

Abstract
Subclinical immunosuppression in chickens is an important but often underestimated factor in the subsequent development of clinical disease. Immunosuppression can be caused by pathogens such as chicken infectious anemia virus, infectious bursal disease virus, reovirus, and some retroviruses (e.g., reticuloendotheliosis virus). Mycotoxins and stress, often caused by poor management practices, can also cause immunosuppression. The effects on the innate and acquired immune responses and the mechanisms by which mycotoxins, stress and infectious agents cause immunosuppression are discussed. Immunoevasion is a common ploy by which viruses neutralize or evade immune responses. DNA viruses such as herpesvirus and poxvirus have multiple genes, some of them host-derived, which interfere with effective innate or acquired immune responses. RNA viruses may escape acquired humoral and cellular immune responses by mutations in protective antigenic epitopes (e.g., avian influenza viruses), while accessory non-structural proteins or multi-functional structural proteins interfere with the interferon system (e.g., Newcastle disease virus).
Keywords: immunoevasion, immunosuppression, avian influenza virus, avian leukosis virus, fowl poxvirus, infectious bursal disease virus, infectious chicken anemia virus, Marek’s disease virus, mycotoxins, Newcastle disease virus, reovirus, reticuloendotheliosis virus
16.1. Introduction
Control of infectious diseases is essential for the production of healthy poultry flocks, and this is generally achieved by extensive vaccination programs in combination with good management practices, including biosecurity measures to reduce the risk of infection. The success of vaccination programs depends on the ability of the birds to mount a vigorous immune response after vaccination. In addition to the innate ability of a particular bird to mount an immune response to a vaccine or an infection, there are numerous external factors influencing the level of protective immunity. The purpose of this chapter is to review the mechanisms and impact of immunosuppression, which can have multiple causes, and the ability of pathogens to counteract immune responses by immunoevasion.
In this chapter, immunosuppression is defined as “A state of temporary or permanent dysfunction of the immune response resulting from insults to the immune system and leading to increased susceptibility to disease,” as originally proposed by Dohms and Saif [1]. with the addition of “and often a suboptimal antibody response,” as suggested by Lütticken [2], to which we add “and suboptimal innate and cell-mediated responses.” Such dysfunction often results from infection of cells of the immune system, leading to their impaired function against the primary and subsequent infections in a non-specific manner. For this chapter, we define immunoevasion as “pathogen-initiated responses counteracting the immune responses to the specific pathogen.” The major difference is that immunosuppression is the consequence of the overall replicative strategy of the causative agent, resulting in increased susceptibility to other pathogens but not necessarily to the causative agent, while immunoevasion is achieved by specific pathogen-encoded determinants, primarily favoring replication of the causative agent. In some instances, infections can result in immunosuppression and immunoevasion—for example, infection with Marek’s disease virus (MDV).
16.2. Immunosuppression
16.2.1. Introduction
Immunosuppression is a major problem for the poultry industry, but actual figures indicating the scale of the problem are difficult to find. Infection with pathogens and/or environmental factors, including management errors, can result in immunosuppression, and interactions between the two usually exacerbate the problem. In an excellent recent review article, Hoerr [3] examined the many environmental and infectious causes of immunosuppression in chickens and turkeys, which are complex and can have a major impact on profitability of broiler and layer flocks (Figure 16.1 ). Environmental stressors include the later period of incubation, hatching and chick handling. Other stressors include suboptimal housing conditions and mycotoxins. Any infection causing clinical disease may result in immunosuppression, but the focus here is on pathogen-induced immunosuppression in the absence of clinical disease.
Figure 16.1.
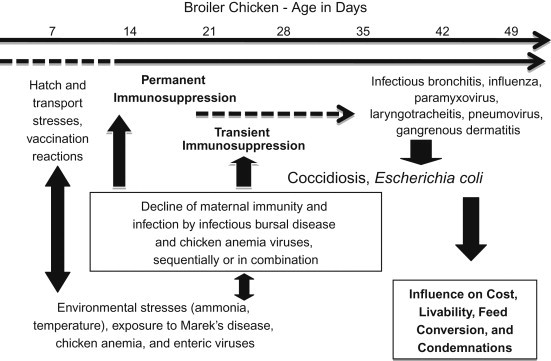
Immunosuppressive interaction in broiler chickens. A high-yield broiler may hatch in an energy-depleted state and be subjected to suboptimal handling and transportation systems, and experience delays in acquiring nutrition, all while processing attenuated live vaccines that produce cytolytic infections. The brooding environment may not meet thermoneutrality, and placement on reused litter creates the potential for exposure to ammonia and potentially immunosuppressive agents (e.g., MD, chicken anemia and enteric viruses). With the decline of maternal immunity, active infection by IBDV can be permanently immunosuppressive at or before 14 days of age and, at least transiently, immunosuppressive when it occurs at an older age. Also, with declining maternal immunity, CIAV can produce a cytolytic infection in the thymus and contribute to the immunosuppression initiated by IBDV. Following this sequence, it is possible for immunocompetency to be compromised in a variable percentage of the flock. This can contribute to increased severity of infections by respiratory viruses and the ability to control secondary respiratory infections by E. coli. Coccidiosis and gangrenous dermatitis may likewise emerge as problems from 25 days of age and older. Collectively, this sequence of events occurs to variable degrees and influences livability, feed conversion, condemnation at processing and total production cost. Each immunosuppressive influence represents a control point to prevent immunosuppression and improve health and welfare.
Source: From Hoerr [3], with permission from the American Association of Avian Pathologists.
Viruses causing immunosuppression and/or clinical disease at any age include MDV, reticuloendotheliosis virus (REV), reovirus and, although controversial, avian leukosis virus (ALV). Other pathogens—for example, chicken infectious anemia virus (CIAV)—may cause clinical disease in young chicks, but the major damage is caused by subclinical infection resulting in immunosuppression. Infectious bursal disease virus (IBDV) also causes clinical disease in young chicks, but results in damage to immune tissue, particularly the bursa of Fabricius and bursa-derived lymphocytes, compromising the host’s ability to mount effective responses upon subsequent infection by other pathogens. Caution is needed, however, in the interpretation of the immunosuppressive effects caused by reovirus, MDV, REV, and IBDV, especially with older studies, because it is not always clear whether these viruses were free of CIAV. For example, Jakowski et al. [4] reported hematopoietic destruction in chickens with Marek’s disease, but it was later learned that tumor material harvested from these birds was positive for CIAV (Wellenstein, personal communication, cited in reference [5]). In addition to viral infections, coccidial infections have been linked to immunosuppression.
16.2.2. Stress-Induced Immunosuppression
Most environmental factors causing immunosuppression are related to management problems such as inadequate water or food supply, ammonia in the houses, temperature stress, “social interactions within a flock,” and so on. Most of these stressors enhance the production of corticosterone. Selection for high versus low corticosterone concentration in blood plasma can influence the degree of stress-induced immunosuppression. In addition, the presence of fungal toxins in feed is an environmental stressor.
It has been recognized for a long time that social stress can exacerbate disease. A classic example is the study by Gross [6] in which MDV-exposed chickens were kept in a socially stressful environment by moving one chicken from one cage into another cage every day. These chickens developed a higher incidence of tumors than MDV-infected chickens kept in a low-stress environment. The effects of social stress on MD were especially enhanced in birds selected for high plasma corticosterone concentrations. Inoculation with chemicals blocking 11-β hydroxylase, which mediates the conversion of deoxycorticosterone to corticosterone in the adrenal glands, reduced the impact of social stress on Marek’s disease and other diseases 6., 7.. Inoculation of chickens with corticosterone using pharmacological doses resulted in a rapid lymphoid depletion in thymus, bursa, and spleen [8]. These authors suggested that bacterial infections (e.g., Escherichia coli) may cause stress-type lesions in the bursa similar to corticosterone-induced lymphoid depletion. Recently, it has been shown that high levels of corticosterone in hens reduce egg production and decrease testosterone and progesterone in the yolk, influencing the quality of the chicks [9]. In addition, embryonic exposure to increased levels of corticosterone can alter juvenile stress responses [10].
Fungal toxins or mycotoxins are often present in feed and can cause clinical disease as well as immunosuppression. The importance of mycotoxins as immunosuppressive toxins and their effects on immune functions have been reviewed by Bondy and Pestka [11] and Hoerr [3]. Aflatoxin B1 (AFB1) is the best known fungal toxin for poultry, although chickens are rather resistant to the toxic effects compared to ducks and turkeys [12]. After ingestion, AFB1 is bioactivated in the liver by cytochrome P450 into the highly toxic component aflatoxin-8,9-epoxide, which can bind to DNA, or after being hydrolyzed, bind to proteins, causing cytotoxicity [13]. The consequences are cell death in the primary lymphoid organs, damage to the intestinal integrity and, especially in broilers, decreased performance.
Antibody and CMI responses are in general decreased 3., 12., although at low levels antibody responses may actually increase during initial exposure [12]. Chicks hatched from hens fed on diets containing aflatoxins also showed impaired immune functions when tested at 2–3 weeks of age, but it is not known how long after hatching the effects last [14]. Fumonisins, trichothecenes (e.g., T2) and ochratoxins (e.g., OTA) have also been implicated as immunotoxicants in chickens, turkeys and ducks. Low levels (15 ppm) of fumonisin B1 (FB1) given to broilers in the feed for 3 weeks reduced macrophage activity, reduced secondary antibody responses to Newcastle disease vaccines, and reduced gene expression of interleukin (IL)-1β, IL-2, interferon (IFN)-α and IFN-γ. This level of FB1 does not cause a decrease in growth performance and is considered an acceptable level in chicken feed [15]. Two toxins belonging to the trichothecenes group, T-2 and deoxynivalenol (DON), have been linked to immunomodulation in chickens 16., 17.. Low doses of T-2 and DON have been linked to increased IgA levels and titers against Newcastle disease virus (NDV), while high doses cause immunosuppression. Broilers fed a diet containing DON and challenged with very virulent IBDV developed more severe effects than broilers fed a diet lacking DON [18].
The interactions between trichothecenes and immune responses are complex [19]. Low doses can actually stimulate immune responses by up-regulation of cytokines, and chemokines, while high doses activate caspases resulting in apoptosis. OTAs are highly toxic in many species, including chickens, and can be found in eggs from OTA-fed chickens. Innate and acquired immune responses are decreased and susceptibility to disease is increased in chickens fed OTA≥1 ppm and in their progeny 20., 21., 22.. For example, OTA at 2 ppm for 14 days significantly increased mortality caused by Salmonella enteric subspecies enterica serovar Gallinarum [23]. Combinations of the different mycotoxins are frequently present in feed, and interactions between them often increase the immunosuppressive effects 24., 25., 26.. The presence of mycotoxins in feed and possible embryonal transmission have important implications and can confound the interpretation of immune dysfunctions in commercial flocks when virally induced immunosuppression occurs.
16.2.3. Coccidia-Induced Immunosuppression
Two groups of coccidian species have been linked to immunosuppression, Cryptosporidium baileyi and Eimeria species, although the evidence is rather limited in both cases. Infection with the latter has been linked to decreased mitogen stimulation responses [27], but antibody responses to T cell-dependent and -independent antigens were not affected [28]. C. baileyi replicates in the epithelial cells of the bursa of Fabricius and the respiratory tract of chickens and is probably more prevalent than diagnostic cases indicate [29]. Oral inoculation of young chickens with high doses of C. baileyi caused histopathological lesions in the epithelium and lamina propia of the bursa [30] and decreased antibody responses to T cell-dependent and -independent antigens 31., 32.. Titers to infectious bronchitis virus (IBV), NDV and avian influenza (AI) vaccine were also decreased after inoculation of chicks with C. baileyi 33., 34., 35.. However, Abbassi et al. 36., 37. found that infection with C. baileyi did not increase Marek’s disease or reduce the efficacy of the MDV vaccine strain CVI988. Interestingly, MDV infection before or after challenge with C. baileyi aggravated the latter. Likewise, MDV vaccination followed 4 days later with C. baileyi challenge-caused respiratory lesions within 6 days (see also Section 16.2.5 MDV).
16.2.4. Virus-Induced Immunosuppression
As mentioned before, we discuss only viruses that may cause immunosuppression independently of clinical symptoms, although the independence may be a temporal feature, as is the case for the tumor-inducing viruses and, depending on the age of challenge, CIAV and IBDV. The following viruses and their effects on the immune responses are discussed: IBDV; CIAV; reovirus; adenovirus; and the three tumor-inducing viruses ALV, REV, and MDV. Detailed information on virus replication, pathogenesis, pathology and immune responses can be found in several chapters in the 12th and 13th editions of Diseases of Poultry 38., 39. and other textbooks on avian diseases.
Infectious Bursal Disease Virus
IBDV, belonging to the Birnaviridae, has been divided into two serotypes, of which only serotype 1 causes immunosuppression and disease in chickens. Several pathotypes are recognized within serotype 1, varying from mild to very virulent. More recently, strains within group 1 have been further subdivided into genetic groups based on restriction enzyme analysis and sequencing. It has been recognized for a long time that serotype 1 strains replicate in B cells expressing Bu-1 and surface immunoglobulin (sIgM) 40., 41., resulting in apoptosis and atrophy of the bursa of Fabricius. The apoptosis of the immature B cells results in severe immunosuppression with impaired antibody responses and increased susceptibility to other pathogens, especially when birds are infected before 3 weeks of age (reviewed 40., 41., 42., 43.).
The genome of IBDV is characterized by a double-stranded (ds)RNA genome consisting of segments A and B. Segment B codes for the viral RNA-dependent RNA polymerase, which, based on recent data, may be involved in pathogenicity [44]. Segment A codes for two structural proteins (VP2 and VP3), an autoprotease (VP4) and a small non-structural peptide (VP5), which partly overlaps with the open reading frame (ORF) coding for VP234 [42]. The polyprotein VP234, but not the mature VP2, VP3 or VP4, arrests B cell division probably by interfering with the cell cycle given that it does not affect cell viability [45].
Using a reverse genetics approach, Yao et al. [46] showed that deletion of VP5 did not prevent virus replication in vivo but did prevent the development of bursal lesions, suggesting a role for VP5 in the pathogenesis. Interestingly, in vitro studies suggest that very early after infection VP5 is anti-apoptotic [47], probably through binding to the p85α regulatory subunit of phosphatidylinositol 3-kinase (PI3K) [48]. Activated PI3K phosphorylates Akt which results in survival of infected cells early during infection. During the later stages of infection, VP5 accumulates in the plasma membrane [49] and causes apoptosis [50]. The induction of apoptosis was recently linked to activation of the Jun NH2-terminal kinase (JNK) [51], although it was not resolved if VP5 or VP2 was responsible for the activation, which was also linked to apoptosis in infected B cells [52]. In addition to the apoptosis of IBDV-infected B cells, non-infected B cells can also undergo apoptosis [53], perhaps caused by the rapid increase of IFN-γ shortly after infection [54]. In summary, the molecular basis for IBDV-induced apoptosis of B cells is the result of a complex set of interactions between the different viral proteins and infected B cells.
It has been known since the 1970s that IBDV infection in chicks younger than 3 weeks of age causes severe damage to the bursa of Fabricius, with depletion of B cells expressing sIgM affecting mostly primary antibody responses. Infection, especially at 1 day of age, also results in a significant decrease of sIg-expressing B cells in spleen and peripheral blood lymphocytes, but does not affect circulating CD4+ and CD8+ T cells 55., 56.. The damage to the bursa is transient, follicles become repopulated with lymphocytes, and tissue architecture is restored, but primary antibody responses remain depressed until at least 7 weeks post-infection (PI). Ultimately however, they also recover. The duration of the recovery process depends on the age at infection and the virulence of the strain 57., 58..
Withers et al. 59., 60. described two types of follicles emerging after recovery from IBDV infection in neonatal chicks: small follicles lacking a distinct cortex and medulla and large follicles with rapidly proliferating B cells and a normal structure. B cells in the large follicles were still capable of undergoing gene conversion and may have been derived from small numbers of surviving bursal stem cells. In contrast, the B cells in the small follicles were considered to be derived from more mature B cells that had already undergone gene conversion. These data suggest that the ability to recover from IBDV-induced suppression of antibody production and diversity is based on the proportion of small versus large follicles developing after infection. Recently, Biró et al. [61] reported that B cell maturation may be negatively affected by IBDV-induced changes in the extracellular matrix of the antigen-trapping regions of the spleen, which may contribute to permanent immunosuppression.
In addition to replication in B cells, IBDV can also infect and replicate in macrophages 62., 63. and mesenchymal stem cells [64]. Khatri et al. [63] found viral RNA and proteins by reverse transcriptase (RT)-PCR and immunohistochemistry, respectively, in bursal macrophages between 1 and 7 days PI. The absolute number of macrophages in the bursa was decreased significantly at 3 and 5 days PI. Similarly, the number of macrophages was also decreased in spleens after IBDV infection [65]. The actual impact on the immune response of these observations is not clear. Pro-inflammatory cytokines, such as interleukin (IL)-6, IL-1β, and CXCLi2, are increased at the transcriptional level in the bursa and spleen, while the anti-inflammatory cytokine transforming growth factor (TGF)-β4 is decreased 63., 65., 66., 67.. These different groups of authors reported somewhat conflicting results for the production of type I IFN and IFN-γ transcription, but the differences could be the result of using different virus strains and/or infecting different age groups. IBDV infection also increased the production of inducible NOS (iNOS) mRNA in bursal macrophages.
Khatri and Sharma [68] showed that the activation of macrophages is through the p38 mitogen-activated protein kinase (MAPK) and nuclear factor (NF)-κβ pathway, but it is not clear whether these pathways are activated through specific viral proteins. The alterations in cytokine transcription are compatible with the inflammation in the bursa during the acute infection. However, it is less clear how these changes play a role in IBDV-associated immunosuppression. It is also important to determine whether the changes occur in IBDV-infected macrophages before they undergo lysis or whether infection and cytokine deregulation occurs in different subpopulations.
Although T cells are not susceptible to infection with IBDV, these cells form an important component of the overall immunopathogenesis of IBD. There is an influx of CD4+ and CD8+ T cells into the bursa between 1 and 10 days PI 57., 69.. The CD4+ cells may also contribute to the production of iNOS by IFN-γ production or other soluble factors stimulating iNOS transcription in macrophages and subsequent nitric oxide (NO) production. NO can contribute to inflammatory lesion development, but may also be involved in down-regulation of splenic T cell responses to mitogens, which is associated with the acute phase of IBDV infection [70]. The infiltrating T cells showed a strong up-regulation of mRNA for several cytolytic molecules (e.g., perforin and granzyme A [71]), suggesting a role in clearance of virus-infected cells and thus a contribution to bursal atrophy.
In conclusion, IBDV infection causes a complex set of interactions between B cells, macrophages, and T cells leading to destruction and subsequent partial recovery of the bursa and long-lasting suppression of primary antibody responses. Recent studies by several groups have provided a better insight in the immunopathogenesis of IBDV in young birds; however, important questions remain. For example: How can we explain the recovery from infection in the face of suppression of primary antibody responses if neutralizing antibodies are the key to recovery? Do we have to postulate that cell-mediated immunity is far more important than previously believed, as suggested by the findings by Raul et al. [71]? Another unresolved question is why mostly young birds develop subclinical disease, while infection of older, antibody-negative birds results in clear-cut pathology including hemorrhages. There may be similarities between the situation in birds with IBDV and that in mice with influenza virus, where the immune response is responsible for most tissue damage, in which case the damage can be reduced by treatment with TGF-β [41].
Chicken Infectious Anemia Virus
CIAV is a small DNA virus of approximately 25 nm currently belonging to the Circoviridae, genus Gyrovirus but most likely to be reclassified as the only genus in the Gyrovirinae of the proposed new family Anelloviridae [72]. It has a single-stranded covalently closed DNA genome of 2.3 kb, which produces one polycistronic transcript coding for three proteins. The virus is extremely resistant to disinfectants and can resist heat treatment at 80°C for 15 minutes. Due to its ubiquitous presence in chicken flocks, its small size, and its resistance to physical and chemical treatments, it can be present as a contaminant in other viruses, especially if these agents are propagated in embryonated chicken eggs. As such, it can become a confounding factor in studies on immunosuppressive properties of other pathogens.
The pathogenesis and immunosuppression caused by CIAV have been reviewed 5., 73., 74.. Infection of chicks may result in clinical disease by vertical transmission, which occurs when hens first become infected during egg production, or by horizontal transmission during the first few weeks of age. However, most chicks are protected against early infection by maternal antibodies, and clinical disease is not frequently seen. Infection after 3 weeks of age is mostly subclinical, but may result in significant immunosuppression. The development of virus-neutralizing (VN) antibodies is essential to curtail virus replication, and immunosuppression caused by IBDV, for example, has been implicated in prolonged replication of CIAV.
The small genome coding for only three proteins, VP1, VP2 and VP3 or apoptin, requires the infection of dividing cells in order to use the cellular machinery for viral DNA replication. VP1 is the capsid and only protein present in the virions, while VP2 has several roles in viral replication. Apoptin causes apoptosis of infected cells, and the mechanisms of the induction of apoptosis have been studied in detail, in part because apoptin may be used as a potential anti-cancer treatment in humans 75., 76.. Dividing cells that are susceptible to infection are hemocytoblasts in the bone marrow, T cell precursors in the thymus, or dividing T cells in response to antigenic stimulation. Infection of hemocytoblasts results in a decrease in erythrocytes, thrombocytes and granulocytes. The loss of the latter two cell types is important because thrombocytes and granulocytes are both important effector cells during bacterial infections and, as a consequence, secondary bacterial infections (e.g., “blue-wing disease”) are frequently associated with CIAV-induced immunosuppression.
Adair et al. [77] reported that CD3+CD8+TCRαβ spleen cells constitute the major CIAV-infected population in the spleen. Recently, Haridy et al. [78] reported that infection in 4-week-old chickens resulted in a moderate loss of CD4+ and CD8+ cells in the spleen and thymus. Infection in 1-day-old chicks causes a more severe depletion of CD4+ and CD8+ cells (e.g., [79]). The effect of virus replication in these cells is especially important when replication of CIAV occurs at the same time that cytotoxic T cells (CTL) are generated in response to vaccination or infection with a second pathogen. Markowksi-Grimsrud and Schat [80] reported the absence of REV-specific CTL 7 days after birds were co-infected with REV and CIAV, at which time CIAV was actively replicating, based on real-time quantitative RT-PCR analysis. Because there was no effect of CIAV infection on transcription of IL-2 or IFN-γ at 7 days PI, it was suggested that the lack of pathogen-specific CTL was caused by CIAV-induced apoptosis of CD8+ cells during the generation of CTL. In contrast to the effect on CTL, natural killer (NK) cells were not affected by CIAV infection [81]. Based on their studies of CIAV infection in MSB-1 cells, Peters et al. [82] suggested that VP2 may also play a role in immunosuppression through down-regulation of major histocompatibility complex (MHC) class I antigens. The importance of this observation for immunosuppression is difficult to evaluate because the assumption is that CIAV-infected cells will become apoptotic.
CIAV-induced immunosuppression has been causally linked to increased incidence of other diseases [5]. For example, infection with CIAV can aggravate infectious bronchitis virus (IBV) -induced disease [83], likely by affecting both CMI and antibody responses. Van Ginkel et al. [84] demonstrated reduced local antibody responses to IBV in the Harderian gland and lacrimal fluids in CIAV-infected chickens. This effect was most likely caused by a decrease in CD4+ Th cells as a consequence of CIAV infection.
The impact of CIAV infection on cytokines has not been well studied, and early investigations relied on bio-assays representing the state of the art at the time (reviewed [85]). More recently, quantitative (q)RT-PCR assays have been used to investigate the effects of CIAV infection on cytokines in relation to virus replication. Unfortunately, the few published results have not included the effect of virus replication prior to 7 days PI, when high levels of virus replication occur 80., 86.. At that time, immunosuppressive effects are already evident with impairment of macrophages [87] and CTL [80], but IFN-γ, IL-2 and IL-1β mRNA levels were not affected [80].
Clearly, additional studies using quantitative RT-PCR assays or enzyme-linked immunoassays (ELISA) are needed to determine the impact of CIAV infection on cytokines starting at 2–3 days PI, because viral antigens can be detected in lymphoid tissues and bone marrow as early as 3–4 days PI [88]. Interestingly, the damage to the thymus and bone marrow was quite extensive 3–12 days PI, although relatively few cells in these organs were positive for viral antigens. The collapse of the thymus is probably the result of damage to the cytokine network needed for T cell maturation. Detailed studies on early cytokine changes during CIAV infection are essential to gain an understanding of the subsequent immunosuppression.
Reovirus
Avian reoviruses belong to the Orthoreovirus genus of the Reoviridae, which have a genome consisting of 10 double-stranded RNA fragments. Avian reovirus infections can cause tenosynovitis and other diseases in chickens, or result in a subclinical infection. Although horizontal transmission is the main route for infection, egg transmission may occur infrequently in chickens 89., 90.. Infections with pathogenic, but not non-pathogenic, strains have been associated with depletion of lymphoid cells in the bursa and thymus and decreased serological responses to inactivated NDV [91]. However, Montgomery et al. [92] were unable to find a significant impact of reovirus infection on antibody responses to NDV, SRBC, and Brucella abortus antigen, although a decrease in relative bursa weight and some lymphocyte depletion in the bursa were noted. The cause of the latter is not clear because reovirus replicates in macrophages but not in B cells 93., 94..
Reovirus infection decreased the responses of peripheral blood monocytes and splenocytes to mitogens at 7 days PI but not afterward 92., 95., 96., 97.. Removal of plastic-adherent cells primed to produce NO restored mitogen responsiveness of T lymphocytes to some degree and suggested that suppressor macrophages were responsible for the immunosuppression 95., 96., 97.. However, NO can also have a beneficial effect by significantly reducing reovirus replication [98]. The pathways of the anti-viral and immunosuppressive actions of NO are very complex and were recently reviewed [99]. The immunosuppressive pathways of NO include modulation of the Th1/Th2 balance through Treg cells and a reduction in T cell proliferation.
The S1 genome segment of avian and mammalian reovirus codes for three open reading frames (ORF), two of which are translated in non-structural proteins. These two proteins have pleiotropic functions, and many of these interfere with cellular signaling pathways [100]. The S1 proteins have been linked to immunosuppression in mammalian reoviruses [101], perhaps through interference with the innate immune response [102]. The avian reovirus-coded S2-coded σA protein has anti-interferon activity, which in addition to immunoevasive properties may contribute to immunosuppression associated with avian reovirus infections [103].
Poult enteritis and mortality syndrome (PEMS) is characterized by diarrhea, growth depression, and immunosuppression with a multifactorial etiology [104]. Turkey reovirus has been associated with some of the aspects of PEMS, including a decrease in thymus and bursa weights [105]. However, this specific reovirus isolate, ARV-CU98, did not infect macrophages or B and T cells [106]. Using a different isolate, NC/SEP-R44/03, in 3-day-old turkeys, Day et al. [107] reported moderate to severe bursal atrophy, a decreased antibody response to NDV, and decreased cutaneous basophil hypersensitivity. It is not clear whether these effects are caused directly by the virus infection or indirectly by interfering with the uptake of nutrients and water.
16.2.5. Tumor Viruses
The three tumor viruses associated with lymphoid tumors, MDV, ALV, and REV, are discussed in more detail in Chapter 19. In this chapter, only the immunosuppressive aspects are reviewed. Extensive reviews of the diseases caused by these viruses are available in the 12th and 13th editions of Diseases of Poultry 38., 39..
Marek’s Disease Virus
MDV, or Gallid herpesvirus 2, belonging to the genus Mardivirus in the subfamily of Alphaherpesvirinae, is causing tumors of T lymphocytes in chickens. It has attracted the attention of virologists and immunologists because highly successful vaccines protecting against the disease became available during the early 1970s. Since then, the virus has evolved from virulent to very virulent (vv) and, more recently, to vv+ pathotypes [108]. This increase in pathogenicity not only has increased the incidence of tumors but also was linked to some new neurological syndromes [109]. In addition, the vv+ strains caused more severe damage to the lymphoid organs than did virulent and vv pathotypes [110], probably leading to increased immunosuppression.
Immunosuppression caused by MDV has been reviewed by Schat [111], and in this chapter we present only the most important aspects of MDV immunosuppression and immunoevasion (see Section 16.4.2). MDV-associated immunosuppression is often divided into an early immunosuppression phase during cytolytic infection and a late immunosuppression phase when the virus is reactivated and tumors develop. One of the key characteristics of early immunosuppression is the destruction of lymphocytes in the lymphoid organs during the first two weeks of infection, causing severe atrophy of the thymus and bursa of Fabricius; this may be permanent or transient depending on the pathotype of MDV.
The model for the pathogenesis of Marek’s disease, as originally outlined by Calnek [112] and Schat [113], provides the basis for our understanding of how MDV causes both B and T cell depletion. Briefly, chickens become infected through the cell-free virus present in feather-follicle dander. The virus is transferred to the lymphoid organs, probably through macrophages, and replicates first in B lymphocytes, causing a productive-restrictive infection in which cell-associated, but not cell-free MDV, is produced and the infected cells are destined to die. This phase is often referred to as the cytolytic phase.
As a consequence of the production of viral antigens and subsequent immune responses, T cells become activated, expressing MHC class II antigens and other activation markers. In contrast to resting T cells, activated T cells are susceptible to MDV infection and become infected. Normally, MDV establishes a latent infection in activated T cells through poorly understood mechanisms [114]. Cytokines [115] and microRNA [116] most likely play a role in the process. Depending on the pathotype of MDV and the genetic resistance of the host, latency may be permanent, temporarily followed by a secondary cytolytic cycle and additional immunosuppression and tumor development, or absent, with continuous virus replication and, frequently, early mortality.
As early as 3 days PI, the expression of IFN-γ is up-regulated [117], which led Schat and Markowski-Grimsrud [115] to suggest that this may up-regulate the expression of IL-8 receptors. The up-regulation of IL-8 receptors is likely an essential step in the transfer of cell-associated MDV from B cells to activated T cells, allowing viral IL-8 produced during the lytic infection [118] to attract activated T cells. Increased cytolytic infection, and thus increased damage to the lymphoid organs associated with some of the vv+ pathotypes, may be caused by a more effective transfer of MDV from B to T cells due to increased levels of vIL-8 production combined with the inability to establish latency after 7 days PI.
Jarosinski et al. [119] reported that vv+ strains produced higher levels of vIL-8 than less virulent strains, but this is likely the consequence of increased virus replication [120] rather than an intrinsic ability to enhance vIL-8 production. The importance of vIL-8 for cytolytic infection is further shown by the fact that tissue culture-attenuated strains have no or very low vIL-8 transcript levels [120] and that deletion of vIL-8 121., 122. or deletion of exon 1 of vIL-8 [123] results in attenuation of cytolytic infection. The reasons for the continued replication of vv+ MDV instead of establishment of latency have not been elucidated. It is of interest that vv+ MDV causes a highly significant up-regulation of pro-inflammatory cytokines as early as 4 days PI in spleen and brain tissues 124., 125.. It is certainly feasible that the distortion in cytokine profile affects the production of cytokines involved in the induction and/or maintenance of latency [126], with the consequence of a prolonged cytolytic infection leading to more profound immunosuppression.
Apoptosis is the most likely mechanism responsible for cell death during cytolytic infection in the lymphoid organs 127., 128. affecting CD4+CD8+ thymocytes. What needs to be resolved is whether MDV-infected thymocytes become apoptotic or whether MDV-induced cytokine deregulation affecting thymocyte maturation causes apoptosis of non-infected cells and a collapse of the thymus architecture [111]. The latter is certainly possible, based on recent evidence that cytokines can be deregulated, especially after infection with vv+ strains. MDV infection can also cause apoptosis in CD4+ peripheral T cells, but CD8+ T cells are apparently not affected [129]. The reasons for the latter are not clear because CD8+ T cells can be infected with and transformed by MDV 130., 131.. Thus far, it has not been resolved which MDV gene products are responsible for immunsuppression. It is likely that the SORF2 gene plays a role, based on two studies using opposing experimental strategies. Deletion mutants lacking several ORFS, including SORF2, caused decreased cytolytic infection without preventing tumor formation [132]. Up-regulation of SORF2 expression through the insertion of the REV long terminal repeat (LTR) resulted in enhanced cytolytic infection in the absence of tumor development [133].
In addition to the destruction of lymphoid tissues, MDV also induces immune suppression through activation of macrophages. These macrophages were able to inhibit mitogen stimulation of T cells obtained from non-infected chickens [134]. Schat and Markowski-Grimsrud [115] suggested that this inhibition was likely the consequence of MDV-induced NO production by the macrophages and was protective rather than immunosuppressive by reducing the pool of activated T cells. The concept of immunosuppression around 7 days PI is conflicting because there is destruction of lymphoid tissue, potential immunoevasion, and the activation of “suppressor” macrophages, while, at the same time, CTL against several MDV proteins are present [115]. In contrast with the more classical strains, vv+ strains may actually infect macrophages [135], and macrophages have been associated with MDV lesions in the brain [136] (and B. L Njaa, K. W. Jarosinski and K. A. Schat, unpublished data). The importance of these observations for immunosuppression is not clear; more likely the macrophages are part of the enhanced pro-inflammatory response in the brain [124].
Late immunosuppression can be caused by lytic infection of lymphoid cells upon reactivation of MDV from latency, causing effects on the immune system similar to those during early lytic infection. In addition, tumor cells may cause immunosuppression (reviewed [111]). However, tumor-induced immunosuppression is the direct consequence of a clinical disease and so is not discussed here.
MDV vaccine strains can also cause immunosuppression. Friedman et al. [137] showed that vaccination with SB-1 (a serotype 2 MDV strain), together with herpesvirus of turkeys (HVT) or CVI988 (a serotype 1 vaccine strain), caused a significant decrease in B lymphocyte activation and antibody production after in vitro stimulation with Salmonella typhimurium or bovine serum albumin (BSA) inoculation, respectively. E. coli-caused mortality was increased in vaccinated chicks independently of the MDV vaccine used, when challenged at 11 or 14 days post-vaccination, but it occurred only in chicks vaccinated with CVI988 when these were challenged at 21 days post-vaccination.
Islam et al. [138] also reported that HVT did not cause increased mortality when chickens were challenged with E. coli at 28 days post-vaccination. However, at 3–10 days HVT caused a significant decrease in B and T lymphocyte numbers. Vaccination with CVI988 precipitated lesions when chicks were challenged with C. baileyi 4 days post-vaccination (see Section 16.2.3). The importance of vaccine-induced immunosuppression is not clear in commercial flocks and is far less important than protection against MDV challenge. The possibility that in ovo vaccination with HVT might induce tolerance was investigated by Zhang and Sharma [139]. Inoculation at 0–14 days of embryonation induced tolerance to HVT but not to BSA. In contrast, vaccination at 18 days of embryonation, which is the appropriate time for in ovo vaccination, did not induce tolerance to HVT.
Avian Leukosis Virus and Reticuloendotheliosis Virus
ALV and REV belong to the Retroviridae. ALV are classified as exogenous or endogenous (or subgroup E) viruses, and the former are further divided in subgroups A, B, C, D, and J. Subgroups A–D are associated with infection of Leghorn-type chickens, but subgroups C and D are of little practical importance for the poultry industry. ALV-J was first described during the late 1980s and is mostly associated with meat-type birds. ALV and REV can also be divided into defective and non-defective (nd) or helper viruses. The defective viruses have often acquired cellular onc genes and cause a rapid onset of tumors and death. These viruses have little importance for immunosuppression because of rapid mortality. The nd viruses can also cause tumors, but transformation occurs through activation of cellular onc genes by the viral long terminal repeats (LTR) [140].
REV and exogenous ALV can be transmitted congenitally or horizontally. Chicks hatched from congenitally infected eggs are tolerant and unable to produce VN antibodies to the virus; they are viraemic, antibody-negative (V+A−) and likely to develop tumors or, in the case of REV, tumors and/or runting and stunting. Horizontal transmission during, or shortly after, hatching can also result in tolerance 140., 141.. This risk is enhanced in chicks when subgroup E or glycoprotein (gp) 85 of subgroup E is expressed during embryonal development [142] or after IBDV infection at 1 day or 6 weeks of age. IBDV infection decreased the frequency of VN antibody-positive chickens and increased virus shedding and viraemia levels significantly, although these effects were dependent on the genetic background of the birds. In contrast, infection with REV at 1 day of age, or MDV at 2 weeks of age, had no significant impact on ALV shedding, viraemia, and antibody development [143]. However, exposure to IBDV at 2 weeks of age did not influence the incidence of ALV-J [144]. The induction of tolerance after congenital infection is virus-specific and is not a general inability to produce antibodies against pathogens other than ALV or REV. For example, chickens congenitally infected with a specific strain of ALV-A were able to produce antibodies to other subgroup A strains [145] and influenza virus, but were partly tolerant to ALV-B [146].
The importance of ALV as an immunosuppressive virus is not clear and probably depends on the subgroup. Fadly et al. [147] and Rup et al. [148] reported that infection with RAV-1, a subgroup A virus, did not suppress antibody responses, mitogen stimulation, or the phytohemagglutinin (PHA) skin test, even in birds developing tumors. In contrast to ALV-A, subgroup B viruses have been linked to suppression of CMI responses. In ovo infection with MAV-2(O) resulted in severe damage to lymphoid organs, suppressed antibody responses, and mitogenic responsiveness [149]. In addition, MAV-2(O) causes a temporary dysfunction of macrophages, suppressing the responses of spleen cells to mitogens 148., 150.. The depressed responses were present as early as 3 days PI and lasted until 21 days PI. Macrophage dysfunction reduced the ability to clear bacteria such as Listeria monocytogenes [151]. A second subgroup B virus, avian erythroblastosis (an acute transforming, defective virus), and its associated helper virus also suppressed T cell responses to mitogens, but in this case the suppression was directly related to T cells and not macrophages [152].
Several groups have shown that ALV-J infection does not affect cell-mediated and humoral immune responses to a significant degree. However, most assays for immune responses and resistance to other diseases were marginally lower in ALV-J-infected chickens than in controls 153., 154., 155., suggesting, but not proving, that immunosuppression may be associated with ALV-J infection. Cui et al. [156] reported that some ALV-J isolates may cause thymus and bursa atrophy and reduced antibody responses to NDV vaccines in chicks when infected at 1 day of age and vaccinated at 7 days. However, the reduced response was not detected when chickens were vaccinated at 30 days of age. Moreover, ALV-J infection did not reduce the antibody response to IBDV vaccine.
REV infection resulted in decreased antibody responses to HVT [157], MDV [158], NDV [159] and H5N1 vaccines [160]. In contrast, Witter et al. 161., 162. found only decreased primary antibody responses to SRBC and Brucella abortus, but no effect on MDV antibodies, although protective immunity to Marek’s disease was decreased. Embryo inoculation with the T strain, but not with the CS strain, also impaired the secondary immune responses to SRBC and Brucella abortus. REV infection increased ALV infection levels in a commercial line [161].
The effect of REV infection on MDV was questioned by Buscaglia et al. [163] because this infection did not impair the establishment of MDV latency or reactivation from latency. These authors suggested that REV may actually reduce the pool of susceptible cells for MDV replication, which would be compatible with the REV-associated atrophy of the lymphoid organ [162]. Filardo et al. [164] suggested that lymphoid atrophy is strain-specific and caused by a combination of env and gag genes.
The immunosuppressive effects of REV infection on cell-mediated immunity have been studied by several groups, mostly using mitogen stimulation assays 161., 163., 165.. The immunosuppressive effects, linked to the development of suppressor cells by Rup et al. 148., 166., are probably the same as Treg cells. Interestingly, Tiwari et al. [167] reported that REV increases the production of TGF-β, which is an important cytokine for the development of Treg cells [168] and in general for the regulation of immune responses [169]. The importance of this type of cell-mediated suppression needs to be further analyzed, especially in view of the development of REV-specific CD8+ CTL starting at 6 days PI 170., 171..
REV-induced immunosuppression may have important practical consequences because REV infection has often contaminated Marek’s disease vaccines 172., 173.. It has been known for a long time that fowlpox viruses (FWPV) can carry replication-competent REV genomes, which may be expressed in vivo 174., 175.. It is therefore surprising that FWPV vaccines are still frequently contaminated with REV 176., 177., suggesting a lack of quality control. The presence of REV in FWPV or FWPV vaccines can result in immunosuppression causing outbreaks of fowlpox, as suggested by Motha and Egerton [178]. Wang et al. [179] compared FWPV vaccine strains with and without REV inserts and wild-type FWPV containing REV. REV-positive FWPV strains, in contrast to those lacking REV, caused decreased antibody titers to BSA and SRBC and decreased responses to PHA-P skin tests and in vitro lymphocyte stimulation.
16.3. Mechanisms of Immunosuppression
In the previous section, three broad categories of immunosuppression causes were identified: corticosteroids in relation to stress, apoptosis and/or necrosis of lymphoid cells, and virus-induced changes in the regulation of immune responses. In many instances, the actual molecular interactions between virus proteins and host cells are poorly identified. To use cytokines as adjuvants to improve vaccines, as suggested by Asif et al. [180], future research needs to focus on a better understanding of the interactions between viruses and cytokine regulation.
16.3.1. Corticosteroids and Stress-Induced Immunosuppression
Dohms and Metz [8] indicated that the mechanisms of stress-induced immunosuppression, especially in birds, were poorly understood. The common pathway for stressors involves the hypothalamic–pituitary–adrenal (HPA) axis and results in the release of glucocorticoids, which are immunosuppressive for many species, including chickens. Treatment of chickens with corticosterones or related glucocorticoids results in lymphopenia and atrophy of the lymphoid organs. Pruett [181] suggested that the interactions between glucocorticoids and the immune system are more complex and, depending on the duration of the stress, can enhance, suppress, or have no effect on immunological variables in mammalian species.
Measuring several immunological parameters, El-Lethey et al. [182] compared the effects of treating chickens with corticosteroids with the effects of a lack of foraging materials. Interestingly, antibody titers against SRBC and tetanus toxoid were decreased after both treatments, but antibodies to human serum albumin were not influenced by either. In addition, cell-mediated immune responses, such as cutaneous response to PHA and delayed-type hypersensitivity (DTH) responses to mycobacterium antigen, were decreased with both treatments. It is also noteworthy that the lack of foraging materials did not significantly enhance plasma corticosterone levels. In conclusion, stress may result in immunosuppression but the actual mechanisms remain elusive.
16.3.2. Apoptosis and/or Necrosis
Virus replication in lymphoid cells is the major cause of cell death for IBDV, CIAV, reovirus, MDV and, to some degree, REV. In most of these infections, apoptosis is the cause of cell death, although necrotic lesions have been reported for MDV [183]. The induction of apoptosis by IBDV VP5 and VP2 was mentioned before, but it is not clear how these proteins actually interact with the apoptotic pathway. Liu and Vakharia [47] showed that IBDV infection in vitro activates effector caspase 3 and the initiation caspase 9, as well as nuclear factor-kappa B (NFκB), resulting in apoptosis late in the infective cycle. These authors suggested that NFκB may be activated through the accumulation of reactive oxygen species, as has been shown for reovirus-induced apoptosis.
CIAV provides another example of viral induction of apoptosis [75]. VP3 (or apoptin) is the major viral protein-inducing apoptosis, although VP2 also may be a minor cause. Early after transfection of MSB-1 cells with a VP3-expressing plasmid, VP3 is present in the cytoplasm and nucleus as fine granular structures. At the time that cells become apoptotic, VP3 forms discrete nuclear structures. The carboxy terminus is positively charged and, based on work with deletion mutants, it is suggested that the positive charge is important for the interaction with the nuclear DNA. Noteborn [5] suggested that this may lead to alterations in the supercoiled organization of the cellular DNA, causing apoptosis or, alternatively, that VP3 acts as a transcriptional regulator of genes involved in the apoptotic process.
Although MDV induces apoptosis, little is known about the viral proteins and mechanisms involved in the induction of apoptosis. Thus far, no proteins preventing or inducing apoptosis have been found in MDV, although in the related HVT Kingham et al. [184] identified an ORF coding for a protein with significant similarity to the putative quail anti-apoptotic gene NR-13. Ross [185] speculated that the phosphorylated polypeptide pp38 was responsible for apoptosis in MDV. Piepenbrink et al. [186] found that pp38 causes decreased production of ATP in mitochondria, which could cause apoptosis.
16.3.3. Virus-Induced Changes in the Regulation of Immune Responses
Thus far, there are few studies describing quantitative changes in cytokine production as a consequence of virus infections in chickens. It is not clear how many of the changes are actually related to immunosuppression rather than those associated with the induction of immune responses. It is clear from the work by Jarosinski et al. [124], using vv+ MDV strains, that strong pro-inflammatory responses may cause pathology. This observation has important consequences for genetic selection for Marek’s disease resistance because the resistant N2a line had a very strong pro-inflammatory response, causing neurological disease with significantly higher levels of cytokine and NO production in the brain. Additional studies on cytokine deregulation leading to immunosuppression are urgently needed using real-time RT-PCR assays and preferably ELISA to examine the impact on transcription and actual production of cytokines.
One of the common immunosuppressive effects described for MDV, reovirus, IBDV, and REV infections is the generation of “suppressor” macrophages. These cells suppress T lymphocyte blastogenesis using mitogen responses and are frequently detected approximately 3–15 days PI. Removal of these cells restores the responsiveness of the T cells (e.g., 96, 134, 167). Interestingly, this immunosuppression has been linked to NO production by macrophages. NO is a highly versatile molecule with immunosuppressive as well as anti-tumor, anti-bacterial and anti-viral activity. It can influence gene expression, probably through the NFκB pathway and, at high levels, can be part of pro-inflammatory pathology (reviewed [187]).
16.4. Immunoevasion
16.4.1. Introduction
As we have learned more about the molecular interactions between viruses and their hosts, it has become apparent that most, if not all, viruses employ immunoevasion mechanisms to survive even the innate immune responses of the host. Those viruses with a persistent life style also deploy a spectrum of subtle, refined mechanisms to survive in the face of potential acquired immune responses. The factors involved in immunoevasion were most easily identified in the large DNA viruses (poxviruses and herpesviruses). With the exception of the molluscipoxviruses, which have no direct relatives in birds but have some similarities with the avian poxviruses, poxviruses cause acute infections; therefore, their immunoevasion mechanisms need only counter (or are only capable of countering) the innate immune responses and possibly delay the acquired responses.
The herpesviruses are able to become latent, a state that requires the expression of very few genes. They do, however, appear to be able to prevent the induction of acquired responses, even though the immune system is regularly open to stimulation by episodic, localized reactivation of latent virus. The recognition that large complicated viruses needed to evolve specific mechanisms to counter innate immune responses arguably drove the suspicion that the smaller, “simple” viruses (often with an RNA genome) must have evolved equivalent mechanisms to survive the same pressures. There are now several examples, particularly in the paramyxoviruses, of small accessory proteins of RNA viruses or of multi-faceted proteins that play a role in immunoevasion, especially in suppressing the type I IFN pathway.
The relative dearth of reagents available for many aspects of avian biology means that detailed study of the mode of action of many immunoevasion mechanisms of avian viruses remains woefully behind that of mammalian viruses. Indeed many of the mechanisms remain presumptive, but the situation should improve with the recent derivation of the draft chicken genome sequence. Recognition of the importance of avian viruses as emerging zoonotic agents—for instance, West Nile virus and H5N1 avian influenza virus—is also likely to drive much more work in this area. We consider immunoevasion mechanisms, both known and postulated, of avian herpesviruses, poxviruses, orthomyxoviruses, paramyxoviruses and reoviruses, as well as review prospects for future work on avian adenoviruses and coronaviruses, orthomyxoviruses and paramyxoviruses.
16.4.2. Immunoevasion Mechanism of the Avian Herpesviruses
Study of the biology of MDV teaches us that this virus uses at least two strategies of immunoevasion, even though no specific mechanisms have as yet been attributed to these observations. Morimura et al. [128] showed that MDV infection down-regulates transcription of CD8. Hunt et al. [188] reported that MHC class I expression was down-regulated in two cells lines, MDV-infected OU2 cells and MDV-transformed MSB-1 cells, treated with 5-bromo-2’deoxyuridine—most likely through a block in the transport of the MHC molecules to the cell surface. Morgan et al. [189], using micro-array analysis, found that MDV up-regulated MHC class I, but this is likely to have been IFN-driven. A mechanism that may favor survival of MDV in its latent state is promotion of the survival of MDV-transformed tumor cells. Whether this is achieved directly by enhanced expression of chicken CD30, previously identified as the AV37 antigen [190], or by the down-regulation of CD28 seen in cells overexpressing CD30 [191] is not clear, nor is the actual viral mechanism of achieving these perturbations in cell surface marker expression.
The herpesviruses have proved a rich hunting ground for those seeking the mediators of immunoevasion, hereafter referred to as immunomodulators, and the avian herpesviruses, such as MDV, prove no exception. The most interesting immunomodulator found in MDV is the IL-8 mimic (vIL-8), identified initially by its sequence homology with host IL-8. More informatively referred by its synonym, eotaxin, IL-8 is a chemokine capable of attracting eosinophils. Deletion of vIL-8 121., 122. or exon 1 of vIL-8 [123] from the RB-1B strain of MDV resulted in a lower frequency of birds with tumors and smaller, less invasive tumors. This suggests that vIL-8 affects virus replication rather than tumorigenesis. The possibility of immunomodulatory functions for vIL-8 cannot be eliminated, but the discovery that it can also be expressed as a fusion protein with the MDV oncoprotein Meq presents the likelihood of a more complicated situation [192].
16.4.3. Immunoevasion Mechanism of the Avian Poxviruses
The avian poxviruses make up one genus (Avipoxvirus) of the Chordopoxvirinae subfamily. The other eight genera comprise only mammalian poxviruses. Recent phylogenetic studies, based on only a couple of conserved genes, reveal that the avipoxviruses show considerable diversity, equivalent to that observed between the Capripoxvirus, Suipoxvirus, Yatapoxvirus and Leporipoxvirus genera of the mammalian poxviruses. They appear to form three major clusters (or clades) of related viruses, one broadly related to FWPV and one to canarypox virus (CNPV), and the other comprising the psittacine poxviruses [193]. Various phylogenenetic studies have indicated that the mammalian poxvirus most closely related to the avipoxviruses is the human Molluscum cotagiosum virus, with which avipoxviruses share many common features of molecular biology; these include key aspects of gene complement and organization, as well as aspects of pathogenesis.
The avipoxviruses have some of the largest DNA viral genomes: up to more than 300 kbp and encoding up to 300 genes. The complete genome sequences of pathogenic and attenuated vaccine strains of FWPV 194., 195. and those of a pathogenic strain of CNPV have been determined [196]. Avipoxviruses cause diseases ranging in severity and mortality from relatively mild cutaneous infections (e.g., fowlpox in chickens) through the more severe diphtheritic infections (also seen in some cases of fowlpox) to disseminated systemic or pneumonia-like infections with high mortality (e.g., CNPV infections). The nature of the infection is probably related to host–virus adaptation, in which immunoevasion mechanisms and especially immunomodulators play an important role. The less severe infections likely reflect long-standing adaptations of virus and host, whereas the more severe probably reflect more recent introductions of viruses to novel hosts.
The derivation of the first sequences brought with it the surprise that FWPV encoded no obvious candidates for modulators of the type-I IFN or IFN-γ pathways or any of the anti-viral effectors that these pathways induce 194., 195., 196.. This was in contrast to the mammalian poxviruses, which encode double-stranded RNA (dsRNA) binding proteins (e.g., E3 of vaccinia virus); mimics of eIF2α, the substrate for activated dsRNA-dependent protein kinase, PKR (e.g., vaccinia virus K3); and secreted, soluble binding proteins for IFN-I and IFN-γ. However, FWPV and CNPV appear to encode mimics of transforming growth factor (TGF)-β, and CNPV encodes an IL-10-like protein. Assuming they act as agonists, both of these candidate immunomodulators are predicted to downgrade the host’s inflammatory responses by stimulation of T regulator cells, thereby protecting the virus-infected cell.
Two groups independently predicted that two different FWPV genes (fpv073 and fpv214) would encode candidate IL-18 binding proteins 194., 195.. Only one of the predicted genes (fpv214) encoded a protein with a conserved IL-18 binding motif. Interestingly, a knockout of this gene in a recombinant FWPV expressing IBDV VP2 resulted in an enhanced CMI response against IBDV when chickens vaccinated with the recombinant were challenged with IBDV. The enhancement was comparable to, but less dramatic than, the enhancement observed when the VP2 recombinant FWPV co-expressed chicken IL18, whether fpv214 was intact or not [197].
The avipoxviruses also encode multiple chemokine-like molecules (e.g., fpv060, fpv061, fpv116, fpv121). These appear to be secreted, although as yet no biochemical activity [194] has been attributed to any of them, partly because of the lack of identified chicken chemokine and chemokine receptor reagents [198]. It is therefore unclear whether they behave as agonists or antagonists and whether they target bona fide chemokine receptors or similar receptors for other host ligands. Avipoxviruses also encode serpentine molecules with seven transmembrane segments resembling G-protein-coupled receptors. These proteins (fpv021, fpv027 and fpv206) are candidate chemokine receptors [194]. Whether they actually bind host chemokines or other host ligands, and whether they are signaling-competent or merely decoy receptors remains to be tested when chicken chemokine reagents become available.
Although no gene encoding IFN-γ-binding protein was predicted from the sequence, a binding protein was identified biochemically by its interaction with recombinant-tagged IFN-γ. It proved to be the product of fpv016 [199]. Type I IFN-binding proteins are not predicted, and it remains to be seen whether alternative approaches will identify genes encoding such proteins. The lack of obvious mechanisms for evasion of type I IFN responses is intriguing, particularly because FWPV is resistant to recombinant chicken type-I IFN at concentrations up to 1000-fold higher than is required to inhibit the chicken cell-adapted vaccinia virus strain MVA [200]. It is also clear that FWPV, unlike other chicken viruses (e.g., IBDV) does not induce chicken type I IFN in CEF culture. Moreover, it can even block the induction of expression of chicken type I interferon β promoter normally seen upon transfection with the dsRNA analog, polyI:C [201].
The avipoxvirus genomes encode many as yet unassigned proteins, which might function in immunoevasion. For instance, one feature that distinguishes avipoxviruses from mammalian poxviruses is that the former encode several families of related proteins. Most notable is the family containing multiple ankyrin repeats. These are found in many types of host proteins, and their presence normally denotes a protein–protein interaction. Up to 31 of the proteins in this family are encoded by FWPV, with 51 (15% of the total gene complement) encoded by the related Canarypox virus [202]. We have no idea of their targets, but presume they are likely to be host proteins and that at least some probably play a role in immunoevasion. Most of these proteins have C-terminal F-box motifs, which in other poxviruses have been shown to interact with the ubiquitin ligase complex via Skp1a and Cullin-1 [203]. By screening chimeric vaccinia MVA-carrying inserts of FWPV genomic DNA for enhanced resistance to chicken type I interferon, Buttigieg et al. [200] recently identified fpv014, an ankyrin repeat protein, as contributing to IFN resistance. They also demonstrated that fpv014 interacted with Cullin-1, but were unable to establish the identitiy of any cellular proteins bound by the ankyrin repeat domain and presumably destined for ubiquitination and likely degradation by the proteasome. Similarly, Laidlaw et al. [201] recently screened a library of FWPV mutants, each carrying a single non-essential gene knockout, for reduced ability to restrict polyI:C-mediated induction of expression of the chicken type I interferon β promoter. They identified another ankyrin repeat protein, fpv012, as contributing to restricting interferon β expression induced by dsRNA, but were also unable to identify the cellular ligand for the ankyrin repeat domain of fpv012.
Identification of two members of this family of proteins from the same virus that appear to modulate chicken IFN responses at different points provides tools to assist ongoing analysis and description of chicken IFN responses, which though broadly similar to those of mammals, clearly differ in significant ways. The best example of this is the absence from chickens, and indeed from all galliforms but not from anseriforms [204], of the cytoplasmic dsRNA sensor RigI, which presumably leaves Mda-5 to perform all cytoplasmic sensing of dsRNA undertaken in mammals by RigI and Mda-5 205., 206., 207.. The complete consequences of this are still not fully apparent or understood.
16.4.4. Immunoevasion Mechanism of the Avian Orthomyxoviruses
Members of the Orthomyxoviridae family are enveloped viruses enclosing eight segments of negative-strand RNA. The best known are of the Influenzavirus A genus, mostly as a consequence of their role in human pandemics. As is now widely appreciated since the emergence of the H5N1 strain, influenza A viruses are primarily an infection of birds, originating in waterfowl, and readily passing to a wide range of birds both wild and domestic.
It was by using influenza virus infection that IFN was discovered in 1957 [208]. Subsequently, it was one of the first viruses for which a resistance mechanism to IFN (activation of host protein p58) was elucidated [209]. Since 1997 and the emergence of the highly pathogenic H5N1 avian influenza virus (AIV), which is capable of causing high mortality in humans, NS1 has been identified as the major viral IFN resistance protein 210., 211., 212., 213.. Moreover, a determinant of the virulence of H5N1 (D92E) has been located on NS1 [214]. These studies have been performed in the context of mammalian IFN responses, which is appropriate because of the clinical threat and practical because the avian IFN response is not fully understood and there is a lack of avian reagents.
Perhaps because of the lack of biochemical assays and reagents, one of the few direct studies of the interaction between AIV and avian IFN was performed with live virus [215]. The study concluded that there was considerable heterogeneity within and between virus populations in their ability to induce and resist avian IFN-I. This heterogeneity was ascribed to their presence in the population of subpopulations that had packaged multiple genome segments. It was presumed that those particles that had packaged multiple segments encoding IFN resistance protein(s) displayed higher resistance, although this characteristic was not necessarily inherited. Such heterogeneity illustrates one of the potential complications of using live-recombinant influenza viruses to dissect out the role of particular molecular determinants of IFN resistance.
Studies with the mammalian system have revealed that NS1 is a very complex, multi-functional protein. It has at least three major functions: (1) binding dsRNA to block IFN, (2) inhibiting host gene expression by preventing mRNA splicing and nuclear export, and (3) enhancing viral mRNA translation. All of these functions merit further investigation in mammalian and avian hosts.
Resistance of influenza virus (influenza A virus unless otherwise specified) to IFN was initially believed to be primarily due to activation of a cellular inhibitor, p58(IPK), or PKR [209]. The mechanism of activation is still not known. Another mechanism involved the ubiquitin-like host protein ISG15, which is one of the most predominant proteins induced by type-I IFN [216]. Influenza B virus strongly induces ISG15, but a specific region of the influenza B virus NS1 protein (NS1B), which includes part of its effector domain, blocks the ability of ISG15 to become covalently linked to its target proteins by inhibiting its UBE1L-mediated activation. The influenza A virus NS1 protein does not bind ISG15, but inhibits its synthesis [217].
Subsequently, however, NS1 was shown to play a major role in resistance to IFN. Specifically, NS1 mutants were able to replicate only in IFN-defective cell lines [211]. NS1, normally 230 amino acids long in influenza A virus, is encoded by virus RNA segment 8. The amino terminus (73 residues) of NS1, which dimerizes [218], is capable of binding dsRNA [219]—albeit at relatively low affinity [217]—thereby preventing activation of the dsRNA-dependent protein kinase, PKR [220]. NS1 also binds poly-adenylated RNA, inhibiting nuclear export of mRNAs [221], and a stem bulge in U6 snRNA, inhibiting pre-mRNA splicing in vitro and in vivo [221]. Both of these activities can down-regulate expression of cellular genes. They can also prevent IFN production by binding to IRF-3, blocking its kinase-mediated activation [213], and by blocking NF-κB activation [222]. Although expression of NS1 alone was reported to induce apoptosis in MDCK and HeLa cells [223], in the context of a viral infection of mammalian or avian cells, its IFN-regulatory activity makes it anti-apoptotic [224]. NS1 selectively enhances translation of viral but not cellular mRNAs by binding eIF4GI, PABP1 and the 5’ UTRs of vmRNAs 225., 226..
It is interesting that the region of NS1 (aa 81–131) binding eIF4GI spans the location of the known virulence mutation (aa 92) and that the region of PABP1 to which NS1 binds is not conserved evolutionarily. The C terminus, or effector domain, down-regulates formation and export of cellular mRNAs by binding to the 30 kDa subunit of CPSF and to PABII 227., 228.. The dsRNA-binding activity of NS1 can be abrogated by mutating two basic residues (R38 and K41 to A). In MDCK cells, virus thus mutated [229] failed to inhibit IFN-β and replicated to lower titers. On passage, a better replicating virus emerged with an S42G mutation that did not improve dsRNA binding but had intermediate virulence in mice.
Recent study of the modulation of avian IFN by AIV has focused on the role of NS1 and of PB2. There are many difficulties with studying the induction of IFN responses in vivo, not least in standardizing the nature of the inducing virus; this can depend on the strain and on its passage history. Penski et al. [230] showed that NS1 deletion mutants of HPAIV (H5N1 and H7N7) induced higher levels of IFN β in CEFs than did parental virus. Liniger et al. [231] showed that NS1 from HPAIV also controls IFN β expression in the chicken macrophage HD-11 cell line. However, NS1 deletion mutants of HPAIV still manage to control IFN β levels in infected chickens [230], suggesting that other mechanisms are involved. Liniger et al. [231] demonstrated that HPAIV PB2 could block induction of IFN β expression in the immortalized chicken fibroblast DF-1 cell line when induced by over-expression of the chicken homologs of VISA/CARDIF or mda-5.
The role of an ESEV motif found at the C-terminus of HPAIV NS1 proteins, but not in seasonal human AIV (which bear the motif RSKV), has been investigated by Zielecki et al. [232] The motif represents a ligand for PDZ domains of cellular proteins. It attenuates virus replication in human, murine, and duck cells but not in CEFs. Nevertheless, it appears to have little influence on virulence in either mice or chickens.
The possibility of using NS1 mutant AIV as live-attenuated vaccines was explored by Steel et al. [233] Viruses with internal deletions of NS1 stimulated higher levels of IFN β expression in mice and were attenuated in chickens. They also stimulated protective immunity against HPAIV challenge (the mutants also contained an E residue at position of PB2 to restrict replication and virulence in mammals).
16.4.5. Immunoevasion Mechanism of the Avian Paramyxoviruses
The Paramyxoviridae family of negative-sense RNA viruses, which includes measles virus, has two well-known avian members: NDV (a member of the Avulavirus genus) and turkey rhinotracheitis virus or avian metapneumovirus (a member of the Metapneumo-virus genus).
There has recently been considerable study of the mechanism by which paramyxoviruses modulate IFN-I, demonstrating the importance of the V proteins in blocking IFN induction and signalling [234]. These proteins are expressed following RNA editing of the mRNA that encodes P protein. Because of the lack of reagents for the avian IFN-I system, this work has barely extended to avian paramyxoviruses. The likely role of the NDV V protein has, however, been demonstrated using genetically modified viruses [235]. Thus, mutant viruses, defective for V protein expression (V− NDV), replicate poorly in embryonated eggs and chicken embryo fibroblasts. This defect can be complemented by transfection of a plasmid-expressing cDNA encoding the V protein into V− NDV-infected cells [235] or by insertion of the influenza virus NS1 gene into the V− NDV. The NS1 gene allows the modified NDV to replicate better in human cells than does the parental NDV, suggesting that the NDV V protein is more effective in modulating the chicken IFN system than the mammalian IFN system (unlike influenza NS1, which modulates both systems effectively).
These differences are likely to be important for the host-range specificities of the two viruses. All examined V proteins of mammalian paramyxoviruses appear to be involved in modulating the mammalian IFN response via interaction with mammalian mda-5 [205]. They can also modulate the induction of expression of the IFN-β promoter by transfected polyI:C in mammalian and avian cells, as can NDV V. However, mda-5 shows species specificity of induction in that mammalian and avian mda-5 can only induce expression of the IFN-β promoter in mammalian and avian cells, respectively [205].
16.4.6. Immunoevasion Mechanism of the Avian Reoviruses
It is clear that dsRNA is a powerful inducer of the anti-viral type-I IFN system. Although the positive-strand RNA viruses, and even poxviruses, generate some dsRNA during their replication and transcription, the dsRNA viruses must be able to prevent recognition of their dsRNA genome by the cell. This is probably largely achieved by ensuring that the genome is never exposed outside of the capsid within the cytoplasm. However, the avian reoviruses, in common with mammalian reoviruses, appear to encode a dsRNA-binding protein that can mask dsRNA from cellular dsRNA-binding proteins such as the dsRNA-dependent protein kinase PKR. The avian reovirus-coded S2-encoded σA protein functions like poxvirus E3 and can actually replace E3 in vaccinia virus [236].
The crystal structure of bacterially expressed σA has now been solved, elucidating the mechanism of its cooperative binding to minimal lengths of 14–18 base pairs of dsRNA [237]. The cellular distribution of σA has also been studied. Although most of it localizes to the cytoplasmic viral factories, some is able to enter the nucleus, apparently independently of the classical nuclear localization signal recognition by importin, and localizes to the nucleoli in a manner dependent on the presence of two arginine residues required for dsRNA binding [238].
16.4.7. Immunoevasion Mechanism of the Avian Birnaviruses
IBDV clearly induces expression of the chicken type I IFN promoter in CEFs [201]. The induction of the IFN-β promoter by virus infection alone is relatively strain-independent, but the ability of IBDV to inhibit transfected polyI:C-mediated induction of the promoter is pathotype-dependent (Laidlaw and Skinner, unpublished), as is the induction of the IFN-β promoter in bursal tissue of IBDV-infected chickens [67]. The viral determinant has not been identified, but a study of IBDV inhibition of TNF-α or Sendai virus-induced IFN expression in human HEK293T cells implicates the viral protease VP4, which is thought to interact with glucocorticoid-induced leucine zipper (GILZ), somehow stimulating its type I IFN suppressive activity. VP4 interacts with GILZ in a yeast-two-hybrid screen and co-localizes with it in the cell, and its IFN-suppressive activity is reduced if GILZ expression is reduced by siRNA [239]. It will be interesting to see whether the same mechanism operates in avian cells.
16.5. Conclusions
Immunosuppression as a consequence of (subclinical) virus infections is a common occurrence with important consequences for the poultry industry. Thus far, most studies describe its effects on immune responses without addressing the mechanistic aspects of the immunosuppression, which is the consequence of the lack of reagents and appropriate techniques. However, during the last few years, and especially since the chicken genome has been sequenced, research has started to focus on interactions between virus infection and effects on the (de)regulation of immune responses. It is expected that rapid progress will be made in the next 5 to 10 years in this area of research.
There has also been a burgeoning interest in the mechanisms by which viruses evade or modulate the innate resistance mechanisms of the host, in particular the IFN-I system. This interest has so far had little impact on the study of avian viruses because of the lack of essential reagents. The recent derivation of the draft chicken genome sequence has opened up easier access to such reagents. It is likely that the coming years will see a rapid increase in the number of studies and hence an increase in our knowledge of the intricacies of avian virus–host systems. Recent developments allowing manipulation of the large and complicated genome of infectious bronchitis virus [240], a coronavirus like the SARS virus, may also help identify additional viral immunomodulators and hopefully elucidate their mode of action 241., 242.. Such technology has been available for some time for avian adenoviruses, but the divergence between mammalian and avian adenoviruses has made it difficult to predict candidate immunomodulators. Hopefully, reannotation of the avian adenovirus CELO genome, which identified three putative surface glycoproteins with immunoglobulin-like folds, will be useful to further analysis [243].
That studies of this type are likely to elucidate new paradigms, which may be relevant even beyond the avian host, is illustrated by the relative dearth in avian herpesviruses and poxviruses of obvious immunomodulators of the type observed in their mammalian cousins. The emergence of West Nile virus and H5N1 AIV as threats to wild birds, farmed poultry, mammalian livestock and companion animals, as well as to humans will, hopefully, ensure the resources needed to understand the complexity of the interactions of these and other viruses with their avian hosts. The hope is that we will be better equipped to control and treat, or possibly even eradicate, such viruses.
References
- 1.Dohms J.E., Saif Y.M. Criteria for evaluating immunosuppression. Avian Dis. 1984;28:305–310. [PubMed] [Google Scholar]
- 2.Lutticken D. Viral diseases of the immune system and strategies to control infectious bursal disease by vaccination. Acta Vet. Hung. 1997;45:239–249. [PubMed] [Google Scholar]
- 3.Hoerr F.J. Clinical aspects of immunosuppression in poultry. Avian Dis. 2010;54:2–15. doi: 10.1637/8909-043009-Review.1. [DOI] [PubMed] [Google Scholar]
- 4.Jakowski R.M., Fredrickson T.N., Chomiak T.W., Luginbuhl R.E. Hematopoietic destruction in Marek’s disease. Avian Dis. 1970;14:374–385. [PubMed] [Google Scholar]
- 5.Schat K.A., Van Santen V. Chicken infectious anemia. In: Saif Y.M., Fadly A.M., Glisson J.R., McDougald L.R., Nolan L.K., Swayne D.E., editors. Diseases of Poultry. 12th ed. Wiley-Blackwell; 2008. pp. 211–235. [Google Scholar]
- 6.Gross W.B. Effect of social stress on occurrence of Marek’s disease in chickens. Am. J. Vet. Res. 1972;33:2275–2279. [PubMed] [Google Scholar]
- 7.Gross W.B. Effect of adrenal blocking chemicals on viral and respiratory infections of chickens. Can. J. Vet. Res. 1989;53:48–51. [PMC free article] [PubMed] [Google Scholar]
- 8.Dohms J.E., Metz A. Stress--mechanisms of immunosuppression. Vet. Immunol. Immunopathol. 1991;30:89–109. doi: 10.1016/0165-2427(91)90011-z. [DOI] [PubMed] [Google Scholar]
- 9.Henriksen R., Groothuis T., Rettenbacher S. Elevated plasma corticosterone decreases yolk testosterone and progesterone in chickens: linking maternal stress and hormone-mediated maternal effects. PLoS One. 2011;6:e23824. doi: 10.1371/journal.pone.0023824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haussmann M.F., Longenecker A.S., Marchetto N.M., Juliano S.A., Bowden R.M. Embryonic exposure to corticosterone modifies the juvenile stress response, oxidative stress and telomere length. Proc. R. Soc. B. 2012;279:1447–1456. doi: 10.1098/rspb.2011.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bondy G.S., Pestka J.J. Immunomodulation by fungal toxins. J. Toxicol. Environm. Health, B. 2000;3:109–143. doi: 10.1080/109374000281113. [DOI] [PubMed] [Google Scholar]
- 12.Yunus A.W., Razzazi-Fazeli E., Bohm J. Aflatoxin B1 in affecting broiler’s performance, immunity, and gastrointestinal tract: a review of history and contemporary issues. Toxins (Basel) 2011;3:566–590. doi: 10.3390/toxins3060566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaz G.J., Murcia H.W., Cepeda S.M. Cytochrome P450 enzymes involved in the metabolism of aflatoxin B1 in chickens and quail. Poultry Sci. 2010;89:2461–2469. doi: 10.3382/ps.2010-00864. [DOI] [PubMed] [Google Scholar]
- 14.Qureshi M.A., Brake J., Hamilton P.B., Hagler W.M., Jr., Nesheim S. Dietary exposure of broiler breeders to aflatoxin results in immune dysfunction in progeny chicks. Poultry Sci. 1998;77:812–819. doi: 10.1093/ps/77.6.812. [DOI] [PubMed] [Google Scholar]
- 15.Cheng Y.H., Ding S.T., Chang M.H. Effect of fumonisins on macrophage immune functions and gene expression of cytokines in broilers. Arch. Anim. Nutr. 2006;60:267–276. doi: 10.1080/17450390600785079. [DOI] [PubMed] [Google Scholar]
- 16.Rezar V., Frankič T., Narat M., Levart A., Salobir J. Dose-dependent effects of T-2 toxin on performance, lipid peroxidation, and genotoxicity in broiler chickens. Poultry Sci. 2007;86:1155–1160. doi: 10.1093/ps/86.6.1155. [DOI] [PubMed] [Google Scholar]
- 17.Yunus A.W., Ghareeb K., Twaruzek M., Grajewski J., Böhm J. Deoxynivalenol as a contaminant of broiler feed: effects on bird performance and response to common vaccines. Poultry Sci. 2012;91:844–851. doi: 10.3382/ps.2011-01873. [DOI] [PubMed] [Google Scholar]
- 18.Dänicke S., Pappritz J., Goyarts T., Xu B., Rautenschlein S. Effects of feeding a Fusarium toxin-contaminated diet to infectious bursal disease virus-infected broilers on the protein turnover of the bursa of Fabricius and spleen. Arch. Anim. Nutr. 2011;65:1–20. doi: 10.1080/1745039x.2010.541191. [DOI] [PubMed] [Google Scholar]
- 19.Pestka J.J., Zhou H.-R., Moon Y., Chung Y.J. Cellular and molecular mechanisms for immune modulation by deoxynivalenol and other trichothecenes: Unraveling a paradox. Toxicol. Lett. 2004;153:61–73. doi: 10.1016/j.toxlet.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 20.Hassan Z.U., Khan M.Z., Khan A., Javed I., Noreen M. In vivo and ex vivo phagocytic potential of macrophages from progeny of breeder hens kept on ochratoxin A (OTA)-contaminated diet. J. Immunotoxicol. 2012;9:64–71. doi: 10.3109/1547691X.2011.635349. [DOI] [PubMed] [Google Scholar]
- 21.Hassan Z.U., Khan M.Z., Saleemi M.K., Khan A., Javed I., Hussain A. Immunological status of White Leghorn chicks hatched from eggs inoculated with ochratoxin A (OTA) J. Immunotoxicol. 2011;8:204–209. doi: 10.3109/1547691X.2011.568020. [DOI] [PubMed] [Google Scholar]
- 22.Hassan Z.U., Khan M.Z., Saleemi M.K., Khan A., Javed I., Noreen M. Immunological responses of male White Leghorn chicks kept on ochratoxin A (OTA)-contaminated feed. J. Immunotoxicol. 2012;9:56–63. doi: 10.3109/1547691X.2011.627393. [DOI] [PubMed] [Google Scholar]
- 23.Gupta S., Jindal N., Khokhar R.S., Gupta A.K., Ledoux D.R., Rottinghaus G.E. Effect of ochratoxin A on broiler chicks challenged with Salmonella gallinarum. Br. Poultry Sci. 2005;46:443–450. doi: 10.1080/00071660500190850. [DOI] [PubMed] [Google Scholar]
- 24.Tessari E.N., Oliveira C.A., Cardoso A.L., Ledoux D.R., Rottinghaus G.E. Effects of aflatoxin B1 and fumonisin B1 on body weight, antibody titres and histology of broiler chicks. Br. Poultry Sci. 2006;47:357–364. doi: 10.1080/00071660600756071. [DOI] [PubMed] [Google Scholar]
- 25.Xue C.Y., Wang G.H., Chen F., Zhang X.B., Bi Y.Z., Cao Y.C. Immunopathological effects of ochratoxin A and T-2 toxin combination on broilers. Poultry Sci. 2010;89:1162–1166. doi: 10.3382/ps.2009-00609. [DOI] [PubMed] [Google Scholar]
- 26.Wang G.H., Xue C.Y., Chen F., Ma Y.L., Zhang X.B., Bi Y.Z., Cao Y.C. Effects of combinations of ochratoxin A and T-2 toxin on immune function of yellow-feathered broiler chickens. Poultry Sci. 2009;88:504–510. doi: 10.3382/ps.2008-00329. [DOI] [PubMed] [Google Scholar]
- 27.Rose M.E., Hesketh P. Infection with Eimeria tenella: modulation of lymphocyte blastogenesis by specific antigen, and evidence for immunodepression. J. Protozool. 1984;31:549–553. doi: 10.1111/j.1550-7408.1984.tb05500.x. [DOI] [PubMed] [Google Scholar]
- 28.Bhanushali J.K., Long P.L. Eimeria tenella infection: does it affect humoral immune responses to heterologous antigens. J. Parasitol. 1985;71:850–852. [PubMed] [Google Scholar]
- 29.McDougald L.R. Cryptosporidiosis. In: Saif Y.M., Fadly A.M., Glisson J.R., McDougald L.R., Nolan N.K., Swayne D.E., editors. Diseases of Poultry. 12th ed. Iowa State Press; Ames, Iowa: 2008. pp. 1085–1091. [Google Scholar]
- 30.Rhee J.K., Kim H.C., Park B.K. Effects of Cryptosporidium baileyi infection on the bursa of Fabricius in chickens. Korean J. Parasitol. 1997;35:181–187. doi: 10.3347/kjp.1997.35.3.181. [DOI] [PubMed] [Google Scholar]
- 31.Rhee J.K., Kim H.C., Park B.K. Effect of Cryptosporidium baileyi infection on antibody response to SRBC in chickens. Korean J. Parasitol. 1998;36:33–36. doi: 10.3347/kjp.1998.36.1.33. [DOI] [PubMed] [Google Scholar]
- 32.Rhee J.K., Yang H.J., Kim H.C. Verification of immunosuppression in chicks caused by Cryptosporidium baileyi infection using Brucella abortus strain 1119-3. Korean J. Parasitol. 1998;36:281–284. doi: 10.3347/kjp.1998.36.4.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhee J.K., Yang H.J., Yook S.Y., Kim H.C. Immunosuppressive effect of Cryptosporidium baileyi infection on vaccination against avian infectious bronchitis in chicks. Korean J. Parasitol. 1998;36:203–206. doi: 10.3347/kjp.1998.36.3.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhee J.K., Kim H.C., Lee S.B., Yook S.Y. Immunosuppressive effect of Cryptosporidium baileyi infection on vaccination against Newcastle disease in chicks. Korean J. Parasitol. 1998;36:121–125. doi: 10.3347/kjp.1998.36.2.121. [DOI] [PubMed] [Google Scholar]
- 35.Hao Y.X., Yang J.M., He C., Liu Q., McAllister T.A. Reduced serologic response to avian influenza vaccine in specific-pathogen-free chicks inoculated with Cryptosporidium baileyi. Avian Dis. 2008;52:690–693. doi: 10.1637/8370-052608-Reg.1. [DOI] [PubMed] [Google Scholar]
- 36.Abbassi H., Coudert F., Dambrine G., Chérel Y., Naciri M. Effect of cryptosporidium baileyi in specific pathogen free chickens vaccinated (CVI988/Rispens) and challenged with HPRS-16 strian of Marek’s disease. Avian Pathol. 2000;29:623–634. doi: 10.1080/03079450020016887. [DOI] [PubMed] [Google Scholar]
- 37.Abbassi H., Dambrine G., Cherel Y., Coudert F., Naciri M. Interaction of Marek’s disease virus and Cryptosporidium baileyi in experimentally infected chickens. Avian Dis. 2000;44:776–789. [PubMed] [Google Scholar]
- 38.Saif Y.M., Fadly A.M., Glisson J.R., McDougald L.R., Nolan N.K., Swayne D.E., editors. Diseases of Poultry. 12th ed. Iowa State Press; Ames Iowa: 2008. [Google Scholar]
- 39.Swayne D.E., Glisson J.R., McDougald L.R., Nair V., Nolan L.K., Suarez D.L., editors. Diseases of Poultry. 13th ed. Wiley-Blackwell; Ames. IA: 2013. [Google Scholar]
- 40.Sharma J.M., Kim I.J., Rautenschlein S., Yeh H.Y. Infectious bursal disease virus of chickens: pathogenesis and immunosuppression. Dev. Comp. Immunol. 2000;24:223–235. doi: 10.1016/s0145-305x(99)00074-9. [DOI] [PubMed] [Google Scholar]
- 41.Williams A.E., Davison T.F. Enhanced immunopathology induced by very virulent infectious bursal disease virus. Avian Pathol. 2005;34:4–14. doi: 10.1080/03079450400025364. [DOI] [PubMed] [Google Scholar]
- 42.Eterradossi N., Saif Y.M. Infectious bursal disease virus. In: Saif Y.M., Fadly A.M., Glisson J.R., McDougald L.R., Nolan N.K., Swayne D.E., editors. Diseases of Poultry. 11th ed. Iowa State Press; Ames, Iowa: 2008. pp. 161–179. [Google Scholar]
- 43.Mahgoub H. An overview of infectious bursal disease. Arch. Virol. 2012;157:2047–2057. doi: 10.1007/s00705-012-1377-9. [DOI] [PubMed] [Google Scholar]
- 44.Le Nouën C., Toquin D., Müller H., Raue R., Kean K.M., Langlois P., Cherbonnel M., Eterradossi N. Different domains of the RNA polymerase of infectious bursal disease virus contribute to virulence. PLoS One. 2012;7:e28064. doi: 10.1371/journal.pone.0028064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peters M.A., Lin T.L., Wu C.C. Infectious bursal disease virus polyprotein expression arrests growth and mitogenic stimulation of B lymphocytes. Arch. Virol. 2004;149:2413–2426. doi: 10.1007/s00705-004-0350-7. [DOI] [PubMed] [Google Scholar]
- 46.Yao K., Goodwin M.A., Vakharia V.N. Generation of a mutant infectious bursal disease virus that does not cause bursal lesions. J. Virol. 1998;72:2647–2654. doi: 10.1128/jvi.72.4.2647-2654.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu M., Vakharia V.N. Nonstructural protein of infectious bursal disease virus inhibits apoptosis at the early stage of virus infection. J. Virol. 2006;80:3369–3377. doi: 10.1128/JVI.80.7.3369-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei L., Hou L., Zhu S., Wang J., Zhou J., Liu J. Infectious bursal disease virus activates the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway by interaction of VP5 protein with the p85α subunit of PI3K. Virology. 2011;417:211–220. doi: 10.1016/j.virol.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 49.Lombardo E., Maraver A., Espinosa I., Fernández-Arias A., Rodriguez J.F. VP5, the nonstructural polypeptide of infectious bursal disease virus, accumulates within the host plasma membrane and induces cell lysis. Virology. 2000;277:345–357. doi: 10.1006/viro.2000.0595. [DOI] [PubMed] [Google Scholar]
- 50.Yao K., Vakharia V.N. Induction of apoptosis in vitro by the 17-kDa nonstructural protein of infectious bursal disease virus: possible role in viral pathogenesis. Virology. 2001;285:50–58. doi: 10.1006/viro.2001.0947. [DOI] [PubMed] [Google Scholar]
- 51.Wei L., Zhu S., Ruan G., Hou L., Wang J., Wang B., Liu J. Infectious bursal disease virus-induced activation of JNK signaling pathway is required for virus replication and correlates with virus-induced apoptosis. Virology. 2011;420:156–163. doi: 10.1016/j.virol.2011.08.027. [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez-Lecompte J.C., Nino-Fong R., Lopez A., Frederick Markham R.J., Kibenge F.S. Infectious bursal disease virus (IBDV) induces apoptosis in chicken B cells. Comp. Immunol. Microbiol. Infect. Dis. 2005;28:321–337. doi: 10.1016/j.cimid.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 53.Jungmann A., Nieper H., Muller H. Apoptosis is induced by infectious bursal disease virus replication in productively infected cells as well as in antigen-negative cells in their vicinity. J. Gen. Virol. 2001;82:1107–1115. doi: 10.1099/0022-1317-82-5-1107. [DOI] [PubMed] [Google Scholar]
- 54.Rauw F., Lambrecht B., van den Berg T. Pivotal role of ChIFN-gamma in the pathogenesis and immunosuppression of infectious bursal disease. Avian Pathol. 2007;36:367–374. doi: 10.1080/03079450701589159. [DOI] [PubMed] [Google Scholar]
- 55.Hirai K., Funakoshi T., Nakai T., Shimakura S. Sequential changes in the number of surface immunoglobulin-bearing B lymphocytes in infectious bursal disease virus-infected chickens. Avian Dis. 1981;25:484–496. [PubMed] [Google Scholar]
- 56.Rodenberg J., Sharma J.M., Belzer S.W., Nordgren R.M., Naqi S. Flow cytometric analysis of B cell and T cell subpopulations in specific-pathogen-free chickens infected with infectious bursal disease virus. Avian Dis. 1994;38:16–21. [PubMed] [Google Scholar]
- 57.Vervelde L., Davison T.F. Comparison of the in situ changes in lymphoid cells during infection with infectious bursal disease virus in chickens of different ages. Avian Pathol. 1997;26:803–821. doi: 10.1080/03079459708419254. [DOI] [PubMed] [Google Scholar]
- 58.Kim I.J., Gagic M., Sharma J.M. Recovery of antibody-producing ability and lymphocyte repopulation of bursal follicles in chickens exposed to infectious bursal disease virus. Avian Dis. 1999;43:401–413. [PubMed] [Google Scholar]
- 59.Withers D.R., Davison T.F., Young J.R. Diversified bursal medullary B cells survive and expand independently after depletion following neonatal infectious bursal disease virus infection. Immunology. 2006;117:558–565. doi: 10.1111/j.1365-2567.2006.02332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Withers D.R., Young Y.R., Davison T.F. Infectious bursal disease virus-induced immunosuppression in the chick is associated with the presence of undifferentiated follicles in the recovering bursa. Viral Immunol. 2005;18:127–137. doi: 10.1089/vim.2005.18.127. [DOI] [PubMed] [Google Scholar]
- 61.Bíró E., Kocsis K., Nagy N., Molnár D., Kabell S., Palya V., Oláh I. Origin of the chicken splenic reticular cells influences the effect of the infectious bursal disease virus on the extracellular matrix. Avian Pathol. 2011;40:199–206. doi: 10.1080/03079457.2011.554797. [DOI] [PubMed] [Google Scholar]
- 62.Burkhardt E., Muller H. Susceptibility of chicken blood lymphoblasts and monocytes to infectious bursal disease virus (IBDV) Arch. Virol. 1987;94:297–303. doi: 10.1007/BF01310722. [DOI] [PubMed] [Google Scholar]
- 63.Khatri M., Palmquist J.M., Cha R.M., Sharma J.M. Infection and activation of bursal macrophages by virulent infectious bursal disease virus. Virus Res. 2005;113:44–50. doi: 10.1016/j.virusres.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 64.Khatri M., Sharma J.M. Susceptibility of chicken mesenchymal stem cells to infectious bursal disease virus. J. Virol. Meth. 2009;160:197–199. doi: 10.1016/j.jviromet.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 65.Palmquist J.M., Khatri M., Cha R.M., Goddeeris B.M., Walcheck B., Sharma J.M. In vivo activation of chicken macrophages by infectious bursal disease virus. Viral Immunol. 2006;19:305–315. doi: 10.1089/vim.2006.19.305. [DOI] [PubMed] [Google Scholar]
- 66.Kim I.J., Karaca K., Pertile T.L., Erickson S.A., Sharma J.M. Enhanced expression of cytokine genes in spleen macrophages during acute infection with infectious bursal disease virus in chickens. Vet. Immunol. Immunopathol. 1998;61:331–341. doi: 10.1016/s0165-2427(97)00135-9. [DOI] [PubMed] [Google Scholar]
- 67.Eldaghayes I., Rothwell L., Williams A., Withers D., Balu S., Davison F., Kaiser P. Infectious bursal disease virus: strains that differ in virulence differentially modulate the innate immune response to infection in the chicken bursa. Viral Immunol. 2006;19:83–91. doi: 10.1089/vim.2006.19.83. [DOI] [PubMed] [Google Scholar]
- 68.Khatri M., Sharma J.M. Infectious bursal disease virus infection induces macrophage activation via p38 MAPK and NF-kappaB pathways. Virus Res. 2006;118:70–77. doi: 10.1016/j.virusres.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 69.Tanimura N., Sharma J.M. Appearance of T cells in the bursa of Fabricius and cecal tonsils during the acute phase of infectious bursal disease virus infection in chickens. Avian Dis. 1997;41:638–645. [PubMed] [Google Scholar]
- 70.Kim I.J., Sharma J.M. IBDV-induced bursal T lymphocytes inhibit mitogenic response of normal splenocytes. Vet. Immunol. Immunopathol. 2000;74:47–57. doi: 10.1016/s0165-2427(00)00160-4. [DOI] [PubMed] [Google Scholar]
- 71.Rauf A., Khatri M., Murgia M.V., Saif Y.M. Expression of perforin–granzyme pathway genes in the bursa of infectious bursal disease virus-infected chickens. Dev. Comp. Immunol. 2011;35:620–627. doi: 10.1016/j.dci.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 72.Biagini, P. (2011). Restructuring and expansion of the family Anelloviridae. <http://talk.ictvonline.org/files/proposals/taxonomy_proposals_general1/m/gen01/default.aspx>.
- 73.Schat K.A. Chicken anemia virus. Curr. Top. Microbiol. Immunol. 2009;331:151–184. doi: 10.1007/978-3-540-70972-5_10. [DOI] [PubMed] [Google Scholar]
- 74.Schat, K. A. and van Santen, V. L. (2013). Chicken infectious anemia. In: Diseases of Poultry (Swayne, D. E., Glisson, J. R., McDougald, L. R., Nair, V., Nolan, L. K. and Suarez, D. L., eds.), pp.248–264. Wiley-Blackwell, Ames, IA.
- 75.Noteborn M.H.M. Chicken anemia virus induced apoptosis: underlying molecular mechanisms. Vet. Microbiol. 2004;98:89–94. doi: 10.1016/j.vetmic.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 76.de Smit M.H., Noteborn H.M. Apoptosis-inducing proteins in chicken anemia virus and TT virus. Curr. Top. Microbiol. Immunol. 2009;331:131–149. doi: 10.1007/978-3-540-70972-5_9. [DOI] [PubMed] [Google Scholar]
- 77.Adair B.M., McNeilly F., McConnell C.D., McNulty M.S. Characterization of surface markers present on cells infected by chicken anemia virus in experimentally infected chickens. Avian Dis. 1993;37:943–950. [PubMed] [Google Scholar]
- 78.Haridy M., Sasaki J., Ikezawa M., Okada K., Goryo M. Pathological and immunohistochemical studies of subclinical infection of chicken anemia virus in 4-week-old chickens. J. Vet. Med. Sci. 2012;74:757–764. doi: 10.1292/jvms.11-0374. [DOI] [PubMed] [Google Scholar]
- 79.Hu L.B., Lucio B., Schat K.A. Depletion of CD4+ and CD8+ T lymphocyte subpopulations by CIA-1, a chicken infectious anemia virus. Avian Dis. 1993;37:492–500. [PubMed] [Google Scholar]
- 80.Markowski-Grimsrud C.J., Schat K.A. Infection with chicken anemia virus impairs the generation of antigen-specific cytotoxic T lymphocytes. Immunology. 2003;109:283–294. doi: 10.1046/j.1365-2567.2003.01643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Markowski-Grimsrud, C. J. and Schat, K. A. (2001). Impairment of cell-mediated immune responses during chicken infectious anemia virus infection In: “Proceedings of the Second International Symposium on Infectious bursal disease and chicken infectious anaemia” Rauischholzhausen, Germany, pp. 395–402.
- 82.Peters M.A., Crabb B.S., Washington E.A., Browning G.F. Site-directed mutagenesis of the VP2 gene of chicken anemia virus affects virus replication, cytopathology and host-cell MHC class I expression. J. Gen. Virol. 2006;87:823–831. doi: 10.1099/vir.0.81468-0. [DOI] [PubMed] [Google Scholar]
- 83.Toro H., van Santen V.L., Li L., Lockaby S.B., van Santen E., Hoerr F.J. Epidemiological and experimental evidence for immunodeficiency affecting avian infectious bronchitis. Avian Pathol. 2006;35:455–464. doi: 10.1080/03079450601028811. [DOI] [PubMed] [Google Scholar]
- 84.Van Ginkel F.W., van Santen V.L., Gulley S.L., Toro H. Infectious bronchitis virus in the chicken Harderian gland and lachrymal fluid: viral load, infectivity, immune cell responses, and effects of viral immunodeficiency. Avian Dis. 2008;52:608–617. doi: 10.1637/8349-050908-Reg.1. [DOI] [PubMed] [Google Scholar]
- 85.Miller M.M., Schat K.A. Chicken infectious anemia virus: an example of the ultimate host-parasite relationship. Avian Dis. 2004;48:734–745. doi: 10.1637/7271-090304R. [DOI] [PubMed] [Google Scholar]
- 86.van Santen V.L., Joiner K.S., Murray C., Petrenko N., Hoerr F.J., Toro H. Pathogenesis of chicken anemia virus: comparison of the oral and the intramuscular routes of infection. Avian Dis. 2004;48:494–504. doi: 10.1637/7155-010904R. [DOI] [PubMed] [Google Scholar]
- 87.McConnell C.D., Adair B.M., McNulty M.S. Effects of chicken anemia virus on macrophage function in chickens. Avian Dis. 1993;37:358–365. [PubMed] [Google Scholar]
- 88.Smyth J.A., Moffett D.A., McNulty M.S., Todd D., Mackie D.P. A sequential histopathologic and immunocytochemical study of chicken anemia virus infection at one day of age. Avian Dis. 1993;37:324–338. [PubMed] [Google Scholar]
- 89.Menendez N.A., Calnek B.W., Cowen B.S. Experimental egg-transmission of avian reovirus. Avian Dis. 1975;19:104–111. [PubMed] [Google Scholar]
- 90.Al-Muffarej S.I., Savage C.E., Jones R.C. Egg transmission of avian reoviruses in chickens: comparison of a trypsin-sensitive and a trypsin-resistant strain. Avian Pathol. 1996;25:469–480. doi: 10.1080/03079459608419156. [DOI] [PubMed] [Google Scholar]
- 91.Sharma J.M., Rosenberger J.K. Infectious bursal disease and reovirus infection of chickens: immune responses and vaccine control. In: Toivanen A., Toivanen P., editors. Vol. II. CRC Press; Boca Raton, FL: 1987. pp. 144–157. (Avian Immunology: Basis and Practice). [Google Scholar]
- 92.Montgomery R.D., Villegas P., Dawe D.L., Brown J. A comparison between the effect of an avian reovirus and infectious bursal disease virus on selected aspects of the immune system of the chicken. Avian Dis. 1986;30:298–308. [PubMed] [Google Scholar]
- 93.Mills J.N., Wilcox G.E. Replication of four antigenic types of avian reovirus in subpopulatons of chicken leukocytes. Avian Pathol. 1993;22:353–361. doi: 10.1080/03079459308418926. [DOI] [PubMed] [Google Scholar]
- 94.von Bülow V., Klasen A. Effects of avian viruses on cultured chicken bone-marrow-derived macrophages. Avian Pathol. 1983;12:179–198. doi: 10.1080/03079458308436162. [DOI] [PubMed] [Google Scholar]
- 95.Pertile T.L., Karaca K., Walser M.M., Sharma J.M. Suppressor macrophages mediate depressed lymphoproliferation in chickens infected with avian reovirus. Vet. Immunol. Immunopathol. 1996;53:129–145. doi: 10.1016/0165-2427(96)05555-9. [DOI] [PubMed] [Google Scholar]
- 96.Pertile T.L., Sharma J.M., Walser M.M. Reovirus infection in chickens primes splenic adherent macrophages to produce nitric oxide in response to T cell-produced factors. Cell. Immunol. 1995;164:207–216. doi: 10.1006/cimm.1995.1163. [DOI] [PubMed] [Google Scholar]
- 97.Neelima S., Ram G.C., Kataria J.M., Goswami T.K. Avian reovirus induces an inhibitory effect on lymphoproliferation in chickens. Vet. Res. Comm. 2003;27:73–85. doi: 10.1023/a:1022014825451. [DOI] [PubMed] [Google Scholar]
- 98.Pertile T.L., Karaca K., Sharma J.M., Walser M.M. An antiviral effect of nitric oxide: inhibition of reovirus replication. Avian Dis. 1996;40:342–348. [PubMed] [Google Scholar]
- 99.Wink D.A., Hines H.B., Cheng R.Y., Switzer C.H., Flores-Santana W., Vitek M.P., Ridnour L.A., Colton C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011;89:873–891. doi: 10.1189/jlb.1010550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu H.-J., Lin P.-Y., Lee J.-W., Hsu H.-Y., Shih W.-L. Retardation of cell growth by avian reovirus p17 through the activation of p53 pathway. Biochem. Biophys. Res. Commun. 2005;336:709–715. doi: 10.1016/j.bbrc.2005.08.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garzelli, C., Onodera, T., Ray, U. R. and Notkins, A. L. The S1 gene from reovirus type 1 is required for immunosuppression. J. Infect. Dis. 152, 640–643. [DOI] [PubMed]
- 102.Boehme K.W., Guglielmi K.M., Dermody T.S. Reovirus nonstructural protein σ1s is required for establishment of viremia and systemic dissemination. Proc. Natl. Acad. Sci. USA. 2009;106:19986–19991. doi: 10.1073/pnas.0907412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Benavente J., Martínez-Costas J. Avian reovirus: structure and biology. Virus Res. 2007;123:105–119. doi: 10.1016/j.virusres.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 104.Barnes H.J., Guy J.S. Poult enteritis-mortality syndrome. In: Saif Y.M., Barnes H.J., Glisson J.R., Fadly A.M., McDougald L.R., Swayne D.E., editors. Diseases of Poultry. 11th ed. Iowa State Press; Ames, IA: 1997. pp. 1171–1180. [Google Scholar]
- 105.Heggen-Peay C.L., Qureshi M.A., Edens F.W., Sherry B., Wakenell P.S., O’Connell P.H., Schat K.A. Isolation of a reovirus from poult enteritis and mortality syndrome (PEMS) and its pathogenicity in turkey poults. Avian Dis. 2002;46:32–47. doi: 10.1637/0005-2086(2002)046[0032:IOARFP]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 106.Heggen-Peay C.L., Cheema M.A., Ali R.A., Schat K.A., Qureshi M.A. Interactions of poult enteritis and mortality syndrome-associated reovirus with various cell types in vitro. Poultry Sci. 2002;81:1661–1667. doi: 10.1093/ps/81.11.1661. [DOI] [PubMed] [Google Scholar]
- 107.Day J.M., Spackman E., Pantin-Jackwood M.J. Turkey origin reovirus-induced immune dysfunction in specific pathogen free and commercial turkey poults. Avian Dis. 2008;52:387–391. doi: 10.1637/8190-120607-Reg. [DOI] [PubMed] [Google Scholar]
- 108.Witter R.L. Increased virulence of Marek’s disease virus field isolates. Avian Dis. 1997;41:149–163. [PubMed] [Google Scholar]
- 109.Gimeno I.M., Witter R.L., Neumann U. Neuropathotyping: a new system to classify Marek’s disease virus. Avian Dis. 2002;46:909–918. doi: 10.1637/0005-2086(2002)046[0909:NANSTC]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 110.Calnek B.W., Harris R.W., Buscaglia C., Schat K.A., Lucio B. Relationship between the immunosuppressive potential and the pathotype of Marek’s disease virus isolates. Avian Dis. 1998;42:124–132. [PubMed] [Google Scholar]
- 111.Schat K.A. Marek’s disease immunosuppression. In: Davison F., Nair V., editors. Marek’s Disease: An Evolving Problem. Academic Press; Amsterdam: 2004. pp. 142–155. [Google Scholar]
- 112.Calnek B.W. Marek’s disease – a model for herpesvirus oncology. Crit. Rev. Microbiol. 1986;12:293–320. doi: 10.3109/10408418509104432. [DOI] [PubMed] [Google Scholar]
- 113.Schat K.A. Marek’s disease: A model for protection against herpesvirus-induced tumours. Cancer Surv. 1987;6:1–37. [PubMed] [Google Scholar]
- 114.Morgan R.W., Xie Q., Cantello J.L., Miles A.M., Bernberg E.L., Kent J., Anderson A. Marek’s disease virus latency. Curr. Top. Microbiol. Immunol. 2001;255:223–243. [PubMed] [Google Scholar]
- 115.Schat K.A., Markowski-Grimsrud C.J. Immune responses to Marek’s disease virus infection. Curr. Top. Microbiol. Immunol. 2001;255:91–120. doi: 10.1007/978-3-642-56863-3_4. [DOI] [PubMed] [Google Scholar]
- 116.Morgan R.W., Burnside J. Roles of avian herpesvirus microRNAs in infection, latency, and oncogenesis. Biochim. Biophys. Acta. 2011;1809:654–659. doi: 10.1016/j.bbagrm.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 117.Xing Z., Schat K.A. Expression of cytokine genes in Marek’s disease virus-infected chickens and chicken embryo fibroblast cultures. Immunology. 2000;100:70–76. doi: 10.1046/j.1365-2567.2000.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Parcells M.S., Lin S.F., Dienglewicz R.L., Majerciak V., Robinson D.R., Chen H.C., Wu Z., Dubyak G.R., Brunovskis P., Hunt H.D., Lee L.F., Kung H.J. Marek’s disease virus (MDV) encodes an interleukin-8 homolog (vIL-8): characterization of the vIL-8 protein and a vIL-8 deletion mutant MDV. J. Virol. 2001;75:5159–5173. doi: 10.1128/JVI.75.11.5159-5173.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jarosinski K.W., O’Connell P.H., Schat K.A. Impact of deletions within the Bam HI-L fragment of attenuated Marek’s disease virus on vIL-8 expression and the newly identified transcript of open reading frame LORF4. Virus Genes. 2003;26:255–269. doi: 10.1023/a:1024447230464. [DOI] [PubMed] [Google Scholar]
- 120.Yunis R., Jarosinski K.W., Schat K.A. Association between rate of viral genome replication and virulence of Marek’s disease herpesvirus strains. Virology. 2004;328:142–150. doi: 10.1016/j.virol.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 121.Cortes P.L., Cardona C.J. Pathogenesis of a Marek’s disease virus mutant lacking vIL-8 in resistant and susceptible chickens. Avian Dis. 2004;48:50–60. doi: 10.1637/7050. [DOI] [PubMed] [Google Scholar]
- 122.Cui X., Lee L.F., Hunt H.D., Reed W.M., Lupiani B., Reddy S.M. A Marek’s disease virus vIL-8 deletion mutant has attenuated virulence and confers protection against challenge with a very virulent plus strain. Avian Dis. 2005;49:199–206. doi: 10.1637/7277-091004. [DOI] [PubMed] [Google Scholar]
- 123.Jarosinski K.W., Schat K.A. Multiple alternative splicing to exons II and III of viral interleukin-8 (vIL-8) in the Marek’s disease virus genome: the importance of vIL-8 exon I. Virus Genes. 2007;34:9–22. doi: 10.1007/s11262-006-0004-9. [DOI] [PubMed] [Google Scholar]
- 124.Jarosinski K.W., Njaa B.L., O’Connell P.H., Schat K.A. Pro-inflammatory responses in chicken spleen and brain tissues after infection with very virulent plus Marek’s disease virus. Viral Immunol. 2005;18:148–161. doi: 10.1089/vim.2005.18.148. [DOI] [PubMed] [Google Scholar]
- 125.Abdul-Careem M.F., Hunter B.D., Sarson A.J., Mayameei A., Zhou H., Sharif S. Marek’s disease virus-induced transient paralysis is associated with cytokine gene expression in the nervous system. Viral Immunol. 2006;19:167–176. doi: 10.1089/vim.2006.19.167. [DOI] [PubMed] [Google Scholar]
- 126.Volpini L.M., Calnek B.W., Sekellick M.J., Marcus P.I. Stages of Marek’s disease virus latency defined by variable sensitivity to interferon modulation of viral antigen expression. Vet. Microbiol. 1995;47:99–109. doi: 10.1016/0378-1135(95)00056-g. [DOI] [PubMed] [Google Scholar]
- 127.Morimura T., Ohashi K., Kon Y., Hattori M., Sugimoto C., Onuma M. Apoptosis and CD8-down-regulation in the thymus of chickens infected with Marek’s disease virus. Arch. Virol. 1996;141:2243–2249. doi: 10.1007/BF01718230. [DOI] [PubMed] [Google Scholar]
- 128.Morimura T., Ohashi K., Kon Y., Hattori M., Sugimoto C., Onuma M. Apoptosis in peripheral CD4+T cells and thymocytes by Marek’s disease virus-infection. Leukemia. 1997;11(Suppl. 3):206–208. [PubMed] [Google Scholar]
- 129.Morimura T., Hattori M., Ohashi K., Sugimoto C., Onuma M. Immunomodulation of peripheral T cells in chickens infected with Marek’s disease virus: involvement in immunosuppression. J. Gen. Virol. 1995;76:2979–2985. doi: 10.1099/0022-1317-76-12-2979. [DOI] [PubMed] [Google Scholar]
- 130.Calnek B.W., Lucio B., Schat K.A., Lillehoj H.S. Pathogenesis of Marek’s disease virus-induced local lesions. 1. Lesion characterization and cell line establishment. Avian Dis. 1989;33:291–302. [PubMed] [Google Scholar]
- 131.Baigent S.J., Ross L.J., Davison T.F. Differential susceptibility to Marek’s disease is associated with differences in number, but not phenotype or location, of pp38+ lymphocytes. J. Gen. Virol. 1998;79:2795–2802. doi: 10.1099/0022-1317-79-11-2795. [DOI] [PubMed] [Google Scholar]
- 132.Parcells M.S., Anderson A.S., Morgan R.W. Retention of oncogenicity by a Marek’s disease virus mutant lacking six unique short region genes. J. Virol. 1995;69:7888–7898. doi: 10.1128/jvi.69.12.7888-7898.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Witter R.L., Li D., Jones D., Lee L.F., Kung H.J. Retroviral insertional mutagenesis of a herpesvirus: a Marek’s disease virus mutant attenuated for oncogenicity but not for immunosuppression or in vivo replication. Avian Dis. 1997;41:407–421. [PubMed] [Google Scholar]
- 134.Lee L.F., Sharma J.M., Nazerian K., Witter R.L. Suppression of mitogen-induced proliferation of normal spleen cells by macrophages from chickens inoculated with Marek’s disease virus. J. Immunol. 1978;120:1554–1559. [PubMed] [Google Scholar]
- 135.Barrow A.D., Burgess S.C., Baigent S.J., Howes K., Nair V.K. Infection of macrophages by a lymphotropic herpesvirus: a new tropism for Marek’s disease virus. J. Gen. Virol. 2003;84:2635–2645. doi: 10.1099/vir.0.19206-0. [DOI] [PubMed] [Google Scholar]
- 136.Barrow A.D., Burgess S.C., Howes K., Nair V.K. Monocytosis is associated with the onset of leukocyte and viral infiltration of the brain in chickens infected with the very virulent Marek’s disease virus strain C12/130. Avian Pathol. 2003;32:183–191. doi: 10.1080/0307985021000071650. [DOI] [PubMed] [Google Scholar]
- 137.Friedman A., Shalem-Meilin E., Heller E.D. Marek’s disease vaccines cause temporary B-lymphocyte dysfunction and reduced resistance to infection in chicks. Avian Pathol. 1992;21:621–631. doi: 10.1080/03079459208418883. [DOI] [PubMed] [Google Scholar]
- 138.Islam A.F., Wong C.W., Walkden-Brown S.W., Colditz I.G., Arzey K.E., Groves P.J. Immunosuppressive effects of Marek’s disease virus (MDV) and herpesvirus of turkeys (HVT) in broiler chickens and the protective effect of HVT vaccination against MDV challenge. Avian Pathol. 2002;31:449–461. doi: 10.1080/0307945021000005824. [DOI] [PubMed] [Google Scholar]
- 139.Zhang Y., Sharma J.M. Immunological tolerance in chickens hatching from eggs injected with cell-associated herpesvirus of Turkey (HVT) Dev. Comp. Immunol. 2003;27:431–438. doi: 10.1016/s0145-305x(02)00117-9. [DOI] [PubMed] [Google Scholar]
- 140.Fadly A.M., Nair V. Leukosis/sarcoma group. In: Saif Y.M., Glisson J.R., McDougald L.R., Nolan L.K., Swayne D.E., editors. Diseases of Poultry. 12th ed. Blackwell Publishing; 2008. pp. 514–568. [Google Scholar]
- 141.Fadly A.M., Zavala G., Witter R.L. Reticuloendotheliosis. In: Saif Y.M., Glisson J.R., McDougald L.R., Nolan L.K., Swayne D.E., editors. Diseases of Poultry. 12th ed. Blackwell Publishing; 2008. pp. 568–588. [Google Scholar]
- 142.Smith E.J., Fadly A.M., Levin I., Crittenden L.B. The influence of ev6 on the immune response to avian leukosis virus infection in rapid-feathering progeny of slow- and rapid-feathering dams. Poultry Sci. 1991;70:1673–1678. doi: 10.3382/ps.0701673. [DOI] [PubMed] [Google Scholar]
- 143.Fadly A.M., Witter R.L., Lee L.F. Effects of chemically or virus-induced immunodepression on response of chickens to avian leukosis virus. Avian Dis. 1985;29:12–25. [PubMed] [Google Scholar]
- 144.Williams S.M., Sellers H.S. Response of white leghorn chickens to infection with avian leukosis virus subgroup J and infectious bursal disease virus. Avian Dis. 2012;56:2–6. doi: 10.1637/9577-101510-reg.1. [DOI] [PubMed] [Google Scholar]
- 145.Meyers P., Qualtiere L.F. The efficacy of chemical bursectomy in chickens with congenital leukosis-virus infection. Immunology. 1976;31:527–532. [PMC free article] [PubMed] [Google Scholar]
- 146.Meyers P., Dougherty R.M. Immunologic reactivity to viral antigens in chickens infected with avian leukosis viruses. J. Natl. Cancer Inst. 1971;46:701–711. [PubMed] [Google Scholar]
- 147.Fadly A.M., Lee L.F., Bacon L.D. Immunocompetence of chickens during early and tumorigenic stages of Rous-associated virus-1 infection. Infect. Immun. 1982;37:1156–1161. doi: 10.1128/iai.37.3.1156-1161.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rup B.J., Hoelzer J.D., Bose H.R., Jr. Helper viruses associated with avian acute leukemia viruses inhibit the cellular immune response. Virology. 1982;116:61–71. doi: 10.1016/0042-6822(82)90403-2. [DOI] [PubMed] [Google Scholar]
- 149.Hirota Y., Martin M.T., Viljanen M., Toivanen P., Franklin R.M. Immunopathology of chickens infected in ovo and at hatching with the avian osteopetrosis virus MAV2-0. Eur. J. Immunol. 1980;10:929–936. doi: 10.1002/eji.1830101208. [DOI] [PubMed] [Google Scholar]
- 150.Cummins T.J., Smith R.E. Association of persistent synthesis of viral DNA with macrophage accessory cell dysfunction induced by avian retrovirus myeloblastosis-associated virus of subgroup B inducing osteopetrosis in chickens. Cancer Res. 1987;47:6033–6039. [PubMed] [Google Scholar]
- 151.Cummins T.J., Orme I.M., Smith R.E. Reduced in vivo nonspecific resistance to Listeria monocytogenes infection during avian retrovirus-induced immunosuppression. Avian Dis. 1988;32:663–667. [PubMed] [Google Scholar]
- 152.Rao A., Kline K., Sanders B.G. Immune abnormalities in avian erythroblastosis virus-infected chickens. Cancer Res. 1990;50:4764–4770. [PubMed] [Google Scholar]
- 153.Stedman N.L., Brown T.P., Brooks R.L., Bounous D.I. Heterophil function and resistance to Staphylococcal challenge in broiler chickens naturally infected with avian leukosis virus subgroup. J. Vet. Pathol. 2001;38:519–527. doi: 10.1354/vp.38-5-519. [DOI] [PubMed] [Google Scholar]
- 154.Landman W.J., Post J., Boonstra-Blom A.G., Buyse J., Elbers A.R., Koch G. Effect of an in ovo infection with a Dutch avian leukosis virus subgroup J isolate on the growth and immunological performance of SPF broiler chickens. Avian Pathol. 2002;31:59–72. doi: 10.1080/03079450120106633. [DOI] [PubMed] [Google Scholar]
- 155.Spackman E., Pope C.R., Cloud S.S., Rosenberger J.K. The effects of avian leukosis virus subgroup J on broiler chicken performance and response to vaccination. Avian Dis. 2003;47:618–626. doi: 10.1637/6091. [DOI] [PubMed] [Google Scholar]
- 156.Cui Z., Sun S., Wang J. Reduced serologic response to Newcastle disease virus in broiler chickens exposed to a Chinese field strain of subgroup J avian leukosis virus. Avian Dis. 2006;50:191–195. doi: 10.1637/7409-071305R1.1. [DOI] [PubMed] [Google Scholar]
- 157.von Bülow V. Immunological effects of reticuloendotheliosis virus as potential contaminant of Marek’s disease vaccines. Avian Pathol. 1977;6:383–393. doi: 10.1080/03079457708418247. [DOI] [PubMed] [Google Scholar]
- 158.Kawamura H., Wakabayashi T., Yamaguchi S., Taniguchi T., Takayanagi N. Inoculation experiment of Marek’s disease vaccine contaminated with a reticuloendotheliosis virus. Natl. Inst. Anim. Health Q. (Tokyo) 1976;16:135–140. [PubMed] [Google Scholar]
- 159.Yoshida I., Sakata M., Fujita K., Noguchi T., Yuasa N. Modification of low virulent Newcastle disease virus infection in chickens infected with reticuloendotheliosis virus. Natl. Inst. Anim. Health Q. (Tokyo) 1981;21:1–6. [PubMed] [Google Scholar]
- 160.Sun S., Cui Z., Wang J., Wang Z. Protective efficacy of vaccination against highly pathogenic avian influenza is dramatically suppressed by early infection of chickens with reticuloendotheliosis virus. Avian Pathol. 2009;38:31–34. doi: 10.1080/03079450802607504. [DOI] [PubMed] [Google Scholar]
- 161.Witter R.L., Lee L.F., Bacon L.D., Smith E.J. Depression of vaccinal immunity to Marek’s disease by infection with reticuloendotheliosis virus. Infect. Immun. 1979;26:90–98. doi: 10.1128/iai.26.1.90-98.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Witter R.L., Smith E.J., Crittenden L.B. Tolerance, viral shedding, and neoplasia in chickens infected with non-defective reticuloendotheliosis viruses. Avian Dis. 1981;25:374–394. [PubMed] [Google Scholar]
- 163.Buscaglia C., Calnek B.W., Schat K.A. Effect of reticuloendotheliosis virus and infectious bursal disease virus on Marek’s disease herpesvirus latency. Avian Pathol. 1989;18:265–281. doi: 10.1080/03079458908418601. [DOI] [PubMed] [Google Scholar]
- 164.Filardo E.J., Lee M.F., Humphries E.H. Structural genes, not the LTRs, are the primary determinants of reticuloendotheliosis virus A-induced runting and bursal atrophy. Virology. 1994;202:116–128. doi: 10.1006/viro.1994.1328. [DOI] [PubMed] [Google Scholar]
- 165.Walker M.H., Rup B.J., Rubin A.S., Bose H.R., Jr. Specificity in the immunosuppression induced by avian reticuloendotheliosis virus. Infect. Immun. 1983;40:225–235. doi: 10.1128/iai.40.1.225-235.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rup B.J., Spence J.L., Hoelzer J.D., Lewis R.B., Carpenter C.R., Rubin A.S., Bose H.R., Jr. Immunosuppression induced by avian reticuloendotheliosis virus: mechanism of induction of the suppressor cell. J. Immunol. 1979;123:1362–1370. [PubMed] [Google Scholar]
- 167.Tiwari R., Bargmann W., Bose H.R., Jr Activation of the TGF-β/Smad signaling pathway in oncogenic transformation by v-Rel. Virology. 2011;413:60–71. doi: 10.1016/j.virol.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 168.Liu V.C., Wong L.Y., Jang T., Shah A.H., Park I., Yang X., Zhang Q., Lonning S., Teicher B.A., Lee C. Tumor evasion of the immune system by converting CD4+CD25− T cells into CD4+CD25+ T regulatory cells: role of tumor-derived TGF-β. J. Immunol. 2007;178:2883–2892. doi: 10.4049/jimmunol.178.5.2883. [DOI] [PubMed] [Google Scholar]
- 169.Letterio J.J., Roberts A.B. Regulation of immune responses by TGF-β. Annu. Rev. Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 170.Lillehoj H.S., Lillehoj E.P., Weinstock D., Schat K.A. Functional and biochemical characterization of avian T lymphocyte antigens identified by monoclonal antibodies. Eur. J. Immunol. 1988;18:2059–2065. doi: 10.1002/eji.1830181228. [DOI] [PubMed] [Google Scholar]
- 171.Weinstock D., Schat K.A., Calnek B.W. Cytotoxic T lymphocytes in reticuloendotheliosis virus-infected chickens. Eur. J. Immunol. 1989;19:267–272. doi: 10.1002/eji.1830190208. [DOI] [PubMed] [Google Scholar]
- 172.Koyama H., Suzuki Y., Ohwada Y., Saito Y. Reticuloendotheliosis group virus pathogenic to chicken isolated from material infected with turkey herpesvirus (HVT) Avian Dis. 1976;20:429–434. [PubMed] [Google Scholar]
- 173.Jackson C.A.W., Dunn S.E., Smith D.I., Gilchrist P.T., Macqueen P.A. Proventriculitis, “nakanuke” and reticuloendotheliosis in chickens following vaccination with herpesvirus of turkeys (HVT) Aust. Vet. J. 1977;53:457–459. doi: 10.1111/j.1751-0813.1977.tb05509.x. [DOI] [PubMed] [Google Scholar]
- 174.Hertig C., Coupar B.E.H., Gould A.R., Boyle D.B. Field and vaccine strains of fowlpox virus carry integrated sequences from the avian retrovirus, reticuloendotheliosis virus. Virology. 1997;235:367–376. doi: 10.1006/viro.1997.8691. [DOI] [PubMed] [Google Scholar]
- 175.Diallo I.S., MacKenzie M.A., Spradbrow P.B., Robinson W.F. Field isolates of fowlpox virus contaminated with reticuloendotheliosis virus. Avian Pathol. 1998;27:60–66. doi: 10.1080/03079459808419275. [DOI] [PubMed] [Google Scholar]
- 176.Awad A.M., Abd El-Hamid H.S., Abou Rawash A.A., Ibrahim H.H. Detection of reticuloendotheliosis virus as a contaminant of fowl pox vaccines. Poultry Sci. 2010;89:2389–2395. doi: 10.3382/ps.2010-00899. [DOI] [PubMed] [Google Scholar]
- 177.Liu Q., Zhao J., Su J., Pu J., Zhang G., Liu J. Full genome sequences of two reticuloendotheliosis viruses contaminating commercial vaccines. Avian Dis. 2009;53:341–346. doi: 10.1637/8579-010609-Reg.1. [DOI] [PubMed] [Google Scholar]
- 178.Motha M.X.J., Egerton J.R. Outbreak of atypical fowlpox in chickens with persistent reticuloendotheliosis viraemia. Avian Pathol. 1987;16:177–182. doi: 10.1080/03079458708436362. [DOI] [PubMed] [Google Scholar]
- 179.Wang J., Meers J., Spradbrow P.B., Robinson W.F. Evaluation of immune effects of fowlpox vaccine strains and field isolates. Vet. Microbiol. 2006;116:106–119. doi: 10.1016/j.vetmic.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 180.Asif M., Jenkins K.A., Hilton L.S., Kimpton W.G., Bean A.G., Lowenthal J.W. Cytokines as adjuvants for avian vaccines. Immunol. Cell. Biol. 2004;82:638–643. doi: 10.1111/j.1440-1711.2004.01295.x. [DOI] [PubMed] [Google Scholar]
- 181.Pruett S.B. Quantitative aspects of stress-induced immunomodulation. Int. Immunopharmacol. 2001;1:507–520. doi: 10.1016/s1567-5769(00)00030-8. [DOI] [PubMed] [Google Scholar]
- 182.El-Lethey H., Huber-Eicher B., Jungi T.W. Exploration of stress-induced immunosuppression in chickens reveals both stress-resistant and stress-susceptible antigen responses. Vet. Immunol. Immunopathol. 2003;95:91–101. doi: 10.1016/s0165-2427(02)00308-2. [DOI] [PubMed] [Google Scholar]
- 183.Schat K.A., Nair V. Marek’s disease. In: Saif Y.M., Fadly A.M., Glisson J.R., McDougald L.R., Nolan L.K., Swayne D.E., editors. Disease of Poultry. 12th ed. Wiley-Blackwell; Ames, IA: 2008. pp. 458–520. [Google Scholar]
- 184.Kingham B.F., Zelnik V., Kopacek J., Majerciak V., Ney E., Schmidt C.J. The genome of herpesvirus of turkeys: comparative analysis with Marek’s disease viruses. J. Gen. Virol. 2001;82:1123–1135. doi: 10.1099/0022-1317-82-5-1123. [DOI] [PubMed] [Google Scholar]
- 185.Ross N.L. T-cell transformation by Marek’s disease virus. Trends Microbiol. 1999;7:22–29. doi: 10.1016/s0966-842x(98)01427-9. [DOI] [PubMed] [Google Scholar]
- 186.Piepenbrink M.S., Li X., O’Connell P.H., Schat K.A. Marek’s disease virus phosphorylated polypeptide pp38 alters transcription rates of mitochondrial electron transport and oxidative phosphorylation genes. Virus Genes. 2009;39:102–112. doi: 10.1007/s11262-009-0372-z. [DOI] [PubMed] [Google Scholar]
- 187.Bruckdorfer R. The basics about nitric oxide. Mol. Aspects Med. 2005;26:3–31. doi: 10.1016/j.mam.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 188.Hunt H.D., Lupiani B., Miller M.M., Gimeno I., Lee L.F., Parcells M.S. Marek’s disease virus down-regulates surface expression of MHC (B Complex) Class I (BF) glycoproteins during active but not latent infection of chicken cells. Virology. 2001;282:198–205. doi: 10.1006/viro.2000.0797. [DOI] [PubMed] [Google Scholar]
- 189.Morgan R.W., Sofer L., Anderson A.S., Bernberg E.L., Cui J., Burnside J. Induction of host gene expression following infection of chicken embryo fibroblasts with oncogenic Marek’s disease virus. J. Virol. 2001;75:533–539. doi: 10.1128/JVI.75.1.533-539.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Burgess S.C., Young J.R., Baaten B.J.G., Hunt L., Ross L.N.J., Parcells M.S., Kumar P.M., Tregaskes C.A., Lee L.F., Davison T.F. Marek’s disease is a natural model for lymphomas overexpressing Hodgkin’s disease antigen (CD30) Proc. Natl. Acad. Sci. USA. 2004;101:13879–13884. doi: 10.1073/pnas.0305789101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Burgess S.C., Davison T.F. Identification of the neoplastically transformed cells in Marek’s disease herpesvirus-induced lymphomas: recognition by the monoclonal antibody AV37. J. Virol. 2002;76:7276–7292. doi: 10.1128/JVI.76.14.7276-7292.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Anobile J.M., Arumugaswami V., Downs D., Czymmek K., Parcells M., Schmidt C.J. Nuclear localization and dynamic properties of the Marek’s disease virus oncogene products Meq and Meq/vIL8. J. Virol. 2006;80:1160–1166. doi: 10.1128/JVI.80.3.1160-1166.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Jarmin S., Manvell R., Gough R.E., Laidlaw S.M., Skinner M.A. Avipoxvirus phylogenetics: identification of a PCR length polymorphism that discriminates between the two major clades. J. Gen. Virol. 2006;87:2191–2201. doi: 10.1099/vir.0.81738-0. [DOI] [PubMed] [Google Scholar]
- 194.Afonso C.L., Tulman E.R., Lu Z., Zsak L., Kutish G.F., Rock D.L. The genome of fowlpox virus. J. Virol. 2000;74:3815–3831. doi: 10.1128/jvi.74.8.3815-3831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Laidlaw S.M., Skinner M.A. Comparison of the genome sequence of FP9, an attenuated, tissue culture-adapted European strain of fowlpox virus, with those of virulent American and European viruses. J. Gen. Virol. 2004;85:305–322. doi: 10.1099/vir.0.19568-0. [DOI] [PubMed] [Google Scholar]
- 196.Tulman E.R., Afonso C.L., Lu Z., Zsak L., Kutish G.F., Rock D.L. The genome of canarypox virus. J. Virol. 2004;78:353–366. doi: 10.1128/JVI.78.1.353-366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Eldaghayes, I. (2005). Use of chicken interleukin-18 as a vaccine adjuvant with a recombinant fowlpox virus fpIBD1, a subunit vaccine giving partial protection against IBDV, PhD Thesis, University of Bristol.
- 198.Jeshtadi A., Henriquet G., Laidlaw S.M., Hot D., Zhang Y., Skinner M.A. In vitro expression and analysis of secreted fowlpox virus CC chemokine-like proteins Fpv060, Fpv061, Fpv116 and Fpv121. Arch. Virol. 2005;150:1745–1762. doi: 10.1007/s00705-005-0560-7. [DOI] [PubMed] [Google Scholar]
- 199.Puehler F., Schwarz H., Waidner B., Kalinowski J., Kaspers B., Bereswill S., Staeheli P. An interferon-g-binding protein of novel structure encoded by the fowlpox virus. J. Biol. Chem. 2003;278:6905–6911. doi: 10.1074/jbc.M207336200. [DOI] [PubMed] [Google Scholar]
- 200.Buttigieg K., Laidlaw S.M., Ross C., Davies M., Goodbourn S., Skinner M.A. Genetic screen of a library of chimeric poxviruses identifies an ankyrin repeat protein involved in resistance to the avian type I interferon response. J. Virol. 2013;87:5028–5040. doi: 10.1128/JVI.02738-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Laidlaw S.M., Robey R., Davies M., Giotis E.S., Ross C., Buttigieg K., Goodbourn S., Skinner M.A. Genetic screen of a mutant poxvirus library identifies an ankyrin repeat protein involved in blocking induction of avian type I interferon. J. Virol. 2013;87:5041–5052. doi: 10.1128/JVI.02736-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sonnberg S., Fleming S.B., Mercer A.A. Phylogenetic analysis of the large family of poxvirus ankyrin-repeat proteins reveals orthologue groups within and across chordopoxvirus genera. J. Gen. Virol. 2011;92:2596–2607. doi: 10.1099/vir.0.033654-0. [DOI] [PubMed] [Google Scholar]
- 203.Sonnberg S., Seet B.T., Pawson T., Fleming S.B., Mercer A.A. Poxvirus ankyrin repeat proteins are a unique class of F-box proteins that associate with cellular SCF1 ubiquitin ligase complexes. Proc. Natl. Acad. Sci. USA. 2008;105:10955–10960. doi: 10.1073/pnas.0802042105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Barber M.R., Aldridge J.R., Jr., Webster R.G., Magor K.E. Association of RIG-I with innate immunity of ducks to influenza. Proc. Natl. Acad. Sci. USA. 2010;107:5913–5918. doi: 10.1073/pnas.1001755107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Childs K., Stock N., Ross C., Andrejeva J., Hilton L., Skinner M., Randall R., Goodbourn S. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology. 2007;359:190–200. doi: 10.1016/j.virol.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 206.Lee C.C., Wu C.C., Lin T.L. Characterization of chicken melanoma differentiation-associated gene 5 (MDA5) from alternative translation initiation. Comp. Immunol. Microbiol. Infect. Dis. 2012;35:335–343. doi: 10.1016/j.cimid.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 207.Liniger M., Summerfield A., Zimmer G., McCullough K.C., Ruggli N. Chicken cells sense influenza A virus infection through MDA5 and CARDIF signaling involving LGP2. J. Virol. 2012;86:705–712. doi: 10.1128/JVI.00742-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lindenmann J., Burke D.C., Isaacs A. Studies on the production, mode of action and properties of interferon. Br. J. Exp. Pathol. 1957;38:551–562. [PMC free article] [PubMed] [Google Scholar]
- 209.Lee T.G., Tomita J., Hovanessian A.G., Katze M.G. Purification and partial characterization of a cellular inhibitor of the interferon-induced protein kinase of Mr 68,000 from influenza virus-infected cells. Proc. Natl. Acad. Sci. USA. 1990;87:6208–6212. doi: 10.1073/pnas.87.16.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bergmann M., Garcia-Sastre A., Carnero E., Pehamberger H., Wolff K., Palese P., Muster T. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J. Virol. 2000;74:6203–6206. doi: 10.1128/jvi.74.13.6203-6206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Garcia-Sastre A., Egorov A., Matassov D., Brandt S., Levy D.E., Durbin J.E., Palese P., Muster T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology. 1998;252:324–330. doi: 10.1006/viro.1998.9508. [DOI] [PubMed] [Google Scholar]
- 212.Hatada E., Saito S., Fukuda R. Mutant influenza viruses with a defective NS1 protein cannot block the activation of PKR in infected cells. J. Virol. 1999;73:2425–2433. doi: 10.1128/jvi.73.3.2425-2433.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Talon J., Horvath C.M., Polley R., Basler C.F., Muster T., Palese P., Garcia-Sastre A. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J. Virol. 2000;74:7989–7996. doi: 10.1128/jvi.74.17.7989-7996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Seo S.H., Hoffmann E., Webster R.G. The NS1 gene of H5N1 influenza viruses circumvents the host anti-viral cytokine responses. Virus Res. 2004;103:107–113. doi: 10.1016/j.virusres.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 215.Marcus P.I., Rojek J.M., Sekellick M.J. Interferon induction and/or production and its suppression by influenza A viruses. J. Virol. 2005;79:2880–2890. doi: 10.1128/JVI.79.5.2880-2890.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bazzigher L., Pavlovic J., Haller O., Staeheli P. Mx genes show weaker primary response to virus than other interferon-regulated genes. Virology. 1992;186:154–160. doi: 10.1016/0042-6822(92)90069-2. [DOI] [PubMed] [Google Scholar]
- 217.Yuan W.K., Krug R.M. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. Embo J. 2001;20:362–371. doi: 10.1093/emboj/20.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wang W.K., Krug R.M. The RNA-binding and effector domains of the viral NS1 protein are conserved to different extents among influenza A and B viruses. Virology. 1996;223:41–50. doi: 10.1006/viro.1996.0453. [DOI] [PubMed] [Google Scholar]
- 219.Hatada E.F., Fukuda R. Binding of influenza A virus NS1 protein to dsRNA in vitro. J. Gen. Virol. 1992;73:3325–3329. doi: 10.1099/0022-1317-73-12-3325. [DOI] [PubMed] [Google Scholar]
- 220.Lu Y., Wambach M., Katze M.G., Krug R.M. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the elF-2 translation initiation factor. Virology. 1995;214:222–228. doi: 10.1006/viro.1995.9937. [DOI] [PubMed] [Google Scholar]
- 221.Qiu Y.K., Krug R.M. The influenza virus NS1 protein is a poly(A)-binding protein that inhibits nuclear export of mRNAs containing poly(A) J. Virol. 1994;68:2425–2432. doi: 10.1128/jvi.68.4.2425-2432.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wang X., Li M., Zheng H., Muster T., Palese P., Beg A.A., Garcia-Sastre A. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J. Virol. 2000;74:11566–11573. doi: 10.1128/jvi.74.24.11566-11573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Schultz-Cherry S., Dybdahl-Sissoko N., Neumann G., Kawaoka Y., Hinshaw V.S. Influenza virus ns1 protein induces apoptosis in cultured cells. J. Virol. 2001;75:7875–7881. doi: 10.1128/JVI.75.17.7875-7881.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zhirnov O.P., Konakova T.E., Wolff T., Klenk H.D. NS1 protein of influenza A virus down-regulates apoptosis. J. Virol. 2002;76:1617–1625. doi: 10.1128/JVI.76.4.1617-1625.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Aragon T., de la Luna S., Novoa I., Carrasco L., Ortin J., Nieto A. Eukaryotic translation initiation factor 4GI is a cellular target for NS1 protein, a translational activator of influenza virus. Mol. Cell. Biol. 2000;20:6259–6268. doi: 10.1128/mcb.20.17.6259-6268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Burgui I., Aragon T., Ortin J., Nieto A. PABP1 and eIF4GI associate with influenza virus NS1 protein in viral mRNA translation initiation complexes. J. Gen. Virol. 2003;84:3263–3274. doi: 10.1099/vir.0.19487-0. [DOI] [PubMed] [Google Scholar]
- 227.Chen Z.K., Krug R.M. Selective nuclear export of viral mRNAs in influenza-virus-infected cells. Trends Microbiol. 2000;8:376–383. doi: 10.1016/s0966-842x(00)01794-7. [DOI] [PubMed] [Google Scholar]
- 228.Nemeroff M.E., Barabino S.M., Li Y., Keller W., Krug R.M. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3’end formation of cellular pre-mRNAs. Mol. Cell. 1998;1:991–1000. doi: 10.1016/s1097-2765(00)80099-4. [DOI] [PubMed] [Google Scholar]
- 229.Donelan N.R., Basler C.F., Garcia-Sastre A. A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J. Virol. 2003;77:13257–13266. doi: 10.1128/JVI.77.24.13257-13266.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Penski N., Haertle S., Rubbenstroth D., Krohmann C., Ruggli N., Schusser B., Pfann M., Reuter A., Gohrbandt S., Hundt J., Veits J., Breithaupt A., Kochs G., Stech J., Summerfield A., Vahlenkamp T., Kaspers B., Staeheli P. Highly pathogenic avian influenza viruses do not inhibit interferon synthesis in infected chickens but can override the interferon-induced antiviral state. J. Virol. 2011;85:7730–7741. doi: 10.1128/JVI.00063-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Liniger M., Moulin H.R., Sakoda Y., Ruggli N., Summerfield A. Highly pathogenic avian influenza virus H5N1 controls type I IFN induction in chicken macrophage HD-11 cells: a polygenic trait that involves NS1 and the polymerase complex. Virol. J. 2012;9:7. doi: 10.1186/1743-422X-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zielecki F., Semmler I., Kalthoff D., Voss D., Mauel S., Gruber A.D., Beer M., Wolff T. Virulence determinants of avian H5N1 influenza A virus in mammalian and avian hosts: role of the C-terminal ESEV motif in the viral NS1 protein. J. Virol. 2010;84:10708–10718. doi: 10.1128/JVI.00610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Steel J., Lowen A.C., Pena L., Angel M., Solorzano A., Albrecht R., Perez D.R., Garcia-Sastre A., Palese P. Live attenuated influenza viruses containing NS1 truncations as vaccine candidates against H5N1 highly pathogenic avian influenza. J. Virol. 2009;83:1742–1753. doi: 10.1128/JVI.01920-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Horvath C.M. Silencing STATs: lessons from paramyxovirus interferon evasion. Cytokine Growth Factor Rev. 2004;15:117–127. doi: 10.1016/j.cytogfr.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 235.Park M.S., Shaw M.L., Munoz-Jordan J., Cros J.F., Nakaya T., Bouvier N., Palese P., Garcia-Sastre A., Basler C.F. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol. 2003;77:1501–1511. doi: 10.1128/JVI.77.2.1501-1511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Gonzalez-Lopez C., Martinez-Costas J., Esteban M., Benavente J. Evidence that avian reovirus sigmaA protein is an inhibitor of the double-stranded RNA-dependent protein kinase. J. Gen. Virol. 2003;84:1629–1639. doi: 10.1099/vir.0.19004-0. [DOI] [PubMed] [Google Scholar]
- 237.Guardado-Calvo P., Vazquez-Iglesias L., Martinez-Costas J., Llamas-Saiz A.L., Schoehn G., Fox G.C., Hermo-Parrado X.L., Benavente J., van Raaij M.J. Crystal structure of the avian reovirus inner capsid protein sigmaA. J. Virol. 2008;82:11208–11216. doi: 10.1128/JVI.00733-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Vazquez-Iglesias L., Lostale-Seijo I., Martinez-Costas J., Benavente J. Avian reovirus σA localizes to the nucleolus and enters the nucleus by a nonclassical energy- and carrier-independent pathway. J. Virol. 2009;83:10163–10175. doi: 10.1128/JVI.01080-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Li Z., Wang Y., Li X., Li X., Cao H., Zheng S.J. Critical roles of glucocorticoid-induced leucine zipper in infectious bursal disease virus (IBDV)-induced suppression of type I interferon expression and enhancement of IBDV growth in host cells via interaction with VP4. J. Virol. 2013;87:1221–1231. doi: 10.1128/JVI.02421-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Casais R., Thiel V., Siddell S.G., Cavanagh D., Britton P. Reverse genetics system for the avian coronavirus infectious bronchitis virus. J. Virol. 2001;75:12359–12369. doi: 10.1128/JVI.75.24.12359-12369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Casais R., Davies M., Cavanagh D., Britton P. Gene 5 of the avian coronavirus infectious bronchitis virus is not essential for replication. J. Virol. 2005;79:8065–8078. doi: 10.1128/JVI.79.13.8065-8078.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Hodgson T., Britton P., Cavanagh D. Neither the RNA nor the proteins of open reading frames 3a and 3b of the coronavirus infectious bronchitis virus are essential for replication. J. Virol. 2006;80:296–305. doi: 10.1128/JVI.80.1.296-305.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Washietl S., Eisenhaber F. Reannotation of the CELO genome characterizes a set of previously unassigned open reading frames and points to novel modes of host interaction in avian adenoviruses. BMC Bioinformatics. 2003;4:55. doi: 10.1186/1471-2105-4-55. [DOI] [PMC free article] [PubMed] [Google Scholar]