

Abstract
The notion about immunity to disease arose from the observation that those who recovered from an apparently contagious disease became resistant to a subsequent similar sickness. Much later it was shown that immunity is transferable by serum. The active serum components were identified to be immunoglobulins (Ig's), called antibodies. The enormous diversity of antibodies specific for distinct viruses, pathogens, and many other antigens is explained by clonal selection, whereby specific B-lymphocyte receptors recognize a particular antigen. The selected B-cells are triggered to undergo replication. A process of further cell differentiation and maturation ensues, leading to secretion of antibodies with high binding affinity toward the triggering antigen. Genes coding for the variable regions (involved in antigen binding) of Ig's are inherited as sets of gene fragments joined to form a complete gene in individual B-cells. This process and further hypermutations ensure the synthesis of diverse high affinity antibodies. The antibodies consist of pairs of light (L) and heavy (H) polypeptide chains. Variations in the constant portion of H-chains lead to production of Ig isotypes (IgM, IgA, IgD, IgE, and IgG (further subdivided into IgG1, IgG2, IgG3 and IgG4)), each having distinct effector functions. Host exposure to viruses leads to the production of antibodies with more than one specificity. Only some of these antibodies, recognizing so-called virus neutralization epitopes, diminish or eliminate virus infectivity. Other virus-specific antibodies play auxiliary roles or are ineffective. Sometimes antibodies cause enhancement of viral diseases or play a role in evasion of the immune system. Many antiviral immunoglobulins are being used for short-term pre-exposure prophylaxis or therapy.
Long-term protective effects can be accomplished only by antibodies elicited by successful vaccination relying on the phenomenon of immunological memory. T-lymphocytes play a major role in initiating and maintaining immunity against subsequent virus exposure. Antibodies are one of the essential features of antigen triggered adaptive immunity. Initial early defense against viruses is provided by components of innate immunity which evolutionarily precedes adaptive immunity, and remains an essential part of defense against pathogens in humans.
Keywords: Adaptive immunity, Antibodies, B-cells, Clonal selection, Glycoproteins, Immunoglobulins, Immunological memory, Innate immunity, Mucosa, Passive immunization, Phylogeny, Receptors, Toll-like receptors (TLRs), Vaccines, Variability
Milestones in History
The primeval notion about immunity to disease arose in Greece about 25 centuries ago from the observation that those who recovered from an apparently contagious disease became resistant to a subsequent similar sickness. The earliest known attempts to intentionally transfer immunity to an infectious disease occurred in China in the tenth century. It involved exposing uninfected people to material from lesions caused by smallpox. This not always successful practice was introduced in the seventeenth century to the Ottoman Empire and subsequently to England and its colonies in North America.
This approach was revolutionized by replacing material from human lesions by that derived from cowpox lesions first in 1774 in England by a farmer, Benjamin Jesty, who used it on his family, and 22 years later by Edward Jenner. Thereafter, the widely used vaccination has been considered as the first example of immunization with a life-attenuated virus. Nevertheless, both the cause of the disease and the success of vaccination remained unexplained.
An understanding started to emerge about a century later when (1) Koch and Pasteur set forth the germ theory of infectious diseases, and Pasteur developed attenuated vaccines against anthrax and rabies; and (2) Von Behring and Kitasato demonstrated that immunity could be transferred by a soluble serum component(s). Several decades later, these components were shown to be antibody immunoglobulins (Heidelberger and Kabat) whose fundamental structure was elucidated by Porter and Edelman in the late 1950s, and further characterized by numerous X-ray crystallography and sequencing studies.
However, several findings indicated that antibodies are not the only mediators of specific immunity. Immune responses corresponding to delayed hypersensitivity and allograft rejections appeared unrelated to the presence of serum antibodies. Immunity could also be transferred by cells from immunized to naive animals (Landsteiner and Chase). It was shown by Gowans and colleagues in 1962 that lymphocytes are essential for immune responses.
Antibodies to very many distinct antigens can be produced upon immunization. Therefore, it was presumed that the diverse antibodies were not preformed but would be generated on demand following antigenic stimuli. In theory, there were two possibilities: that an antigen either directs or selects for the formation of a specific antibody. Further studies fully supported the clonal selection theory (Burnet, Jerne, and Talmage). The repertoire of diverse antibodies undergoing a process of maturation leading to high-affinity antibodies is generated by somatic rearrangements and hypermutation of immunoglobulin genes (Tonegawa).
Although it became evident that both immunoglobulins and cells are essential for immunity, the details of the process whereby antigens and viruses elicit immune responses were contributed to by results of studies in transplantation biology and notably the discovery of the major histocompatibility complex (MHC; HLA molecules on human cells) (Benaceraff, Dausset, and Snell). Their role in presentation of antigen fragments and interactions of specific antibody producing B-cells with T-cells, as well as the function of cytokines, chemokines, and adhesion molecules will be discussed in later sections of this article.
Immunoglobulins, lymphocytes, MHC molecules, and antigen receptors are all components of adaptative (acquired) immunity. Components of innate immunity, responding rapidly to an invading pathogen, play a key role in initiating and orchestrating the adaptive response (Janeway).
The major milestones in the history of immunology and their time lines are summarized in Table 1 .
Table 1.
History of immunology timeline
| 1798 | Smallpox vaccination | Edward Jenner |
| 1876 | Validation of germ theory of disease by discovering bacterial basis of anthrax | Robert Koch |
| 1879 | Chicken cholera vaccine development | Louis Pasteur |
| 1890 | Discovery of diphtheria ‘antitoxin’ in blood | Emil Von Behring and Shibasaburo Kitasato |
| 1882 | Isolation of the tubercle bacillus | Robert Koch |
| 1883 | Delayed type hypersensitivity | Robert Koch |
| 1884 | Phagocytosis; cellular theory of immunity | Elie Metchnikoff |
| 1891 | Proposal that antibodies are responsible for immunity | Paul Ehrlich |
| 1891 | Passive immunity | Emil Roux |
| 1894 | Complement and antibody activity in bacteriolysis | Jules Bordet |
| 1900 | A, B, and O blood groups | Karl Landsteiner |
| 1901 | Cutaneous allergic reaction | Maurice Arthus |
| 1903 | Opsonization by antibody | Almroth Wright and Stewart Douglas |
| 1907 | Discipline of immunochemistry founded | Svante Arrhenius |
| 1910 | Viral immunology theory | Peyton Rous |
| 1917 | Haptens discovered | Karl Landsteiner |
| 1921 | Cutaneous reactions | Carl Prausnitz and Heinz Kustner |
| 1924 | Reticuloendothelial system | Ludwig Aschoff |
| 1939 | Discovery that antibodies are gamma globulins | Elvin Kabat |
| 1942 | Adjuvants | Jules Freund and Katherine McDermott |
| 1942 | Cellular transfer of sensitivity in guinea pigs (anaphylaxis) | Karl Landsteiner and Merill Chase |
| 1944 | Immunological hypothesis of allograft rejection | Peter Medawar |
| 1948 | Demonstration of antibody production in plasma B-cells | Astrid Fagraeus |
| 1949 | Distinguishing self vs. nonself and its role in maintaining immunological unresponsiveness (tolerance) to self | Macfarlane Burnet and Frank Fenner |
| 1952 | Discovery of agammaglobulinemia (antibody immunodeficiency) | Ogden Bruton |
| 1953 | Immunological tolerance hypothesis | Rupert Billingham, Leslie |
| Brent, Peter Medawar, and Milan Hasek | ||
| 1955–59 | Clonal selection theory | Niels Jerne, David Talmage, and Macfarlane Burnet |
| 1957 | Discovery of interferon | Alick Isaacs and Jean Lindenmann |
| 1958 | Identification of first autoantibody and first recognition of autoimmune disease | Henry Kunkel |
| 1959–62 | Elucidation of antibody structure | Rodney Porter and Gerald Edelman |
| 1959 | Lymphocytes as the cellular units of clonal selection; discovery of lymphoid circulation | James Gowans |
| 1961 | Discovery of thymus involvement in cellular immunity | Jacques Miller |
| 1968 | Recognition of B- and T-cells in immunodeficiencies | Robert Good |
| 1968 | Distinction of bone marrow- and thymus-derived lymphocyte populations; discovery or T- and B-cell collaboration | Jacques Miller and Graham Mitchell |
| 1965 | Demonstration of allelic exclusion in B-cells | Benvenuto Pernis |
| 1968–70 | Elaboration of two-signal model of lymphocyte activation | Peter Bretscher and Melvin Cohn |
| 1970 | Discovery of membrane immunoglobulins | Benvenuto Pernis and Martin |
| Raff | ||
| 1971 | Recognition of hypervariable regions in Ig chains | Elvin Kabat |
| 1972 | Elucidation of the major histocompatibility complex | Baruj Benacerraf, Jean Dausset, and George Snell |
| 1974 | Discovery of MHC restriction | Rolf Zinkernagel and Peter Doherty |
| 1974 | HLA-B27 predisposes to an autoimmune disease | Derek Brewerton |
| 1975 | Monoclonal antibodies used in genetic analysis | Georges Kohler and Cesar Milstein |
| 1976 | First demonstration of cross-priming | Michael Bevan |
| 1978 | Direct evidence for somatic rearrangement in immunoglobulin genes | Susumu Tonegawa |
| 1978 | Recognition that dendritic cells are distinctive and highly potent antigen-presenting cells | Ralph Steinman |
| 1979 | Discovery of leukocyte adhesion molecules and their role in lymphocyte trafficking | Eugene Butcher |
| 1984–87 | Identification of genes for the T-cell antigen receptor | Mark Davis, Leroy Hood, Stephen Hedrick, and Gerry Siu |
| 1987 | Crystal structure of MHC peptide solved | Pam Bjorkman, Jack Strominger, and Don Wiley |
| 1989 | Emerging field of innate immunity, infectious nonself model of immune recognition (‘stranger hypothesis’) | Charlie Janeway |
| 1994 | Danger hypothesis of immune responsiveness | Polly Matzinger |
| 1986 | Discovery of T-helper subsets | Tim Mossmann and Bob Coffman |
| 1989 | Discovery of first chemokines | Edward Leonard, Teizo Yoshimura, and Marco Baggiolini |
| 1991 | Discovery of the first costimulatory pathway (CD28/B7) for T-cell activation | Kevin Urdahl and Mark Jenkins |
| 1992 | Cloning of CD40 ligand and recognition of its role in T-cell-dependent B-cell activation | Armitage, Spriggs, Lederman, Chess, Noelle, Aruffo, et al. |
| 1996–7 | Discovery of the role of Toll and Toll-like receptors in immunity | R. Medzhitov, CA. Janeway, Jr., and J. Hoffmann |
Reprinted with permission from Steven Greenberg, MD, from http://www.columbia.edu/itc/hs/medical/pathophys/immunology/readings/ConciseHistoryImmunology.pdf.
Immunoglobulin Phylogeny
The adaptive immune system, based on clonally diverse repertoires of antigen recognition molecules, arose in jawed vertebrates (gnasthostomes) about 500 million years ago. Homologs of human immunoglobulins, T-cell antigen receptors (TCRs), MHC I and MHC II molecules, and recombination activating genes (RAGs) have been identified in all classes of gnasthostomes. This has provided evolutionary advantages allowing the recognition of potentially lethal pathogens, including viruses, and the initiation of protective responses against them (Figure 1 ), and represented an add-on to a preexisting innate immune system. For immunoglobulins, somatic variation was further expanded through class switching, gene conversion, and somatic hypermutation. Jawless vertebrates (agnathans) were shown to also have an adaptive immune system based on recombinatorial assembly of genetic units different from those of gnasthostomes, to generate a diverse repertoire of lymphocytes, each with distinct receptors (Figure 2 ). The assembly relies on highly variable leucine-rich repeat (LRR) protein modular units.
Figure 1.
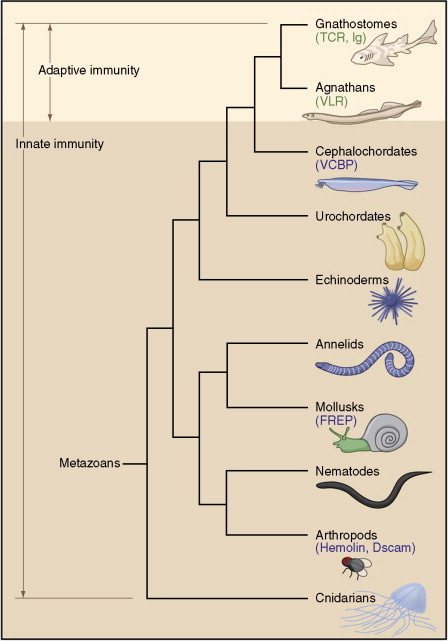
Phylogenetic tree indicating theoretical evolutionary relationships of metazoans and the emergence of adaptive immunity in conjunction with innate immunity. Families of immune molecules, other than Toll-like receptors (TLRs), are indicated in blue: chitin-binding domain-containing proteins (VCBPs), fibrinogen-related proteins (FREPs), hemolin and Down's syndrome cell adhesion molecule (Dscam). The recombinatorial based immune receptors are indicated in green: T-cell receptors (TCRs), immunoglobulins (Ig's), and variable lymphocyte receptors (VLRs). Reprinted from Cooper MD and Alder N (2006) The evolution of adaptive immune systems. Cell 124: 815–822, with permission from Elsevier.
Figure 2.
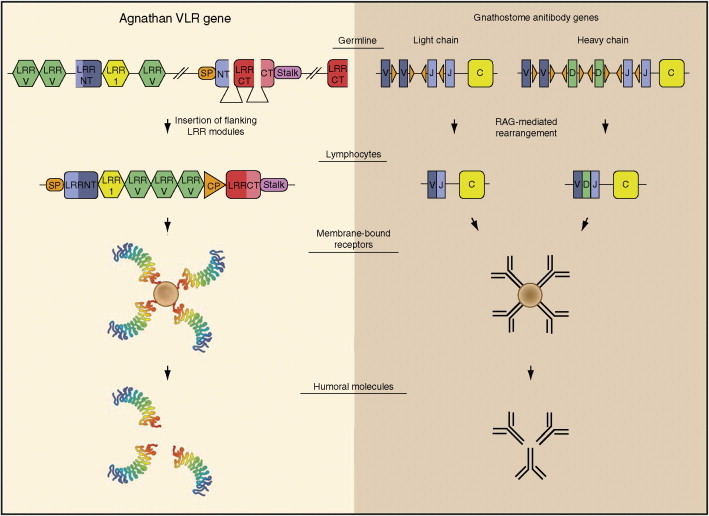
Two recombinatorial systems used for generating diverse antigen receptors in vertebrates. The figure compares the assembly of leucine-rich repeat (LRR) modular genetic units in agnathan lymphocytes to generate variable lymphocyte receptor (VLR) genes vs. the rearrangement of Ig gene segments in gnathostome B-lymphocytes to generate diverse antibody genes. Variable 24-amino-acid LRR (LRRV, green); N-terminal capping LRR (LRRNT, blue); variable 18-amino-acid LRR (LRR1, yellow); signal peptide (SP, orange); first six amino acids of LRRNT (NT, light blue); C-terminal capping LRR (LRRCT, red); last nine amino acids of LRRCT (CT, pink); variable 13 amino acid connecting peptide (CP, orange); and invariant VLR stalk (Stalk, purple). The small orange triangles adjacent to the representative V (blue), D (green), and J (light blue) gene segments represent recombination signal sequences (RSSs). The C (yellow) indicates the immunoglobulin constant region. Reproduced from Cooper MD and Alder N (2006) The evolution of adaptive immune systems. Cell 124: 815–822, with permission from Elsevier.
Most gnasthotomes generate a large part of their immuglobulin diversity in ways similar to those in humans. However, exceptions have occurred. Two of these, relevant to passive immunotherapy and development of diagnostics using the corresponding immunoglobulins, are mentioned here. (1) Only three classes of H-chain genes, corresponding to IgM, IgY, and IgA, exist in chicken. Class switching occurs from IgM to either IgY or IgA. IgY is considered to be the ancestor of ‘present-day’ IgG and IgE. Unlike human IgG, IgY does not bind to cellular Fc receptors and to proteins A and G, respectively, and does not activate the complement system. (2) H-chain antibodies devoid of light chains occur in the Camelidae species (camels, dromedaries, and llamas). Single-domain fragments from such antibodies react with specific antigens with high affinity constants (nanomolar range). The binding involves longer than average third variable loops of the antibody molecules. These unique antibodies and their antigen-specific fragments are expected to become important for biotechnological and medical applications, including intrabodies (intracellular antibodies).
Innate Immunity
The innate immune system provides an early defense against pathogens and alerts the adaptive immune system when initial invasion by a pathogen has occurred. The innate system predates evolutionarily the adaptive system and relies on antimicrobial peptides and on germline-encoded pattern-recognition receptors (PRRs). Antimicrobial peptides can damage enveloped viruses and interfere with processes involved in fusion of viruses with target cell membranes. PRRs recognize microbial (viral) components defined as pathogen-associated molecular patterns (PAMPs). Different PRRs are specific for distinct PAMPs, have distinct expression patterns, activate different signaling pathways, thereby eliciting distinct responses against invading pathogens. PRRs can be subdivided into two classes: Toll-like receptors (TLRs) and non-Toll-like innate immune proteins.
Toll was originally identified as a differentiation protein in Drosophila, and later (1996) shown to play a role in defense against fungal infection. Searching the entire DNA sequence database for similarities with the coding sequence for Toll resulted in identification of Toll-like sequences, and ultimately in discovery of at least 10 functional TLRs in humans. TLRs are type 1 integral membrane glycoproteins the extracellular domain of which contains several LRR motifs and a cytoplasmic signaling domain homologous to that of the interleukin-1 receptor (IL-1R). TLRs 2, 3, 4, 7, 8, and 9 recognize PAMPs characteristic for viruses. Viral DNAs rich in CpG motifs are recognized by TLR9, leading to activation of pro-inflammatory cytokines and type 1 interferon (IFN) secretion. TLR7 and TLR8, expressed within the endosomal membrane, are specific for viral single-stranded RNA (ssRNA). Double-stranded RNA (dsRNA), generated during viral infection as an intermediate for ssRNA viruses or during transcription of viral DNA, is recognized by TLR3. TLR3 is expressed in dendritic cells (DCs) and in a variety of epithelial cells, including airway, uterine, corneal, vaginal, cervical, biliary, and intestinal cells. Cervical mucosal epithelial cells also express functional TLR9, suggesting that TLR3 and TLR9 provide an antiviral environment for the lower female reproductive tract. Some viral envelope glycoproteins are recognized by TLR2 and TLR4, each expressed at the cell surface, leading to production of pro-inflammatory cytokines. In general, the engagement of TLRs by microbial PAMPs triggers signaling cascades leading to the induction of genes involved in antimicrobial host defenses. This includes the maturation and migration of DCs from sites where infection occurs to lymphoid organs where DCs can initiate antigen-specific immune responses. Triggering distinct TLRs elicits different cytokine profiles and different immune responses. Engagement of TL3 and TRL 4, respectively, upregulates polymeric immunoglobulin receptor expression on cells. Thus, bridges between innate and adaptive immune responses are established.
TLRs are expressed either at the cell surface or in lysosomal/endosomal membranes. Therefore, they would not recognize pathogens that succeeded in invading the cytosolic compartment. These pathogens are detected by a variety of cytoplasmic PRRs. They include retinoic acid inducible protein (RIG-1) with a helicase domain recognizing viral dsRNA, and a related protein, MDA5. Other proteins which may be involved in innate antiviral immunity include a triggering receptor on myeloid cells (TREM-1), myeloid C-type lectins and siglecs, recognizing sialic acid. One of the elements of both innate and adaptative immunity is the complement system. Some viruses or virus-infected cells can directly activate the complement cascade in the absence of antiviral antibodies.
Many viruses are endowed with properties subverting innate immune responses. Vaccinia virus produces proteins suppressing TLR- and IL-1R-induced signaling cascades. Paramyxoviruses produce proteins which associate with MDA5, and thus inhibit dsRNA-induced activation processes. Adenoviruses avoid immune surveillance by TRL9. A nonstructural protein of hepatitis C virus blocks signaling by RIG-1 and MDA5. Marburg and Ebola viruses, members of the family Filoviridae, elicit direct activation of TREM-1 on neutrophils. This can lead to vigorous inflammatory responses contributing to fatal hemorrhagic fevers in infected humans. Thus, some viruses have developed strategies to overcome either innate or adaptive (see next section) immune responses.
Adaptive Immunity
Adaptive immunity is a complex anticipatory system triggered by exposure to antigens, including viruses. Its hallmarks are selectivity, diversification, specificity, and memory. The principal effector molecules of the system are antigen-binding receptors (immunoglobulin (Ig) and T-cell receptors (TCR)). The following simplified overview (see Figure 3 ) will be limited to Ig's and humoral immune responses.
Figure 3.
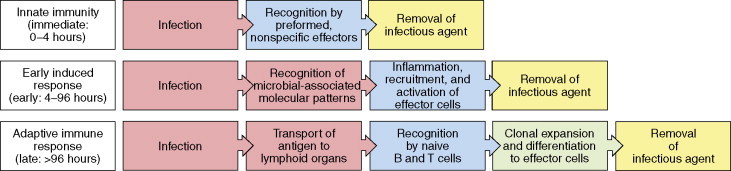
Three phases of a response to an initial infection. Copyright 2005 from Janeway CA, Jr., Travers P, Walport M, and Shlomchik MJ (2005)Immunobiology. The Immune System in Health and Disease, 6th edn, figure 2.1, p. 37. New York: Garland Science. Reproduced by permission of Garland Science/Taylor & Francis LLC.
Differentiation of B-cells into antibody-generating plasma cells occurs through distinct pathways. Two pathways lead to rapid IgM and IgA antibody production by B-1 cells against T-cell-independent antigens. These antibodies have low antigen-binding constants, and immunological memory does not evolve. The third pathway involves clonal selection. The B-cells synthesize IgM and IgD low affinity antibodies which are expressed as antigen receptors on the surface of cells. Each B-cell produces a different receptor recognizing a distinct epitope. Encounter with the appropriate epitope on an antigen elicits cell division, and generation of selected clones with identical receptor specificities. The clones further differentiate into specific antibody-secreting cells. The remarkable diversity of antibodies is attributable to the fact that genes coding for Ig variable regions are inherited as sets of gene fragments, each encoding a portion of the variable region of a particular Ig polypeptide chain. The fragments are joined together to form a complete gene in individual lymphocytes. The joining process involves addition of DNA sequences to the ends of fragments to be joined, thus increasing diversity. Further diversity arises from the assembly of each Ig protein from pairs of H- and L-chains, each B-cell producing only one kind of either chain (Figure 4 ). In addition, the assembled genes for Ig's mutate rapidly when B-cells are activated by binding an antigen. These hypermutations lead to new receptor variants representing the process of affinity maturation of an immune response. Moreover, B-cells and their progeny can produce an additional variation by altering the constant part of the H chain due to gene rearrangements (=Ig isotype and subclass switching) (Figure 5, Figure 6, Figure 7 ). These antibodies have identical paratopes but distinct effector functions (complement activation, phagocytosis, transcytosis, etc.). As a consequence of the aforementioned processes, there are B-lymphocytes of at least 100 million distinct specificities in every human individual at any given time.
Figure 4.
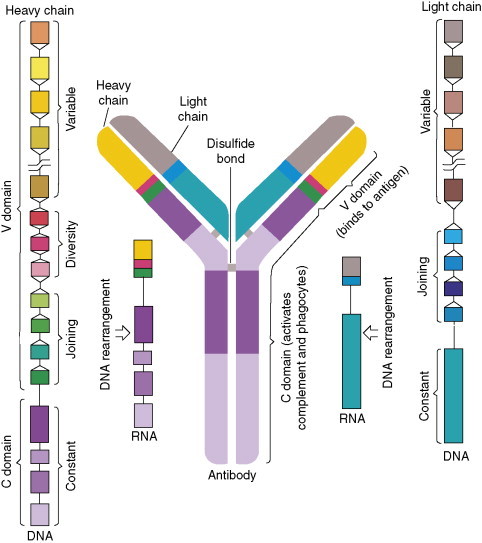
Antibody molecule consisting of pairs of H- and L-chains each encoded by genes assembled from different DNA segments. The segments rearrange to generate genes for chains that are distinct in each B-cell. The joining is variable so that the gene segments can encode the estimated 100 million specific antibodies each human is capable of producing. Reprinted from Janeway CA, Jr. (1993) How the immune system recognizes invaders. Scientific American (Sep.): 73–79, with permission of Scientific American and Ian Worpole (artist).
Figure 5.
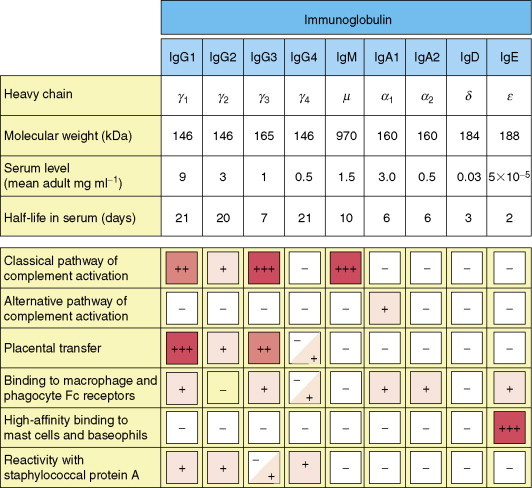
Properties of human immunoglobulin isotypes. The molecular mass of IgM corresponds to that of a pentamer. IgE is associated with immediate hypersensitivity. When attached to mast cells, it has a much higher half-life than in plasma. Copyright 2005 and permission as shown in legend for Figure 3 (source = figure 4.17).
Figure 6.
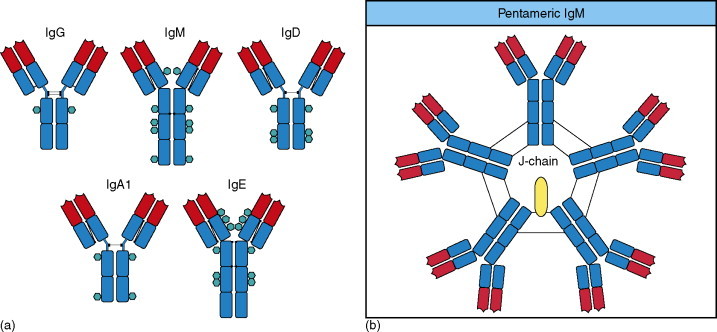
(a) Structural organization of human immunoglobulin isotype monomers. Both IgM and IgE lack a hinge region and each contains an additional heavy chain domain. Disulfide bonds linking the chains are indicated by black lines. N-linked glycans are shown as turquoise hexagons. (b) Pentameric IgM is associated with an additional polypeptide, the J-chain. The monomers are cross-linked by disulfide bonds to each other and to the J-chain. Copyright 2005 and permission as shown in legend for Figure 3 (source figures 4.18 and 4.23).
Figure 7.
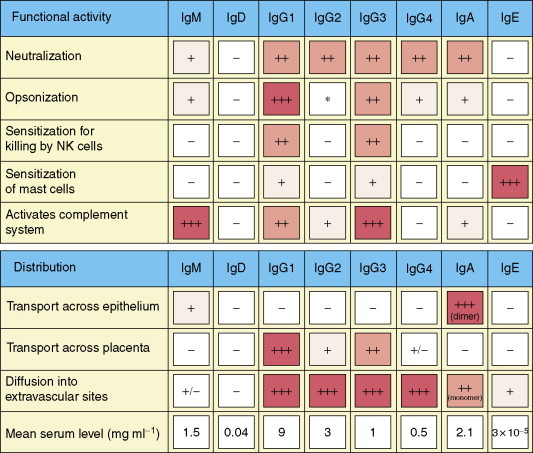
Functions and distribution of human immunoglobulin isotypes. Copyright 2005 and permission as shown in legend for Figure 3 (source figure 9.19).
The majority of Ig is produced in mucosa-associated tissues, predominantly in the intestine, rather than in the bone marrow, spleen, and lymph nodes, in the form of IgA. This harmonizes with the fact that mucosal surfaces represent the predominant sites for entry of pathogens, including viruses. Coincidentally, it has been demonstrated that the gut is the major site for HIV-1 replication and depletion of CD4+ cells. There are two subclasses of IgA, IgA1 and IgA2, which occur in monomeric, dimeric, tetrameric, and polymeric (pIgA) forms. A distinguishing feature of secretory IgA is its association with another glycoprotein, the secretory component (SC) (Figure 8 ). SC is also the extracellular portion (which can be generated by proteolytic cleavage) of an integral epithelial cell membrane protein, the polymeric Ig receptor (pIgR) mediating transcytosis of pIgA and IgM. The latter process allows neutralization of pathogens within intracellular vesicular compartments.
Figure 8.
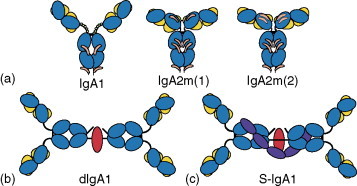
Structural organization of different molecular forms of human IgA. Heavy chains are shown in mid-blue, light chains in yellow, J-chain in red, and the secretory component (SC) in navy blue. (a) Monomeric mIgA. N-linked glycans are shown in orange, and O-linked glycans by small green circles. (b) Dimeric IgA1. (c) Dimeric secretory IgA1 (S-IgA1). For clarity, glycans are not shown for (b) and (c). Reprinted from Woof JM and Mestecky J (2005) Mucosal immunoglobulins. Immunological Reviews 206: 64–82, figure 1, with permission from Blackwell Publishing.
An essential aspect of adaptive immunity is its ability to recall past encounters with a pathogen for decades or even an entire lifetime. This fundamental feature is the foundation of successful vaccination. Germinal center-derived memory B-cells have the following attributes: antigen specificity; hypermutated Ig variable gene segments; and ability to bestow immunological memory following their adoptive transfer to immunologically naïve recipients. The CD27 surface antigen is a marker for memory B-cells.
The multifaceted performance of B-cells is the result of a thoroughly orchestrated ensemble involving CD4+ helper T-cells, DCs, MHC class II antigens, cell-differentiation antigens (CDs), cytokines, etc. In summary, in addition to occupancy of the Ig receptor, B-cells must interact with antigen-specific T-cells. The T-cells, through specific TCRs, recognize peptide fragments generated from the antigen internalized by the B-cell, and displayed on the surface of the B-cell as a peptide–MHC class II complex. Helper T-cells stimulate the B-cell following binding of the CD40 ligand on the T-cell to CD40 on the B-cell; the interaction of tumor necrosis factor (TNF)–TNF receptor family ligand pairs; and the release of specific cytokines. Further details of these interactions are shown in Figure 9, Figure 10, Figure 11 .
Figure 9.
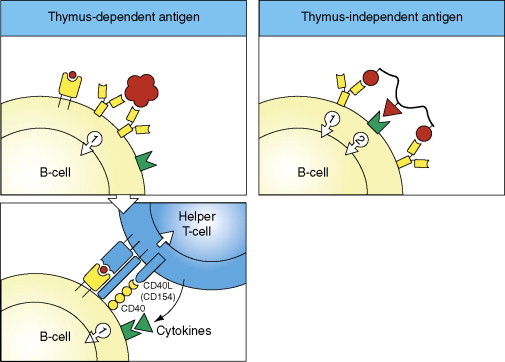
T-helper-cell-dependent initiation of the humoral immune response (two left panels). The first signal required for B-cell activation is delivered by binding of antigen (virus) (large red particle) to Ig cell receptors corresponding to monomeric IgM. Internalization and degradation of the antigen, and complex formation of the resulting peptide(s) (small red circle) with MHC class II molecules on the B-cell, allow the second signal to be delivered, that is, by the interaction between CD40 on the B-cell and the CD40 ligand (=CD154) on the CD4+ helper T-cell, and the engagement of the T-cell receptor (TCR) with the peptide-MHC class II complex on the B-cell. The activation is promoted by binding of cytokines to their specific receptors (see Figure 10). For comparison (right panel), in case of T-helper-cell-independent antigens, the second signal can be delivered by the antigen itself, either through binding of a part of the antigen to a receptor of the innate immune system (e.g., TLRs; green), or by extensive cross-linking of the membrane IgM by a polymeric antigen. Copyright 2005 and permission as shown in legend for Figure 3 (source figure 9.2).
Figure 10.
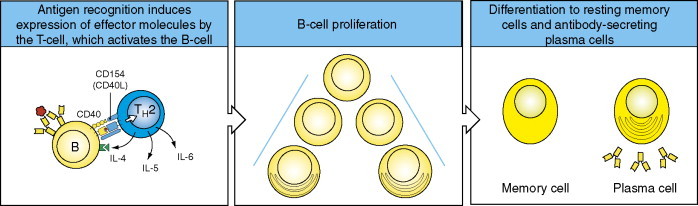
Antigen recognition induces the expression of B-cell stimulatory interleukins IL-4, IL-5, and IL-6 (and/or others) by the T-cell, driving the proliferation and differentiation of B-cells into antibody-secreting plasma cells. Activated B-cells can alternatively become memory B-cells. Copyright 2005 and permission as shown in legend for Figure 3 (source figure 9.3).
Figure 11.
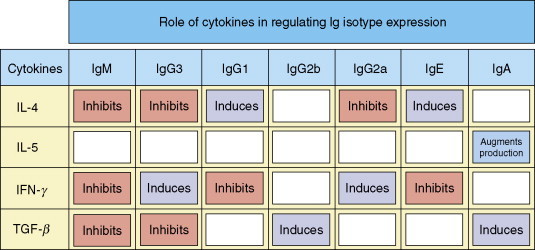
Role of cytokines in regulating Ig isotype switching. The individual cytokines either induce (violet) or inhibit (red) the production of particular Ig isotypes. IFN, interferon; TGF, transforming growth factor. Copyright 2005 and permission as shown in legend for Figure 3 (source figure 9.7).
The intricacies of all these tightly coordinated events seem to minimize the possibility that ‘rationally designed’ synthetic vaccines will be able to successfully recreate the specificity of virus neutralization B-cell epitopes or neotopes.
Biological Functions of Antiviral Antibodies
The surface of viruses is represented by a mosaic cluster of protein or glycoprotein subunits. The subunits correspond to a single or two or more species. The pattern of repetitiveness is usually a key factor responsible for the efficiency of early and rapid B-cell responses, potent IgM antibody production, and efficient downstream antibody class switching. However, the immune response is not restricted to antigenic sites (epitopes and neotopes) on the surface of viruses. Virus particles, following initial infection or provided as a vaccine, also separate into constituent parts. Consequently, unassembled surface subunits (their epitopes and cryptotopes) and internal virus components become exposed to the immune system, ultimately resulting in the production of antibodies having multiple specificities. Only some of these are directed against intact viruses, and may have virus-neutralizing properties. The formation of antibodies with distinct specificities may not be simultaneous but rather sequential. Especially in case of some not directly cytopathic viruses (hepatitis B and C, lymphocytic choriomeningitis, and HIV-1, prone to elicit a chronic carrier state), virus-neutralizing antibodies (VNAbs) appear with a delay after non-neutralizing antibodies. The latter may function as a ‘decoy’ if they are targeted like VNAb to virus surface epitopes.
Virus-Neutralizing and Protective Antibodies
VNAbs are crucial for protection against reinfection by a virus the VNAbs are specific for. Protection by efficacious vaccines correlates closely with in vitro determined VNAb titers of sera from immunized individuals. Protection by passive immunization relies on VNAb recognizing neutralization epitopes (or neotopes) on the virus surface. Coating of virus particles by antibodies is necessary but not always sufficient for virus neutralization. The effectiveness of virus neutralization correlates with the rate of antibody binding to critical epitopes and is augmented by slow dissociation of the formed antigen–antibody complexes. These kinetic parameters can be determined experimentally.
Spatial adaptive complementarity, electrostatic interactions, hydrogen bonds, and van der Waals forces contribute to the binding. Experimentally determined virus neutralization can depend on the target cells used. Virus neutralization is a multihit process and is successful when the number of unencumbered viral molecules, essential for initiation of the virus replicative cycle, is brought below a minimum threshold level. The mechanism of neutralization depends on processes obligatory for reproduction of a particular virus, and may involve the following steps: attachment to cell receptors; post-attachment events, internalization (endocytosis); fusion with cell membranes or endosomal vesicles; uncoating and/or intracellular localization; and enzymatic activities (e.g., transcription). Direct occupancy by VNAb of cell receptor binding sites on the virus surface might not be obligatory for neutralization. Steric hindrance or induction of deleterious conformational changes may be sufficiently effective. A unique feature of several anti-HIV-1 human monoclonal VNAb, having distinct specificities, is a very long finger-like third complementarity determining region of the immunoglobulin heavy chain allowing access to a recessed critical site on HIV-1 gp120. Such feature is rare in the human immunoglobulin repertoire but is common in the Ig's of Camelidae.
The mechanism and magnitude of VNAb neutralizing effects are influenced by their immunoglobulin isotype and subtype which affect interactions with complement, Fc receptors, and transcytosis through mucosal epithelia. Non-neutralizing virus-surface-binding antibodies sometimes enhance the effectiveness of VNAb, limit viral spread in the early phases of infection, and contribute to its suppression through antibody-mediated cellular cytotoxicity (ADCC). Antibodies directed to epitopes (or neotopes) on distinct surface proteins may act synergistically in virus neutralization.
The principle that VNAbs specific for virus surface components provide protection against disease is not absolute. The flavivirus nonstructural protein NS1 elicits a protective immune response against yellow fever, dengue, and tick-borne encephalitis viruses. The paratope binding site containing F(ab′)2 fragments are ineffective. Thus the immunoglobulin Fc portion is obligatory for the protective effect.
Antibody-Dependent Enhancement of Viral Diseases
Some viruses make use of antiviral antibodies to gain entry into target cells thus widening cell receptor usage to initiate infection. The infectious virus–antibody complexes rely upon the Fc portion of IgG antibodies to gain entry into monocytes/macrophages and granulocyte through Fc (FcR) or complement receptors (CR) expressed on these cells.The characteristic feature of the viruses is their propensity to establish persistent infections and their antigenic diversity. Antibody-dependent enhancement (ADE) has been demonstrated to occur in vitro with members of the families Bunyaviridae, Coronaviridae, Flaviviridae, Orthomyxoviridae, Paramyxoviridae, Retroviridae, Rhabdoviridae, and Togaviridae. A link between in vitro ADE and clinical manifestations cannot be always established. A relationship between ADE and disease exacerbation has been observed for dengue, measles, yellow fever, and respiratory syncytial viruses (RSVs). ADE may occur in children infected at a time when the level of transferred maternal antiviral antibodies declines to insufficient levels. ADE could represent an obstacle for development of vaccines, as has been the case for anti-RSV vaccines. A vaccine consisting of formaldehyde-treated measles virus hemagglutinin (ineffective to elicit antibodies to the virus fusion protein) induced antibodies causing ADE and led to aggravated atypical disease following infection with measles virus.
FcR- and CR-independent ADE was shown to occur following binding to HIV-1 of antibodies eliciting conformational changes in the gp120 envelope glycoprotein, allowing direct virus binding to cellular co-receptors while bypassing the primary binding to the primary CD4 cell receptor.
Immune System Evasion by Viruses
Persistence in an infected host and repeated reactivation of many viruses rely on several specific evasion strategies of adaptive immunity. Thus common protective responses are redirected or altered to the advantage of the infectious agent. This includes antiviral antibody responses and involves (1) specific paratope–epitope interactions (Fab fragments) and (2) effector mechanisms mediated by the Fc portion of antibodies.
The first mechanism is provided by genetically determined amino acid replacements leading to changes of virus epitopes (or neotopes) involved in virus neutralization. The sites of these escape mutations are usually on the viral surface, result in structural changes in antibody/virus contact sites, and lead to much less favorable kinetic parameters for antibody binding or completely abrogate binding. Presentation of new glycan chains on enveloped viruses or elimination of these chains due to mutations of N-glycosylation sites may cause substantial epitope alterations. The rate of escape mutation appearance is promoted by error-prone replication of the viral genome. Antibodies with new specificities must be produced to bring the mutant viruses under control. The process is repeated, and if not successful, persistent infection is established. A similar process leads to evasion from T-cell-mediated protective responses.
Secondly, several viruses bypass clearance processes facilitated by the Fc portion of bound antibodies by encoding and expressing Fc receptor analogs. Subversion of the complement cascade provides another way how to block clearance of cell-free virus and infected cells.
Additional scenarios for escaping immune surveillance include: interference with MHC class I restricted antigen presentation involving also inhibition of MHC class I cell surface expression or synthesis of viral MHC class I homologs; blocking MHC class II restricted antigen presentation; downregulation of cellular CD4 or its degradation; interference with cytokine effector functions; and other strategies.
Immunoglobulins for Passive Immunization against Human Viruses
Transfer of immunity from an immune donor to an unprotected recipient by serum is one of the early landmarks in the history of immunology (Von Behring). The active serum components have later been identified as immunoglobulins. The half-life of immunoglobulin isotypes in serum is 6–21 days (Figure 5). Consequently, administered antivirus immunoglobulins can provide only short term prophylactic and therapeutic benefits, respectively, unless they are administered repeatedly or incorporated into a slow-release medical device. The most common applications are pre-exposure (travel, protection against community-wide infection(s), medical professionals, immunosuppressed individuals, combination with live vaccines to minimize their potential side effects) and post-exposure prophylaxis (passive immunization may provide immediate protection while the benefits from vaccination are delayed). Local mucosal applications of immunoglobulins ‘as needed’ appear promising against perinatal and sexual transmission, respectively, of several viruses (e.g., herpesviruses and HIV-1).
The immunoglobulins are isolated from serum of individuals pre-screened for high levels of antibodies against a particular virus or from vaccinated individuals. The immunoglobulins are purified, treated to remove or inactivate infectious agents which might be present in the pooled serum source, notwithstanding rigorous screening of the individual sera entering the pool. The products are further processed depending on their intended intramuscular or intravenous applications. All these immunoglobulins are polyclonal with respect to the ‘indicated’ virus and contain other antibodies originally present in the pooled sera. Oral administration of antibodies produced in bovine colostrums or chicken yolk has been recently suggested.
Alternatively, monoclonal antibodies (mAbs) specific for epitopes, known to elicit virus-neutralizing and protective immune responses, are used. They are prepared using hybridoma technologies, immortalized human peripheral B-cells, transgenic mice and bacteriophage expression libraries. If derived from animal species, the mAbs are ‘humanized’ using recombinant DNA techniques by replacing amino acid sequences outside the antigen-binding sites with sequences corresponding to human immunoglobulins. By an in vitro directed evolution process allowing manipulation of antigen-binding kinetics, mAb variants having much faster antigen association rates and much slower dissociation rates can be produced. Such antibodies have a much improved capacity to neutralize the target virus and may have a great clinical potential. Their production in high yield in plants offers an economically advantageous approach applicable to both IgG and secretory IgA antibodies.
While polyclonal antibodies from human sera provide immunological diversity, mAbs are highly specific for a single virus epitope. Potential alterations of such epitopes may generate virus neutralization escape mutants and decrease or eliminate the effectiveness of mAb prophylactics/therapeutics. Polyclonal antibody preparations prepared from pooled sera are not uniform and vary with the source of serum pools.
This problem can be overcome by the development of human recombinant antigen-specific polyclonal antibodies by a novel Sympress technology (Symphogen, Lyngby, Denmark).
Immunoglobulins, either already in clinical use or in development, are directed against one of the following viruses: hepatitis A and B; cytomegalovirus; rabies; respiratory syncytial virus; smallpox; vaccinia; varicella zoster; measles; mumps; rubella; parvovirus B19; Epstein–Barr virus; herpes simplex; tick-borne encephalitis; poliovirus; Hantavirus; West Nile virus; rotavirus; poliovirus; HIV-1; Ebola virus; and severe acute respiratory syndrome-associated coronavirus.
Unlike vaccines, passive immunization can rapidly deliver protective levels of antibodies directly to susceptible mucosal sites where many virus infections are initiated. Secretory IgA because of its polyvalency and relative stability may have advantages over IgG for passive immunization at these sites.
Antibodies function also as immunomodulators which can bridge innate and acquired, and cellular and humoral immune responses, respectively. Infected host cells can be targeted by linking anticellular toxins to antiviral antibodies or by bispecific antibodies in which one Fab fragment of the antibody is virus specific and the other one recognizes a host cell component or receptor.
Research on antibody-mediated immunity and understanding of antibody-based prophylaxis and therapies for virus diseases have provided a foundation for research on and development of antivirus vaccines.
Vaccines against Human Viral Diseases
Vaccination is the most successful medical intervention against viral diseases. Vaccines prevent or moderate illnesses caused by virus infection in an individual and prevent or diminish virus transmission to other susceptible persons, thus contributing to herd immunity. This effect is expected to be long term, depends on establishment of immunological memory at both the B- and T-cell levels, and may require consecutive or repeated vaccinations. The effectiveness of vaccination might be diminished or compromised for viruses occurring in the form of simultaneous quasi-species or undergoing time-dependent changes of antigenic properties (antigenic drift and antigenic shift). However, in some cases, vaccination predisposes to aggravated disease elicited by infection with a virus identical or related to that used for vaccination, that is, antibody enhancement of virus infection occurs (dengue and respiratory syncytial viruses and HIV-1).
The following categories of vaccines can be distinguished (Table 2 ): (1) live attenuated; (2) whole virus (inactivated); (3) glycoprotein subunit vaccines; and (4) protein vaccines based on recombinant DNA technologies. One vaccine in category (3), hepatitis B surface antigen (HBsAg), is derived from plasma of hepatitis B virus carriers. It is remarkable that vaccines in categories (3) and (4) correspond to multi-subunit self-assembled particles having antigenic specificities closely similar or identical to those expressed on the surface of virus particles. On the other hand, individual virus protein or glycoprotein subunit molecules or their peptide fragment have been less suitable candidates for vaccine development because of insufficient similarities with intact viruses.
Table 2.
Past and present vaccines against human viral diseases
| Live attenuated | Killed whole virus | Glycoprotein subunit | Genetically engineered |
|---|---|---|---|
| Influenza (nasal) | Influenza | Influenza | Papillomavirus |
| Rabies | Rabies | Hepatitis B | Hepatitis B |
| Poliovirus | Poliovirus | ||
| Yellow fever | Japanese encephalitis | ||
| Measles | Tick-borne encephalitis | ||
| Mumps | Hepatitis A | ||
| Rubella | |||
| Adenovirus | |||
| Varicella zoster | |||
| Rotavirus | |||
| Smallpox (vaccinia) |
Vaccine formulations require the incorporation of an immunological adjuvant to enhance their immunogenicity. Adjuvants are designed to optimize antigen delivery and presentation, enhance the maturation of antigen-presenting dendritic cells, and induce immunomodulatory cytokines.
Currently, most vaccines are administered parenterally using syringes with needles.The procedure is disliked by many, and is questionable for mass vaccination programs in developing countries. Therefore, efforts are being made to produce vaccines which can be delivered onto mucosal surfaces, that is, mostly orally or nasally. In addition, high-workload needle-free injection devices are being developed. At this juncture, the bifurcated needle, developed by Benjamin Rubin over 40 years ago, must be mentioned. It proved to be essential for the successful campaign to eradicate smallpox worldwide.
Veterinary Vaccines
The development and use of veterinary vaccines has the following aims: cost-effective prevention and control of virus diseases in animals; induce herd immunity; improve animal welfare and food production for human consumption; decrease the usage of veterinary drugs, thereby minimizing their environmental impact and food contamination; and decrease the incidence of zoonoses (e.g., infections by avian influenza, rabies, West Nile, Rift Valley fever viruses, respectively).
Research, development, and production of some veterinary vaccines have been on the forefront of the general field of vaccinology. The foot-and-mouth disease virus vaccine was the first one produced at an industrial scale (Frenkel method). A vaccinia-rabies virus G protein recombinant vaccine was among the first biotechnology-based vaccines licensed. The world's first DNA vaccine (against West Nile virus in horses) was approved by the US Department of Agriculture in July 2005. DIVA (Differentiating Infected from Vaccinated Animals; also termed marker) veterinary recombinant vaccines and companion diagnostic tests have been developed. They can be applied to programs to control and eradicate virus infections. These are examples to be considered for the development of human vaccines. The latter will require rigorous evaluations for safety and efficacy which are more difficult to obtain than in veterinary settings. A list of licensed veterinary vaccines is shown in Table 3 .
Table 3.
Current veterinary vaccines
| Species | Live | Killed | Recombinant | DNA |
|---|---|---|---|---|
| Avian | Encephalomyelitis | Encephalomyelitis (fowl pox vector) | ||
| Influenza | Influenza (fowl pox vector) | |||
| Pneumovirus | Paramyxovirus | |||
| Polyomavirus | ||||
| Reovirus | ||||
| Bursal disease | Bursal disease | Bursal disease (Marek's disease vector) | ||
| Marek's disease | Marek's disease (Marek's disease vector) | |||
| Fowl pox | Fowl pox | |||
| Newcastle disease | Newcastle disease Bronchitis | Newcastle disease (fowl pox vector) | ||
| Anemia | ||||
| Laryngotracheitis | Laryngotracheitis (fowl pox vector) | |||
| Duck enteritis | ||||
| Duck hepatitis | ||||
| Canary pox | ||||
| Feline | Calicivirus | Calicivirus immunodeficiency virus | ||
| Leukemia virus | Leukemia virus (canarypox vector) | |||
| Infectious peritonitis | ||||
| Rhinotracheitis | Rhinotracheitis | |||
| Panleukopenia | Panleukopenia | |||
| Rabies | Rabies (canary pox vector) | |||
| Canine | Adenovirus 2 | |||
| Parvovirus | Parvovirus | |||
| Coronavirus | Coronavirus | |||
| Parainfluenza | Parainfluena (canary pox vector) | |||
| Canine distemper | Canine distemper (canary pox vector) | |||
| Measles | ||||
| Hepatitis | ||||
| Rabies | Rabies | |||
| Sheep and goat | Bluetongue | |||
| Ovine ecthyma | ||||
| Poxviruses | ||||
| Louping ill | ||||
| Equine | Influenza | Influenza | Influenza (canary pox vector) | |
| Rhinopneumonitis | Rhinopneumonitis | |||
| Rotavirus | ||||
| Arteritis | ||||
| African horse sickness | African horse sickness | |||
| Encephalomyelitis | ||||
| West Nile virus Flavivirus chimera | West Nile virus | West Nile virus (canary pox vector) | West Nile virus | |
| Bovine | Respiratory syncytial virus | Respiratory syncytial virus | ||
| Rhinotracheitis | Rhinotracheitis | |||
| Diarrhea | Diarrhea | |||
| Bronchitis | Bronchitis | |||
| Parainfluenza 3 | ||||
| Rotavirus | Rotavirus | |||
| Coronavirus | Coronavirus | |||
| Herpes 1 | Herpes 1 | |||
| Foot-and-mouth disease | Foot-and-mouth disease | |||
| Rinderpest | ||||
| Porcine | Pseudorabies | Pseudorabies | ||
| Enterovirus | Influenza | |||
| Parvovirus | Circovirus | |||
| Rotavirus | Rotavirus | |||
| Transmissible gastroenteritis | Transmissible gastroenteritis | |||
| Reproductive and respiratory syndrome | Reproductive and respiratory Syndrome | |||
| Hog cholera | Hog cholera |
Several of the described vaccines are being administered as combination vaccines.
See also
AIDS: Vaccine Development; Antigen Presentation; Antigenic Variation; Antigenicity and Immunogenicity of Viral Proteins; Cytokines and Chemokines; Diagnostic Techniques: Serological and Molecular Approaches; DNA Vaccines; Immune Response to Viruses: Cell-Mediated Immunity; Neutralization of Infectivity; Vaccine Production in Plants; Vaccine Strategies
Further Reading
- Ahmed R., editor. Immunological memory. Vol. 211. 2006. pp. 5–337. (Immunological Reviews). [Google Scholar]
- Burton D.R., editor. Antibodies in viral infection. Vol. 260. 2001. pp. 1–300. (Current Topics in Microbiology and Immunology). [Google Scholar]
- Casadevall A., Dadachova E., Pirofsky L.A. Passive antibody therapy for infectious diseases. Nature Reviews Microbiology. 2004;2:695–703. doi: 10.1038/nrmicro974. [DOI] [PubMed] [Google Scholar]
- Cooper M.D., Alder N. The evolution of adaptive immune systems. Cell. 2006;124:815–822. doi: 10.1016/j.cell.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Frank S.A. Princeton University Press; Princeton: 2002. Immunology and Evolution of Infectious Disease. [PubMed] [Google Scholar]
- Hangartner L., Zinkernagel R.M., Hengartner H. Antiviral antibody responses: The two extremes of a wide spectrum. Nature Reviews Immunology. 2006;6:231–243. doi: 10.1038/nri1783. [DOI] [PubMed] [Google Scholar]
- Janeway C.A., Jr. How the immune system recognizes invaders. Scientific American. 1993:73–79. doi: 10.1038/scientificamerican0993-72. (Sep.) [DOI] [PubMed] [Google Scholar]
- Janeway C.A., Jr., Travers P., Walport M., Shlomchik M.J. The Immune System in Health and Disease. 6th edn. Garland Science; New York: 2005. Immunobiology. [Google Scholar]
- Levine M.P., Kaper J.B., Rappuoli R., Liu M., Good M.F., editors. New Generation Vaccines. 3rd edn. Dekker; New York: 2004. [Google Scholar]
- O'Neill L.A. Immunity's early-warning system. Scientific American. 2005:38–45. (Jan.) [PubMed] [Google Scholar]
- Paul W.E., editor. Fundamental Immunology. 5th edn. Lippincott Williams and Wilkins; Philadelphia: 2003. [Google Scholar]
- Plotkin S.A., Orenstein W.A. 4th edn. Saunders–Elsevier; Philadelphia: 2004. Vaccines. [Google Scholar]
- Pulendran B., Ahmed R. Translating innate immunity into immunological memory: Implications for vaccine development. Cell. 2006;124:849–863. doi: 10.1016/j.cell.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Van Regenmortel M.H.V. Reductionism and the search for structure–function relationships in antibody molecules. Journal of Molecular Recognition. 2002;15:240–247. doi: 10.1002/jmr.584. [DOI] [PubMed] [Google Scholar]
- Weissman I.L., Cooper M.D. How the immune system develops. Scientific American. 1993:65–71. doi: 10.1038/scientificamerican0993-64. (Sep.) [DOI] [PubMed] [Google Scholar]
- Woof J.M., Mestecky J. Mucosal immunoglobulins. Immunological Reviews. 2005;206:64–82. doi: 10.1111/j.0105-2896.2005.00290.x. [DOI] [PubMed] [Google Scholar]