

Abstract
The host–vector–pathogen interactions are governed since their origin by a subtle balance between attack and transmission strategies and defense mechanisms. Molecular dialogues are established between a pathogen and its host, these ones will generate a mutual selection pressure that will result in profound changes in expression of host and pathogen genes. In the post-genomic era, proteomics is in its infancy, but represents a promising tool to fill the blanks that still exist in biology despite the sequencing of the genomes of many organisms. This chapter presents the interest in proteomics toward a better understanding of host–vector–pathogen cross talks through a synthetic review of the available data generated by proteomics studies, discussing the pitfalls of current approaches, and presenting new approaches to decipher from molecular interactions with population proteomics leading to biomarkers discovery for diagnosis, therapy, and vaccine purposes.
Keywords: Host–parasite interactions, Parasito-proteomics, Pathogeno-proteomics, Population proteomics, Proteomics
1. Introduction
Living organisms are constantly exposed to pathogens. In any environment, a molecular war begins when a host encounters a pathogen. In many host–pathogen associations, the molecular war is in progress a long time ago. Nevertheless, a disease as an outcome of a pathogen attack remains an exception rather than a rule. Most host species have acquired strategies by selective pressure to mislead the pathogen and to win the fight during their cross talk (i.e., molecular dialogue). However, many pathogen species have acquired strategies by selective pressure to bypass the host defenses to win the molecular war and to ensure the completion of their life cycle. Pathogens remain a significant threat to any host species. Critical to the mitigation of this threat is the ability to rapidly detect, respond to, treat, and contain the pathogen transmission. Since many centuries, some scientific fields (i.e., agroecology, evolutionary ecology, evolutionary medicine, biochemistry, microbiology, medicine, veterinary medicine, immunology, and molecular biology) have surveyed host–parasite interactions to improve our understanding of pathogenic diseases and to prevent pathogen transmission in host populations.
During the course of human history, pathogenic diseases have seriously affected many societies worldwide. In Europe, one of the most dramatic disease events was the great plague pandemic of the mid-14th century.1, 2 Notably, pathogenic diseases are a leading cause of premature death in the world. Pathogenic diseases result from an intimate relationship between a host and a pathogen which involves molecular “cross talk.” Clearly, elucidation of this complex molecular dialogue between host and pathogen is desirable in order to improve our understanding of pathogen virulence, to develop pathogen-specific host biomarkers, and to define novel therapeutic and vaccine targets. Proteomics applications to decipher host–parasite interactions are in their infancy in spite of important technological and scientific advances since the post-genomic era, and should lead to new insights on host specificity and on the evolution of pathogen virulence. In this chapter, we present the interest of proteomics to survey host–pathogen interactions, a synthetic review of previous proteomics studies, the pitfalls of the current approach in surveys, new conceptual approaches to decipher host–parasite interactions, a new avenue to decipher the cross-talk diversity involved in trophic interactions in a habitat (i.e., the population proteomics), and a 5-year view for future prospects on proteomics and host–pathogen interactions.
2. Interest of Proteomics to Study Host–Pathogen Interactions
Since the start of the genomic era in the early 1990s, many parasitologists and molecular biologists are confident that complete sequencing of the genome of the partners in host–pathogen associations for pathogens with simple life cycle (i.e., one host) and in host–vector–pathogen associations for pathogens with complex life cycle (i.e., at least two hosts) will enable the total understanding of the molecular mechanisms involved in most of the pathogenic diseases and will contribute to find new drugs for treating them3, 4; insufficient progress has been achieved in the control of such diseases as malaria and sleeping sickness, despite decades of intensive genomic projects on host–pathogen interactions, vaccines, and chemotherapeutics. Pathogens continue to be a major cause of morbidity and mortality in humans and domestic livestock, especially in developing countries.5, 6, 7, 8, 9
Until now, many parasitologists and molecular biologists have focused their studies on DNA analyses based on the central dogma of molecular biology—that is to say, the general pathway for the expression of genetic information stored in DNA. Although the basic blueprint of life is encoded in DNA, the execution of the genetic plan is carried out by the activities of proteins. The fabric of biological diversity is therefore protein based, and natural selection acts at the protein level.10 At the end of the 20th century, it had become clear to many parasitologists and molecular biologists that knowing genome sequences, while technically mandatory, was not in itself enough to fully understand complex biological events, such as the immune response of a host to a pathogen infection or the molecular strategies used by pathogens to thwart the host defenses during their interaction.11, 12, 13, 14, 15
The evolution of any given species has tremendously increased complexity at the level of pre- (gene splicing, mRNA editing) and posttranslational (phosphorylation, glycosylation, acetylation, and so on) gene–protein interaction. The genomics era has revealed that: (1) DNA sequences may be “fundamental,” but can provide little information on the dynamic processes within and between host and a parasite during their physical and molecular interaction11, 12; (2) the correlation between the expressed “transcriptome” (i.e., total mRNA transcription pattern) and the levels of translated proteins is poor16, 17, 18; and (3) a single gene can produce different protein products.13, 14, 18 Moreover, the structure, function, abundance, and even the number of proteins in an organism cannot yet be predicted from the DNA sequence alone.11, 17, 19 Also, posttranslational modifications, such as phosphorylation and glycosylation, are often extremely important for the function of many proteins, although most of these modifications cannot yet be predicted from genomic or mRNA sequences.17 Thus, the biological phenotype of an organism is not directly related to its genotype (i.e., DNA sequences).
Epigenetic systems control and modify gene expression. Almost all the elements of epigenetic control systems are proteins.19 The cells of an organism are reactive systems in which information flows not only from genes to proteins but in the reverse direction as well.3 The proteome is the genome operating system by which the cells of an organism react to environmental signals.19 It comprises an afferent arm, the cytosensorium (i.e., many cellular proteins are sensors, receptors, and information transfer units from environmental signals) and an efferent arm, the cytoeffectorium (i.e., in cells, reaction of the genome via regulation of either individual proteins or a group of proteins in response to environmental changes).
Proteomics is the study of the proteome. In a broad sense, the proteome (i.e., the genome operating system) means all the proteins produced by a cell or tissue. Proteomics will contribute to bridge the gap between our understanding of genome sequence and cellular behavior. Proteomics offers an excellent way to study the reaction of the host and pathogen proteomes (i.e., genome-operating systems) during their complex biochemical cross talk.20, 21 Using the first generation proteomics approach, two-dimensional electrophoresis (2-DE), and mass spectrometry (MS), posttranslational modifications of host and pathogen proteins (such as phosphorylation, glycosylation, acetylation, and methylation) in reaction to their interaction can be detected. Such modifications are vital for the correct activity of numerous proteins and are being increasingly recognized as a major mechanism in cellular regulation. Although 2-DE offers a high-quality approach for the study of host and/or pathogen proteomes, during the post-genomic era several proteomics approaches (e.g., bottom-up; top-down) and quantitative proteomics strategies have been developed, which complement classical 2-DE (see Fig. 11.1 ).17, 22, 23, 24, 25, 26 Table 11.1 presents a comparison of the most popular proteomics tools.
Figure 11.1.
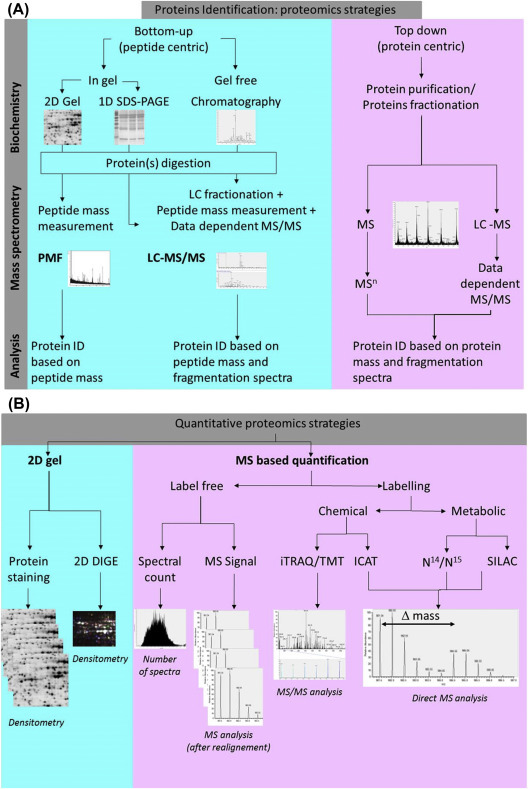
Deciphering of host–parasite cross talk with proteomics. Bottom-up and Top-down approaches (A); and quantitative proteomics strategies (B).
Table 11.1.
A Comparison of Proteomics Tools
| Name of Technique | Separation | Quantification | Identification of Candidate Protein Spots | Hydrophobic Proteins | Requirement for Protein Identification | Potential for Discovering New Proteins | Detection of Specific Isoforms | Relative Assay Time | Cost to Acquire and to Use |
|---|---|---|---|---|---|---|---|---|---|
| 2-DE | Electrophoresis: IEF PAGE |
Densitometry of stains | Mass spectrometry (PMF; MS/MS) | Dependent on detergents used | No | Yes | Yes | Moderate | Cheap |
| 2-DIGE | Electrophoresis: IEF PAGE |
Densitometry of Cy3- and Cy5-labeled proteins normalize to Cy2 | Mass spectrometry (PMF; MS/MS) | Dependent on detergents used | No | Yes | Yes | Moderate | Expensive |
| MuDPIT | LC–LC of peptides | None | Mass spectrometry (MS/MS) | Theoretically better than electrophoresis but not systematically examined | No | Yes | Yes | Rapid | Moderate |
| ICATTM | LC of peptides | Through use of heavy and light tags | Mass spectrometry (MS/MS) | No better than 2-DE | No | Yes | No | Rapid | Moderate |
| iTRAQ | LC of peptides | Labeling with isobaric mass tags | Mass spectrometry (MS/MS) | No better than 2-DE | No | Yes | Yes | Rapid | Expensive |
| SILAC | LC of peptides | Metabolic labeling with enriched stable isotope of amino acids ([13C615N4]arginine and/or [13C615N2] lysine). | Mass spectrometry (MS/MS) | No better than 2-DE | No | Yes | Yes | Rapid | Moderate |
| SELDI-TOF MS | Binding of proteins based on their chemical and physical characteristics | Comparison of MS peaks | Requires series of samples or coupling to second MS instrument | Moderate | No | Yes | No | Rapid | Expensive |
| Protein arrays | Antibody-based chips (binding to affinity reagent) | Densitometry of binding | Binding to particular affinity reagent | Unknown | Yes | No | Yes | Rapid | Cheap |
2-DE, two-dimensional electrophoresis; 2-DIGE, two-dimensional difference in gel electrophoresis; ICAT, isotope coded affinity tags; iTRAQ, isobaric tag for relative and absolute quantification; LC, liquid chromatography; LC–LC, tandem liquid chromatography; MS/MS, tandem mass spectrometry; MuDPIT, multidimensional protein identification technology; PAGE, polyacrylamide gel electrophoresis; PMF, peptide mass fingerprint; SELDI-TOF MS, spectrum enhanced laser desorption ionization-time of flight mass spectrometry; SILAC, stable isotope labeling by amino acids in cell culture.
3. Retrospective Analysis of Previous Proteomics Studies
The host–pathogen cross talks reflect the balance of host defenses and pathogen virulence mechanisms. Post-genomic technology promises to revolutionize many fields in biology by providing enormous amounts of genetic data from model and nonmodel organisms. Proteomics is a case point and promises to bridge the gap between our understanding of genome sequences and cellular behavior involved in host–pathogen interactions. Proteomics offers the possibility to characterize host–pathogen interactions from a global proteomic view. To date, most proteomics surveys on host–parasite interactions have focused on cataloguing protein content of pathogens and identifying virulence-associated proteins or proteomic alterations in host response to a pathogen. Also, many parasitologists and molecular biologists have used proteomics to find pathogen-specific host biomarkers for rapid pathogen detection and characterization of host–pathogen cross talks during the infection process. In this section, a synthetic retrospective of previous proteomics studies on host–pathogen interactions and some pitfalls of these surveys are presented.
3.1. Deciphering of the Molecular Strategies Involved in Parasite Immune Evasion
To elude the vigilance of the immune system of a host, particularly mammals, a causative microorganism must actually act as a double agent. Indeed, the broad immunity has a natural or innate and adaptive component. Innate immunity constitutes the first antimicrobial defense and rapidly induces soluble mediators, such as complement, inflammatory cytokines, and chemokines, together with effector cells, such as macrophages and natural killers, in order to control or delay the spreading of the infectious agent. Then a specific response of adaptive immunity will take place to eliminate pathogens that would have survived innate immune response.27 These immune selective pressures have conducted pathogens to develop mechanisms to modulate and alter host responses or to evade phagocytosis. As a result of these host–pathogen interactions, protein expression profiles of the host immune system (susceptibility/tolerance factors) and of the pathogen (virulence/pathogenicity factors) are mutually modified.28, 29, 30
Depending on the pathogen type (virus, bacteria, fungi, and unicellular or multicellular parasites), strategies of interactions will be different and the subversion of the host immune responses will exhibit specificities at the protein level (for reviews see Refs. 20, 31, 32). In fact, these molecular dialogues and conflicts can be seen as a chess game between the host immune cell populations and the pathogen populations, in which the pathogen plays with the whites (i.e., it starts the game). Because of differences in host–pathogen organisms' size and ratio, leading to size differences of respective proteomes, the pathogen proteome could be considered as overwhelmed by the host proteome during the interactions. But in terms of immune evasion, this is not limiting because the immune system works on a qualitative basis, which constitutes a second advantage for the pathogen that can induce large-scale damages with low amounts of molecules. By contrast, this represents not only one major limitation to characterize host–pathogen interactions, but also a challenging perspective for proteomics technology. This is why retrospectively proteomics studies were mainly conducted to evidence pathogenic virulence and pathogenicity factors.33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43
Independent of the proteomics workflow used for analysis, parasite immune evasion could be illustrated by at least three strategies that are commonly widespread among pathogens: (1) immune evasion based on antigenic variation, (2) inhibition of adaptive immunity activation systems, and (3) host mimicry. In African trypanosomes, the antigenic variation of the variant surface glycoprotein (VSG) constituting the surface coat of the parasite is well described.44 But as in proteomics study, the parasite population, which has switched the VSG, is so poorly represented that it goes undetected, and therefore always keeps one step ahead of host immune responses. Also in trypanosomatids, Leishmania amastigotes, which establish within macrophage (a major immune effector cell), developed the ability to degrade class II major histocompatibility molecules to prevent Th1-type immunity to be induced.45 Another protozoan parasite, Toxoplasma gondii, generates its parasitophorous vacuole with elements of the plasma membrane from the targeted host cells, thus using the host “self” to evade immune recognition.46 These few examples actually perfectly illustrate how difficult it is to decipher, at the protein level during interactions, the pathogen molecular components involved in immune evasion. However, a new quantitative proteomics tools, the SILAC (stable isotope labeling by amino acids in cell culture) lately allowed the detection of 148 proteins of a microsporidian parasite during the kinetics of a host infection.47 Among these proteins, many are involved in parasite proliferation, and an overrepresentation of putative secreted effectors proteins was observed. Finally, this SILAC survey also suggests that this microsporidia species could use a transposable element as a lure strategy to escape the host innate immune system. Advances in proteomics offer challenging perspectives to decipher the molecular war in host–pathogen interactions.26
3.2. Host Proteome Responses to Parasite Infection
While it seems obvious to say that when a pathogen will infect a host, the later will react by expressing molecules that can be characterized by clinical proteomics, it is surprising how few studies are devoted to this research. Yet the discovery of biomarkers signing an infected state from a healthy state is the heart of the Infectious Disease Research,48, 49 and expression proteomics has quickly developed to characterize the differential expression of proteins encoded by a particular gene and their posttranslational modifications in biological fluids and tissues.50, 51, 52 In characterizing the host proteome responses to a pathogen infection, different levels of analysis have to be considered: soluble biomarkers expressed in biological fluids (e.g., serum, saliva, urine, and cerebrospinal fluid), tissue biomarkers indicative of an organ response and cellular biomarkers indicative of a cell-type response (e.g., immune cells).
Interestingly, the majority of the proteomics studies on host response to infection were performed on viral deregulation of host cells proteome ex vivo.53, 54, 55, 56, 57, 58, 59, 60, 61 These works allowed to characterize at the molecular level the overall modifications in protein profiles of the target cells, and were of high interest to the better understanding of the pathogen influence on its host. In bacteria, studies have evaluated the mode of action of known toxins or bacterial components on host cells.62, 63 Concerning parasites, ex vivo experiments on host–parasite interactions have highlighted molecular details of manipulation strategies suffered by target cells during toxoplasmosis Chagas' disease or malaria.64, 65, 66 Curiously, few works directly focused on the subversion of the immune system, mainly through monocyte/macrophage deregulation.67, 68
As a paradox, the most striking studies on host proteome response to parasite infection were performed on arthropod (infectious diseases vectors)–parasite interactions. Probably because the parasite induced a strong phenotype modification,69, 70 particularly in the case of insect behavior manipulation.71, 72 Although few in number, taken together, these pioneering analyses of the response of the proteome of the host to a pathogen pave the way for the dynamic analysis of host–pathogen interactions. These approaches deserve to be strengthened and extended to all infectious diseases to increase and improve knowledge of the molecular dialogue and conflict that govern host–pathogen interactions.
On the other hand, the clinical aspect is important in infectious diseases, a number of studies have sought to characterize more comprehensively the proteome response of the host to infection in biological fluids, with a purpose diagnosis. One interesting pioneering study was performed in rabbits and allowed to detect intra-amniotic infection by proteomic-based amniotic fluid analysis.73 For human diseases or those of livestock, the biological fluid, which should enable the detection of infection, linked to host proteome response, in the host serum. Several studies performed on this biological sample have allowed discriminating host-commensal from host–pathogen interactions in Candida albicans 74 and determining the immunome of pathogens.75, 76 Moreover, in African trypanosomiasis, proteomics analysis of the serum not only was indicative of the host response to infection, but also was promising for characterizing disease progression toward neurological disorder.77, 78 This illustrates how proteomics will help in considering at different analytical levels the host proteome response to a pathogen infection, with the prospect of benefits in improving diagnostics and therapeutics.
3.3. Biomarkers Linked to Infection Process by a Pathogen Using SELDI-TOF-MS Technology
High-throughput proteomic technology offers promise for the discovery of disease biomarkers and have extended our ability to unravel proteomes. In this section, we focus on the Surface-enhanced laser desorption time of flight mass spectrometry (SELDI-TOF-MS) technology. This MS-based method requires a minimal amount of sample for analysis and allows the rapid high-throughput analysis of complex protein samples.79 SELDI-TOF-MS differs from conventional matrix–assisted laser desorption ionization (MALDI)-TOF-MS because the target surfaces, to which the proteins and matrices are applied to, are coated with various chemically active ProteinChip surfaces (ion exchange, immobilized metal affinity capture, and reverse phase arrays). Therefore, it is possible to fractionate proteins within a mixture, or particular classes of proteins, on the array surface prior to analysis. As with MALDI, different matrices can be used to facilitate the ionization and desorption of proteins from the SELDI array surface.80
This technology was initially applied to the discovery of early diagnostic or prognostic biomarkers of cancer.81, 82, 83 Subsequently, this technology was used to discover fluid or tissue protein biomarkers for infectious diseases, such as HIV-1,84, 85, 86, 87, 88, 89 hepatitis B and C viruses,66, 90, 91, 92, 93 severe acute respiratory syndrome94 and BK virus,95 African trypanosomiasis,78, 96 infection of Artemia by cestodes,97 tuberculosis,98 bacterial endocarditis,99 and Helicobacter pylori infection.100
Certain individuals are resistant to HIV-1 infection, despite repeated exposure to the virus. The analysis of resistance to HIV infection is one of the research avenues, which has the hope of resulting in the development of a more effective treatment or a successful preventive vaccine against HIV infection. However, the molecular mechanism underlying resistance in repeatedly HIV-1-exposed, uninfected individuals (EU) is unclear. A complementary transcriptome and SELDI-TOF-MS analyses have been performed on peripheral blood T cells, plasma or serum from EU, their HIV-1-infected sexual partners, and healthy controls.86 This study detected a specific biomarker associated with innate host resistance to HIV infection, as an 8.6-kDa A-SAA cleavage product.
In the same vein, understanding the virus–host interactions that lead to patients with acute hepatitis C virus (HCV) infection to viral clearance is a key toward the development of more effective treatment and prevention strategies. SELDI-TOF-MS technology has been used to compare, at a proteomic level, plasma samples, respectively, from donors who had resolved their HCV infection after seroconversion, from donors with chronic HCV infection, and from unexposed healthy donors.92 A candidate marker of about 9.4 kDa was found to be higher in donors with HCV clearance than in donors with chronic infection. This biomarker was identified by nanoLC-Q-TOF-MS/MS as Apolipoprotein C-III and validated by Western Blot analysis. Among the most strongly upregulated genes in Dengue virus–infected Aedes aegypti salivary glands, one study identified a gene belonging to the cecropin family. The overexpression of this antimicrobial peptide was confirmed using the SELDI-TOF-MS technique.101
4. Toward New Conceptual Approaches to Decipher the Host–Parasite Interactions for Parasites With Simple or Complex Life Cycle
One main goal of “parasite-proteomics” surveys is to find proteins for use as pathogen-specific host biomarkers and to decipher the host–pathogen cross talks. Some papers emphasize that a significant number of surveys were done with a nonrigorous experimental design and without a conceptual approach to disentangle a general host proteome response from a specific host proteome response during the interaction with a pathogen.12, 20, 30, 43, 102 A new attitude is essential to improve the reliability of proteomics data on host–pathogen interactions. Lately, some conceptual approaches have been proposed to researchers working on host–pathogen interactions to improve the reliability of “parasite-proteomics” results and to stimulate the creation of proteomic database with a holistic view of host–pathogen interactions. Thus, in this section, three new avenues to decipher host–pathogen interactions for any pathogen species (i.e., with simple or complex life cycle) are presented.
4.1. A Holistic Approach to Disentangle the Host and Parasite Genome Responses During Their Interactions
Some proteomics studies have shown common features in the innate response of plants, insects, and mammals.103, 104, 105, 106 The plant defense response is mediated by disease-resistance genes (R genes), which are abundant throughout the genome and confer resistance to many microorganisms, nematodes, and/or insects. R genes of several families of plants studied to date show homology with the Drosophila receptor Toll and the mammalian interleukin-1 receptor. In addition, plants, invertebrates, and vertebrates produce a class of peptides called “defensins,” which are pathogen-inducible.103 Some peptides and/or proteins used by phytophagous or animal parasites to modify the genome expression of their host share many structural and functional homologies. Thus, for example, phytoparasitic root-knot nematodes of the genus Meloidogyne secrete substances into their plant hosts in order to make a giant cell used as a feeding site.107, 108 A similar system is observed for the zooparasite, Trichinella spiralis (Stichosomida: Trichinellidae).109 Furthermore, the injection of a peptide isolated from nematode secretions to either plant protoplasts or human cells enhances cell division.110 The mechanism is not yet well known, but protein induction is considered as a strong possibility.
These days, many data are obtained by genomic and proteomics projects concerned with host–parasite interactions. Nevertheless, as mentioned earlier, generally little effort is made to elaborate such projects with respect to a holistic view of the goal to increase knowledge concerning immune responses of a host along with the biochemical cross talk between host and pathogen/parasite. Thus far, “parasito-proteomics” studies are in their infancy but have already led to new insights concerning molecular pathogenesis and microorganism identification.111, 112 However, many “parasito-proteomics” studies have been done with powerful tools, but without a conceptual approach to disentangle the host and parasite genome responses during their interactions.
Lately, a new holistic approach was proposed to parasitologists and molecular biologists based on evolutionary concepts of the immune response of a host to an invading parasite (for more details see Ref. 20). For instance, this new conceptual approach enables the classification of the host genomic response to infection by a parasite according to the immune mechanisms used (constitutive versus induced) and the degree of specificity. From an evolutionary-ecological point of view, host immune responses to a particular parasite can be plotted on a chart according to the immune mechanisms used (constitutive versus induced) and degree of specificity. The first axis of the defense chart refers to the immune mechanisms employed by the host with the two extreme cases: (1) a constitutive immune mechanism used by the host to rapidly impair the invasion by a parasite and (2) an induced immune mechanism, which has the advantage of avoiding a costly defense system, yet has the disadvantage that the parasite might escape host control.15 The second axis of the defense chart refers to the degree of specificity of the host immune response.
Whatever the tactics used and the degree of specificity, the host genome ensures the adequate operation of the immune response via the proteome (genome operating system). For each immune tactic, many proteins are implicated. Consequently, any researcher in parasito-proteomics working with the immune defense chart will be able to categorize the host genome reaction for any given parasite at any given time. Also, for the pathogen, from an evolutionary-ecological point of view, parasite molecular strategies used to counteract host immune system can be plotted on a chart according to the infection mechanisms used (constitutive versus induced) and degree of specificity. This type of approach should be as much hypothesis generating for parasito-proteomics as for evolutionary ecology itself.
Lately, pioneer proteomics studies on parasite-induced alteration of host behavior (widespread transmission strategy among pathogens) have been carried out on six arthropod host–parasite associations: two orthoptera–hairworm associations, two insect vector–pathogen associations, and two gammarid–parasite associations.113 These “parasito-proteomics” studies were based on the conceptual approach suggested by Biron et al.20, 21 Thus, in each study, many biological treatments have been effected to control the potential confusion resulting from proteins that are nonspecific to the manipulative process and to find the protein potentially linked with host behavioral changes. Also, for each study, to limit the possible effects of multiple infection and/or host sex-specific factors on the host proteome response, only monoinfected host males were used for the proteomics analysis. These “parasito-proteomics” surveys on the parasitic manipulation hypothesis showed that proteomic tools and the conceptual approach suggested by Biron et al.20, 21 are sensitive enough to disentangle host proteome alterations, and also the parasite proteome alterations linked to many factors, such as the circadian cycle, the parasitic status, parasitic emergence, the quality of a habitat, and the manipulative process.
4.2. Pathogeno-Proteomics: A New Avenue to Decipher Host–Vector–Pathogen Interactions
Relationships between pathogens and their hosts and vectors depend on a molecular dialogue tightly regulated. The reciprocal influence of a pathogen with its host or vector will affect the level of their genomes and their expression, respectively.30 Variability and cross-regulation increase from genomic DNA (mutations, rearrangement, methylations, and so on) through RNA transcripts (initiation, splicing, maturation, editing, stability, and so on) to functional proteins (initiation, folding, posttranslational modifications, localization, function, and so on). Pathogeno-proteomics is a new approach to decipher host–vector–pathogen interactions, which integrates modifications at all analytical levels (genome, transcriptome, proteome: whole cell content, and secretome: naturally excreted–secreted molecules) through the analysis of their end-products' profile (Fig. 11.2 ). The concept is based on a management with drawers of the analytic workflow, from the determination of number of experimental treatments and design of the biological material preparation to the dedicated proteomics and bioinformatics tools needed to answer a research question in cell immunobiology (directly involved in host–pathogen interactions (Fig. 11.3 )) but also in ecology and evolution, population's biology, and adaptive processes.10, 30, 114 Moreover, it has been proved that the results of this type of integrated approach has a concrete impact on the discovery of the causes of infectious diseases, as well as on improving the diagnosis, vaccine development, and rational drug design.115, 116, 117 Despite a theoretical aspect,118 the pathogeno-proteomics concept brought new insights into important aspects of cell signaling119 and molecular medicine.120, 121 As an example, proteomics and bioinformatics tools enable the formulation of relevant biological hypothesis on why part of the fungal population is killed while a significantly high percentage survives in C. albicans–macrophage interactions,122 leading to addition of a specific database for studying C. albicans–host interactions.123 Direct applications in terms of discovery of antifungal drug targets or design of new effective antibacterial vaccines become reality.40, 124 Other studies have also highlighted the pathogenic changes in the brain of SIV-infected monkeys,125 adaptive metabolic changes in Trypanosoma cruzi and Trypanosoma congolense,126, 127 or molecular biomarkers of intestinal disorder induced by H. pylori or Tritrichomonas muris.1, 128 Subsequently, the use of model organisms interacting with infectious agent of medical importance emphasized the complexity and pathogen specificity of the worm's immune response.129 Taken together, these examples demonstrate the potential of the concept of pathogeno-proteomics and promote this new research avenue.
Figure 11.2.

Pathogeno-proteomics: integrating analytical levels in host–vector–pathogen interactions.
Figure 11.3.

A new biological entity named host–pathogen interactome corresponding to complete set of protein–protein interactions existing between all the proteins of a host and a pathogen during their interaction.
5. Population Proteomics: An Emerging Discipline to Study Host–Parasite Interactions
The host susceptibility to a pathogen and/or the pathogen virulence are often fluctuating within a host population even when infected hosts are collected in the same habitat and at the same time. This host phenotypic variability can be caused by three factors: (1) host genotype and/or pathogen genotype, (2) different environmental experiences (e.g., habitat fragmented in microclimates), and (3) host coinfection by pathogens (i.e., competition or mutualism among coinfecting pathogens within hosts). What are the host–pathogen cross talks at individual and population scales in a habitat? Is it possible to detect and to decipher the host proteome variability within a habitat for the molecular mechanisms and for the protein networks involved in the host–pathogen interactions? In this section, a new emerging discipline in proteomics, the population proteomics, and its prospects are presented with results of some pioneer studies on this topic, especially in human population proteomics.
5.1. Prospects With Population Proteomics for Any Living Organisms
One limiting factor for the first generation of proteomics tools (e.g., 2-DE) is the amount of proteins required to study the host and/or pathogen proteome expression(s) during their interactions. Most surveys in “parasite-proteomics” were done by pooling many individuals for any treatment (e.g., infected and noninfected hosts) required to answer a query. Thus, with this kind of experimental protocol, no data can be acquired on the interindividual variation in expression of host and pathogen proteomes during their cross talk. New proteomics tools and methods have been developed as 2D-LC/MS that can permit to study the interindividual variation of molecular cross talk in host–pathogen associations.130, 131, 132
At the beginning of the century, Dobrin Nedelkov proposed a new scientific field in proteomics: the population proteomics.130 Population proteomics was defined as the study of protein diversity in human populations, or more specifically, targeted investigation of human proteins across and within populations to define and understand protein diversity with the main aim to discover disease-specific protein modulations.133 Biron et al.114 have proposed to broaden the “population proteomics” concept to all living organisms with the aims to complement the population genetics and to offer a new avenue to decipher the cross talk diversity involved in trophic interactions in a habitat since the execution of the genetic plan is carried out by the activities of proteins and natural selection acts at the protein level.10, 134
The apparent separation between genomics and proteomics that leads to different perspective on the same ecological reality is a fundamental limitation that needs to be overcome if complex processes, such as adaptation, pathogen virulence, and host susceptibility, are to be understood. Population proteomics coupled with population genetics has a great potential to resolve issues specific to the ecology, the evolution of natural populations, the dynamic of host susceptibility to pathogens, the evolution of pathogen virulence, and the range of host genotypes that can be infected with a given pathogen genotype in host–parasite interactions. Some perspectives for the population proteomics are resumed in Fig. 11.4 . Even if we are yet far from this “promised land,” a better understanding of the information contained in proteomics markers should permit an impressive amount of information to be gathered on the past as well as current environmental conditions experienced by a given population of a species, something that could be summarized as “show me your proteome and I will tell you who you are, where you are from, and where you should go from here.”
Figure 11.4.

Potential of population proteomics as an emerging discipline in proteomics.
Lately, pioneer surveys on population proteomics have been carried out with classical proteomic tools (i.e., 2-DE and MS) (1) to determine the genetic variability between species and between populations of a given species,135, 136, 137 (2) to identify biochemical signatures linked to particular habitat and/or environmental conditions,138, 139 and (3) for phylogenetic studies.130, 140 Nedelkov et al.141, 142 have investigated the human plasma proteins diversity by using approaches similar to enzyme-linked immunosorbent assay but utilizing MS as method of detection.133 These pioneer results should help not only to discover disease-specific protein modulations but also to find pathogen-specific protein biomarkers. The next subsection presents in detail the Nedelkov' results on protein diversity in human populations.
5.2. Human Population Proteomics
Human population proteomics deciphers protein diversity in human populations. In a broader term, human population proteomics can be compared to human population genomics, where individuals are interrogated with the aim of cataloguing common genetic variants and determining how they are distributed among people within populations and among populations in different parts of the world.143, 144, 145 Although human population proteomics cannot (yet) claim such outreach and goals, it has the potential to become an important proteomics subdiscipline as the tools and approaches that enable it become more embraced and practiced.
Human population proteomics does not engage the study of entire proteomes because it is very likely that, for a specific cell or tissue proteome, there is no definitive set and number of proteins that is common to all within a group or a larger population. Instead, human population proteomics focuses on interrogation of a selected number of proteins but from a large number of individuals, to delineate the distribution of specific protein modifications within these subpopulations. Hence, targeted protein analysis approaches utilizing MS as detection method are employed. MS measures a unique feature of each fully expressed protein—its molecular mass. Changes in the protein structure resulting from structural modifications are reflected in its molecular mass and can be detected via MS, without a priori knowledge of the modification. The MS methods utilized in human population proteomics must be capable of analyzing hundreds, if not thousands of samples per day, with high reproducibility and sensitivity. Hence, top-down MS approaches utilizing affinity ligands are the most likely methods of choice for population proteomics.144 Surface-immobilized ligands can be utilized to affinity-retrieve a protein of interest from a biological sample, after which the protein (with or without the affinity ligand) is introduced into a mass spectrometer. One of the first affinity MS methods developed was mass spectrometric immunoassay (MSIA).146 The approach combines targeted protein affinity-extraction with rigorous characterization using MALDI-TOF MS (Fig. 11.5 ). Protein(s) are extracted from a biological sample with the help of affinity pipettes derivatized with polyclonal antibodies. The proteins are eluted from the affinity pipettes with a MALDI matrix, and are MS-analyzed. Enzymatic digestion, if needed, is performed on the MALDI target itself. Specificity and sensitivity, as in traditional immunoassays, are dictated by the affinity-capture reagents—the antibodies.
Figure 11.5.

Schematics of the mass spectrometric immunoassay (MSIA) approach.
However, a second measure of specificity is incorporated in the resulting mass spectra, wherein each protein registers at specific m/z value. During data analysis, the major signal in the mass spectrum that corresponds to the targeted protein is initially evaluated; it should be within a reasonable range (e.g., error of measurement of <0.05%) from the value of the empirically calculated mass obtained from the sequence of the protein deposited in the Swiss-Prot databank. Once this mass value is confirmed (or observed to be shifted), the presence of protein modifications is noted by the appearance of other signals in the mass spectra (usually in the vicinity of the native protein peaks), or by mass shifts of the major protein signal. Modifications can be tentatively assigned by accurate measurement of the observed mass shifts (from the wild-type protein signals and/or in silico calculated mass) and knowledge of the protein sequence and possible modifications. The identity of the modifications is then verified using proteolytic digestion and mass mapping approaches in combination with high-performance MS.
In an initial study of human protein diversity using MS methods of detection, 25 plasma proteins from a cohort of 96 healthy individuals were investigated via MS immunoassays.147 The protocol and an example of the data generated for one of the proteins, transthyretin (TTR), are outlined in Fig. 11.6 . The TTR MSIA assays were performed in parallel on the 96 human plasma samples using affinity pipettes derivatized with anti-TTR antibody. Following MS analysis, data matrix containing all tentatively assigned modifications was assembled. Then, peptide-mapping experiments were performed on selected number of samples to identify the specific modifications and finalize the modifications database. The data for all 25 proteins is presented in Fig. 11.7 , which lists the modifications observed for 18 of the 25 proteins studied (modifications were not observed for 7 proteins), and shows the frequency of each modification in the 96 samples cohort. A total of 53 protein variants were observed for these 18 proteins, stemming from posttranslational modifications and point mutations. The largest number of posttranslationally modified protein variants was found to be C- or N-terminal truncated protein isoforms. Deglycosylation, oxidation, and cysteinylation were also observed among several of the proteins. Among the point mutations detected for four of the proteins, notable was the high incidence of point mutations for apolipoprotein E and TTR, which is consistent with genomic studies that have found these proteins to be highly polymorphic. The overall frequency of the modifications in the 96 samples cohort was wide ranged. Fourteen modifications were observed in all 96 samples, suggesting that they must be regarded as wild-type protein forms. Others, such as most of the point mutations, were present in only few of the samples. Overall, 23 of the modifications were observed in more than 65% of the samples, and 20 in less than 15% of the 96 samples analyzed. Upon further data analysis, and taking into the consideration the gender, age, and ethnicity of the individuals who provided the samples, it was determined that the Gly6Ser mutation in TTR was detected only in individuals of Caucasian origin, which is consistent with existing knowledge about the occurrence of this common non-amyloidogenic population polymorphism in Caucasians.148, 149 Another correlation was observed in regard to interprotein variations in specific individuals: all seven individuals for which carbohydrate-deficient transferrin was detected were also characterized with deglycosylated antithrombin III.
Figure 11.6.

An outline of a population proteomics approach using TTR as an example. m/z, mass-to-charge ratio; TTR, transthyretin.
Figure 11.7.
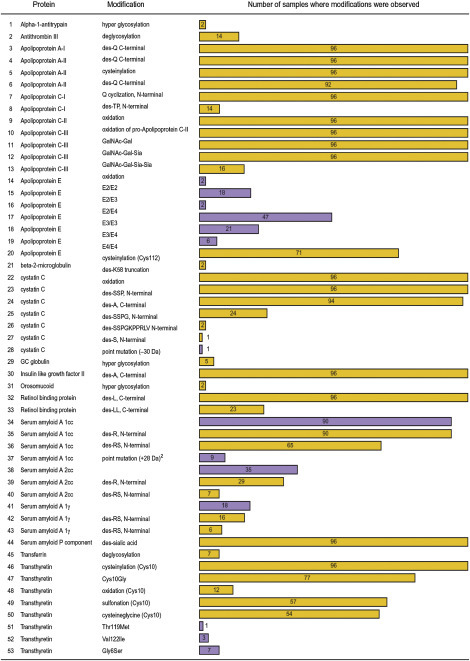
Modifications observed in 18 of the 25 proteins analyzed from 96 human plasma samples.
Following this small-scale protein diversity study, a second study of human protein diversity was carried out wherein the number of samples was greatly expanded in order to get an accurate view of the distribution of some of the protein modifications in the general population.142 Thousand individuals from four geographical regions in the United States (California, Florida, Tennessee, and Texas) were selected, and the protein modifications for beta-2-microglobulin (b2m), cystatin C (cysC), retinol-binding protein (RBP), transferrin (TRFE), and TTR were delineated (in the 96 samples study, these five proteins accounted for 19 of the 53 protein variants observed). The results of the study are summarized in Fig. 11.8 , which lists the protein modifications observed and the frequency of each in the 1000 samples cohort. A total of 27 protein modifications (20 posttranslational modifications and 7 point mutations) were detected, with various frequencies in the cohort of samples. Variants resulting from oxidation were observed most frequently, along with single amino acid truncations. Least frequent were variants arising from point mutations and extensive sequence truncations. In total, 6 modifications were observed with high frequency (present in >80% of the samples), 5 were of medium frequency (20–50% of the samples), and 16 were low-frequency modifications observed in <7% of the samples. Nine of the low-frequency modifications were not observed in the 96 individuals study. Thus, by increasing the size of the population, it became possible to detect these low-occurrence protein modifications. When the frequencies of the modifications in the two studies were compared, an excellent correlation was obtained. For example, in both cohorts about 7% of the individuals were characterized with carbohydrate-deficient transferrin. Upon further data analysis based on the gender, age, and geographical origin of the individuals who provided the samples, it was determined that the samples obtained from California contained significantly less protein modifications than the samples obtained from Florida, Tennessee, and Texas, even though the samples from all four states were collected in the same way within a 3-month window in the spring of 2005, and stored under identical conditions until analysis. Correlations were also made in regard to the gender distribution of two protein modifications. Carbohydrate-deficient transferrin was observed in about 1% of the females and about 10% of the males in the 1000 cohort. Carbohydrate-deficient transferrin is an FDA-approved clinical biomarker for alcoholism, and this gender correlation can partially be explained by the higher prevalence of alcohol dependence in males than in females. The second gender correlation was related to cystatin C: all 10 of the cystatin C point mutations were found in males.
Figure 11.8.
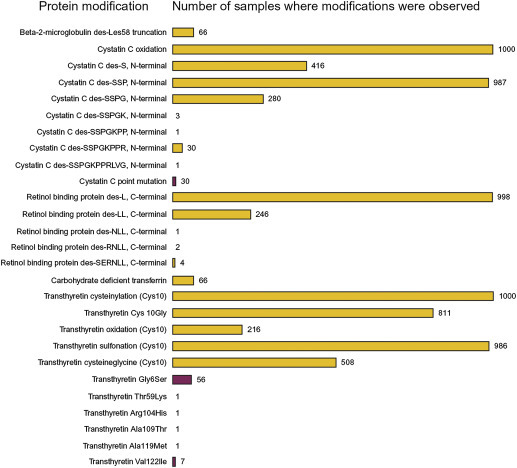
Modifications observed for five proteins studied from 1000 human plasma samples.
Two conclusions can be made from these two systematic studies of protein modifications and variants. First, MS is capable of detecting structural protein modifications, and, when coupled to immunoaffinity separations, it can be employed in a high-throughput systematic study of human protein diversity. Second, the human protein diversity is far more complex than the variation observed at the genetic level. While it might be premature to declare the human proteins variation “the next big thing,” it is reasonable to predict that assessing human proteome variations among and within populations will be a paramount effort that can facilitate biomarker discovery. Such endeavor would represent a paradigm shift in proteomics with significant clinical and diagnostic implications, as protein variations, quantitative and qualitative, begin to be associated with specific diseases.
6. Conclusion
From the dawn of human evolution to the influenza and HIV/AIDS pandemics of the 20th and early 21st centuries, infectious diseases have continued to emerge and re-emerge with great ferocity and, by so doing, seriously affect populations as well as challenge our abilities to fight the responsible agents. Over the past decade, strains of many common pathogens have continued to develop resistance to the drugs that once were effective against them. In the battle against pathogens, humankind has created new mega-technologies, such as massive sequencing, proteomics, and bioinformatics, but without conceptual approaches based on the evolutionary concepts. Parasite genome sequences do not of themselves provide a full explanation of the biology of an organism and on the molecular war involved in host–pathogen associations. Since the 1990s, proteomic tools have been successfully employed in a large number of studies to find and identify proteins involved in biological phenomena, for example, immunity, host–parasite interactions, and so on. Even so, many studies have, as outlined earlier, revealed pitfalls in the approaches used. Thus, whatever the new technological advancements, it is apparent that parasitologists and molecular biologists should attempt to improve their experimental design. This new attitude will surely improve the reliability of the data deriving from proteomics studies and will open the way for an enhanced comprehension of many biological mechanisms. In this chapter, new ways based on evolutionary concepts are suggested to enable further elucidation of the molecular complexities of host–pathogen genome interactions. These new ways could help to increase the knowledge about the molecular war involved in host–pathogen associations taking into account the environmental factors.
References
- 1.Achtman M., Morelli G., Zhu P., Wirth T., Diehl I., Kusecek B. Microevolution and history of the plague bacillus, Yersinia pestis. Proc Natl Acad Sci USA. 2004;101:17837–17842. doi: 10.1073/pnas.0408026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watts S. Yale University Press; New Haven, CT: 1997. Epidemics and history: disease, power and Imperialism. [Google Scholar]
- 3.Hochstrasser D.F. Proteome in perspective. Clin Chem Lab Med. 1998;36:825–836. doi: 10.1515/CCLM.1998.146. [DOI] [PubMed] [Google Scholar]
- 4.Degrave W.M., Melville S., Ivens A., Aslett M. Parasite genome initiatives. Int J Parasitol. 2001;3:532–536. doi: 10.1016/s0020-7519(01)00144-8. [DOI] [PubMed] [Google Scholar]
- 5.Ouma J.H., Vennervald B.J., Butterworth A.E. Morbidity in schistosomiasis: an update. Trends Parasitol. 2001;17:117–118. doi: 10.1016/s1471-4922(00)01877-8. [DOI] [PubMed] [Google Scholar]
- 6.Ryan E.T. Malaria: epidemiology, pathogenesis, diagnosis, prevention, and treatment-an update. Curr Clin Top Infect Dis. 2001;21:83–113. [PubMed] [Google Scholar]
- 7.Guzman M.G., Kouri G. Dengue: an update. Lancet Inf Dis. 2002;2:33–42. doi: 10.1016/s1473-3099(01)00171-2. [DOI] [PubMed] [Google Scholar]
- 8.Gelfand J.A., Callahan M.V. Babesiosis: an update on epidemiology and treatment. Curr Infect Dis Rep. 2003;5:53–58. doi: 10.1007/s11908-003-0065-z. [DOI] [PubMed] [Google Scholar]
- 9.WHO. Global Health Observatory (GHO) Data. http://www.who.int/gho/malaria/epidemic/deaths/en/; 2016.
- 10.Karr T.L. Application of proteomics to ecology and population biology. Heredity. 2008;100:200–206. doi: 10.1038/sj.hdy.6801008. [DOI] [PubMed] [Google Scholar]
- 11.Barret J., Jefferies J.R., Brophy P.M. Parasite proteomics. Parasitol Today. 2000;16:400–403. doi: 10.1016/s0169-4758(00)01739-7. [DOI] [PubMed] [Google Scholar]
- 12.Ashton P.D., Curwen R.S., Wilson R.A. Linking proteome and genome: how to identify parasite proteins. Trends Parasitol. 2001;17:198–202. doi: 10.1016/s1471-4922(00)01947-4. [DOI] [PubMed] [Google Scholar]
- 13.Fell D.A. Beyond genomics. Trends Genet. 2001;17:680–682. doi: 10.1016/s0168-9525(01)02521-5. [DOI] [PubMed] [Google Scholar]
- 14.Fields S. Proteomics in genomeland. Science. 2001;291:1221–1224. doi: 10.1126/science.291.5507.1221. [DOI] [PubMed] [Google Scholar]
- 15.Schmid-Hempel P., Ebert D. On the evolutionary ecology of specific immune defence. Trends Ecol Evol. 2003;18:27–32. [Google Scholar]
- 16.Anderson L., Seilhaver J. A comparison of selected mRNA and protein abundance in human liver. Electrophoresis. 1997;18:533–537. doi: 10.1002/elps.1150180333. [DOI] [PubMed] [Google Scholar]
- 17.Gygi S.P., Rochon Y., Franza B.R., Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maniatis T., Tasic B. Alternative pre-mRNA splicing and proteome expression in metazoans. Nature. 2002;418:236–243. doi: 10.1038/418236a. [DOI] [PubMed] [Google Scholar]
- 19.Anderson N.G., Anderson N.L. Twenty years of two-dimensional electrophoresis: past, present and future. Electrophoresis. 1996;17:443–453. doi: 10.1002/elps.1150170303. [DOI] [PubMed] [Google Scholar]
- 20.Biron D.G., Moura H., Marché L., Hughes A.L., Thomas F. Towards a new conceptual approach to ‘Parasitoproteomics’. Trends Parasitol. 2005;2005(21):162–168. doi: 10.1016/j.pt.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 21.Biron D.G., Joly C., Galéotti N., Ponton F., Marché L. The proteomics: a new prospect for studying parasitic manipulation. Behav Process. 2005;68:249–253. doi: 10.1016/j.beproc.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 22.Fung E.T., Thulasiraman V., Weinberger S.R., Dalmasso E.A. Protein biochips for differential profiling. Curr Opin Biotechnol. 2001;12:65–69. doi: 10.1016/s0958-1669(00)00167-1. [DOI] [PubMed] [Google Scholar]
- 23.Lopez M.F., Pluskal M.G. Protein micro- and macroarrays: digitizing the proteome. J Chromatogr B. 2003;787:19–27. doi: 10.1016/s1570-0232(02)00336-7. [DOI] [PubMed] [Google Scholar]
- 24.Wu C.C., MacCoss M.J., Howell K.E., Yates J.R., III A method for the comprehensive proteomics analysis of membrane proteins. Nat Biotechnol. 2003;21:532–538. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]
- 25.Bischoff R., Luider T.M. Methodological advances in the discovery of protein and peptide disease markers. J Chromatogr B. 2004;803:27–40. doi: 10.1016/j.jchromb.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 26.Chetouhi C., Panek J., Bonhomme L., El Alaoui H., Texier C., Langin T. Cross-talk in host–parasite associations: what do past and recent proteomics approaches tell us? Inf Genet Evol. 2015;33:84–94. doi: 10.1016/j.meegid.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 27.Roitt I.M., Delves P.J. Blackwell Publishing, Inc; 2001. Essential immunology. [Google Scholar]
- 28.Zhang C.G., Chromy B.A., McCutchen-Maloney S.L. Host-pathogen interactions: a proteomic view. Expert Rev Proteomics. 2005;2:187–282. doi: 10.1586/14789450.2.2.187. [DOI] [PubMed] [Google Scholar]
- 29.Coiras M., Camafeita E., López-Huertas M.R., Calvo E., López J.A., Alcamí J. Application of proteomics technology for analyzing the interactions between host cells and intracellular infectious agents. Proteomics. 2008;8:852–873. doi: 10.1002/pmic.200700664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holzmuller P., Grébaut P., Brizard J.P., Berthier D., Chantal I., Bossard G. “Pathogeno-proteomics”: towards a new approach of host-vector-pathogen interactions. Ann NY Acad Sci. 2008;1149:66–70. doi: 10.1196/annals.1428.061. [DOI] [PubMed] [Google Scholar]
- 31.Walduck A., Rudel T., Meyer T.F. Proteomic and gene profiling approaches to study host responses to bacterial infection. Curr Opin Microbiol. 2004;7:33–38. doi: 10.1016/j.mib.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 32.Viswanathan K., Früh K. Viral proteomics: global evaluation of viruses and their interaction with the host. Expert Rev Proteomics. 2007;4:815–829. doi: 10.1586/14789450.4.6.815. [DOI] [PubMed] [Google Scholar]
- 33.Ouellette M., Olivier M., Sato S., Papadopoulou B. Studies on the parasite Leishmania in the post-genomic era. Med Sci. 2003;19:900–909. doi: 10.1051/medsci/20031910900. [DOI] [PubMed] [Google Scholar]
- 34.Texier C., Brosson D., El Alaoui H., Méténier G., Vivarès C.P. Post- genomics of microsporidia, with emphasis on a model of minimal eukaryotic proteome: a review. Folia Parasitol. 2005;52:15–22. doi: 10.14411/fp.2005.003. [DOI] [PubMed] [Google Scholar]
- 35.Van Hellemond J.J., van Balkom B.W., Tielens A.G. Schistosome biology and proteomics: progress and challenges. Exp Parasitol. 2007;117:267–274. doi: 10.1016/j.exppara.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 36.Liu N., Song W., Wang P., Lee K., Chan W., Chen H. Proteomics analysis of differential expression of cellular proteins in response to avian H9N2 virus infection in human cells. Proteomics. 2008;8:1851–1858. doi: 10.1002/pmic.200700757. [DOI] [PubMed] [Google Scholar]
- 37.Bird D.M., Opperman C.H. The secret(ion) life of worms. Genome Biol. 2009;10:205. doi: 10.1186/gb-2009-10-1-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steuart R.F. Proteomic analysis of Giardia: studies from the pre- and post-genomic era. Exp Parasitol. 2010;124:26–30. doi: 10.1016/j.exppara.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 39.Weiss L.M., Fiser A., Angeletti R.H., Kim K. Toxoplasma gondii proteomics. Expert Rev Proteomics. 2009;6:303–313. doi: 10.1586/epr.09.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jagusztyn-Krynicka E.K., Dadlez M., Grabowska A., Roszczenko P. Proteomic technology in the design of new effective antibacterial vaccines. Expert Rev Proteomics. 2009;6:315–330. doi: 10.1586/epr.09.47. [DOI] [PubMed] [Google Scholar]
- 41.Premsler T., Zahedi R.P., Lewandrowski U., Sickmann A. Recent advances in yeast organelle and membrane proteomics. Proteomics. 2009;9:4731–4743. doi: 10.1002/pmic.200900201. [DOI] [PubMed] [Google Scholar]
- 42.Bhavsar A.P., Auweter S.D., Finlay B.B. Proteomics as a probe of microbial pathogenesis and its molecular boundaries. Future Microbiol. 2010;5:253–265. doi: 10.2217/fmb.09.114. [DOI] [PubMed] [Google Scholar]
- 43.Holzmuller P., Grébaut P., Cuny G., Biron D.G. Tsetse flies, trypanosomes, humans and animals: what is proteomics revealing about their crosstalks? Expert Rev Proteomics. 2010;7:113–126. doi: 10.1586/epr.09.92. [DOI] [PubMed] [Google Scholar]
- 44.Morrison L.J., Marcello L., McCulloch R. Antigenic variation in the African trypanosome: molecular mechanisms and phenotypic complexity. Cell Microbiol. 2009;11:1724–1734. doi: 10.1111/j.1462-5822.2009.01383.x. [DOI] [PubMed] [Google Scholar]
- 45.Antoine J.C., Prina E., Courret N., Lang T. Leishmania spp.: on the interactions they establish with antigen-presenting cells of their mammalian hosts. Adv Parasitol. 2004;58:1–68. doi: 10.1016/S0065-308X(04)58001-6. [DOI] [PubMed] [Google Scholar]
- 46.Plattner F., Soldati-Favre D. Hijacking of host cellular functions by the Apicomplexa. Annu Rev Microbiol. 2008;62:471–487. doi: 10.1146/annurev.micro.62.081307.162802. [DOI] [PubMed] [Google Scholar]
- 47.Panek J., El Alaoui H., Mone A., Urbach S., Demettre E., Texier C. Hijacking of host cellular functions by an intracellular parasite, the microsporidian Anncaliia algerae. PLoS One. 2014;9:e100791. doi: 10.1371/journal.pone.0100791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.te Pas M.F., Claes F. Functional genomics and proteomics for infectious diseases in the post-genomics era. Lancet. 2004;363:1337. doi: 10.1016/S0140-6736(04)16082-0. [DOI] [PubMed] [Google Scholar]
- 49.Azad N.S., Rasool N., Annunziata C.M., Minasian L., Whiteley G., Kohn E.C. Proteomics in clinical trials and practice: present uses and future promise. Mol Cell Proteomics. 2006;5:1819–1829. doi: 10.1074/mcp.R600008-MCP200. [DOI] [PubMed] [Google Scholar]
- 50.Fournier I., Wisztorski M., Salzet M. Tissue imaging using MALDI-MS: a new frontier of histopathology proteomics. Expert Rev Proteomics. 2008;5:413–424. doi: 10.1586/14789450.5.3.413. [DOI] [PubMed] [Google Scholar]
- 51.Hood B.L., Malehorn D.E., Conrads T.P., Bigbee W.L. Serum proteomics using mass spectrometry. Methods Mol Biol. 2009;520:107–128. doi: 10.1007/978-1-60327-811-9_8. [DOI] [PubMed] [Google Scholar]
- 52.Wilm M. Quantitative proteomics in biological research. Proteomics. 2009;9:4590–4605. doi: 10.1002/pmic.200900299. [DOI] [PubMed] [Google Scholar]
- 53.Zheng X., Hong L., Shi L., Guo J., Sun Z., Zhou J. Proteomics analysis of host cells infected with infectious bursal disease virus. Mol Cell Proteomics. 2008;7:612–625. doi: 10.1074/mcp.M700396-MCP200. [DOI] [PubMed] [Google Scholar]
- 54.Liu H.C., Hicks J., Yoo D. Proteomic dissection of viral pathogenesis. Dev Biol. 2008;132:43–53. doi: 10.1159/000317143. [DOI] [PubMed] [Google Scholar]
- 55.Lee S.R., Nanduri B., Pharr G.T., Stokes J.V., Pinchuk L.M. Bovine viral diarrhea virus infection affects the expression of proteins related to professional antigen presentation in bovine monocytes. Biochim Biophys Acta. 2009;1794:14–22. doi: 10.1016/j.bbapap.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 56.Sun J., Jiang Y., Shi Z., Yan Y., Guo H., He F. Proteomic alteration of PK-15 cells after infection by classical swine fever virus. J Proteome Res. 2008;7:5263–5269. doi: 10.1021/pr800546m. [DOI] [PubMed] [Google Scholar]
- 57.Pastorino B., Boucomont-Chapeaublanc E., Peyrefitte C.N., Belghazi M., Fusaï T., Rogier C. Identification of cellular proteome modifications in response to West Nile virus infection. Mol Cell Proteomics. 2009;8:1623–1637. doi: 10.1074/mcp.M800565-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vester D., Rapp E., Gade D., Genzel Y., Reichl U. Quantitative analysis of cellular proteome alterations in human influenza A virus-infected mammalian cell lines. Proteomics. 2009;9:3316–3327. doi: 10.1002/pmic.200800893. [DOI] [PubMed] [Google Scholar]
- 59.Antrobus R., Grant K., Gangadharan B., Chittenden D., Everett R.D., Zitzmann N. Proteomic analysis of cells in the early stages of herpes simplex virus type-1 infection reveals widespread changes in the host cell proteome. Proteomics. 2009;9:3913–3927. doi: 10.1002/pmic.200900207. [DOI] [PubMed] [Google Scholar]
- 60.Zhang X., Zhou J., Wu Y., Zheng X., Ma G., Wang Z. Differential proteome analysis of host cells infected with porcine circovirus type 2. J Proteome Res. 2009;8:5111–5119. doi: 10.1021/pr900488q. [DOI] [PubMed] [Google Scholar]
- 61.Zhang L., Jia X., Zhang X., Sun J., Peng X., Qi T. Proteomic analysis of PBMCs: characterization of potential HIV-associated proteins. Proteome Sci. 2010;8:12. doi: 10.1186/1477-5956-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuhn J.F., Hoerth P., Hoehn S.T., Preckel T., Tomer K.B. Proteomics study of anthrax lethal toxin-treated murine macrophages. Electrophoresis. 2006;27:1584–1597. doi: 10.1002/elps.200500747. [DOI] [PubMed] [Google Scholar]
- 63.Shui W., Gilmore S.A., Sheu L., Liu J., Keasling J.D., Bertozzi C.R. Quantitative proteomic profiling of host-pathogen interactions: the macrophage response to Mycobacterium tuberculosis lipids. J Proteome Res. 2009;8:282–289. doi: 10.1021/pr800422e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Teixeira P.C., Iwai L.K., Kuramoto A.C., Honorato R., Fiorelli A., Stolf N. Proteomic inventory of myocardial proteins from patients with chronic Chagas' cardiomyopathy. Braz J Med Biol Res. 2006;39:1549–1562. doi: 10.1590/s0100-879x2006001200005. [DOI] [PubMed] [Google Scholar]
- 65.Nelson M.M., Jones A.R., Carmen J.C., Sinai A.P., Burchmore R., Wastling J.M. Modulation of the host cell proteome by the intracellular apicomplexan parasite Toxoplasma gondii. Infect Immun. 2008;76:828–844. doi: 10.1128/IAI.01115-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu Y., Nelson M.M., Quaile A., Xia D., Wastling J.M., Craig A. Identification of phosphorylated proteins in erythrocytes infected by the human malaria parasite Plasmodium falciparum. Malar J. 2009;8:105. doi: 10.1186/1475-2875-8-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oura C.A., McKellar S., Swan D.G., Okan E., Shiels B.R. Infection of bovine cells by the protozoan parasite Theileria annulata modulates expression of the ISGylation system. Cell Microbiol. 2006;8:276–288. doi: 10.1111/j.1462-5822.2005.00620.x. [DOI] [PubMed] [Google Scholar]
- 68.Fischer J., West J., Agochukwu N., Suire C., Hale-Donze H. Induction of host chemotactic response by Encephalitozoon spp. Infect Immun. 2007;75:1619–1625. doi: 10.1128/IAI.01535-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Biron D.G., Marché L., Ponton F., Loxdale H.D., Galéotti N., Renault L. Behavioural manipulation in a grasshopper harbouring hairworm: a proteomics approach. Proc R Soc Lond B. 2005;272:2117–2126. doi: 10.1098/rspb.2005.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rachinsky A., Guerrero F.D., Scoles G.A. Differential protein expression in ovaries of uninfected and Babesia-infected southern cattle ticks, Rhipicephalus (Boophilus) microplus. Insect Biochem Mol Biol. 2007;37:1291–1308. doi: 10.1016/j.ibmb.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 71.Lefèvre T., Thomas F., Ravel S., Patrel D., Renault L., Le Bourligu L. Trypanosoma brucei brucei induces alteration in the head proteome of the tsetse fly vector Glossina palpalis gambiensis. Insect Mol Biol. 2007;16:651–660. doi: 10.1111/j.1365-2583.2007.00761.x. [DOI] [PubMed] [Google Scholar]
- 72.Lefevre T., Thomas F., Schwartz A., Levashina E., Blandin S., Brizard J.P. Malaria Plasmodium agent induces alteration in the head proteome of their Anopheles mosquito host. Proteomics. 2007;7:1908–1915. doi: 10.1002/pmic.200601021. [DOI] [PubMed] [Google Scholar]
- 73.Klein L.L., Freitag B.C., Gibbs R.S., Reddy A.P., Nagalla S.R., Gravett M.G. Detection of intra-amniotic infection in a rabbit model by proteomics-based amniotic fluid analysis. Am J Obstet Gynecol. 2005;193:1302–1306. doi: 10.1016/j.ajog.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 74.Pitarch A., Nombela C., Gil C. Proteomic profiling of serologic response to Candida albicans during host-commensal and host-pathogen interactions. Methods Mol Biol. 2009;470:369–411. doi: 10.1007/978-1-59745-204-5_26. [DOI] [PubMed] [Google Scholar]
- 75.Sakolvaree Y., Maneewatch S., Jiemsup S., Klaysing B., Tongtawe P., Srimanote P. Proteome and immunome of pathogenic Leptospira spp. revealed by 2DE and 2DE-immunoblotting with immune serum. Asian Pac J Allergy Immunol. 2007;25:53–73. [PubMed] [Google Scholar]
- 76.Ju J.W., Joo H.N., Lee M.R., Cho S.H., Cheun H.I., Kim J.Y. Identification of a serodiagnostic antigen, legumain, by immunoproteomic analysis of excretory-secretory products of Clonorchis sinensis adult worms. Proteomics. 2009;9:3066–3078. doi: 10.1002/pmic.200700613. [DOI] [PubMed] [Google Scholar]
- 77.Papadopoulos M.C., Abel P.M., Agranoff D., Stich A., Tarelli E., Bell B.A. A novel and accurate diagnostic test for human African trypanosomiasis. Lancet. 2004;363:1358–1363. doi: 10.1016/S0140-6736(04)16046-7. [DOI] [PubMed] [Google Scholar]
- 78.Agranoff D., Stich A., Abel P., Krishna S. Proteomic fingerprinting for the diagnosis of human African trypanosomiasis. Trends Parasitol. 2005;21:154–157. doi: 10.1016/j.pt.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 79.De Bock M., de Seny D., Meuwis M.A., Chapelle J.P., Louis E., Malaise M. Challenges for biomarker discovery in body fluids using SELDI-TOF-MS. J Biomed Biotechnol. 2010:906082. doi: 10.1155/2010/906082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Merchant M., Weinberger S.R. Recent advancements in surface-enhanced laser desorption/ionization-time of flight-mass spectrometry. Electrophoresis. 2000;21:1164–1177. doi: 10.1002/(SICI)1522-2683(20000401)21:6<1164::AID-ELPS1164>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 81.Petricoin E.F., Liotta L.A. SELDI-TOF-based serum proteomic pattern diagnostics for early detection of cancer. Curr Opin Biotechnol. 2004;15:24–30. doi: 10.1016/j.copbio.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 82.Yang S.Y., Xiao X.Y., Zhang W.G., Zhang L.J., Zhang W., Zhou B. Application of serum SELDI proteomic patterns in diagnosis of lung cancer. BMC Cancer. 2005;20(5):83. doi: 10.1186/1471-2407-5-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xiao Z., Prieto D., Conrads T.P., Veenstra T.D., Issaq H.J. Proteomic patterns: their potential for disease diagnosis. Mol Cell Endocrinol. 2005;230:95–106. doi: 10.1016/j.mce.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 84.Luo X., Carlson K.A., Wojna V., Mayo R., Biskup T.M., Stoner J. Macrophage proteomic fingerprinting predicts HIV-1-associated cognitive impairment. Neurology. 2003;60:1931–1937. doi: 10.1212/01.wnl.0000064396.54554.26. [DOI] [PubMed] [Google Scholar]
- 85.Sun B., Rempel H.C., Pulliam L. Loss of macrophage-secreted lysozyme in HIV-1-associated dementia detected by SELDI-TOF mass spectrometry. AIDS. 2004;18:1009–1012. doi: 10.1097/00002030-200404300-00008. [DOI] [PubMed] [Google Scholar]
- 86.Missé D., Yssel H., Trabattoni D., Oblet C., Lo Caputo S., Mazzotta F. IL-22 participates in an innate anti-HIV 1 host-resistance network through acute-phase protein induction. J Immunol. 2007;178:407–415. doi: 10.4049/jimmunol.178.1.407. [DOI] [PubMed] [Google Scholar]
- 87.Luciano-Montalvo C., Ciborowski P., Duan F., Gendelman H.E., Meléndez L.M. Proteomic analyses associate cystatin B with restricted HIV-1 replication in placental macrophages. Placenta. 2008;29:1016–1023. doi: 10.1016/j.placenta.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Toro-Nieves D.M., Rodriguez Y., Plaud M., Ciborowski P., Duan F., Pérez Laspiur J. Proteomic analyses of monocyte derived macrophages infected with human immunodeficiency virus type 1 primary isolates from Hispanic women with and without cognitive impairment. J Neurovirol. 2009;15:36–50. doi: 10.1080/13550280802385505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wiederin J., Rozek W., Duan F., Ciborowski P. Biomarkers of HIV-1 associated dementia: proteomic investigation of sera. Proteome Sci. 2008;17:7–8. doi: 10.1186/1477-5956-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Poon T.C., Hui A.Y., Chan H.L., Ang I.L., Chow S.M., Wong N. Prediction of liver fibrosis and cirrhosis in chronic hepatitis B infection by serum proteomic fingerprinting: a pilot study. Clin Chem. 2005;51:328–335. doi: 10.1373/clinchem.2004.041764. [DOI] [PubMed] [Google Scholar]
- 91.Kanmura S., Uto H., Kusumoto K., Ishida Y., Hasuike S., Nagata K. Early diagnostic potential for hepatocellular carcinoma using the SELDI ProteinChip system. Hepatology. 2007;45:948–956. doi: 10.1002/hep.21598. [DOI] [PubMed] [Google Scholar]
- 92.Molina S., Misse D., Roche S., Badiou S., Cristol J.P., Bonfils C. Identification of apolipoprotein C-III as a potential plasmatic biomarker associated with the resolution of hepatitis C virus infection. Proteomics Clin Appl. 2008;2:751–761. doi: 10.1002/prca.200800020. [DOI] [PubMed] [Google Scholar]
- 93.Fujita N., Sugimoto R., Motonishi S., Tomosugi N., Tanaka H., Takeo M. Patients with chronic hepatitis C achieving a sustained virological response to peginterferon and ribavirin therapy recover from impaired hepcidin secretion. J Hepatol. 2008;49:702–710. doi: 10.1016/j.jhep.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 94.Pang R.T., Poon T.C., Chan K.C., Lee N.L., Chiu R.W., Tong Y.K. Serum proteomic fingerprints of adult patients with severe acute respiratory syndrome. Clin Chem. 2006;52:421–429. doi: 10.1373/clinchem.2005.061689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jahnukainen T., Malehorn D., Sun M., Lyons-Weiler J., Bigbee W., Gupta G. Proteomic analysis of urine in kidney transplant patients with BK virus nephropathy. J Am Soc Nephrol. 2006;17:3248–3256. doi: 10.1681/ASN.2006050437. [DOI] [PubMed] [Google Scholar]
- 96.Stiles J.K., Whittaker J., Sarfo B.Y., Thompson W.E., Powell M.D., Bond V.C. Trypanosome apoptotic factor mediates apoptosis in human brain vascular endothelial cells. Mol Biochem Parasitol. 2004;133:229–240. doi: 10.1016/j.molbiopara.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 97.Sánchez M.I., Thomas F., Perrot-Minnot M.J., Biron D.G., Bertrand-Michel J., Missé D. Neurological and physiological disorders in Artemia harboring manipulative cestodes. J Parasitol. 2009;95:20–24. doi: 10.1645/GE-1550.1. [DOI] [PubMed] [Google Scholar]
- 98.Liu Q., Chen X., Hu C., Zhang R., Yue J., Wu G. Serum protein profiling of smear-positive and smear-negative pulmonary tuberculosis using SELDI-TOF mass spectrometry. Lung. 2010;188:15–23. doi: 10.1007/s00408-009-9199-6. [DOI] [PubMed] [Google Scholar]
- 99.Fenollar F., Goncalves A., Esterni B., Azza S., Habib G., Borg J.P. A serum protein signature with high diagnostic value in bacterial endocarditis: results from a study based on surface-enhanced laser desorption/ionization time-of-flight mass spectrometry. J Infect Dis. 2006;194:1356–1366. doi: 10.1086/508429. [DOI] [PubMed] [Google Scholar]
- 100.Wu M.S., Chow L.P., Lin J.T., Chiou S.H. Proteomic identification of biomarkers related to Helicobacter pylori-associated gastroduodenal disease: challenges and opportunities. J Gastroenterol Hepatol. 2008;23:1657–1661. doi: 10.1111/j.1440-1746.2008.05659.x. [DOI] [PubMed] [Google Scholar]
- 101.Luplertlop N., Surasombatpattana P., Patramool S., Dumas E., Wasinpiyamongkol L., Saune L. Induction of a peptide with activity against a broad spectrum of pathogens in the Aedes aegypti salivary gland, following infection with Dengue virus. PLoS Pathog. 2011;7(1):e1001252. doi: 10.1371/journal.ppat.1001252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tastet C., Bossis M., Gauthier J.P., Renault L., Mugniéry D. Meloidogyne chitwoodi and M. fallax protein variation assessed by two-dimensional electrophoregram computed analysis. Nematology. 1999;1:201–214. [Google Scholar]
- 103.Broekaert W.F., Terras F.R.G., Cammue B.P.A., Osborn R.W. Plants defensins: novel antimicrobial peptides as components of the host defense system. Plant Physiol. 1995;108:1353–1358. doi: 10.1104/pp.108.4.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rock F.L., Hardiman G., Timans J.C., Kastelein R.A., Bazan J.F. A family of human receptors structurally related to Drosophilla Toll. Proc Natl Acad Sci USA. 1998;95:588–593. doi: 10.1073/pnas.95.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cao H., Baldini R.L., Rahme L.G. Common mechanism for pathogens of plant and animals. Annu Rev Phytopath. 2001;39:259–284. doi: 10.1146/annurev.phyto.39.1.259. [DOI] [PubMed] [Google Scholar]
- 106.Taylor J.E., Hatcher P.E., Paul N.D. Crosstalk between plant responses to pathogens and herbivores: a view from the outside in. J Exp Bot. 2003;55:159–168. doi: 10.1093/jxb/erh053. [DOI] [PubMed] [Google Scholar]
- 107.Abad P., Favery B., Ross M.N., Castagnone-Sereno P. Root-knot nematode parasitism an host response: molecular basis of a sophisticated interaction. Mol Plant Pathol. 2003;4:217–224. doi: 10.1046/j.1364-3703.2003.00170.x. [DOI] [PubMed] [Google Scholar]
- 108.Doyle E.A., Lambert K.N. Meloidogyne javanica chorismate mutase 1 alters plant cell development. Mol Plant Microbe Interact. 2003;16:123–131. doi: 10.1094/MPMI.2003.16.2.123. [DOI] [PubMed] [Google Scholar]
- 109.Jasmer D.P. Trichinella spiralis infected skeletal muscle cells arrest in G2/M and cease muscle gene expression. J Cell Biol. 1993;121:785–793. doi: 10.1083/jcb.121.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goverse A., De Engler J.A., Verhees J., Van der Krol S., Helder J.H., Gheysen G. Cell cycle activation by plant parasitic nematodes. Plant Mol Biol. 2000;43:747–776. doi: 10.1023/a:1006367126077. [DOI] [PubMed] [Google Scholar]
- 111.Moura H., Ospina M., Woolfitt A.R., Barr J.R., Visvesvara G.S. Analysis of four human microsporidian isolates by MALDI-TOF mass spectrometry. J Eukaryot Microbiol. 2003;50:156–163. doi: 10.1111/j.1550-7408.2003.tb00110.x. [DOI] [PubMed] [Google Scholar]
- 112.Biron D.G., Agnew P., Marché L., Renault L., Sidobre C., Michalakis Y. Proteome of Aedes aegypti larvae in response to infection by the intracellular parasite Vavraia culicis. Int J Parasitol. 2005;35:1385–1397. doi: 10.1016/j.ijpara.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 113.Lefèvre T., Adamo S.A., Biron D.G., Missé D., Hughes D., Thomas F. Invasion of the body snatchers: the diversity and evolution of manipulative strategies in host–parasite interactions. In: Webster J.P., Rollinson D., Hay S.I., editors. vol. 68. Academic Press; London: 2009. pp. 46–84. (Advances in parasitology). [DOI] [PubMed] [Google Scholar]
- 114.Biron D.G., Loxdale H.D., Ponton F., Moura H., Marché L., Brugidou C. Population proteomics: an emerging discipline to study metapopulation ecology. Proteomics. 2006;6:1712–1715. doi: 10.1002/pmic.200500423. [DOI] [PubMed] [Google Scholar]
- 115.Doytchinova I.A., Taylor P., Flower D.R. Proteomics in vaccinology and immunobiology: an informatics perspective of the immunone. J Biomed Biotechnol. 2003;2003:267–290. doi: 10.1155/S1110724303209232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bansal A.K. Bioinformatics in microbial biotechnology–a mini review. Microb Cell Fact. 2005;4:19. doi: 10.1186/1475-2859-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chautard E., Thierry-Mieg N., Ricard-Blum S. Interaction networks: from protein functions to drug discovery. A review. Pathol Biol. 2009;57:324–333. doi: 10.1016/j.patbio.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 118.Kint G., Fierro C., Marchal K., Vanderleyden J., De Keersmaecker S.C. Integration of 'omics' data: does it lead to new insights into host-microbe interactions? Future Microbiol. 2010;5:313–328. doi: 10.2217/fmb.10.1. [DOI] [PubMed] [Google Scholar]
- 119.Kleppe R., Kjarland E., Selheim F. Proteomic and computational methods in systems modeling of cellular signaling. Curr Pharm Biotechnol. 2006;7:135–145. doi: 10.2174/138920106777549722. [DOI] [PubMed] [Google Scholar]
- 120.Ahram M., Petricoin E.F. Proteomics discovery of disease biomarkers. Biomark Insights. 2008;23:325–333. doi: 10.4137/bmi.s689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ostrowski J., Wyrwicz L.S. Integrating genomics, proteomics and bioinformatics in translational studies of molecular medicine. Expert Rev Mol Diagn. 2009;6:623–630. doi: 10.1586/erm.09.41. [DOI] [PubMed] [Google Scholar]
- 122.Diez-Orejas R., Fernández-Arenas E. Candida albicans-macrophage interactions: genomic and proteomic insights. Future Microbiol. 2008;3:661–681. doi: 10.2217/17460913.3.6.661. [DOI] [PubMed] [Google Scholar]
- 123.Vialás V., Nogales-Cadenas R., Nombela C., Pascual-Montano A., Gil C. Proteopathogen a protein database for studying Candida albicans—host interaction. Proteomics. 2009;9:4664–4668. doi: 10.1002/pmic.200900023. [DOI] [PubMed] [Google Scholar]
- 124.Tournu H., Serneels J., Van Dijck P. Fungal pathogens research: novel and improved molecular approaches for the discovery of antifungal drug targets. Curr Drug Targets. 2005;6:909–922. doi: 10.2174/138945005774912690. [DOI] [PubMed] [Google Scholar]
- 125.Pendyala G., Trauger S.A., Kalisiak E., Ellis R.J., Siuzdak G., Fox H.S. Cerebrospinal fluid proteomics reveals potential pathogenic changes in the brains of SIV-infected monkeys. J Proteome Res. 2009;5:2253–2260. doi: 10.1021/pr800854t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Grébaut P., Chuchana P., Brizard J.P., Demettre E., Seveno M., Bossard G. Identification of total and differentially expressed excreted-secreted proteins from Trypanosoma congolense strains exhibiting different virulence and pathogenicity. Int J Parasitol. 2009;10:1137–1150. doi: 10.1016/j.ijpara.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 127.Roberts S.B., Robichaux J.L., Chavali A.K., Manque P.A., Lee V., Lara A.M. Proteomic and network analysis characterize stage-specific metabolism in Trypanosoma cruzi. BMC Syst Biol. 2009;16(3):52. doi: 10.1186/1752-0509-3-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kashiwagi A., Kurosaki H., Luo H., Yamamoto H., Oshimura M., Shibahara T. Effects of Tritrichomonas muris on the mouse intestine: a proteomic analysis. Exp Anim. 2009;58:537–542. doi: 10.1538/expanim.58.537. [DOI] [PubMed] [Google Scholar]
- 129.Bogaerts A., Beets I., Temmerman L., Schoofs L., Verleyen P. Proteome changes of Caenorhabditis elegans upon a Staphylococcus aureus infection. Biol Direct. 2010;17(5):11. doi: 10.1186/1745-6150-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.LaCount D.J., Vignali M., Chettier R., Phansalkar A., Bell R., Hesselberth J.R. A protein interaction network of the malaria parasite Plasmodium falciparum. Nature. 2005;438:103–107. doi: 10.1038/nature04104. [DOI] [PubMed] [Google Scholar]
- 131.Navas A., Albar J.P. Application of proteomics in phylogenetic and evolutionary studies. Proteomics. 2004;4:299–302. doi: 10.1002/pmic.200300603. [DOI] [PubMed] [Google Scholar]
- 132.Predel R., Wegener C., Russell W.K., Tichy S.E., Russell D.H., Nachman R.J. Peptidomics of CNS-associated neurohemal systems of adult Drosophila melanogaster: a mass spectrometric survey of peptides from individuals’ flies. J Comp Neurol. 2004;474:379–392. doi: 10.1002/cne.20145. [DOI] [PubMed] [Google Scholar]
- 133.Brand S., Hahner D., Ketterlinus R. Protein profiling and identification in complex biological samples using LC-MALDI. Drug Plus Int. September 2005:6–8. [Google Scholar]
- 134.Nedelkov D. Population proteomics: investigation of protein diversity in human populations. Proteomics. 2008;8:779–786. doi: 10.1002/pmic.200700501. [DOI] [PubMed] [Google Scholar]
- 135.Cieslak A., Ribera I. Aplicaciones de proteómica en ecología y evolución. Ecosistemas. 2009;18:34–43. [Google Scholar]
- 136.Chevalier F., Martin O., Rofidal V., Devauchelle A.D., Barteau S., Sommerer N. Proteomic investigation of natural variation between Arabidopsis ecotypes. Proteomics. 2004;4:1372–1381. doi: 10.1002/pmic.200300750. [DOI] [PubMed] [Google Scholar]
- 137.Diz A.P., Skibinski D.O.F. Evolution of 2-DE protein patterns in a mussel hybrid zone. Proteomics. 2007;7:2111–2120. doi: 10.1002/pmic.200600954. [DOI] [PubMed] [Google Scholar]
- 138.Valcu C.M., Lalanne C., Muller-Starck G., Plomion C., Schlink K. Protein polymorphism between 2 Picea abies populations revealed by 2-dimensional gel electrophoresis and tandem mass spectrometry. Heredity. 2008;99:364–375. doi: 10.1093/jhered/esn007. [DOI] [PubMed] [Google Scholar]
- 139.Thiellement H., Bahrman N., Damerval C., Plomion C., Rossignol M., Santoni V. Proteomics for genetic and physiological studies in plants. Electrophoresis. 1999;20:2013–2026. doi: 10.1002/(SICI)1522-2683(19990701)20:10<2013::AID-ELPS2013>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 140.Pedersen K.S., Codrea M.C., Vermeulen C.J., Loeschcke V., Bendixen E. Proteomic characterization of a temperature-sensitive conditional lethal in Drosophila melanogaster. Heredity. 2010;104:125–134. doi: 10.1038/hdy.2009.132. [DOI] [PubMed] [Google Scholar]
- 141.Dorus S., Busby S.A., Gerike U., Shabanowitz J., Hunt D.F., Karr T.L. Genomic and functional evolution of the Drosophila melanogaster sperm proteome. Nat Genet. 2006;38:1440–1445. doi: 10.1038/ng1915. [DOI] [PubMed] [Google Scholar]
- 142.Nedelkov D., Kiernan U.A., Niederkofler E.E., Tubbs K.A., Nelson R.W. Investigating human plasma proteins diversity. Proc Natl Acad Sci USA. 2005;102:10852–10857. doi: 10.1073/pnas.0500426102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nedelkov D., Phillips D.A., Tubbs K.A., Nelson R.W. Investigation of human protein variants and their frequency in the general population. Mol Cell Proteomics. 2007;6:1183–1187. doi: 10.1074/mcp.M700023-MCP200. [DOI] [PubMed] [Google Scholar]
- 144.Nedelkov D., Tubbs K.A., Niederkofler E.E., Kiernan U.A., Nelson R.W. High- throughput comprehensive analysis of human plasma proteins: a step toward population proteomics. Anal Chem. 2004;76:1733–1737. doi: 10.1021/ac035105+. [DOI] [PubMed] [Google Scholar]
- 145.Nedelkov D. Mass spectrometry-based immunoassays for the next phase of clinical applications. Expert Rev Proteomics. 2006;3:631–640. doi: 10.1586/14789450.3.6.631. [DOI] [PubMed] [Google Scholar]
- 146.Nedelkov D., Kiernan U.A., Niederkofler E.E., Tubbs K.A., Nelson R.W. Population proteomics: the concept, attributes, and potential for cancer biomarker research. Mol Cell Proteomics. 2006;5:1811–1818. doi: 10.1074/mcp.R600006-MCP200. [DOI] [PubMed] [Google Scholar]
- 147.Nelson R.W., Krone J.R., Bieber A.L., Williams P. Mass-spectrometric immunoassay. Anal Chem. 1995;67:1153–1158. doi: 10.1021/ac00103a003. [DOI] [PubMed] [Google Scholar]
- 148.Nedelkov D. Population proteomics: addressing protein diversity in humans. Expert Rev Proteomics. 2005;2:315–324. doi: 10.1586/14789450.2.3.315. [DOI] [PubMed] [Google Scholar]
- 149.Connors L.H., Lim A., Prokaeva T., Roskens V.A., Costello C.E. Tabulation of human transthyretin (TTR) variants. Amyloid. 2003;10:160–184. doi: 10.3109/13506120308998998. [DOI] [PubMed] [Google Scholar]