

Abstract
As obligate parasites of cellular processes, viruses must take over cellular macromolecular machinery. It is also becoming clear that viruses routinely control intracellular signaling pathways through the direct or indirect control of kinases and phosphatases. This control of cellular phosphoproteins is important to promote a variety of viral processes, from control of entry to nuclear function to the stimulation of viral protein synthesis. This review focuses on the takeover of the cellular phosphatidylinositol-3-kinase (PI3K)/Akt signaling pathway by a variety of retroviruses, DNA viruses, and RNA viruses, highlighting the functions ascribed to virus activation of PI3K and Akt activity. This review also describes the role that the PI3K/Akt pathway plays in the host response, noting that it that can trigger anti- as well as proviral functions.
Keywords: Virus replication, Innate immune response, Tumorigenesis, PI3K, Akt, mTOR, Translational control, Stress response, Apoptosis
Abbreviations
- 4E-BP1
eIF4E binding protein 1
- CMV
cytomegalovirus
- DNA
deoxyribonucleic acid
- EGFR
epidermal growth factor receptor
- FAK
focal adhesion kinase
- FOXO1
forkhead box O1
- HSV1
herpes simplex virus 1
- IL-1
interleukin-1
- IL-6
interleukin-6
- IL-8
interleukin-8
- JEV
Japanese encephalitis virus
- mTOR
mammalian target of rapamycin
- mTORC2
mTOR complex 2
- NF-κB
nuclear factor kappa B
- NSP1
nonstructural protein 1
- P70S6k
p70 ribosomal protein S6 kinase
- PI3K
phosphatidyl inositol 3 kinase
- PP2A
protein phosphatase 2A
- RNA
ribonucleic acid
- rpS6
ribosomal protein S6
- RSV
respiratory syncytial virus
- RTK
receptor tyrosine kinase
- SARS
severe acute respiratory syndrome
- SV40
simian virus 40
I. Introduction
Viral disease is one of the most significant human health concerns in modern medicine, manifesting as a broad range of acute infections, chronic diseases, and virus-associated malignancies. Viruses such as human immunodeficiency virus (HIV), hepatitis C, influenza, rotavirus, chikungunya, and dengue affect millions of people each year worldwide. The emergence of novel strains of H1N1 flu1 and previously unrecognized pathogens such as severe acute respiratory syndrome (SARS) coronavirus2 and SFTS bunyavirus3, 4 has highlighted the threats of viral emergence and pandemic with which humanity regularly contends. This, along with the emergence of antiviral resistance among viruses such as influenza, highlights the need to better understand virus replication and the virus–host interaction to identify critical restriction points that can be targets for antiviral drug development and vaccine generation.
One controlling aspect of viral pathogenesis and replication is their dependence on the cells that they infect. Unlike other microbial pathogens such as bacteria and fungi which are capable of autonomous growth, viruses are completely dependent on cellular processes for replication. It has long been recognized that viruses are dependent upon the use of cellular machinery for protein synthesis: nucleotide and protein building blocks, ribonucleic acid (RNA) and deoxyribonucleic acid (DNA) polymerases, and membranes. More recently, it has become clear that viruses also rely upon intracellular communication.5, 6 Particularly, viruses appear to be adept at capturing protein phosphorylation cascades for their own use.
Most, if not all, viruses require protein phosphorylation in order to replicate. This would initially suggest that viruses would gain a replication edge by generally promoting protein phosphorylation. However, simply promoting all cellular protein phosphorylation is not likely to be advantageous to viruses, as the activity of some host kinases (and the resultant phosphorylation events) are known to antagonize viral replication. Thus, a nonspecific increase in phosphorylation would trigger an antiviral response. Viruses have a need for specificity in activating host kinases and phosphatases. Simply stated, viruses need to activate signaling pathways that promote their replication7, 8, 9 while blocking or avoiding the activation of antiviral signaling pathways.10
Controlling kinase activity can promote protein translation,11 increase metabolic activity, drive cell division, and inhibit cellular apoptosis. All of these are advantageous for virus replication. In addition, many viruses produce virus proteins that require direct phosphorylation in order to properly function but make no kinase that will carry out this phosphorylation.9, 10 This dictates that they must recruit kinase activity from their host.
It is therefore not surprising that viruses have developed mechanisms to control cellular signaling through regulating kinase and phosphatase signaling cascades. What is surprising is the commonality of host pathways that are targeted. In theory, any number of kinases might be important for the replication of any particular virus, but this possibility of each virus using different kinases is not actually observed. Rather, as is emphasized below, one signaling pathway has emerged as a common player in the replication of many different viruses—the phosphatidylinositol-3-kinase (PI3K)/Akt phosphorylation cascade.
The repeated identification of the PI3K/Akt pathway as a central player in virus replication has underscored the importance of this pathway as a central regulator of cell health and metabolism. As a central regulator of multiple cellar processes that control translation, metabolism, and cell death, active PI3K/Akt signaling can fulfill many viral “needs.” This is not to suggest that this signaling pathway only provides positive feedback for the virus. Studies of virus interaction with this pathway have also shown that the pathway is not simply a “proviral” kinase when activated, but that it also acts as part of the host response to viral infection. This suggests that PI3K/Akt signaling represents a two-faced player in interactions between the virus and cell, able to both promote viral replication and be an active part of the immune response that will eventually quash virus replication. This review will describe the different approaches that viruses have taken to dominate the PI3K/Akt signaling pathway. The viruses chosen in this review highlight the varied manner in which this pathway is manipulated and turned to the advantage of the invader, while also pointing out how the pathway also plays a role in host defense.
II. Overview of the PI3K/Akt Signaling Pathway
The PI3K/Akt signal transduction cascade is a classical phosphorylation cascade that utilizes tyrosine, lipid, and serine–threonine phosphorylation to transduce external signals to internal responses (see Fig. 1 for a schematic diagram). Normally, signaling through this pathway is initiated by the stimulation of a receptor tyrosine kinase (RTK) by a cytokine or a growth factor at the cell surface. Activation of the RTK recruits and activates PI3K. PI3K is a heterodimer consisting of a p110 catalytic and a p85 regulatory subunit.12 PI3K is responsible for converting phosphatidylinositol 4,5-bisphosphate (PIP2) to 3,4,5-triphosphorylated phosphoinositide (PIP3). PIP3-rich membrane domains serve as docking sites for proteins that contain a pleckstrin homology domain such as Akt.
Fig. 1.

Overview of the PI3K/Akt pathway. The diagram illustrates the two subunits of PI3K (p85 and p110) bound to an activated receptor tyrosine kinase (classical examples are the epidermal growth factor and the insulin receptor). Active PI3K catalyzes the addition of a single phosphate to the 3 position of PIP4,5-bisphosphate to make PIP3. This triphosphorylated inositol serves as an affinity ligand for the pleckstrin homology domains of the Akt kinase PDK1 and Akt, with the binding of PIP3 by the Akt PH domain serving to uncover the activation domain of Akt. Phosphorylation by PDK1 occurs at position T308, and a second activating phosphorylation occurs at S473. Active Akt will phosphorylate various downstream protein targets that control cell growth (AMPK, p21, and GSK3 are illustrated) and translational control (mTOR and its downstream effectors p70S6K and 4E-BP1 are illustrated) and act to suppress apoptosis (inhibitory phosphorylation of BAD and FOXO1 are illustrated).
The recruitment of inactive Akt protein to PIP3-rich areas of the plasma membrane results in a conformational change that exposes the activation loop of Akt.13 Akt's activating kinase, phosphoinositide-dependent protein kinase (PDK1)14, is also recruited to PIP3 microdomains. PDK1 phosphorylates Akt on threonine 308 (Thr308) of the exposed activation loop, activating Akt and leading to a second phosphorylation of Akt at serine 473 (Ser473) by a kinase presumed to be mTORC2 that further potentiates kinase activity.13, 15, 16, 17
Activated Akt can control the signaling of several key pathways supporting different types of cellular functions (Fig. 1). The PI3/Akt signaling pathway promotes cell growth, cell survival, and tumorigenesis through the phosphorylation and inactivation of cell cycle kinase inhibitors such as p21 and the inactivation of transcription factors that can inhibit cell cycle progression and promote apoptosis such as the forkhead family of proteins (e.g., FOXO118). Akt also can block the actions of metabolically repressive kinases such as AMPK19 and so can drive the up regulation of metabolic activity in the cell.20
Akt also promotes cellular translation through GSK3 phosphorylation and the activation of mammalian target of rapamycin complex 1 (mTORC1), a kinase complex that activates ribosomal S6 kinase and inhibits the translational repressor eIF4E-BP1 (4E-BP1).21 Akt activation of mTORC1 is indirect. Akt phosphorylates and represses the hamartin and tuberin complex (TSC1 and TSC222). Inhibition of TSC1/TSC2 allows Rheb-mediated activation of mTORC1 complex kinase activity. MTORC1 phosphorylates and activates the ribosomal protein S6 kinase (p70S6K) and inactivates the translation suppressor 4EBP1, leading to increased translation.
Akt activity antagonizes apoptotic signaling. This is accomplished through direct actions such as inhibiting pro-apoptotic factors (e.g., phosphorylation and inhibition of BAD,23 a pro-apoptotic member of the BCL-2 family) and through more indirect actions, such as the activation of the transcription factor nuclear factor kappa B (NF-κB)24 and, as mentioned above, the inactivation of FOXO1. With actions that influence multiple phosphoprotein cascades involved in different core aspects of cell function, PI3K/Akt signaling pathway is a prime candidate for a “hub” kinase whose activation can have a diverse set of consequences, all of which are centered on promoting cell survival and increasing the metabolic capacity of the cell.
III. Virus Modulation of PI3K/Akt Signaling
With the central nature of the PI3K/Akt pathway in controlling cell functions, it stands to reason that the control of such a signaling “hub” would greatly benefit an invading virus that is dependent upon cellular functions. Extensive research supports this hypothesis, finding viral domination of this pathway in various contexts.5, 25, 26 To illustrate the different ways in which viruses attack and control the pathway, this review will explain different examples of how viruses co-opt the host signaling apparatus through the PI3K/Akt pathway for selfish gain. The review will also highlight how viral activation of this pathway can contribute to the host's response to viral infection. This review will classify virus interactions with the PI3K/Akt pathway based on the aspect(s) of PI3K/Akt signaling from which each virus benefits. This approach is complementary to other reviews which have taken the approach of classifying the interaction based on virus families.5, 26
The first examples presented here highlight viruses that are suggested to have “simple” interaction paradigms. For the purpose of this review, a “simple” interaction is one where the virus or virus family appears to activate the PI3K/Akt pathway to benefit from just one aspect of its cellular control, such as the promotion of cell cycle progression, or the inhibition of apoptosis. Following this, examples of viruses that benefit from several of the outcomes of PI3K/Akt signaling will be discussed.
A. Simple Paradigms of Viruses That Interact with PI3K/Akt
1. Viral Proteins That use Molecular Mimicry to Activate the PI3K/Akt Signaling Axis
Several viral proteins are known to directly activate the PI3K/Akt signaling axis through molecular mimicry or replacement (Fig. 2 ). These viral proteins helped in elucidating the initial understanding of the PI3K/Akt signaling cascade. The paradigm example v-akt was originally described as a retroviral oncogene encoded by the AKT8 mouse transforming retrovirus.27 The kinase was described as a fusion protein with the viral gag (matrix protein). It was quickly recognized that the protein had a cellular homolog,28 which was named c-akt. This protein was later determined to be the same as the cellular kinases that had been characterized under the names of PKB29 and RAC-PK.30 Though the role of v-akt in the life cycle of the AKT8 virus was never established, v-akt is capable of transforming cells, suggesting that the virus utilized the v-akt protein to drive cell cycle progression. The finding that a cellular kinase had been essentially directly incorporated into the viral genome as a fusion protein with the viral gag protein was the first demonstration of the importance of this kinase for viral replication.
Fig. 2.
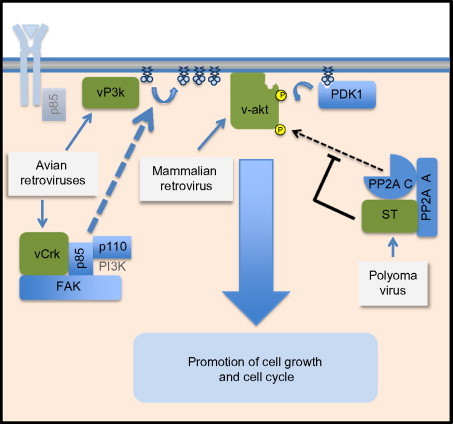
Viral mimicry of PI3K pathway members. The diagram illustrates steps at which viral proteins either replace or actively modulate Akt signaling. Shown are the vPI3K (p110 subunit) produced by the avian retrovirus A16, and the vAkt protein produced by the mammalian AKT8 retrovirus which directly activates Akt signaling. Also shown is vCrk, which stimulates Akt signaling in a focal adhesion kinase (FAK)-dependent manner. Illustrated on the right-hand side of the figure is the SV40 small t (smt) protein. The smt protein binds the PP2A/C heterodimer and inactivates Akt dephosphorylation, providing an indirect but potent activation of Akt kinase activity. The actions of all these proteins can drive cell proliferation in a manner that is dependent on Akt-phosphorylated proteins.
2. Oncoretrovirus Control of PI3K/Akt Signaling
The control of the PI3K/Akt signaling pathway by molecular replacement extends to several other viruses, notably avian sarcoma viruses. The AV16 sarcoma virus encodes for a PI3K homolog, named vP3K,31 which encodes a homolog of the cellular catalytic subunit of PI3K (p110). Like v-Akt, vP3K acts to provide constitutively active PI4,5-bisphosphate kinase activity without the need for the p85 regulatory subunit, generating high levels of PIP3. This serves to activate Akt,32 which phosphorylates and inactivates the FOXO1 transcription repressor33 and promotes cellular translation through the phosphorylation of 4E-BP1.34 These actions remove a cell-cycle brake, allowing proliferative signals to dominate.
A slightly different approach to activation of the PI3K/Akt pathway activation is taken by the CT10 and AV1 viruses. These viruses each encode a homolog to the cellular Crk protein.35, 36, 37 vCrk activates the PI3K/Akt signaling pathway through the recruitment of PI3K to the focal adhesion kinase.38 This activity leads to the transformation of vCrk-infected cells39 presumably by mechanisms similar to those described for vP3K (activation of cellular translation and inactivation of transcription factors such as FOXO1).
In all of these examples, a consistent theme is evident—retroviruses have repeatedly captured host proteins that can activate the PI3K/Akt pathway. It is thought that the cell proliferation induced by these proteins is critical for the replication of these viruses, as the breakdown of the nuclear envelope during the cell cycle gives these retroviruses access to cellular DNA and thereby allows the virus to integrate within this DNA. Further, proliferating cells are more likely to activate retroviral gene expression to stimulate virus production. However, this may not be the only role for the PI3K/Akt pathway in the retrovirus life cycle, and more recent research into viruses, such as HIV, suggests that the virus may benefit from inducing Akt activity to prevent premature apoptosis.40
B. Potentiation of Akt Signaling Through the Direct Control of Phosphatase Activity
1. Polyomavirus Control of Akt Signaling
Viruses have also adopted approaches of inducing PI3K/Akt pathway signaling by attacking phosphatase activity. As is true in most kinase cascades, the activity of Akt signaling can be decreased through phosphatase-mediated dephosphorylation of Akt activation domains. Conversely, kinase signaling can be potentiated by decreasing phosphatase activity. For PI3K/Akt signaling, the cellular phosphatase PP2A is at least partially responsible for the dephosphorylation and inactivation of Akt.41, 42 By decreasing the activity of PP2A, more Akt will remain in the active state. This approach to activating Akt signaling has been adopted by the genus Polyomaviridae. Simian virus 40 (SV40) is the prototype example of a polyomavirus that stimulates Akt activation by inhibiting the dephosphorylation of Akt.43 To do this, the SV40 small t antigen of SV40 (SV40ST) binds the cellular phosphatase PP2A that is responsible for a large fraction of Akt phosphatase activity.44 PP2A is a heterotrimeric protein comprising a scaffolding subunit (A), a catalytic subunit (C), and a regulatory subunit (B). There are many variants of the B subunit, and each is believed to be responsible for directing the phosphatase catalytic subunit directly to a single substrate (such as Akt) or set of substrates. SV40ST displaces the B subunit from the A and C PP2A subunits, inhibiting much of the phosphatase activity which would normally dampen or reverse Akt and mTOR activation (see Fig. 2). The resulting potentiation of Akt signaling helps drive cell proliferation, moving the cell into S phase, where virus DNA synthesis is initiated.
This modulation of PP2A to alter PI3K/Akt signaling is utilized by other viruses in the polyomavirus genus as well, including BK virus,45 mouse polyoma virus,46 and JC virus.47 In addition, polyomaviruses can indirectly stimulate the PI3K/Akt pathway through additional mechanisms,48, 49, 50 indicating that these viruses attack the PI3K/Akt pathway at multiple points to ensure pathway activation.
2. Papillomavirus Control of Akt Signaling
A similar mechanism of activating Akt signaling (inhibition of PP2A activity that increases Akt activation) has been proposed for the E7 oncoprotein of papillomavirus HPV.51 The HPV E7 protein is critical for the maintenance of cellular DNA synthesis in HPV-infected cells,52 which is a necessity, as HPV requires the host cell DNA synthesis machinery for its replication.53 Thus, activation of Akt to remove brakes on cell DNA synthesis (i.e., FOXO) is of clear value to virus replication. This action is also important in the cases where the E7 gene is incorporated into host DNA (not part of the normal virus life cycle). Under these circumstances, Akt signaling may promote malignant transformation,51 thereby underscoring a potential link between viral control of cellular phosphorylation for its own means and the induction of cancer.
3. Virus Activation of Akt to Promote an Antiapoptotic Signal
In the examples presented above, virus takeover of the Akt signaling pathway was associated with the induction of cellular proliferation. In many examples, though, viruses that activate the PI3K/Akt pathway do not benefit from the promotion of cellular proliferation or cellular transformation. For many viruses, the PI3K/Akt pathway can provide the short-term benefit of keeping the infected cell alive through the suppression of apoptotic signals. The critical nature of PI3K/Akt-mediated suppression of apoptosis is best illustrated by viruses that cause acute infections, such as the RNA viruses.
For most RNA viruses, their life cycle is carried out in the cytoplasm of their host cell. These viruses therefore have minimal requirements for nuclear components or cell cycle progression, and in many cases block nuclear functions and cell-cycle progression.54, 55, 56 Thus, PI3K/Akt-mediated promotion of cell division or alteration of transcription does not play an essential role in virus replication. Instead, phosphorylation of cytoplasmic proteins in order to delay apoptosis keeps the host cell alive until viral progeny can be produced.
4. Activation of Akt Signaling by Paramyxoviruses
The trigger for PI3K/Akt activation in infection with paramyxoviruses often occurs early in infection. As an example, respiratory syncytial virus (RSV), a significant pediatric pathogen, activates PI3K/Akt signaling at the level of PI3K activation.57 Pathway stimulation appears to involve the activation of sphingosine kinase, which, in turn, leads to the phosphorylation and activation of Akt through a mechanism that may involve the extracellular signaling of sphingosine 1 phosphate.58 Akt activity is critical for delaying cellular apoptosis during RSV infection.57
Similarly, the genetically related Sendai virus, human parainfluenza virus 5 (HPIV5), and HPIV3 have also been shown59 or suggested59, 60 to activate PI3K and Akt early in infection, an action associated with delaying apoptosis and potentially activating the viral polymerase. For all of these viruses, signaling through the Akt pathway allows the completion of the viral replication cycle. Inhibition of PI3K or Akt signaling leads to faster apoptosis59 and, in some reports, reduced virus growth.60 In the case of Sendai virus, Akt activation may also play a role in the ability of this virus to establish a persistent infection in cells in tissue culture.59
5. Activation of Akt Signaling by Picornaviruses
Viruses in the picornavirus family are also reported to activate PI3K/Akt signaling. For both poliovirus and the common-cold-causing rhinovirus, viral attachment and entry serve as the trigger for PI3K/Akt activity.61 For poliovirus, the exact trigger is unknown; for rhinovirus, binding of the virus to its receptor recruits the tyrosine kinase Syk, which interacts directly with the p85 subunit of PI3K and activates PI3K/Akt signaling.62, 63 It is likely that the activation of Akt is important for avoiding cell death in the case of both viruses. An important role for Akt in forestalling apoptosis has been shown in the case of poliovirus, where the Akt phosphorylation of Ask1 limits the activity of its downstream effector Jnk1 and thereby decreases apoptotic signals.
Cardioviruses also appear to manipulate Akt activity to limit apoptosis in infected cells. For both encephalomyocarditis virus (EMCV) and coxsackievirus, viral infection is associated with the stimulation of PI3K/Akt activity.64 Unlike poliovirus and rhinovirus infection, the activation of PI3K does not appear to be entry dependent.64 Activation is important for suppression of apoptosis and promotion of virus replication. For coxsackievirus, Akt activation results in the stimulation of NF-κB activity, which promotes cell survival.65
6. Activation of Akt Signaling by Reoviruses
PI3K/Akt activation to promote the survival of infected cells is also seen during infection of cells with double-stranded RNA viruses of the reovirus family. As an example, rotavirus stimulates PI3K/Akt activity within the first 2 h of infection.66 Through the use of both mutant virus and viral protein overexpression approaches, it has been shown that Akt activation is carried out by the rotavirus nonstructural protein 1 (NSP1).67 The activation of Akt by NSP1 can occur even in the absence of other viral components and involves a direct interaction between NSP1 and PI3K (see Fig. 3 ), suggesting that the protein alters kinase activity. The mechanism of this targeting is currently unknown. Activation of Akt has also been suggested for the avian reovirus A1133.68 The activation of Akt in rotavirus-infected cells suppresses apoptosis to allow the completion of the virus life cycle.69 Akt signaling in infected cells is also required for the expression of integrin proteins66 which can strongly promote both survival signaling and virus attachment, suggesting that Akt activation may send survival signals and promote viral attachment in infected cells.
Fig. 3.
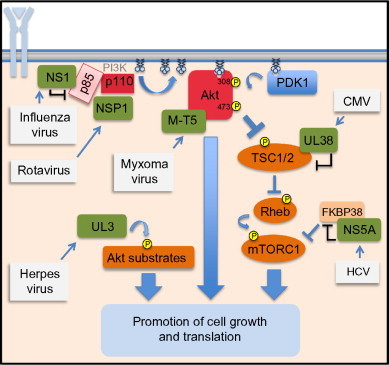
Virus proteins that directly interact with the Akt signaling pathway. Illustrated on the left-hand side of the diagram are activation of PI3K by the influenza NS1 protein through direct interaction with the p85 subunit and the activation of Akt by the rotavirus NSP1 protein. Illustrated in the center of the image are the myxomavirus Akt activator M-T5 and the herpes virus kinase UL3, a viral ortholog of the Akt kinase. Illustrated on the right-hand side of the diagram is action of the hepatitis C protein NS5A to bind FKBP38, relieving an inhibitory regulation to activate mTOR. Also illustrated is action of UL38, which inhibits TSC2, also relieving an inhibitory regulation to activate mTOR.
7. Flavivirus Activation of PI3K/Akt Signaling
Like many viruses, the flaviviruses dengue virus and Japanese encephalitis virus70 stimulate PI3K/Akt activation within the first few hours of infection. For these viruses, the mechanism of activation is unknown, but activation appears to be transient70 and curiously does not seem to impact virus replication in any fashion. Hepatitis C, another flavivirus, has not been reported to activate either PI3K or Akt but has been shown to promote cell survival through the activation of the downstream effector, mTOR kinase.71 This activation is facilitated by the viral nonstructural protein NS5A, which has the capacity to bind directly to FKBP38, a negative regulator of mTOR72 (see Fig. 3). The association of FKBP38 with mTORC1 is blocked by NS5A binding, thereby stimulating mTORC1 activity, promoting translation, and limiting apoptosis. This mechanism of targeting mTORC1 by relieving an inhibitor action is seen in the actions of a cytomegalovirus (CMV) virus protein, UL38 (see below).
C. Viruses That Rely on Multiple Signaling Endpoints of the PI3K/Akt Pathway
As described in the introduction, the PI3K/Akt pathway acts as a signaling “hub” triggering a branching phosphorylation cascade that alters multiple cellular processes. The viruses described above appear to benefit primarily from one aspect of “hub” activation or one subset of the phosphoproteins. This is not true of all viruses. Some viruses benefit from more than one signaling arm of the PI3K/Akt pathway. Three examples of this more complex virus/host interaction illustrate this well: the domination of signaling by herpes viruses, poxviruses, and the influenza virus.
1. Herpes Viruses Benefit from the Stimulation of Translation Through PI3K/Akt Signaling
The herpes virus family consists of several viruses that have each taken differing approaches to achieve the same result: preservation of a favorable environment for both viral transcription and viral translation. The life cycle of these viruses is particularly pertinent to their interaction with the PI3K/Akt pathway, as herpes viruses have both a lytic phase, where they actively replicate in cells, and a latent phase, where they are maintained as episomes in the host genome. This dual life cycle means the virus requires different cell environments at different times. During latent infection, the virus requires little from the host, while during the lytic phase, many host factors and a high metabolic throughput are desirable.
2. Epstein-Barr Virus Activation of PI3K/Akt Signaling
One example of how herpes viruses interact with PI3K/Akt signaling is the Epstein–Barr virus (EBV). EBV is associated with Burkitt lymphoma/leukemia and other lymphoproliferative disorders. EBV establishes a latent stage in which most viral proteins are not produced and replication is dormant. In this phase, virus-infected cells produce a latency protein LMP2A. LMP2A promotes Akt phosphorylation, provides a general antiapoptotic signal, and also promotes c-MYC translation through translational upregulation caused by mTOR activation.73 The development of EBV-driven Burkitt lymphoma/leukemia is dependent upon the constitutive expression of c-MYC, which modulates the expression of target genes that encode many cellular processes including cell growth, division, and apoptosis. LMP2A protects cells that express c-MYC from apoptosis through the upregulation of antiapoptotic genes.74
LMP1, another latency-associated protein produced by the EBV genome, also activates the Akt pathway.75 For LMP1, the activation of Akt activity is associated with a nuclear function—the inhibition of DNA damage repair—and not protein synthesis promotion. This suggests the LMP1 and LMP2 proteins of EBV target different pools of PI3K and Akt to control the function of this signaling pathway in different cellular compartments.
The activity of Akt is also targeted in the lytic phase of EBV replication. Expression of BRLF1, a protein that induces lytic replication of EBV, results in the activation of Akt in overexpression studies, suggesting that pathway PI3K activity is an important signal for the emergence of the virus from latency.76 Consistent with this, inhibition of PI3K signaling blocks EBV reactivation from a latent state.
This dual use of the PI3K/Akt signaling for both latent and lytic replication reveals an interesting paradigm for cellular signaling in virus infection. Akt activity appears to be a basic requirement for the survival of EBV-infected cells, but cannot in itself be a trigger for reactivation. Akt activity is much more likely to be a basal maintenance factor. Further research will undoubtedly uncover true triggers that cooperate with the PI3K/Akt pathway to drive reactivation. In the interim, the apparent requirement for PI3K/Akt signaling to maintain a latent state and help drive reactivation has identified an Achilles heel of EBV-infected cells. Targeting EBV-infected cells for death by adding inhibitors of this pathway are showing initial promise as a way to inhibit EBV + tumor growth.77
3. Herpes Virus Activation of PI3K/Akt Signaling
Unlike EBV, the activation of the PI3K/Akt phosphorylation cascade in herpes simplex virus 1 infection is transient.78 The activation has been proposed to be due to the actions of the viral VP11/12 protein,79 though virus binding to the cell surface has also been proposed as the activating trigger.80 Despite the transient nature of Akt activation,78 downstream effectors of Akt such as 4E-BP1 remain phosphorylated throughout virus infection.81 The preservation of the phosphorylated state appears to be the responsibility of the viral kinase UL3. UL3 is capable of phosphorylating multiple Akt substrates including TSC2, GSK3, and the transcription factor FOXO. UL3 appears to stimulate the assembly of translation initiation factor complexes that drive the translation of viral mRNAs, though additional actions seem likely81 as the phosphorylation of multiple Akt substrates suggest UL3's kinase activity impacts multiple signaling pathways (Fig. 3). This host kinase mimicry is functionally analogous to that seen for the retroviruses described above, where a virus acquires or evolves a kinase that phosphorylates the same substrates as Akt. The HSV example is distinct in that UL3 is not homologous to Akt at a protein sequence level.
4. Cytomegalovirus Activation of PI3K/Akt Signaling
Unlike EBV and HSV, CMV is a herpes virus family member that has not been shown to directly or indirectly activate PI3K or Akt. Instead, CMV targets the downstream effector mTOR through a viral protein/host protein interaction strategy. CMV encodes a protein, named UL38,82 that interacts directly with the tuberous sclerosis protein 2 (TSC2), a protein that is involved in suppressing mTOR activity. Binding of UL38 acts to “silence” the TSC2 inhibition of mTOR activity (Fig. 3), resulting in an increase in mTOR substrate phosphorylation.82, 83 This action phenocopies Akt activation, as Akt will directly phosphorylate and inhibit TSC2. By producing UL38, CMV replaces this function of Akt activation. Further, the UL38 inactivation of TSC2 also blocks the ability of the metabolic sensor AMPK to modulate mTOR activity by activating TSC2. In CMV-infected cells, expression of UL38 acts to preserve the mTOR arm of the PI3K/Akt pathway, an action that promotes viral translation and replication.
Similar to the multifactor targeting of the PI3K/Akt pathway seen in EBV-infected cells, it has also been reported that CMV-infected cells show a change in the substrate specificities of the mTOR complexes mTORC1 and mTORC2.84 These changes are not associated with the UL38 protein but may be important for a change in substrate specificities that accompany a localization of mTORC1 to the replication centers of CMV in infected cells.85 These findings suggest that there are mechanisms of mTOR control in addition to UL38-mediated activation of mTOR.
5. Poxvirus Utilizes PI3K/Akt Activity to Drive Translation, and Viral Assembly
Poxviruses are some of the most genetically complex viruses known, expressing more than 200 genes from a DNA genome, including viral homologs of many cellular proteins. While a homolog of PI3K or Akt is not present, signaling through the PI3K/Akt pathway is critical for virus replication. For vaccinia, cowpox,86 and myxoma viruses,87 PI3K/Akt activity is rapidly induced following productive infection. This activation appears to play at least three distinct roles during poxvirus infection. One is the suppression of apoptosis, which has been suggested to be important to allow proper viral replication.86 The second is the promotion of assembly of the eIF4F translation initiation factor complex through mTOR-mediated inhibition of 4E-BP1 function. This promotes virus mRNA translation.88, 89 PI3K/Akt activity appears to be especially critical for allowing the transcription of mRNAs that encode for proteins involved in the assembly and budding of poxviruses.86 The third role for PI3K/Akt is at a late stage of viral infection, where it is important for the proper progression of morphogenesis.89, 90 Addition of PI3K inhibitors during infection or deletion of the p85 alpha and beta subunits of PI3K altered multiple aspects of vaccinia replication.90 Of note were a decrease in late gene expression and the progression of the virus particle from an immature form to the intracellular mature virus.
At least one poxvirus, the rabbitpox virus (myxoma), produces a protein dedicated to harnessing and activating Akt. This protein, named M-T5, is an ankyrin-repeat protein. It was recently shown that this protein binds directly to Akt (see Fig. 3) and promotes its activation and signaling in a manner that is analogous to the cellular protein PikeA.87, 91 M-T5 not only activates Akt but also forces relocalization of nuclear Akt to the cytoplasm.92 Removal of M-T5 from the myxomavirus genome results in a markedly attenuated virus that is incapable of causing lethal disease in rabbits.93 This highlights the importance of Akt activity for poxvirus pathogenesis. How Akt activation supports the pathogenesis of virus replication in an animal model is not fully resolved. Also unknown at this point is whether other poxviruses such as smallpox or monkeypox have proteins analogous to M-T5 that function to activate PI3K/Akt signaling.
6. Influenza Benefits from Entry Assistance and Suppression of Apoptosis Through PI3K/Akt Pathway Activation
As one of the most significant endemic pathogens confronting humanity, influenza virus represents a virus that has repeatedly shown its ability to infect and adapt. As a virus that produces fewer than a dozen proteins, it is not surprising that host factors aid in influenza replication. What is striking about influenza is the extent to which the virus appears to utilize Akt-related signaling throughout the viral life cycle. Binding of influenza virus to the cell surface leads to the activation of Akt, potentially through clustering of RTKs such as the epidermal growth factor receptor (EGFR).94 While a role for PI3K in flu virus entry seems clear, other signaling pathways (e.g., the ras signaling pathway) also appear to cooperate with the PI3K/Akt pathway to promote virus entry.95
The importance of PI3K/Akt signaling for influenza continues after infection. Following virus entry, influenza A virus directly activates Akt signaling96, 97 through the binding of the viral NSP1 to the P85 subunit of PI3K96, 98 (Fig. 3). NS1 binds to p85 at a coiled-coil region of p85,99, 100 relieving contacts between p85 and p110 subunit of PI3K that normally inhibit activity. The resulting activation of PI3K dramatically stimulates Akt activity in influenza-infected cells. For some strains of flu, it has been shown that NS1 will also bind the cellular adaptor Crk, an action which further potentiates PI3K/Akt signaling.101 Functionally, this activation of Akt signaling has been proposed to lead to the inhibition of apoptosis and to allow the preservation of cell integrity during the viral replication cycle.102, 103 However, this may not be universally true. Viruses lacking NS1 function are stronger inducers of apoptosis during infection, but stimulation of Akt activity in viruses that lack the NS1 protein does not inhibit apoptosis. This suggests the true role for Akt activation during the replication of influenza is yet to be defined.
D. The Other Side of the Coin: Akt in the Antiviral Response
The PI3K/Akt pathway is also a signaling pathway that is utilized in the cellular defense against invading pathogens. As described above, phosphorylation signals are the backbone of the antiviral response. Akt signaling is increasingly recognized to play a role in this response.104 This is particularly true for the host response to interferon, where PI3K and Akt signaling potentiates the translation of interferon-stimulated genes through the actions of mTOR and p70S6K which stimulate interferon-responsive gene translation.105, 106 Akt also plays a role in stimulating the production of inflammatory cytokines by contributing to the activation of the NF-κB transcription factor.57
There are many examples of virus-mediated activation of Akt signaling leading to the activation of host innate immune genes such as inflammatory cytokines. Akt activation results in the increased production of proinflammatory cytokines IL-6 and IL-1β in avian reovirus infected cells.68 In cells infected with RSV, the stimulation of Akt activity leads to the expression of inflammatory cytokines such as interleukin-8 (IL-8) during infection.57, 107, 108 EMCV also stimulates inflammatory cytokine production through the activation of the PI3K/Akt pathway.109
Influenza-infected cells produce IL-8 and RANTES in a PI3K/Akt-dependent manner,110 and virus activation of Akt potentiates the expression of interferon-β.97 Influenza virus-infected macrophages can be kept alive by chemokine receptor-5-mediated activation of the Akt pathway, which allows the infected cell to clear other infected cells.111 Thus activation of Akt signaling can act as a double-edged sword during infection, promoting viral infection and replication and driving the expression of host danger signals that can recruit immune cells to the site of infection. Thus, though many viruses appear to activate the PI3K/Akt pathway, this activation also seems to carry with it some negative consequences. One approach to avoiding these negative consequences is to inactivate this signaling pathway upon infection.
E. Virus Suppression of Akt Signaling
With the importance of Akt signaling in the host antiviral response, it comes as little surprise that some viruses appear to avoid the use of the PI3K/Akt pathway altogether and instead appear to inhibit its function. This is a stark departure from the many examples of viruses that activate the signaling pathway, but underscores that while activation is common, it is not a requirement for virus replication.
1. Measles Virus Inhibition of PI3K/Akt Signaling
The first example of virus inhibition of PI3K/Akt signaling was described for measles virus, a pediatric pathogen whose hallmark of infection is the suppression of immune function. This immune suppression occurs through the decreased proliferation and responsiveness of lymphocytes.112, 113 While it is not fully understood how measles induces immune cell quiescence, recent studies point to a strong role of the PI3K/Akt pathway in this effect. Both in vivo and in vitro, the attachment of measles virus to the surface of T cells, can force Akt dephosphorylation in uninfected cells (Fig. 4 ). This inactivation of PI3K/Akt signaling is not a consequence of infection but merely requires virus binding to a cell surface receptor.114 The receptor that is responsible for signaling this downregulation is currently unidentified, but following the binding of measles virus particles to the cell surface, there is both a decrease in PI3K and Akt signaling and an upregulation of the inositol phosphatase SIP110115 that lowers the overall level of PIP3 in the cell membrane. This promotes long-term suppression of PI3K/Akt signaling. The artificial stimulation of Akt activity overcomes measles-induced immunosuppression, suggesting that the inhibition of Akt is a central cause of measles virus-induced immune suppression.
Fig. 4.
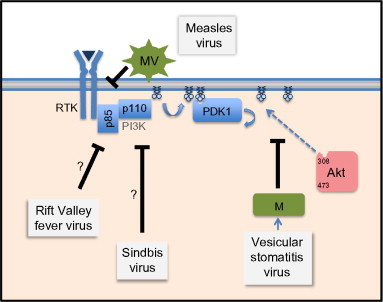
Virus inactivation of Akt signaling. Four different viral approaches are shown. Measles virus suppresses Akt signaling from outside the cell through binding to a currently undefined receptor to block the accumulation of PIP3. The matrix protein of vesicular stomatitis virus blocks Akt activation through an alteration of Akt movement. PIP3 accumulation occurs in the presence of VSV infection, but phosphorylation of Akt is suppressed. Both the Rift Valley fever virus and Sindbis virus (an alphavirus) inhibit Akt activity during infection, but through a currently undefined mechanism (illustrated with a question mark).
In addition to the effect of virus particle binding on Akt signaling, active infection of cells by measles virus continues to block Akt activity even after entry. This suppression of a central cell-viability pathway has no demonstrable negative effect on measles replication.116 Thus, unlike most viruses (even related viruses such as RSV and HPIV5), measles replication appears to be insensitive to the activity of the PI3K/Akt pathway and does not require the antiapoptotic protection that could be afforded by activating this pathway.
2. Rhabdovirus Inhibition of PI3K/Akt Signaling
A different mechanism of Akt suppression is seen upon infection by another RNA virus, vesicular stomatitis virus (VSV). VSV is a veterinary pathogen that is being developed both as an oncolytic agent117 and as a platform for effective vaccines.118 VSV replication is unaffected by Akt signaling.119 As is seen in measles virus, infection of cells with VSV results in a rapid inhibition of Akt signaling in infected insect120 and mammalian cells.121 This inhibition, which extends to inhibit the downstream components mTOR,122 4E-BP1,123 and rpS6,124 makes cells insensitive to normal Akt-stimulating factors such as insulin and epidermal growth factor and is due in large part to the actions of the viral matrix protein (Fig. 4).121 Unlike measles, VSV does not block Akt activity through inhibition of PI3K signaling. Instead, the virus blocks the activating phosphorylation of Akt by inhibiting the phosphorylation of Akt at the plasma membrane through the actions of the viral matrix protein.
3. Bunyavirus Inhibition of PI3K/Akt Signaling
The inhibition of PI3K/Akt signaling is also noted in cells infected with Rift Valley fever virus (RVFV). Recent phosphoproteome analysis of RVFV-infected cells showed that infection led to the dephosphorylation of the Akt-phosphorylated sites of FOXO1 and GSK3β.125 The authors also observed a dephosphorylation of IRS1, an insulin-receptor adaptor that can facilitate PI3K/Akt activation. While the proteomics analysis did not identify Akt phosphorylation changes, the results are very much in line with RVFV inhibition of the Akt pathway. It is not clear how this virus evokes that inhibition of signaling at this point, but the analysis is consistent with the interpretation that it is downregulating RTK activity.
4. Alphavirus Inhibition of PI3K/Akt Signaling
Recent studies of alphaviruses show that these viruses also appear to downregulate the PI3K/Akt signaling pathway. Analysis of cells infected with the prototype alphavirus, Sindbis virus, showed that the virus downregulates PI3K/Akt activity. This downregulation of Akt phosphorylation had little effect on virus replication, and mTOR inhibitors did not significantly alter the replication of Sindbis virus.126 This shows that alphaviruses are also capable of replicating without the contribution of these signaling pathways, which is directly analogous to what is seen in measles and VSV replication. The inhibition of PI3K/Akt signaling by Sindbis is likely relevant to the pathogenesis of alphaviruses, as these viruses are known to be neurotropic, and Akt activity is an important survival factor for these cells.127
5. Akt Antagonism as Antiviral Therapy?
The startling regularity with which viruses appear to utilize the Akt pathway has not gone unnoticed in the antiviral drug discovery field. There have been many suggestions that Akt inhibitors or inhibitors of downstream effectors such as mTOR might be used as either specific or general antiviral compounds, targeting steps of the virus lifestyle ranging from fusion and entry128 to transcription inhibition60 to triggering cell death of infected cells.40 However, given the important role of Akt in promoting immune cell responsiveness114, 129 and the negative effect of PI3K/Akt inhibition on host immune cell function seen during measles virus infection, such an approach seems likely to dampen the host immune response. This latter conclusion is supported by reports showing that the PI3K/Akt pathway controls interferon production in dendritic cells130, 131 and that it is important for controlling an autophagy-based antiviral response.120 This suggests that such an approach is unlikely to succeed as a broad-spectrum approach. However, for viruses that activate PI3K/Akt signaling to maintain a latent infection such as the EBV, inhibiting signaling through this pathway appear to be successful in limiting downstream effects of latency, such as cellular transformation.132 This suggests that there may be limited situations in which this approach will be successful.
The PI3K/Akt signaling pathway itself may represent an evolutionary “tug-of-war” for viruses. If the PI3K/Akt signaling pathway is just as essential for host response and survival as it is for viral function, inactivation is not an effective response for the infected host. Similarly, for viruses that have adapted to activate the pathway and that rely on the downstream effect of this activation, they must also contend with “unwelcome” results of such pathway activation, including the stimulation of cytokine production. This suggests that PI3K/Akt signaling is part of the evolutionary “red queen race” between virus and host, where virus attempts at controlling the host cell are often thwarted by host evolution.
IV. Conclusion and Outlook
While it has long been recognized that viruses are wholly dependent upon the use of cellular machinery for macromolecular events such as protein synthesis, it is now clear that viruses additionally rely upon cellular phosphoproteins and signaling cascades to mold the cellular environment to their advantage. The PI3K/Akt phosphorylation cascade is an emerging paradigm for how these interactions occur. Many viruses have incorporated methods to activate PI3K/Akt signaling in order to forestall apoptosis, drive the cell cycle, and thereby support their replication processes. Somewhat paradoxically, other viruses have evolved an opposite approach, short-circuiting the PI3K/Akt signaling axis. In this latter case, viruses appear to benefit from the repression of the host antiviral response that comes from blocking Akt signaling. Why do different viruses, some from the same families, take different approaches? This is a question that will need to be answered in future research.
It is likely that a better understanding of the importance of viral control of the PI3K/Akt signaling will be gained when the Akt substrates that are vital for viral replication are defined. Some significant progress has been made already. The phosphorylation of host factors such as the mTOR substrate 4E-BP is important for the robust translation of many viral messages, and it is likely that other Akt substrates that stimulate translation will also be sown to be important. It is likely that still more Akt-regulated phosphoproteins play a role in viral processes. Perhaps the most obvious but most poorly understood is the possibility that viral proteins themselves serve as substrates for Akt or effectors such as mTOR and GSK3. Recent work has suggested that Akt may phosphorylate the phosphoprotein of several negative-stranded RNA viruses60 and that it may also control calicivirus polymerase function.133 Future discoveries will likely greatly expand the number of viral substrates and more clearly point out the viral dependencies on this signaling axis.
Acknowledgments
The authors would like to thank Rachel Fearns and Erin Hodges for helpful conversations, Rahm Gummuluru for insight regarding retroviruses and lentiviruses, and Rebecca Connor for significant help with editing and revision. We apologize that a number of references could not be incorporated because of space consideration.
References
- 1.Neumann G., Noda T., Kawaoka Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature. 2009;459:931–939. doi: 10.1038/nature08157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frieman M., Baric R. Mechanisms of severe acute respiratory syndrome pathogenesis and innate immunomodulation. Microbiol Mol Biol Rev. 2008;72:672–685. doi: 10.1128/MMBR.00015-08. Table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feldmann H. Truly emerging - a new disease caused by a novel virus. N Engl J Med. 2011;364(16):1561–1563. doi: 10.1056/NEJMe1102671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu X.J., Liang M.F., Zhang S.Y., Liu Y., Li J.D., Sun Y.L. Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med. 2011;364:1523–1532. doi: 10.1056/NEJMoa1010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buchkovich N.J., Yu Y., Zampieri C.A., Alwine J.C. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat Rev Microbiol. 2008;6:266–275. doi: 10.1038/nrmicro1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munter S., Way M., Frischknecht F. Signaling during pathogen infection. Sci STKE. 2006;2006:re5. doi: 10.1126/stke.3352006re5. [DOI] [PubMed] [Google Scholar]
- 7.Hale B.G., Randall R.E., Ortin J., Jackson D. The multifunctional NS1 protein of influenza A viruses. J Gen Virol. 2008;89:2359–2376. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- 8.Langland J.O., Jacobs B.L. The role of the PKR-inhibitory genes, E3L and K3L, in determining vaccinia virus host range. Virology. 2002;299:133–141. doi: 10.1006/viro.2002.1479. [DOI] [PubMed] [Google Scholar]
- 9.Xiang Y., Condit R.C., Vijaysri S., Jacobs B., Williams B.R., Silverman R.H. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J Virol. 2002;76:5251–5259. doi: 10.1128/JVI.76.10.5251-5259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berro R., Pedati C., Kehn-Hall K., Wu W.L., Klase Z., Even Y. CDK13, a new potential human immunodeficiency virus type 1 inhibitory factor regulating viral mRNA splicing. J Virol. 2008;82:7155–7166. doi: 10.1128/JVI.02543-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arias C., Walsh D., Harbell J., Wilson A.C., Mohr I. Activation of host translational control pathways by a viral developmental switch. PLoS Pathog. 2009;5:e1000334. doi: 10.1371/journal.ppat.1000334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogt P.K., Hart J.R., Gymnopoulos M., Jiang H., Kang S., Bader A.G. Phosphatidylinositol 3-kinase: the oncoprotein. Curr Top Microbiol Immunol. 2010;347:79–104. doi: 10.1007/82_2010_80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calleja V., Alcor D., Laguerre M., Park J., Vojnovic B., Hemmings B.A. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol. 2007;5:e95. doi: 10.1371/journal.pbio.0050095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol. 1998;10:262–267. doi: 10.1016/s0955-0674(98)80149-x. [DOI] [PubMed] [Google Scholar]
- 15.Facchinetti V., Ouyang W., Wei H., Soto N., Lazorchak A., Gould C. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27:1932–1943. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andjelkovic M., Alessi D.R., Meier R., Fernandez A., Lamb N.J., Frech M. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- 17.Galetic I., Andjelkovic M., Meier R., Brodbeck D., Park J., Hemmings B.A. Mechanism of protein kinase B activation by insulin/insulin-like growth factor-1 revealed by specific inhibitors of phosphoinositide 3-kinase—significance for diabetes and cancer. Pharmacol Ther. 1999;82:409–425. doi: 10.1016/s0163-7258(98)00071-0. [DOI] [PubMed] [Google Scholar]
- 18.Biggs W.H., 3rd, Meisenhelder J., Hunter T., Cavenee W.K., Arden K.C. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahn-Windgassen A., Nogueira V., Chen C.C., Skeen J.E., Sonenberg N., Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280:32081–32089. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- 20.Robey R.B., Hay N. Is Akt the “Warburg kinase”?—Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19:25–31. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wullschleger S., Loewith R., Hall M.N. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 22.Gao X., Zhang Y., Arrazola P., Hino O., Kobayashi T., Yeung R.S. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol. 2002;4:699–704. doi: 10.1038/ncb847. [DOI] [PubMed] [Google Scholar]
- 23.Ramaswamy S., Nakamura N., Vazquez F., Batt D.B., Perera S., Roberts T.M. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci USA. 1999;96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kane L.P., Shapiro V.S., Stokoe D., Weiss A. Induction of NF-[kappa]B by the Akt/PKB kinase. Curr Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 25.Cooray S. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J Gen Virol. 2004;85:1065–1076. doi: 10.1099/vir.0.19771-0. [DOI] [PubMed] [Google Scholar]
- 26.Norman K.L., Sarnow P. Herpes Simplex Virus is Akt-ing in translational control. Genes Dev. 2010;24:2583–2586. doi: 10.1101/gad.2004510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bellacosa A., Testa J.R., Staal S.P., Tsichlis P.N. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- 28.Bellacosa A., Franke T.F., Gonzalez-Portal M.E., Datta K., Taguchi T., Gardner J. Structure, expression and chromosomal mapping of c-akt: relationship to v-akt and its implications. Oncogene. 1993;8:745–754. [PubMed] [Google Scholar]
- 29.Coffer P.J., Woodgett J.R. Molecular cloning and characterisation of a novel putative protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur J Biochem. 1991;201:475–481. doi: 10.1111/j.1432-1033.1991.tb16305.x. [DOI] [PubMed] [Google Scholar]
- 30.Jones P.F., Jakubowicz T., Pitossi F.J., Maurer F., Hemmings B.A. Molecular cloning and identification of a serine/threonine protein kinase of the second-messenger subfamily. Proc Natl Acad Sci USA. 1991;88:4171–4175. doi: 10.1073/pnas.88.10.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang H.W., Aoki M., Fruman D., Auger K.R., Bellacosa A., Tsichlis P.N. Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science. 1997;276:1848–1850. doi: 10.1126/science.276.5320.1848. [DOI] [PubMed] [Google Scholar]
- 32.Aoki M., Batista O., Bellacosa A., Tsichlis P., Vogt P.K. The akt kinase: molecular determinants of oncogenicity. Proc Natl Acad Sci USA. 1998;95:14950–14955. doi: 10.1073/pnas.95.25.14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aoki M., Jiang H., Vogt P.K. Proteasomal degradation of the FoxO1 transcriptional regulator in cells transformed by the P3k and Akt oncoproteins. Proc Natl Acad Sci USA. 2004;101:13613–13617. doi: 10.1073/pnas.0405454101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aoki M., Blazek E., Vogt P.K. A role of the kinase mTOR in cellular transformation induced by the oncoproteins P3k and Akt. Proc Natl Acad Sci USA. 2001;98:136–141. doi: 10.1073/pnas.011528498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reichman C.T., Mayer B.J., Keshav S., Hanafusa H. The product of the cellular crk gene consists primarily of SH2 and SH3 regions. Cell Growth Differ. 1992;3:451–460. [PubMed] [Google Scholar]
- 36.Tsuchie H., Chang C.H., Yoshida M., Vogt P.K. A newly isolated avian sarcoma virus, ASV-1, carries the crk oncogene. Oncogene. 1989;4:1281–1284. [PubMed] [Google Scholar]
- 37.Matsuda M., Mayer B.J., Hanafusa H. Identification of domains of the v-crk oncogene product sufficient for association with phosphotyrosine-containing proteins. Mol Cell Biol. 1991;11:1607–1613. doi: 10.1128/mcb.11.3.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akagi T., Murata K., Shishido T., Hanafusa H. v-Crk activates the phosphoinositide 3-kinase/AKT pathway by utilizing focal adhesion kinase and H-Ras. Mol Cell Biol. 2002;22:7015–7023. doi: 10.1128/MCB.22.20.7015-7023.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akagi T., Shishido T., Murata K., Hanafusa H. v-Crk activates the phosphoinositide 3-kinase/AKT pathway in transformation. Proc Natl Acad Sci USA. 2000;97:7290–7295. doi: 10.1073/pnas.140210297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chugh P., Bradel-Tretheway B., Monteiro-Filho C.M., Planelles V., Maggirwar S.B., Dewhurst S. Akt inhibitors as an HIV-1 infected macrophage-specific anti-viral therapy. Retrovirology. 2008;5:11. doi: 10.1186/1742-4690-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao T., Furnari F., Newton A.C. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 42.Arroyo J.D., Hahn W.C. Involvement of PP2A in viral and cellular transformation. Oncogene. 2005;24:7746–7755. doi: 10.1038/sj.onc.1209038. [DOI] [PubMed] [Google Scholar]
- 43.Yuan H., Veldman T., Rundell K., Schlegel R. Simian virus 40 small tumor antigen activates AKT and telomerase and induces anchorage-independent growth of human epithelial cells. J Virol. 2002;76:10685–10691. doi: 10.1128/JVI.76.21.10685-10691.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang S.I., Lickteig R.L., Estes R., Rundell K., Walter G., Mumby M.C. Control of protein phosphatase 2A by simian virus 40 small-t antigen. Mol Cell Biol. 1991;11:1988–1995. doi: 10.1128/mcb.11.4.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liacini A., Seamone M.E., Muruve D.A., Tibbles L.A. Anti-BK virus mechanisms of sirolimus and leflunomide alone and in combination: toward a new therapy for BK virus infection. Transplantation. 2010;90:1450–1457. doi: 10.1097/TP.0b013e3182007be2. [DOI] [PubMed] [Google Scholar]
- 46.Andrabi S., Gjoerup O.V., Kean J.A., Roberts T.M., Schaffhausen B. Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc Natl Acad Sci USA. 2007;104:19011–19016. doi: 10.1073/pnas.0706696104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bollag B., Hofstetter C.A., Reviriego-Mendoza M.M., Frisque R.J. JC virus small T antigen binds phosphatase PP2A and Rb family proteins and is required for efficient viral DNA replication activity. PLoS One. 2010;5:e10606. doi: 10.1371/journal.pone.0010606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Utermark T., Schaffhausen B.S., Roberts T.M., Zhao J.J. The p110alpha isoform of phosphatidylinositol 3-kinase is essential for polyomavirus middle T antigen-mediated transformation. J Virol. 2007;81:7069–7076. doi: 10.1128/JVI.00115-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu Y., Alwine J.C. Interaction between simian virus 40 large T antigen and insulin receptor substrate 1 is disrupted by the K1 mutation, resulting in the loss of large T antigen-mediated phosphorylation of Akt. J Virol. 2008;82:4521–4526. doi: 10.1128/JVI.02365-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu Y., Alwine J.C. Human cytomegalovirus major immediate-early proteins and simian virus 40 large T antigen can inhibit apoptosis through activation of the phosphatidylinositide 3′-OH kinase pathway and the cellular kinase Akt. J Virol. 2002;76:3731–3738. doi: 10.1128/JVI.76.8.3731-3738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pim D., Massimi P., Dilworth S.M., Banks L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene. 2005;24:7830–7838. doi: 10.1038/sj.onc.1208935. [DOI] [PubMed] [Google Scholar]
- 52.Cheng S., Schmidt-Grimminger D.C., Murant T., Broker T.R., Chow L.T. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev. 1995;9:2335–2349. doi: 10.1101/gad.9.19.2335. [DOI] [PubMed] [Google Scholar]
- 53.McLaughlin-Drubin M.E., Münger K. The human papillomavirus E7 oncoprotein. Virology. 2009;384:335–344. doi: 10.1016/j.virol.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin G.Y., Lamb R.A. The paramyxovirus simian virus 5 V protein slows progression of the cell cycle. J Virol. 2000;74:9152–9166. doi: 10.1128/jvi.74.19.9152-9166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gustin K.E. Inhibition of nucleo-cytoplasmic trafficking by RNA viruses: targeting the nuclear pore complex. Virus Res. 2003;95:35–44. doi: 10.1016/S0168-1702(03)00165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lyles D.S. Cytopathogenesis and inhibition of host gene expression by RNA viruses. Microbiol Mol Biol Rev. 2000;64:709–724. doi: 10.1128/mmbr.64.4.709-724.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thomas K.W., Monick M.M., Staber J.M., Yarovinsky T., Carter A.B., Hunninghake G.W. Respiratory syncytial virus inhibits apoptosis and induces NF-kappa B activity through a phosphatidylinositol 3-kinase-dependent pathway. J Biol Chem. 2002;277:492–501. doi: 10.1074/jbc.M108107200. [DOI] [PubMed] [Google Scholar]
- 58.Monick M.M., Cameron K., Powers L.S., Butler N.S., McCoy D., Mallampalli R.K. Sphingosine kinase mediates activation of extracellular signal-related kinase and Akt by respiratory syncytial virus. Am J Respir Cell Mol Biol. 2004;30:844–852. doi: 10.1165/rcmb.2003-0424OC. [DOI] [PubMed] [Google Scholar]
- 59.Peters K., Chattopadhyay S., Sen G.C. IRF-3 activation by Sendai virus infection is required for cellular apoptosis and avoidance of persistence. J Virol. 2008;82:3500–3508. doi: 10.1128/JVI.02536-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun M., Fuentes S.M., Timani K., Sun D., Murphy C., Lin Y. Akt plays a critical role in replication of nonsegmented negative-stranded RNA viruses. J Virol. 2008;82:105–114. doi: 10.1128/JVI.01520-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Autret A., Martin-Latil S., Brisac C., Mousson L., Colbere-Garapin F., Blondel B. Early phosphatidylinositol 3-kinase/Akt pathway activation limits poliovirus-induced JNK-mediated cell death. J Virol. 2008;82:3796–3802. doi: 10.1128/JVI.02020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bentley J.K., Newcomb D.C., Goldsmith A.M., Jia Y., Sajjan U.S., Hershenson M.B. Rhinovirus activates interleukin-8 expression via a Src/p110beta phosphatidylinositol 3-kinase/Akt pathway in human airway epithelial cells. J Virol. 2007;81:1186–1194. doi: 10.1128/JVI.02309-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lau C., Wang X., Song L., North M., Wiehler S., Proud D. Syk associates with clathrin and mediates phosphatidylinositol 3-kinase activation during human rhinovirus internalization. J Immunol. 2008;180:870–880. doi: 10.4049/jimmunol.180.2.870. [DOI] [PubMed] [Google Scholar]
- 64.Esfandiarei M., Luo H., Yanagawa B., Suarez A., Dabiri D., Zhang J. Protein kinase B/Akt regulates coxsackievirus B3 replication through a mechanism which is not caspase dependent. J Virol. 2004;78:4289–4298. doi: 10.1128/JVI.78.8.4289-4298.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Esfandiarei M., Boroomand S., Suarez A., Si X., Rahmani M., McManus B. Coxsackievirus B3 activates nuclear factor kappa B transcription factor via a phosphatidylinositol-3 kinase/protein kinase B-dependent pathway to improve host cell viability. Cell Microbiol. 2007;9:2358–2371. doi: 10.1111/j.1462-5822.2007.00964.x. [DOI] [PubMed] [Google Scholar]
- 66.Halasz P., Holloway G., Turner S.J., Coulson B.S. Rotavirus replication in intestinal cells differentially regulates integrin expression by a phosphatidylinositol 3-kinase-dependent pathway, resulting in increased cell adhesion and virus yield. J Virol. 2008;82:148–160. doi: 10.1128/JVI.01980-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bagchi P., Dutta D., Chattopadhyay S., Mukherjee A., Halder U.C., Sarkar S. Rotavirus nonstructural protein 1 suppresses virus-induced cellular apoptosis to facilitate viral growth by activating the cell survival pathways during early stages of infection. J Virol. 2010;84:6834–6845. doi: 10.1128/JVI.00225-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin P.Y., Liu H.J., Liao M.H., Chang C.D., Chang C.I., Cheng H.L. Activation of PI 3-kinase/Akt/NF-kappaB and Stat3 signaling by avian reovirus S1133 in the early stages of infection results in an inflammatory response and delayed apoptosis. Virology. 2010;400:104–114. doi: 10.1016/j.virol.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 69.Halasz P., Holloway G., Coulson B.S. Death mechanisms in epithelial cells following rotavirus infection, exposure to inactivated rotavirus or genome transfection. J Gen Virol. 2010;91:2007–2018. doi: 10.1099/vir.0.018275-0. [DOI] [PubMed] [Google Scholar]
- 70.Lee C.-J., Liao C.-L., Lin Y.-L. Flavivirus activates phosphatidylinositol 3-kinase signaling to block caspase-dependent apoptotic cell death at the early stage of virus infection. J Virol. 2005;79:8388–8399. doi: 10.1128/JVI.79.13.8388-8399.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peng L., Liang D., Tong W., Li J., Yuan Z. Hepatitis C virus NS5A activates the mammalian target of rapamycin (mTOR) pathway, contributing to cell survival by disrupting the interaction between FK506-binding protein 38 (FKBP38) and mTOR. J Biol Chem. 2010;285:20870–20881. doi: 10.1074/jbc.M110.112045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bai X., Ma D., Liu A., Shen X., Wang Q.J., Liu Y. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science. 2007;318:977–980. doi: 10.1126/science.1147379. [DOI] [PubMed] [Google Scholar]
- 73.Moody C.A., Scott R.S., Amirghahari N., Nathan C.O., Young L.S., Dawson C.W. Modulation of the cell growth regulator mTOR by Epstein-Barr virus-encoded LMP2A. J Virol. 2005;79:5499–5506. doi: 10.1128/JVI.79.9.5499-5506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bultema R., Longnecker R., Swanson-Mungerson M. Epstein-Barr virus LMP2A accelerates MYC-induced lymphomagenesis. Oncogene. 2009;28:1471–1476. doi: 10.1038/onc.2008.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen Y.R., Liu M.T., Chang Y.T., Wu C.C., Hu C.Y., Chen J.Y. Epstein-Barr virus latent membrane protein 1 represses DNA repair through the PI3K/Akt/FOXO3a pathway in human epithelial cells. J Virol. 2008;82:8124–8137. doi: 10.1128/JVI.00430-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Darr C.D., Mauser A., Kenney S. Epstein-Barr virus immediate-early protein BRLF1 induces the lytic form of viral replication through a mechanism involving phosphatidylinositol-3 kinase activation. J Virol. 2001;75:6135–6142. doi: 10.1128/JVI.75.13.6135-6142.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cen O., Longnecker R. Rapamycin reverses splenomegaly and inhibits tumor development in a transgenic model of Epstein-Barr virus-related Burkitt's lymphoma. Mol Cancer Ther. 2011;10:679–686. doi: 10.1158/1535-7163.MCT-10-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Benetti L., Roizman B. Protein kinase B/Akt is present in activated form throughout the entire replicative cycle of deltaU(S)3 mutant virus but only at early times after infection with wild-type herpes simplex virus 1. J Virol. 2006;80:3341–3348. doi: 10.1128/JVI.80.7.3341-3348.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wagner M.J., Smiley J.R. Herpes simplex virus requires VP11/12 to activate Src family kinase-phosphoinositide 3-kinase-Akt signaling. J Virol. 2011;85:2803–2812. doi: 10.1128/JVI.01877-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.MacLeod I.J., Minson T. Binding of herpes simplex virus type-1 virions leads to the induction of intracellular signalling in the absence of virus entry. PLoS One. 2010;5:e9560. doi: 10.1371/journal.pone.0009560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chuluunbaatar U., Roller R., Feldman M.E., Brown S., Shokat K.M., Mohr I. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev. 2010;24:2627–2639. doi: 10.1101/gad.1978310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Moorman N.J., Cristea I.M., Terhune S.S., Rout M.P., Chait B.T., Shenk T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe. 2008;3:253–262. doi: 10.1016/j.chom.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kudchodkar S.B., Yu Y., Maguire T.G., Alwine J.C. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J Virol. 2004;78:11030–11039. doi: 10.1128/JVI.78.20.11030-11039.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kudchodkar S.B., Yu Y., Maguire T.G., Alwine J.C. Human cytomegalovirus infection alters the substrate specificities and rapamycin sensitivities of raptor- and rictor-containing complexes. Proc Natl Acad Sci USA. 2006;103:14182–14187. doi: 10.1073/pnas.0605825103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Clippinger A.J., Maguire T.G., Alwine J.C. Human cytomegalovirus infection maintains mTOR activity and its perinuclear localization during amino acid deprivation. J Virol. 2011;85:9369–9376. doi: 10.1128/JVI.05102-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soares J.A., Leite F.G., Andrade L.G., Torres A.A., De Sousa L.P., Barcelos L.S. Activation of the PI3K/Akt pathway early during vaccinia and cowpox virus infections is required for both host survival and viral replication. J Virol. 2009;83:6883–6899. doi: 10.1128/JVI.00245-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang G., Barrett J.W., Stanford M., Werden S.J., Johnston J.B., Gao X. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc Natl Acad Sci USA. 2006;103:4640–4645. doi: 10.1073/pnas.0509341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zaborowska I., Walsh D. PI3K signaling regulates rapamycin-insensitive translation initiation complex formation in vaccinia virus-infected cells. J Virol. 2009;83:3988–3992. doi: 10.1128/JVI.02284-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hu N., Yu R., Shikuma C., Shiramizu B., Ostrwoski M.A., Yu Q. Role of cell signaling in poxvirus-mediated foreign gene expression in mammalian cells. Vaccine. 2009;27:2994–3006. doi: 10.1016/j.vaccine.2009.02.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McNulty S., Bornmann W., Schriewer J., Werner C., Smith S.K., Olson V.A. Multiple phosphatidylinositol 3-kinases regulate vaccinia virus morphogenesis. PLoS One. 2010;5:e10884. doi: 10.1371/journal.pone.0010884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Werden S.J., Barrett J.W., Wang G., Stanford M.M., McFadden G. M-T5, the ankyrin repeat, host range protein of myxoma virus, activates Akt and can be functionally replaced by cellular PIKE-A. J Virol. 2007;81:2340–2348. doi: 10.1128/JVI.01310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Werden S.J., Lanchbury J., Shattuck D., Neff C., Dufford M., McFadden G. The myxoma virus m-t5 ankyrin repeat host range protein is a novel adaptor that coordinately links the cellular signaling pathways mediated by Akt and Skp1 in virus-infected cells. J Virol. 2009;83:12068–12083. doi: 10.1128/JVI.00963-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mossman K., Lee S., Barry M., Boshkov L., McFadden G. Disruption of M-T5, a novel myxoma virus gene member of poxvirus host range superfamily, results in dramatic attenuation of myxomatosis in infected European rabbits. J Virol. 1996;70:4394–4410. doi: 10.1128/jvi.70.7.4394-4410.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Eierhoff T., Hrincius E.R., Rescher U., Ludwig S., Ehrhardt C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog. 2010;6:12585–12593. doi: 10.1371/journal.ppat.1001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fujioka Y., Tsuda M., Hattori T., Sasaki J., Sasaki T., Miyazaki T. The Ras-PI3K signaling pathway is involved in clathrin-independent endocytosis and the internalization of influenza viruses. PLoS One. 2011;6:e16324. doi: 10.1371/journal.pone.0016324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hale B.G., Jackson D., Chen Y.H., Lamb R.A., Randall R.E. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc Natl Acad Sci USA. 2006;103:14194–14199. doi: 10.1073/pnas.0606109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ehrhardt C., Marjuki H., Wolff T., Nurnberg B., Planz O., Pleschka S. Bivalent role of the phosphatidylinositol-3-kinase (PI3K) during influenza virus infection and host cell defence. Cell Microbiol. 2006;8:1336–1348. doi: 10.1111/j.1462-5822.2006.00713.x. [DOI] [PubMed] [Google Scholar]
- 98.Shin Y.K., Liu Q., Tikoo S.K., Babiuk L.A., Zhou Y. Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J Gen Virol. 2007;88:13–18. doi: 10.1099/vir.0.82419-0. [DOI] [PubMed] [Google Scholar]
- 99.Hale B.G., Batty I.H., Downes C.P., Randall R.E. Binding of influenza A virus NS1 protein to the inter-SH2 domain of p85 suggests a novel mechanism for phosphoinositide 3-kinase activation. J Biol Chem. 2008;283:1372–1380. doi: 10.1074/jbc.M708862200. [DOI] [PubMed] [Google Scholar]
- 100.Hale B.G., Kerry P.S., Jackson D., Precious B.L., Gray A., Killip M.J. Structural insights into phosphoinositide 3-kinase activation by the influenza A virus NS1 protein. Proc Natl Acad Sci USA. 2010;107:1954–1959. doi: 10.1073/pnas.0910715107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Heikkinen L.S., Kazlauskas A., Melen K., Wagner R., Ziegler T., Julkunen I. Avian and 1918 Spanish influenza a virus NS1 proteins bind to Crk/CrkL Src homology 3 domains to activate host cell signaling. J Biol Chem. 2008;283:5719–5727. doi: 10.1074/jbc.M707195200. [DOI] [PubMed] [Google Scholar]
- 102.Ehrhardt C., Wolff T., Pleschka S., Planz O., Beermann W., Bode J.G. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J Virol. 2007;81:3058–3067. doi: 10.1128/JVI.02082-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhirnov O.P., Klenk H.D. Control of apoptosis in influenza virus-infected cells by up-regulation of Akt and p53 signaling. Apoptosis. 2007;12:1419–1432. doi: 10.1007/s10495-007-0071-y. [DOI] [PubMed] [Google Scholar]
- 104.Kaur S., Katsoulidis E., Platanias L.C. Akt and mRNA translation by interferons. Cell Cycle. 2008;7:2112–2116. doi: 10.4161/cc.7.14.6258. [DOI] [PubMed] [Google Scholar]
- 105.Kaur S., Sassano A., Joseph A.M., Majchrzak-Kita B., Eklund E.A., Verma A. Dual regulatory roles of phosphatidylinositol 3-kinase in IFN signaling. J Immunol. 2008;181:7316–7323. doi: 10.4049/jimmunol.181.10.7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaur S., Sassano A., Dolniak B., Joshi S., Majchrzak-Kita B., Baker D.P. Role of the Akt pathway in mRNA translation of interferon-stimulated genes. Proc Natl Acad Sci USA. 2008;105:4808–4813. doi: 10.1073/pnas.0710907105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mastronarde J.G., He B., Monick M.M., Mukaida N., Matsushima K., Hunninghake G.W. Induction of interleukin (IL)-8 gene expression by respiratory syncytial virus involves activation of nuclear factor (NF)-kappa B and NF-IL-6. J Infect Dis. 1996;174:262–267. doi: 10.1093/infdis/174.2.262. [DOI] [PubMed] [Google Scholar]
- 108.Mastronarde J.G., Monick M.M., Hunninghake G.W. Oxidant tone regulates IL-8 production in epithelium infected with respiratory syncytial virus. Am J Respir Cell Mol Biol. 1995;13:237–244. doi: 10.1165/ajrcmb.13.2.7626291. [DOI] [PubMed] [Google Scholar]
- 109.Freudenburg W., Moran J.M., Lents N.H., Baldassare J.J., Buller R.M., Corbett J.A. Phosphatidylinositol 3-kinase regulates macrophage responses to double-stranded RNA and encephalomyocarditis virus. J Innate Immun. 2009;2:77–86. doi: 10.1159/000243785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Guillot L., Le Goffic R., Bloch S., Escriou N., Akira S., Chignard M. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J Biol Chem. 2005;280:5571–5580. doi: 10.1074/jbc.M410592200. [DOI] [PubMed] [Google Scholar]
- 111.Tyner J.W., Uchida O., Kajiwara N., Kim E.Y., Patel A.C., O'Sullivan M.P. CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat Med. 2005;11:1180–1187. doi: 10.1038/nm1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Griffin D.E. Measles virus-induced suppression of immune responses. Immunol Rev. 2010;236:176–189. doi: 10.1111/j.1600-065X.2010.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Avota E., Gassert E., Schneider-Schaulies S. Measles virus-induced immunosuppression: from effectors to mechanisms. Med Microbiol Immunol. 2010;199:227–237. doi: 10.1007/s00430-010-0152-3. [DOI] [PubMed] [Google Scholar]
- 114.Avota E., Avots A., Niewiesk S., Kane L.P., Bommhardt U., ter Meulen V. Disruption of Akt kinase activation is important for immunosuppression induced by measles virus. Nat Med. 2001;7:725–731. doi: 10.1038/89106. [DOI] [PubMed] [Google Scholar]
- 115.Avota E., Harms H., Schneider-Schaulies S. Measles virus induces expression of SIP110, a constitutively membrane clustered lipid phosphatase, which inhibits T cell proliferation. Cell Microbiol. 2006;8:1826–1839. doi: 10.1111/j.1462-5822.2006.00752.x. [DOI] [PubMed] [Google Scholar]
- 116.Carsillo M., Kim D., Niewiesk S. Role of AKT kinase in measles virus replication. J Virol. 2010;84:2180–2183. doi: 10.1128/JVI.01316-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stojdl D.F., Lichty B., Knowles S., Marius R., Atkins H., Sonenberg N. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 118.Geisbert T.W., Bausch D.G., Feldmann H. Prospects for immunisation against Marburg and Ebola viruses. Rev Med Virol. 2010;20:344–357. doi: 10.1002/rmv.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dunn E.F., Fearns R., Connor J.H. Akt inhibitor Akt-IV blocks virus replication through an Akt-independent mechanism. J Virol. 2009;83:11665–11672. doi: 10.1128/JVI.01092-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shelly S., Lukinova N., Bambina S., Berman A., Cherry S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity. 2009;30:588–598. doi: 10.1016/j.immuni.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dunn E.F., Connor J.H. Dominant inhibition of Akt/protein kinase B signaling by the matrix protein of a negative-strand RNA virus. J Virol. 2011;85:422–431. doi: 10.1128/JVI.01671-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Alain T., Lun X., Martineau Y., Sean P., Pulendran B., Petroulakis E. Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production. Proc Natl Acad Sci USA. 2010;107:1576–1581. doi: 10.1073/pnas.0912344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Connor J.H., Lyles D.S. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J Virol. 2002;76:10177–10187. doi: 10.1128/JVI.76.20.10177-10187.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Minami K., Tambe Y., Watanabe R., Isono T., Haneda M., Isobe K. Suppression of viral replication by stress-inducible GADD34 protein via the mammalian serine/threonine protein kinase mTOR pathway. J Virol. 2007;81:11106–11115. doi: 10.1128/JVI.01063-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Popova T.G., Turell M.J., Espina V., Kehn-Hall K., Kidd J., Narayanan A. Reverse-phase phosphoproteome analysis of signaling pathways induced by Rift valley fever virus in human small airway epithelial cells. PLoS One. 2010;5:e13805. doi: 10.1371/journal.pone.0013805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mohankumar V., Dhanushkodi N.R., Raju R. Sindbis virus replication, is insensitive to rapamycin and torin1, and suppresses Akt/mTOR pathway late during infection in HEK cells. Biochem Biophys Res Commun. 2011;406:262–267. doi: 10.1016/j.bbrc.2011.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yuan J., Lipinski M., Degterev A. Diversity in the mechanisms of neuronal cell death. Neuron. 2003;40:401–413. doi: 10.1016/s0896-6273(03)00601-9. [DOI] [PubMed] [Google Scholar]
- 128.Sharma N.R., Mani P., Nandwani N., Mishra R., Rana A., Sarkar D.P. Reciprocal regulation of AKT and MAP kinase dictates virus-host cell fusion. J Virol. 2010;84:4366–4382. doi: 10.1128/JVI.01940-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hand T.W., Cui W., Jung Y.W., Sefik E., Joshi N.S., Chandele A. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc Natl Acad Sci USA. 2010;107:16601–16606. doi: 10.1073/pnas.1003457107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Guiducci C., Ghirelli C., Marloie-Provost M.A., Matray T., Coffman R.L., Liu Y.J. PI3K is critical for the nuclear translocation of IRF-7 and type I IFN production by human plasmacytoid predendritic cells in response to TLR activation. J Exp Med. 2008;205:315–322. doi: 10.1084/jem.20070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cao W., Manicassamy S., Tang H., Kasturi S.P., Pirani A., Murthy N. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat Immunol. 2008;9:1157–1164. doi: 10.1038/ni.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nepomuceno R.R., Balatoni C.E., Natkunam Y., Snow A.L., Krams S.M., Martinez O.M. Rapamycin inhibits the interleukin 10 signal transduction pathway and the growth of Epstein Barr virus B-cell lymphomas. Cancer Res. 2003;63:4472–4480. [PubMed] [Google Scholar]
- 133.Eden J.S., Sharpe L.J., White P.A., Brown A.J. Norovirus RNA-dependent RNA-polymerase is phosphorylated by an important survival kinase, Akt. J Virol. 2011;85:10894–10898. doi: 10.1128/JVI.05562-11. [DOI] [PMC free article] [PubMed] [Google Scholar]