed different synthetic approaches to potential active molecules. Two antiinfluenza drugs, Zanamivir (7: GlaxoSmithKlein’s Relenza)9 and Oseltamivir phosphate (19: Gilead’s Tamiflu, marketed by Roche),10 inhibit NA by strongly binding its active site. The IC50 values of these drugs are in the nanomolar range. Oseltamivir phosphate is an orally active prodrug, active in the corresponding carboxylic acid form, whereas Zanamivir has low bioavailability and is administered by inhalation. These drugs are considered to be effective to treat H5N1 influenza. Although these molecules are relatively small, the development of a practical synthesis that can provide the quantity required worldwide is highly challenging. Some of the literature-proposed synthetic strategies for Oseltamivir phosphate are based on DA strategies.
The Roche purpose of process chemistry is to provide the target compound on a large scale (ton scale) as efficiently as possible without using toxic and hazardous reagents. A successful DA route in this direction is shown in Scheme 1.4 . This synthesis started with a racemic DA reaction between furan and ethyl acrylate. The reaction proceeded in the presence of 1 equiv. ZnCl2 affording the thermodynamically more stable exo-20 as major product (ratio: 9:1). Enzymatic resolution of 20 allowed access to the desired (R) isomer 21 with 97% ee at 75% conversion (20% yield). Compound 21 was converted into the endo aziridine 22 by treatment with diphenylphosphoryl azide through [3+2] cycloaddition. After transesterification at the phosphate moiety and aziridine-opening with pentan-3-ol, compound 23 was obtained and submitted to hydrolysis of the phosphoryl amide and hydrochloride formation to give 24. This latter can be converted into 19 by aziridine formation and introduction of an amino functionality (Scheme 1.4).11
Scheme 1.4.
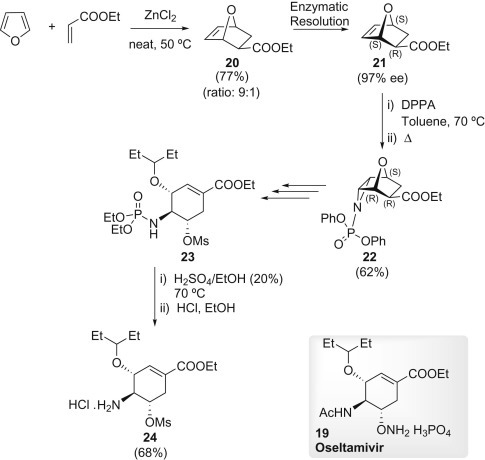
Synthetic strategy for the ethyl (3R,4S,5S)-4-amino-5-[(methylsulfonyl)oxy]-3-(pentan-3-yloxy)cyclohex-1-ene-1-carboxylate 24.
In 2006, Corey et al. reported a concise azide-free synthesis of 19 starting from the catalyzed enantioselective DA reaction developed in his group (Scheme 1.5 ).12 The DA reaction between butadiene and trifluoroethyl acrylate proceeded at room temperature in a sealed tube without solvent in the presence of 10 mol% triflimide-activated oxazaborolidine catalyst 25, giving the cyclohexene derivative 26 in 97% yield and with 97% ee. The excellent enantioselectivity was explained by a transition state model, in which butadiene approached the activated dienophile from the Re-face opposite the shielding phenyl group of the catalyst. The chiral ligand was recovered efficiently and ammonolysis of 26 followed by conversion into the N-Boc-imide 27 (oxalyl chloride and tBuOH) allowed for the successive bromo-lactamization performed in the presence of LiOtBu and NBS, affording 28. After elimination of HBr with Diazabicycloundecene (DBU), allylic bromination followed by ethanolysis under basic conditions produced dienyl ester 29. Regio- and stereoselective bromoamidation of the C3 C4 double bond was achieved by the originally developed method using N-bromoacetamide in the presence of a catalytic amount of SnBr4 as an activator of the Br+ donor in acetonitrile as solvent. The reaction proceeded through bromonium ion formation with attractive interactions between the carbonyl oxygen of the Boc group and Br+, giving 30.
Scheme 1.5.

Synthesis of Oseltamivir phosphate 19.
The conversion of 30 into Oseltamivir phosphate (19) was straightforward. Treatment of 30 with tetrabutylammonium hexamethyldisilazide produced an aziridine intermediate; the three-membered ring-opening was accomplished through a Lewis acid-catalyzed (Cu2+) reaction with pentan-3-ol. The cleavage of the Boc group was conducted with H3PO4, giving 19. Besides all the described chemical transformations, it can be clearly seen that the pivotal step of Corey’s synthesis is the catalytic enantioselective DA reaction affording the enantiomerically enriched starting compound 26.
The Shibasaki and Kanai’s third-generation synthesis of 19 started from the DA reaction between the (buta-1,3-dien-1-yloxy)trimethylsilane and fumaroyl chloride, run at room temperature over 2 h.13 After the reaction was complete, TMSN3 and 4-Dimethylamino pyridine (DMAP) were added to the mixture, and the corresponding acylazide was formed. On quenching of the reaction with HCl, desilylation took place and compound 31 was obtained in 55% yield. Although the DA reaction afforded a 2:1 (endo/exo) mixture of diastereomers, the undesired exo isomer selectively decomposed during the acidic cleavage of the trimethylsilyl (TMS) ether. A Curtius rearrangement was conducted by heating a tBuOH solution of acylazide 31 at reflux. In this process, two nitrogen atoms at C-4 and C-5 were differentiated, affording 32. Selective hydrolysis of the cyclic carbonate with LiOH and subsequent N-acetylation produced 33, which was oxidized to enone 34 under modified Moffat conditions with isobutyric anhydride (Scheme 1.6 ).
Scheme 1.6.

Synthesis of Oseltamivir phosphate 19 via azido-strategy.
The use of the sterically bulky anhydride in this step was essential in order to avoid O-acylation. At this stage, the enantiomers were separated by chiral HPLC and the Ni-mediated conjugate addition of TMSCN, followed by α-bromination, elimination of HBr, and stereoselective reduction with LiAl-(OtBu)3H, afforded 35 the same intermediate as in the second-generation synthesis performed by the authors.
Fukuyama’s synthesis of 19, reported in 2007, started with a novel asymmetric DA reaction between the dihydropyridine derivative 36 and acrolein, promoted by MacMillan catalyst (R)-5-benzyl-2,2,3-trimethylimidazolidin-4-one (Scheme 1.7 ), affording the bicyclic aldehyde 37.14 Kraus oxidation followed by bromo-lactonization produced 38 in 26% yield. The moderate yield was attributed to the asymmetric DA reaction, but 38 was easily purified through aqueous acid/base partition and crystallization. Chemically and enantiomerically pure 38 was obtained without column chromatography. The excellent enantioselectivity in the DA reaction can be explained by the model15 originally proposed by MacMillan: the diene 36 would approach the activated dienophile from the side opposite to the bulky benzyl group located on the catalyst. After the Cbz group of 38 had been exchanged for a Boc group, oxidation with a catalytic amount of RuO2·nH2O and a stoichiometric amount of NaIO4 afforded an imide intermediate. The RuO2·nH2O was recovered for reuse by quenching of the reaction with iPrOH and filtration. Ammonolysis of the lactone followed by O-mesylation produced amide 39, which was subjected to Hofmann rearrangement in the presence of PhI(OAc)2 and allyl alcohol to give an allyl carbamate. Treatment with NaOEt afforded aziridine 40 through ethanolysis of the imide, aziridination, and elimination of HBr in one-pot fashion. An aziridine-opening reaction with pentan-3-ol, cleavage of the Boc group with trifluoroacetic acid (TFA), N-acetylation, deprotection of C-5 amine, and phosphate salt formation produced 19.16
Scheme 1.7.
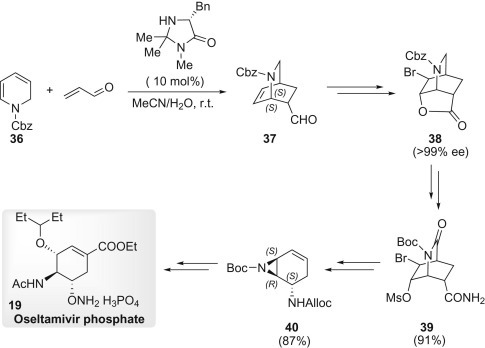
Synthesis of Oseltamivir phosphate 19.
Although the yield of the catalytic asymmetric DA reaction requires further improvement, Fukuyama’s 14-step synthesis is within the practical range thanks to the following points: (1) the starting material is the inexpensive pyridine, (2) the number of column purifications is minimal, and (3) safe and common reagents are used for synthesis.
Although Oseltamivir (19) is a relatively small molecule, developing a practical synthesis that can satisfy worldwide demand in an environmentally friendly and safe way is quite challenging. Roche’s current process synthesis relies on naturally occurring shikimic acid as the starting material. There is no doubt, however, that artificial asymmetric catalysts can provide potentially more straightforward starting materials. The synthetic routes developed by three academic groups (the Corey, Shibasaki and Kanai, and Fukuyama groups) support this viewpoint.17 New synthetic methodologies allow chemists to design conceptually new strategies and, in this sense, improvement in synthetic efficiency is intimately related to progress in synthetic methodologies. Another important research vector is to identify molecules with improved pharmacological effects. Like other drugs, Oseltamivir phosphate is not perfect. The emergence of an Oseltamivir-resistant influenza virus and possible side effects on the human nervous system are two major concerns relating to Oseltamivir. Flexible synthetic routes should allow for the synthesis of analogs in order to address these problems. Hence, organic synthesis can actively contribute to answer biologic questions.
Before a safe and effective vaccine is available to protect the possible pandemic flu, NA inhibitors are the only therapy we have. Because of the oral bioavailability and good toleration, Roche’s Tamiflu is recommended as the best choice for therapeutics and prophylaxis of influenza. The spread of the avian virus H5N1 makes the situation of Tamiflu supply and demand increasingly serious. It should be rational to further optimize current manufacturing processes based on increasing the supply of shikimic and quinic acids through improved isolation, fermentation, or synthesis. On the other hand, new synthetic routes that do not involve complex natural products as precursors, but rather cheap and widely available commodity chemicals, should also be developed. Some academic chemists have reported significant results of experimental study. Discovery of new functional molecules is an extremely important task for synthetic chemists.18
Regarding the contrast to influenza viruses, an interesting short-step synthesis of a model core structure associated with halenaquinone and related natural compounds19 is reported for the preparation of the furan-fused tetracyclic compound 43 that has a notable antiviral activity and was concisely synthesized on the basis of o-quinodimethane chemistry (Scheme 1.8 ). Again, the pivotal step is represented by the intramolecular DA reaction of the quinodimethane intermediate 42 with the furan moiety that acts as dienophile.
Scheme 1.8.

Intramolecular DA reaction of the quinodimethane intermediate 42. MIC values for 43 and 44 given in parentheses. DA, Diels–Alder.
The Severe Acute Respiratory Syndrome (SARs) of the congeners of 43 were investigated, aiming at the discovery of new candidates for antiviral drugs. As a preliminary study, the antiviral activity was surveyed using the assay method of HA titers. Several derivatives having more potent antiviral activity than the lead compound 43 were obtained and, especially, the Triisopropylsilyl (TIPS) derivative 44 was revealed to have the lowest minimum inhibitory concentration (MIC) value and good therapeutic index.20 These results prompted the authors to investigate the possibility of revealing dihydrofuran-fused compounds as a new class of antiinfluenza agents possessing a novel structural characteristic.21 The inhibitory activity of the some compounds including 43 and 44 was investigated against viral growth in Madin–Darby canine kidney (MDCK) cells using influenza A/Aichi/2/68 (H3N2 subtype) virus strain at 10 mM drug concentration. The results showed that several compounds inhibited the virus growth and could have potential as new antiinfluenza agents. In particular, some modified structure of 43 exhibited potent activity, suppressing the virus proliferation up to ca. 30% of control.
In summary, influenza being a long-standing health problem, the developments of NA inhibitors promise new hopes for virus treatment. Tamiflu, the phosphate salt of Oseltamivir (19), is an orally available NA inhibitor designed to have a cyclohexene scaffold to mimic the structure of the oxonium-like intermediate 47 in the enzymatic cleavage of the terminal sialoside from cell receptors (Scheme 1.9 ). Many new synthetic procedures of 19 without using shikimic acid have been explored because current industrial manufacturing may have shortages of shikimic acid for global supply during influenza pandemics. In these shikimate-independent syntheses, several different approaches can be utilized to construct the multiple substituted cyclohexene ring: (1) use of existing six-membered rings, (2) construction by DA reactions, (3) construction by intramolecular aldol, Dieckmann and Wittig reactions, (4) construction by tandem Michael–Wittig reactions, (5) construction by olefin metathesis, and (6) construction by Claisen rearrangement. Phosphonate group is generally used as a bioisostere of carboxylate in drug design. The syntheses of Tamiphosphor (49) and Zanaphosphor (50), which are the phosphonate congeners of 19 and 7, were accomplished and shown to possess potent inhibitory activities against avian and human influenza viruses, including the Oseltamivir-resistant strain, according to the NA inhibition, cell-based assay, and mice experiments.
Scheme 1.9.
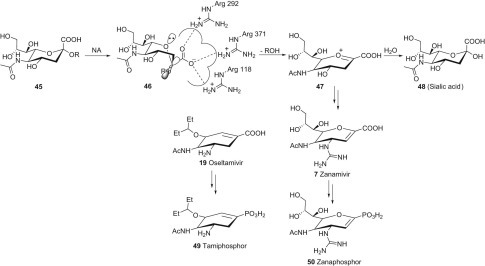
Enzymatic process toward NA inhibitor syntheses. NA, Neuraminidase.
For the first time, phosphonate monoalkyl esters are proved to be real active drugs, but not prodrugs of the parental phosphonic acids. A review covers the design, synthesis, and biological activity of phosphonate compounds as effective antiinfluenza agents.22
From the mechanistic point of view, the synthetic approach just reported finds useful applications in other syntheses. In fact, the o-quinodimethane intramolecular cycloaddition was applied for the total synthesis of estrane type of steroid.23 For example, the first asymmetric total synthesis of (+)-chenodeoxycholic acid was achieved via the intramolecular cycloaddition of the o-quinodimethane 51 as a key step (Scheme 1.10 ).24
Scheme 1.10.

Intramolecular synthesis of (+)-Chenodeoxycholic acid.
More flexible and efficient routes to both aromatic and nonaromatic steroids were planned on the basis of this type of reaction sequence. In this route, the trans-benzoperhydroindane 53 was set as a key compound for preparing either A-trienic or nonaromatic steroids (54 or 55, respectively) by easy manipulation of its benzene ring. In turn, 53 could be accessed by intramolecular cycloaddition of the o-quinodimethane 52, generated in situ by thermolysis of benzocyclobutene 51 (Scheme 1.10).
As a typical example of the application of this methodology, the synthesis of (±)-19-norspironolactone 57 is shown in Scheme 1.11 .25 The synthetic strategy is characterized by the one-step creation of B, C, D, and E rings in a stereoselective manner via intramolecular [4+2] cycloaddition reaction of the o-quinodimethanes 56, and then A-ring formation followed by functionalization of the C-7 position. These results show that all the investigated reactions proceed with high stereoselectivity, leading to the preferred formation of the trans–anti isomers. Thus it seems possible that the high stereoselectivity might reflect the severe steric interactions present in the endo transition states T3 and T4 and the exo transition state T2 in comparison with the exo transition state T1.
Scheme 1.11.

Transition Structures (TSs) for the intramolecular synthesis of (±)-19-norspironolactone 57.
Thus we could obtain information about the stereochemical outcome of the DA cycloaddition reactions of the olefinic o-quinodimethanes 56 that have asymmetrically substituted tertiary chiral centers.26
The facility of the thermally allowed conrotatory 4π-electrocyclic ring opening of cyclobutenes is known to depend on the electronic nature of the substituents on the cyclobutene ring.27 In the course of the studies of the substituent effect on this reaction, the exceptionally facile ring opening reaction of cyclobutenes was observed when arylsulfinyl, arylsulfonyl, and diphenylphosphinyl substituents are located on the cyclobutene structures. All the reactions can be carried out in Triisopropylsilyl (THF) at −30°C for 10 min, using n-butyllithium as base, and were found to proceed in moderate to high yields.28 The usefulness of small ring compounds for the syntheses of various pharmacologically important compounds and the novel reaction modes specific to these ring systems have been largely demonstrated. These findings may further serve as a means for the development of new reactions and synthetic methodologies.29
The application of DA cycloaddition reactions to the synthesis of antiviral compounds also finds in the hetero version of these [4+2] processes a wide panorama of valuable reactions. Here we have focused reader attention to some specific cases that are able to open a wider range of opportunities to access the target compounds.
It has been known for some time that aminosugar derivatives that inhibit glycoprotein processing have potential activity against HIV.30 These naturally occurring aminodeoxysugars derive either from piperidines, pyrrolidines, pyrrolizines, or octahydroindolizines, and are deprived of the anomeric OH group. Compounds 63a and 63b, which are derivatives of (+)-amino erythrose, fall within the pyrrolidine group.
The synthesis of these compounds relies upon HDA cycloaddition reactions of nitrosocarbonyl intermediates.31 In a previous publication Tschamber et al. showed that nitrosodienophiles react face selectively with the convex α-side of type 59 azetidinodiazepines to give the direct and the inverse cycloadducts of type 60 in high yields (Scheme 1.12 ). These results agree with the previously described high face-selectivity of the DA process. The acylnitrosodienophile (i.e., benzyloxycarbonylnitroso dienophile) was prepared in situ from the corresponding hydroxamic acid 58, according to some known procedures, and reacted at once with the diene component to afford 60.31 A primary amino group was introduced on 60 by azide elimination and then acetylation. Catalytic osmylation in the presence of the cooxidant N-methylmorpholine N-oxide (NMO) gave in excellent yield the expected cis diols of type 61. These were transformed into the corresponding acetonide derivatives (i.e., 62). One-pot hydrogenolyses (H2/Pd/C) of the single N–O bond and of the benzyloxycarbonyl moiety triggered molecular rearrangements, which led ultimately to pyrrolidine derivatives 63a,b. The formation of 63 results from a mechanistically straightforward multistep sequence: after hydrogenolysis of the O–Bn and of the N–O bonds, followed by decarboxylation, the hemiaminal functionality breaks up, leading to an aldehyde that condenses at once with the primary NH2-C(1) amine. The ensuing Δ1 pyrroline is then hydrogenated (Pd/C) to give pyrrolidine 63. This multistep mechanism is akin to the one observed during the catalytic hydrogenolysis of a trihydroxytetrahydroxazine, which led also to a dihydroxypyrrolidine.
Scheme 1.12.

Nitrosocarbonyl mediated synthesis of pyrrolidine derivatives 63a,b.
Compounds 63a and 63b were tested for their anti-HIV activity. These compounds were evaluated in two separate experiments in duplicate in a primary screen against HIV (strain GB 8) in JM cells (3 d assay). Activity was measured by syncytium formation and cytotoxicity in an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium (MTT) assay, using castanospermine as reference compound. In these assays 63a and 63b (hydrochlorides) showed some antiviral activity, which was less pronounced than that of castanospermine.32
Ulosonic acids are a group of naturally occurring carbohydrates that are involved in many biologically important processes and have attracted considerable attention in the last few years.33 The seven carbon-atom analog in this series, 3-deoxy-d-arabino-hept-2-ulosonic acid (DAH), is formed in plants by stereoselective condensation of phosphoenolpyruvate with D-erythrose 4-phosphate mediated by DAHP synthase, and it has been shown that DAH is a key intermediate in the biosynthesis of aromatic amino acids from glucose (Shikimate pathway). It is therefore not surprising that compounds of this type have become targets of many synthetic endeavors. In 1999, Wu et al. proposed an efficient methodology to the synthesis of the ethyl esters of 2,6-anhydro-3-deoxy-d-gluco-heptanoic acid 68a and 2,6-anhydro-3-deoxy-d-allo-heptanoic acid 68b using the chiral catalyst salen–Co(II) complex in the HDA reaction that led to significant improvement in both the yield and the selectivities.34
The key step in the synthesis involved the efficient elaboration of the silyl enol ether 65, which was prepared from d-glyceraldehyde acetonide (Scheme 1.13 ). The synthesis is based on the HDA cycloaddition reaction between the diene 64 and the ethyl glyoxylate in the presence of salen–Co(II) complex to afford 65 in 65% yield. Hydroxylation of 65 with a catalytic amount of OsO4 (10 mol%) and 2 equiv of NMO in acetone–water (3:1) at room temperature for 9 h provided a 4:1 mixture of 66 in 90% yield. The diastereoselectivity, however, was unacceptably low and separation of the products was very difficult. The asymmetric dihydroxylation on 65 was conducted by treatment with AD-mix-β35 at 0°C or room temperature, affording only 10% conversion of the starting material, even after prolonged reaction time (7 d). Better conversion was later achieved by employing larger amounts of K2OsO2(OH)4 (5 mol%); a single diastereoisomer 66 could be obtained in 84% yield. The excellent diastereoselectivity was attributed to the matched interactions between the substrate chirality and that of the chiral ligand.
Scheme 1.13.

Synthetic strategy for Zanamivir 7 and related compounds.
Reduction of α-hydroxy ketone 66 with NaBH4 at 0°C in ethanol provided the trans-diol in 94% yield. The hydroxyl groups were then protected as acetates, followed by selective hydrolysis of terminal O-isopropylidene acetals and the oxidative cleavage of the exposed glycol with periodic acid. The following by reduction of the resulting terminal aldehyde with NaBH4 gave compound 67a in 85% (two steps) isolated yield. Finally, deprotection of 67a with K2CO3/EtOH afforded ethyl 2,6-anhydro-3-deoxy-d-gluco-heptanoate (68a). Treatment of 66 with hindered lithium tri-t-butoxy aluminum hydride in THF at 0°C for 10 h gave a 2:1 mixture of cis- and trans-isomers in 80% yield. Both the selectivity and the yield were improved (cis/trans=3:1, 90% yield) when the reaction temperature was lowered to −15°C. Protection of the 4,5-dihydroxy as an isopropylidene acetal (91% yield), oxidation and isopropylidene removal by reaction with Dowex 50W (H+) in ethanol/water (9:1) at 50°C afforded the ethyl 2,6-anhydro-3-deoxy-d-allo-heptanoate (67b) quantitatively.36 These esters were useful precursors to the simple analogs of the antiinfluenza agent GG167 (i.e., Zanamivir, 7).37 GG167 (4-guanidino-2,4-dideoxy-2,3-dehydro-N-acetylneuraminic acid, 7) is a known viral NA (sialidase) inhibitor that, following intranasal administration in ferrets, is at least 100–1000 times more effective than ribavirin and amantadine against influenza A and B viruses. It retains its activity even when treatments are delayed until 24 h postinfection and has no effect on the serum antibody response to infection.38 The method for the stereoselective introduction of hydroxy groups at C-4 and C-5 illustrated in this work may be also applicable to syntheses of other members in this series of compounds. Some further applications of this strategy in the syntheses of Kdn, Neu5Ac, GG167 analogs were investigated on the basis of the excellent HDA strategy.
Another promising NA inhibitor known as BCX-1812 (RWJ-270201) is currently under evaluation in clinical trials (Scheme 1.14 ).39 This compound demonstrated notable selectivity and potency against NA for a wide range of influenza A and B viruses. It has been emphasized that the relative positions of the interacting functional groups are essential for the NA inhibitory potency, rather than the absolute positions of the central rings. The stereoselective synthesis of tetrasubstituted cyclopentane of BCX-1812 was described by Miller et al., emphasizing the utility of acylnitroso cycloaddition chemistry.31 As the starting material to synthesize NA inhibitor, the racemic oxazanorbornene derivative (±)-69 protected by a Boc group was synthesized (Scheme 1.14). Reductive N–O bond cleavage of (±)-69 using Mo(CO)6 and NaBH4 gave the allylic alcohol (±)-70, which was converted to its ethyl carbonate by treatment with ethyl chloroformate. The ethyl carbonate moiety was replaced by the nitromethyl group by reaction with MeNO2 and a catalytic amount of Pd(0), to furnish the primary nitro compound that was transformed into the carboxylic acid by employing a mixture of NaNO2 and AcOH in Dimethylformamide (DMF), followed by treatment with a catalytic amount of (TMS)-Cl in MeOH to give the corresponding methyl ester (±)-71. While subsequent elaboration described later was carried out on racemic (±)-71, either enantiomer could be prepared starting from the corresponding antipodes of (±)-70, available from enzymatic resolutions. The authors utilized a nitrile oxide [3+2] dipolar cycloaddition for incorporation of key additional framework components and functionality. The 1,4-cis-relationship of the rather bulky Boc-protected amine and methyl ester groups of (±)-71 was anticipated to induce selective reaction of the nitrile oxide from the opposite face. However, while the regiochemistry of the reaction was less predictable, precedent indicated that it would proceed as desired.
Scheme 1.14.

Nitrosocarbonyl mediated synthesis of BCX-1812 precursor 74.
Nitrile oxides can be derived from the corresponding oximes in situ, and it has been generally known that nitrile oxides bearing a wide range of substituents are capable of participating in [3+2] dipolar cycloaddition reactions (Scheme 1.14). As the precursor for [3+2] dipolar cycloaddition with compound (±)-71, oxime 72 containing diethyl units was prepared from the commercially available aldehyde and hydroxylamine (94% yield). The reaction of dipolarophile (±)-71 and oxime 72, which was transformed into the corresponding nitrile oxide dipole in situ in the presence of NaOCl and Et3N, induced a [3+2] dipolar cycloaddition to provide bicyclic isoxazoline (±)-73. As confirmed by X-ray crystallographic analysis, the desired isomer (±)-73 was the only isolated product despite the possibility for the generation of other unwanted stereo- and regioisomers. The isoxazoline ring of (±)-73 was then subjected to hydrogenolysis in MeOH containing PtO2 and an equivalent amount of HCl at 40 psi. Subsequent acetylation furnished compound (±)-74 as precursor of the final product BCX-1812.
Upon the removal of the Boc protecting group, compound (±)-74 was then converted to the corresponding protected guanidino derivative by treatment with 1,3-bis(t-Boc)-2-methyl-2-thiopseudourea in the presence of HgCl2. Subsequent basic hydrolysis with 1 N NaOH afforded the corresponding acid in 95% yield. Cleavage of the Boc protecting groups using TFA in CH2Cl2 in the presence of Et3SiH as t-butyl cation scavenger furnished the desired final product BCX-1812 in 78% yield.40
Moving from sugars or pseudo-sugars to alkaloids, the E-ring decarboxylated camptothecin (75) analogs, nothapodytines A and B, were isolated from Nothapodytes foetida.41 Nothapodytine B (mappicine ketone) is an oxidized derivative of natural alkaloid mappicine (76), showing significant cytotoxicity in the human KB cell line (Fig. 1.4 ).42
Figure 1.4.

Structures of camptothecin and mappicine.
Since nothapodytine B (mappicine ketone) has been identified as an antiviral agent with selective activities against herpes simplex virus (HSV)-1 and HSV-2, this family of alkaloids has received considerable attention by organic chemists. The limited supplies of mappicine ketone made it necessary to develop novel synthetic methodologies for its preparation. An example is offered by the novel synthetic approach to mappicine (76), employing the intramolecular HDA reaction as a key step. The proposed synthesis by Toyota, Komori, and Ihara relies upon the chemistry of 1-azabutadienes, although little was known about the practical applications of HDA reactions of 1-azabutadiene derivatives for the construction of polycyclic natural alkaloids.43
The starting compound for the synthesis of mappicine (76) is the quinoline-azide 77 (Scheme 1.15 ). The amide formation reaction was carried out to literature reported procedures: the reduction of 77 with wet triphenylphosphine followed by the condensation with fumaric acid monoethyl ester in the presence of BOP and Hunig’s base gave the unsaturated amide 78 in 67% overall yield. Intramolecular HDA reaction of 78 led to the cycloadduct 79 (76%), which was treated with aqueous hydrogen bromide to provide the ethyl ester (93% yield) in a single step. The electron-withdrawing substituent group (ethyl ester in 79) did not impede the cycloaddition; on the contrary, it promoted the autoxidation of the corresponding cycloadduct. Finally, the ethyl ester was subjected to transesterification reaction with methanol in the presence of concentrated H2SO4 to afford the product 80. Compound 80 had already been transformed into nothapodytine B and mappicine (76) by Kametani et al. by reduction of the ester moiety.44
Scheme 1.15.

Synthesis of mappicine 76.
In nearly the same years, American research groups proposed an alternative route to the same target. The key N-sulfonyl-1-aza-1,3-butadiene 81 was required for use in the LUMOdiene-controlled HDA reaction. Treatment of 81 with 1,1-dimethoxy-1-propene at room temperature (12 h, benzene) led to the formation of the sensitive [4+2] cycloadduct 82. Notably, the deliberate incorporation of the noncomplementary C4 electron-withdrawing substituent resulted in an HDA cycloaddition that proceeded at 25°C, presumably by lowering the diene LUMO without altering the inherent [4+2] cycloaddition regioselectivity (Scheme 1.16 ).
Scheme 1.16.

Enantioselective synthesis of mappicine 76.
Due to the expected sensitivity of the HDA adduct to hydrolysis, subsequent aromatization of the crude adduct (t-BuOK, THF, −35°C, 30 min) provided the methoxy-pyridine derivative in good yields (65%) without intermediate isolations. Presumably, aromatization proceeds by initial base-catalyzed elimination of methanesulfinic acid, which is facilitated by the C4–CO2Me substitution, followed by elimination of methanol.45 Addition of EtMgBr in the presence of a tertiary amine (EtMgBr, Et3N, toluene, −10°C, 4 h, 79%) proceeded cleanly to give the corresponding ethyl ketone 83 without competitive tertiary alcohol formation by virtue of tertiary amine-promoted ketone enolization. The final step required the cyclization to form the C ring. This was accomplished in one operation by treatment of 83 with a saturated solution of HBr(g) in CF3CH2OH (80°C, 24 h) followed by the addition of K2CO3 (25°C, 1 h) to provide the Nothepodytine B (mappicine ketone) directly without workup and isolation of the intermediate. This approach worked beautifully to give the ketone in 88% overall yield, and the final product proved identical in all respects with the properties reported for authentic material. Reduction of Nothepodytine B (mappicine ketone) with NaBH4 as first described by Kametani and later by Kingsbury and Comins provided (±)-mappicine (76).44., 46. Additionally, reduction of Nothepodytine B (mappicine ketone) with (S)-BINAL-H47 provided (S)-(−)-mappicine (73%, 99.9% ee), which exhibited a CD spectrum identical with that described for naturally occurring material confirming the original absolute configuration assignment.48 The cytotoxic evaluation (L1210 cell line) of 76 in both (+) and (−) forms are only weakly cytotoxic (IC50=23 and 13 μM, respectively).49
HIV protease inhibitors (PIs) continue to be an essential component of the arsenal in the fight against HIV infection and AIDS. The viral enzyme has the unusual ability to cleave peptide bonds between phenylalanine and proline, and inhibitors incorporating a hydroxyethylamine transition-state mimetic of Phe-Pro such as Saquinavir (inset of Scheme 1.17 ) are very potent and selective inhibitors of HIV protease. The reported synthesis does not allow ready substitution of the carbocyclic ring, a region that could potentially lead to enhanced potency and better pharmacokinetic properties of inhibitors. We have now developed a chiral DA route, which allows ready synthesis of a range of substituted carbocyclic mimetics of scissile Phe-Pro. The dienophile 84 did not react with the 2-(trimethylsilyloxy)-1,3-butadiene under thermal conditions, but reacted smoothly under Lewis acid catalysis to give, after work-up, a 4:1 mixture of diastereomeric trans-ketones in 83% yield from which the required isomer 85 was easily separable (Scheme 1.17).
Scheme 1.17.

Synthetic strategy to Saquinavir.
Oxazolidinone 85 was thus converted to the acid by treatment with lithium hydroxide/hydrogen peroxide and thence to the required tert-butylamide 86 via the N-hydroxysuccinimide ester.
The equatorial and axial alcohol 87a and 87b could be obtained, respectively, by using l-selectride in excellent yields and selectivities. They also could be readily converted to a range of ethers by treatment with sodium hydride in DMF at 0°C and the corresponding alkyl halide. The axial alcohol 87b could also be converted to the saturated carbocycle 88 by transformation to the mesylate, elimination, and reduction.50 Following elaboration of the carbocyclic ring, the Boc and acetonide protecting groups were easily removed using dichloromethane/TFA containing a few drops of water to give amines suitable for further elaboration to inhibitors of HIV protease.
(−)-Virantmycin (96) (Scheme 1.18 ) is an unusual chlorinated member of the large class of tetrahydroquinoline alkaloids. It was isolated from a strain of Streptomyces nitrosporeus by Omura et al.51 and was found to be a potent DNA and RNA virus inhibitor as well as an antifungal agent.52 Several total syntheses53 of racemic (±)-Virantmycin, natural (−)-Virantmycin, and the unnatural (+)-antipode were proposed, stressing the importance and the promising pharmacological profile of the substance. Various synthetic approaches toward the construction of tetrahydroquinoline ring systems are available, including insertion, ring expansion, and ring contraction reactions. However, one of the most versatile and convergent strategies for the construction of tetrahydroquinoline alkaloids is the HDA reaction of o-azaxylylenes, which are formed by a 1,4-elimination from a suitable benzylic halide precursor and suitable dienophiles. One key fragment for our synthetic route toward Virantmycin is arylthiocarbamate 91 (Scheme 1.18). This substance has been known for more than 110 years, but synthetic methodologies for its reliable and high-yield formation are scarce. The classical method developed by Paal and Laudenheimer, heating 2-aminobenzyl alcohol (89) in carbon disulfide at reflux for several hours without additional solvent, leads to formation of the desired product 91, via the intermediate 90, in a maximum yield of 30%, which can be raised to about 40% if the reaction is performed in a sealed vial. A second possibility includes the reaction of 2-azidobenzyl alcohol in the presence of triphenylphosphane in a Staudinger-type reaction leading to an iminophosphorane. This intermediate can be reacted with carbon disulfide in a sealed vial at elevated temperature to yield the cyclized product 91 in a yield of 53%.
Scheme 1.18.

Synthetic strategy for the preparation of (−)-Virantmycin96.
The allylic alcohol 92 was then allowed to react in an Appel-type reaction with cyclic thiocarbamate 91. Using our standard conditions, the reaction proceeded smoothly, but purification of the product proved to be cumbersome, as the reaction byproduct triphenylphosphane sulfide cannot be easily removed by column chromatography. Exposure of the dissolved crude reaction product in Dichlromethane (DCM) and treatment with trifluoroacetic anhydride synthesize the desired carbamate 93 in 83% isolated yield due. Having completed the synthesis of advanced carbamate intermediate 93, the stage was set for the key [4+2] cycloaddition: dry cesium carbonate (several hours at <0.1 Torr and 100°C) was added to a solution of 93 in DCM and stirring for 4 d at room temperature smoothly provided tricyclus 94 in a yield of 85%. The completion of the synthesis could be achieved by aromatic iodination using iodine monochloride in DCM, which provided the desired compound as a single regioisomer 95 in a yield of 90%. The synthesis of the advanced precursor 95 was completed in four consecutive steps starting from 91 in an overall yield of 49%. The remaining steps of the total synthesis toward (±)-Virantmycin were already performed.54
The identification of biologically active organic compounds is of utmost importance both for basic research and drug development. Besides the classical approach of identifying compounds by in vitro screens (i.e., by binding to an isolated protein), high-content cell-based screens have proven to be a powerful complementary approach.55 The proposal to employ the frameworks of natural product classes as evolutionary selected and biologically prevalidated starting points in structural space for compound collection development represent an interesting research route, developing a tree-like classification of the natural product frameworks identified in nature. The biology-oriented synthesis of compound collections derived from these natural product frameworks provides a promising strategy in the search of new biologically active agents and can be efficiently carried out on polymer-bound substrates, as they allow rapid diversification without intermediate purification steps. Natural product-derived and -inspired compound collections have been synthesized on the solid support in a number of cases.56 However, in order to meet the stereochemical demands of natural product frameworks, the use of enantioselective transformations on the solid phase presents a highly attractive though rarely addressed approach. Scheme 1.19 reports the enantioselective solid-phase synthesis of a natural product-inspired α,β-unsaturated δ-lactone collection and its investigation in cell-based screens, monitoring cell cycle progression, and viral entry into cells. The α,β-unsaturated δ-lactone motif belongs to the most frequently occurring scaffolds in nature and is the characteristic underlying structural element of antiproliferative agents, immunosuppressants, and inhibitors for different enzymes, in particular protein phosphatases. For the development of an asymmetric solid-phase synthesis of α,β-unsaturated δ-lactones, the enantioselective oxa HDA reaction between aldehydes and electron-rich 1-alkoxydienes 99 immobilized on the polymeric carrier is the key in stereodifferentiating transformation. 1-Alkoxydienes 99 were synthesized on the solid support starting from a Wang bromo-resin 97 and subjected to enantioselective cycloaddition with ethyl glyoxylate 100 as the reactive heterodienophile (Scheme 1.19).
Scheme 1.19.

Solid phase synthesis of lactone 103.
The enantioselective HDA reaction with ethyl glyoxylate proceeded with 90%–95% ee in the presence of 50 mol% Ti-(R)-BINOL complex. The absolute and relative configurations of the cycloadducts were determined after release of the lactones from the resin and comparison of their specific rotations and characteristic Nuclear Magnetic Resonance (NMR) data with the corresponding values reported in literature. Through simple transformations, the esters 101 could be elaborated into the resin-bound final compounds of type 102. After cleavage from the resin, compounds 103 were tested for their biological activity; the synthesized compound collection was subjected to the aforementioned cell-based screens, monitoring their influence on cell cycle progression. In this screen, BSC-1 cells (from African green monkey) cultured in 384-well plates were treated for 8 h with 30 μM of the synthesized compounds. This cell line was chosen due to its proven advantage in assessing the influence of chemical compounds on the cell cycle.57 Treated cells were fixed with formaldehyde and stained for chromatin and the actin- and microtubule cytoskeleton. Microscopic analysis revealed that several of the investigated compounds influenced the microtubule cytoskeleton in dividing and/or nondividing cells.58
In the previous paragraph we saw that Tamiflu (Oseltamivir phosphate, 19) has been recognized as a key weapon in the combat of a new type of influenza, and its importance and demand are now increasing. Tamiflu was first developed by Gilead Science and is synthesized and marketed by Roche. Its production is performed starting from shikimic acid, a natural product, so that development of a new synthesis from nonnatural readily available chemical feedstock is required. The synthesis of Tamiflu was investigated by many groups in the world and the total synthesis was accomplished by Corey, Shibasaki, and others.59 To construct such a polyfunctionalized cyclohexene unit, an HDA approach between pyrroles and acetylene was investigated by a Japanese research group. The synthesis started from the HDA reaction of N-Boc-pyrrole 104 (Scheme 1.20 ). A solution of bromoacetylene carboxylate (105) in N-Boc-pyrrole (104) was heated at 90°C for 39 h and the DA adduct, 7-tert-butyl 2-methyl 3-bromo-7-azabicyclo[2.2.1]hepta-2,5-diene-2,7-dicarboxylate (106), was isolated in 57% yield. Treatment of 106 with mCPBA gave 5,6-epoxide 107 in 46% yield. The oxidation took place in a stereoselective manner and exo-107 was prepared as a single isomer. The use of oxone for the conversion resulted in the formation of a mixture of isomers. The authors succeeded in improving the yield of 107 up to 64% by repeating the reaction procedure twice. The reduction of 107 and basic hydrolysis yielded the corresponding carboxylic acid, that then opened the epoxide to give tricyclic lactone alcohol 108 in 89% yield in one step. Other synthetic steps were required to convert the lactone 108 into the enco-epoxide 109, obtained in 85% yield.
Scheme 1.20.

N-Boc pyrrole-based synthesis of Oseltamivir 19.
The conversion of 109 into the cyclohexene-epoxide 110 was greatly enhanced when Hexa-methylphosphoramide (HMPA) was used as cosolvent of the reaction with Lithium Diisopropylamide (LDA) in THF at −78°C, yielding 110 up to 62% yield in the presence of 1.5 equivalents of HMPA.
The final stage of the synthesis was performed in the following way (Scheme 1.20). Installation of the 3-pentyloxy group was achieved by treatment with 3-pentanol in the presence of BF3OEt2 to obtain compound 111 in 54% yield. The epimerization at the hydroxyl group was then carried out by oxidation-reduction pathway. Thus the conversion to the corresponding ketone was achieved by treatment with the Dess–Martin periodinane (DMP). The reduction with NaBH4 smoothly gave 112 in 91% yield. The diastereoselectivity was about 3:1. The conversion of 112 to Tamiflu 19 has already been reported and thus concludes the formal synthesis of Tamiflu from readily available organic feedstock. Overall yield was 5.4%, and all the steps proceeded in a sufficiently stereoselective manner.
The 2-azabicyclo[2.2.2]octanes (isoquinuclidines) are found widely in natural products such as iboga-type indole alkaloids, which have varied and interesting biological properties, and vinca alkaloids such as vinblastine and vincristin, which possess isoquinuclidines with the aspidosperma portion.60 It has been indicated that ibogaine reduces cravings for alcohol and other drugs by means of its ability to boost the levels of a growth factor known as glial cell line–derived neurotrophic factor. In addition, it was also shown that the isoquinuclidines can be used as the synthetic intermediate for the synthesis of Oseltamivir phosphate (19). Furthermore, isoquinuclidines are also valuable intermediates in the synthesis of other alkaloids and in medicinal chemistry. It is therefore meaningful to establish an effective asymmetric synthetic methodology for chiral isoquinuclidines. A well-established route to the chiral ring system is through the asymmetric HDA reaction of 1,2-dihydropyridines with dienophiles. However, only a few examples of employing organometal catalysts or organocatalysts have been reported. Despite the obvious advantages of the catalytic enantioselective version using an organocatalyst, to the best of our knowledge, only one example employing a MacMillan catalyst has been reported for the organocatalytic asymmetric version of this reaction, which was used as the key reaction for the efficient practical total synthesis of Tamiflu by Fukuyama et al.61 The effectiveness of acrolein derivative 114 was investigated using superior catalyst 115 (Scheme 1.21 ) in the presence of the aza-diene 113. The reaction was carried out at 0°C in the presence of 10 mol% of 115 to give the DA adduct 116 in 83% yield, >99% ee.
Scheme 1.21.

Enantioselective synthesis of Oseltamivir 19.
This is the first example of an enantioselective DA reaction of 1,2-dihydropyridine with substituted dienophile using an organocatalyst. Reduction with NaBH4 led to the alcohol 117, which was finally converted into the desired target 19.62
We have already seen that Sialic acids are closely related to carboxylated sugars widespread in mammalian glycoproteins and glycolipids. They are typically found at the outermost end of glycan chains of all kind of cells and are involved in a variety of physiochemical and pathological processes. This class of sugars has structural and modulatory roles and serves as components of binding site for various pathogens and toxins; in addition, another important function they play is decorating microbial pathogens, allowing them to evade host immunity.63 Influenza virus is subdivided into three serologically distinct types—A, B, and C—all of which recognize sialic acid and its derivatives as their functional receptors. Sialidase is the influenza virus surface enzyme recognizing the N-acetylneuraminic acid (Neu5Ac) moiety, which is typically associated as α-linked terminal saccharidic unit of mammalian glycoconjugates.64 In the search for new sialidase-inhibitor antiinfluenza drugs, the synthesis of a new class of exo enitols obtained from Neu5Ac and their use as electron-rich dienophiles in chemo-, regio-, and stereoselective HDA reactions with α,α′-dioxothiones as electron-poor dienes, to form diasteromerically pure β-Neu5Ac derivatives, structurally attractive for the development of new inhibitors of sialidase, was developed by Italian and Greek research groups.65
The methyl ester 118 was converted at room temperature into the tert-butyldimethylsilyl triflate (TBDMSOTf) to afford the tris silyl derivative 119 in 81% yield (Scheme 1.22 ). The direct reduction of 119 by treatment with sodium borohydride in isopropanol and the subsequent oxidation to the lactone 120 was realized with sodium periodate in methanol/water as solvent (40% yield calculated over two steps). The desired exo enitol 121 was finally obtained by reaction of 120 with Tebbe reagent in pyridine. Compound 121 was isolated in 67% yield as single diasteroisomer.
Scheme 1.22.

Synthesis of spiroketal 127.
The heterodienes selected to react with the dienophile 121 were obtained treating the commercially available β-ketoester 122 with phthalimidosolfenyl chloride (PhtNSCl) and pyridine under reported conditions.65 The α,α′-dioxothione 124 was formed under mild conditions from the phtalimido derivative and trapped in situ by the exo enitol 121 in an inverse electron-demand DA reaction. The cycloaddition reaction was totally chemo- and regioselective and the cycloadduct 125 was obtained as single diasteroisomer. The ethyl ester 126, isolated as pure α anomer, was hydrolyzed with lithium hydroxide in THF/H2O as solvent, to afford, in high yield, the completely deprotected spiroketal 127 as a diasteromerically pure compound.
To obtain a putative binding mode of the newly synthesized compounds within the NA active site, docking calculations were conducted for compounds 127 and the 4-fluorobenzene derivative 128. Their ethyl ester precursors were not considered in this study, since they are expected to be readily hydrolyzed by endogenous esterases in vivo. The crystallographic structure of the complex between sialic acid (α-Neu5Ac) and the influenza virus sialidase was employed in order to make a direct comparison between 127, 128 and the interactions of Neu5Ac with the active site residues (Fig. 1.5 ).
Figure 1.5.

(A) Crystallographic structure of α-Neu5Ac complex with influenza virus sialidase from PDB ID 2BAT with the active site residues represented as surface (left) and sticks (right). (B) and (C) illustrate the predicted conformations of 127 and 128, respectively, and the interacting active site residues of sialidase. Protein side-chain atoms are labeled.
From: Richichi, B.; Lunghi, C.; Papakyriakou, A.; Francesconi, O.; Nativi, C. Pure Appl. Chem. 2013, 85, 1803–1811.
Two models for each compound were prepared, having the pyranose ring either in chair or boat configurations. Bound conformations retained all intermolecular interaction with the NA active site residues (Fig. 1.5A), displaying only minimal differences at the glycerol moiety. The estimated mean free energy of binding for Neu5Ac was −5.8 kcal/mol, corresponding to an inhibition constant of ~55 μM. Analysis of the docking results obtained for compounds 127 and 128 revealed a high number of distinct conformational clusters, many of which adopted a completely different binding mode in comparison with Neu5Ac. As a consequence, most of the hydrogen-bonding interactions displayed by Neu5Ac and the active site residues of NA are lost. On the other hand, docking of 127 and 128 in the same boat configuration as that of the bound Neu5Ac exhibited two conformational clusters that approximate the binding mode of Neu5Ac. For compound 127, the highest energy conformation of the top ranked cluster (15%), with a mean free energy of binding of −5.0 kcal/mol, is shown in Fig. 1.5B. In this binding mode, the carboxylate group is slightly shifted with respect to that of sialic acid (<1 Å), thus maintaining the hydrogen-bonding network with Arg118, Arg292, and Arg371. However, for the proper accommodation of the oxathiine moiety inside the active site, the pyranose ring of 127 is rotated by ~45° and shifted toward residue Ile222 by ~3 Å, which results in loss of the hydrophobic contacts with Trp178. Compound 127 is predicted to interact with most of the residues displayed by Neu5Ac–sialidase complex, albeit with a lower affinity that is mainly due to a loss of ~1.4 kcal/mol in the electrostatic energy term. As for compound 127, the p-fluorophenyl substituent gives rise to a larger shift of the oxathiine ring toward Arg118, thus losing contact with Arg292 (Fig. 1.5C). In this binding mode, the p-fluorophenyl group does not display contacts with any protein residue. In addition, the interaction between C4-OH of 128 and Glu119 is not present, and Glu276 exhibits only van der Waals contacts with the glycerol side-chain. For these reasons, compound 128 is predicted to bind sialidase with an even lower affinity (mean free energy of binding of −4.1 kcal/mol). Conclusively, the oxathiine ring of the spiroketal compounds might displace the pyranose ring from the crystallographic position displayed by sialic acid and its derivatives, though proper functionalization of the spiroketal scaffold could compensate for the interaction with key amino acid residues of NA. The new spiroketal scaffolds could represent a novel class of sialidase inhibitors.
The 5-aryl/heteryl/idene-4-thiazolidinone derivatives constitute an important series of biologically active compounds and important intermediates in modern medicinal and synthetic organic chemistry by possessing synthetic multifunctionality and bioactivity in diverse pharmacological areas.66
The synthetic strategy was based on 5-ethoxymethylidene-4-thioxo-2-thiazolidinone (5-ethoxymethylideneisorhodanine) as heterodiene in HDA reaction. Nevertheless, this type of HDA reaction is rarely used and only with a limited number of dienophiles, such as acrylic and maleic acid derivatives and β-nitrostyrene.67
HDA reaction of 129 with 1,4-naphthoquinone (Scheme 1.23 ) afforded 131 through (1) the intermediate dehydrogenation in the presence of hydroquinone excess, (2) the formation of an additional endocyclic double bond, and (3) EtOH elimination.
Scheme 1.23.

Synthetic strategy toward compounds 133 and 135a,b.
Furthermore, the HDA reaction of acetylene derivatives as dienophiles in [4+2]-cyclocondensation with 129 passed with ethanol elimination and simultaneous conjugated double-bond rearrangement. Thus reaction of 129 with propynoic acid afforded the corresponding 2-oxo-2H-thiopyrano[2,3-d]-1,3-thiazole derivative 133. [4+2]-Cyclocondensation of 129 with aroylacrylic acids yielded regioselectively the 6-aryl-2-oxo-3,5-dihydro-2H-thiopyrano[2,3-d]-1,3-thiazol-6-yl methanones 135a,b.
The synthesized compounds 131, 133, and 145a,b were tested for the in vitro anticancer activity on 60 human tumor cell lines at concentration of 10 μM. Derivatives 145a,b, besides low mean levels of tumor cells growth inhibition, have shown selectivity for leukemia cells. The compound 131 was characterized by high levels of growth inhibition of cancer cell lines SK-MEL-5 (melanoma), OVCAR-3 (ovarian cancer), and MDA-MB-435 (breast cancer). Moreover, compounds 131 and 133 were also tested for antiviral activity according to the antimicrobial acquisition and coordinating facility screening program. The studied compounds did not show significant activity against viruses flu A (H1N1), flu A (H3N2), flu A (H5N1), flu B, measles, parainfluenza virus, respiratory syncytial virus A and SARS, but compound 131 did possess moderate activity against coronavirus SARS.
Azabicyclic compounds are remarkable subunits of alkaloid natural products such as ibogaine, dioscorine, and coronaridine.68 Among them isoquinuclidine ring systems are of eminent importance because of their potential as bioactive compounds or pharmaceuticals. There are also numerous natural compounds (terpenes and alkaloids) and drugs that contain purely carbocyclic scaffolds. Particularly, derivatives of several bridged carbobicyclic compounds with an exocyclic imine group were reported as being highly active against Trypanosoma b. rhodesiense and the resistant K1 strain of Plasmodium falciparum.69
Several approaches to azabicyclo compounds are known in literature but reactions that enable a convenient direct and metal-free route to complex bridged carbo- and heterobicycles using malononitrile and aldehyde as strikingly simple starting compounds have never been investigated. Because aldehydes are more electrophilic and simple starting materials, Scheme 1.24 reports the use of some aldehydes (1 equiv) with malononitrile 2 (1.5 equiv) in the presence of imidazole at 15 mol% loading.
Scheme 1.24.

Malonodinitrile synthetic applications to azabicyclo compounds syntheses.
The synthetic attention was directed to the artemisinin-derived aldehydes as substrates. Since the seminal discovery in 1972 of artemisinin (natural 1,2,4-trioxane sesquiterpene) from Artemisia annua L., by Youyou Tu (Nobel Prize 2015),70 diverse varieties of artemisinin-derived dimers and hybrids have been synthesized. Natural product hybrid molecules often possess strikingly improved or new biological activities compared with those of their parent compounds, as in the examples of different highly potent artemisinin-derived hybrids with antimalarial, antiviral, and anticancer activities.71 Thus predicting the potential of these new transformations to structurally diverse and complex scaffolds of value to medicinal chemistry, artemisinin-derived hybrids were prepared. Scheme 1.24 shows two reactions of enantiomerically pure artemisinin-based aldehydes 136 and 138 with malononitrile in the presence of imidazole (15 mol%). Delightfully, new artemisinin-derived hybrids 137a,b were synthesized in 52% overall yield, in 5:1 ratio. Hybrids 139a,b were synthesized in 38% overall yield; a 3:1 ratio of corresponding products was formed.
Fig. 1.6 reports the complex mechanism suggested by the authors supported by Density Functional Theory (DFT) calculations as well as by some Mass Spectrometry (MS) investigations that allowed detection of some synthetic intermediates.
Figure 1.6.

Suggested mechanism chart.
Modified from: Bock, C. M.; Parameshwarappa, G.; Bönisch, S.; Neiss, C.; Bauer, W.; Hampel, F.; Görling, A.; Tsogoeva, S. B. Chem. Eur. J. 2016, 22, 5189–5197.
The obtained hybrids were investigated from the biological point of view. Remarkably, hybrids 139a,b display superior potency against human cytomegalovirus (EC50 of 0.071 μm and 0.260 μm, respectively) compared with that of their parent compound artemisinin (EC50>10 μm) and are even more active than clinically used antiviral agent ganciclovir.72
Thiazolidinone-based molecules are attractive targets in the rational design of drug-like compounds, which possess antiinflammatory, antioxidant, antitumor, choleretic, diuretic, and other activities. In addition, 4-thiazolidinones are known as inhibitors of PRL-3 and JSP-1 phosphatases, HCV NS3 protease, antiapoptic proteins complex Bcl-XL-BH3, TNFα-TNFRc-1 complex, Ras farnesyl transferase. Among thiazolidinone derivatives, 5-arylidene-4-thiazolidinones are of wide interest due to their diverse biological activity and clinical applications.73 In previous studies thiopyrano[2,3-d]thiazoles were reported as potential antitripanosomal, antioxidant, anticancer, and antiinflammatory agents. A series of novel thiopyrano[2,3-d]thiazole-6-carbaldehydes were prepared and found active for antitumor and antiviral compounds. The synthesis of these compounds is based upon [4+2]-cyclization with acrolein via HDA reaction.
The synthesis of target thiopyrano[2,3-d]thiazoles followed the general pathway outlined in Scheme 1.25 for a specific compound. First, the starting 5-arylidene-4-thioxo-2-thiazolidinones 142 were obtained by Knoevenagel condensation of 4-thioxo-2-thiazolidinone 141 with several aldehydes in the presence of EDDA as the base catalyst. The reactivity of the sulfur atom at the 4-position of 5-ylidene-4-thioxo-2-thiazolidinones allows it to be used as a highly active heterodiene component in HDA reactions.
Scheme 1.25.

Thiazolidinone approach to compound 143.
The synthesized compound 143 was evaluated for antiviral activity. Regarding Epstein–Barr virus, it was found that compound 143 showed excellent activity (EC50=0.07 μM, SI=3279) by VCA ELISA test and possessed moderate activity against HSV-1 (SI=31) and Varicella zoster virus (SI=34) by viral Chlorinated Polyethylene (CPE) test.74
The design, synthesis, and evaluation of a new class of exceptionally potent HIV-1 PIs was reported by American and Japanese research groups in a study where the structure design strategies are based upon promoting extensive interactions in the active site of HIV-1 protease. The authors have examined and compared X-ray structural variations of critical active site interactions of numerous HIV-1 PIs bound to both wild-type and mutant proteases.75 Therefore, a promising inhibitor design strategy would be the maximization of the active site interactions and particularly the promotion of a robust network of hydrogen-bonding interactions with backbone atoms of wild-type HIV-1 protease.76 As depicted in Fig. 1.7 , the design model to combat drug resistance involves the design of P2 and P2′ ligands that would form robust hydrogen bonds with polar groups in S2 and S2′ regions. Furthermore, the synthetic plan requires filling the hydrophobic S1 and S1′ subsites with hydrophobic P1 and P1′ ligands.
Figure 1.7.

Proposed model for the design of PIs to combat drug resistance. The model suggests the formation of robust hydrogen bonds by the P2 and P2′ ligands simultaneously in both the S2 and S2′ subsites. The transition-state hydroxyl group binds to catalytic aspartates, and the P1 and P1′ ligands fill in the S1 and S1′ subsites. PI, Protease inhibitor.
From: Ghosh, A. K.; Venkateswara Rao, K.; Nyalapatla, P. R.; Osswald, H. L.; Martyr, C. D.; Aoki, M.; Hayashi, H.; Agniswamy, J.; Wang, Y.-F.; Bulut, H.; Das, D.; Weber, I. T.; Mitsuya, H. J. Med. Chem. 2017, 60, 4267–4278.
Among many possible classes of transition-state binders, the incorporation of an (R)-hydroxyethylamine sulfonamide isostere can be performed readily varying P1 and P1′ substituents to provide the nonpeptide drug-like scaffold. Hence, the design, synthesis, and X-ray structural studies of a new class of PIs incorporating an unprecedented 6–5–5 ring-fused crown-like tetrahydropyranofura as the P2 ligand with the (R)-hydroxyethylsulfonamide isostere is proposed on the basis of a DA reaction of readily available ethyl vinyl boronate 144 and cyclopentadiene at −78°C, in the presence of the in situ generated catalyst 145, providing the corresponding cycloadduct that was oxidized with H2O2 in the presence of KHCO3, leading to the alcohol 146 in 80% yield over two steps in gram scale (Scheme 1.26 ).
Scheme 1.26.
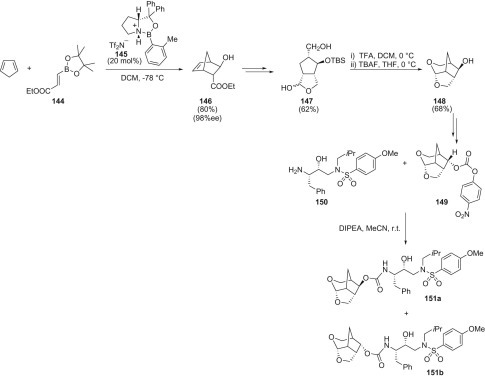
Enantioselective synthesis of compounds 151a,b.
Optical purity of alcohol 146 was determined to be 98% ee by chiral HPLC analysis. Alcohol 146 was converted to bicyclic acetal 147 in a three-step sequence, which was obtained in 62% yield. The lactol mixture was treated with TFA in DCM to afford, after removal of the TBS group with tetrabutylammonium fluoride, the alcohol 148 in 68% yields over two steps. This exo-alcohol 148 was converted to endo-alcohol by Dess–Martin oxidation followed by reduction of the resulting ketone with NaBH4 in MeOH in 76% yield over two steps. Both exo- and endo-alcohols were converted to the corresponding mixed activated carbonate derivatives of type 149 in excellent yields. The final step to P2 ligands stands on the 4-methoxybenzenesulfonamide 150 coupling with the activated carbonates of the endo- and exo-(crown-like)-THF of type 149 in the presence of diisopropylethylamine to furnish inhibitors 151a and 151b in good yields.
The examination of the preliminary model of endo-(crown-like)-THF containing inhibitor 151b and exo-(crown-like)-THF-derived inhibitor 151a indicated that the acetal oxygens in the endo-derivative are suitably positioned to form hydrogen bonds with Asp30 and Asp29 amide NHs. Also, the tricyclic scaffold of inhibitor 151b appeared to fill the hydrophobic pocket in the S2 site more effectively than inhibitor 151a. Inhibitor 151b containing endo-crn-THF ligand and a 4-methoxybenzenesulfonamide as P2′ ligand exhibited an enzyme inhibitory K i of 14 pM, compared to inhibitor 151a containing exo-(crown-like)-THF ligand (Ki of 10.8 nM).
The X-ray crystal structure of the wild type HIV-1 protease cocrystallized with inhibitor 151b was refined. It contains the protease dimer and the inhibitor bound to HIV-1 protease in two orientations related by a 180° rotation with 55%/45% relative occupancies (Fig. 1.8 ).77
Figure 1.8.

Inhibitor 151b-bound X-ray structure of HIV-1 protease (PDB code 5ULT). The major orientation of the inhibitor is shown. The inhibitor carbon atoms are shown in gray, water molecules are black spheres, and the hydrogen bonds are indicated by dotted lines.
From: Ghosh, A. K.; Venkateswara Rao, K.; Nyalapatla, P. R.; Osswald, H. L.; Martyr, C. D.; Aoki, M.; Hayashi, H.; Agniswamy, J.; Wang, Y.-F.; Bulut, H.; Das, D.; Weber, I. T.; Mitsuya, H. J. Med. Chem. 2017, 60, 4267–4278.
The inhibitor is bound in the active site cavity by forming a series of hydrogen bonding interactions and numerous weaker CH···O interactions with the main chain atoms of HIV-1 protease. The major conformation of the inhibitor forms hydrogen bonding interactions of its urethane NH with the carbonyl oxygen of Gly27. The inhibitor also forms tetracoordinated water-mediated interactions connecting the inhibitor carbonyl oxygen and sulfonamide oxygen with the amides of Ile50 and Ile50′ in the flaps. Furthermore, the p-methoxy group of the P2′-sulfonamide forms a hydrogen bond with the amide NH of Asp30′ as well as with its side chain carboxyl group.
We move now to the field of terpenes. In 1972 Hesp et al. reported the isolation and structure characterization of the tetracyclic diterpene Aphidicolin 157 (Scheme 1.27 ), produced by the fungus Cephalosporium aphidicolia.78 Aphidicolin 157 shows marked activity against Herpes simplex Type I virus both in vitro and in the rabbit eye. The synthetic approach relies upon the condensation of the cyclohexanone derivative 152 with 2-bromo-acrolein in the presence of LDA to afford 153 that is converted into the intermediate 154 through a critical Heck cyclization.
Scheme 1.27.

Synthetic strategy to Aphidicolin 157.
From 154, via Claisen condensation and Wacker oxidation, the diene 155 is obtained in 62% yield, leading to the product of the intramolecular DA reaction 156, in 81% yield. In few other steps, 156 is converted into the target compound 157.79
It is well-known that a number of natural products contain a quinone moiety. They are antibiotics such as mitomycin C and anthracyclicnes and show remarkable biological activities. Toddacoumaquinone 160 is a unique coumarin-naphtoquinone dimer isolated from Toddalia asiatica and its synthesis was planned through a DA cycloaddition of the diene 158 with the quinone 159 (Scheme 1.28 ).
Scheme 1.28.

Synthesis of quinone 160.
Compound 160 was tested for antiviral activities against HSV-1 and HSV-2 using acyclovir as control compound and showed a weak activity against both virus strains.80
The leaves of the rare littoral plant Stemodia maritima L. (family Scrophulariaceae) have long been used as a folk medicine in the Caribbean Islands for the treatment of venereal disease. Attracted by the reported medicinal properties, Manchand et al. carried out phytochemical investigations on this plant in 1973, isolating and characterizing two structurally unique tetracyclic diterpenes, stemodin and stemodinone 165 (Scheme 1.29 ).81 Stemodane diterpenes, by virtue of their unique tetracyclic carbon skeleton, their alleged medicinal properties of S. maritima, and their close structural resemblance with aphidicolin, prompted a considerable number of synthetic investigations on this diterpene family.
Scheme 1.29.

Synthesis of stemodinone 165.
The planned synthetic route proceeds via the dienol ether 162, which was derived from 161 by the treatment of LDA in the presence of trimethylsilyl chloride (TMSCl). Thus, the crude ether 162, without further purification, was converted to the bicyclic enedione 163 via DA reaction with 3-butyn-2-one, followed by desilylation of the resulting silyl enol ether. The bicyclic seven-membered lactone 163 was then easily isomerized to the cis-fused five-membered lactone 164, having the desired stereochemical feature at C-10, by the Pd(0)-catalyzed lactone migration reaction. The final compound 165 was obtained in few more steps in the racemic form.82
Ascochlorin (174) is an antiviral antibiotic obtained from the filter cake of the fermented broth of Ascochyta viciae Libert. It has a strong inhibitory effect on viral growth in cultured cells.83 The compound features are three stereogenic centers, a sesquiterpene unit, a 1,3-diene system, and an aromatic unit having an aldehyde, a methyl, a chloride, and two hydroxy substituents. Due to its high biological activity, the ascochlorin family has attracted the attention of synthetic chemists. Among the total syntheses of (±)-ascochlorin reported in the literature, an example of asymmetric DA reaction, followed by cuprate, and Julie olefination reactions as the key strategies in the total synthesis of this biologically active natural compound is reported. For the synthesis of a sesquiterpene unit, the tigloylsultam 166 was allowed to react with 1,3-butadiene to give the desired DA adduct 167 in 85% yield and in 96% de. The chiral auxiliary was easily removed by treatment of cycloadduct 167 with LiOH in the presence of H2O2, which gave cyclohexenecarboxylic acid 169 in 94% yield and the recovered sultam auxiliary 168 in 80% yield (Scheme 1.30 ).
Scheme 1.30.

Synthetic strategy of Ascochlorin 174.
Standard iodolactonization methodology was then applied to the crude carboxylic acid 169. The reaction yielded the desired iodolactone 170 in 98% yield. Cleavage of the lactone unit with LiAlH4 and oxidation of the allylic alcohol with MnO2 afforded the desired unsaturated ketone. Reduction first, and oxidation of the resulting alcohol by Swern oxidation reaction conditions or by TPAP8 cleanly gave aldehyde 171. Julie olefination with 172 afforded the desired compound 173, precursor of 174.84
In 1988, efforts to discover novel microbial metabolites led to the isolation of new classes of natural products, called pradimicins and benanomicins.85 Pradimicins A, B, and C and were shown to be active against various fungi, yeasts, and viruses.86 The unique biological activity that this class of compounds exhibits and the ongoing need for antifungal and antiviral treatments make it a valuable target in synthetic chemistry. The structures of the pradimicins and benanomicins consist of an amino acid, a disaccharide, and a benzo[a]naphthacenequinone core. There are approximately 20 congeners in this family to date, and their specific structures vary according to their amino acid and disaccharide components. Pradimicin A 181 (Scheme 1.31 ), one of the first and most abundant congeners isolated, was initially subjected to biological testing, but was claimed to have limited solubility in water. To overcome this issue, testing was also performed with some analogs, which have different and more water-soluble amino acid and disaccharide functionalities.
Scheme 1.31.

Synthetic strategy to Pradimicin A 181.
Pradimicins have shown activity against numerous pathogens, including systemic fungal infections and, potentially more importantly, against HIV. Scheme 1.31 shows an alternative route to Pradimicin A 181 that starts with protection of the alcohol 175 as TIPS derivative 176, followed by deprotection of the triple bond to give the alkyne 177. MW promoted DA cycloaddition reaction with Dimethylacetylenedicarboxylate (DMAD) afforded in 1 min the cycloaddutct 179, subsequently oxidized with DDQ to 180, the precursor of the final compound 181.87
Panduratin A (188) is the prototypical member of a family of related cyclohexenyl natural products and is isolated as a racemic mixture from Boesenbergia rotunda, a perennial herb found in Southeast Asia (Scheme 1.32 ).88 The plant has been used in traditional medicine to treat conditions such as diarrhea, asthma, indigestion, dysentery, and many others. Studies have shown that these extracts have anti-HIV, antibacterial, antifungal, and antitumor activity. The synthesis proposed by an Australian research group relies upon the DA cycloaddition of methyl cinnamate (182) and (E)-ocimene (183), performed under high pressure conditions (19 kbar), to afford the regioisomeric panduratins 184a and 184b in a 1:2.9 ratio. Subsequent oxidation of alcohol, derived from 184a by reduction with LiAlH4, with the DMP provided aldehyde 185 as the branching point toward the final target. Addition of the organolithium reagent derived from bromobenzene 186 followed by DMP oxidation gave methoxymethyl ether (MOM)-protected ketone 187. Removal of the MOM groups with acidic methanol cleanly afforded Panduratin A (188).89
Scheme 1.32.

Synthesis of Panduratin A 188.
The regiochemical outcome of the DA cycloaddition between methyl cinnamate (182) and (E)-ocimene (183) was in accord with an analysis based on computationally derived FMOs (Fig. 1.9 ).
Figure 1.9.

Atomic orbital coefficients of the 2p orbitals of methyl cinnamate (182), and (E)-ocimene (183) calculated at the B3LYP/6-31G(d) level of theory.
From: Pasfield, L. A.; de la Cruz, L.; Ho, J.; Coote, M. L.; Otting, G.; McLeod, M. D. Asian J. Org. Chem. 2013, 2, 60–63.
Alignment of the larger diene HOMO (2Pz atomic) coefficient with that of the corresponding dienophile LUMO coefficient predicts formation of ester 184b as the major regioisomer.
Wickerol A (194) proved to be highly active against the type A/H1N1 influenza virus with an IC50 of 0.07 μg/mL. The natural product features a unique tetracyclic carbon skeleton that comprises two adjacent quaternary carbons and seven or eight stereocenters, respectively, two of which are quaternary as well. Although the 6–5–6–6 skeleton of the Wickerols contains only five- and six-membered primary rings, the molecule is remarkably strained due to numerous syn pentane interactions. Despite the formidable challenge posed by the Wickerols, only a single synthetic study was disclosed.90 In 2017 an asymmetric synthesis of Wickerol A (194) was proposed as a testament to the power of catalysis in cycloadditions and the many pleasant and unpleasant surprises that we encounter when working with complex, sterically congested molecules. Key intermediate 191, which contains all carbons of the Wickerols except two, could be accessed with a DA addition of known enone 189 to diene 190. The synthesis commenced with the DA cycloaddition of the achiral enone 189 to the chiral diene 190 (Scheme 1.33 ). The intermolecular cycloaddition represented a major challenge because both partners, though electronically matched, are sterically very hindered. Initially, thermal or high pressure conditions were explored, but unsuccessfully.
Scheme 1.33.

Synthetic strategy to Wickerol A 194.
Lewis acid activation was found to be the solution of the problem. Jung’s conditions, employing a mixture of AlBr3/AlMe3 as catalyst, gave the desired endo-product 191 as single diastereomer.91 Both the quaternary and the neopentyl stereocenters were set with the correct simple and induced diastereoselectivity. Presumably, the enone approaches the diene via an endo-transition state and the methyl group provides less steric hindrance than the siloxy methyl moiety. Having set the correct configuration, the subsequent modification conducted to the compound 192, which led to the ring system of Wickerol A (194) complete with the introduction of the missing methyl groups. The methyl group on the D-ring could be added with complete diastereoselectivity by exposure of 193 to methyl lithiocyanocuprate in the presence of BF3·Et2O. Given the steric hindrance at the neopentyl β-position and the introduction of additional syn-pentane strain, it is remarkable that this reaction proceeded at all. Wickerol A (194) was then obtained through a sequence of reaction required for the insertion of another methyl group.92
A research group of Belgian authors demonstrated that both d- and l-cyclohexenylguanine are highly potent and selective antiherpes virus (HSV-1, HSV-2, VZV, CMV) agents. Their activity profile is similar to that of the known antiviral drugs acyclovir and ganciclovir, and they represent the most potent antiviral nucleosides containing a six-membered carbohydrate mimic that have ever been reported. Remarkably, the antiviral activity profiles of both enantiomers are very similar. The enantioselective synthesis of the d- and l-enantiomers of these compounds starting from (R)-carvone was already described.93 However, the synthesis is long and time-consuming, and it is not suited for the preparation of large amounts of the final products that are needed for full biological evaluation. This fact is one of the major drawbacks of this type of synthetic approach: the need of high amounts of products because of the length of the synthetic route causing low final chemical yields. The Belgian authors set up a novel synthetic route to overcome these problems based on the DA reaction between the dienophile 195 and the Danishefsky’s diene 196 in the presence of hydroquinone at 180°C for 1.5 h (Scheme 1.34 ).
Scheme 1.34.

Synthesis of nucleoside analog 201.
Adduct 197 was obtained in 62% yield as a 4:1 mixture of the endo- and exo-adducts. The highly functionalized adduct 197 is sensitive to acid and moisture and was immediately reduced with excess LiAlH4, giving the triol 198 in 66% yield. This reaction involves the reduction of two ester groups and concomitant rearrangement of the silyloxyenol ether to the enone intermediate, which is then further reduced to 198. Reaction of triol 198 with benzaldehyde dimethyl acetal in the presence of para-Toluensulfoni acid (PTSA) at room temperature for 1 d afforded the benzylidene acetal 199 (70% yield). Introduction of the purine base moiety was performed by the reaction with 2-amino-6-chloropurine under Mitsunobu reaction conditions to generate the chloropurine 200 with inversion of the configuration at C1. The N7-isomer (14%) of 200 was also isolated. Finally, acidic hydrolysis of 200 gave the racemic (±)-201 in 27% overall yield starting from 199.94
The example just reported is a typical case of carbocyclic nucleoside synthesis. Carbanucleosides are modified nucleosides in which a methylene group has replaced the oxygen atom of the furanose ring.95 Due to the absence of a glycosidic linkage, carbanucleosides are chemically more stable and not subjected to the phosphorylases that cleave the N-glycosidic linkage in conventional nucleosides. Many compounds of this class with potent and selective biological activity were identified, including carbovir and abacavir, which are currently used therapeutically as anti-HIV agents.96 An important aspect of the anti-HIV therapy is the suppression of viral replication in the brain; in this regard an enhanced lipophilicity of potential drugs is likely to be advantageous. The modification of some structures by fusion of a carbocyclic or heterocyclic rings to the carbasugar moiety increases the lipophilicity (and hence the access to the central nervous system, a major reservoir of HIV),97 maintaining at the same time the rigidity imposed by the C2′–C3′ double bond. The synthesis of carbocyclic nucleosides containing a fused isoxazoline ring and lacking a methylene group in the side chain of the carbocyclic unit was suggested as a new class of analogs.98 Nucleosides lacking a methylene group in the side chain have been reported and in some cases display reduced cytotoxicity. The synthetic route that was exploited is based on the HDA cycloaddition of dienes with transient acyl nitroso moieties,31 affording privileged structures for the synthesis of carbocyclic nucleosides. The HDA cycloadduct N-benzoyl-2,3-oxazanorborn-5-ene 202 was obtained from freshly distilled cyclopentadiene that quantitatively trapped the nitrosocarbonyl benzene generated in situ in good yields through the mild oxidation of benzonitrile oxide Ph-CNO (BNO) with a slight excess of NMO according to the reported procedure (Scheme 1.35 ).31
Scheme 1.35.

Nitrosocarbonyl approach to nucleoside analogs 207a,b.
The HDA cycloadduct 202 was found to be an excellent dipolarophile toward BNO addition, affording the two regioisomeric cycloadducts 203a,b, which were easily isolated in quantitative yields by chromatographic separation in a 3:2 regioisomeric ratio. A proper tuning of the subsequent synthetic steps had to be done for the scale-up necessary for the synthesis of nucleosides in the due amount for the biological tests. The ease detachment of the benzoyl group from the cycloadducts 203a,b was achieved through NaOH/MeOH treatment at room temperature for 24 h, affording the fairly stable hydroxylamines. The reaction was conducted by adding a slight excess of NaOH, added portionwise in 2–3 h, to a methanol solution of cycloadducts 203a,b, stirred until complete disappearance of the starting materials. The hydrogenolytic cleavage of the N—O bond of the hydroxylamines was performed by the reaction with Pd/C 10% in ethyl acetate at room temperature for 3 h.
Hence, the aminols 204a,b were obtained and submitted to the transformations into the desired nor-nucleosides through the linear construction of the purine and pyrimidine rings. The synthesis of the purine-nucleosides requires the two steps. The first one is the condensation of the regioisomeric aminols 204a,b with the 5-amino-4,6-dichloropyrimidine to give the pyrimidine derivatives of type 205a,b. This condensation is indeed the most difficult step in many reported syntheses, which attained only low yields (around 50%). Better yields of 205a,b were achieved by increasing the reaction temperature gradually up to the boiling point of the solvent and working in sealed tubes. The pyrimidine derivatives 205a,b were obtained in 79% yields for both regioisomeric compounds. The conversion of the pyrimidine derivatives 205a,b into the chloropurine compounds 206a,b, was achieved by treatment with ethyl orthoformate and a catalytic amount of pTsOH. The final chlorine replacement method with selected amines was conducted upon heating MeOH solutions of 205a,b at 50°C in the presence of an excess NH3 or some primary or secondary amines affording the adenine derivatives 207a,b in more than 92% yields, which have been submitted to the biological tests. Compounds 207a,b were evaluated for their inhibitory activity against a wide variety of viruses, including HSV-1, vaccinia virus, vesicular stomatitis virus, parainfluenza-3 virus, reovirus-1, sindbis virus, coxsackie virus B4, Punta Toro virus, and respiratory syncytial virus.
All synthesized compounds showed no specific antiviral effects, however some differences between the regioisomeric nucleosides can be noted. Regioisomers of type b, involving a phenyl group distal to the heterocyclic base, display better response than regioisomers of type a, which have proximal phenyl and heterocyclic base. Moreover, all compounds were evaluated for antiviral activity against HIV-1 (strain IIIB) and HIV-2 (strain Rob) in MT-4 cell culture and none of these compounds showed inhibitory activity at concentration up to 400 μg/mL. The modest antiviral activity showed from these compounds could be likely linked to the lack of substrate activity for cellular and/or viral nucleoside kinases, or alternatively, the lack of recognition of the compounds by the viral DNA or RNA polymerases.99
The same strategy was applied for the synthesis of isoxazolino-carbocyclic nor-nucleosides incorporating an anthracene moiety through nitrosocarbonyl intermediates chemistry.31 A variety of analogs were attained, starting from the already reported stereodefined heterocyclic aminols through the linear construction of purine heterocyclic rings. The synthesis hinges on the exo selective 1,3-dipolar cycloaddition of the stable anthracenenitrile oxide to the N-benzoyl-2,3-oxazanorborn-5-ene 202 and simple elaborations of the regiorisomeric cycloadducts 208a,b (Scheme 1.36 ). The synthetic route of Scheme 1.35 was here applied again and the chloropurine compounds 209a,b were prepared.100
Scheme 1.36.

Synthetic strategy anthracene-substituted nucleoside analogs 210a,b.
A selection of nucleoside primary and secondary amines derivatives 210a,b were initially tested for their inhibitory activity against a variety of viruses, including hepatitis B and C, human papillomavirus (HPV), as well as influenza viruses of type A and B. Modest antiviral activities were observed in hepatitis assays while the activities in the cases of influenza viruses were almost negligible. Good antiviral activity was found for compound of type 210b (R=H; R′=Et), with no cellular toxicity at the dose tested in the case of HPV. It was reported that the sulfonyl derivative 211 is one of the most active compounds against HPV proposed in literature (Fig. 1.10 ) and displays its antiviral activity by an allosteric mechanism: it is a hyperbolic competitive inhibitor of the ATPase activity of HPV6 E1 and also inhibits its helicase activity.101
Figure 1.10.

Structure of sulfonyl derivative 211 and structure overlap with compound 210b.
Fig. 1.10 shows the sulfonyl structure 211 (orange shading) optimized through DFT calculations along with that of compound 210b (R=H; R′=Et) and the overlap between them obtained through Root Means Squared (RMS) fitting of the aromatic moieties. As we can see, there is a nice overlap of the anthracene and naphthalene aromatic rings while the second phenyl ring of the diphenyl moiety crosses the purine ring directing the sulfonyl group in the same position where the ethyl group is located. Moreover, the isoxazoline ring nicely overlaps the amide group. The larger activity shown by those compounds, which bear a naphthyl residue, could suggest a possible mechanism based on DNA-intercalation. Some isoxazolidinyl polycyclic aromatic hydrocarbons have been proposed as DNA-intercalating antitumor agents and molecular modeling studies on these structures confirm the degree of binding when a polycyclic aromatic residue is linked to an isoxazoline heterocyclic ring.102
These investigations were completed by the synthesis of isoxazolino-carbocyclic anthracene nor-nucleosides, prepared through nitrosocarbonyl chemistry, containing uracil residues, tested for their inhibitory activity against some viruses, such as HSV-1 and HSV-2, Zoster virus, and hepatitis B and C. The activities were almost negligible in most of the cases. Again, a remarkable antiviral activity was found for a specific regioisomer 212 with no cellular toxicity at 1–100 μM dose concentration in the case of HPV.103 Regarding HPV, its complex replication machinery begins with the specific binding of E2 to the viral DNA and the recruitment of protein E1, which rapidly oligomerizes to a hexamer (around the nucleic acid) displaying its helicase activity. With this mechanism in mind, the binding affinity of the uracil derivative 212 was evaluated to the HPV11 E2 protein (Fig. 1.11 ). Flexible molecular docking of the uracil derivative 212 on this receptor gives a docking score that is comparable and even higher than the ones obtained through the self-docking of the crystallographic inhibitor. As Fig. 1.11 shows, 212 is anchored at the receptor pocket involved in the protein–protein interaction (PPI) through the isoxazoline ring and the histidine His32 residue.104
Figure 1.11.

Structure of uracil derivative 212.
From: Memeo, M. G.; Lapolla, F.; Maga, G.; Quadrelli, P. Tetrahedron Lett. 2015, 56, 1986–1990.
The anthracene and the uracil substituted cyclopentanol moieties are perfectly complementary to the pocket involved in the PPI, and, in particular, the anthracene substituent shades a hydrophobic pocket involved in the specific E2–E1 interaction. A second round of molecular docking calculations was conducted on the specific sequence of DNA recognized by viral E2. Strong indications were obtained that 212 can specifically bind the DNA minor groove. The anticonfiguration between the uracil and anthracene substituents allows for the 212 backbone adaptations on the minor DNA groove. The interactions of the uracil ring as well as the cyclopentane hydroxyl group with the viral DNA are established with the heterobases of the nucleic acid, reasonably inhibiting in this way the DNA replication machinery. The last docking calculations were carried out on the viral helicase protein E1. In this case low docking scores along with lack of specific interactions suggests the low probability that E1 represents the biological target.
Upon changing the nitrile oxide structure, the regioisomeric cycloadducts 213a,b of the bromonitrile oxide to the N-benzoyl-2,3-oxazanorborn-5-ene 202 were easily prepared and elaborated into a novel class of uracil nor-nucleoside derivatives (Scheme 1.37 ). In the key synthetic step represented by the reductive N–O bond cleavage, an unusual double ring opening afforded the aminol intermediates 214a,b containing a β-hydroxynitrile structure. By adapting known protocols, the aminols entered the linear construction of uracil rings.105
Scheme 1.37.

Bromonitrile oxide strategy to nucleoside analogs 216a,b.
The novel nucleosides 216a,b were found structurally similar to a potent antiviral compound, brivudin (BVDU), and molecular modeling and docking selected one of the two regioisomeric structures as a promising candidate for antiviral tests, due to the nice level of binding with the thymidine kinase (TK), the enzyme involved in virus replication. In order to verify the stability of the poses and to better simulate the induced-fit effect of the ligands on the enzyme, optimization and MD simulations were performed on the complexes of 216a,b within TK. The superimposition of 216a,b with BVDU is shown in Fig. 1.12 .106
Figure 1.12.

(A) Overlay of BVDU with 216a and 216b after 5 ns of MD simulations, starting from the best docking solutions. The residues of the active site shown belong to the complex with BVDU. (B) Predicted binding mode of compound 216a after MD simulation. (C) Predicted binding mode of compound 216b after MD simulation. Hydrogen bonds are shown as dashed lines. BVDU, Brivudin.
From: Savion, M.; Memeo, M. G.; Bovio, B.; Grazioso, G.; Legnani, L.; Quadrelli, P. Tetrahedron. 2012, 68, 1845–1852.
In the case of compound 216a, the stabilizing pairwise interaction of the nitrogen basis with Gln125 is maintained, some interactions are lost, and some new interactions are observed. Adaptation of 216a into the TK binding site seems to be driven by the proximity of the CN group to the side chain of Tyr101, which causes a change in the hydrogen bond network found for the cyclopentane moiety. Actually, the OH at C3′ remains in contact with the side chain of Tyr101, while the OH at C4′ establishes an interaction with Glu225. Moreover, the lone pair of the latter hydroxyl group accepts a hydrogen bond from the charged side chain of Arg222, a residue in turn blocked by salt bridges with Glu225 and Glu83. Conversely, in the case of compound 216b, only one of the two hydrogen bonds of the nucleobase with Gln125 is maintained. Moreover, the OH groups at C2′ and C4′ of the cyclopentane ring interacts with Glu225 and Glu83, respectively. Furthermore, it is worthy pointing out that both compounds 216a,b, compared with BVDU, lack the bromovinyl chain with the consequent loss of the related hydrophobic interactions that might exert an anchoring effect in the pocket of the enzyme shaped by Trp88, Tyr132, Arg163, and Ala167.
On pursuing the studies on antiviral compounds and in the search of novel small molecules with potential biological activity, some nucleoside analogs were prepared and found to be moderately active against human herpes and varicella viruses or strongly active against HPV as well as influenza A virus H1N1 by taking advantage of the synthetic protocol relied upon the nitrosocarbonyl chemistry.31
Pyridine and quinoline hydroximoyl chlorides 219a–d were prepared according to the literature procedures (Scheme 1.38 ). The selected aldehydes 217a–d were converted in high yields into the corresponding oximes 218a–d following the classical methods and from the latter, the desired hydroximoyl chlorides were obtained upon chlorination with chlorine gas.107
Scheme 1.38.

Nitrosoxcarbonyl strategy to nucleoside analogs 224 and 225.
The in situ generation of the nitrile oxides 220a–d is required in the present cases since none of the nitrile oxides at hand displayed any stability at room temperature, possibly in the solid state, for a long time. The mild oxidation of these 1,3-dipoles with NMO is conducted one-pot in the presence of the required trapping diene (freshly distilled cyclopentadiene or 1,3-cyclohexadiene) to afford the nitrosocarbonyl HDA cycloadducts 222a–d and 223a–d. The derivatives 224a–d and 225a–d, obtained through N–O bond reductive cleavage, were tested against a variety of viruses but positive results were obtained only in the case of the respiratory influenza A H1N1 virus. The majority of the compounds were found to be inactive, but in a single case 225d the EC50 values are just 10 times those of the reference compound while the SI50 values must be increased by the same factor.
One more time, HPV finds in the nitrosocarbonyl chemistry a valuable synthetic tool to achieve a remarkably active target against this virus. A fleeting heterocyclic nitrosocarbonyl 230 derived from the corresponding nitrile oxide is at work in a short-cut synthesis of 4-bromo-N-[(1R*,4S*)-4-hydroxy-2-cyclohexen-1-yl]-2-thiazolecarboxamide 232 (Scheme 1.39 ). The synthetic strategy is based on HDA cycloaddition of the in situ generated nitrile oxide 229 to 1,3-cyclohexadiene followed by mild reductive cleavage of the N–O bond in the cycloadduct 231.
Scheme 1.39.

Bromothiazole nucleoside analog synthesis 232.
The new 2-thiazolecarboxamide derivative 232 is obtained in good yields and the results of the in vitro antiviral tests indicated that the product was active against HPV virus. Moreover, some structural evidence shines some light on future perspectives on the application of pericyclic reactions or the synthesis of biological active molecules. The findings demonstrated that the synthetic methodology works well for the preparation of heterocyclic substituted novel compounds that displayed interesting and promising activity against viruses and in particular against HPV. The results clearly showed that compound 232 was inefficient against herpes, hepatitis, and influenza viruses. An exception emerged against HPV-11. Compound 232 showed an EC50 value <1 along with a CC50 value >100. The secondary screening demonstrated that compound 232 was found less effective in raft cultures than in primary assay in HEK 293 cells. The EC50/90 values are higher than those of the reference compound U0126, as well as the CC50 value. The SI50/90 values however are close to the reference.108
The identical strategy but using a different starting material is at work in another example of HPV active molecule, a new adenine derivative, prepared through the chemistry of nitrosocarbonyl intermediates.31 The synthesis relies upon the functionalization of adenine by inserting an ester moiety, to be transformed into the corresponding hydroxamic acid 233 (Scheme 1.40 ). The oxidation conducted in situ in the presence of cyclopentadiene afforded the HDA cycloadduct 235, further elaborated to give the target nucleoside analog 236.
Scheme 1.40.

Nitrosocarbonyl strategy to nucleoside analog.
This single product displayed a selective high activity against HPV. The EC50 and EC90 values are remarkably lower than the values collected for Cidofovir used as a control drug. The EC50 is 39 times lower while the CC50 value is 97.11 μM.109
A number of synthetic carbocyclic nucleosides, where the furan ring is replaced by a carbocyclic ring, was found to display important therapeutic properties. The US FDA has in fact approved Abacavir for HIV-1 treatment and Entecavir for chronic HBV infections (Fig. 1.13 ). Highly oxygenated troponoids have been identified as lead compounds for HBV infections. To date, structure-function studies on these molecules have been limited due to a scarcity of synthetic methods for their preparation. A successful adaptation of a [5+2] oxidopyrilium cycloaddition/ring-opening synthesis was applied to prepare HBV antagonists such as the 2,3,7-trihydroxy-5-methylcyclohepta-2,4,6-trien-1-one shown in Fig. 1.13.
Figure 1.13.

Structures of abacavir and entecavir; [5+2] cycloaddition/ring opening reaction.
Adapted from: Hirsch, D. R.; et al. Org. Biomol. Chem. 2018, 16, 62–69.
Analogously, compounds containing tricyclic spacer displayed interesting antiviral activities. An interesting synthetic approach to these systems is shown in Scheme 1.41 : furan and ethyl fumarate were allowed to react to afford the DA cycloadduct 237 that is reduced with LiAlH4 to give the diol 238. Treatment of the diol with benzyl azidoformate allowed for the intramolecular cyclization to give, after deprotection of the amino group, the aminol 239, which was condensed with the commercially available 4,6-dichlro-5-amino-pyrimidine to give the adduct 240. Standard acid-catalyzed orthoformate cyclization led to the 6-chloropurine nucleoside 241 that was found active against Coxsackie virus (EC50 1.86 and CC50 50 μM).110
Scheme 1.41.

Synthetic strategy to nucleosidic compound 241.
The same authors followed a similar strategy to obtain the DA cycloadduct 242 (Scheme 1.42 ) from furan and acrolein, even in very low yields. Upon functionalization of the C C double bond, the exo regioisomeric diols 243a,b were obtained and converted into the endo ones. The stereochemical inversion was needed to apply the Mitsunobu protocol for the insertion of the 6-chloropurine ring, leading to the nucleosides 245a,b in very good yields.
Scheme 1.42.

Synthetic strategy to compounds 245a,b.
Both the products were tested against the Coxsackie virus displaying the following activities: 245a, EC50 1.40 and CC50 >50 μM; 245b, EC50 2.21 and CC50 >50 μM.111
This group of Czech and Belgian researchers is actively involved in the synthesis of novel nucleosides. The preparation of novel 1′-homonucleoside derivatives locked in a West conformation by 1′,4′-bridge, consisting of annulated benzene or naphthalene ring, was illustrated. The crucial step of the synthesis was DA reaction of an appropriate aryne with a suitable furane derivative. Antiviral properties of novel compounds were studied and slight activity against HCV was detected in several compounds, although largely correlated with cytotoxicity.112
Three nucleosides 253, 255a,b showed screening results suggestive of having weak in vitro antiviral activity against Coxsackie B3 virus. Adenosine 253 (concentration=1 μg/mL) showed 4.9% of inhibition and thymidine 255b (concentration=1 μg/mL) showed 5.5% of inhibition and higher concentration (concentration=100 μg/mL) led to highly toxic uridine 255a (concentration=1 μg/mL), which gave the best result, showing more than 12.3% of inhibition. Their synthesis is shown in Scheme 1.43 ; the starting point was the lactol 246, as a good candidate for the Wittig reaction with allyltriphenylphosphonium bromide to introduce the diene functionality in 247.113
Scheme 1.43.

Synthesis of uracil derivative 255a,b.
After screening with different bases, potassium tert-butoxide (KOtBu) in THF gave the best yield of triene of 88% with a Z to E ratio of 4:1. The mixture of trienes was subjected to oxidation with IBX as an oxidant and furnished trienone 247 in 87% yield. Trienone 247 was highly unstable and polymerized quickly after standing in vacuum. Trienone 247 underwent the Intra Molecular Diels-Alder (IMDA) reaction in toluene, in the presence of methylene blue, in a sealed tube to give hydrindenone 248 in good yield. Hydrindenone 248 was first reduced with NaBH4 to the corresponding alcohols in a 1:4 ratio. One of them could be oxidized back and protected to give a silyl ether that was epoxidized to give 249. After the reduction of the epoxide, the newly formed hydroxyl group was acetylated and the silyl protection removed to afford 250, further converted through azide chemistry into the amine 251. The linear approach was then adopted to synthesize the target molecules. Reaction of amine 251 with 5-amino-4,6-dichloropyrimidine furnished pyrimidine 252 in miserable yield. Standard orthoformate cyclization afforded the adenine derivative 253. Conversely, condensation with freshly prepared isocyanates (R=H, Me) obtained the ureas 254a,b, and were subsequently cyclized to the uracil derivative 255a and the timine derivative 255b.
Antivirals and 1,3-dipolar cycloaddition reactions
1,3-Dipoles represent a valuable class of reacting intermediates frequently used in the synthesis of antiviral compounds. Examples of application of the chemistry of these in situ generated reacting species have been already accounted in previous pages, where some nitrile oxides were at work in the synthesis of some biological active molecules.98., 99., 100., 103., 105., 107., 108., 109. Their roles were for both the introduction of suitable residues necessary to display the correct antiviral activity and that of starting material for the generation of other reacting species, such as the nitrosocarbonyl intermediates,31 which represented the key intermediates for the synthesis of the target compounds.
This section reports some remarkable examples of the use of some of the most frequently used 1,3-dipoles in the synthesis of antiviral compounds. Not all the 1,3-dipoles will be reviewed, just those successfully employed for the preparation of biologically active molecules; in vitro data will be also reported here.
One of the most frequently used 1,3-dipoles is azide. The versatility of this 1,3-dipole is well known since its use in “click chemistry” as well as for the preparation of triazoles, valuable heterocycles in the synthesis of biologically active compounds. Organic azides belong to the propargyl-allenyl category of dipoles and since the discovery of triazole formation from phenyl azide and dimethyl acetylenedicarboxylate in 1893, they found large success in the construction of heterocyclic frameworks and core structures of natural and bioactive compounds.114
A simple case is shown in Scheme 1.44 where treatment of 4-acetoxybutylbromide with sodium azide in anhydrous DMF gave the 4-acetoxybutylazide in 96% yield. The 6-(methylthio)-9-(prop-2-yn-1-yl)-9H-purine was reacted with the azido-compound via 1,3-dipolar cycloaddition reaction, in anhydrous toluene under reflux, to afford a mixture of two possible regioisomers.
Scheme 1.44.

Azido-strategy to compound 258.
The regiosiomer 256 was obtained in 89%. It is well known from the literature115 that the addition of azides to unsymmetrical acetylenes is determined by steric and electronic factors. In general, such addition tends to give mainly the isomers with electron-withdrawing groups at the 4-position and electron-releasing groups at 5-position. Thus, after separation of the regioisomers, treatment with methanol saturated with ammonia in a sealed reacting vessel at 100°C led to the carboacyclic nucleoside 257. The carboacyclic nucleoside 258 was synthesized in 73% yield via treatment of 257 with CH3ONa/CH3OH at room temperature. Other compounds were prepared, but only compounds 157 and 258 have a low activity, in vitro, against parainfluenza virus type 3 and reovirus type 1, respectively (MIC50=240 μg/mL, MCC>400 μg/mL). Neither anti-HIV-1(IIIB) nor anti-HIV-2(ROD) activity was observed in MT-4 cells for the products at hand.116
Some 5-(1,2,3-triazol-1-ylmethyl)uridine derivatives of type 260,261 were synthesized via the 1,3-dipolar cycloaddition of a 5-azidomethyluridine derivative 259 with substituted mono- and di-substituted acetylenes (Scheme 1.45 ).
Scheme 1.45.

Azidomethyluridine synthetic applications to nucleoside analogues.
The antiviral activities of these compounds against hepatitis A virus (HAV, MBB cell culture-adapted strain) and HSV-1 were tested. For the antiviral activity against HAV-27 it is noted that at both concentrations tested, 10 and 20 μg/105 cells, compounds of type 260 revealed the highest antiviral activity in this series, and compounds of type 261 revealed high activity at 10 μg/105 cells using amantadine (C*) as a control. For the antiviral activity against HSV-1 the results revealed that compounds of type 260 showed the highest effect at 10 μg/105 cells, while compounds of type 261 showed moderate activity.117
The synthesis and antiviral evaluation of a series of C5-(1,4- and 1,5-disubstituted-1,2,3-triazolo)-nucleoside derivatives of type 264 was described by Agrofoglio et al. The key steps of these syntheses are the regioselective Huisgen’s 1,3-dipolar cycloaddition reactions, using either copper-catalyzed azide-alkyne cycloaddition (CuAAC) or ruthenium-catalyzed azide-alkyne cycloaddition (RuAAC) under microwave activation starting from a triple bond functionalized and protected nucleoside structure 262 and a series of substituted azides 263 (Scheme 1.46 ).
Scheme 1.46.

Synthesis of analog 264.
Some compounds among the synthesized series possess activity against HSV-1 and HSV-2, varicella-zoster virus, human cytomegalovirus, and vaccinia virus. Their cytostatic activities were determined against murine leukemia cells, human T-lymphocyte cells, and cervix carcinoma cells. Compounds were also evaluated on a wide panel of RNA viruses, including Vesicular stomatitis virus, influenza viruses type A (H1N1 and H3N2) and B in MDCK cell cultures, parainfluenza-3 virus, reovirus-1, Sindbis virus, and Punta Toro virus in Vero cell cultures and Vesicular stomatitis, Coxsackie B4, and respiratory syncytial virus, however with no specific antiviral effect. Some cellular activity was observed, which most likely implies that the compounds are able to penetrate into cells. Some of the synthesized compounds showed activities against several DNA viruses in the micromolar range. Whereas the 1,4-regioisomers exhibited antiviral activity, it is interesting to note that the (1,5)-regioisomers were found inactive against this wide panel of viruses and display no toxicity. In order to design compounds with a significantly increased potency, it will be necessary to study their phosphorylation by human and viral kinases; this process would be greatly aided by a cocrystal structure of the best compounds bound in the enzyme active sites. Also, it will be of interest to investigate the properties of their monophosphorylated derivatives against thymidylate synthase and their triphosphorylated derivatives against the viral and cellular DNA polymerases to reveal the mechanism of biological action.118 Copper-catalyzed 1,3-dipolar cycloaddition reaction between azido-ristocetin aglycon and various propargyl glycosides were used to prepare new sugar derivatives of ristocetin. Some of these compounds were found to be active against Gram-positive bacteria and showed favorable antiviral activity against the H1N1 subtype of influenza A virus.119
O6-(Benzotriazol-1H-yl)guanosine and its 2′-deoxy analog can be readily converted to the O 6-allyl derivatives of type 265 that upon diazotization with t-BuONO and TMS-N3 yield the C-2 azido derivatives of type 266 (Scheme 1.47 ).
Scheme 1.47.

Synthesis of nucleosides 268 and 270.
American and Belgian researchers have previously analyzed the solvent dependent azide·tetrazole equilibrium of C-6 azidopurine nucleosides, and in contrast to these, the O 6-allyl C-2 azido nucleosides appear to exist predominantly in the azido form, relatively independent of solvent polarity.120 Consistent with the presence of the azido functionality, each neat C-2 azide displayed a prominent IR band at 2126-2130 cm−1. A screen of conditions for the ligation of the azido nucleosides with alkynes showed that CuCl in t-BuOH/H2O is optimal, yielding C-2 1,2,3-triazolyl nucleosides 267 in 70%–82% yields. Removal of the silyl groups with Et3N·3HF followed by deallylation with PhSO2Na/Pd(PPh3)4 gave the C-2 triazolylinosine nucleosides 268-270. Products were desilylated for biological assays. The two C-2 triazolyl adenosine analogs demonstrated pronounced antiproliferative activity in human ovarian and colorectal carcinoma cell cultures. When evaluated for antiviral activity against a broad spectrum of DNA and RNA viruses, some of the C-2 triazolylinosine derivatives showed modest inhibitory activity against cytomegalovirus. Compound 270, however, displayed a somewhat more pronounced activity as anti-CMV (EC50 73 μM).121
The efficient synthesis of a new series of polyhydroxylated dibenzyl ω-(1H-1,2,3-triazol-1-yl)alkylphosphonates as acyclic nucleotide analogs of type 273 was described by Belgian and Polish researchers starting from dibenzyl ω-azido-(polyhydroxy)alkylphosphonates 271 and selected alkynes 272 under microwave irradiation (Scheme 1.48 ).
Scheme 1.48.

Synthesis of analogs 273.
Selected O,O-dibenzylphosphonate acyclonucleotides were transformed into the respective phosphonic acids. All compounds were evaluated in vitro for activity against a broad variety of DNA and RNA viruses and for cytostatic activity against murine leukemia L1210, human T-lymphocyte CEM, and human cervix carcinoma HeLa cells. Compound 273(Tymine) exhibited antiviral activity against Influenza A H3N2 subtype (EC50=20 μM-visual CPE score; EC50=18 μM-MTS method; MCC>100 μM, CC50>100 μM) in MDCK cell cultures, and 273(Uracil-Bz) was active against vesicular stomatitis virus and respiratory syncytial virus in HeLa cells (EC50=9 and 12 μM, respectively). Moreover, compound 273(BenzoUracil-Bz) showed activity against both HSV-1 and HSV-2 in HEL cell cultures (EC50 = 2.9 and 4 μM, respectively) and feline herpes virus in Crandell–Rees feline kidney (CRFK) cells (EC50 = 4 μM), but at the same time it exhibited cytotoxicity toward uninfected cells (MCCP4 lM). Several other compounds have been found to inhibit proliferation of L1210, CEM as well as HeLa cells with IC50 in the 4–50 μM range.122
A new series of 1-amino-3-(1H-1,2,3-triazol-1-yl)propylphosphonates (R)- and (S)-277 were obtained from enantiomerically pure (R)- and (S)-1-tert-butoxycarbonyl (Boc)-amino-3-azidopropylphosphonates 276 and N-propargylated nucleobases in good yields (Scheme 1.49 ).
Scheme 1.49.

Synthesis of derivatives 277.
The synthesis stands on the tosylation of the alcohol 274 and azido functionalization of 275. The cycloaddition to the N-propargylated nucleobases afforded the protected compounds 277.
All 1,2,3-triazolylphosphonates (R)- and (S)-277 were evaluated for their activities against a broad range of DNA and RNA viruses. Compound (R)-277 (B=3-acetylindole) was moderately active against vesicular stomatitis virus in HeLa cell cultures (EC50=45 μM). In addition, (S)-277 (B=adenine), (R)-277 (B=N3-Bz-benzuracil), (R)-277 (B=3-acetylindole), and (R)-277 (B=5,6-dimethylbenzimidazole) were cytotoxic toward CRFK cells (CC50=2.9, 45, 72, and 96 μM, respectively). The cited compounds 277 were also slightly cytostatic to different tumor cell lines.123
The same authors of the previous work prepared a new series of 4-substituted [(1,2,3-triazol-1-yl)acetamido]methylphosphonates of type 280 as acyclic nucleotide analogs, synthesized from diethyl (2-chloroacetamido)methylphosphonate 278 via azidation followed by 1,3-dipolar cycloaddition with selected alkynes derived from natural nucleobases or their mimetics (Scheme 1.50 ).
Scheme 1.50.

Synthesis of compound 280.
All compounds were tested for their antiviral activities against DNA and RNA viruses as well as for cytostatic activity or cytotoxicity. Among all tested compounds, [(1,2,3-triazol-1-yl)acetamido]-methylphosphonate 280 bearing a methyl-imidazole substituent showed activity against the vesicular stomatitis virus (EC50=45 μM) in HeLa cell cultures.124
Nucleoside analogs are commonly used in HIV/AIDS therapy and as a first line therapeutics for many tumor types. After entering through the membrane, nucleoside analogs are phosphorylated by intracellular nucleoside kinases into active 5′-mono-, di-, and triphosphates. The drug resistance to nucleoside analogs refers to nucleoside transporter deficiency, reduced nucleoside kinase activity, overexpression of multidrug resistance proteins, or modifications in apoptotic pathways.125 A conceivable strategy to avoid resistance is to use a prephosphorylated nucleoside directly as a drug that can thus bypass intracellular phosphorylation. However, nucleoside triphosphates (NTPs) are very poorly internalized by cells. Attempts to get around this problem by masking the phosphate groups by turning them into phosphate esters have not been successful until recently.126 The fully substituted di- and triphosphate esters become unstable. Difficulties in the development of lipophilic NTP prodrugs arise due to hydrolytical instability of the pyrophosphate bond, while in the natural NTPs this bond is kinetically stable due to negative charges that slow down cleavage by nucleophiles.
An alternative to the lipophilic prodrug approach could be in using drug delivery systems such as liposomes, nanogels, nanoparticles, and so on, which could potentially solve the problem of poor cell absorption of NTPs. SiO2 nanoparticles represent a simple and versatile method for the preparation of SiO2-dNTP conjugates. There are several examples when nucleoside analogs were originally approved as antiviral agents and then it has been shown that they display promising anticancer properties. Thus conjugates of antiviral phosphorylated nucleoside analogs may also possess potential antitumor activity. Some Russian researchers prepared triphosphates of anti-HIV nucleoside drugs lamivudine and zalcitabine with γ-alkynyl phosphoramidate group, obtained their conjugates with SiO2 nanoparticles (SiO2-dNTP) via Cu(I)-catalyzed azide-alkyne cycloaddition, and assessed their antiviral and cytotoxic properties. Scheme 1.51 shows the synthetic route.
Scheme 1.51.

SiO2 nanoparticles synthesis.
The pivotal step is represented by the insertion of a triple bond on the phosphorylated derivative 282 to give 283 that undergoes 1,3-dipolar cycloaddition with the azide 284 to afford the final compound 285. The conjugates of phosphorylated lamivudine and dideoxycytidine (zalcitabine) showed higher potency than the parent nucleosides. The conjugate of phosphorylated azidothymidine (AZT) was less active against HIV-1 than the parent nucleoside probably because of the replacement of its 3′-azido group by 1,2,3-triazole ring. These results show an opportunity for using SiO2 nanoparticles as a transport for delivering phosphorylated nucleosides to cells in order to increase their efficiency as antiviral and anticancer drugs.127 Nonobligate chain terminating nucleosides with a linear substituent (azido or ethynyl group) at the 4′ position represent an important class of compounds in antiviral discovery, particularly against HCV and HIV. 3′- AZT-derived 1,2,3-triazoles can be potent inhibitors of HIV-1. To gauge the medicinal chemistry impact of functionalizing the 4′-linear substituent and possibly generate novel antiviral nucleoside scaffolds, azide-alkyne cycloaddition reactions with 4′-AZT were investigated. Fig. 1.14 shows the structures of families of thymidine and triazole derivatives.
Figure 1.14.

Structures of triazole-thymidine and azido-triazole derivatives.
Antiviral screening identified a few triazole analogs moderately active against HIV-1 (18%–62% inhibition at 10 mM) and/or influenza A virus (15%–50% inhibition at 10 mM), and none active against West Nile virus or HCV. These results suggest that the linear 4′ azido group of ADRT may be essential for target binding and that its chemical manipulation could largely compromise antiviral potency.128 Azido-triazole derivatives were found to be potent anti-HBV agents.127b
The use of multivalent carbohydrate compounds to block cell-surface lectin receptors is a promising strategy to inhibit the entry of pathogens into cells and could lead to the discovery of novel antiviral agents. One of the main problems with this approach, however, is that it is difficult to make compounds of an adequate size and multivalency to mimic natural systems such as viruses. Hexakis adducts of [60]fullerene are useful building blocks in this regard because they maintain a globular shape at the same time as allowing control over the size and multivalency. Water-soluble tridecafullerenes decorated with 120 peripheral carbohydrate subunits, so-called superballs, were prepared through the collaboration of several European research groups and could be synthesized efficiently from hexakis adducts of [60]fullerene in one step by using copper-catalyzed azide-alkyne cycloaddition click chemistry (Fig. 1.15 ).
Figure 1.15.

Syntheses of the tridecafullerenes using CuAAC click chemistry. The core fullerene (endowed with 12 alkyne groups) is joined to the peripheral fullerene-based compounds by click chemistry. Reagents and conditions: (1) CuBr·S(CH3)2, sodium ascorbate, Cu(0), DMSO, 25°C, 48 h, 73%–79%; (2) CuSO4·5H2O, sodium ascorbate, THF/H2O, 80°C (MW), 2 h, 76%. CuAAC, Copper-catalyzed azide-alkyne cycloaddition.
Modified from: Munoz, A.; Sigwalt, D.; Iliescas, B. M.; Luczkowiak, J.; Rodriguez-Perez, L.; Nierengarten, I.; Holler, M.; Remy, J.-S.; Buffet, K.; Vincent, S. P.; Rojo, J.; Delgado, R.; Nierengarten, J.-F.; Martin, N. Nat. Chem. 2016, 8, 50–57.
Infection assays show that these superballs are potent inhibitors of cell infection by an artificial Ebola virus with half-maximum inhibitory concentrations in the subnanomolar range.129
In the previous example we have seen the application of a CuAAC methodology for the synthesis of antiviral compounds. There is considerable attention directed at chemically modifying nucleic acids with robust functional groups in order to alter their properties. Since the breakthrough of CuAAC, there have been several reports describing the synthesis and properties of novel triazole-modified nucleic acid derivatives for potential downstream DNA- and RNA-based applications. A review will focus on highlighting representative novel nucleic acid molecular structures that have been synthesized via the click azide-alkyne cycloaddition. Many of these derivatives show compatibility for various applications that involve enzymatic transformation, nucleic acid hybridization, molecular tagging, and purification and gene silencing. The details of these applications can be found in these manuscripts where the chemistry of triazoles seems to be really promising for future developments of antiviral and anticancer compounds.130., 131.
In the field of macromolecular structures, distinct and well-defined glycopolymers for deciphering the biological roles of natural bioactive polysaccharides were prepared from Fucose monomers, chemically synthesized and decorated with specific sulfation patterns including unsulfate, monosulfate, disulfate, and trisulfate groups (Fig. 1.16 ).
Figure 1.16.

Synthesis of fucoidan-mimetic glycopolymers.
From: Fan, F.; Cai, C.; Wang, W.; Gao, L.; Li, J.; Li, J.; Gu, F.; Sun, T.; Li, J.; Li, C.; Yu, G. ACS Macro Lett. 2018, 7, 330–335.
The six fucoidan-mimetic glycopolymers were successfully fabricated through Microwave (MW)-assisted ring-opening metathesis polymerization in an emulsion system. Three glycopolymers associated with 2-O-sulfation exhibited better inhibitory effects on the H1N1 virus, while glycopolymers with monosulfate groups were more effective against the H3N2 virus. These findings would promote the development of novel antiinfluenza A virus drugs based on natural fucoidans.132
Another class of 1,3-dipoles that has been extensively used in the synthesis of antiviral compounds is constituted of nitrones. Nitrones are arguably the most useful tools because of their ability to generate nitrogen- and oxygen-based functionality and to introduce chiral centers stereoselectively. They were first prepared by Beckmann in 1890 and several methods are known for their preparation, apt to fit the synthetic requirements to be used in the planned syntheses. They constitute a valuable entry to isoxazolidines; these heterocyclic rings are a remarkable alternative to sugar moieties in nucleosides and they mimic the spacer of natural compounds. The facility of preparation of these 1,3-dipoles and the variety of substituents that can be placed on the 1,3-dipolar structures determine the success of these reactive species in the synthesis of new biologically active compounds.
Among the numerous examples reported in literature, it is worth mentioning the use of unconventional nitrones bearing phosphonate substituents leading to a new class of truncated phosphonated oxazanucleosides. The phosphonate nitrone 286 undergoes 1,3-dipolar cycloaddition reaction with the ethyl 2-acetoxyacrylate in THF under reflux to afford the isoxazolidine 287 as minor component of the diasteroisomeric mixture (80% yield). Upon adapting the Vorbrüggen protocol, uracil, 5-fluorouracil, and thymine were inserted on the isoxazolidine skeleton to give the final compounds 288a-c (Scheme 1.52 ).
Scheme 1.52.

Nitrone synthesis of isoxazolidines.
The biological tests indicate that these anomers 288a-c inhibit the reverse transcriptase of Avian Moloney virus (AMV), human T-lymphotropic virus (HTLV-1), and HIV. In particular, the 5-fluorouracil derivative 288c is the more promising compound, acting on Abelson Murine Leukemia virus and HIV at the concentration of 1 and 10 nM, respectively. The level of the inhibitory activity of this product toward HTLV-1 and HIV was 10-fold higher than that of tenofovir and similar to that of AZT. Moreover, this compound does not show any cytotoxicity according to 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay.
The 1,3-dipolar cycloaddition to vinyl acetate led also to the diasteromers of type 289 in 62% yield. Insertion of thymine and 5-fluorouracil led to the nucleosides 290a,b that were found to inhibit completely the reverse transcriptase of the AMV and HIV at concentration 1 nM, at a level comparable with that of tenofovir (1 nM) and 10-fold lower than AZT (10 nM). Moreover, MTS assays indicate a very low toxicity (CC50 > 500 μM) in comparison with AZT (CC50 12.14 μM). These and many other applications of phosphonate nitrones have been reviewed133 and the processes studied from the mechanistic point of view; the cycloadditions, in fact, showed a complete regioselectivity and a nearly exclusive cis stereoselectivity. M062X/6-31G(d,p) calculations were conducted to rationalize the regio- and the stereochemical outcome.134
A novel series of 5-arylcarbamoyl-2-methylisoxazolidin-3-yl-3-phosphonates of type 293A,B were synthesized via the 1,3-dipolar cycloaddition reaction of the N-methyl-C-(diethoxyphosphoryl)nitrone 286 with N-substituted naphthalimide acrylamides 292A,B prepared from the amine 291A,B upon functionalization with acryloyl chloride in presence of triethylamine (Scheme 1.53 ).
Scheme 1.53.

Nitrone synthetic strategy to compound 293.
All cis- and trans-isoxazolidine phosphonates obtained were assessed for antiviral activity against a broad range of DNA and RNA viruses. Isoxazolidines trans-293A and trans-293B exhibited the highest activity (EC50=8.9 μM) toward cytomegalovirus. Compounds cis- and trans-293A as well as cis- and trans-293B were found potent against HSV and vaccinia viruses (EC50 in the 45–58 μM range). Antiproliferative evaluation of all obtained isoxazolidines revealed promising activities toward tested cancer cell lines with IC50 in the 1.1–19 μM range.135
Another class of 1,3-dipole that found attention in the synthesis of nucleoside analogs is that of nitrile oxides. Although less used in the synthesis of nucleoside because of their high instability and difficulties for their preparation, some cases are reported in which the choice of the proper substituent can solve selectivity problems and facilitate the access to biologically active molecules.
A series of novel isoxazoline-linked pseudodisaccharide derivatives 298 were regiospecifically synthesized by the 1,3-dipolar cycloaddition reaction of α-allyl-C-glycopyranoside 297 and sugar-derived nitrile oxide 296 with good yields. The nitrile oxide was prepared starting from the aldehyde 284 upon conversion into the corresponding oxime 295 and chlorination with N-chlorosuccinimide (NCS) followed by in situ generation of the 1,3-dipole in the presence of the suitable dipolarophile 297 (Scheme 1.54 ).
Scheme 1.54.

Synthetic strategy to compound 298.
The structure of the compound 298 were characterized by NMR spectroscopy and MS spectrometry and confirmed by the X-ray crystallographic analysis. The glycosidase inhibitory activities of 298 and other compounds were determined with hydrolytic reactions of glycosidases using acarbose as a control. However, the compound 298 showed a selectively moderate inhibition to β-glucosidase; the inhibition of 298 was 20% at the concentration of 2.7 μmol/mL. HIV-RT and antitumor activity were also preliminarily evaluated. Some of these types of cycloadducts exhibited potent inhibitory activity to HIV-RT.136
The introduction of a second heteroatom into the furanose ring would result in several new classes of nucleoside analogs. The presence of multiheteroatoms in the five-membered ring is desired to retain the required conformation for the recognition between the nucleoside and the target enzyme (reverse transcriptase). The use of oxygen or sulfur atoms has produced several diheteroatom-substituted analogs and some of the dioxolanyl or oxathiolanyl nucleosides are potent virus inhibitors.137 It has been synthetically challenging to introduce both oxygen and nitrogen into the five-membered ring while keeping a glycosidic bond. The N–O compounds such as dihydroisoxazolidine display a mutual induction effect of two heteroatoms exerting several desired properties. First, the nitrogen atom becomes less basic to prevent its protonation; therefore, the synthesized compounds are expected to be stable at physiological pH. Second, the oxygen atom may also become less nucleophilic, making the glycosidic bond more resistant to acidic hydrolysis. The use of nitrile oxides in the preparation of antiviral derivatives via 1,3-dipolar cycloaddition reactions represents a valuable method to achieve these targets.138
[2+2] Cycloaddition reaction for antiviral compounds
Nucleosides are structurally defined compounds where the spacer often plays a key role in recognition and in the biological activity outcome. Cycloaddition reactions offer a variety of methods for the synthesis of nucleoside and in particular regarding the spacer structure. Within the frame of cyclic spacers and given the most frequently encountered five- and six-membered ring structures, the cyclobutane moiety received considerable attention due to the broad spectrum of antiviral and anticancer compounds that were discovered. Moreover, cyclobutane amino acids and dipeptides were found in nature, some of them with interesting properties as antibiotics and antivirals.
The cyclobutane structural unit can be easily prepared through the photochemical [2+2] cycloaddition reaction or on the use of suitable precursors bearing the preformed cyclobutane ring. Absolute chirality can be raised from chiral precursors obtained from natural sources or by the use of chiral catalysts. Functionalization of the carbocyclic ring is also a challenging task, crucial for the synthesis of polyfunctionalized nucleosides and amino acids.
In the search for therapeutic agents against HSV and HIV, much attention was focused on the preparation of analogs of natural nucleosides as antimetabolites. Such analogs were modified in the carbohydrate portion of the molecule, the spacer, and in the base portion. Examination of molecular models suggested that a cyclobutane ring might serve as a surrogate for the oxetan ring of oxetanocin A. This observation led to the synthesis of the racemic carbocyclic analogs of oxetanocin A, compounds 302 and 303 that exhibited broad-spectrum activity against HSV and HIV (Scheme 1.55 ). They were considered promising agents for AIDS treatment.
Scheme 1.55.

[2+2] Photochemical approach to nucleoside analogs 302 and 303.
For the synthesis of these analogs, many preparative methods for optically active cyclobutane derivatives were developed. Some reported syntheses resort to a racemic or enantiomerically pure substituted cyclobutanone as the key intermediate. Furthermore, the nucleoside strategy may rely on a convergent synthesis or a linear approach. In Scheme 1.55, the 4-(((tert-butyldimethylsilyl) oxy)methyl)pyridin-2(1H)-one 299 was irradiated at 300 nm and the intermediate 300 was obtained as a result of a [2+2] photochemical ring closure. Synthetic elaboration of 300 led to the protected cyclobutane-amine 301 that is the starting point for the linear construction of adenine and guanine, respectively, to afford the compounds 302 and 303. This represents just a simple example of the way these compounds can be easily obtained; many other approaches are reported in literature, not only regarding the preparation of antiviral compounds but also for the synthesis of amino acids, peptides, and other biologically active molecules.139
Acknowledgment
Financial support by the University of Pavia and MIUR (PRIN 2011, CUP: F11J12000210001) is gratefully acknowledged. We also thank “VIPCAT – Value Added Innovative Protocols for Catalytic Transformations” project (CUP: E46D17000110009) for valuable financial support.
References
- 1.Domingo L.R., Ríos-Gutiérrez M., Pérez P. Molecules. 2016;21:748–770. doi: 10.3390/molecules21060748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Domingo L.R. Molecules. 2016;21:1319–1334. doi: 10.3390/molecules21101319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Nishiwaki N., editor. Methods and Applications of Cycloaddition Reactions in Organic Syntheses. John Wiley & Sons, Inc.; 2014. [Google Scholar]; (b) Maretina I.A., Ionin B.I., editors. Alkynes in Cycloadditions. John Wiley & Sons, Ltd; 2013. ; p 310. [Google Scholar]; (c) Hassner A., editor. Vol. 13. Springer-Verlag Berlin Heidelberg; 2008. Synthesis of Heterocycles via Cycloadditions II; p. 212. (Topics in Heterocyclic Chemistry). [Google Scholar]; (d) Hassner A., editor. Vol. 12. Springer-Verlag Berlin Heidelberg; 2008. Synthesis of Heterocycles via Cycloadditions I; p. 226. (Topics in Heterocyclic Chemistry). [Google Scholar]; (e) Kobayashi S., Jorgensen K.A., editors. Cycloaddition Reactions in Organic Synthesis. Wiley-VCH; 2002. ; p 332. [Google Scholar]; (f) Padwa A., Pearson W.H., editors. Vol. 59. Wiley, J. and Sons: Chichester; UK: 2002. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; p. 940. (Chemistry of Heterocyclic Compounds). [Google Scholar]; (g) Harmata M., editor. Vol. 6. JAI Press Inc.; 1999. p. 249. (Advances in Cycloaddition). [Google Scholar]; (h) Lautens M., editor. Vol. 4. JAI Press Inc.; 1997. p. 210. (Advances in Cycloaddition). [Google Scholar]; (i) Curran D.P., editor. Vol. 3. JAI Press Inc.; 1993. p. 210. (Advances in Cycloaddition). [Google Scholar]; (j) Curran D.P., editor. Vol. 2. JAI Press Inc.; 1990. p. 219. (Advances in Cycloaddition). [Google Scholar]; (k) Zefirov N.S., Kazimirchik I.V., Lukin K.A., editors. Cycloaddition of Dichlorocarbene to Olefins; Izd. Nauka; 1985. ; p 152. [Google Scholar]; (l) Padwa A., editor. Vol. 2. John Wiley and Sons; 1984. p. 704. (1,3-Dipolar CycloadditionChemistry). [Google Scholar]; (m) Padwa A., editor. Vol. 1. John Wiley and Sons.; 1984. p. 817. (1,3-Dipolar Cycloaddition Chemistry). [Google Scholar]; (n) Muller L.L., Hamer J., editors. The Formation of Three and Four-Membered Heterocycles. Interscience Publishers; 1967. 1, 2-Cycloaddition Reactions. ; p 362. [Google Scholar]; (o) Ulrich H., editor. Vol. 9. Academic Press; 1967. p. 364. (Organic Chemistry: A Series of Monographs). [Google Scholar]; (p) Hamer J., editor. Vol. 8. Academic Press; 1966. 1,4-Cycloaddition Reactions: The Diels-Alder Reaction in Heterocyclic Syntheses; p. 530. (Organic Chemistry: A Series of Monographs). [Google Scholar]; (q) Vrabel M., Carrell T., editors. Cycloadditions in Bioorthogonal Chemistry. Springer; 2016. Topics in Current Chemistry Collections. ; p 107. [Google Scholar]
- 4.Rezza, G. Virus. Scienze della Vita; Treccani: Roma, 2007; pp 611–618. For a cutting-edge view on antiviral compounds, see De Clerq, E. Med. Chem. Rev. 2013, 33, 1249–1277.
- 5.(a) Colman P.M. In: The Influenza Viruses: Influenza Virus Neuraminidase, Enzyme and Antigen. Krug R.M., editor. Plenum; New York: 1989. pp. 175–218. [Google Scholar]; (b) Colman P.M. Protein Sci. 1994;3:1687–1696. doi: 10.1002/pro.5560031007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) See also Kovac, P., Ed. Carbohydrate Chemistry; CRC Press: Boca Raton, FL, 2012; Vol. 1, pp 239–258, Chp 26–28.
- 6.Zhang L., Williams M.A., Mendel D.B., Escarpe P.A., Chen X., Wang K.-Y., Graves B.J., Lawton G., Kim C.U. Bioorg. Med. Chem. Lett. 1999;9:1751–1756. doi: 10.1016/s0960-894x(99)00280-2. [DOI] [PubMed] [Google Scholar]
- 7.Kerrigan S.A., Smith P.W., Stoodley R.J. Tetrahedron Lett. 2001;42:4709–4712. [Google Scholar]
- 8.Kerrigan S.A., Pritchard R.G., Smith P.W., Stoodley R.J. Tetrahedron Lett. 2001;42:7687–7690. [Google Scholar]
- 9.von Itzstein M., Wu W.-Y., Kok G.B., Pegg M.S., Dyason J.C., Jin B., Phan T.V., Smythe M.L., White H.F., Oliver S.W., Colman P.M., Varghese J.N., Ryan D.M., Woods J.M., Bethell R.C., Hotham V.J., Cameron J.M., Penn C.R. Nature. 1993;363:418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kim C.U., Lew W., Williams M.A., Liu H., Zhang L., Swaminathan S., Bischofberger N., Chen M.S., Mendel D.B., Tai C.Y., Laver W.G., Stevens R.C. J. Am. Chem. Soc. 1997;119:681–690. doi: 10.1021/ja963036t. [DOI] [PubMed] [Google Scholar]; (b) Farina V., Brown J.D. Angew. Chem. Int. Ed. 2006;45:7330–7334. doi: 10.1002/anie.200602623. For a recent review, see. [DOI] [PubMed] [Google Scholar]
- 11.(a) Abrecht S., Harrington P., Iding H., Karpf M., Trussardi R., Wirz B., Zutter U. Chimia. 2004;58:621–629. [Google Scholar]; (b) Abrecht S., Federspiel M.C., Estermann H., Fischer R., Karpf M., Mair H.-J., Oberhauser T., Rimmler G., Trussardi R., Zutter U. Chimia. 2007;61:93–99. [Google Scholar]
- 12.(a) Yeung Y.-Y., Hong S., Corey E.J. J. Am. Chem. Soc. 2006;128:6310–6311. doi: 10.1021/ja0616433. [DOI] [PubMed] [Google Scholar]; (b) Ryu D.H., Corey E.J. J. Am. Chem. Soc. 2003;125:6388–6390. doi: 10.1021/ja035393r. [DOI] [PubMed] [Google Scholar]
- 13.(a) Yamatsugu K., Kamijo S., Suto Y., Kanai M., Shibasaki M. Tetrahedron Lett. 2007;48:1403–1406. [Google Scholar]; (b) Mita T., Fukuda N., Roca F.X., Kanai M., Shibasaki M. Org. Lett. 2007;9:259–262. doi: 10.1021/ol062663c. [DOI] [PubMed] [Google Scholar]
- 14.Satoh N., Akiba T., Yokoshima S., Fukuyama T. Angew. Chem. Int. Ed. 2007;46:5734–5736. doi: 10.1002/anie.200701754. [DOI] [PubMed] [Google Scholar]
- 15.(a) Ahrendt K.A., Borths C.J., MacMillan D.W.C. J. Am. Chem. Soc. 2000;122:4243–4244. [Google Scholar]; (b) Lelais G., Mac-Millan D.W.C. Aldrichimica Acta. 2006;39:79–87. [Google Scholar]
- 16.Moriarty R.M., Chany C.J., II, Vaid R.K., Prakash O., Tuladhar S.M. J. Org. Chem. 1993;58:2478–2482. [Google Scholar]
- 17.Shibasaki M., Kanai M. Eur. J. Org. Chem. 2008:1839–1850. [Google Scholar]
- 18.Gong J., Xu W. Curr. Med. Chem. 2008;15:3145–3159. doi: 10.2174/092986708786848497. [DOI] [PubMed] [Google Scholar]
- 19.Matsuya Y., Qin H., Nagaoka M., Nemoto H. Heterocycles. 2004;62:207–211. [Google Scholar]
- 20.Mosmann T. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 21.Matsuya Y., Sasaki K., Ochiai H., Nemoto H. Biorg. Med. Chem. 2007;15:424–432. doi: 10.1016/j.bmc.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 22.(a) Shie J.-J., Fang J.-M. J. Chin. Chem. Soc. 2014;61:127–141. [Google Scholar]; (b) Grondal C., Jeanty M., Enders D. Nat. Chem. 2010;2:167–178. doi: 10.1038/nchem.539. [DOI] [PubMed] [Google Scholar]
- 23.(a) Kametani T., Nemoto H., Ishikawa H., Shiroyama K., Fukumoto K. J. Am. Chem. Soc. 1976;98:3378–3379. doi: 10.1021/ja00427a057. [DOI] [PubMed] [Google Scholar]; (b) Kametani T., Nemoto H., Ishikawa H., Shiroyama K., Matsumoto H., Fukumoto K. J. Am. Chem. Soc. 1977;99:3461–3466. doi: 10.1021/ja00452a045. [DOI] [PubMed] [Google Scholar]; (c) Kametani T., Matsumoto H., Nemoto H., Fukumoto K. J. Am. Chem. Soc. 1978;100:6218–6220. [Google Scholar]
- 24.Kametani T., Suzuki K., Nemoto H. J. Am. Chem. Soc. 1981;103:2890–2891. [Google Scholar]
- 25.Nemoto H., Fujita S., Nagai M., Fukumoto K., Kametani T. J. Am. Chem. Soc. 1988;110:2931–2938. [Google Scholar]
- 26.Nickisch K., Bittler D., Casals-Stenzel J., Laurent H., Nickolson R., Nishino Y., Petzoldt K., Wiechert R.J. J. Med. Chem. 1985;28:546–550. doi: 10.1021/jm50001a002. [DOI] [PubMed] [Google Scholar]
- 27.Jefford C.W., Bernardinelli G., Wang Y., Spellmeyer D.C., Buda A., Houk K.N. J. Am. Chem. Soc. 1992;114:1157–1165. [Google Scholar]
- 28.(a) Kametani T., Tsubuki M., Nemoto H., Suzuki K. J. Am. Chem. Soc. 1981;103:1256–1258. [Google Scholar]; (b) Nemoto H., Suzuki K., Tsubuki M., Minemura K., Fukumoto K., Kametani T., Furuyama H. Tetrahedron. 1983;39:1123–1132. [Google Scholar]
- 29.Nemoto H. Chem. Pharm. Bull. 2007;55:961–974. doi: 10.1248/cpb.55.961. [DOI] [PubMed] [Google Scholar]
- 30.Karplas A., Fleet G.W.J., Dwek R.A., Petursson S., Namgoong S.K., Ramsden N.G., Jacob G.S., Rademacher T.W. Proc. Natl. Acad. Sci. U.S.A. 1988;85:9229–9233. doi: 10.1073/pnas.85.23.9229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Memeo M.G., Quadrelli P. Chem. Rev. 2017;117:2108–2200. doi: 10.1021/acs.chemrev.6b00684. For a review on nitrosocarbonyl intermediates see. [DOI] [PubMed] [Google Scholar]
- 32.Tschamber T., Craig C.J., Muller M., Streith J. Tetrahedron. 1996;52:6201–6214. [Google Scholar]
- 33.(a) Corfield A.P., Schauer R. In: Schauer R., editor. Vol. 10. Springer Verlag: Wien; New York: 1982. p. 5. (Sialic Acids: Chemistry, Metabolism and Function in Cell Biology Monographs). [Google Scholar]; (b) Schauer R. Adv. Carbohydr. Chem. Biochem. 1982;40:131. doi: 10.1016/s0065-2318(08)60109-2. [DOI] [PubMed] [Google Scholar]; (c) Ng S.S., Dain J. In: Biological Roles of Sialic Acid. Rosenberg R.A., Schengrund C.L., editors. Plenum Press; New York: 1976. p. 59. [Google Scholar]; (d) Jeanloz R.W., Codington J.F. In: Biological Roles of Sialic Acid. Rosenberg R.A., Schengrund C.L., editors. Plenum Press; New York: 1976. p. 201. [Google Scholar]
- 34.Hu Y.-J., Huang X.-D., Yao Z.-J., Wu Y.-L. J. Org. Chem. 1998;63:2456–2461. doi: 10.1021/jo971186w. [DOI] [PubMed] [Google Scholar]
- 35.Hashiyama T., Morikava K., Sharpless K.B. J. Org. Chem. 1992;57:5067–5068. [Google Scholar]
- 36.Li L.-S., Wu Y., Wu Y.-L. J. Carbohydr. Res. 1999;18:1067–1077. [Google Scholar]
- 37.Smith P.W., Sollis S.L., Howes P.D., Cherry P.C., Cobley K.N., Taylor H., Whittington A.R., Scicinski J., Bethell R.C., Taylor N., Skarzynski T., Cleasby A., Singh O., Wonacott A., Varghese J., Colman P. Bioorg. Med. Chem. Lett. 1996;6:2931–2936. [Google Scholar]
- 38.Ryan D.M., Ticehurst J., Dempsey M.H. Antimicrob. Agents Chemother. 1995;39:2563–2564. doi: 10.1128/aac.39.11.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.(a) Sorbera L.A., Graul A., Castaner J. Drugs Future. 2000;25:249–251. [Google Scholar]; (b) Chand P., Kotian P.L., Dehghani A., El-Kattan Y., Lin T.-H., Hutchison T.L., Babu Y.S., Bantia S., Elliott A.J., Montgomery J.A. J. Med. Chem. 2001;44:4379–4392. doi: 10.1021/jm010277p. [DOI] [PubMed] [Google Scholar]
- 40.Mineno T., Miller M.J. J. Org. Chem. 2003;68:6591–6596. doi: 10.1021/jo034316b. [DOI] [PubMed] [Google Scholar]
- 41.Wu T.S., Chan Y.Y., Leu Y.L., Chern C.Y., Chen C.F. Phytochemistry. 1996;42:907–908. [Google Scholar]
- 42.Wu T.S., Leu Y.L., Hsu H.C., Ou L.F., Chen C.C., Chen C.F., Ou J.C., Wu Y.C. Phytochemistry. 1995;39:383–385. doi: 10.1016/0031-9422(94)00901-5. [DOI] [PubMed] [Google Scholar]
- 43.Toyota M., Komori C., Ihara M. J. Org. Chem. 2000;65:7110–7113. doi: 10.1021/jo000816i. [DOI] [PubMed] [Google Scholar]
- 44.Kametani T., Takeda H., Nemoto H., Fukumoto K. J. Chem. Soc. Perkin Trans. 1. 1975:1825–1828. [Google Scholar]
- 45.Boger D.L., Hüter O., Mbiya K., Zhang M. J. Am. Chem. Soc. 1995;117:11839–11849. [Google Scholar]
- 46.(a) Kametani T., Takeda H., Nemoto H., Fukumoto K. Heterocycles. 1975;3:167–170. [Google Scholar]; (b) Comins D.L., Saha J.K. J. Org. Chem. 1996;61:9623–9624. [Google Scholar]; (c) Josien H., Curran D.P. Tetrahedron. 1997;53:8881–8886. [Google Scholar]
- 47.Noyori R., Tomino I., Tanimoto Y., Nishizawa M. J. Am. Chem. Soc. 1984;106:6709–6716. [Google Scholar]
- 48.Govindachari T.R., Ravindranath K.R., Viswanathan N. J. Chem. Soc. Perkin Trans. 1. 1974:1215–1217. [PubMed] [Google Scholar]
- 49.Boger D.L., Hong J. J. Am. Chem. Soc. 1998;120:1218–1222. [Google Scholar]
- 50.Crackett P., Demont E., Eatherton A., Frampton C.X.S., Gilbert J., Kahn I., Redshaw M.S., Watson W. Synlett. 2004:679–683. [Google Scholar]
- 51.Omura S., Nakagawa A., Hashimoto H., Oiwa R., Iwai Y., Hirano A., Shibukawa N., Kojima Y. J. Antibiot. 1980;33:1395–1396. doi: 10.7164/antibiotics.33.1395. [DOI] [PubMed] [Google Scholar]
- 52.Nakagawa A., Iwai Y., Hashimoto H., Miyazaki N., Oiwa R., Takahashi Y., Hirano A., Shibukawa N., Kojima Y., Omura S. J. Antibiot. 1981;34:1408–1415. doi: 10.7164/antibiotics.34.1408. [DOI] [PubMed] [Google Scholar]
- 53.(a) Hill M.L., Raphael R.A. Tetrahedron Lett. 1986;27:1293–1296. [Google Scholar]; (b) Hill M.L., Raphael R.A. Tetrahedron Lett. 1990;46:4587–4594. [Google Scholar]; (c) Morimoto Y., Matsuda F., Shirahama H. Synlett. 1991;3:201–203. [Google Scholar]; (d) Morimoto Y., Matsuda F., Shirahama H. Tetrahedron. 1996;52:10609–10630. [Google Scholar]; (e) Steinhagen H., Corey E.J. Org. Lett. 1999;1:823–824. doi: 10.1021/ol9908575. [DOI] [PubMed] [Google Scholar]
- 54.Keck D., Vanderheiden S., Bräse S. Eur. J. Org. Chem. 2006;21:4916–4923. [Google Scholar]
- 55.Stockwell B.R. Nature. 2004;432:846–854. doi: 10.1038/nature03196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berg T. Angew. Chem. Int. Ed. 2005;44:5008–5011. doi: 10.1002/anie.200500721. [DOI] [PubMed] [Google Scholar]
- 57.Mayer T.U., Kapoor T.M., Haggarty S.J., King R.W., Schreiber S.L., Mitchtison T.J. Science. 1999;286:971–974. doi: 10.1126/science.286.5441.971. [DOI] [PubMed] [Google Scholar]
- 58.Leßmann T., Leuenberger M.G., Menninger S., Lopez-Canet M., Müller O., Hümmer S., Bormann J., Korn K., Fava E., Zerial M., Mayer T.U., Waldmann H. Chem. Biol. 2007;14:443–451. doi: 10.1016/j.chembiol.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 59.Kamimura A., Nakano T. J. Org. Chem. 2010;75:3133–3136. doi: 10.1021/jo1002856. and references therein. [DOI] [PubMed] [Google Scholar]
- 60.(a) Buchi G., Coffen D.L., Kocsis K., Sonnet P.E., Ziegler F.E. J. Am. Chem. Soc. 1966;88:3099–3109. [Google Scholar]; (b) Marazano C., LeGoff M., Fourrey J., Das B.C. J. Chem. Soc. Chem. Commun. 1981:389–391. [Google Scholar]; (c) Raucher S., Bray B.L. J. Org. Chem. 1987;109:442–446. [Google Scholar]; (d) Reding M.T., Fukuyama T. Org. Lett. 1999;1:973–976. [Google Scholar]
- 61.(a) Satoh N., Akiba T., Yokoshima S., Fukuyama T. Angew. Chem. Int. Ed. 2007;46:5734–5736. doi: 10.1002/anie.200701754. [DOI] [PubMed] [Google Scholar]; (b) Satoh N., Akiba T., Yokoshima S., Fukuyama T. Tetrahedron. 2009;65:3239–3245. [Google Scholar]
- 62.Nakano N., Osone K., Takeshita M., Kwon E., Seki C., Matsuyama H., Takanoa N., Koharia Y. Chem. Commun. 2010;46:4827–4829. doi: 10.1039/c0cc00110d. [DOI] [PubMed] [Google Scholar]
- 63.Vimr E.R., Kalivoda K.A., Deszo E.L., Steenbergen S.M. Microbiol. Mol. Biol. Rev. 2004;68:132–153. doi: 10.1128/MMBR.68.1.132-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wagner R., Matrosovich M., Klenk H.D. Rev. Med. Virol. 2002;12:159–166. doi: 10.1002/rmv.352. [DOI] [PubMed] [Google Scholar]
- 65.Richichi B., Lunghi C., Papakyriakou A., Francesconi O., Nativi C. Pure Appl. Chem. 2013;85:1803–1811. [Google Scholar]
- 66.(a) Singh S.P., Parmar S.S., Raman K., Stenberg V.I. Chem. Rev. 1981;81:175–203. [Google Scholar]; (b) Theocharis S., Margeli A., Kouraklis G. Curr. Med. Chem. Anticancer Agents. 2003;3:239–251. doi: 10.2174/1568011033482431. [DOI] [PubMed] [Google Scholar]; (c) Theocharis S., Margeli A., Vielh P., Kouraklis G. Cancer Treat. Rev. 2004;30:545–554. doi: 10.1016/j.ctrv.2004.04.004. [DOI] [PubMed] [Google Scholar]; (d) Sing W.T., Lee C.L., Yeo S.L., Lim S.P., Sim M.M. Bioorg. Med. Chem. Lett. 2001;11:91–94. doi: 10.1016/s0960-894x(00)00610-7. [DOI] [PubMed] [Google Scholar]; (e) Rao A., Balzarini J., Carbone A., Chimirri A., De Clercq E., Monforte A.M., Monforte P., Pannecouque C., Zappalà M. Farmaco. 2004;59:33–39. doi: 10.1016/j.farmac.2003.09.001. [DOI] [PubMed] [Google Scholar]; (f) Murphy G.J., Holder J.C. Trends Pharmacol. Sci. 2000;21:469–474. doi: 10.1016/s0165-6147(00)01559-5. [DOI] [PubMed] [Google Scholar]; (g) Bailey C.J. Trends Pharmacol. Sci. 2000;21:259–265. doi: 10.1016/s0165-6147(00)01506-6. [DOI] [PubMed] [Google Scholar]
- 67.Atamanyuk D., Zimenkovsky B., Atamanyuk V., Lesyk R. Synth. Commun. 2014;44:237–244. [Google Scholar]
- 68.(a) Cappendijk S.L.T., Dzoljic M.R. Eur. J. Pharmacol. 1993;241:261–265. doi: 10.1016/0014-2999(93)90212-z. [DOI] [PubMed] [Google Scholar]; (b) Page C.B., Pinder A.R. J. Chem. Soc. 1964:4811–4816. [Google Scholar]; (c) Delorenzi J.C., Attias M., Gattass C.R., Andrade M., Rezende C., da Cunha Pinto A., Henriques A.T., Bou-Habib D.C., Saraiva E.M.B. Antimicrob. Agents Chemother. 2001;45:1349–1354. doi: 10.1128/AAC.45.5.1349-1354.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.(a) Seebacher W., Schlapper C., Brun R., Kaiser M., Saf R., Weis R. Eur. J. Pharm. Sci. 2005;24:281–289. doi: 10.1016/j.ejps.2004.11.003. [DOI] [PubMed] [Google Scholar]; (b) Seebacher W., Schlapper C., Brun R., Kaiser M., Saf R., Weis R. Eur. J. Med. Chem. 2006;41:970–977. doi: 10.1016/j.ejmech.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 70.Tu Y. Nat. Med. 2011;17:1217–1220. doi: 10.1038/nm.2471. [DOI] [PubMed] [Google Scholar]
- 71.(a) Reiter C., Fröhlich T., Zeino M., Marschall M., Bahsi H., Leidenberger M., Friedrich O., Kappes B., Hampel F., Efferth T., Tsogoeva S.B. Eur. J. Med. Chem. 2015;97:164–172. doi: 10.1016/j.ejmech.2015.04.053. [DOI] [PubMed] [Google Scholar]; (b) Reiter C., Fröhlich T., Gruber L., Hutterer C., Marschall M., Voigtländer C., Friedrich O., Kappes B., Efferth T., Tsogoeva S.B. Bioorg. Med. Chem. 2015;23:5452–5458. doi: 10.1016/j.bmc.2015.07.048. [DOI] [PubMed] [Google Scholar]
- 72.Bock C.M., Parameshwarappa G., Bönisch S., Neiss C., Bauer W., Hampel F., Görling A., Tsogoeva S.B. Chem. Eur. J. 2016;22:5189–5197. doi: 10.1002/chem.201504798. [DOI] [PubMed] [Google Scholar]
- 73.(a) Geronikaki A.A., Lagunin A.A., Hadjipovlou-Litina D.I., Eleftheriou P.T., Filimonov D.A., Poroikov V.V., Alam I., Saxena A.K. J. Med. Chem. 2008;51:1601–1609. doi: 10.1021/jm701496h. [DOI] [PubMed] [Google Scholar]; (b) Shin M.H., Ke F.Y. Bioorg. Med. Chem. Lett. 2004;12:4633–4643. doi: 10.1016/j.bmc.2004.06.033. [DOI] [PubMed] [Google Scholar]; (c) Lesyk R.B., Zimenkovsky B.S. Curr. Org. Chem. 2004;8:1547–1577. [Google Scholar]; (d) Ahn J.H., Kim S.J., Park W.S., Cho S.Y., Ha J.D., Kim S.S., Kang S.K., Jeong D.G., Jung S.-K., Lee S.-H., Kim H.M., Park S.K., Lee K.H., Lee C.W., Ryu S.E., Choi J.-K. Bioorg. Med. Chem. Lett. 2006;16:2996–2999. doi: 10.1016/j.bmcl.2006.02.060. [DOI] [PubMed] [Google Scholar]; (e) Liu W., Bulgaru A., Haigentz M., Stein C.A., Perez-Soler R., Mani S. Curr. Med. Chem. Anticancer Agents. 2003;3:217–223. doi: 10.2174/1568011033482459. [DOI] [PubMed] [Google Scholar]; (f) Lee J., Kim J., Koh J.S., Chung H.H., Kim K.H. Bioorg. Med. Chem. Lett. 2006;16:1954–1956. doi: 10.1016/j.bmcl.2005.12.074. [DOI] [PubMed] [Google Scholar]
- 74.Lozynskyi A., Golota S., Zimenkovsky B., Atamanyuk D., Gzella A., Lesyk R. Phosphorus, Sulfur Silicon. 2016;191:1245–1249. [Google Scholar]
- 75.Ghosh A.K., Chapsal B.D., Weber I.T., Mitsuya H. Acc. Chem. Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 76.Ghosh A.K., Sridhar P.R., Leshchenko S., Hussain A.K., Li J., Kovalevsky A.Y., Walters D.E., Wedekind J., Grum-Tokars V., Das D., Koh Y., Maeda K., Gatanaga H., Weber I.T., Mitsuya H. J. Med. Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 77.Ghosh A.K., Venkateswara Rao K., Nyalapatla P.R., Osswald H.L., Martyr C.D., Aoki M., Hayashi H., Agniswamy J., Wang Y.-F., Bulut H., Das D., Weber I.T., Mitsuya H. J. Med. Chem. 2017;60:4267–4278. doi: 10.1021/acs.jmedchem.7b00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.(a) Bundret K.M., Dalziel W., Hesp B., Jarvis J.A.J., Neidle S. J. Chem. Soc. Chem. Commun. 1972:1027–1028. [Google Scholar]; (b) Dalziel W., Hesp B., Stevenson K.M., Jarvis J.A. J. Chem. Soc. Perkin Trans. 1. 1973:2841–2851. [Google Scholar]
- 79.(a) Toyota M., Nishikawa Y., Fukumoto K. Tetrahedron Lett. 1994;35:6495–6498. [Google Scholar]; (b) Toyota M., Nishikawa Y., Seishi T., Fukumoto K. Tetrahedron. 1994;50:10183–10192. [Google Scholar]
- 80.(a) Ishikawa T., Kotake K.-I., Ishii H. Chem. Pharm. Bull. 1995;4:1039–1041. doi: 10.1248/cpb.43.1039. [DOI] [PubMed] [Google Scholar]; (b) Desimoni G., Faita G., Quadrelli P. Chem. Rev. 2013;113:5924–5988. doi: 10.1021/cr4000732. [DOI] [PubMed] [Google Scholar]
- 81.Manchand P.S., White J.D., Wright H., Clardy J. J. Am. Chem. Soc. 1973;95:2705–2706. [Google Scholar]
- 82.Tanaka T., Murakami K., Kanda A., Patra D., Yamamoto S., Satoh N., Kim S.-W., Rahman S.M.A., Ohno H., Iwata C. J. Org. Chem. 2001;66:7107–7112. doi: 10.1021/jo015808w. [DOI] [PubMed] [Google Scholar]
- 83.(a) Tamura G., Suziki S., Takatsuki K.A., Arima K. J. Antibiot. 1968;21:539–544. doi: 10.7164/antibiotics.21.539. [DOI] [PubMed] [Google Scholar]; (b) Takatsuki A., Tamura G., Arima K. Appl. Microbiol. 1969;17:825–829. doi: 10.1128/am.17.6.825-829.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dogan O., Oppolzer W. Turk. J. Chem. 2001;25:273–281. [Google Scholar]
- 85.Tsunakawa M., Nishio M., Ohkuma H., Tsuno T., Konishi M., Naito T., Oki T., Kawaguchi H. J. Org. Chem. 1989;54:2532–2536. [Google Scholar]
- 86.Kakushima M., Sawada Y., Nishio M., Tsuno T., Oki T. J. Org. Chem. 1989;54:2536–2539. [Google Scholar]
- 87.Zilke L., Hall D.G. Eur. J. Org. Chem. 2012;22:4153–4163. [Google Scholar]
- 88.Win N.N., Awale S., Esumi H., Tezuka Y., Kadota S. Chem. Pharm. Bull. 2008;56:491–496. doi: 10.1248/cpb.56.491. [DOI] [PubMed] [Google Scholar]
- 89.Pasfield L.A., de la Cruz L., Ho J., Coote M.L., Otting G., McLeod M.D. Asian J. Org. Chem. 2013;2:60–63. [Google Scholar]
- 90.Ye D.K.J., Richard J.-A. Tetrahedron Lett. 2014;55:2183–2186. [Google Scholar]
- 91.(a) Lee E., Song H.Y., Kang J.W., Kim D.-S., Jung C.-K., Joo J.M. J. Am. Chem. Soc. 2002;124:384–385. doi: 10.1021/ja017265d. [DOI] [PubMed] [Google Scholar]; (b) Jung M.E., Ho D., Chu H.V. Org. Lett. 2005;7:1649–1651. doi: 10.1021/ol050361p. [DOI] [PubMed] [Google Scholar]; (c) Jung M.E., Guzaev M. J. Org. Chem. 2013;78:7518–7526. doi: 10.1021/jo400909t. [DOI] [PubMed] [Google Scholar]
- 92.Liu S.-A., Trauner D. J. Am. Chem. Soc. 2017;139:9491–9494. doi: 10.1021/jacs.7b05046. [DOI] [PubMed] [Google Scholar]
- 93.(a) Wang J., Froeyen M., Hendrix C., Andrei G., Snoeck R., De Clercq E., Herdewijn P. J. Med. Chem. 2000;43:736–745. doi: 10.1021/jm991171l. [DOI] [PubMed] [Google Scholar]; (b) Wang J., Herdewijn P. J. Org. Chem. 1999;64:7820–7827. [Google Scholar]; (c) Wang J., Busson R., Blaton N., Rozenski J., Herdewijn P. J. Org. Chem. 1998;63:3051–3058. [Google Scholar]
- 94.Wang J., Morral J., Hendrix C., Herdewijn P. J. Org. Chem. 2001;66:8478–8482. doi: 10.1021/jo015924z. [DOI] [PubMed] [Google Scholar]
- 95.(a) Agrofoglio L., Challand S.R. Acyclic, Carbocyclic and l-Nucleosides. Kluver Academic Publishers; Dordrecht, The Netherlands: 1998. [Google Scholar]; (b) Crimmins M.T. Tetrahedron. 1998;54:9229–9272. [Google Scholar]; (c) Ueda T. In: Townsend L.B., editor. Vol. 1. Plenum Press; New York: 1988. (Chemistry of Nucleosides and Nucleotides). Chapter 1. [Google Scholar]
- 96.Vince R., Hua M. J. Med. Chem. 1990;33:17–21. doi: 10.1021/jm00163a004. [DOI] [PubMed] [Google Scholar]
- 97.Belmonte L., Barré P., de Bracco M.M.E., Ruibal-Ares B.H. Curr. Med. Chem. 2003;10:303–312. doi: 10.2174/0929867033368358. [DOI] [PubMed] [Google Scholar]
- 98.Quadrelli P., Scrocchi R., Caramella P., Rescifina A., Piperno A. Tetrahedron. 2004;60:3643–3651. [Google Scholar]
- 99.Quadrelli P., Martinez Vazquez N., Scrocchi R., Corsaro A., Pistarà V. Sci. World J. 2014;2014:492178. doi: 10.1155/2014/492178. Article ID 492178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moggio Y., Legnani L., Bovio B., Memeo M.G., Quadrelli P. Tetrahedron. 2012;68:1384–1392. [Google Scholar]
- 101.(a) White P.W., Faucher A.-M., Massariol M.-J., Welchner E., Rancourt J., Cartier M., Archambault J. Antimicrob. Agents Chemother. 2005;49:4834–4842. doi: 10.1128/AAC.49.12.4834-4842.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Faucher A.-M., White P.W., Brochu C., Grand-Maitre C., Rancourt J., Fazal G. J. Med. Chem. 2004;47:18–21. doi: 10.1021/jm034206x. [DOI] [PubMed] [Google Scholar]; (c) Yoshino M. Biochem. J. 1987;248:815–820. doi: 10.1042/bj2480815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Johnson J.A., Gangemi J.D. Antimicrob. Agents Chemother. 1999;43:1198–1205. doi: 10.1128/aac.43.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Memeo M.G., Lapolla F., Maga G., Quadrelli P. Tetrahedron Lett. 2015;56:1986–1990. [Google Scholar]
- 104.(a) D’Abramo C.M., Archambault J. Open Virol. J. 2011;5:80–95. doi: 10.2174/1874357901105010080. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim S.S., Tam J.K., Wang A.F., Hegde R.S. J. Biol. Chem. 2000;275:31245–31254. doi: 10.1074/jbc.M004541200. [DOI] [PubMed] [Google Scholar]; (c) Wang Y., Coulombe R., Cameron D.R., Thauvette L., Massariol M.-J., Amon L.M., Fink D., Titolo S., Welchner E., Yoakim C., Archambault J., White P.W. J. Biol. Chem. 2004;279:6976–6985. doi: 10.1074/jbc.M311376200. [DOI] [PubMed] [Google Scholar]
- 105.Savion M., Memeo M.G., Bovio B., Grazioso G., Legnani L., Quadrelli P. Tetrahedron. 2012;68:1845–1852. [Google Scholar]
- 106.De Clercq E. Biochem. Pharmacol. 2004;68:2301–2315. doi: 10.1016/j.bcp.2004.07.039. [DOI] [PubMed] [Google Scholar]
- 107.Al-Saad D., Memeo M.G., Quadrelli P. Sci. World J. 2014:10 p. Article ID 472373. [Google Scholar]
- 108.Al-Saad D., Memeo M.G., Quadrelli P. Lett. Org. Chem. 2016;13:757–763. [Google Scholar]
- 109.Al-Saad D., Memeo M.G., Quadrelli P. Chem. Select. 2017;2:10340–10346. [Google Scholar]
- 110.Hrebabecky H., Dracinsky M., De Palma A.M., Neyts J., Holy A. Collect. Czech. Chem. Commun. 2009;74:469–485. [Google Scholar]
- 111.Hrebabecky H., Dracinsky M., De Palma A.M., Neyts J., Holy A. Collect. Czech. Chem. Commun. 2009;74:487–502. [Google Scholar]
- 112.Dejmek M., Hrebabecky H., Dracinsky M., De Palma A.M., Neyts J., Leyssen P., Mertlikova-Kaiserova H., Nenka R. Collect. Czech. Chem. Commun. 2011;76:1549–1566. doi: 10.1016/j.bmcl.2011.05.070. [DOI] [PubMed] [Google Scholar]
- 113.Shing T.K.M., Wong A.W.H., Li H., Liu Z.F., Chan P.K.S. Org. Biomol. Chem. 2014;12:9439–9445. doi: 10.1039/c4ob01763c. [DOI] [PubMed] [Google Scholar]
- 114.Pokorny J., Borkova L., Urban M. Curr. Med. Chem. 2018;25:636–658. doi: 10.2174/0929867324666171009122612. [DOI] [PubMed] [Google Scholar]
- 115.Alvarez R., Velasquez S., San-félix A., Aquaro S., De Clercq E., Perno C.F., Karlsson A., Balzarini J., Camarasa M.J. J. Med. Chem. 1994;37:4185–4194. doi: 10.1021/jm00050a015. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 116.Moukha-Chafiq O., Taha M.L., Lazrek H.B., Vasseur J.J., Pannecouque C., Witvrouw M., De Clercq E. Nucleosides Nucleotides Nucleic Acids. 2001;20:1811–1821. doi: 10.1081/NCN-100107192. [DOI] [PubMed] [Google Scholar]
- 117.Abdel-Rahmana A.A.H., Wada T. Z. Naturforsch. 2009;64:163–166. doi: 10.1515/znc-2009-3-402. [DOI] [PubMed] [Google Scholar]
- 118.Montagu A., Roy V., Balzarini J., Snoeck R., Andrei G., Agrofoglio L.A. Eur. J. Med. Chem. 2011;46:778–786. doi: 10.1016/j.ejmech.2010.12.017. [DOI] [PubMed] [Google Scholar]
- 119.Pinter G., Bereczki I., Batta G., Otvos R., Sztaricskai F., Roth E., Ostorhazi E., Rozgonyi F., Naesens L., Szarvas M., Boda Z., Herczegh P. Bioorg. Med. Chem. Lett. 2010;20:2713–2717. doi: 10.1016/j.bmcl.2010.03.080. [DOI] [PubMed] [Google Scholar]
- 120.Lakshman M.K., Singh M.K., Parrish D., Balachandran R., Day B.W. J. Org. Chem. 2010;75:2461–2473. doi: 10.1021/jo902342z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lakshman M.K., Kumar A., Balachandran R., Day B.W., Andrei G., Snoeck R., Balzarini J. J. Org. Chem. 2012;77:5870–5883. doi: 10.1021/jo300628y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Glowacka I.E., Balzarini J., Andrei G., Snoeck R., Schols D., Piotrowska D.G. Bioorg. Med. Chem. 2014;22:3629–3641. doi: 10.1016/j.bmc.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Glowacka I.E., Balzarini J., Piotrowska D.G. Arch. Pharm. Chem. Life Sci. 2014;347:496–505. doi: 10.1002/ardp.201300471. [DOI] [PubMed] [Google Scholar]
- 124.Glowacka I.E., Balzarini J., Wroblewski A.E. Arch. Pharm. Chem. Life Sci. 2014;347:506–514. doi: 10.1002/ardp.201300468. [DOI] [PubMed] [Google Scholar]
- 125.Senanayake T.H., Warren G., Wei X., Vinogradov S.V. J. Controlled Release. 2013;167:200–209. doi: 10.1016/j.jconrel.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schulz T., Balzarini J., Meier C. ChemMedChem. 2014;9:762–775. doi: 10.1002/cmdc.201300500. [DOI] [PubMed] [Google Scholar]
- 127.(a) Vasilyeva S.V., Shtil A.A., Petrova A.S., Balakhnin S.M., Achigecheva P.Y., Stetsenko D.A., Silnikov V.N. Bioorg. Med. Chem. 2017;25:1696–1702. doi: 10.1016/j.bmc.2017.01.038. [DOI] [PubMed] [Google Scholar]; (b) Liu Y., Peng Y., Lu J., Wang J., Ma H., Song C., Liu B., Qiao Y., Yu W., Wu J., Chang J. Eur. J. Med. Chem. 2018;143:137–149. doi: 10.1016/j.ejmech.2017.11.028. [DOI] [PubMed] [Google Scholar]
- 128.Vernekar S.K.V., Qiu L., Zacharias J., Geraghty R.T.J., Wang Z. Med. Chem. Commun. 2014;5:603–608. [Google Scholar]
- 129.Munoz A., Sigwalt D., Iliescas B.M., Luczkowiak J., Rodriguez-Perez L., Nierengarten I., Holler M., Remy J.-S., Buffet K., Vincent S.P., Rojo J., Delgado R., Nierengarten J.-F., Martin N. Nat. Chem. 2016;8:50–57. doi: 10.1038/nchem.2387. [DOI] [PubMed] [Google Scholar]
- 130.Efthymiou T., Gong W., Desaulniers J.-P. Molecules. 2012;17:12665–12703. doi: 10.3390/molecules171112665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Saini M.S., Dwivedi J. Int. J. Pharm. Sci. Res. 2013;4:2866–2879. [Google Scholar]
- 132.Fan F., Cai C., Wang W., Gao L., Li J., Li J., Gu F., Sun T., Li J., Li C., Yu G. ACS Macro Lett. 2018;7:330–335. doi: 10.1021/acsmacrolett.8b00056. [DOI] [PubMed] [Google Scholar]
- 133.Giofré S.V., Romeo R., Chiacchio U., Romeo G., Chiacchio M.A. Mini-Rev. Org. Chem. 2015;12:249–257. [Google Scholar]
- 134.Giofré S.V., Romeo R., Garozzo A., Cicero N., Campisi A., Lanza G., Chiacchio M.A. ARKIVOC. 2015;vii:253–269. [Google Scholar]
- 135.Kokosza K., Andrei G., Schols D., Snoeck R., Piotrowska D.G. Bioorg. Med. Chem. 2015;23:3135–3146. doi: 10.1016/j.bmc.2015.04.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang P., Wei C., Wang E., Wanga W., Liu M., Yin Q., Chen H., Wang K., Li X., Zhang J. Carbohydr. Res. 2012;351:7–16. doi: 10.1016/j.carres.2011.11.025. [DOI] [PubMed] [Google Scholar]
- 137.(a) Kim H.O., Schinazi R.F., Shanmuganathan K., Jeong L.S., Beach J.W., Nampalli S., Cannon D.L., Chu C.K. J. Med. Chem. 1993;36:519–634. doi: 10.1021/jm00057a001. [DOI] [PubMed] [Google Scholar]; (b) Schinazi R.F., Gosselin G., Faraj A., Korba B.E., Liotta D.C., Chu C.K., Mathé C., Imbach J.-L., Sommadossi J.-P. Antimicrob. Agents Chemother. 1994;38:2172–2174. doi: 10.1128/aac.38.9.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jin H., Siddigui M.A., Evans C.A., Tse H.L.A., Mansour T.S. J. Org. Chem. 1995;60:2621–2623. [Google Scholar]; (d) Liang C., Lee D.W., Newton M.G., Chu C.K. J. Org. Chem. 1995;60:1546–1553. [Google Scholar]
- 138.Gi H.-J., Xiang Y., Schinazi R.F., Zhao K. J. Org. Chem. 1997;62:88–92. doi: 10.1021/jo961779r. [DOI] [PubMed] [Google Scholar]
- 139.Ortuño R.M., Moglioni A.G., Moltrasio G.Y. Curr. Org. Chem. 2005;9:237–259. and references therein. [Google Scholar]