

Abstract
An accurate virus diagnosis invariably requires laboratory testing of clinical specimens for the presence of virus, viral antigens, or specific antibodies. The past few decades have seen a major revolution in the operation of virus diagnostic laboratories and in their role in clinical patient management. Virus isolation has been largely replaced by sensitive nucleic acid detection assays and the measurement of specific antibodies at a very high level of sensitivity and specificity.
Keywords: Virus diagnosis, viral antigen, electron microscopy, nucleic acid hybridization, virus isolation, serum antibodies, serology
Arriving at an accurate virus diagnosis on clinical information alone is unreliable in most cases. Exceptions include the setting of known epidemics where large numbers of cases may present with a similar clinical picture; during the smallpox eradication campaign, photographs of the distinctive rash of smallpox were distributed to allow field workers to institute immediate containment measures. However, accurate virus diagnosis nearly always requires laboratory testing. The past few decades have seen a major revolution in the operation of virus diagnostic laboratories, and their role in the clinical management of patients has become vital. This has been driven by a number of factors:
-
1.
The development of molecular technologies and ways to make these accessible for diagnostic laboratories;
-
2.
The plethora of new antiviral agents and a more sophisticated understanding of how these should be used; and
-
3.
An increased awareness of the clinical value of, and demand for, prompt information about viral loads, viral sequence data, and antiviral resistance information.
The modern virus diagnostic laboratory is characterized by high test throughputs, rapid turnaround times, and a close liaison with clinical staff. Many of the older and slower diagnostic approaches such as animal inoculation, virus isolation in cell culture, and serological demonstration of a four-fold rise in antibody titer, are now of minor importance, if practiced at all.
Rationale for Performing Laboratory Virus Diagnosis
-
1.
The appropriate management of the patient depends on knowledge that follows from the diagnosis. Specific management does not always include, or stop at, chemotherapy. A rapid, specific, and accurate diagnosis can often avoid the need for further unnecessary tests, limit unnecessary or continuing antibiotic use (e.g., in cases of meningitis), ensure quicker management of patients, allow a more informed prognosis, and save on costs. Furthermore, specific active management measures may be indicated. The management of needlestick injuries to medical staff, screening of donated organs before transplantation, and application of blood isolation precautions for particular surgical procedures, are all highly dependent on the testing for blood-borne viruses. Cesarean section may be advisable if a woman has primary genital herpes at the time of delivery. Special care and education are required for a baby with congenital defects attributable to rubella or cytomegalovirus. Pregnancy termination is recommended if rubella is diagnosed in the first trimester of pregnancy.
-
2.
Use of antiviral agents usually requires accurate laboratory identification of the infecting virus. In some chronic diseases, for example, those caused by HIV and hepatitis B virus, decisions about when to initiate therapy require a full understanding of clinical and laboratory findings. Drug combinations appropriate for each patient are selected and later modified based on the drug resistance profile of the infecting virus strain. Results of virus load testing are used to monitor the response to therapy and guide changes to therapy.
-
3.
Infections may demand public health measures to prevent spread to others. For instance, blood banks routinely screen for HIV and hepatitis B and C viruses that may be present in blood donated by symptomless carriers. Since herpes simplex virus type 2 is readily transmissible to sexual partners, in some settings contact tracing helps protect sexual partners. Nosocomial infections (e.g., varicella, measles), often in epidemic form, may create havoc in a leukemia ward of a children’s hospital, unless hyperimmune IgG is promptly administered to potential contacts following diagnosis of the sentinel case. Documentation of a novel strain of influenza virus may herald the start of a major epidemic against which vulnerable older members of the community should be immunized. Positive identification of a particular arbovirus in a case of encephalitis enables authorities to promulgate warnings and initiate appropriate mosquito control measures. Introduction of a dangerous exotic disease demands containment and surveillance, and so on.
-
4.
Surveillance of viral infections may shed light as to the significance, natural history, and prevalence of a virus in the community, allowing control measures to be designed, control priorities to be established, and immunization programs to be monitored and evaluated.
-
5.
Continuous surveillance of a community may provide evidence of new epidemics, new diseases, new viruses, or new virus–disease associations. New viruses and new virus–disease associations continue to be discovered every year. It should be stressed that over 90% of all the human viruses known today were completely unknown at the end of World War II. Opportunities are legion for astute clinicians as well as pathologists, virologists, and epidemiologists to be instrumental in such discoveries.
The traditional approaches to laboratory diagnosis of viral infections have been (1) direct detection in patient material of virions, viral antigens, or viral nucleic acids, (2) isolation of virus in cultured cells, followed by identification of the isolate, and (3) detection and measurement of antibodies in the patient’s serum (serology). In recent decades direct detection methods, capable of providing a definitive answer in less than 24 hours, have undergone major advancement, whereas virus serology has become restricted to particular purposes only. Virus isolation is now used infrequently outside research or specialist areas. A summary of the major strengths and limitations of the several alternative approaches to the diagnosis of viral infections is given in Table 10.1 .
Table 10.1.
Advantages and Disadvantages of Various Diagnostic Methods
| Diagnostic Method | Advantages | Disadvantages/Problems |
|---|---|---|
| Virus isolation | Produces further material for study of agent | Slow, time-consuming, can be difficult and expensive |
| Usually highly sensitive | Selection of cell type, etc., may be critical | |
| “Open-minded” | Useless for non-viable virus or for non-cultivable agents | |
| Direct observation by electron microscopy | Rapid | Relatively insensitive |
| Detects viruses that cannot be grown in culture | Cumbersome for large numbers of samples | |
| Detects non-viable virus | Limited to a few infections | |
| “Open-minded” | ||
| Serological identification of virus or antigen, for example, EIA | Rapid and sensitive | Not applicable to all viruses |
| Provides information on serotypes | Interpretation may be difficult | |
| Readily available, often as diagnostic kits | Not as sensitive as PCR | |
| Targeted to a specific agent | ||
| Detection of viral genomes by PCR | Rapid, very sensitive | High sensitivity may lead to detection of non-relevant co-infections |
| Potentially applicable to all viruses incl. non-cultivable | Risk of DNA contamination | |
| Reagents (primers) for additional viruses easily made | Needs good quality control | |
| Good quantitation of load | Targeted to a specific agent | |
| Can be multiplexed | ||
| Antibody seroconversion (acute and convalescent sera) | Useful if appropriate samples for direct detection cannot be obtained, or to exclude a particular infection retrospectively | Slow, late (retrospective) |
| Interpretation may be difficult | ||
| Targeted to a specific agent | ||
| IgM serology | Rapid | False positives may occur |
| Targeted to a specific agent |
The best diagnostic methods should satisfy the criteria of: speed, simplicity, sensitivity, specificity, and cost. Standardized diagnostic reagents of reliable quality are widely available commercially, assays have been miniaturized to conserve reagents, and instruments have been developed to automate the barcoding and dispensing of samples, diluting and rinsing, reading the tests, and computerized analysis and delivery of the results. Moreover, a veritable cascade of commercial kits for nucleic acid tests, antigen and antibody assays is now available, together with the relevant equipment to allow automated high-throughput assays and ongoing quality assurance. Solid-phase enzyme immunoassays (EIAs) and real-time PCR technology, in particular, have revolutionized diagnostic virology and are now methods of choice for a wide range of indications.
Collection, Packaging, and Transport of Samples
The chance of successful diagnosis by virus isolation or direct detection of virus depends critically on the attention given by the attending physician to the collection of the sample. Clearly, the clinical sample must be taken from the right place at the right time. The right time is as soon as possible after the patient first presents, because virus is usually present in maximum concentration at about the time symptoms first develop, and then declines during the ensuing days. Samples taken as a last resort after days or weeks of empirical treatment (e.g., antibiotic therapy) are almost invariably useless.
The site from which the specimen is collected will depend on the clinical symptoms and signs, together with knowledge of the pathogenesis of the suspected disease. As a general rule the epithelial surface that constitutes the portal of entry and the primary site of viral replication is usually the best site for obtaining samples (Table 10.2 ). The key specimen in respiratory infections as well as in many generalized infections, is a nasal or throat swab, or in the case of a young child a nasopharyngeal aspirate in which mucus is drawn from the back of the nose and throat into a mucus trap using a vacuum pump. A sample of feces is also essential in enteric and many generalized infections of the gastrointestinal tract.
Table 10.2.
Specimens Appropriate for Laboratory Diagnosis of Various Clinical Syndromes
| Syndrome | Specimen |
|---|---|
| Respiratory | Nasal or throat swab; nasopharyngeal aspirate; sputum |
| Enteric | Feces |
| Genital | Genital swab, urine |
| Eye | Conjunctival (and/or corneal) swab |
| Skin | Vesicle fluid/swab/scraping; biopsy solid lesion |
| Central nervous system | Cerebrospinal fluid; feces (enteroviruses) |
| Generalized | Throat swaba; fecesa; blood leukocytesa |
| Autopsy/biopsy | Relevant organ |
| Any | Blood for serologyb |
Depending on known or presumed pathogenesis.
Blood is allowed to clot, then serum kept for assay of antibody.
Swabs may be taken from the genital tract, from the eye, or from vesicular skin lesions. Some viruses responsible for systemic infections can be isolated from blood (plasma or leukocytes). Cerebrospinal fluid (CSF) may yield virus in cases of meningitis or encephalitis. Biopsy or autopsy specimens may be taken by needle or knife from any part of the body for virus isolation, or snap-frozen for immunofluorescence. Obviously, tissue taken at autopsy or biopsy for the purpose of virus isolation must not be placed in formalin or any other fixative. In the case of many generalized viral diseases it may not be obvious as to what specimen is required. As a rough working rule it can be said that at a sufficiently early stage in the disease, virus can usually be isolated from a throat swab, feces, or blood. In all cases, it is essential to capture sufficient patient material on the (preferably moistened) swab; negative diagnoses are to be expected if a medical attendant fails to collect enough material from the patient.
Because of the lability of many viruses, specimens intended for virus isolation must always be kept cold and moist. Immediately after collection the swab should be swirled around in a small screw-capped bottle containing virus transport medium. This medium consists of a buffered balanced salt solution, to which has been added protein (e.g., gelatin or albumin) to protect the virus against inactivation, and antibiotics to prevent the multiplication of bacteria and fungi. (If it is at all probable that the specimen will also be used for attempted isolation of bacteria, rickettsia, chlamydia, or mycoplasmas, the collection medium must not contain antibiotics—the portion used for virus isolation can be treated with antibiotics later.) The swab stick is then broken off aseptically into the fluid, the cap is tightly fastened and secured with adherent tape to prevent leakage, and the bottle is labeled with the patient’s name, date of collection, and nature of specimen. This is then dispatched immediately to the laboratory, accompanied by a completed laboratory request form which must include an adequate clinical history, a provisional diagnosis, request for a particular test, and the date of onset of the illness.
If a transit time of more than an hour or so is anticipated the container should be sent refrigerated (but not frozen), with cold packs (4°C) or ice in a thermos flask or styrofoam box. International or interstate transport of specimens, particularly in hot weather, generally requires that the container be packed in dry ice (solid CO2) to maintain the virus in a frozen state. Governmental and International Air Transport Association (IATA) regulations relating to the transport of biological materials require precautions such as double-walled containers with absorbent padding in case of breakage. Permits must be obtained from the appropriate authorities for interstate and international transportation. Especially, hazardous samples are subject to additional regulations, differing in different countries.
Nucleic acid tests have revolutionized diagnostic virology. The sensitivity of nucleic acid assays means that previously unsuitable specimens can now be used (e.g., deep nasal swabs or throat swabs can be examined for respiratory viruses instead of requiring nasal pharyngeal aspirates or tracheal aspirates); in addition, the quality of the collection is less important (self-collection is suitable for human papilloma virus testing) and transport is less critical as the nucleic acid in viral particles is relatively stable (e.g., respiratory samples in VTM can be sent through the mail, and dried blood spots can be used). Nucleic acid tests have increased the range of detected viruses, but the down side is that testing is specific for the suspected virus, with the consequence that variants may be missed unless the assays are designed to be inclusive.
Direct Identification of Virus, Viral Antigen, or Viral Genome
Direct detection of virus material in a patient sample has become the method of choice for a very large number of different infections. These methods have the advantage of speed, as they do not rely on virus culture or a rise in antibody titer. The sensitivity and specificity of antigen detection methods have increased greatly through use of high-quality reagents and solid phase reagent supports allowing ease of washing and signal detection; the sensitivity of nucleic acid detection has advanced from early dot blot and Southern blot assays to the widespread and reliable use of PCR.
Direct Detection of Virions by Electron Microscopy
The morphology of most viruses is sufficiently characteristic to allow assigning many viruses to the correct family by appearance in the electron microscope. Moreover, viruses that have never been cultured may nevertheless be recognized. During the 1970s electron microscopy was the means to the discovery in feces of several new groups of previously non-cultivated viruses. The human rotaviruses, caliciviruses, astroviruses, hepatitis A virus, and previously unknown types of adenoviruses and coronaviruses were all initially identified in this way.
The most widely used procedure is negative staining, where virus-containing fluid is placed on a carbon or formvar-coated grid usually with prior clarification and concentration by ultracentrifugation; virions adhere to the surface and become “negatively stained” when an electron-dense fluid such as sodium phosphotungstate is added and surrounds the virions. There are several variations on the method, mainly to remove excess salts and proteins which affect the translucency of the specimen in the microscope. Characteristic virion size and structural details provide useful information toward virus identification when routine diagnostic approaches fail. Pre-incubation of the virus specimen with specific antibody can be used to assist in virus identification—antibody-coated virions appear fuzzier than controls (see Fig. 33.1), and the procedure (immune electron microscopy) can also be used for quantitating specific antibody.
A second procedure is thin-section electron microscopy of fixed tissue sections. This is used particularly in a research setting, for example, where viral-like inclusions suggest a viral etiology or where the pathology of a particular disease is being investigated.
The biggest limitation of electron microscopy as a diagnostic tool is its low sensitivity; specimens need to contain at least 106 virions per milliliter or milligram for there to be any chance to detect the virus by electron microscopy. Such levels are often surpassed in feces and vesicle fluid, but not in respiratory mucus. The method can be quick for one or several samples only, but is impractical for large batch testing.
Detection of Viral Antigens
A great variety of different immunological assays can be used to detect viral antigens in patient samples, using well-characterized reagents containing known antibody specificities. Where the antigen-containing reagent is defined, these same assay techniques can be used similarly to detect unknown antibodies (see below).
Enzyme Immunoassay (EIA)
The introduction of EIA, also known as ELISA (for enzyme-linked immunosorbent assay), revolutionized diagnostic virology in the days before the development of polymerase chain reaction (PCR), and still has widespread specific uses. EIAs can be designed in different formats to detect antigen or antibody. The high sensitivity of the method means that less than 1ng of viral antigen per milliliter can be detected in specimens taken directly from a patient.
The development of solid-phase assays was a major technical advance; the available test configurations are almost limitless, and a wide variety of direct, indirect, and reversed assays have been developed for diagnosing viral infections. The critical principle is that all the components making up the layers of the reaction are known and defined, with the exception of the “unknown” component that may be present in the specimen. For the solid-phase format, the “capture” antibody can be attached (by simple adsorption or by covalent bonding) to a solid substrate, typically the wells of polystyrene or polyvinyl microtiter plates, or polystyrene beads, this format aiding considerably the rinsing of the solid surface between applications of reagents.
The simplest format is the direct EIA (Fig. 10.1 ). Virus and soluble viral antigens from the specimen are allowed to adsorb to the captured antibody. After unbound antigen has been washed away, an enzyme-labeled antiviral antibody (the “detector” antibody) is added. (Various enzymes can be linked to antibody; horseradish peroxidase and alkaline phosphatase are the most commonly used.) After a final washing step, readout is based on the color change that follows addition of an appropriate organic substrate for the particular enzyme. The colored product of the action of the enzyme on the substrate should be clearly seen by eye. The test can be made quantitative by serially diluting the antigen to obtain an endpoint, or by using spectrophotometry to measure the amount of enzyme-conjugated antibody bound to the captured antigen. Sensitivity can be enhanced using the high binding affinity of avidin for biotin; antibody is conjugated to biotin, a reagent of low Mr that gives reproducible labeling and does not alter the antigen-binding capacity of the detector antibody. The antigen–antibody complex is identified by adding avidin-labeled enzyme, followed by enzyme substrate (Fig. 10.3, right). To further increase sensitivity, high-energy substrates are available which release fluorescent, chemiluminescent, or radioactive products that can be identified in very small amounts.
Figure 10.1.

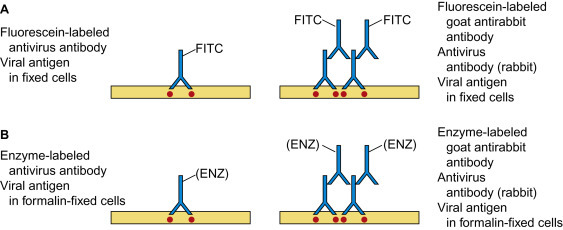
Enzyme immunoassays (EIA or ELISA) for detection of virus and/or viral antigen. Left: Direct method. Right: Indirect method, using biotinylated antivirus antibody, followed by enzyme- (e.g., peroxidase)- labeled avidin. In each case an enzyme substrate is then added to develop a color reaction. Note the immobilization of the capture antibody on a solid support to facilitate subsequent washing steps.
Reproduced from MacLachlan, N.J., Dubovi, E.J., 2011. Veterinary Virology, fourth ed., Academic Press, with permission.
Figure 10.3.
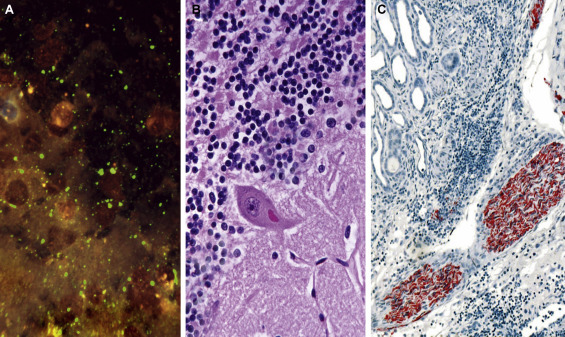
Diagnosis of rabies. (A) Direct immunofluorescence on impression smear of fox brain (possible human contact), showing prominent brilliant apple-green masses of viral antigen (Negri bodies of light microscopy) and punctate “dust,” against a dark background. FITC-labeled anti-rabies globulin, UV microscopy. (B) Histopathology on human postmortem brain section. Finding Negri bodies (shown here in the cytoplasm of a Purkinje cell in the cerebellum) often requires careful searching. H&E. (C) Immunohistochemistry used in rabies research and in special diagnostic situations, for example, for postmortem diagnosis in patients infected via organ transplants. Here viral antigen fills the axoplasm of peripheral nerves. Human rabies, biotin-conjugated anti-rabies globulin, avidin-alkaline phosphatase, and naphthol fast-red method.
Indirect immunoassays are widely used because of a somewhat greater sensitivity and the avoidance of the need to label each antiviral antibody in the laboratory repertoire. In one protocol, the detector antibody is unlabeled, and a further layer, labeled (species-specific) anti-immunoglobulin, is added as the “indicator” antibody; of course, the viral antibodies constituting the capture and detector antibodies must be raised in different animal species. Alternatively, the detector antibodies can be labeled with biotin and a positive reaction indicated using enzyme-labeled avidin. In another method, labeled staphylococcal protein A, which binds to the Fc moiety of IgG of most mammalian species, can be used as the indicator in indirect immunoassays.
Monoclonal viral antibodies (mAbs) are frequently used in immunoassays as detector antibodies, but the use of mAbs as capture antibodies is limited because of the restricted epitope repertoire recognized by such cloned antibody molecules and thus antigen capture is less stable. The obvious advantages of mAbs are that these represent the well-characterized, purified, monospecific antibody of a defined class, recognizing only a single epitope, as well as being free of “natural” and other extraneous antibodies against host antigens or adventitious agents. Monoclonal antibodies also make a vital contribution as reference reagents, being easily manufactured in large quantities. It is important to select mAbs of high affinity, but not of such high specificity that some strains of the virus being sought might be missed in the assay. Indeed, the specificity of the assay can be pre-determined by selecting a mAb directed at an epitope that is either confined to a particular viral serotype or common to all serotypes within a given species or genus.
Radioimmunoassay and Time-Resolved Fluoroimmunoassay
Radioimmunoassay (RIA) predates EIA. The only significant difference is that the label is not an enzyme but a radioactive isotope such as 125I, and the bound antibody is measured in a gamma counter. RIA is a highly sensitive and reliable assay that lends itself well to automation, but the cost of the equipment and the health hazard of working with radioisotopes argue against its use in small laboratories.
Time-resolved fluorescence immunoassay (TR-FIA) is a non-isotopic immunoassay, in which the indicator antibody is labeled with a fluorophore (a europium chelate). Following excitation by light, the fluorophore emits fluorescence of a different wavelength that can be measured in a time-lapse fluorometer. The method is as sensitive as RIA and has the advantage of a stable fluorophore, but it requires costly equipment that can be contemplated only by laboratories with a large throughput of specimens.
Latex Particle Agglutination
Perhaps the simplest of all immunoassays is the agglutination by antigen of small latex beads previously coated with antiviral antibody. The test can be read by eye within minutes. Not surprisingly, diagnostic kits based on this method have become popular with small laboratories and with medical practitioners. However, these assays suffer from both low sensitivity and low specificity. Thus, false negatives occur unless large numbers of virions are present, and therefore this assay for antigen tends to be restricted to examination of feces. False positives, on the other hand, can occur commonly with fecal specimens.
Immunochromatography
Immunochromatography or lateral flow tests involve the migration of an antigen, or antigen–antibody complexes, through a support, for example, nitrocellulose film, filter paper, or agarose. In a typical format, a labeled antibody is allowed to react with the unknown antigen, the antigen–antibody complex migrates by capillary action through the solid support, and a second antibody embedded in the solid support allows detection of the complexes if antigen is present. Positive and negative controls are included to ensure that individual tests are valid. Immunochromatography tests are available for measuring viral antigens such as HIV p24, dengue NS1, influenza A and B, RSV, etc. and are of particular value for rapid point-of-care testing where rapid results are needed and access to equipment is limited.
Detection of Antigen in Tissues
Viral (or non-viral) antigens can be detected in fixed or frozen tissue sections, or in exfoliated cells, using the same principles as above, where the cells or tissues on a glass slide fill the role of the solid support. Commonly used indicators include fluorescein, horseradish peroxidase, and alkaline phosphatase. The approach can be valuable in virus diagnosis, for example, by detecting viral antigens in exfoliated cells or tissue samples, and also in studying disease pathogenesis should the presence of viral material be matched to particular organs or histopathology.
Once antibody is labeled with a fluorochrome, the antigen–antibody complex emits light of a particular longer wavelength when excited by short wavelength light. This can be visualized as fluorescence in an ordinary microscope after light of all other wavelengths is filtered out. The sensitivity of the method is generally too low to detect complexes of fluorescent antibody with virions or soluble antigen; hence, the antigen in the test typically takes the form of virus-infected cells. There are two main variants of the technique.
Direct Immunofluorescence
For direct immunofluorescence, a frozen tissue section, or an acetone-fixed cell smear, or monolayer on a coverslip, is incubated with fluorescein-tagged antiviral antibody (Fig. 10.3, A, left). Unbound antibody is then washed away, and the cells are viewed by light microscopy using a powerful ultraviolet/blue light source. The apple-green light emitted from the specimen is revealed (against a black background) by incorporating filters into the eyepieces so that all the blue and ultraviolet incident light is adsorbed (Fig. 10.2 ).
Figure 10.2.
Detection of viral antigens in tissues or cell smears by immunofluorescence (A) and immunohistochemistry (B). In each case, the direct method is shown on the left, and the indirect method on the right.
Reproduced from MacLachlan, N.J., Dubovi, E.J., 2011. Veterinary Virology, fourth ed., Academic Press (Figure 5.6), with permission.
Indirect Immunofluorescence
Indirect (“sandwich”) immunofluorescence differs in that the antiviral antibody is untagged, but fills the role of the meat in the sandwich. It binds to antigen and is itself recognized by fluorescein-conjugated anti-immunoglobulin (Fig. 10.3 , A, right). The high affinity of avidin for biotin can also be exploited in immunofluorescence, by coupling biotin to antibody and fluorescein to avidin.
In the diagnostic setting, immunofluorescence has proved to be of great value in the early identification of viral antigens in infected cells taken from patients with diseases known to have a relatively small number of possible etiological agents. There is little difficulty in removing partly detached infected cells from the mucous membrane of the upper respiratory tract, genital tract, eye, or from the skin, simply by swabbing or scraping the infected area with reasonable firmness. Cells are also present in mucus aspirated from the nasopharynx; aspirated cells must be extensively washed by centrifugation to remove mucus before fixation and staining. Respiratory infections with paramyxoviruses, orthomyxoviruses, adenoviruses, and herpesviruses are particularly amenable to rapid diagnosis by immunofluorescence. Immunofluorescence can also be applied to infected tissue, for example, brain biopsies for the diagnosis of such lethal diseases as herpes simplex encephalitis or measles SSPE, or at necropsy, for the verification of rabies in the brain of animals captured after biting humans (Fig. 10.3).
Immunoperoxidase Staining
An alternative method of locating and identifying viral antigen in infected cells is to use antibody coupled to horseradish peroxidase; subsequent addition of hydrogen peroxide together with a benzidine derivative forms a colored insoluble precipitate. The advantages of the method are that the preparations are permanent and only an ordinary light microscope is needed. Endogenous peroxidase, present in a number of tissues, particularly leukocytes, can produce false positives, but this problem can be circumvented by proper technique and adequate controls. Other enzymes, for example, alkaline phosphatase, can also be used as indicator systems (Fig. 10.4 ).
Figure 10.4.

Immunohistochemistry showing West Nile virus antigens in a brain tissue section from a fatal case of encephalitis. Cortical neurons are particularly involved. Paraffin-embedded section. Biotin-conjugated West Nile virus specific hyperimmune globulin conjugated with avidin-alkaline phosphatase; naphthol fast-red method. Counterstained with hematoxylin.
Reproduced from Sherif Zaki, U.S. Centers for Disease Control and Prevention, with permission.
Detection of Viral Nucleic Acids
Nucleic acid detection is now widely applied by use of polymerase chain reaction (PCR) assays, combined with advances in oligonucleotide synthesis, standardized automated procedures for nucleic acid extraction, and real-time detection of PCR products. Rapid advances in nucleic acid sequencing technology and the availability of sequence databases have greatly enhanced analysis of the results obtained. Isothermal RNA and DNA amplification assays are also readily available. Signal amplification assays were used previously but have been superseded.
Nucleic Acid Hybridization
From the time of the development of the first nucleic acid hybridization techniques, a variety of test formats were applied to viral nucleic acid detection. Early approaches usually involved a hybridization reaction between an immobilized nucleic acid target and a labeled probe, washing away unbound probe, followed by subsequent detection of bound probe. These include dot-blot assays using nucleic acid-containing samples immobilized onto filters; Southern blot hybridization, where viral nucleic acids are separated by electrophoresis according to molecular weight, blotted to a filter and detected by hybridization; and in situ hybridization applied to infected tissue sections or exfoliated cells. Real-time PCRs nowadays often use hybridization in solution as part of the product detection strategy.
Traditionally, radioactive isotopes such as 32P and 35S were used to label nucleic acids or oligonucleotides used as probes for hybridization tests, with the signal being read by counting in a spectrometer or by autoradiography. These have been largely replaced by non-radioactive labels. Some of these (e.g., fluorescein or peroxidase) produce a signal directly, whereas others (e.g., biotin or digoxigenin) act indirectly by binding another labeled ligand that then emits the signal. Biotinylated probes can be combined with various types of readouts, for example, an avidin-based EIA. Chemiluminescent substrates, such as luminol, have also been widely exploited.
Polymerase Chain Reaction
PCR (Fig. 10.5 ) constitutes one of the greatest advances in molecular biology. It enables a single copy of any gene sequence to be enzymatically amplified in vitro at least a million-fold within a few hours. Thus viral DNA extracted from a very small number of virions or infected cells can be amplified to the point where it can be readily identified. PCR can also be used to detect viral RNA by including a preliminary step in which reverse transcriptase is used to convert RNA to DNA. It is not necessary or usual to amplify the whole genome, but it is necessary to know at least sufficient nucleotide sequence in order to synthesize two oligonucleotide primers, usually about 20 residues in length, that hybridize to opposite strands of the target DNA and flank the region one chooses to amplify. The two primers (sometimes known as “forward” and “reverse” primers) provide the DNA polymerase with an initiation point to which additional nucleotides can be attached, and also attach the reaction to the specific DNA target region. Primers can be synthesized containing attached ligands or molecular tags, thereby generating tagged DNA product molecules to facilitate further analysis. Computer programs can be used to design optimum primer sets and to predict optimal PCR reaction conditions, for example, time/temperature/ionic conditions. Where the target might be expected to show variability at a particular site, “degenerate” primers containing different bases at that site can be synthesized to ensure all variants are detected.
Figure 10.5.
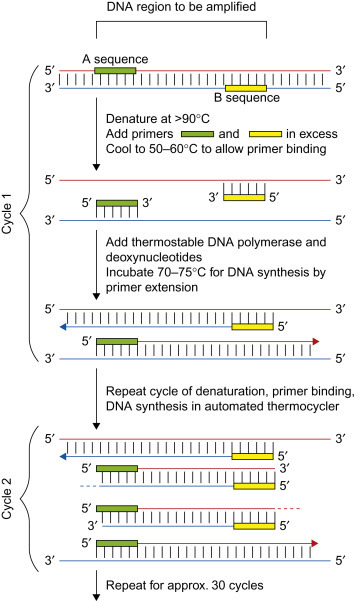
Principle of DNA sequence amplification using the polymerase chain reaction. The method relies on thermal cycling, that is, cycles of repeated heating and cooling to melt and re-anneal DNA along with enzymatic replication of the targeted melted strands of DNA. Primers (short DNA fragments) containing sequences complementary to each end of the target region of the DNA of interest must first be synthesized. Then, the two strands of the DNA double helix are physically separated at a high temperature in a process called DNA melting. In the second step, the temperature is lowered and the two DNA strands become templates for DNA synthesis. In the presence of heat-resistant DNA polymerase and deoxynucleotide triphosphates, two new copies of the desired region of the DNA are produced. As PCR progresses through cycles of melting, annealing, and extension, the DNA generated is itself used as a template for replication, setting in motion a chain reaction in which the DNA template is exponentially amplified. After the first few cycles, virtually all the templates consist of just the short region chosen for amplification by the choice of primers. After 30 cycles, this region has been amplified many million fold. Many different variations of the PCR method have been developed to perform a wide array of genetic manipulations.
Courtesy of Holmes, I.H., Strugnell, R., reproduced from MacLachlan, N.J., Dubovi, E.J., 2011. Veterinary Virology, fourth ed., Academic Press, with permission.
The process is carried out under carefully controlled conditions of temperature, ionic strength, primer concentration, and nucleotide concentration. There are three main steps: (1) melting the target DNA at 95°C, (2) cooling to around 50–60°C to allow binding of two oligonucleotide primers, and (3) synthesis by extension from the oligonucleotide primers of two DNA strands located between and including the two primers, thus generating complementary copies of the original (plus and minus) target strands. The synthesis reaction is catalyzed by DNA polymerase added to the reaction. The primer extension products from the first cycle act as templates in a second reaction, and the cycle of melting, primer binding, and primer extension are repeated many times. The number of DNA copies increases exponentially. Thus after 30 cycles the number of DNA copies, beginning with a single copy of the target sequence, is theoretically 230 or 1 billion, and in practice over 1 million can be achieved. Forty cycles can be completed in less than an hour depending on reaction volumes and method, and by a suitable automated thermal cycling device. The amplified DNA may be detected either as a stained band of correct molecular weight on agarose gel electrophoresis, or by hybridization with labeled DNA or RNA probes culminating in any of a wide variety of readout systems. However the development of real-time detection systems (see below) has revolutionized the speed and quantitative accuracy of the method and greatly helped reduce false-positive reactions due to contamination.
Selection of the most suitable pair of primers is a matter of critical importance. Each may be chosen as highly specific for a particular virus strain or, alternatively, to represent consensus sequences within a gene known as conserved within a given family or genus. By probing for a conserved gene, such as that for RNA polymerase, it is even possible to discover a previously unknown agent. Reactions must be carried out under very carefully controlled conditions of ionic strength, temperature, primer concentration, and nucleotide concentration. Deviations can result in non-specific amplification.
PCR technology has undergone numerous major developments since its first description in 1983. These include:
-
1.
Use of the heat-stable DNA polymerase (Taq) of Thermus aquaticus, an organism naturally found in hot springs. Since this makes it no longer necessary to replenish the enzyme between cycles, the reaction tubes can remain sealed, and it also allows the use of higher annealing temperatures thereby increasing reaction specificity by reducing false-positive reactions due to mismatching;
-
2.
Use of “nested” PCR reactions, whereby a second amplification is carried out with primers internal to the initial target after the initial PCR amplification of a target sequence. This can greatly increase sensitivity, albeit at the potential cost of increasing the risk of false- positive reactions;
-
3.
Due to the detection of trace amounts of DNA sequence with extreme sensitivity, early PCR results suffered from regular problems of environmental contamination with DNA products leading to false-positive results. The procedure developed a reputation for unreliability, and laboratories went to heroic efforts to prevent reaction contamination, including stringent physical segregation of the different steps in the procedure. This has become far less of a problem with recent advances, in particular the use of real-time detection methods;
-
4.
Real-time PCR testing. This method for detecting PCR products required the development of a thermocycler with an inbuilt fluorimeter allowing measurement of newly synthesized PCR products in the reaction tube as were accumulating, that is, in real time. The reaction products can be detected by dyes that fluoresce when bound non-specifically to any double-stranded DNA, for example, SYBR Green; or by sequence-specific reporter probes (EgTaqMan) containing a fluorescent reporter at one end and a fluorescence quencher at the other, that bind specifically to newly synthesized target sequences leading to loss of quenching and emission of a quantitative fluorescence signal. As the reaction progresses, the intensity of the fluorescent signal is continually monitored and plotted, generating curves the position of which reflects the original concentration of target. This approach has largely replaced other methods for product detection because it does not require the reaction tube to be opened once the reaction has started, thereby minimizing contamination, it achieves excellent quantitative measurements, is more sensitive than earlier detection systems, and if a sequence-specific reporter probe is used it has improved specificity because only the target sequence and no other non-specific DNA molecules is detected;
-
5.
Development of more accurate quantitation. Real-time detection methods allow generation of standard curves the position of which corresponds to the original concentration of target, and an unrelated copy number control can be included in the reaction to detect any non-specific inhibition of the enzyme reaction. With a number of infections, the clinical interpretation and management are now dependent on regular quantitation of the patient’s “viral load”;
-
6.
Use of multiplex PCR. Once different fluorescent probes became available for PCR product detection, it became possible to include different sets of primers bearing different fluorescent labels for different targets in the same reaction, and thereby to detect a number of different products simultaneously. This is exploited in a number of standard protocols for specific sites, for example, respiratory virus infections (Fig. 10.6 ).
Figure 10.6.
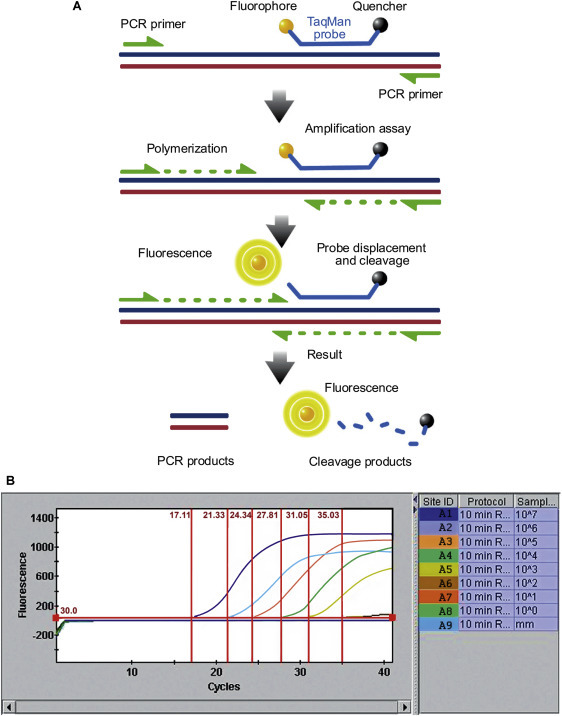
(A) TaqMan probe chemistry mechanism. These probes rely on the 5′-3′ nuclease activity of Taq DNA polymerase to cleave a dual-labeled probe during hybridization to the complementary target sequence. (B) Real-time quantitative PCR data. Reaction curves for a test run are used to assess assay conditions, using dilutions of an RNA transcript (copy number control) of a cloned segment of canine pneumovirus. The vertical lines represent the Ct value, which is the number of PCR cycles required for the fluorescent signal to cross the threshold value. TaqMan1 probe was labeled with FAM (6-carboxy fluorescein; the reporter dye) at the 5′ end and BHQ (Black Hole Quencher; the quencher) at the 3′ end.
Reproduced from MacLachlan, N.J., Dubovi, E.J., 2011. Veterinary Virology, fourth ed., Academic Press, with permission.
Isothermal amplification methods are variations of PCR that do not require high (95°C) temperature cycling to melt the newly synthesized DNA product to allow binding of a new round of primers. These methods rely on enzymes to displace the strands and include Loop Mediated Isothermal Amplification (LAMP), Strand Displacement Amplification (SDA), Helicase-Dependent Amplification (HAD), and Nicking Enzyme Amplification Reaction (NEAR). These methods have the potential advantage in being extremely fast and do not require thermal cycling equipment. Several examples are available as approved diagnostic tests.
Due to the speed, sensitivity, and versatility of PCR methods for detecting and quantifying viral genomes, this approach is now a significant part of the routine work of the diagnostic laboratory, and many commercial test systems are available. Methods such as antigen detection and virus isolation tend to require different reagent preparation expertise and methods specific for each individual virus, whereas PCR technology is more universally applicable because its specificity relies primarily on the chosen primer sequences. The procedure is invaluable when dealing with (1) viruses that cannot be cultured satisfactorily, (2) specimens that contain predominantly inactivated virus, as a result of prolonged storage or transport, (3) where quantitative results are needed, or (4) to avoid exposing laboratory staff to culturing larger quantities of a biohazard category 3 or 4 agent. Furthermore, it is easy to make new PCR reagents for new viruses, and thereby rapidly expand the capacity for testing.
Problems that may be encountered with PCR include (1) some clinical samples that may contain factors that inhibit the activity of the DNA polymerase, leading to falsely low or negative results. This can be detected by testing in parallel a sample control that contains a standard unrelated DNA target or “copy number control,” (2) the possibility of contamination of the reaction with extraneous DNA not originating from the patient although nowadays it is much less of an issue, (3) as with all methods that are specific for a particular virus or viruses (in contrast to “open-minded” procedures such as virus isolation or electron microscopy), PCR will only detect those agents that are recognized by the primer set and test conditions. Thus, variant viruses may be missed if these sequences are not amplified or the sequences amplified are not detected in the product assay system used, (4) the extreme sensitivity of PCR means that low levels of chronic, latent, or concurrent infecting genomes unrelated to the patient’s clinical condition, may be detected.
As with all diagnostic procedures, before finally ascribing a laboratory finding to a patient’s condition, the patient’s history and physical findings, knowledge of disease pathogenesis, and clinical acumen, must all be brought into play. For example, varicella zoster virus DNA has been detected as a contaminant in a genital herpes simplex virus infection—the PCR giving a strong HSV2 but weak VZV reaction because of the primers used.
Microarray Technologies
Another technological advance that is impacting the field of diagnostics is the use of microarrays or microchips for nucleic acid detection. The microchip is a solid support matrix onto which have been “printed” spots, each containing one of several hundred to several thousand unique oligonucleotides. These oligonucleotides can represent conserved sequences from virtually all viruses represented in the various genetic databases, or can be customized to represent only viruses from a given specific disease syndrome, such as acute respiratory disease in children. The basis of the technology is the capture by these oligonucleotides of randomly amplified labeled nucleic acid sequences from clinical specimens. The binding of a labeled sequence is detected by laser scanning of the chip and software programs that assess the strength of the binding. From the map position of the reacting oligonucleotides, the software identifies the virus in the clinical sample. This type of test was used to initially determine that the virus responsible for severe acute respiratory syndrome (SARS) was a coronavirus. With knowledge of the oligonucleotide sequences that bind an unknown virus, primers can be made to eventually determine the entire nucleotide sequence of the virus. The low cost of oligonucleotide synthesis, development of laser scanning devices, nucleic acid amplification techniques, and software development have made this technology available in specialized reference laboratories, increasingly more common in national diagnostic laboratory networks.
Microarray technology has been used as a research tool for some years, for example, for studying comprehensive gene expression in cells and tissues. It is now being applied increasingly for monitoring host responses to infections and for assessment of the development of antiviral drug resistance. It has been used to map immune response gene expression in hepatitis C-infected liver tissue, and changes in leukocyte gene expression in patients with sepsis. It is now also being directly applied in the diagnostic arena. For example, commercial microarrays are available for the simultaneous detection of a large panel of respiratory viruses, by reacting the product of a multiplex PCR and RT-PCR reaction with a solid chip or liquid bead platform carrying oligonucleotide probes for multiple viruses. Similar approaches have been described for the simultaneous detection and typing of human papillomavirus sequences in routine clinical specimens. The transfer of this technology to the routine diagnostic laboratory still presents challenges, not least due to the requirements for a rapid turn-around, high-throughput, quality-assured procedure for the diagnostic setting, and the unpredictability and heterogeneity of clinical samples. Nevertheless it is likely that application of microarray technology will eventually lead to another transformation of the diagnostic laboratory.
Next-Generation Deep Sequencing
“Deep” sequencing, or “next-generation” sequencing refers to novel techniques of DNA sequencing directly from DNA fragments without the need for cloning in vectors, allowing the generation of enormous amounts of sequence data at high speed and low cost from a single run. A number of systems are commercially available, the approach has yielded enormous benefit in various research fields, and its application to diagnostic virology is now being explored. Different platforms have different advantages and pitfalls, for example, differences in read length and read depth, and access to a capable bioinformatics resource is essential to ensure that data analysis can be done appropriately. For an unbiased or broad-based analysis, all nucleic acids in a clinical sample can be sequenced on a multiplex platform; however, because host sequences can frequently outnumber infecting virus sequences by 105 or more, prior selective enrichment of virus sequences can be achieved, for example, by hybrid capture using PCR-derived capture probes, by prior PCR amplification of suspected virus sequences in the unknown target sample, or by physical methods of partial purification, for example, based on the nuclease resistance of encapsidated viral genomes.
This approach is already being applied to many purposes in virology, for example:
-
1.
Discovering new pathogens in undiagnosed illness or outbreaks (e.g., the coronavirus causing Middle East respiratory syndrome [MERS] first reported in Saudi Arabia in 2012), the novel highly divergent rhabdovirus Bas-Congo virus (BASV), and the novel polyoma viruses HPYV9 and Merkel cell polyomavirus (MCPyV);
-
2.
Retrospective diagnosis of undiagnosed illness, for example, encephalitis, using stored autopsy samples;
-
3.
Screening vaccines for contaminants;
-
4.
Analysis of the quasi-species sequence composition of the viruses in a clinical sample, including detection of minor variants with new pathogenic implications, for example, drug resistance;
-
5.
Investigation of the diversity and evolution of particular viral genomes;
-
6.
Studies of the human virome in health and disease.
Full application of deep sequencing technology to virus diagnosis has not yet been achieved, and deep sequencing methods are likely to complement existing methods rather than replace them. Costs have plummeted enormously, now allowing the technology to move from large institutions to the more widespread diagnostic setting. However many development challenges remain, including the huge computing and data analysis requirements that are created by the generation of enormous amounts of data.
Virus Isolation
In recent years, virus isolation has changed from its role as the “gold standard” of the diagnostic laboratory, to occupying more of a research or historical role. Newer techniques, particularly sensitive antigen EIAs and PCRs, do not suffer from the drawbacks of virus isolation that included high cost, slowness, false negatives due to virus being inactivated during transport or complexed with antibody, cell lines losing sensitivity, and increased concerns about live virus transmission to staff. Many diagnostic laboratories no longer try to isolate viruses in cell culture; virus isolation in cell culture is mostly limited to research laboratories or where there is a need to grow up large amounts of virus. Even bulk antigen production is now often done preferentially by expression of recombinant antigens, and unknown agents can be sought using molecular techniques such as random-priming PCR.
Similarly, techniques for animal inoculation, for example, suckling mice, and egg inoculation procedures, are only performed where a laboratory has a specialist need, for example, influenza virus to be used in vaccine production is grown in embryonated hens’ eggs.
For successful virus isolation in cell culture, the following steps are followed.
-
1.
Care must be given to maintaining a cold chain at 4°C from the patient to the laboratory, but with care not to freeze the sample as some viruses are labile to freezing.
-
2.
Samples may need preparation by treatment with antibiotics, clarification of debris, or disruption of mucus.
-
3.
Appropriate cell lines for the virus(es) sought are chosen and inoculated, taking care that the cells are metabolically healthy; that cells are being grown in an appropriate cell culture medium and serum concentration, and the cultures are in the appropriate state for the virus(es) being sought, that is, confluent or actively dividing; at the appropriate temperature, that is, 37° or 33° (for respiratory viruses); and that the laboratory periodically checks the cell lines being used for their continuing sensitivity to support growth of reference viruses. Specially designed cell lines may express particular virus receptors to broaden specificity or indicator systems for easier detection of the presence of virus.
-
4.
Evidence that a new virus has been isolated may come from routine reading of monolayers for cytopathic effects (CPEs), which may be distinctive for a particular virus. Alternatively, monolayers may be screened by a variety of techniques including antigen staining, hemadsorption, electron microscopy, or PCR. Blind passage of material not showing a CPE may be advisable to identify slow-growing agents, and regular comparison with uninoculated control cells is essential to distinguish viral CPE from degeneration of uninfected cell monolayers.
-
5.
Finally, a variety of methods may be needed to obtain final identification of the virus that has been isolated, sometimes including detailed strain identification or sequencing.
For further detailed description of virus isolation, the reader is encouraged to consult more detailed sources, for example, national reference laboratories or information distributed by the US Centers for Disease Control and Prevention (http://www.cdc.gov) and the World Health Organization (http://www.who.int) (Fig. 10.7 ).
Figure 10.7.
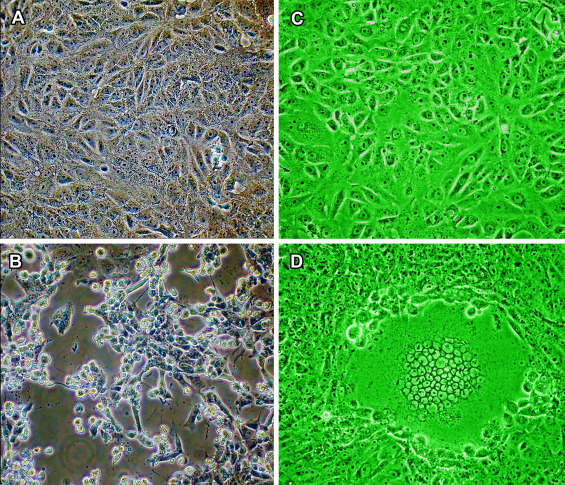
Cytopathic effect of viruses in cell cultures. (A) Confluent monolayer of uninfected Vero cells. (B) Vero cells infected with the SARS coronavirus (SARS-CoV) 24 hours post-infection. Massive cytopathic effect (CPE), with cells rounded up and detaching from the substrate. (C) Confluent monolayer of uninfected Vero cells. (D) Vero cells infected with herpes simplex virus 1 (HSV-1) at 24 hours post-infection. A single large syncytium of at least 50 cells (massed nuclei) which is unstable and later progresses to a necrotic/apoptotic CPE. Since most diagnostic specimens contain a much lower titer of infectious virus, CPE is usually more asynchronous, beginning with few foci involving few cells and taking longer to achieve widespread infection. Phase contrast microscopy (C, D) monochromatic green light provides slightly better resolution.
(A), (B) from Institute for Medical Virology, University Hospital, Frankfurt, with permission.
Further Characterization of Viruses Detected
For most routine diagnostic purposes it is usually not necessary to determine the exact strain or sequence of the virus that has been detected. On the other hand, there are certain situations when this information can be crucial for patient management, or for epidemiological or public health purposes. Serological methods including EIAs and neutralization of virus growth can be used to identify different serotypes, assisted by the availability of appropriate monoclonal antibodies (Fig. 10.8 ). Different influenza virus isolates are characterized and compared by comparing hemagglutination-inhibition titers using standard antisera.
Figure 10.8.
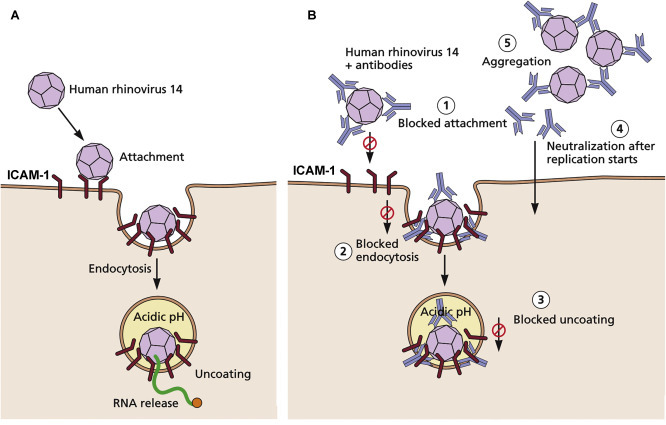
Virus neutralization, as exemplified by the interactions of neutralizing antibodies with human rhinovirus 14. (A) The normal route of infection. The virus attaches to the ICAM-1 receptor and enters by endocytosis. As the internal pH of the endosome decreases, the particle uncoats and releases its RNA genome into the cytoplasm. (B) Possible mechanisms of neutralization of the virus by antibodies. At least five mechanisms of neutralization have been supported by experimental evidence: (1) blocked attachment—binding of antibody molecules to virus results in steric interference with virus-receptor binding, (2) blocked endocytosis—antibody molecules binding to the capsid can alter the capsid structure, affecting the process of endocytosis, (3) blocked uncoating—antibodies bound to the incoming particle fix the capsid in a stable conformation so that pH-dependent uncoating is not possible, (4) blocked uncoating inside the cell—antibodies themselves may be taken up by endocytosis and interact with virions inside the cell after infection starts, (5) aggregation—because all antibodies are divalent, they can aggregate virus particles, facilitating their destruction by phagocytes. Any or all of these mechanisms may operate in quantitative neutralization assays. Virus neutralization assays may be used to measure the titer of neutralizing antibody in clinical samples, by reacting a standard virus inoculum with different dilutions of serum. There are many other variations, for example, known antisera to different virus serotypes may be used to identify the serotype of a new virus isolate; tests may be run in cell cultures with cytopathic effect or plaque endpoints, or in animals when necessary, and many steps may be automated.
Adapted from Smith, T.J., et al., 1995. Semin Virol 6:233–242. Then from Flint, S.J., et al., 2016. Principles of Virology: pathogenesis and control, fourth ed., Washington, DC: ASM Press, with permission.
Complete nucleotide sequencing of a viral genome is not often attempted for diagnostic purposes at present although it probably will be increasingly widespread over the next few years. However, obtaining a partial sequence from PCR products is widely used and has replaced earlier molecular approaches such as restriction mapping of DNA products and oligonucleotide fingerprinting. For example, the resistance of a patient’s circulating HIV to the commonly used anti-retroviral drugs is predicted from the pattern of resistance-associated point mutations in the sequence of the viral polymerase. This can be determined from the sequence of the patient’s HIV PCR product, which is then compared with commercially available sequence databases to correlate polymerase sequence with drug resistance phenotype. The result is used to plan the patient’s individualized drug regimen or to modify a drug regimen to which the patient’s virus has become resistant. Because different rabies virus sequence variants have segregated according to geographical region and animal host species, sequencing of rabies virus recovered from a patient can be used to determine the origin of the infection. In order to investigate an apparent cluster of HIV patients who may or may not have acquired their infection from a common source, for example, a single medical practice or surgeon or a common sexual partner, partial sequences of their viruses can be compared with the usual random sequence variation seen in that population, to assess the likelihood that this truly represents common source infection.
Measurement of Serum Antibodies
The identification of unknown viruses or viral antigens using panels of antibodies of known specificity has already been discussed. Any of the above serological techniques may be employed the other way around, using panels of known antigens to identify unknown antibodies. The basic principles of those most widely used today are set out in Table 10.3 (Fig. 10.9 ).
Table 10.3.
Serological Procedures Used in Virology
| Technique | Principle |
|---|---|
| Enzyme immunoassay | Patient antibody binds to antigen; enzyme-labeled anti-Ig binds to antibody; washing; added substrate changes color |
| Radioimmunoassay | Patient antibody binds to antigen; radiolabeled anti-Ig binds to antibody; washing and counting |
| Western blot | Virus is disrupted, proteins separated by gel electrophoresis and transferred (blotted) onto nylon membrane; antibodies in test serum bind to viral proteins; labeled anti-Ig binds to particular bands; revealed by EIA or autoradiography |
| Latex particle agglutination | Patient antibody agglutinates antigen-coated latex particles |
| Virus neutralization | Patient antibody neutralizes virus infectivity; readout involves CPE inhibition or plaque reduction in cell culture, or protection of animals |
| Hemagglutination inhibition | Patient antibody inhibits agglutination of red blood cells by virus or antigen |
| Immunofluorescence | Patient antibody binds to intracellular antigen; fluorescein-labeled anti-Ig binds; fluoresces by UV microscopy |
| Immunodiffusion | Antibodies and soluble antigens produce visible lines of precipitate in a gel |
| Complement fixation | Antigen–antibody complex binds complement, which is thereafter unavailable for lysis of sheep RBC in the presence of antibody to RBC |
Figure 10.9.
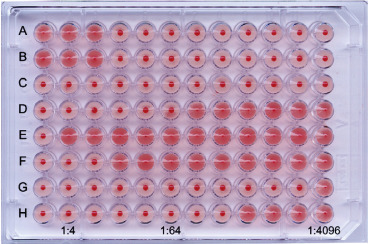
Hemagglutination inhibition (HI) test for detecting antibodies specific for influenza virus (H3N8, one of the highly pathogenic viruses that are of concern should these gain increased human-to-human transmissibility capability). Sera are (A) treated to remove non-specific agglutinins and non-specific inhibitors of agglutination. (B) Treated sera are serially diluted (twofold) in buffered saline in rows of wells of 96-well microtiter plates. (C) A volume of influenza virus containing 4 hemagglutinin units is added to each well and plates are incubated for 30 minutes. (D) A standard volume of turkey red blood cells (0.5% suspension) is added to each well. (E) The plates are incubated and read when control wells show complete red blood cell settling (button). Where virus agglutinates the erythrocytes, these form a shield pattern; however, where enough antibody is present to coat the viral HA, hemagglutination is inhibited, and the erythrocytes settle to form a button on the bottom of the cup. Rows (A), (B): Titration of the influenza virus used in the test, confirming that the correct amount (4 units) has been added to test wells. Row (C): Red blood cell control. Rows (D–H): Serially diluted test sera, with the first well in each row serving as a serum control for non-specific hemagglutination. Results: HI titers are the reciprocal of the last dilution of the serum showing inhibition of agglutination by test virus. Row (D): HI titer = 64; row (E): HI titer <4; row (F): HI titer = 8; row (G): HI titer = 2048; row (H): HI titer = 256.
Reproduced from MacLachlan N.J. and Dubovi E.J. (2011), Veterinary Virology, fourth ed., Academic Press, with permission.
Diagnosis of Acute Infection by Demonstrating a Rise in Antibody
Following the traditional approach, “paired sera” are taken from the patient, the “acute-phase” serum sample as early as possible in the illness and the “convalescent-phase” sample at least 10 to 14 days later. Blood is collected in the absence of anticoagulants and given time to clot, and then the serum is separated. Acute and convalescent serum samples should be tested simultaneously. For certain tests that measure inhibition of some biological function of a virus, for example, virus neutralization or hemagglutination-inhibition, the sera must first be inactivated by heating at 56°C for 30 minutes and sometimes treated by additional methods, in order to destroy various types of non-specific inhibitors of infectivity or hemagglutination, respectively. Prior treatment of the serum is not generally required for assays that simply measure antigen–antibody binding, such as EIA, RIA, Western blot, latex agglutination, immunofluorescence, or immunodiffusion.
The paired sera are then titrated for antibodies using any of a wide range of available serological techniques. Demonstration of a significant rise in antibody titer (conventionally, a four-fold rise when titrating the test serum as two-fold dilutions) is taken as proof of “seroconversion,” that is, acute infection with the agent in question. In practice, there are many subtleties in the interpretation of results. For example, the timing of the collection of the paired sera in relation to the date of onset of illness must be carefully assessed to allow for inappropriate sample timing. Moreover, an individual with immunity due to prior infection or immunization may undergo a second or reinfection, often of a subclinical nature, but showing a rise in antibody titer. It is obvious that in reality, two well-timed paired sera to allow demonstration of a four-fold rise in titer, are often not available. Attempts are sometimes made to infer that a recent infection has occurred, on the basis of one serum sample containing a relatively high antibody titer; indeed laboratories may issue guidelines about what titer of antibody in a single serum may imply recent infection. In practice this is often unreliable, as the peak antibody titer seen in the convalescent stage of an infection can vary significantly from person to person.
Immunoglobulin M Class-Specific Antibody Assays
A rapid diagnosis can be made on the basis of a single acute-phase serum by demonstrating virus-specific antibody of the IgM class. Because IgM antibodies appear early after infection but decrease to low levels within 1 to 2 months and generally disappear altogether within 3 months, the presence of these antibodies is indicative of either recent or chronic infection. Moreover, if present in a newborn baby, these antibodies are diagnostic of intrauterine infection, because maternal IgM antibodies, unlike IgG, do not cross the placenta. All of the immunoassays described above can readily be rendered IgM class-specific; EIA and immunofluorescence are the most generally useful. Typical indirect assays for virus-specific IgM antibodies are depicted in Fig. 10.10 .
Figure 10.10.
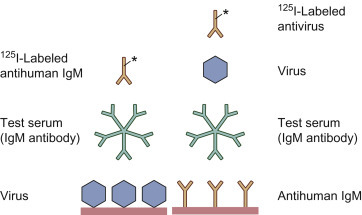
Radioimmunoassay for IgM class antibody. In the simpler configuration (left), virus-specific IgM antibody in the test serum reacts with viral antigen immobilized on the solid phase, and the IgM antibody is then detected by labeled anti-IgM antibody. This configuration can lead to false-positive results due to rheumatoid factor, or false-negative results due to blocking by competing antiviral IgG antibody in the test serum. In the alternative configuration (right), IgM antibody is captured from the test serum by immobilized anti-IgM, and any specific antiviral IgM is detected by progressive incubation with viral antigen and then labeled antiviral antibody. Enzyme-labeled antibodies have now largely replaced I125-labeled antibodies as a preferred indicator system.
A particular problem with IgM antibody assays is interference by the so-called rheumatoid factor, which is antibody, mainly of the IgM class, directed against the constant region (Fc) of normal IgG. Though first described in rheumatoid arthritis and other autoimmune diseases, rheumatoid factor is in fact prevalent in many infectious diseases, and it is found in the majority of congenitally infected neonates. Rheumatoid factor produces false positives in IgM immunoassays, because it binds to antiviral (as well as normal) IgG antibodies in human serum, forming IgM–IgG complexes. Thus, any antiviral IgG antibodies in the sample, which will also bind to the immobilized antigen, may then capture rheumatoid factor present, which will in turn be detected by the anti-human IgM antibodies employed as detector antibody in the assay format depicted in the left-hand side of Fig. 10.10.
In order to minimize the impact of rheumatoid factor, it is advisable to employ a reverse (or capture) IgM assay (Fig. 10.10, right). In its simplest form monoclonal anti-human IgM is used as the capture antibody and labeled virus as the detector/indicator. More commonly, unlabeled virus is the detector, then labeled monoclonal antiviral IgG antibodies, or perhaps better still F(abʹ)2 antibody fragments, are added as indicator.
Another pitfall with IgM antibody assays is the potential for cross-reaction between closely related viruses. This is because antibodies produced early in an immune response, particularly IgM antibodies, are typically more broadly reacting until B cell affinity maturation has taken place. For example, infection with a dengue virus may produce IgM antibodies that cross-react with a number of related flaviviruses, and it may be necessary to titrate the serum against a panel of antigens, or test later IgG samples, to identify the particular flavivirus involved. False-negative IgM results are occasionally encountered in infants, in some respiratory infections, and during reactivation of latent herpesviruses. Despite these pitfalls, IgM antibody capture assays are an important first-line diagnostic approach for some infections, for example, hepatitis A. Class-specific immunoassays can also be designed to measure specific antiviral antibodies of the IgA, IgE, or IgG class, or of any given IgG subclass.
Immunoblotting (“Western Blotting”)
Western blotting allows simultaneous but independent detection of antibodies against a number of different proteins present in a particular virus preparation. There are four key steps to western blotting (Fig. 10.11 ). First, concentrated virus is solubilized and the constituent proteins separated into discrete bands according to molecular mass (M r) by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Second, the separated proteins are electrophoretically transferred (“blotted”) onto a nitrocellulose sheet in order to immobilize the separated polypeptides and to make these available for reaction with antibodies. Third, the test serum is allowed to react with the viral proteins on the nitrocellulose sheet and unbound components are washed away, Finally, any bound antibody is demonstrated using an enzyme-labeled anti-species antibody. Thus, immunoblotting permits the demonstration of antibodies to some or all of the proteins of any given virus preparation, and can be used to monitor the development of antibodies to different antigens at different stages of infection. The technique has played a crucial role as the ultimate confirmatory test for samples giving an initial reaction in screening EIAs for anti-HIV antibody; experience has taught which western blotting patterns are likely to represent true anti-HIV antibodies, and which represent likely false-positive reactions in the initial EIA.
Figure 10.11.

Principles of western blotting for identification of antibodies or antigens. (1) A virus-containing sample is digested with the anionic detergent sodium dodecylsulfate (SDS) and electrophoresed on a polyacrylamide slab gel (PAGE), which separates the different viral proteins according to Mr. (2) The bands of viral protein are then transferred (“blotted”) onto a nitrocellulose membrane by capillary transfer or usually by electrophoresis in a different plane to immobilize the polypeptides. (3) The unoccupied areas of the membrane are blocked (“quenched”) by saturation with a suitable protein, then washed, dried, and cut into strips which can be used to test individual patient sera. Each test serum or plasma (“primary” antibody) is then incubated with one strip to enable antibodies to bind to the individual viral proteins. Following rinsing, bound antibody is detected by the addition of enzyme-labeled anti-human immunoglobulin (“secondary” antibody). Following another wash, the bands are revealed by the addition of a substrate chosen to produce an insoluble colored product. This technique may be used with a preparation of known antigens, for example, an infected cell lysate, having been separated on a gel, to examine a serum sample containing unknown antibodies; alternatively, a known antiserum can be used to detect those antigens in an unknown sample that react after running on a gel.
Reproduced from Flint, S.J., et al., Principles of Virology, second ed., ASM Press, with permission.
Western blotting is also an important research tool for characterizing viral antigen mixtures, using the antibody reagent rather than the antigen as the “known” component in the reaction.
Applications of Serology
Serological tests are used for many important purposes apart from diagnosing or excluding acute or chronic infection (see Box 10.1 ). These assays are valuable in many contexts because these provide indications of both clinical and subclinical infections, thereby giving a truer record of total number of infections. The finding of antibodies to a virus in a single sample carries very different clinical implications depending on the virus (see Box 10.2 ). The screening for immunity is very useful to document successful immunization in an individual, and to check the coverage and efficacy of vaccination in a population. In addition, testing the susceptibility of the close contacts of an individual with a potentially dangerous infection allows for the protection of at-risk (non-immune) individuals by segregation or immunization, and provides a baseline for subsequent monitoring as to the course of infection.
Box 10.1. Applications of Serological Tests.
-
1.
Diagnose acute infection by demonstrating a fourfold rise in titer of viral antibodies
-
2.
Diagnose acute infection by demonstrating the presence of specific IgM antibodies
-
3.
Estimate prevalence of a persistent infection in a population, for example, what percentages have persistent CMV infection
-
4.
Estimate incidence of an infection over a time period, for example, the rate of new hepatitis C infections
-
5.
Evaluate immunization programs, for example, measles, hepatitis B
-
6.
Assess level of herd immunity, for example, influenza
-
7.
Assess exposure resulting from occupation, lifestyle— whether certain activities are associated with increased rates of infection?
-
8.
Use age-specific prevalence patterns to analyze transmission efficiencies and possible transmission routes, for example, age-specific acquisition of EBV antibody
-
9.
Determine whether an apparently new infection in man has actually been already circulating subclinically
-
10.
Search for an animal reservoir of an apparently new zoonotic infection, for example, which non-human species show antibodies to SARS or Ebola viruses
Box 10.2. Significance of a Single Positive Result for Antibody with Different Viruses.
-
●
Anti-HBs=immune, not infectious
-
●
Anti-HIV=infected and infectious
-
●
Anti-HCV=past infection, and ~60–80% chance of being still infected
-
●
Anti-HSV=still infected, intermittently infectious by salivary route but not blood transfusion
-
●
Anti-HSV type 2=as above, +the site of primary infection is most likely to be genital
-
●
Anti-CMV, anti-EBV=still infected, potentially infectious (by salivary contact or blood transfusion)
-
●
Antibody to rubella virus=immune to second clinical attack; may undergo asymptomatic reinfection but the fetus is not at risk
-
●
Antibody to an exotic virus (e.g., Lassa fever, Ebola) in an acutely sick, recently returned traveler=likely to be acute infection
Sensitivity and Specificity
The interpretation and performance of any diagnostic assay are judged by two essential criteria, specificity and sensitivity. The sensitivity of a given test is a measure of the percentage of those with the disease (or infection) in question who are identified as positive by that test. For example, a particular EIA used to screen a population for HIV antibody may display a sensitivity of 98%, that is, of every 100 infected people, 98 will be correctly detected and 2 will be missed (the false-negative rate equals 2%). In contrast, the specificity of a test is a measure of the percentage of those without the disease (or infection) who yield a negative result. For example, the same EIA for HIV antibody may have a specificity of 97%, that is, of every 100 uninfected people, 97 will be correctly diagnosed as clear but 3 will be incorrectly scored as infected (the false-positive rate equals 3%).
Whereas sensitivity and specificity are fixed percentages intrinsic to the particular diagnostic assay, the predictive value of an assay is greatly affected by the prevalence of the disease (or infection) involved. Thus, if the above EIA is used to screen a high-risk population with an HIV prevalence of 50%, the predictive value of the assay will be high; however, if it is used to screen blood donors with a known HIV prevalence of 0.1%, the great majority of the 3.1% who register as positive will in fact be false positives and will require follow-up with a confirmatory test of a much higher specificity. This striking illustration draws attention to the importance of selecting diagnostic assays with a particular objective in mind. A test with high sensitivity is required when the aim is to screen for a serious infection, the diagnosis of which must not be missed; a test with high specificity is also required for confirmation that the diagnosis is correct.
The sensitivity of a given immunoassay is really a measure of its ability to detect small amounts of either antibodies or antigen. For instance, EIA, RIA, and neutralization assays generally display substantially higher sensitivity than immune electron microscopy, immunofluorescence, or the older serological methods of complement fixation and immunodiffusion. Improvements in sensitivity are achieved by miniaturization of assays, use of purified reagents, sensitive instrumentation, and signal amplification. On the other hand, the specificity of an immunoassay is a measure of the test’s capacity to discriminate; thus it is influenced mainly by the purity of the key reagent, that is, antigen when testing for antibodies, or antibodies when testing for antigen. Use of cloned antigens or synthetic peptides will usually improve the specificity of antibody detection. Note that use of signal amplification to increase the sensitivity of a test is very likely to also increase the signal due to background or false-positive reactions, that is, the signal:noise ratio may not be improved.
Interpretation of Laboratory Results
The isolation and identification of a particular virus, or demonstration of infection by serological methods, in a patient with a given disease, are not necessarily relevant to the patient’s immediate clinical problem. Concurrent subclinical infection with a virus unrelated to the illness in question is not uncommon. In attempting to interpret whether a virus that has been isolated is related to a patient’s disease, one must be guided by the following considerations. First, the site from which the virus was isolated is important. For example, one would be quite confident about the etiological significance of herpes simplex virus isolated from a sample of brain tissue, or of mumps virus isolated from the CSF of a patient with meningitis, because these sites are usually sterile without any normal bacterial or viral flora. On the other hand, an echovirus recovered from the feces, or herpes simplex virus from the throat, may not necessarily be causing any pathology as such viruses are often associated with unapparent infections. Second, knowledge that the virus and the disease in question are often causally associated engenders confidence that the isolate is significant.
In addition to the obvious issue of diagnosing the cause of a patient’s illness, it is very important to remember that laboratory diagnostic methods are used to answer many other significant clinical and public health questions (see Box 10.3 ).
Box 10.3. Information that May Be Sought from Laboratory Virus Testing.
-
1.
Is the patient infected with a particular virus?
-
2.
If so, which strain or serotype is involved?
-
3.
What is the level of replication or virus load?
-
4.
Is the patient infectious to others?
-
5.
Which site(s) of the body are involved?
-
6.
What is the stage or duration of the infection?
-
7.
What is the type or extent of the immune response?
-
8.
What are the implications for therapy or prevention of infection of others?
-
9.
Has the patient been infected in the past?
-
10.
Is the patient now immune?
A number of very important practical issues should not be forgotten as these can seriously interfere with the reliability of laboratory results for a given clinical situation. These include:
-
1.
Contamination of the sample with extraneous viruses or nucleic acids, unrelated to the disease being investigated. This may occur at any stage in the diagnostic chain, from collection of the sample through to the final stage of the test procedure;
-
2.
False-positive and false-negative results may occur with any test, for numerous reasons including sensitivity and specificity;
-
3.
Mix-up of samples, labeling errors. Laboratories frequently build in system checks to detect such errors.
Frequently, laboratories will include a requirement for confirmation by repeat testing in the case of results that are subject to particular significance or unreliability.
Laboratory Safety
Although virology can be considered one of the less hazardous human occupations compared with building, mining, or driving a car, it is also true that many cases of serious illness and over 700 deaths from laboratory-acquired infection have been recorded over the years, particularly from togaviruses, flaviviruses, arenaviruses, and filoviruses. Potentially hazardous procedures are described in Table 10.4 . In many countries, including the United States, European Union, Canada, and Australia, pathogens are classified into categories 1 to 4 based on properties of pathogenicity, transmissibility, host range, the availability of treatment and vaccination, etc. These categories are endorsed by the World Health Organization. An appropriate level of containment and handling practice is specified for each risk category, ranging from Biosafety level 1 (BSL 1)—standard microbiological practice—for category 1 organisms, through to BSL 4—maximum security facilities and containment protocols—for category 4 viruses and organisms. In research laboratories, work usually involves known agents where the hazards are predictable and safety procedures appropriate to the level of hazard can be applied. However, diagnostic laboratories accept human samples of unknown infective status that are in a practical sense an extension of the patient. These may contain HIV, hepatitis B or C virus, or other common or exotic infective agents. The usual approach is to regard all unknown biological specimens as potentially infectious, and to apply a level of containment appropriate to the majority of agents likely to be present, for example, BSL2; individual samples from known or suspected cases of greater hazard, for example, Ebola, are handled at a higher containment level once these are identified. Similarly, material from laboratory or wild animals, particularly primates, may contain unknown pathogens with a danger to humans.
Table 10.4.
Laboratory Hazards
| Route of Hazard | Procedure or Source of Hazard |
|---|---|
| Aerosol | Homogenization (e.g., of tissue in blender) |
| Centrifugation | |
| Ultrasonic vibration | |
| Broken glassware | |
| Pipetting | |
| Ingestion | Mouth pipetting |
| Eating or smoking in laboratory | |
| Inadequate washing/disinfection of hands | |
| Skin penetration | Needle stick injury |
| Hand cut by broken glassware | |
| Leaking container contaminating hands | |
| Animal bite | |
| Pathologist handling infected organs | |
| Receptionist handling blood-stained request form | |
| Splash into eye | Break in pressurized tubing |
| Sudden liquid spillage |
For all containment levels, good laboratory technique is an essential aspect for avoiding laboratory hazards. Use of elaborate safety equipment, negative air pressure management, etc. will not compensate for sloppy or unsafe practices. Rigorous aseptic technique must be practiced. Mouth pipetting is banned. Laboratory coats must be worn at all times, gloves must be used for handling potentially infectious materials, and special care taken with sharp objects. Rigorous attention must be given to sterilization, where possible by autoclaving, of all potentially infectious waste as well as used equipment. Special arrangements for the disposal of “sharps” are essential. Spills are cleaned up with an appropriate chemical disinfectant. Staff should be immunized against such diseases as hepatitis B and poliomyelitis as a matter of routine and, when vaccines are available, against more exotic agents in those special laboratories handling such viruses. Limitations should be placed on the type of work undertaken by pregnant or immunosuppressed employees.
It is also important to inactivate the infectivity of any particularly dangerous viruses employed as antigens in serological tests other than virus neutralization (e.g., arenaviruses, rhabdoviruses, togaviruses, or flaviviruses). This can be done, without destroying antigenicity, by γ-irradiation, or by photodynamic inactivation with ultraviolet light in the presence of psoralen or related compounds. Viral proteins produced by recombinant DNA technology provide a simple, standardized, and safe alternative to whole virus as antigen in many serological tests.
Further Reading
- Jerome K.R., editor. Lennette’s Laboratory Diagnosis of Viral Infections. fourth ed. CRC Press; Boca Raton, Florida: 2010. ISBN-10: 142008495X. [Google Scholar]
- Kaslow R.A., Stanberry L.R., LeDuc J.W. Viral Infections of Humans, Epidemiology and Control. fifth ed. Springer; New York: 2014. ISBN 978-1-4899-7447-1. [Google Scholar]