

Abstract
Background
Klippel-Feil syndrome (KFS) represents a rare anomaly characterized by congenital fusion of the cervical vertebrae. The underlying molecular etiology remains largely unknown because of the genetic and phenotypic heterogeneity.
Methods
We consecutively recruited a Chinese cohort of 37 patients with KFS. The clinical manifestations and radiological assessments were analyzed and whole-exome sequencing (WES) was performed. Additionally, rare variants in KFS cases and controls were compared using genetic burden analysis.
Results
We primarily examined rare variants in five reported genes (GDF6, MEOX1, GDF3, MYO18B and RIPPLY2) associated with KFS and detected three variants of uncertain significance in MYO18B. Based on rare variant burden analysis of 96 candidate genes related to vertebral segmentation defects, we identified BAZ1B as having the highest probability of association with KFS, followed by FREM2, SUFU, VANGL1 and KMT2D. In addition, seven patients were proposed to show potential oligogenic inheritance involving more than one variants in candidate genes, the frequency of which was significantly higher than that in the in-house controls.
Conclusions
Our study presents an exome-sequenced cohort and identifies five novel genes potentially associated with KFS, extending the spectrum of known mutations contributing to this syndrome. Furthermore, the genetic burden analysis provides further evidence for potential oligogenic inheritance of KFS.
Keywords: Klippel-Feil syndrome, Whole-exome sequencing, Genetic burden analysis, Genetic mutational spectrum, Oligogenic inheritance
Background
Klippel-Feil syndrome (KFS), which was first reported by Maurice Klippel and Andre Feil in 1912, is a complex skeletal disorder characterized by congenital fusion of two or more cervical vertebrae [1]. The incidence of KFS has been estimated as approximately 1/40000–1/42000, with a slight female predominance by approximately 3:2 [2, 3]. However, the true incidence of KFS may be higher than reported because of heterogeneity in phenotypic expression and a lack of large-scale population screening studies. Based on retrospective studies of cervical computed tomography (CT) in 2917 patients and 131 patients with cervical spondylotic myelopathy, the incidence of KFS may be as high as 1/172 to 5/131 [4, 5].
The main etiology of KFS is the impaired development of the cervical vertebrae, which leads to the improper segmentation of the cervical spine. Although KFS can occur as an isolated malformation, it is often associated with a variety of congenital diseases and other systemic malformations, including scoliosis, Sprengel deformity, urinary malformations, gastrointestinal malformations, hearing impairment, congenital heart defects and various neurological anomalies [1, 6–8]. In addition, congenital fusion deformity of the cervical vertebrae often alters the kinematics of the cervical spine in ways that may accelerate degenerative changes throughout the region. Therefore, patients with KFS usually complain of neck and back pain, respiratory failure, and decreased mobility and may be at risk for death in severe cases due to a lack of early prediction methods.
Recent studies have suggested that the failed vertebral segmentation underlying KFS is caused by defective somitogenesis in the cervical region [9]. Autosomal dominant and recessive inheritance patterns have both been reported in families with KFS. According to Online Mendelian Inheritance in Man (OMIM), mutations in GDF6 (MIM: 601147), MEOX1 (MIM: 600147), GDF3 (MIM: 606522), MYO18B (MIM: 607295) and RIPPLY2 (MIM: 609891) have been associated with KFS [10–15]. Nevertheless, the basis of genetic predisposition to KFS is largely unknown due to the rarity of the disease; only a small number of KFS cases can be explained by the five specific pathogenic genes mentioned above [16]. To further decipher the molecular basis of KFS at the exome level, we herein investigate the molecular findings of WES among a cohort of 37 KFS patients/families, and further analyze rare variants by using a genetic burden method.
Methods
Cohort collection
From January 2016 to April 2018, we consecutively recruited 37 patients of Han Chinese ethnicity at Peking Union Medical College Hospital (PUMCH) who had been diagnosed with KFS under DISCO (Deciphering disorders Involving Scoliosis and COmorbidities, DISCO http://discostudy.org/) project. Demographic information, physical examination results, clinical symptoms on presentation, and a detailed medical history were obtained. Radiological assessments including anteroposterior, lateral neutral, and flexion-extension plain radiographs; CT; and magnetic resonance imaging (MRI) were performed on each patient to give a prior clinical diagnosis. Radiographic parameters of interest, such as the segmentation of congenitally fused vertebrae, the Samartzis classification of KFS [8], cervical scoliosis, and sagittal cervical alignment, were also recorded. All radiographic evaluations were conducted by trained spine surgeons, and the clinical review was performed by an alternate observer blinded to the radiographic assessment. None of the investigators were involved in the direct care of the patients.
Prior to study participation, written informed consent was provided by each participant. The study was approved by the Department of Scientific Research and Ethics Committee of PUMCH in China.
Genomic DNA preparation and whole-exome sequencing
Genomic DNA for 37 KFS patients (14 cases along with their healthy parents) was extracted from peripheral blood lymphocytes. WES was performed on peripheral blood DNA for all participants. DNA samples were prepared in Illumina libraries and then underwent whole-exome capture with the SureSelect Human All Exon V6 + UTR r2 core design (91 Mb, Agilent, USA), followed by sequencing on the Illumina HiSeq 4000 platform in 150-bp paired-end reads mode (Illumina, San Diego, CA, USA).
Variant annotation and interpretation
WES data processing was performed with the in-house developed PUMP (Peking Union Medical college hospital Pipeline) [17]. Interpretation of single-nucleotide variants (SNVs) and insertions/deletions (indel variant alleles) were adapted from the American College of Medical Genetics and Genomics (ACMG) guidelines [18]. Computational prediction tools (Genomic Evolutionary Rate Profiling [GERP] [19], Combined Annotation Dependent Depletion [CADD] [20], Sorting Intolerant Form Tolerant [SIFT] [21], Polyphen-2 [22], and MutationTaster [23]) were used to predict the conservation and pathogenicity of candidate variants. All variants were compared to publicly available databases such as the 1000 Genomes Project (http://www.internationalgenome.org/), the NHLBI GO Exome Sequencing Project (ESP) Exome Variant Server (http://evs.gs.washington.edu/EVS/), and the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org/). The in-house control database consisted of WES data from 534 unrelated Chinese individuals with no apparent skeletal anomalies from DISCO project.
Mutational burden analysis
A criterion of the internal (combined case and control population) and external (ExAC, and gnomAD) was used to filter for rare genuine variants with a minor allele frequency (MAF) < 0.001.
After filtering, we performed an initial analysis focusing on variants in reported pathogenic genes (GDF6, MEOX1, GDF3, MYO18B and RIPPLY2) related to KFS. Additionally, 96 genes that were biologically related to vertebral segmentation defects according to previous reports or the Human Phenotype Ontology (HPO; https://hpo.jax.org/), NCBI Gene (https://ncbi.nlm.nih.gov/gene/) and OMIM (http://omim.org/) databases were added to the candidate gene list (Table S1).
To extend the spectrum of known mutations contributing to KFS, we performed a second-stage analysis to search for exome-wide rare variants in 96 candidate genes related to vertebral segmentation defects using the genetic burden analysis [24].
Statistical analysis
Mean comparison of relevant features was conducted using Student’s t-test. One-tailed P-values of < 0.05 were considered statistically significant. Fisher’s exact test was used for genetic burden analysis.
Results
Cohort information
The study cohort consisted of 37 unrelated KFS patients of Han Chinese ethnicity, including 14 cases with unaffected parents (trios) and 23 singleton cases (Table S2). There were 20 males (54.1%) and 17 females (45.9%), with a mean age at diagnosis of 12.6 ± 5.3 years. Detailed demographic information is presented in Table 1.
Table 1.
Demographic and clinical characteristics of the KFS cohort
| Parameter | KFS cohort (n = 37) |
|---|---|
| Age in years, mean (range) | 12.6 (5–26) |
| Sex, n (%) | |
| Men | 20 (54.1) |
| Women | 17 (45.9) |
| Level of fusion, n (%) | |
| C1–C2 | 3 (8.1) |
| C2–C3 | 15 (40.5) |
| C3–C4 | 14 (37.8) |
| C4–C5 | 10 (27.0) |
| C5–C6 | 13 (35.1) |
| C6–C7 | 21 (56.8) |
| C7–T1 | 10 (27.0) |
| KFS classification, n (%) | |
| Type I | 15 (40.5) |
| Type II | 5 (13.5) |
| Type III | 17 (45.9) |
| Comorbidities, n (%) | |
| Intraspinal anomalies | 9 (24.3) |
| Extraskeletal anomalies | 12 (32.4) |
| Clinical manifestations, n (%) | |
| Limited cervical ROM | 17 (45.9) |
| Short neck | 10 (27.0) |
| Low posterior hairline | 6 (16.2) |
| Clinical triad | 4 (10.8) |
| Torticollis | 4 (10.8) |
Abbreviation: ROM range of motion
Clinical features and radiographic parameters
Among the 37 patients with KFS, there was a mean of 2.3 congenital fusion levels (range 1–7), which included various regions of the cervical spine. The most commonly fused segments were C6–C7, C2–C3, and C3–C4, which occurred in 56.8%, 40.5%, and 37.8% of the patients, respectively (Fig. 1). According to the KFS classification criteria by Samartzis [8], 15 (40.5%) patients were Type I, with a single congenitally fused cervical segment; five (13.5%) patients were Type II, demonstrating multiple noncontiguous congenitally fused cervical segments; and 17 (45.9%) patients were Type III, showing multiple contiguous congenitally fused cervical segments.
Fig. 1.
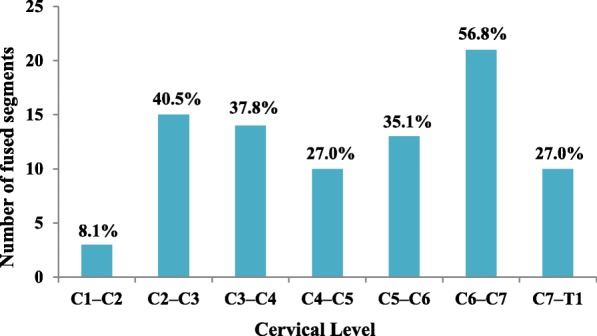
The number and prevalence (%) of fused segments in our KFS cohort. The figure illustrates the distribution of fused cervical levels among the cervical segments
The KFS patients presented with varied intraspinal and extraskeletal anomalies. There were nine patients (24.3%) manifesting intraspinal deformities, including syringomyelia, diastematomyelia, and tethered cord, and 12 patients (32.4%) presented with extraskeletal manifestations such as various cardiac defects and malformation of the urinary and digestive systems. KFS has been characterized as a clinical triad of short neck, low posterior hairline, and limited cervical range of motion (ROM). In our cohort, limited cervical ROM was the most common clinical feature (17 patients, 45.9%), followed by short neck (10 patients, 27.0%) and low posterior hairline (six patients, 16.2%). However, 43.2% of the patients did not show any typical manifestation of the clinical triad; furthermore, only 10.8% of the patients showed all three signs in the clinical triad at the same time. Detailed characteristics and radiographic parameters are shown in Table 1.
Variants of known KFS-related genes
After WES data processing and variant interpretation, we firstly examined filtered rare variants for potential causative variants in five reported genes associated with KFS. As a result, 3 variants were found in MYO18B (Table 2): a splice region mutation (c.2695 + 3A > G) and two missense variants (c.662 T > C and c.5020G > A) were identified in patients CS132, CS1015 and CS1049, respectively. The c.2695 + 3A > G variant was found in patient CS132, who was affected by cervical vertebral fusion (C2–C6), short neck, low posterior hairline, limited cervical ROM and protruding ears but no rib or spinal cord abnormalities. Moreover, congenital cervical fusions combined with congenital heart disease were found in both CS1015 and CS1049. The variant c.662 T > C (p. Leu221Pro) in exon 4 was identified in proband CS1015, who presented with a single vertebral fusion level at C6–C7 and no severe clinical manifestations in the cervical spine. However, the c.5020G > A (p. Gly1674Arg) variant was observed in exon 31 from patient CS1049, who had long-segment cervical fusion (C1-T1), a prominent clinical triad and torticollis.
Table 2.
MYO18B variants and clinical features of patients
| Identifier | CS132 | CS1015 | CS1049 |
|---|---|---|---|
| Sex/age (years) at diagnosis | F/12 | M/5 | F/7 |
| Mutation information | |||
| Variant type | Splice region | Missense | Missense |
| Zygosity | Heterozygous | Heterozygous | Heterozygous |
| Chr_Position | 22_26219648 | 22_26164545 | 22_26299670 |
| Variant nomenclature | c.2695 + 3A > G | c.662 T > C (p.Leu221Pro) | c.5020G > A (p.Gly1674Arg) |
| ExAC AF | 0.000017 | 0.000034 | 0.000013 |
| gnomAD AF | Novel | 0.0000324 | 0.00003232 |
| In-house exome database AF | 0.0029 | Novel | Novel |
| Clinical features | |||
| Fused levels | C2–C6 | C6–C7 | C1-T1 |
| Comorbidities | Protruding ears | Tetralogy of Fallot, congenital solitary kidney | Patent foramen ovale |
| Clinical manifestations | Clinical triad | None | Clinical triad, torticollis |
| Classification | Type III | Type I | Type III |
| Other vertebral and costal abnormalities | None | None |
Rib fusion Sacrococcygeal agenesis |
MYO18B, an unconventional class XVIII myosin, is mainly expressed in human cardiac and skeletal muscle and plays putative roles in diverse human syndromes and cellular processes [25]. Consistent with the reported phenotypes of myocardial defects in MYO18B-deficient mice, two of our patients suffered from cardiac deformities. A homozygous nonsense mutation in MYO18B (c.6905C > A: p.S2302*) was identified in two unrelated patients who came from consanguineous families and exhibited a similar phenotype of KFS, myopathy, short stature, and facial dysmorphism [14], and moreover, compound heterozygous frameshift variants (c.6768delG: p.Leu2257SerfsTer16 and c.6660_6670delATTAGAACCTG: p.Arg2220SerfsTer74) in MYO18B were reported in a medulloblastoma patient with a previous diagnosis of KFS [26]; nonetheless, there are still no reports on pathogenic missense variants in MYO18B associated with KFS due to the limited sample size.
Genetic burden analysis of candidate genes related to vertebral segmentation defects
Based on the rare variant filtering of the candidate gene list (Table S1), we found that the average number of rare variants per individual in the KFS cohort (n = 33) was significantly higher than that in control individuals (n = 202) (p < 0.0001) (Table S3), which suggests that the candidate genes were relevant to the KFS etiology.
Furthermore, in the gene-based burden analysis, we compared the exome-wide frequency of rare coding variants for each candidate gene in KFS cases and controls. Among the genes associated with the KFS cohort compared to controls, the five with the lowest P-values were BAZ1B (P = 0.00000002), FREM2 (P = 0.0003), SUFU (P = 0.0004), VANGL1 (P = 0.0072) and KMT2D (P = 0.0214) (Table 3).
Table 3.
Genetic burden analysis for rare variant frequencies in KFS cases and controls
| KFS cases (n) | Variant alleles | Control subjects (n) | Variant alleles | p-value | |
|---|---|---|---|---|---|
| BAZ1B | 37 | 3 | 534 | 1 | 0.00000002 |
| FREM2 | 37 | 3 | 534 | 5 | 0.0003 |
| SUFU | 37 | 2 | 534 | 2 | 0.0004 |
| VANGL1 | 37 | 2 | 534 | 4 | 0.0072 |
| KMT2D | 37 | 3 | 534 | 11 | 0.0214 |
Variants identified in potential KFS-associated genes
We identified three qualified variants of BAZ1B from three patients. The in-frame insertion variant c.3804_3821dupGGAGGAGGAGGAAGAAGA (p. Glu1268_Glu1273dup) in BAZ1B was identified in CS63. The patient was a 6-year-old female with complaints of restricted neck motion and torticollis. Radiological evaluation detected fusion of the cervical vertebrae at C6-T1, scoliosis, diastematomyelia, syringomyelia, and polycystic kidney disease. BAZ1B encodes a member of a bromodomain protein family that is involved in chromatin-dependent regulation of transcription. Deletion of this gene has been reported in Williams-Beuren syndrome (WBS), a developmental disorder featuring multiple skeletal deformities such as scoliosis, hallux valgus, little-finger clinodactyly, fusion of the cervical spine and Chiari I malformation [27]. Although BAZ1B has no previously identified connection to KFS, the present finding may represent a novel disease association. In addition, two other rare missense variants of BAZ1B, namely, c.527A > G (p.Lys176Arg) and c.1364G > A (p.Arg455Gln), were also identified. Both patients suffered from single-segment cervical fusion and only mild clinical manifestations, despite multiple intraspinal deformities such as diastematomyelia, tethered cord and syringomyelia.
A heterozygous missense variant of c.8479C > T (p. Arg2827Cys) in the FREM2 gene was detected, with an ExAC allele frequency of 0.000016. This variant was predicted as pathogenic by the functional prediction programs SIFT, Polyphen-2 and MutationTaster, with a GERP score of 3.79 and a CADD score of 18.07. It was reported that homozygosity for a splice site mutation in the FREM2 gene was associated with Fraser syndrome and that compound heterozygosity for a missense mutation was related to cryptophthalmos [28, 29].
Three other rare heterozygous missense variants c.13364G > A (p.Arg4455His), c.3074C > T (p.Ser1025Leu), c.8939C > T (p.Ala2980Val) in KMT2D were identified in CS488, CS1162 and CS1210. These three patients suffered from multiple contiguous fused cervical levels: C2–C7, C2–C7, and C5-T1, respectively. The protein encoded by KMT2D has been shown to be a transcriptional regulator of the beta-globin and estrogen receptor genes and is reported to be a cause of Kabuki syndrome [30–32]. Therefore, the significant association of KMT2D with KFS involving multiple fused segments (Type III) may represent an expansion of the known phenotype associated with this gene.
Two other rare missense variants in FREM2 and variants of the associated genes VANGL1 and SUFU are shown in Table 4.
Table 4.
Information on rare variants of five novel associated genes
| Patient | Gene symbol | Variant type | Zygosity | Chr_Position | Ref transcript | Variant nomenclature | GERP++ score | CADD score | ExAC AF-total |
|---|---|---|---|---|---|---|---|---|---|
| CS63 | BAZ1B | In-frame insertion | Het | 7_72861616 | NM_032408.3 | c.3804_3821dupGGAGGAGGAGGAAGAAGA (p.Glu1268_Glu1273dup) | – | – | 0.000016 |
| CS216 | BAZ1B | Missense | Het | 7_72912871 | NM_032408.3 | c.527A > G (p.Lys176Arg) | 5.26 | 17.03 | 0.0000082 |
| CS519 | BAZ1B | Missense | Het | 7_72892427 | NM_032408.3 | c.1364G > A (p.Arg455Gln) | 5.56 | 11.17 | 0.000016 |
| CS193 | FREM2 | Missense | Het | 13_39452441 | NM_207361.4 | c.8842 T > C (p.Tyr2948His) | 0.572 | 5.468 | 0.000033 |
| CS1049 | FREM2 | Missense | Het | 13_39450454 | NM_207361.4 | c.8479C > T (p.Arg2827Cys) | 3.79 | 18.07 | 0.000016 |
| OS1056 | FREM2 | Missense | Het | 13_39424205 | NM_207361.4 | c.6410A > T (p.Tyr2137Phe) | 5.79 | 17.11 | 0.0000083 |
| CS488 | KMT2D | Missense | Het | 12_49425124 | NM_003482.3 | c.13364G > A (p.Arg4455His) | 5.57 | 13.45 | 0.000058 |
| CS1162 | KMT2D | Missense | Het | 12_49444297 | NM_003482.3 | c.3074C > T (p.Ser1025Leu) | 3.4 | 6.896 | 0.000059 |
| CS1210 | KMT2D | Missense | Het | 12_49432200 | NM_003482.3 | c.8939C > T (p.Ala2980Val) | 2.77 | 11.24 | 0.000017 |
| CS63 | SUFU | Missense | Het | 10_104375107 | NM_016169.3 | c.1105G > A (p.Val369Ile) | 5.21 | 12.54 | 0.000091 |
| CS132 | SUFU | Splice region | Het | 10_104375165 | NM_016169.3 | c.1157 + 6C > T | – | – | 0.000033 |
| CS1130 | VANGL1 | Synonymous | Het | 1_116206326 | NM_138959.2 | c.249G > A (p.Ser83=) | – | – | 0.00012 |
| CS927 | VANGL1 | Missense | Het | 1_116227985 | NM_138959.2 | c.1151C > G (p.Pro384Arg) | 5.44 | 25.8 | 0.0000083 |
Abbreviations: Het heterozygous, Chr chromosome, ExAC Exome Aggregation Consortium
Potential oligogenic inheritance
Among qualified variants, seven patients (18.9%) showed potential involvement of multiple associated variations in different genes (Table 5), which represented an increased ratio compared with that of the in-house controls (30/534) (P = 0.00148). Specifically, an in-frame insertion variant of BAZ1B, a splice acceptor variant of GRIP1, and two missense variants of SUFU and TBX6 were detected in patient CS63, and there were three patients carrying three variants in different candidate genes and another three patients carrying two variants in different genes. Additionally, MYO18B, SUFU, and BAZ1B, mutations of which were probably causative of KFS, were identified in two or more patients. This finding suggests the possibility of an oligogenic pathogenesis pattern.
Table 5.
Potential oligogenic inheritance in seven KFS patients
| Patient | Gene symbol | Variant type | Variant nomenclature |
|---|---|---|---|
| CS63 | BAZ1B | In-frame insertion | c.3804_3821dupGGAGGAGGAGGAAGAAGA (p.Glu1268_Glu1273dup) |
| GRIP1 | Splice acceptor | c.1043-1G > A | |
| SUFU | Missense | c.1105G > A (p.Val369Ile) | |
| TBX6 | Missense | c.499C > T (p.Arg167Cys) | |
| CS132 | MYO18B | Splice region | c.2695 + 3A > G |
| SUFU | Splice region | c.1157 + 6C > T | |
| WNT7A | Missense | c.83C > T (p.Ser28Leu) | |
| CS587 | FUZ | Missense | c.819C > A (p.Asp273Glu) |
| MAP3K7 | Missense | c.1115G > A (p.Arg372His) | |
| POR | Missense | c.1798C > T (p.Arg600Trp) | |
| CS676 | CHD7 | Synonymous | c.4008C > T (p.Ile1336=) |
| FRAS1 | Missense | c.7423G > A (p.Glu2475Lys) | |
| CS519 | BAZ1B | Missense | c.1364G > A (p.Arg455Gln) |
| COG1 | Missense | c.739C > T (p.His247Tyr) | |
| CS1015 | ANKRD11 | Missense | c.6067G > T (p.Ala2023Ser) |
| HOXD13 | Missense | c.814G > A (p.Val272Ile) | |
| MYO18B | Missense | c.662 T > C (p.Leu221Pro) | |
| CS1049 | FREM2 | Missense | c.8479C > T (p.Arg2827Cys) |
| MYO18B | Missense | c.5020G > A (p.Gly1674Arg) |
Discussion
KFS is a relatively rare disorder characterized by congenital synostosis of the cervical vertebrae because of a segmentation or formation defect. However, articles related to KFS are limited and mostly use individual case reports or small case series. Therefore, the 37-patient cohort in the current study is a relatively large reported KFS cohort for assessing clinical features and radiologic parameters and for identifying molecular findings by WES.
The clinical manifestations of KFS show substantial heterogeneity, and the typical clinical features are defined as a triad of short neck, low posterior hairline, and limited neck ROM. However, as found in our case cohort, the complete clinical triad was noted in only 10.8% of patients. Moreover, 43.2% of our KFS cohort did not present with any of these findings, which was consistent with a single-center retrospective study of 31 patients by Samartzis in 2016 [33]. Further analysis with imaging data demonstrated that 73.3% of the 15 Type I KFS patients exhibited none of the three clinical triad signs; on the other hand, all four patients presenting with clinical triad findings were considered Type III. KFS patients with multiple contiguous congenitally fused cervical levels are confirmed to be at a significantly increased risk of severe clinical manifestations. As such, proper precautionary measures and close follow-up should be suggested in KFS patients, especially those regarded as Type III.
Although the pathogenesis of the developmental defect underlying KFS is thought to be attributable to the paraxial mesoderm and somites at the embryonic stage, which may result from mutations or disruptions in genes regulating segmentation and resegmentation, the underlying etiology of KFS is still inconclusive [2, 34, 35]. Previous studies suggested that mutations in GDF6 [10, 36] and GDF3 [13] were associated with autosomal dominant KFS. In addition, a truncating mutation in MEOX1 [11, 12, 37] and a homozygous nonsense mutation in MYO18B [14, 38] were identified in autosomal recessive KFS families. Using WES, a homozygous frameshift mutation (c.299delT: p. L100fs) in RIPPLY2 was confirmed as a novel gene for autosomal recessive KFS in a consanguineous family [15, 39]. In this study, by performing WES on 37 KFS patients, we investigated rare variants in known KFS-related genes, and only three variants in MYO18B were detected. However, these novel missense variants in MYO18B are still of uncertain significance. Therefore, the etiology of the disease has not been fully explained by the reported mutations associated with KFS.
Gene-based burden analysis has grown into a new approach for discovering genes associated with rare disorders, in which the mutational burden of qualifying variants is compared between case and control subjects for each gene [40, 41]. As a result, we identified five genes (BAZ1B, FREM2, VANGL1, SUFU and KMT2D) with a significantly increased number of predicted damaging exonic variants in the KFS cohort compared with in-house controls in genetic burden analysis. Pathogenic mutations in BAZ1B are frequently associated with Williams-Beuren syndrome, which is a neurodevelopmental disorder characterized by craniofacial dysmorphology, cardiovascular problems, renal abnormalities and musculoskeletal abnormalities [27]. Mutations in VANGL1 have previously been associated only with caudal regression syndrome, a rare neural tube defect disease with an abnormal development of the vertebral column and spinal cord. Furthermore, a rare case report described a female patient with Williams-Beuren syndrome combined with caudal regression syndrome. In addition to the typical developmental abnormalities, fusion of L4–5 and sacrococcygeal agenesis were reported based on radiologic evaluation [42]. These data reinforced the probability of an important role for BAZ1B and VANGL1 in the pathogenesis of congenital vertebral fusion, broadening the spectrum of known disorders related to these genes.
Missense variants and splice region mutations of SUFU were identified in two patients. Aside from the common Joubert syndrome phenotypes such as hyperpnoea, eye movements and developmental retardation, calvarial bone formation has been linked to SUFU mutations. Although SUFU has not been associated with congenital cervical fusion deformities, it has been reported that SUFU is the main negative regulator of the Sonic Hedgehog pathway, and ablation of SUFU inhibits the proliferation of osteoprogenitor cells, resulting in failure of calvarial bone formation [43, 44]. Nevertheless, the relationship between SUFU and the KFS phenotype still requires further study.
Oligogenic inheritance, characterized by the concurrent effect of two or more distinct genes on the resulting phenotype, was subsequently confirmed to apply to other skeletal deformities such as arthrogryposis [45]. Based on the results from previous studies and our findings, the etiology of congenital cervical fusion is unlikely to be fully explained by a monogenetic model for a fraction of patients. It is possible that mutation burden and combinatorial effects of rare variants in genes that interact genetically in the same biological pathways modify the phenotype of KFS due to synergistic or counteracting effects.
Conclusions
In conclusion, our study presents clinical features and WES findings from a large cohort of KFS patients to date. Beyond identifying known candidate genes, our analysis highlights five novel rare variants associated with cervical congenital fusion among KFS patients through genetic burden analysis. These results indicate a highly significant enrichment of predicted damaging genes and the potential oligogenic inheritance of KFS.
Supplementary information
Additional file 1 Table S1. List of candidate genes associated with vertebral segmentation defects as well as related diseases Table S2. Participants’ demographic and clinical characteristics Table S3. Gene burden analysis of rare variants of candidate genes between KFS cases and in-house controls
Acknowledgments
The author would like to thank the patients and their family members for their help and informed consent.
Abbreviations
- KFS
Klippel-Feil syndrome
- WES
Whole-exome sequencing
- ROM
Range of motion
- WBS
Williams-Beuren syndrome
- MRI
Magnetic resonance imaging
- CT
Computed tomography
- OMIM
Online Mendelian Inheritance in Man
- SNVs
Single-nucleotide variants
- ACMG
American College of Medical Genetics and Genomics
- GERP
Genomic Evolutionary Rate Profiling
- CADD
Combined Annotation Dependent Depletion
- SIFT
Sorting Intolerant Form Tolerant
- ESP
Exome Sequencing Project
- ExAC
Exome Aggregation Consortium
Authors’ contributions
ZL, SZ, SC, YZ, and LW performed the study. YN, HW, JH, SW, GL, XL and JC participated in the experiment and data collection/interpretation for the study. YT, ZW, and JZ conceived of the study and participated in its design. YW and NW were responsible for coordination, data collection/interpretation and proofreading of the final manuscript. The final version of the manuscript has been reviewed and approved by all the authors for publication.
Funding
This research was funded in part by the National Natural Science Foundation of China (81822030 to N.W., 81572097 and 81871746 to Y.W., 81772299 and 81930068 to Z.W., 81672123 and 81972037 to J.Z., 81902178 to S.W., 81902271 to G.L.), Beijing Natural Science Foundation (7191007 to Z.W.), CAMS Initiative Fund for Medical Sciences (2016-I2M-3-003 to N.W., 2016-I2M-2-006 and 2017-I2M-2-001 to Z.W.), National Key Research and Development Program of China (No. 2018YFC0910506 to N.W. and Z.W.).
Availability of data and materials
All data analyzed during this study is included in this published article. The raw data is available at the corresponding author upon request.
Ethics approval and consent to participate
All subjects provided informed consent to take part in the study. Written informed consent was obtained from the parents/guardians of the minors included in this study (minors are considered anyone under the age of 16). The study was approved by the Department of Scientific Research and Ethics Committee of PUMCH in China.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yipeng Wang, Email: ypwang@vip.126.com.
Nan Wu, Email: dr.wunan@pumch.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12891-020-03229-x.
References
- 1.Hensinger R, Lang J, MacEwen G. Klippel-Feil syndrome; a constellation of associated anomalies. J Bone Joint Surg Am. 1974;56:1246–1253. doi: 10.2106/00004623-197456060-00018. [DOI] [PubMed] [Google Scholar]
- 2.Tracy MR, Dormans JP, Kusumi K. Klippel-Feil Syndrome. Clin Orthop Relat Res. 2004;424:183–190. doi: 10.1097/01.blo.0000130267.49895.20. [DOI] [PubMed] [Google Scholar]
- 3.Brown MW, Templeton AW, Hodges FJ. The incidence of acquired and congenital fusions in the cervical spine. Am J Roentgenol Radium Ther Nucl Med. 1964;92:1255–1259. [PubMed] [Google Scholar]
- 4.Gruber J, Saleh A, Bakhsh W, Rubery PT, Mesfin A. The prevalence of Klippel-Feil syndrome: a computed tomography-based analysis of 2,917 patients. Spine Deformity. 2018;6:448–453. doi: 10.1016/j.jspd.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Nouri A, Tetreault L, Zamorano JJ, Mohanty CB, Fehlings MG. Prevalence of Klippel-Feil syndrome in a surgical series of patients with cervical Spondylotic myelopathy: analysis of the prospective, multicenter AOSpine North America study. Global Spine J. 2015;5:294–299. doi: 10.1055/s-0035-1546817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xue X, Shen J, Zhang J, Tian Y, Zhao H, Wang Y, et al. Klippel-Feil syndrome in congenital scoliosis. Spine (Phila Pa 1976) 2014;39:E1353–E1358. doi: 10.1097/BRS.0000000000000587. [DOI] [PubMed] [Google Scholar]
- 7.Mesfin A, Bakhsh WR, Chuntarapas T, Riew KD. Cervical scoliosis: clinical and radiographic outcomes. Global Spine J. 2016;6:7–13. doi: 10.1055/s-0035-1554776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samartzis DD, Herman J, Lubicky JP, Shen FH. Classification of congenitally fused cervical patterns in Klippel-Feil patients: epidemiology and role in the development of cervical spine-related symptoms. Spine. 2006;31:E798–E804. doi: 10.1097/01.brs.0000239222.36505.46. [DOI] [PubMed] [Google Scholar]
- 9.Cho W, Shepard N, Arlet V. The etiology of congenital scoliosis: genetic vs. environmental-a report of three monozygotic twin cases. European spine journal : official publication of the European spine society, the European spinal deformity society, and the European section of the cervical spine research. Society. 2018;27:533–537. doi: 10.1007/s00586-018-5604-2. [DOI] [PubMed] [Google Scholar]
- 10.Tassabehji M, Fang ZM, Hilton EN, McGaughran J, Zhao Z, de Bock CE, et al. Mutations in GDF6 are associated with vertebral segmentation defects in Klippel-Feil syndrome. Hum Mutat. 2008;29:1017–1027. doi: 10.1002/humu.20741. [DOI] [PubMed] [Google Scholar]
- 11.Bayrakli F, Guclu B, Yakicier C, Balaban H, Kartal U, Erguner B, et al. Mutation in MEOX1 gene causes a recessive Klippel-Feil syndrome subtype. BMC Genet. 2013;14:95. doi: 10.1186/1471-2156-14-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohamed JY, Faqeih E, Alsiddiky A, Alshammari MJ, Ibrahim NA, Alkuraya FS. Mutations in MEOX1, encoding mesenchyme homeobox 1, cause Klippel-Feil anomaly. Am J Hum Genet. 2013;92:157–161. doi: 10.1016/j.ajhg.2012.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye M, Berry-Wynne KM, Asai-Coakwell M, Sundaresan P, Footz T, French CR, et al. Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum Mol Genet. 2010;19:287–298. doi: 10.1093/hmg/ddp496. [DOI] [PubMed] [Google Scholar]
- 14.Alazami AM, Kentab AY, Faqeih E, Mohamed JY, Alkhalidi H, Hijazi H, et al. A novel syndrome of Klippel-Feil anomaly, myopathy, and characteristic facies is linked to a null mutation in MYO18B. J Med Genet. 2015;52:400–404. doi: 10.1136/jmedgenet-2014-102964. [DOI] [PubMed] [Google Scholar]
- 15.Karaca E, Yuregir OO, Bozdogan ST, Aslan H, Pehlivan D, Jhangiani SN, et al. Rare variants in the notch signaling pathway describe a novel type of autosomal recessive Klippel-Feil syndrome. Am J Med Genet Part A. 2015;167a:2795–2799. doi: 10.1002/ajmg.a.37263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chacón-Camacho O, Camarillo-Blancarte L, Pelaez-González H, Mendiola J, Zenteno JC. Klippel-Feil syndrome associated with situs inversus: description of a new case and exclusion of GDF1, GDF3 and GDF6 as causal genes. Eur J Med Genet. 2012;55:414–417. doi: 10.1016/j.ejmg.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Wang K, Zhao S, Liu B, Zhang Q, Li Y, Liu J, et al. Perturbations of BMP/TGF-β and VEGF/VEGFR signalling pathways in non-syndromic sporadic brain arteriovenous malformations (BAVM) J Med Genet. 2018;55:675–684. doi: 10.1136/jmedgenet-2017-105224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11:1–9. doi: 10.1038/nprot.2015.123. [DOI] [PubMed] [Google Scholar]
- 22.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 24.Blue GM, Ip E, Walker K, Kirk EP, Loughran-Fowlds A, Sholler GF, et al. Genetic burden and associations with adverse neurodevelopment in neonates with congenital heart disease. Am Heart J. 2018;201:33–39. doi: 10.1016/j.ahj.2018.03.021. [DOI] [PubMed] [Google Scholar]
- 25.Schieffer KM, Varga E, Miller KE, Agarwal V, Koboldt DC, Brennan P, et al. Expanding the clinical history associated with syndromic Klippel-Feil: a unique case of comorbidity with medulloblastoma. Eur J Med Genet. 2019;62:103701. doi: 10.1016/j.ejmg.2019.103701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berger J, Berger S, Li M, Currie PD. Myo18b is essential for sarcomere assembly in fast skeletal muscle. Hum Mol Genet. 2017;26:1146–1156. doi: 10.1093/hmg/ddx025. [DOI] [PubMed] [Google Scholar]
- 27.Lalli MA, Jang J, Park JH, Wang Y, Guzman E, Zhou H, et al. Haploinsufficiency of BAZ1B contributes to Williams syndrome through transcriptional dysregulation of neurodevelopmental pathways. Hum Mol Genet. 2016;25:1294–1306. doi: 10.1093/hmg/ddw010. [DOI] [PubMed] [Google Scholar]
- 28.Shafeghati Y, Kniepert A, Vakili G, Zenker M. Fraser syndrome due to homozygosity for a splice site mutation of FREM2. Am J Med Genet Part A. 2008;146a:529–531. doi: 10.1002/ajmg.a.32091. [DOI] [PubMed] [Google Scholar]
- 29.Yu Q, Lin B, Xie S, Gao S, Li W, Liu Y, et al. A homozygous mutation p.Arg2167Trp in FREM2 causes isolated cryptophthalmos. Hum Mol Genet. 2018;27:2357–2366. doi: 10.1093/hmg/ddy144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong C, Umar M, Bartoletti G, Gahankari A, Fidelak L, He F. Expression pattern of Kmt2d in murine craniofacial tissues. Gene Expr Patterns. 2019;34:119060. doi: 10.1016/j.gep.2019.119060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roma D, Palma P, Capolino R, Figà-Talamanca L, Diomedi-Camassei F, Lepri FR, et al. Spinal ependymoma in a patient with kabuki syndrome: a case report. BMC Med Genet. 2015;16:80. doi: 10.1186/s12881-015-0228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Faundes V, Malone G, Newman WG, Banka S. A comparative analysis of KMT2D missense variants in kabuki syndrome, cancers and the general population. J Hum Genet. 2019;64:161–170. doi: 10.1038/s10038-018-0536-6. [DOI] [PubMed] [Google Scholar]
- 33.Samartzis D, Kalluri P, Herman J, Lubicky JP, Shen FH. "clinical triad" findings in pediatric Klippel-Feil patients. Scoliosis Spinal Disord. 2016;11:15. doi: 10.1186/s13013-016-0075-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giampietro PF, Raggio CL, Blank RD, McCarty C, Broeckel U, Pickart MA. Clinical, genetic and environmental factors associated with congenital vertebral malformations. Mol Syndromol. 2013;4:94–105. doi: 10.1159/000345329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rolfe RA, Bezer JH, Kim T, Zaidon AZ, Oyen ML, Iatridis JC, et al. Abnormal fetal muscle forces result in defects in spinal curvature and alterations in vertebral segmentation and shape. J Orthop Res. 2017;35:2135–2144. doi: 10.1002/jor.23518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei A, Shen B, Williams LA, Bhargav D, Gulati T, Fang Z, et al. Expression of growth differentiation factor 6 in the human developing fetal spine retreats from vertebral ossifying regions and is restricted to cartilaginous tissues. J Orthop Res. 2016;34:279–289. doi: 10.1002/jor.22983. [DOI] [PubMed] [Google Scholar]
- 37.Dauer MVP, Currie PD, Berger J. Skeletal malformations of Meox1-deficient zebrafish resemble human Klippel-Feil syndrome. J Anat. 2018;233:687–695. doi: 10.1111/joa.12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ajima R, Akazawa H, Kodama M, Takeshita F, Otsuka A, Kohno T, et al. Deficiency of Myo18B in mice results in embryonic lethality with cardiac myofibrillar aberrations. Genes Cells. 2008;13:987–999. doi: 10.1111/j.1365-2443.2008.01226.x. [DOI] [PubMed] [Google Scholar]
- 39.McInerney-Leo AM, Sparrow DB, Harris JE, Gardiner BB, Marshall MS, O'Reilly VC, et al. Compound heterozygous mutations in RIPPLY2 associated with vertebral segmentation defects. Hum Mol Genet. 2015;24:1234–1242. doi: 10.1093/hmg/ddu534. [DOI] [PubMed] [Google Scholar]
- 40.Nicolaou N, Pulit SL, Nijman IJ, Monroe GR, Feitz WF, Schreuder MF, et al. Prioritization and burden analysis of rare variants in 208 candidate genes suggest they do not play a major role in CAKUT. Kidney Int. 2016;89:476–486. doi: 10.1038/ki.2015.319. [DOI] [PubMed] [Google Scholar]
- 41.Clarke CM, Fok VT, Gustafson JA, Smyth MD, Timms AE, Frazar CD, et al. Single suture craniosynostosis: identification of rare variants in genes associated with syndromic forms. Am J Med Genet A. 2018;176:290–300. doi: 10.1002/ajmg.a.38540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singer G, Schalamon J, Ainoedhofer H, Petek E, Kroisel PM, Höllwarth ME. Williams-Beuren syndrome associated with caudal regression syndrome and coagulopathy--a case report. J Pediatr Surg. 2005;40:e47–e50. doi: 10.1016/j.jpedsurg.2005.07.048. [DOI] [PubMed] [Google Scholar]
- 43.Li J, Cui Y, Xu J, Wang Q, Yang X, Li Y, et al. Suppressor of fused restraint of hedgehog activity level is critical for osteogenic proliferation and differentiation during calvarial bone development. J Biol Chem. 2017;292:15814–15825. doi: 10.1074/jbc.M117.777532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Mori R, Romani M, D'Arrigo S, Zaki MS, Lorefice E, Tardivo S, et al. Hypomorphic recessive variants in SUFU impair the sonic hedgehog pathway and cause Joubert syndrome with Cranio-facial and skeletal defects. Am J Hum Genet. 2017;101:552–563. doi: 10.1016/j.ajhg.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pehlivan D, Bayram Y, Gunes N, Coban Akdemir Z, Shukla A, Bierhals T, et al. The genomics of Arthrogryposis, a complex trait: candidate genes and further evidence for Oligogenic inheritance. Am J Hum Genet. 2019;105:132–150. doi: 10.1016/j.ajhg.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 Table S1. List of candidate genes associated with vertebral segmentation defects as well as related diseases Table S2. Participants’ demographic and clinical characteristics Table S3. Gene burden analysis of rare variants of candidate genes between KFS cases and in-house controls
Data Availability Statement
All data analyzed during this study is included in this published article. The raw data is available at the corresponding author upon request.