

Abstract
The new technologies of genomics, proteomics, and molecular immunology have led to an impressive increase in our knowledge of all aspects of virology, providing insights that can guide new vaccine concepts. When developing a new vaccine, the choice of approach is made very much on a case-by-case basis, and for a given virus is driven by knowledge its pathogenesis, serotype diversity, antigenic variation, immune evasion mechanisms, latency and route of transmission. High importance is given to what type of immunity arises as a result of natural infection and whether the pathogen can cause persistent and/or repeated infections in a single host. This article discusses the medical need for new human viral vaccines and focuses on the strategies available for their development and some of the challenges posed by the more difficult targets.
Keywords: Antigenic diversity, Live-attenuated, Inactivated, Influenza vaccine production, Immune responses, Multiple serotype vaccines, Vaccine, Vaccine development, Vaccine vectors
Introduction
To develop a new viral vaccine, get it licensed, and bring it to market is a lengthy, complex, and very expensive task. It requires a detailed knowledge of all aspects of the virus, especially its structure, epidemiology, pathology, and immunobiology, and demands a close collaboration between fundamental scientists, regulatory authorities, and industrial scientists and engineers. Completely new vaccines against human viruses appear infrequently on the market and the cost and complexity of their development has escalated with time, mainly due to the increased regulatory pressures to have highly defined products and to ensure complete clinical safety and high efficacy. Over the past 20 years or so, six new human virus vaccines have been developed for licensure in major markets; these are hepatitis B, Japanese encephalitis, hepatitis A, varicella (recently with a zoster formulation), and, over the past year, rotavirus, and human papilloma virus (HPV). The introduction of HPV vaccines is a technological triumph that offers to dramatically reduce the incidence of cervical cancer worldwide in the long term. The newest rotavirus vaccines seem, so far, to be free from the complications that led to the withdrawal of the earlier Rotashield vaccine in 1999 and promise to be very effective in reducing the burden of rotavirus diarrhea worldwide. In spite of this impressive progress, however, virus vaccine development has not accelerated over recent decades, and there remains a significant list of human virus diseases of widespread prevalence for which there are no vaccines available. These include human immunodeficiency virus (HIV), hepatitis C, hepatitis E, Eptein–Barr virus, herpes simplex virus (HSV), cytomegalovirus (CMV), respiratory syncytial virus (RSV), and parainfluenza viruses (PIV) (see Table 1 ).
Table 1.
Human viruses causing disease with important medical need for which no vaccines are available
| Adenoviruses | Human immunodeficiency virus (HIV) |
| Chikungunya | Human metapneumovirus (hMPV) |
| Cytomegalovirus (CMV) | |
| Dengue | Norwalk virus |
| Enterovirus 71 (EV71) | Parainfluenza virus (PIV) |
| Epstein–Barr virus (EBV) | Parvovirus B19 |
| Hantavirus | Respiratory syncytial virus (RSV) |
| Hepatitis C (HCV) | Rhinovirus |
| Hepatitis E (HEV) | SARS |
| Herpes simplex virus (HSV) | West Nile virus |
In the veterinary world, the list of successes is significantly more impressive, and both the time cost of developing a new vaccine can be substantially reduced by the ability to reach proof of concept through direct challenge experiments, often with the wild-type virus. In this arena, the requirements are often very different to those for human vaccines, especially for vaccines of production animals where ease of administration and cost per dose are paramount. Recognizing that many approaches and outstanding fundamental challenges are similar for human and veterinary virus vaccines, this article focuses principally on the former with occasional reference to veterinary vaccines where they illustrate particular concepts.
Fueled by the new technologies of genomics, proteomics, and molecular immunology, the past 20 years have seen an impressive increase in our knowledge of all aspects of virology, providing insights to guide new vaccine concepts. The biological properties of viruses influencing choice of strategy include pathogenesis, serotype diversity, antigenic variation, immune evasion mechanisms, latency, and route of transmission. New vaccine candidates have been described for a good number of the viruses listed in Table 1, and although many of these are in an early, pre-proof-of-concept stage, some are substantially developed and offer realistic prospects of licensure over the next decade. The most promising of these, based on pre-clinical and clinical results obtained so far, are dengue, hepatitis E, and HSV and CMV. Significant challenges remain however, and really promising candidate vaccines against pathogens such as HIV, hepatitis C, and infant RSV remain elusive. This article focuses on the strategies available to develop new viral vaccines and discusses some of the challenges posed by the more difficult targets.
Types of Vaccines
There are multiple possible approaches to the development of a viral vaccine that can be generally described as follows:
-
1.
Killed whole or split virus vaccines. This approach requires that the virus can be grown to high titer in cell culture or other scalable medium such as hens eggs; that the virus can be successfully and completely inactivated using an agent such as formaldehyde or B-propiolactone without destroying immunogenicity; that, from an industrialization perspective, the immunogenic dose is low to modest with respect to virus yield (in the 10 μg range) and that the killed whole or split particle elicits protective immunity. This approach has had excellent successes in the form of vaccines such as inactivated polio vaccine (IPV), hepatitis A (HAV) vaccine, and influenza.
-
2.
Subunits or single proteins prepared by recombinant DNA methods and fermentation processes in cell culture. This approach may work well when a single protein can provide immunity and where the expression system allows appropriate folding and processing of the viral protein.
-
3.Live-attenuated vaccines. There are several approaches possible.
-
a.Use of a closely related animal virus that is not well adapted for efficient and widespread replication in humans and therefore does not cause disease, but nevertheless provokes an immune response that protects against the corresponding human virus. The best known example of this is the use of vaccinia virus to vaccinate against smallpox, but a similar approach has been used for rotaviruses, with genetic reassortment to confer appropriate antigenicity.
-
b.Development of an empirically attenuated human virus by multiple passages in tissue culture, typically of nonhuman origin, and/or passage in animals. Attenuation is usually achieved by the accumulation of a number of mutations that affect the efficient functioning at normal body temperature of various genes or gene products, thereby reducing virulence. Replication competence, however, is maintained at a sufficient level to stimulate a protective immune response. Although there are some drawbacks to this approach from a safety perspective (e.g., a risk of reversion), this method has provided a bedrock of vaccinology over many decades and has worked well for viruses such as polio – oral polio vaccine (OPV), mumps, measles, rubella, and yellow fever.
-
c.Live-attenuated vaccines prepared by knowledge-based manipulation of the viral genome. There are several examples of candidate vaccines in this category including HSV and influenza.
-
a.
-
4.
Vectored or chimeric virus approaches. This is where an existing virus vaccine can be modified genetically to carry genes encoding antigens from a foreign virus. The chimeric vaccine should retain the attenuation and growth characteristics of the parent vaccine strain but stimulate immunity against the foreign virus.
-
5.
Naked DNA. This is where a DNA encoding viral antigens plus appropriate expression control sequences is administered directly to the recipient. Expression of the DNA leads to an immune response against the antigens encoded.
Principles of Vaccine Development and Examples
When developing a new vaccine, the choice of approach is made very much on a case-by-case basis, and for a given virus is driven by knowledge of its biology, structure, antigenic diversity, and pathogenesis. High importance should be given to what type of immunity arises as a result of natural infection and whether the pathogen can cause persistent and/or repeated infections in a single host. Experiments in animal models may also allow the dissection of the immune response to identify correlates of protection. The use of primates in particular can be useful if the disease produced is similar to that observed in humans. However, many viruses are highly host specific and may have evolved strategies to evade immune responses that may also be host specific (such as recruitment of downregulators of complement fixation). Care must therefore be taken as results in animal models may not be entirely reproducible in the natural host.
Killed Vaccines
Evidence that circulating antibodies are sufficient to provide immunity may come, for example, from the observation that the disease is modified or exacerbated in immune deficiencies such as hypo-gammaglobulinaemia and/or that passive immune globulin can protect against infection and disease. The latter observation was made for several viruses prior to vaccine development, including hepatitis A, suggesting that the key to vaccine development in these cases would be the stimulation of a strong humoral immune response. Such responses can often be adequately provided by killed vaccines, and so this was an obvious choice of approach. The strategy will also be influenced by the successes and failures with closely related viruses (either human or animal) that have similarities in epidemiology, pathogenesis, and mode of transmission. Thus, for HAV, the successful paradigm of the inactivated polio virus vaccine from the same virus family (Picornaviridae) provided further confidence that a killed whole virus particle approach would be effective. Indeed, inactivated HAV vaccines were developed successfully on this basis by several companies in the early to mid-1990s. Current HAV vaccines are prepared by propagating the virus in an approved cell substrate, human fibroblasts or human diploid cell culture MRC-5, purification, inactivation using formalin, and adjuvanted with aluminum hydroxide. The vaccines are given parenterally, as a two-dose series, 6–18 months apart.
A further example of a killed vaccine is rabies vaccine. Several WHO-recommended inactivated rabies vaccines are available currently. They are all similar in being whole virus, used after inactivation by β-propiolactone, and purification and concentration by ultracentrifugation and/or ultrafiltration. The vaccines are required to have a protective potency defined as >2.5 international units (IU). The potency is determined by the National Institute of Health (NIH) potency test which is based on assays using intra-cerebral challenge of previously immunized mice. The main differences between the rabies vaccines currently available lies in the cell culture used for production. The cell cultures used for the WHO-recommended vaccines are MRC-5 cells for purified human diploid cell vaccine (HDCV), Vero cells for purified Vero cell culture rabies vaccine (PVRV), and primary duck or chicken embryo fibroblasts for purified duck embryo vaccine (PDEV) and purified chick embryo culture vaccine (PCECV), respectively.
When a new virus emerges that poses a severe threat to human health such as the human coronavirus which caused severe acute respiratory syndrome (SARS) in 2002, it is necessary to start working immediately to develop a vaccine. The huge challenge in such a scenario is time. Generally, to develop a vaccine from basic research through animal studies, clinical lot development, analytical test development, clinical trials, industrial scaleup, and licensure takes 8–12 years. These timelines can be compressed in case of extreme urgency, but this compression is not unlimited. Although experience on existing vaccines can be exploited, all processes and procedures need to be evaluated, validated, and implemented. Working with a BMBL Section III Laboratory Biosafety Level 3 (BSL 3) agent, such as human SARS coronavirus, is not exceptional for vaccine manufacturers, but many precautions need to be taken in regards to equipment and laboratory practices, as little was known about the SARS virus in the early stages. The choice of the vaccine approach was indeed influenced by the time factor. If the virus grows well in cell culture, an inactivated viral vaccine is usually the option of choice as it is the fastest to accomplish. Fortunately, in case of the human SARS coronavirus, the virus did grow well on Vero cells, was efficiently inactivated either by formol or β-propiolactone, and appeared to be very immunogenic in several animal models as well as in human beings. After 2003, no more human SARS cases were observed and the development of such vaccines has generally been put on hold for the present.
Recombinant Protein Vaccines
As discussed above, a further simple strategy, particularly when only antibodies are required, is to use recombinant DNA methods to express a single surface structural protein of the virus in a host–vector system such as Escherichia coli or one of several yeast species. This approach may be adopted when the virus cannot be propagated efficiently in culture, making a killed vaccine approach impossible, when the inactivation process may diminish immunogenicity, or when a focused immune response against a specific protein is required. This approach has proved very successful for hepatitis B, where the vaccine is composed of particulate complexes of the virus surface glycoprotein HBsAg produced in yeast. These particles mimic virus-like particles produced during natural infection and induce a highly protective and long-lasting immune response. This vaccine has been on the market since 1992 as a three-dose series of injection, each containing 5–20 μg HBsAg. A new version of this vaccine has recently been licensed with a formulation containing 20 μg HBsAg, adjuvanted with monophosphoryl lipid A (MPL) and alum, which reduces the incidence of nonresponders compared to a population vaccinated with the licensed vaccine.
The recently introduced human papilloma virus vaccines have also been developed using the recombinant protein approach (see below). But as with killed vaccine approaches, one size does not fit all, and for many viruses the approach of recombinant protein expression has not proved successful for a variety of reasons. For example, many viruses have a complex structure that cannot easily be reproduced in foreign hosts at high yield, particularly when the final structure is formed from several conformationally interdependent proteins. Incorrectly folded or immature proteins may not elicit functional, protective antibodies. Second, for some viruses, immune responses to several proteins together may be necessary to provide complete protection against disease. Third, recombinant proteins administered conventionally are generally poor at providing cellular responses of a Th1 profile that may be necessary for protection against viruses such as HIV, hepatitis C virus (HCV), and members of the herpes family. Consequently, for many viruses, live-attenuated approaches or more complex production systems and/or methods of delivery are required as discussed below.
Live-Attenuated Vaccines
Live-attenuated vaccines are used to prevent diseases such as yellow fever, polio, mumps, measles, and rubella. They are based on viral strains that have lost their virulence, but are still capable of replicating sufficiently well to provoke a protective immune response. They cause infection but without inducing the clinical manifestations, eliciting a humoral as well as cellular immune response. Historically, the attenuation was obtained by passage in animals. The first demonstration of attenuation of a virus in cell culture was that of the yellow fever virus by Lloyd and Theiler. The attenuation resulted from a prolonged passage on cultures of chick embryo tissue. Another example is that of the development of the oral polio vaccines by Albert Sabin in the 1950s. Wild-type strains of each of the three serotypes were passaged in monkey testicular tissue both in vitro and in vivo, while testing in monkeys was performed by intracerebral inoculation at various stages of passage. Eventually, strains were selected that were unable to induce paralysis in animals. The number of passages and cloning steps required to achieve the desired level of attenuation varied between the serotypes. These vaccines are still routinely used in many countries of the world and have been the principal tool with which the WHO has pursued its campaign of global polio eradication.
The advent of genome sequencing and recombinant DNA techniques in the 1980s allowed the key mutations conferring attenuation, empirically introduced by Sabin's passages, to be identified. In addition to temperature sensitivity mutations affecting protein structure, all three attenuated strains had in common, point mutations in the 5′ noncoding regions which affected the stability of RNA secondary structure believed to be important for interaction with host factors and for internal entry of ribosomes.
Rotavirus
As discussed above, live-attenuated vaccines may also be based on a closely related animal virus. The rotavirus vaccine licensed by Merck in 2006 is based on the bovine WC3 rotavirus. The original monovalent bovine strain is naturally attenuated for human beings, but does not induce protective immunity. To improve the effectiveness of the strain for human use, reassortant strains were prepared which contained genes encoding capsid proteins from the most common human serotypes on a background of the bovine strain. The present vaccine contains five single gene reassortants, each containing a gene for a capsid protein from human serotypes G1, G2, G3, G4, and PIA (Figure 1 ). A three-dose regimen with 2.0–2.8 × 106 infectious units per reassortant, administered orally beginning at age 6–12 weeks with a 4–10 week interval between doses, provides 70% protection against both mild and severe rotavirus diarrhea.
Figure 1.

Rotavirus reassortant to generate oral live virus vaccine. RotaTeq is a polyvalent vaccine consisting of five human-bovine reassortants: four G serotypes (G1, G2, G3, G4) representing 80% of the G strains circulating worldwide, and one P serotype representing >75% of the P strains circulating worldwide. Reprinted by permission from Macmillan Publishers Ltd: Nature Medicine (Buckland BC (2005) The process development challenge for a new vaccine. Nature Medicine 11: S16–S19.), copyright (2005).
Nowadays, for some viruses, attenuated viral vaccines can be designed on a more rational basis, by specifically targeting virulence factor functions that may not be essential for virus replication, especially in cell culture, but are necessary in vivo to counter host innate defense mechanisms. An example here is the NS1 gene of influenza A viruses. This protein is able to downregulate interferon production in the virus-infected cell and some deletion mutants of NS1 lack this function and are therefore much more easily controlled by the host interferon response and are thereby attenuated. The augmented interferon (IFN) response provoked by such viruses may also have the advantage of providing stronger immune stimulation resulting in increased immune responses. So far, such strains have only been tested in animal models but they offer promise as future influenza vaccine strains.
A drawback of many live-attenuated vaccines is that they require a cold chain from point of production to point of use and this may pose logistical difficulty, especially in developing countries. Also, the safety of live-attenuated viral vaccines is under constant scrutiny because of the risk, albeit small, that the mutations conferring attenuation will revert to wild type, allowing the virus to become virulent again. This is the reason why some live-attenuated vaccines are not recommended for immunosuppressed patients.
Viral Vectors
If neither the killed nor the attenuated vaccine approach is appropriate or feasible, one can consider the use of a viral vector. In this case, an attenuated virus is used as a backbone carrying immunogenic proteins of the virus of interest. In general, the viral proteins chosen are the membrane and/or envelope proteins as these proteins are presented on the outside of the virus particle and recognized by the immune system. An example of a viral vector is the yellow virus vaccine strain 17D. This vaccine strain was developed in the 1930s, since which time over 400 million people have been immunized with this vaccine. The strategy here is to use the 17D vaccine as a vector to deliver the two structural proteins, the premembrane PRE-M and envelope proteins from closely related flaviviruses. The resulting chimeric virus needs to be viable and to replicate efficiently in an acceptable cell substrate for vaccine production. The chimerivax dengue virus approach (by Acambis in collaboration with sanofi pasteur) has been developed using the PRE-M and envelope genes of wild-type clinical isolates. The technology involved is illustrated in Figure 2 . The yellow fever virus genome has been cloned as an infectious cDNA. This infectious cDNA is manipulated to remove the PRE-M and E-Genes of yellow fever virus and exchange them for the coat protein genes of each of the dengue virus serotypes. Thus four individual chimerivax cDNAs are constructed. Transcribing these cDNAs to RNA provides infectious RNA with which to transfect cells in culture. Thus, the resulting virus is a heterologous virus containing the immunizing antigens of dengue virus with the replicative engine of the yellow fever 17D vaccine. Chimeric dengue viruses are expected to mimic the biological properties of yellow fever 17D which has the excellent characteristics of providing minimal reactogenicity and lifelong immunity. The candidate vaccines have been characterized in preclinical models, neurovirulence in mice, viremia and immunogenicity in monkeys, and shown to have desirable characteristics. Moreover, when the four constructs are mixed and administered to monkeys, seroconversion against all four dengue serotypes appears to occur simultaneously in most infected animals. Moreover, the antibodies generated seem to be functional in that they neutralize dengue viruses in plaque reduction tests. The chimerivax dengue viruses grow well in culture and are well suited to industrial scaleup. In human volunteers, the chimeric viruses are safe and well tolerated and elicit specific immune responses against the different dengue serotypes. These strains therefore provide excellent candidates for further development.
Figure 2.
A DNA copy of the genome of yellow fever virus (blue) is manipulated to replace the prM and E genes by those of a related flavivirus such as Dengue (red). Transfection of mRNA transcribed from the resulting cDNA produces a ‘chimeric virus’ in cell culture.
DNA
Since the early 1990s, there has been considerable interest in the possible use of naked DNA as a vaccine delivery method. Naked DNA has the advantage that it can be taken up by cells and express the viral protein encoded. Depending on the conditions, this expression can be mid-to long term, thereby providing a substantial stimulation of the immune response. DNA vaccinology has apparently worked well in mice, but, so far, results in humans have been mainly disappointing requiring milligram amounts of DNA. Delivery of the DNA on colloidal gold, however, seems to offer a better prospect of success, as reported recently for hepatitis B virus and influenza. Regulatory issues concerning the use of DNA vaccines and its possible insertion integration into chromosomal genes are potential drawbacks to this type of approach, especially for use in prophylactic vaccination in infants. Further work is needed on safety issues before it can be seriously considered as a means to vaccinate populations.
The Challenge of Antigenic Variation
Antigenic Variation
Antigenic variation is displayed by a number of important pathogenic viruses and poses a particular problem for vaccine developers. The variation may be manifest in different ways depending on the virus’ natural biology. Thus, for some viruses such as foot-and-mouth disease virus (FMDV), rhinoviruses, and HPV, multiple antigenic variants or serotypes co-circulate, sometimes with particular geographical patterns or ecological niches. Individual strains may show antigenic drift, presumably generating new serotypes over time. The rate of drift and the generation of new serotypes are not well understood for these viruses but may involve genetic recombination as well as cumulative mutational change. Other viruses may show a different pattern of antigenic variation: for example, influenza A viruses circulate as a limited number of subtypes (currently two in humans, H1 and H3), and each of these accumulate antigenic changes over several seasons (antigenic drift), escaping the most recently generated population-based immunity as they evolve. Occasionally, a new subtype may emerge (antigenic shift) either through genetic reassortment of a human strain with an avian strain, or possibly through direct evolution from an avian strain. Generally new subtypes displace existing subtypes as was the case when H2 emerged to displace H1 in 1957 and when H3 displaced H2 in 1968. For reasons that are not clear, this displacement did not occur when H1 reemerged in 1976 and since then there have been two influenza A subtypes co-circulating in the human population. This type of ‘longitudinal’ antigenic variation is clearly different from that of the multiple serotype viruses discussed above, and is generally more tractable in terms of vaccine development. Yet a different pattern is observed with HIV and to some extent HCV, where a limited number of genetic clades may contain very many different antigenic variants and where longitudinal variation to escape recently generated immune responses occurs within a single persistently infected individual. This type of natural biology generates a plethora of antigenic variants that can co-circulate. Providing immunological protection against all of these is an immense challenge for vaccine developers.
Multiple Serotype Vaccines
So what are the strategies available to develop vaccines against antigenically variable viruses? Most straightforwardly, one can generate simple killed vaccines against the currently circulating strains as discussed above and use these where the virus is prevalent. This strategy has had some success in the case of FMDV, where the geographical range of the virus may be (at least partially) restricted by regulations on movement of susceptible farm animals, and the vaccines are cheap and quick to prepare. However, even though simple killed vaccines have been shown to work for individual serotypes of rhinoviruses, it is difficult to imagine that this approach would be effective for this virus where, presumably because of widespread human contact and international travel, there seems to be a freer global circulation of multiple and unpredictable serotypes each winter. Moreover, for most people, rhinovirus infections are relatively trivial and therefore the balance of medical need versus industrial feasibility/commercial attractiveness of preparing vaccines in advance against multiple strains does not favor such a strategy. It would perhaps be a different matter if it were possible to design a simple rhinovirus vaccine that provided cross-protection against all serotypes.
The case of HPV is more manageable because of the small number of serotypes associated with severe disease. Thus, in this case, a significant impact on disease can be made by vaccinating against just a few of the many different genotypes. Both currently available vaccines contain HPV types 16 and 18 to vaccinate against cervical cancer, and the Merck vaccine contains, in addition, HPV types 6 and 11 to vaccinate against genital warts (condylomata accuminata). The absence of efficient cell culture systems for papillomaviruses has required the development of eukaryotic expression systems to produce virus-like particles composed of the L1 capsid protein, which are highly immunogenic. Future, second-generation vaccines will likely incorporate one or more of the additional highly or moderately oncogenic serotypes, such as 31, 33, and 35.
Influenza Annual Vaccination
For influenza, the limited number of circulating subtypes in any particular season makes it possible to adopt a strategy of annual vaccination. Thus, current influenza vaccines are trivalent, containing strains of H1 and H3 of influenza A and an influenza B strain. The strategy of annual vaccination is not without risk and requires a high level of international cooperation on disease surveillance and strain isolation, construction of high-yielding seed viruses, preparation of reagents for formulation, and industrial production. Following strain selection and recommendation by WHO, vaccine production has to occur over a very tight time schedule to ensure that vaccine is ready for the following winter (Figure 3 ). Occasionally problems arise such as a mismatch between the selection of a particular vaccine strain and the virus that eventually circulates during the following winter. This may compromise the effectiveness of that particular component of the vaccine. Other potential problems include less than optimal growth of the high growth reassortant seed at the industrial scale, leading to less vaccine being produced, and the lateness of seeds or reagents impacting on prompt delivery of vaccine for the flu season. The vast majority of influenza vaccine used currently is partially purified, killed whole or split virus prepared in embryonated hen's eggs, formulated to 15 μg of hemagglutinin (HA) of each strain, and provides generally good protection that correlates well with the induction of virus neutralizing or hemagglutination-inhibiting antibodies. However, vaccines prepared using different technologies are arriving on the market or are in advanced stages of development. These include live-attenuated (cold-adapted) strains, licensed by MedImmune in the USA in 2003, influenza-recombinant surface protein (hemagglutinin) produced in a baculovirus expression system from Protein Sciences, and inactivated virus vaccines prepared in cell cultures such as Vero, Madin–Darby canine kidney (MDCK), or PerC-6. This exploration of alternative technologies in recent years has been fueled by several criteria, perhaps the most important of which is greater and more flexible scaleup capability. The recent concerns over the possible emergence of an H5 pandemic has focused attention on the present worldwide limits in capacity, most notably in a situation of ‘surge’ demand, and governments have responded by providing the industry with incentives to increase capacity and diversify methods of production. A further response to this concern has been ‘dose sparing’ clinical studies on influenza vaccines adjuvanted with alum and other, proprietary adjuvants. These studies, using an H5N1 strain, suggest that it may be possible to reduce the vaccine dose from 2 × 90 μg HA nonadjuvanted, to 2 × 30 μg HA adjuvanted with aluminum hydroxide or even as low as 3.75 μg HA adjuvanted with new proprietary adjuvants. Moreover, there have been renewed suggestions that it may be possible to develop vaccines with a broader and perhaps multiseasonal protective effect by stimulating cellular immune responses, particularly against nucleoprotein (NP), a claim currently made for the live-attenuated approach, and even a universal flu vaccine, for example, based on the well-conserved M2 virus surface protein. Animal challenge experiments, especially using multiple arrays of the M2 protein, have been encouraging to date and suggest it may be possible to provoke a much stronger response against M2 than that induced by natural infection. Whether such a response will provide solid protection in humans however remains to be established.
Figure 3.
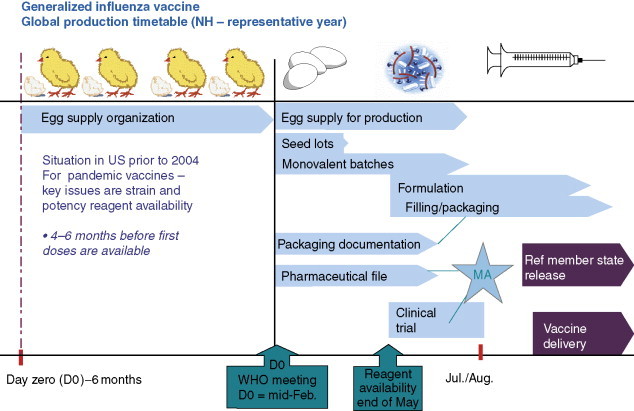
Approximate time schedule for the production of annual influenza vaccine from embryonated hens eggs.
HIV Approaches
The vast array of antigenic variants of HIV and HCV renders the approaches discussed above extremely difficult for these types of viruses. For HIV in particular, many strategies have been tried, so far without significant success. Early on in the AIDS pandemic the focus was on the use of simple recombinant surface glycoproteins, aimed at inducing neutralizing antibodies in the hope that even limited protection against homologous or closely related strains would provide a proof of concept that could be built upon. Unfortunately, this type of approach was not successful and a large phase III clinical trial carried out by Vaxgen using E. coli, which produced gp120, was not able to provide convincing evidence of protection even against strains closely related to that present in the vaccine formulation. Subsequently, there has been considerable effort on the induction of cellular responses, especially CD8 cytotoxic T lymphocytes, and more recently on a balanced cellular response to include CD4 effector mechanisms. The objective here is to provide the means for the immune system to launch an immediate attack on the first cells to become infected following exposure to the virus. Ideally, such an attack would prevent the primary viremia by eliminating the virus before it becomes established in the body. However, more realistically, there is evidence from primate studies that strong preexisting T-cell responses can control the primary viremia and reduce the viral set point during the asymptomatic phase. A low viral set point is associated with slow or no progression to AIDS. In addition, the HIV evades immune surveillance by actively downregulating the major histocompatibility complex I (MHCI) molecules on the surface of infected cells by the HIV nef protein. Vaccination strategies to produce cellular responses have mainly used vectors such as the vaccinia virus strains MVA and NYVAC, canarypox, adenoviruses, or naked DNA, either with multiple doses of a single type of construct or in heterologous prime-boost strategies. Antigens delivered have ranged from substantial regions of gag-pol-env of HIV to multiple copies in a ‘string of beads’ format of defined T-cell epitopes presented by common human leukocyte antigen (HLA) haplotypes. In general, these studies have not delivered T-cell responses of sufficient magnitude to be strongly encouraging, although one such strategy based on canarypox delivery of gag-pol-env antigens, followed by boosting with a recombinant env protein, has progressed to a clinical phase III study. Although many commentators have expressed doubt about whether this approach will show efficacy, it may generate useful information on the role of cellular responses in controlling HIV loads.
Most recently, HIV vaccine efforts have again turned to the induction of neutralizing antibodies, this time aimed specifically at epitopes that have been defined by studying unusual but highly informative broadly neutralizing monoclonal antibodies. The fact that such antibodies exists is highly encouraging from a vaccine perspective. The concepts here are based on the notion that certain conserved but crucial regions of gp120 or gp41 are naturally poorly immunogenic, either because they are relatively hidden in the conformationally folded protein or are shielded by strongly immunogenic noncritical domains or by glycosylation, or because they are only transiently exposed during the structural rearrangements that accompany cell binding and virus penetration. It is argued that because antibodies against these regions are neutralizing they will be protective if they can be generated prophylactically with sufficient avidity and at sufficient titer. This ‘cryptic epitope’ idea has been discussed for several viruses over many years and is akin to that mentioned above for influenza M2, in that the objective is to generate a far stronger response against a particular antigen or epitope than that resulting from natural infection. Such antibodies, once induced, will need to have the kinetic properties necessary to effectively neutralize the virus in vivo. So far there are no examples among virus vaccines that prove this concept. For HIV gp160, the particular construction, presentation, and formulation of molecules able to raise high-titer antibodies against these conserved regions (many of which are imprecisely defined) are far from obvious. Nevertheless, the induction of prophylactic immune responses of this type is certainly worth detailed investigating in detail, given the magnitude of the HIV problem. The challenge of developing a vaccine against HIV however remains immense.
Conclusions and Perspectives
The easy viral vaccine targets of significant medical importance have been done. The viruses against which we do not have vaccines today are either of regionalized or sporadic importance medically and the incentives to develop them have not been sufficiently large or they are viruses that pose significant challenges in terms of their biological characteristics. Thus, HIV and HCV pose challenges because of their antigenic variation and the fact that natural immune responses are unable to protect and/or eliminate the virus. RSV poses challenges because of the immunopotentiation of pathogenesis that, for infants, must be avoided at all costs.
Nevertheless, there are grounds for optimism. A new generation of adjuvants, making it possible to selectively orientate immune responses toward Th1 or Th2 as necessary, promises the possibility of being able to ‘improve on nature’ in terms of immune response provoked by the viral antigens. New developments in vectors and virus ‘chimeras’ offer promise for vaccines such as dengue and perhaps RSV and parainfluenza viruses and targeted modification of immunomodulatory genes may offer prospects of new vaccines against herpes family viruses. Fundamental studies on virus pathogenesis, epidemiology, and immunobiology are greatly aided by new technologies such as genomics and proteomics and it is likely that the improved understanding will increase the technical and scientific feasibility of developing new viral vaccines in the years ahead.
See also
AIDS: Vaccine Development; Antigenic Variation; Antigenicity and Immunogenicity of Viral Proteins; DNA Vaccines; Immune Response to Viruses: Antibody-Mediated Immunity; Neutralization of Infectivity; Vaccine Production in Plants; Vaccine Safety
Further Reading
- Buckland B.C. The process development challenge for a new vaccine. Nature Medicine. 2005;11:S16–S19. doi: 10.1038/nm1218. [DOI] [PubMed] [Google Scholar]
- Garber D.A., Silvestri G., Feinberg M.B. Prospects for an AIDS vaccine; Three big questions, no easy answers. Lancet. 2004;4:379–413. doi: 10.1016/S1473-3099(04)01056-4. [DOI] [PubMed] [Google Scholar]
- Koelle D., Corey L. Recent progress in herpes simplex virus immunobiology and vaccine research. Clinical Microbiological Reviews. 2003;16(1):69–113. doi: 10.1128/CMR.16.1.96-113.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin S.A., Rupprecht C.E., Koprowski H. Rabies vaccine. In: Plotkin S.A., Orenstein W.A., editors. Vaccines. 4th edn. Saunders; Philadelphia: 2005. pp. 1011–1038. [Google Scholar]