

Publisher Summary
Viruses first infect their host, then spread, and then damage their target tissues. To ensure their perpetuation, viruses must be transmitted to other susceptible individuals—that is, they must be shed with secretions or excretions into the environment, be taken up by another host or a vector, or be passed congenitally from mother to offspring. Viruses have developed a remarkable variety of strategies to ensure their own survival. Individual viruses cause their associated diseases through a considerable variety of distinct pathogenic mechanisms. Viruses differ greatly in their virulence, but even in a population infected by a particular virus strain, there are usually striking differences in the outcome of infection of individual animals. Similarly, there is much variation among viruses of the same species, and the determinants of viral virulence are often multigenic, meaning that several viral genes contribute to the virulence of individual viruses. The determinants of host resistance/susceptibility are usually multifactorial and include not only a variety of host factors but environmental ones as well. There is wide variation in the virulence of viruses, ranging from those that almost always cause inapparent infections, to those that usually cause disease, to those that usually cause death. Meaningful comparison of the virulence of viruses requires that factors such as the infecting dose of the virus and the age, sex, and condition of the host animals and their immune status be equal; however, these conditions are never met in nature, where heterogeneous, outbred animal populations are the rule and the dynamics of exposure and viral infection are incredibly varied.
Viral infection is not synonymous with disease, as many viral infections are subclinical (i.e., asymptomatic, inapparent), whereas others result in disease of varying severity that is typically accompanied by characteristic clinical signs in the affected host (Figure 3.1 ). Amongst many other potentially contributing factors, the outcome of the virus—host encounter is essentially the product of the virulence of the infecting virus on the one hand and the susceptibility of the host on the other. The term virulence is used as a quantitative or relative measure of the pathogenicity of the infecting virus—that is, a virus is said to be either pathogenic or non-pathogenic, but its virulence is stated in relative terms (“virus A is more virulent than virus B” or “virus strain A is more virulent in animal species Y than species Z”). The terms pathogenicity and virulence refer to the capacity of a virus to cause disease in its host, and are unrelated to the infectivity or transmissibility (contagiousness) of the virus.
Figure 3.1.
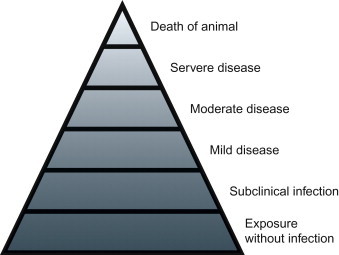
The iceberg concept of viral infection and diseases.
For viruses to cause disease they must first infect their host, spread to (and within) and damage target tissues. To ensure their perpetuation, viruses must then be transmitted to other susceptible individuals—that is, they must be shed with secretions or excretions into the environment, be taken up by another host or a vector, or be passed congenitally from mother to offspring. Viruses have developed a remarkable variety of strategies to ensure their own survival. Similarly, individual viruses cause their associated diseases through a considerable variety of distinct pathogenic mechanisms.
Interplay of Viral Virulence and Host Resistance, or Susceptibility Factors in Expression of Viral Diseases
Viruses differ greatly in their virulence, but even in a population infected by a particular virus strain there are usually striking differences in the outcome of infection of individual animals. Similarly, there is much variation amongst viruses of the same species and the determinants of viral virulence are often multigenic, meaning that several viral genes contribute to the virulence of individual viruses. Similarly, the determinants of host resistance/susceptibility are usually multifactorial, and include not only a variety of host factors but environmental ones as well.
The advent and application of molecular technologies has facilitated mapping of virulence determinants in the genome of many viruses (e.g., by whole-genomic sequencing of virus strains, and manipulation of molecular clones), as well as the location of resistance/susceptibility determinants in the genome of experimental animals. Virus strain differences may be quantitative, involving the rate and yield of virus replication, lethal dose, infectious dose, the number of cells infected in a given organ, or they may be qualitative, involving organ or tissue tropism, extent of host-cell damage, mode and efficacy of spread in the body, and character of the disease they induce.
Assessment of Viral Virulence
There is wide variation in the virulence of viruses, ranging from those that almost always cause inapparent infections, to those that usually cause disease, to those that usually cause death. Meaningful comparison of the virulence of viruses requires that factors such as the infecting dose of the virus and the age, sex, and condition of the host animals and their immune status be equal; however, these conditions are never met in nature, where heterogeneous, outbred animal populations are the rule and the dynamics of exposure and viral infection are incredibly varied. Hence, subjective and vague terminology may be used to describe the virulence of particular viruses in domestic and wild animals. Precise measures of virulence are usually derived only from assays in inbred animals such as mice. Of course, such assays are only feasible for those viruses that grow in mice, and care must always be exercised in extrapolating data from mice to the host species of interest.
The virulence of a particular strain of virus administered in a particular dose, by a particular route, to a particular age and strain of laboratory animal may be assessed by determining its ability to cause disease, death, specific clinical signs, or lesions. The dose of the virus required to cause death in 50% of animals [lethal dose 50 (LD50)] has been a commonly used measure of virulence, but is now passing out of favor in the research arena for ethical reasons. For example, in the susceptible BALB/c strain of mouse, the LD50 of a virulent strain of ectromelia virus is 5 virions, as compared with 5000 for a moderately attenuated strain and about 1 million for a highly attenuated strain. Viral virulence can also be measured in experimental animals by determining the ratio of the dose of a particular strain of virus that causes infection in 50% of individuals [infectious dose 50 (ID50)] to the dose that kills 50% of individuals (the ID50:LD50 ratio). Thus, the ID50 of a virulent strain of ectromelia virus in BALB/c mice is 2 virions and the LD50 about 5 virions, whereas for resistant C57BL strain mice the ID50 is the same but the LD50 is 1 million virions. The severity of an infection, therefore, depends on the interplay between the virulence of the virus and the resistance of the host. Viral virulence also can be estimated through assessment of the severity, location, and distribution of gross, histologic, and ultrastructural lesions in affected animals.
Determinants of Viral Virulence
The advent of molecular biology has facilitated determination of the genetic basis of virulence of many viruses, along with other important aspects of their replication. Specifically, the role of potential determinants of virulence identified by genetic sequence comparison of viruses of defined virulence can be confirmed unequivocally by manipulation of molecular clones of the virus in question. This “reverse genetics” strategy utilizing molecular (infectious) clones was first widely employed using complementary DNA (cDNA) copies of the entire genome of simple positive-strand RNA viruses such as alphaviruses and picornaviruses, as RNA transcribed from full-length cDNA copies (clones) of these viruses is itself infectious after transfection into cells. The virion RNA of negative-sense RNA viruses such as rhabdoviruses is not infectious, but infectious virus can be recovered from cDNA clones if the necessary proteins are also produced in cells transfected with full-length RNA transcripts. Even the considerable logistical challenges posed by RNA viruses with segmented genomes (such as influenza viruses, bunyaviruses, arenaviruses, and reoviruses) have been overcome, and molecular clones of these viruses are now used for reverse genetic manipulation. It is also now possible to specifically manipulate the genomes of even the very large DNA viruses as artificial chromosomes. Of necessity, most experimental work has been carried out in inbred laboratory animals, although molecular clones of a substantial number of pathogenic animal viruses have now been evaluated in their respective natural animal hosts. It is apparent from these reverse genetic studies that several viral genes can contribute to the virulence of individual viruses, as described under each virus family in Part II of this book.
Viruses exhibit host and tissue specificity (tropism), usually more than is appreciated clinically. Mechanistically, the organ or tissue tropism of the virus is an expression of all the steps required for successful infection, from the interaction of virus attachment molecules and their cellular receptors to virus assembly and release (see Chapter 2). Organ and tissue tropisms also involve all stages in the course of infection in the whole host animal, from the site of entry, to the major target organs responsible for the clinical signs, to the site involved in virus release and shedding.
Caution should be exercised in attributing characteristics of viral epidemics or epizootics solely to the virulence of the causative virus, as there typically is considerable variation in the response of individual infected animals, both within and between animal species. For example, during the epizootic of West Nile virus infection that began in North America in 1999, approximately 10% of infected horses developed neurological disease (encephalomyelitis) and, of these, approximately 30–35% died. Neuroinvasive disease was even less common in humans infected with this same strain of West Nile virus, whereas infected corvids (crows and their relatives) almost uniformly developed disseminated, rapidly fatal infections.
Determinants of Host Resistance/Susceptibility
As just described for West Nile virus, genetic differences in host resistance/susceptibility to viral infections are most obvious when different animal species are compared. Viral infections tend to be less pathogenic in their natural host species than in exotic or introduced species. For instance, myxoma virus produces a small benign fibroma in its natural host, which are wild rabbits of the Americas (Sylvilagus spp.), but an almost invariably fatal generalized infection in the European rabbit, Oryctolagus cuniculus. Likewise, zoonotic (transmitted from animal to human) infections caused by arenaviruses, filoviruses, and many arboviruses are severe in humans but mild or asymptomatic in their reservoir animal hosts.
The innate and adaptive immune responses to particular viral infections differ greatly from one individual to another (see Chapter 4). Studies with inbred strains of mice have confirmed that susceptibility to specific viruses may be associated with particular major histocompatibility antigen haplotypes, presumably because of their central role in directing the nature of the adaptive immune response generated to the infecting virus. Similarly, studies with genetically modified mice have unequivocally confirmed the critical role of innate immune responses, especially those associated with the interferon system, in conferring antiviral resistance and protection.
Expression of critical receptors on target cells is a fundamental determinant of host resistance/susceptibility to a particular virus. The more conserved or ubiquitous the receptor, the wider the host range of the virus that exploits it; for example, rabies virus, which uses sialylated gangliosides in addition to the acetylcholine receptor, has a very wide host range, but infection is restricted narrowly to a few host cell types, including myocytes, neurons, and salivary gland epithelium. Changes in viral attachment proteins can lead to the emergence of variant viruses with different tropism and disease potential. For example, porcine respiratory coronavirus arose from transmissible gastroenteritis virus, which is strictly an enteric pathogen, through a substantial deletion in the gene encoding the viral spike protein that mediates virus attachment. This change affected the tropism of the virus as well as its transmissibility.
Physiologic Factors Affecting Host Resistance/Susceptibility
In addition to innate and adaptive immune responses, a considerable variety of physiologic factors affect host resistance/susceptibility to individual viral diseases, including age, nutritional status, levels of certain hormones, and cell differentiation.
Viral infections tend to be most serious at both ends of life—in the very young and the very old. Rapid physiologic changes occur during the immediate postpartum period and resistance to the most severe manifestations of many intestinal and respiratory infections builds quickly in the neonate. Maturation of the immune system is responsible for much of this enhanced, age-related resistance, but physiologic changes also contribute. Malnutrition can also potentially impair immune responsiveness in adults, but it often difficult to distinguish adverse nutritional effects from other factors found in animals living in very adverse environments.
Certain infections, particularly herpesvirus infections, can be reactivated during pregnancy, leading to abortion or perinatal infection of the progeny of infected dams. The fetus itself is uniquely susceptible to a number of different viral infections.
Cellular differentiation and the stage of the cell cycle may affect susceptibility to infection with specific viruses. For example, parvoviruses replicate only in cells that are in the late S phase of the cell cycle, so the rapidly dividing cells of bone marrow, intestinal epithelium, and the developing fetus are vulnerable. The rapidly dividing, often migratory cell populations that occur during embryogenesis in the developing fetus are exquisitely susceptible to infection and injury by a number of viruses, notably several highly teratogenic viruses that infect the developing central nervous system.
Almost all viral infections are accompanied by fever. In classic studies of myxoma virus infection in rabbits, it was shown that increasing body temperature increased protection against disease, whereas decreasing temperature increased the severity of infection. Blocking the development of fever with drugs (e.g., salicylates) increased mortality. Similar results have been obtained with ectromelia and coxsackievirus infections in mice. In contrast, fever does not accompany viral infection in certain poikilotherms (e.g., fish), in which this response is probably of no or lesser selective advantage.
The immunosuppressive effects of increased concentrations of corticosteroids, whether endogenous or exogenous in origin, can reactivate latent viral infections or exacerbate active mild or subclinical viral infections, such as those caused by herpesviruses. This mechanism probably contributes to the increased incidence of severe viral infections that occurs in settings in which animals are transported or brought into crowded environments, such as animal shelters and feedlots. Products of host inflammatory and innate immune responses also probably contribute to the transient immunosuppression and other general signs that can accompany viral infections.
Mechanisms of Viral Infection and Virus Dissemination
At the level of the cell, infection by viruses (see Chapters 1 and 2Chapter 1Chapter 2) is quite different from that caused by bacteria and other microorganisms, whereas at the level of the whole animal and animal populations there are more similarities than differences. Like microorganisms, viruses must gain entry into their host’s body before they can exert their pathogenic effects; entry of virus into the host can occur through any of a variety of potential routes, depending on the properties of the individual virus (Figure 3.2 ; Table 3.1 ).
Figure 3.2.
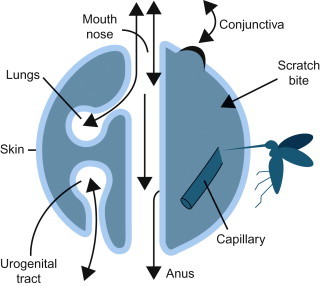
The surfaces of the body in relation to the entry and shedding of viruses.
(Courtesy of C. A. Mims.)
Table 3.1.
Obligatory Steps in Viral Infection
| Step in Infection Process | Requirement for Virus Survival and Progression of Infection |
|---|---|
| Entry into host and primary virus replication | Evade host’s natural protective and cleansing mechanisms |
| Local or general spread in the host, cell and tissue tropism, and secondary virus replication | Evade immediate host defenses and natural barriers to spread; at the cellular level the virus takes over necessary host-cell functions for its own replication processes |
| Evasion of host inflammatory and immune defenses | Evade host inflammatory, phagocytic, and immune defenses long enough to complete the virus transmission cycle |
| Shedding from host | Exit host body at site and at concentration needed to ensure infection of the next host |
| Cause damage to host | Not necessary, but this is the reason we are interested in the virus and its pathogenetic processes |
Routes of Virus Entry
Viruses are obligate intracellular parasites that are transmitted as inert particles. To infect its host, a virus must first attach to and infect cells at one of the body surfaces, unless these potential barriers are bypassed by parenteral inoculation via a wound, needle, or the bite of an arthropod or vertebrate. Cedric Mims represented the animal body as a set of surfaces, each covered by a sheet of epithelial cells separating host tissues from the outside world (Figure 3.2). The skin that covers the animal body externally has a relatively impermeable outer layer of keratin, whereas the mucosal epithelial lining of the respiratory tract and much of the gastrointestinal and urogenital tracts lacks this protective layer. Similarly, in and around the eyes, the protective keratinized layer of skin is replaced by the non-keratinized epithelial lining of the conjunctiva and cornea. Each of these sites is the target for invasion by specific viruses. In animals without significant areas of keratinized epithelium (e.g., fish), the skin and gills serve as an extensive mucosal surface that is the initial site of infection with many viruses.
Entry via the Respiratory Tract
The mucosal surfaces of the respiratory tract are lined by epithelial cells that can potentially support the replication of viruses, so defenses are necessary to minimize the risk of infection. The respiratory tract from the nasal passages to the distal airways in the lungs is protected by the “mucociliary blanket,” which consists of a layer of mucus produced by goblet cells that is kept in continuous flow by the coordinated beating of cilia on the luminal surface of the epithelial cells that line the nasal mucosa and airways. The airspaces (alveoli) are protected by resident alveolar macrophages. The distance to which inhaled particles penetrate into the respiratory tract is inversely related to their size, so that larger particles (greater than 10 μm in diameter) are trapped on the mucociliary blanket lining the nasal cavity and airways and small particles (less than 5 μm in diameter) can be inhaled directly into the airspaces, where they are ingested by alveolar macrophages. Most inhaled virions are trapped in mucus and then carried by ciliary action from the nasal cavity and airways to the pharynx, and then swallowed or coughed out.
The respiratory system is also protected by the innate and adaptive immune mechanisms that operate at all mucosal surfaces (see Chapter 4), including specialized lymphoid aggregates [e.g., nasal associated lymphoid tissue (NALT) and tonsils- and bronchus-associated lymphoid tissue (BALT)] that occur throughout the respiratory tree. Despite its protective mechanisms, however, the respiratory tract is perhaps the most common portal of virus entry into the body. Viruses can infect the host via the respiratory tract by first attaching to specific receptors on epithelial cells within the mucosa, thus avoiding clearance by either the mucociliary blanket or phagocytic cells. After invasion, some viruses remain localized to the respiratory system, or spread from cell to cell to invade other tissues, whereas many others become widely disseminated via lymphatics and/or the bloodstream.
Entry via the Gastrointestinal Tract
A substantial number of viruses (enteric viruses) are spread to susceptible hosts by ingestion of virus-contaminated food or drink. The mucosal lining of the oral cavity and esophagus (and forestomachs of ruminants) is relatively refractory to viral infection, with the notable exception of that overlying the tonsils, thus enteric viral infections typically begin within the mucosal epithelium of the stomach and/or intestines. The gastrointestinal tract is protected by several different defenses, including acidity of the stomach, the layer of mucus that tenaciously covers the mucosa of the stomach and intestines, the antimicrobial activity of digestive enzymes as well as that of bile and pancreatic secretions, and innate and adaptive immune mechanisms, especially the activity of defensins and secretory antibodies such as immunoglobulin (Ig) A, the latter produced by B lymphocytes in the gastrointestinal mucosa and mucosal associated lymphoid tissues. Despite these protective mechanisms, enteric infection is characteristic of certain viruses that first infect the epithelial cells lining the gastrointestinal mucosa or the specialized M cells that overlie intestinal lymphoid aggregates (Peyer’s patches).
In general, viruses that cause purely enteric infection, such as rotaviruses and enteroviruses, are acid and bile resistant. However, there are acid- and bile-labile viruses that cause important enteric infections; for example, coronaviruses such as transmissible gastroenteritis virus are protected during passage through the stomach of young animals by the buffering action of suckled milk. Not only do some enteric viruses resist inactivation by proteolytic enzymes in the stomach and intestine, their infectivity is actually increased by such exposure. Thus cleavage of an outer capsid protein by intestinal proteases enhances the infectivity of rotaviruses and some coronaviruses. Rotaviruses, coronaviruses, toroviruses, and astroviruses are all major causes of viral diarrhea in animals, whereas the great majority of enteric infections caused by enteroviruses and adenoviruses are asymptomatic. Parvoviruses, morbilliviruses, and many other viruses can also cause gastrointestinal infection and diarrhea, but only after reaching cells of the gastrointestinal tract in the course of generalized (systemic) infection after viremic spread.
Entry via the Skin
The skin is the largest organ of the body, and its dense outer layer of keratin provides a mechanical barrier to the entry of viruses. The low pH and presence of fatty acids in skin provide further protection, as do various other components of innate and adaptive immunity, including the presence of migratory dendritic cells (Langerhans cells) within the epidermis itself. Breaches in skin integrity such as insect or animal bites, cuts, punctures, or abrasions predispose to viral infection, which can either remain confined to the skin, such as the papillomaviruses, or disseminate widely. Deeper trauma can introduce viruses into the dermis and subcutis, where there is a rich supply of blood vessels, lymphatics, and nerves that can individually serve as routes of virus dissemination. Generalized infection of the skin, such as occurs in lumpy skin disease, sheeppox, and others, is the result, not of localized cutaneous infection but of systemic viral spread via viremia.
One of the most efficient ways by which viruses are introduced through the skin is via the bite of arthropods, such as mosquitoes, ticks, Culicoides spp. (hematophagous midges), or sandflies. Insects, especially flies, may act as simple mechanical vectors (“flying needles”); for example, equine infectious anemia virus is spread among horses, rabbit hemorrhagic disease virus and myxoma virus are spread among rabbits, and fowlpox virus among chickens in this way. However, most viruses that are spread by arthropods replicate in their vector. Viruses that are both transmitted by and replicate in arthropod vectors are called arboviruses.
Infection can also be acquired through the bite of an animal, as in rabies, and introduction of a virus by skin penetration may be iatrogenic—that is, the result of veterinary or husbandry procedures. For example, equine infectious anemia virus has been transmitted via contaminated needles, twitches, ropes, and harnesses, and orf virus and papillomaviruses can be transmitted via ear tagging, tattooing, or virus-contaminated inanimate objects (fomites).
Entry via Other Routes
Several important pathogens (e.g., several herpesviruses and papillomaviruses) are spread through the genital tract. Small tears or abrasions in the penile mucosa and the epithelial lining of the vagina may occur during sexual activity and facilitate transmission of venereal virus. The conjunctiva, although much less resistant to viral invasion than the skin, is constantly cleansed by the flow of secretion (tears) and mechanical wiping by the eyelids; some adenoviruses and enteroviruses gain entry at this site, and a substantial number of viruses can be experimentally transmitted by this route.
Host Specificity and Tissue Tropism
The capacity of a virus to infect cells selectively in particular organs is referred to as tropism (either cell or organ tropism), which is dependent on both viral and host factors. At the cellular level, there must be an interaction between viral attachment proteins and matching cellular receptors. Although such interactions are usually studied in cultured cells, the situation is considerably more complex in vivo. Not only do some viruses require several cellular receptors/co-receptors (see Chapter 2), some viruses utilize different receptors on different cells; for example, the cell attachment glycoprotein of human immunodeficiency virus can bind several receptors (including CD4, CXCR4 and CCR5), which allows it to infect both T lymphocytes and macrophages. Expression of receptors can be dynamic; for example, it has been shown experimentally that animals treated with neuraminidase have substantial protection against intranasal infection with influenza virus that lasts until the neuraminidase-sensitive receptors have regenerated. Receptors for a particular virus are usually restricted to certain cell types in certain organs, and only these cells can be infected. In large part, this accounts for both the tissue and organ tropism of a given virus and the pathogenesis of the disease caused by the virus.
The presence of critical receptors is not the only factor that determines whether the cell may become infected—intracellular factors that exert their effect subsequent to virus attachment, such as viral enhancers, may also be required for productive infection. Viral enhancers are gene activators that increase the efficiency of transcription of viral or cellular genes; specifically, they are short, often tandem-repeated sequences of nucleotides that may contain motifs representing DNA-binding sites for various cellular or viral site-specific DNA-binding proteins (transcription factors). Viral enhancers augment binding of DNA-dependent RNA polymerase to promoters, thereby accelerating transcription. Because many of the transcription factors affecting individual enhancer sequences in viruses are restricted to particular cells, tissues, or host species, they can determine the tropism of viruses and can act as specific virulence factors. The genomic DNA of papillomavirus contains such enhancers, which are active only in keratinocytes and, indeed, only in the subset of these cells in which papillomavirus replication occurs. Enhancer sequences have also been defined in the genomes of retroviruses and several herpesviruses, amongst others, where they also appear to influence tropism by regulating the expression of viral genes in specific cell types.
Mechanisms of Viral Spread and Infection of Target Organs
Virus replication may be restricted to the body surface through which the virus entered—for example, the skin, respiratory tract, gastrointestinal tract, genital tract, or conjunctiva. Alternatively, the invading virus may breach the epithelial barrier and be spread through the blood (hematogenous spread), lymphatics, or nerves to cause a generalized infection, or infection in a specific site such as the central nervous system (brain and spinal cord).
In pioneering experiments in 1949, Frank Fenner used ectromelia virus (the agent of mousepox) as a model system that first revealed the sequence of events leading to systemic infection and disease. Groups of mice were inoculated in the footpad of a hind limb, and at daily intervals their organs were titrated to determine the amount of virus present. Fenner showed that, during the incubation period, infection spread through the mouse body in a step-wise fashion (Figure 3.3 ). The virus first replicated locally in tissues of the footpad and then in the draining lymph nodes. Virus produced in these sites then gained entry into the bloodstream, causing a primary viremia, which brought the virus to its initial target organs (organ tropism), especially the spleen, lymph nodes, and the liver. This stage of infection was accompanied by the development of focal necrosis, first in the skin and draining lymph nodes in the inoculated hind limb and then in the spleen and liver. Within days there was extensive necrosis in both the spleen and liver, and rapid death. However, this was not the entire pathogenetic sequence because, to complete the viral life cycle, shedding and infection of the next host had to be explained. Fenner found that the virus produced in the target organs—that is, the spleen and liver—caused a secondary viremia that disseminated virus to the skin and mucosal surfaces. Infection in the skin caused a macular and papular rash from which large amounts of virus were shed, leading to contact exposure of other mice. Fenner’s studies with ectromelia virus stimulated similar studies that have defined the pathogenesis of many other viral infections.
Figure 3.3.
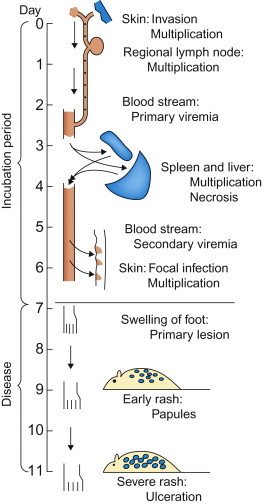
Frank Fenner’s classic study of the pathogenesis of ectromelia (mousepox) viral infection. This was the first study ever done using serial (daily) titration of the viral content of organs and tissues, and the model for many studies that have since advanced knowledge of the pathogenesis of systemic viral infections.
[From F. Fenner. Mousepox (infectious ectromelia of mice): a review. J. Immunol.63, 341–373 (1949), with permission.]
Local Spread on Epithelial Surfaces
Viruses first replicate in epithelial cells at the site of entry and produce a localized infection, often with associated virus shedding directly into the environment from these sites. The spread of infection along epithelial surfaces occurs by the sequential infection of neighboring cells, which, depending on the individual virus, may or may not precede spread into the adjacent subepithelial tissues and beyond.
In the skin, papillomaviruses and poxviruses such as orf virus remain confined to the epidermis, where they induce localized proliferative lesions, whereas other poxviruses such as lumpy skin disease virus, spread widely after cutaneous infection. Viruses that enter the body via the respiratory or intestinal tracts can quickly cause extensive infection of the mucosal epithelium, thus diseases associated with these infections progress rapidly after a short incubation period. In mammals, there is little or no productive invasion of subepithelial tissues of the respiratory tract after most influenza and parainfluenza virus infections, or in the intestinal tract following most rotavirus and coronavirus infections. Although these viruses apparently enter lymphatics and thus have the potential to spread, they usually do not do so, because appropriate viral receptors or other permissive cellular factors such as cleavage-activating proteases or transcription enhancers are restricted to epithelial cells, or because of other physiological constraints.
Restriction of viral infection to an epithelial surface should never be equated with any lack of virulence or disease severity. Although localized, injury to the intestinal mucosa caused by rotaviruses and coronaviruses can result in severe and, especially in neonates, even fatal diarrhea. Similarly, influenza virus infection can cause extensive injury in the lungs, leading to acute respiratory distress syndrome and possibly death.
Subepithelial Invasion and Lymphatic Spread
A variety of factors probably contribute to the ability of some viruses to breach the epithelial barrier and to invade the subepithelial tissues, including (1) targeted migration of virus within phagocytic leukocytes, specifically dendritic cells and macrophages, and (2) directional shedding of viruses from the infected epithelium (see Chapter 2). Dendritic cells are abundant in the skin and at all mucosal surfaces, where they constitute a critical first line of immune defense, both innate and adaptive (see Chapter 4). Migratory dendritic cells (such as Langerhans cells in the skin) “traffic” from epithelial surfaces to the adjacent (draining), regional lymph node, and infection of these cells may be responsible for the initial spread of alphaviruses, bluetongue and other orbiviruses, and feline and simian human immunodeficiency viruses, amongst many others. Directional release of virus into the lumen of the respiratory or intestinal tracts facilitates local spread to the surface of contiguous epithelial cells and immediate shedding into the environment, whereas shedding from the basolateral cell surface of epithelial cells potentially facilitates invasion of subepithelial tissues and subsequent virus dissemination via lymphatics, blood vessels, or nerves.
Many viruses that are widely disseminated in the body following infection at epithelial surfaces are first carried to the adjacent (local) lymph nodes through the afferent lymphatic drainage (Figure 3.4 ). Within the draining lymph node, virions may be inactivated and processed by macrophages and dendritic cells so that their component antigens are presented to adjacent lymphocytes to stimulate adaptive immune responses (see Chapter 4). Some viruses, however, replicate efficiently in macrophages (e.g., many retroviruses, orbiviruses, canine distemper virus and other morbilliviruses, arteriviruses such as porcine reproductive and respiratory syndrome virus, and some herpesviruses), and/or in dendritic cells and lymphocytes. From the regional lymph node, virus can spread to the bloodstream in efferent lymph, and then quickly be disseminated throughout the body, either within cells or as cell-free virions. Blood-filtering organs, including the lung, liver, and spleen, are often target organs (tropism) of viruses that cause disseminated infections.
Figure 3.4.
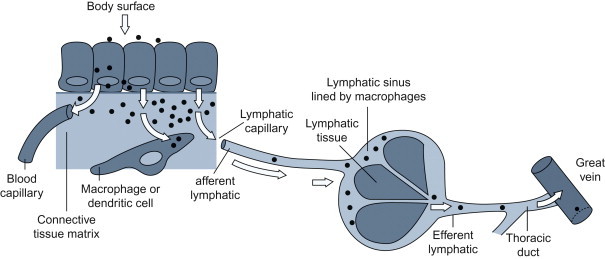
Subepithelial invasion and lymphatic spread of infection.
(Adapted from the work of C. A. Mims.)
Normally, there is a local inflammatory response at the site of viral invasion, the severity of which reflects the extent of tissue damage. Inflammation leads to characteristic alterations in the flow and permeability of local blood vessels, as well as leukocyte trafficking and activity; some viruses take advantage of these events to infect cells that participate in this inflammatory response, which in turn can facilitate spread of these viruses either locally or systemically. Local inflammation may be especially important to the pathogenesis of arthropod-transmitted viruses because of the marked reaction at the site of virus inoculation induced by the bite of the arthropod vector.
Spread via the Bloodstream: Viremia
The blood is the most effective vehicle for rapid spread of virus through the body. Initial entry of virus into the blood after infection is designated primary viremia, which, although usually inapparent clinically, leads to the seeding of distant organs—as exemplified in Fenner’s pioneering studies of ectromelia virus infection. Virus replication in major target organs leads to the sustained production of much higher concentrations of virus, producing a secondary viremia (Figure 3.5 ) and infection in yet other parts of the body that ultimately results in the clinical manifestations of the associated disease.
Figure 3.5.
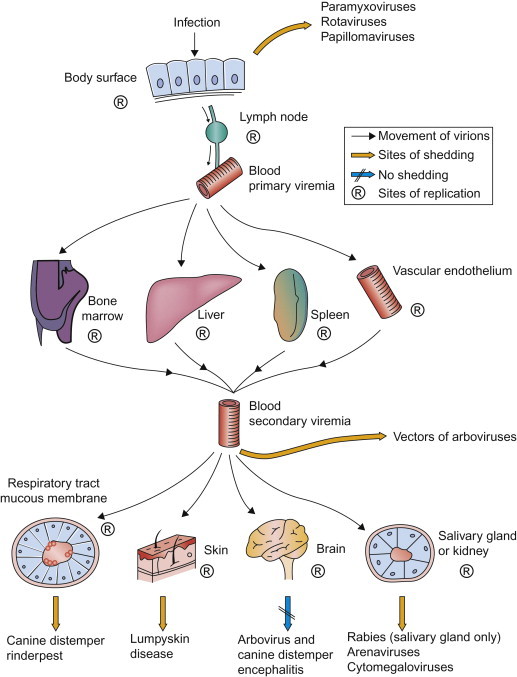
The role of viremia in the spread of viruses through the body, indicating sites of replication and important routes of shedding of various viruses.
(Adapted from the work of C. A. Mims and D. O. White.)
In the blood, virions may circulate free in the plasma or may be contained in, or adsorbed to, leukocytes, platelets, or erythrocytes. Parvoviruses, enteroviruses, togaviruses, and flaviviruses typically circulate free in the plasma. Viruses carried in leukocytes, generally lymphocytes or monocytes, are often not cleared as readily or in the same way as viruses that circulate in the plasma. Specifically, cell-associated viruses may be protected from antibodies and other plasma components, and they can be carried as “passengers” when leukocytes that harbor the virus emigrate into tissues. Individual viruses exhibit tropism to different leukocyte populations; thus monocyte-associated viremia is characteristic of canine distemper, whereas lymphocyte-associated viremia is a feature of Marek’s disease and bovine leukosis. Erythrocyte-associated viremia is characteristic of infections caused by African swine fever virus and bluetongue virus. The association of bluetongue virus with erythrocytes facilitates both prolonged viremia by delaying immune clearance, and infection of the hematophagous (blood feeding) Culicoides midges that serve as biological vectors of the virus. A substantial number of viruses, including equine infectious anemia virus, bovine viral diarrhea virus, and bluetongue virus, associate with platelets during viremia—an interaction that might facilitate infection of endothelial cells. Neutrophils, like platelets, have a very short lifespan; neutrophils also possess powerful antimicrobial mechanisms and they are rarely infected, although they may contain phagocytosed virions.
Virions circulating in the blood are removed continuously by macrophages, thus viremia can typically be maintained only if there is a continuing introduction of virus into the blood from infected tissues or if clearance by tissue macrophages is impaired. Although circulating leukocytes can themselves constitute a site for virus replication, viremia is usually maintained by infection of the parenchymal cells of target organs such as the liver, spleen, lymph nodes, and bone marrow. In some infections, such as African horse sickness virus and equine arteritis virus infections of horses, viremia is largely maintained by the infection of endothelial cells and/or macrophages and dendritic cells. Striated and smooth muscle may also be an important site of replication of some certain viruses.
There is a general correlation between the magnitude of viremia generated by blood-borne viruses and their capacity to invade target tissues, thus the failure of some attenuated vaccine viruses to generate a significant viremia may account for their lack of tissue invasiveness. Certain neurotropic viruses are virulent after intracerebral inoculation, but avirulent when given peripherally, because they do not attain viremia titers sufficient to facilitate invasion of the nervous system. The capacity to produce viremia and the capacity to invade tissues from the bloodstream are thus two different properties of a virus. For example, some strains of Semliki Forest virus (and certain other alphaviruses) have lost the capacity to invade the central nervous system while retaining the capacity to generate a viremia equivalent in duration and magnitude to that produced by neuroinvasive strains.
Viruses that circulate in blood, especially those that circulate free in plasma, encounter, amongst many others, two cell types that exert especially important roles in determining the subsequent pathogenesis of infection: macrophages and vascular endothelial cells.
Virus Interactions with Macrophages
Macrophages are bone marrow-derived mononuclear phagocytic cells that are present in all compartments of the body, including those that occur “free” in plasma (monocytes) or the pulmonary airspaces (alveolar macrophages), and those that are present in all tissues, including the subepithelial connective tissues beneath mucosal surfaces, fixed tissue macrophages such as osteoclasts (bone), microglia (central nervous system), and those that line the sinusoids of the lymph nodes and liver, spleen, bone marrow, etc. Together with dendritic cells, macrophages have a critical role in antigen processing and presentation to other immune cells that is central to the initiation of adaptive immune responses (see Chapter 4). They also initiate innate immune responses because of their ability to detect the presence of pathogen-associated molecular patterns (PAMPs) through specific receptors—for example, Toll-like receptors. Macrophages are heterogeneous in their functional activity, which can vary markedly depending on their location and state of activation; even in a given tissue or site there are subpopulations of macrophages that differ in phagocytic activity and in susceptibility to viral infection. The various kinds of interactions that can occur between macrophages and virions may be described in relation to Kupffer cells, the macrophages that line the sinusoids of the liver, as shown in Figure 3.6 . Not shown in this model is tissue invasion via carriage of virus inside monocytes/macrophages that emigrate through the walls of small blood vessels—sometimes referred to as the “Trojan Horse” mechanism of invasion, which is especially important in the pathogenesis of lentivirus infections.
Figure 3.6.
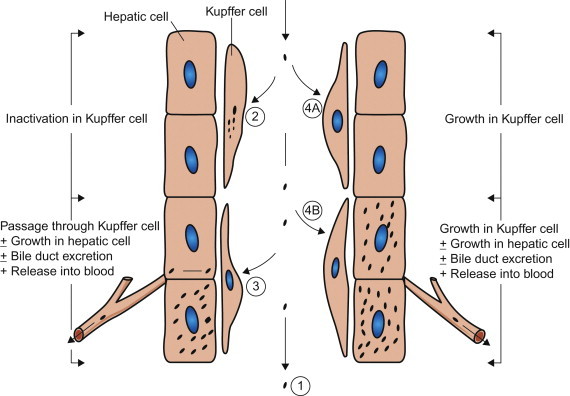
Types of interaction between viruses and macrophages, exemplified by Kupffer cells, the macrophages that line the sinusoids in the liver. (1) Macrophages may fail to phagocytose virions; e.g., in Venezuelan equine encephalitis virus infection this is an important factor favoring prolonged high viremia. (2) Virions may be phagocytosed and destroyed: because the macrophage system is so efficient, viremia can be maintained only if virions enter the blood as fast as they are removed. (3) Virions may be phagocytosed and then transferred passively to adjacent cells (hepatocytes in the liver); e.g., in Rift Valley fever virus infection, the virus replicates in hepatocytes and causes severe hepatitis—the virus produced in the liver sustains the high viremia. (4) Virions may be phagocytosed by macrophages and then may replicate in them: (4A) with some viruses, such as lactate dehydrogenase elevating virus in mice, only macrophages are infected and progeny from that infection are the source of the extremely high viremia; (4B) more commonly, as in infectious canine hepatitis, the virus replicates in both macrophages and hepatocytes, producing severe hepatitis.
(Adapted from the work of C. A. Mims and D. O. White.)
Differences in virus–macrophage interactions may account for differences in the virulence of individual strains of the same virus, and differences in host resistance. Although macrophages are inherently efficient phagocytes, this capacity is even further enhanced after their activation by certain microbial products and cytokines such as interferon-γ. Macrophages also have Fc receptors and C3 receptors that further augment their ability to ingest opsonized virions, specifically those virions that are coated with antibody or complement molecules. Viruses in many families are capable of replicating in macrophages, thus opsonization of virions by antibody can actually facilitate antibody-mediated enhancement of infection, which may be a major pathogenetic factor in human dengue and several retrovirus infections.
Viral infection can itself lead to transcriptional activation of macrophages and dendritic cells, with production of inflammatory and vasoactive mediators such as tissue necrosis factor that contribute to the pathogenesis of viral diseases, particularly hemorrhagic viral fevers such as Ebola and bluetongue.
Virus Interactions with Vascular Endothelial Cells
The vascular endothelium with its basement membrane and tight cell junctions constitutes the blood–tissue interface and a barrier for particles such as virions. Parenchymal invasion by circulating virions depends on crossing this barrier, often in capillaries and venules, where blood flow is slowest and the vascular wall is thinnest. Virions may move passively between or through endothelial cells and the basement membrane of small vessels, be carried within infected leukocytes (Trojan horse mechanism), or infect endothelial cells and “grow” their way through this barrier, with infection of the luminal aspect of the cell and release from the basal aspect. This subject has been studied most intensively in relation to viral invasion of the central nervous system, but it also applies to secondary invasion of many tissues during generalized infections.
Infection of endothelial cells is also important to the pathogenesis of viral diseases characterized by vascular injury that results in widespread hemorrhage and/or edema, the so-called hemorrhagic viral fevers. Virus-induced endothelial injury leads to coagulation and vascular thrombosis and, if widespread, disseminated intravascular coagulation (DIC). However, it is likely that inflammatory and vasoactive mediators produced by virus-infected macrophages and dendritic cells, such as tissue necrosis factor, also contribute to the pathogenesis of vascular injury in hemorrhagic viral fevers.
Spread via Nerves
Although infection of the central nervous system can occur after hematogenous spread, invasion via the peripheral nerves is also an important route of infection—for example, in rabies, Borna disease, and several alphaherpesvirus infections (e.g., B virus encephalitis, pseudorabies, and bovine herpesvirus 5 encephalitis). Herpesvirus capsids travel to the central nervous system in axon cytoplasm and, while doing so, also sequentially infect the Schwann cells of the nerve sheath. Rabies virus and Borna disease virus also travel to the central nervous system in axon cytoplasm, but usually do not infect the nerve sheath. Sensory, motor, and autonomic nerves may be involved in the neural spread of these viruses. As these viruses move centripetally, they must cross cell–cell junctions. Rabies virus and pseudorabies virus are also known to cross at synaptic junctions (Figure 3.7 ).
Figure 3.7.
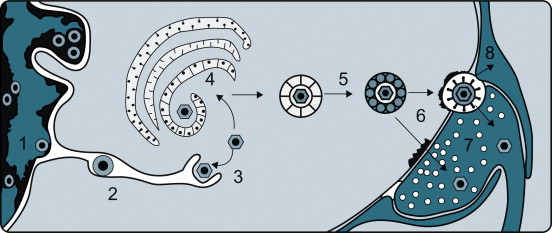
Events leading to the passage of pseudorabies virus across the junction between nerve cells on its centripetal intra-axonal transit to the brain. (1) Virions replicate in the nucleus of a peripheral nerve cell, acquiring an envelope as they bud from the inner lamella of the nuclear envelope. (2) Virions traverse the endoplasmic reticulum. (3) Virions are subsequently released into the cytoplasm after a fusion event between the virion envelope and endoplasmic reticulum membrane. (4) Virions acquire another envelope at the Golgi apparatus. (5) Virions are transported across the cytoplasm in vacuoles. (6) Virions enter the next neuron by fusion of the viral envelope and plasma membrane at a synaptic terminus. (7) Virions, now without their envelope, are carried centrally by retrograde axoplasmic flow, reaching the cell body and nucleus of the neuron, where further replication occurs. The process continues, eventually bringing the virus to the brain, where necrotizing encephalitis follows. (8) Some virions invade and replicate in the Schwann cells of the myelin sheaths surrounding neurons, thereby amplifying the amount of virus available to invade neurons.
[From J. P. Card, L. Rinaman, R. B. Lynn, B. H. Lee, R. P. Meade, R. R. Miselis, and L. W. Enquit. Pseudorabies virus infection of the rat central nervous system: ultrastructural characterization of virus replication, transport and pathogenesis. J. Neurosci.13, 2515–2539 (1993), with permission.]
In addition to passing centripetally from the body surface to the sensory ganglia and from there to the brain, herpesviruses can move through axons centrifugally from ganglia to the skin or mucous membranes. This is the same phenomenon that occurs after reactivation of latent herpesvirus infections and in the production of recrudescent epithelial lesions.
Rabies virus, Borna disease virus, respiratory mouse hepatitis virus, some togaviruses, and certain other viruses are able to use olfactory nerve endings in the nares as sites of entry. They gain entry in the special sensory endings of the olfactory neuroepithelial cells, cause local infection and progeny virus (or subviral entities containing the viral genome) then travel in axoplasm of olfactory nerves directly to the olfactory bulb of the brain.
Mechanisms of Virus Shedding
Shedding of infectious virions is crucial to the maintenance of infection in populations (see Chapter 6). For viruses that replicate only at epithelial surfaces, exit of infectious virions usually occurs from the same organ system involved in virus entry (e.g., the respiratory or gastrointestinal system; Figure 3.2). In generalized viral infections, shedding can occur from a variety of sites (Figure 3.5), and some viruses are shed from several sites. The amount of virus shed in an excretion or secretion is important in relation to transmission. Very low concentrations may be irrelevant unless very large volumes of infected material are involved; however, some viruses occur in such high concentrations that a minute quantity of virus-laden secretion or excretion can readily lead to transmission to the next animal host. Enteric viruses are in general more resistant to inactivation by environmental conditions than respiratory viruses; especially when suspended in water, such viruses can persist for some time.
Viruses such as influenza and the pneumoviruses that typically cause localized infection and injury of the respiratory tract are shed in mucus and are expelled from the respiratory tract during coughing or sneezing. Viruses are also shed from the respiratory tract in several systemic infections. Enteric viruses such as rotaviruses are shed in the feces, and the more voluminous the fluid output the greater is the environmental contamination they cause. A few viruses are shed into the oral cavity from infected salivary glands (e.g., rabies virus and cytomegaloviruses) or from the lungs or nasal mucosa during infection of the respiratory system. Salivary spread depends on activities such as licking, nuzzling, grooming, or biting. Virus shedding in saliva may continue during convalescence or recurrently thereafter, especially with herpesviruses.
The skin is an important source of virus in diseases in which transmission is by direct contact or via small abrasions: papillomaviruses and some poxviruses and herpesviruses employ this mode of transmission. Although skin lesions are produced in several generalized diseases, in only a few is virus actually shed from the skin lesions. However, in vesicular diseases such as foot-and-mouth disease, vesicular stomatitis, and swine vesicular disease, the causative viruses are produced in great quantities in vesicles within the mucosa and skin of affected animals; virus is shed from these lesions after the vesicles rupture. Localization of virus in the feather follicles is important in the shedding of Marek’s disease virus by infected chickens.
Urine, like feces, tends to contaminate food sources and the environment. A number of viruses (e.g., infectious canine hepatitis virus, foot-and-mouth disease viruses, and arenaviruses) replicate in tubular epithelial cells in the kidney and are shed in urine. Viruria is prolonged and common in equine rhinitis A virus infection and life-long in arenavirus infections of reservoir host rodents; it constitutes the principal mode of contamination of the environment by these viruses.
Several viruses that cause important diseases of animals are shed in the semen and are transmitted during coitus; for example, equine arteritis virus can be shed for months or years in the semen of apparently healthy carrier stallions, long after virus has been cleared from other tissues. Similarly, viruses that replicate in the mammary gland are excreted in milk, which may serve as a route of transmission—for example, caprine arthritis–encephalitis virus, mouse mammary tumor virus, and some of the tick-borne flaviviruses. In salmonid fish, the fluid surrounding eggs oviposited during spawning may contain high concentrations of viruses such as infectious hemopoietic necrosis virus, which is an important mode of virus transmission in both hatchery and wild fish populations.
Although not “shedding” in the usual sense of the word, blood and tissues from slaughtered animals must be considered important sources of viral contagion. Virus-laden blood is also the basis for transmission when it contaminates needles and other equipment used by veterinarians and others treating or handling sick animals. Similarly, the use of virus-contaminated fetal bovine serum can result in similar contamination of biological products.
Virus Infection Without Shedding
Many sites of virus replication might be considered “dead ends” from the perspective of natural spread; however, replication at these sites can indirectly facilitate virus transmission as, for instance, carnivores and omnivores may be infected by consuming virus-laden meat or tissues. Similarly, classical swine fever (hog cholera), African swine fever, and vesicular exanthema of swine viruses have been previously translocated to different regions and countries through feeding garbage containing contaminated pork scraps. The epizootic of bovine spongiform encephalopathy (mad cow disease) in the United Kingdom was spread widely amongst cattle by the feeding of contaminated meat and bone meal containing bovine offal that included nervous tissue.
Many retroviruses are not shed at all, but instead are transmitted directly in the germ plasm or by infection of the avian egg or developing mammalian embryo. Despite the lack of horizontal transmission, these vertically transmitted viruses accomplish the same ends as those shed into the environment—that is, transmission to new hosts and perpetuation in nature.
Mechanisms of Viral Injury and Disease
The outcome of a viral infection is dependent on the ability of the infecting virus to infect, colonize and then cause tissue- or organ-specific injury in the host, in addition to its ability to avoid clearance by the host’s innate and adaptive immune responses (see Chapter 4). After successful infection, viruses can cause disease in their hosts either by direct injury to target cells or by inducing immune or inflammatory responses that themselves mediate tissue injury and cause disease.
Virus–Cell Interactions
An appreciation of the potential adverse outcomes of infection in the individual cell is key to understanding the impact of viral infection in complex tissues and organs—and, indeed, the whole host animal. As described in the preceding section, cellular tropism of viruses is determined by the presence of appropriate cellular receptors and, frequently, cell-type specific transcription factors (enhancers). Viruses typically encode genes that modulate host-cell functions for their own benefit and, of course, the host has elaborate innate defenses to restrict viral functions. Thus the viral and cellular factors that influence the outcome of infection are often in delicate balance and easily shifted one way or the other.
Virus infection can cause a wide variety of potentially deleterious changes in the many different kinds of cells that occur in the animal host. The disruption of cellular functions, the induction of cell death or transformation, or the activation of an inappropriate immune response are all potentially manifested as disease by the infected host (Figure 3.8 ). Although virus-induced changes at the cellular, subcellular, and molecular levels are most commonly studied in cultured cells, additional insight has been gained through the use of explant and organ cultures, transplantation of infected cells and tissues back into experimental animals, and the extensive recent use of genetically modified laboratory animals in conjunction with molecular clones of individual viruses.
Figure 3.8.
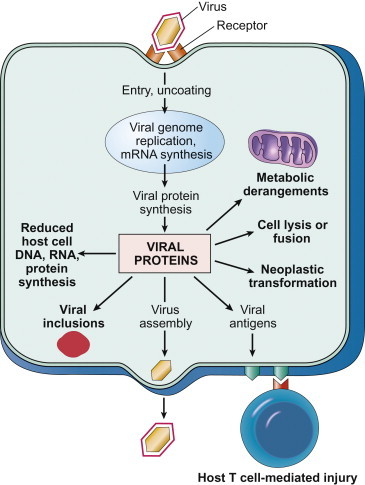
Potential mechanisms by which viruses cause injury to cells.
[From Robbins & Cotran Pathologic Basis of Disease, V. Kumar, A. K. Abbas, N. Fausto, J. Aster, 8th ed., p. 343. Copyright © Saunders/Elsevier (2010), with permission.]
Types of Virus–Cell Interaction
Viral infections may be cytocidal (cytolytic, cytopathic) or non-cytocidal, and productive or non-productive (abortive)—that is, not all infections lead to cell death or the production and release of new virions. However, critical changes can occur in virus-infected cells regardless of whether the infection is productive or not. Certain kinds of cells are permissive—that is, they support complete replication of a particular virus—whereas others are non-permissive—that is, virus replication may be blocked at any point from virus attachment through to the final stages of virion assembly and release, and this outcome can be determined by either cellular factors, such as the presence of specific proteolytic enzymes or cellular transcription enhancers, or viral factors, such as the deletion in defective interfering particles of key genes required for virus replication.
Some of the most important of all non-productive virus–cell interactions are those associated with persistent infections or latent viral infections, which will be described in a subsequent section. The term persistent infection simply describes an infection that lasts a long time, considerably beyond the interval when infection normally would be expected to be cleared. The term latent infection describes a specific type of persistent infection that “exists but is not exhibited”—that is, an infection in which infectious virions are not formed. In either case, the virus or its genome is maintained indefinitely in the cell, either by the integration of the viral nucleic acid into the host-cell DNA or by carriage of the viral nucleic acid in the form of an episome, and the infected cell survives and may divide repeatedly; in some instances persistently infected cells never release virions, whereas in others the infection may become productive when induced by an appropriate stimulus, such as the periodic reactivation and virus shedding associated with many latent herpesvirus infections. Persistent or latent infections with oncogenic viruses may also lead to cell transformation, as described later in this chapter. The various types of interaction that can occur between virus and cell are summarized in Table 3.2 and in Figure 3.8.
Table 3.2.
Types of Virus–Cell Interaction
| Type of Infection | Effects on Cell | Production of Infectious Virions | Examples |
|---|---|---|---|
| Cytocidal | Morphologic changes in cells (cytopathic effects); inhibition of protein, RNA, and DNA synthesis; cell death | Yes | Alphaherpesviruses, enteroviruses, reoviruses |
| Persistent, productive | No cytopathic effect; little metabolic disturbance; cells continue to divide; may be loss of the special functions of some differentiated cells | Yes | Pestiviruses, arenaviruses, rabies virus, most retroviruses |
| Persistent, non-productive | Usually nil | No, but virus may be induceda | Canine distemper virus in brain |
| Transformation | Alteration in cell morphology; cells can be passaged indefinitely; may produce tumors when transplanted to experimental animals | No, oncogenic DNA viruses | Polyomavirus, adenoviruses |
| Yes, oncogenic retroviruses | Murine, avian leukosis and sarcoma viruses |
By co-cultivation, irradiation, or chemical mutagens.
Cytocidal Changes in Virus-Infected Cells
Cytopathic viruses kill the cells in which they replicate, by preventing synthesis of host macromolecules (as described below), by producing degradative enzymes or toxic products, or by inducing apoptosis (see Chapter 4). After inoculation of a cytopathic virus into a monolayer of cultured cells, the first round of virus replication yields progeny virions that spread through the medium to infect both adjacent and distant cells; all cells in the culture may eventually become infected. The resulting cell damage is known as a cytopathic effect (CPE). Cytopathic effects can usually be observed by low-power light microscopy of unstained cell cultures (Figure 3.9 ). The nature of the cytopathic effect is often characteristic of the particular virus involved, and is therefore an important preliminary clue in the identification of clinical isolates in the diagnostic laboratory (see Chapters 2 and 5Chapter 2Chapter 4Chapter 5).
Figure 3.9.
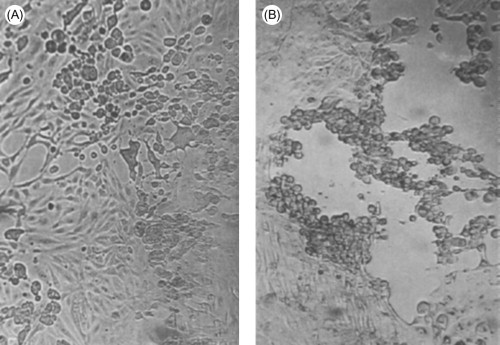
Cytopathic effects produced by different viruses. The cell monolayers are shown as they would normally be viewed in the laboratory, unfixed and unstained. (A) Typical cytopathology of an enterovirus: rapid rounding of cells, progressing to complete cell lysis. (B) Typical cytopathology of a herpesvirus: focal areas of swollen rounded cells. Magnification: ×60.
(Courtesy of I. Jack.)
So many pathophysiologic changes occur in cells infected with cytopathic viruses that the death of the cell usually cannot be attributed to any particular event; rather, cell death may be the final result of the cumulative action of many insults. Nevertheless, a variety of specific mechanisms have been identified that might in the future be potentially targeted for therapeutic intervention. General mechanisms of virus-induced cell injury and death (Figure 3.8) include:
Inhibition of Host-Cell Nucleic Acid Synthesis is an inevitable consequence of viral inhibition of host-cell protein synthesis and its effect on the cellular machinery of DNA replication. Some viruses, especially the large DNA viruses, use specific mechanisms to promote their own synthetic processes through production of virus-encoded regulatory proteins.
Inhibition of Host-Cell RNA Transcription occurs during replication of viruses in several different families, including poxviruses, rhabdoviruses, reoviruses, paramyxoviruses, and picornaviruses. In some instances, this inhibition may be the indirect consequence of viral effects on host-cell protein synthesis that decrease the availability of transcription factors required for RNA polymerase activity. Certain viruses encode specific transcription factors to regulate the expression of their own genes, and these factors sometimes modulate the expression of cellular genes as well. For example, herpesviruses encode proteins that bind directly to specific viral DNA sequences, thereby regulating the transcription of viral genes.
Inhibition of Processing of Host-Cell Messenger RNAs occurs during replication of vesicular stomatitis viruses, influenza viruses, and herpesviruses, through interference with the splicing of cellular primary mRNA transcripts that are needed to form mature mRNAs. In some instances, spliceosomes are formed, but subsequent catalytic steps are inhibited. For example, a protein synthesized in herpesvirus-infected cells suppresses RNA splicing and leads to reduced amounts of cellular mRNAs and the accumulation of primary mRNA transcripts.
Inhibition of Host-Cell Protein Synthesis while viral protein synthesis continues is a characteristic of many viral infections. This shutdown is particularly rapid and profound in picornavirus infections, but it is also pronounced in togavirus, influenzavirus, rhabdovirus, poxvirus, and herpesvirus infections. With some other viruses, the shutdown occurs late in the course of infection and is more gradual, whereas with non-cytocidal viruses, such as pestiviruses, arenaviruses, and retroviruses, there is no dramatic inhibition of host-cell protein synthesis, and no cell death. The mechanisms underlying the shutdown of host-cell protein synthesis are varied, including those just described in addition to the production of viral enzymes that degrade cellular mRNAs, the production of factors that bind to ribosomes and inhibit cellular mRNA translation, and the alteration of the intracellular ionic environment favoring the translation of viral mRNAs over cellular mRNAs. Most importantly, some viral mRNAs simply outcompete cellular mRNAs for cellular translation machinery by mass action—the large excess of viral mRNA outcompetes cellular mRNA for host ribosomes. Viral proteins may also inhibit the processing and transport of cellular proteins from the endoplasmic reticulum, and this inhibition may lead to their degradation. This effect is seen in lentivirus and adenovirus infections.
Cytopathic Effects of “Toxic” Viral Proteins reflect the accumulation of large amounts of various viral components in the cell late in infection. It was previously believed that cytopathic effect was simply a consequence of the intrinsic toxicity of these proteins, but most cell damage probably represents the supervening of virus replication events on cellular events. Hence, the list of “toxic proteins” has been shortened, but some remain. For example, the toxicity of adenovirus penton and fiber proteins appears to be direct and independent of adenovirus replication.
Interference with Cellular Membrane Function can affect the participation of cellular membranes in many phases of virus replication, from virus attachment and entry, to the formation of replication complexes, to virion assembly. Viruses may alter plasma membrane permeability, affect ion exchange and membrane potential, or induce the synthesis of new intracellular membranes or the rearrangement of previously existing ones. For example, a generalized increase in membrane permeability occurs early during picornavirus, alphavirus, reovirus, rhabdovirus, and adenovirus infections.
Enveloped viruses specifically direct the insertion of their surface glycoproteins, including fusion proteins, into host-cell membranes as part of their budding process, often leading to membrane fusion and syncytium formation. Syncytia are a conspicuous feature of infection of cell monolayers by lentiviruses, coronaviruses, paramyxo-viruses, respiroviruses, morbilliviruses, pneumoviruses, henipaviruses and some herpesviruses, which result from the fusion of an infected cell with neighboring infected or uninfected cells (Figure 3.10 ). Such multinucleate syncytia (syn. multinucleated giant cells) may also occur in the tissues of animals infected with these viruses; for example, in horses infected with Hendra virus and cattle infected with respiratory syncytial virus. Syncytia may represent an important mechanism of spread of viruses in tissues: fusion bridges may allow subviral entities, such as viral nucleocapsids and nucleic acids, to spread while avoiding host defenses. Cell membrane fusion is mediated by viral fusion proteins or fusion domains on other viral surface proteins. For example, the fusion activity of influenza viruses is carried on the hemagglutinin spikes, whereas the fusion activity of paramyxoviruses such as parainfluenza virus 3 is carried on separate spikes composed of fusion (F) protein. At high multiplicity of infection, paramyxoviruses may cause a rapid fusion of cultured cells without any requirement for virus replication; this phenomenon occurs simply as a result of the action of fusion protein activity of input virions as they interact with plasma membranes.
Figure 3.10.
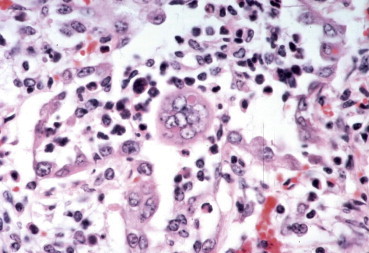
Syncytial cell with an intracytoplasmic inclusion in the lung of a calf infected with bovine respiratory syncytial virus.
(Courtesy of M. Anderson, University of California, Davis.)
Cells in monolayer cultures infected with influenza viruses, paramyxoviruses, and togaviruses, all of which bud from the plasma membrane, acquire the ability to adsorb erythrocytes. This phenomenon, known as hemadsorption (Figure 3.11 ), is the result of incorporation of viral spike glycoprotein into the plasma membrane of infected cells, which then serves as a receptor for ligands on the surface of erythrocytes. The same glycoprotein spikes are responsible for hemagglutination in vitro—that is, the agglutination of erythrocytes. Although hemadsorption and hemagglutination are not known to play a part in the pathogenesis of viral diseases, both phenomena are used in laboratory diagnostics (see Chapter 5).
Figure 3.11.
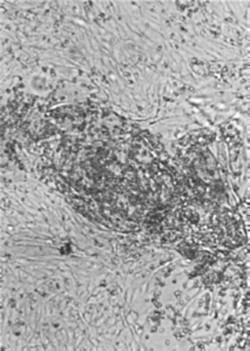
Hemadsorption: erythrocytes adsorb to infected cells that have incorporated hemagglutinin into the plasma membrane. The cell monolayers are shown as they would normally be viewed in the laboratory, unfixed and unstained. Magnification: ×60.
(Courtesy of I. Jack.)
Viral proteins (antigens) inserted into the host-cell plasma membrane may also constitute targets for specific humoral and cellular immune responses that cause the lysis of the cell. This may happen before significant progeny virus is produced, thus slowing or arresting the progress of infection and hastening recovery (see Chapter 4). Alternatively, in some instances the immune response may cause immune-mediated tissue injury and disease. Viral antigens may also be incorporated in the membrane of cells transformed by viruses, and play an important role in immune-mediated resolution, or regression—of viral papillomas, for example.
Changes in cell shape are characteristic of many viral infections of cultured cells. Such changes are caused by damage to the cytoskeleton, which is made up of several filament systems, including microfilaments (e.g., actin), intermediate filaments (e.g., vimentin), and microtubules (e.g., tubulin). The cytoskeleton is responsible for the structural integrity of the cell, for the transport of organelles through the cell, and for certain cell motility activities. Particular viruses may damage specific filament systems: for example, canine distemper virus, vesicular stomatitis viruses, vaccinia virus, and herpesviruses cause a depolymerization of actin-containing microfilaments, and enteroviruses induce extensive damage to microtubules. Such damage contributes to the drastic cytopathic changes that precede cell lysis in many infections. The elements of the cytoskeleton are also employed by many viruses in the course of their replication: in virus entry, in the formation of replication complexes and assembly sites, and in virion release.
Non-Cytocidal Changes in Virus-Infected Cells
Non-cytocidal viruses usually do not kill the cells in which they replicate. On the contrary, they often cause persistent infection during which infected cells produce and release virions but overall cellular metabolism is little affected. In many instances, infected cells even continue to grow and divide. This type of interaction can occur in cells infected with several kinds of RNA viruses, notably pestiviruses, arenaviruses, retroviruses, and some paramyxoviruses. Nevertheless, with few exceptions (e.g., some retroviruses), there are slowly progressive changes that ultimately lead to cell death. In the host animal, cell replacement occurs so rapidly in most organs and tissues that the slow fallout of cells as a result of persistent infection may have no effect on overall function, whereas terminally differentiated cells such as neurons, once destroyed, are not replaced, and persistently infected differentiated cells may lose their capacity to carry out specialized functions.
Viruses such as the pestiviruses, arenaviruses, Bornavirus, and retroviruses that do not shut down host-cell protein, RNA, or DNA synthesis and that do not rapidly kill their host cells, can produce important pathophysiologic changes in their hosts by affecting crucial functions that are associated neither with the integrity of cells nor their basic housekeeping functions. Damage to the specialized functions of differentiated cells may still affect complex regulatory, homeostatic, and metabolic functions, including those of the central nervous system, endocrine glands, and immune system.
Ultrastructural Changes in Virus-Infected Cells
Electron microscopy is useful for evaluation of changes in virus-infected cells. Early changes in cell structure often are dominated by proliferation of various cell membranes: for example, herpesviruses cause increased synthesis, even reduplication, of nuclear membranes; flaviviruses cause proliferation of the endoplasmic reticulum; picornaviruses and caliciviruses cause a distinctive proliferation of vesicles in the cytoplasm; and many retroviruses cause peculiar fusions of cytoplasmic membranes. Other ultrastructural changes that are prominent in many viral infections include disruption of cytoskeletal elements, mitochondrial damage, and changes in the density of the cytosol. Late in the course of infection, many cytolytic viruses cause nuclear, organelle, and cytoplasmic rarefaction and/or condensation, with terminal loss of host-cell membrane integrity. In many instances the inevitability of cell death is obvious, but in others host-cell functional loss is subtle and cannot be attributed to particular ultrastructural changes. In non-cytolytic infections, most functional losses cannot be attributed to damage that is morphologically evident. Specific examples reflecting the range of host-cell changes occurring in virus-infected cells are included in many of the chapters in Part II of this book.
In addition to changes directly attributable to virus replication, most virus-infected cells also show non-specific changes, very much like those induced by physical or chemical insults. The most common early and potentially reversible change is cloudy swelling, a change associated with increasing permeability of the cellular membranes leading to swelling of the nucleus, distention of the endoplasmic reticulum and mitochondria, and rarefaction of the cytoplasm. Later in the course of many viral infections the nucleus becomes condensed and shrunken, and cytoplasmic density increases. Cell destruction can be the consequence of further loss of osmotic integrity and leakage of lysosomal enzymes into the cytoplasm. This progression is consistent with the so-called common terminal pathway to cell death.
Virus-Mediated Tissue and Organ Injury
The severity of a viral disease is not necessarily correlated with the degree of cytopathology produced by the causative virus in cells in culture. Many viruses that are cytocidal in cultured cells do not produce clinical signs in vivo (e.g., many enteroviruses), whereas some that are non-cytocidal in vitro cause lethal disease in animals (e.g., retroviruses and rabies virus). Further, depending on the organ affected, cell and tissue damage can occur without producing clinical signs of disease—for example, a large number of hepatocytes (liver cells) may be destroyed in Rift Valley fever in sheep without significant clinical signs. When damage to cells does impair the function of an organ or tissue, this may be relatively insignificant in a tissue such as skeletal muscle, but potentially devastating in organs such as the heart or the brain. Likewise, virus-induced inflammation and edema are especially serious consequences in organs such as the lungs and central nervous system.
Mechanisms of Viral Infection and Injury of Target Tissues and Organs
The mechanisms by which individual viruses cause injury to their specific target organs are described in detail under individual virus families in Part II of this book, thus the objective of this section is to provide a brief overview of potential pathogenic mechanisms that viruses can use to cause injury in their target tissues.
Viral Infection of the Respiratory Tract
Viral infections of the respiratory tract are extremely common, especially in animals housed in crowded settings. Individual viruses exhibit tropism for different levels of the respiratory tract, from the nasal passages to the pulmonary airspaces (terminal airways and alveoli), but there is considerable overlap. Tropism of respiratory viruses is probably a reflection of the distribution of appropriate receptors and intracellular transcriptional enhancers, as well as physical barriers, physiological factors, and immune parameters. For example, bovine rhinoviruses replicate in the nasal passages because their replication is optimized at lower temperatures, whereas bovine respiratory syncytial virus preferentially infects epithelial cells lining the terminal airways; thus rhinoviruses may cause mild rhinitis, whereas respiratory syncytial virus is the cause of bronchiolitis and bronchointerstitial pneumonia. Some viruses cause injury to the type I or type II pneumocytes lining the alveoli, either directly or indirectly; if extensive, injury to type I pneumocytes leads to acute respiratory distress syndrome, whereas injury to type II pneumocytes delays repair and healing in the affected lung.
Influenza viruses replicate in both the nasal passages and airways of infected mammals, but influenza virus infection is typically confined to the lung because of the requirement for hemagglutinin cleavage by tissue-specific proteases. However, highly virulent influenza viruses such as the current Eurasian–African H5N1 virus can spread beyond the lungs to cause severe generalized (systemic) infection and disease. The ability of this virus to escape the lung may be related to its tropism to type I pneumocytes that line alveoli, and its ability to cause systemic disease may reflect that its hemagglutinin can be cleaved by ubi-quitous proteases that are present in many tissues. Similarly in birds, high-pathogenicity avian influenza viruses have several basic amino acids at the hemagglutinin cleavage site, which expands the range of cells capable of producing infectious virus because cleavage can be affected intracellularly by ubiquitous endopeptidase furins located in the trans-Golgi network. In contrast, the hemagglutinin protein of low pathogenicity avian influenza viruses is cleaved extracellularly by tissue-restricted proteases that are confined to the respiratory and gastrointestinal tracts (see Chapter 21).
Regardless of the level of the respiratory tree that is initially infected, viral infection typically leads to local cessation of cilial activity, focal loss of integrity of the lining mucus layer, and multifocal destruction of small numbers of epithelial cells (Figure 3.12 ). Initial injury is followed by progressive infection of epithelial cells within the mucosa, and inflammation of increasing severity, with exudation of fluid and influx of inflammatory cells. Fibrin-rich inflammatory exudate and necrotic cellular debris (degenerate neutrophils and sloughed epithelium) then accumulate in the lumen of the affected airways or passages, with subsequent obstruction and, in severe cases, increasing hypoxia and respiratory distress. The mucosa is quickly regenerated in animals that survive, and adaptive immune responses clear the infecting virus and prevent reinfection for variable periods of time (depending on the particular virus).
Figure 3.12.
(A) Avian influenza virus infection in the respiratory tract of a chicken. The normal side-by-side position of columnar epithelial cells has been replaced by cuboidal cells without cilia, several of which exhibit massive virus budding from their apical surface. Thin-section electron microscopy. Magnification: ×10,000. (B, C) Scanning electron micrographs showing desquamating cells in an influenza-virus-infected mouse trachea and the adherence of Pseudomonas aeruginosa. Bar: 2 μm. (B) Normal mouse trachea showing a single bacterium (arrow) on a serous cell. (C) Microcoliny of P. aeruginosa adhering to a residual epithelial cell on an otherwise denuded surface.
[B, C: Courtesy of P. A. Small, Jr.]
In addition to their direct adverse consequences, viral infections of the respiratory tract often predispose animals to secondary infections with bacteria, even those bacteria that constitute the normal flora in the nose and throat. This predisposition can result from interference with normal mucociliary clearance as a consequence of viral injury to the mucosa, or suppression of innate immune responses. For example, cellular expression of Toll-like receptors is depressed in the lung after influenza virus infection, and thus convalescent animals may be less able to quickly recognize and neutralize invading bacteria. This potential synergy between respiratory viruses and bacteria is compounded by overcrowding of animals as occurs during shipping and in feedlots and shelters.
Viral Infection of the Gastrointestinal Tract
Infection of the gastrointestinal tract can be acquired either by ingestion of an enteric virus (e.g., rotaviruses, coronaviruses, astroviruses, toroviruses) of which infection is confined to the gastrointestinal tract or as a consequence of generalized hematogenous spread of a systemic viral infection such as with certain parvoviruses (e.g., feline panleukopenia, canine parvovirus), pestiviruses (e.g., bovine viral diarrhea virus), and morbilliviruses (e.g., canine distemper, rinderpest). Enteric virus infections usually result in rapid onset of gastrointestinal disease after a short incubation period, whereas systemic infections have a longer incubation period and are typically accompanied by clinical signs that are not confined to dysfunction of the gastrointestinal tract.
Virus-induced diarrhea is a result of infection of the epithelial cells (enterocytes) lining the gastrointestinal mucosa. Rotaviruses, astroviruses, coronaviruses, and toroviruses characteristically infect the more mature enterocytes that line the intestinal villi, whereas parvoviruses and pestiviruses infect and destroy the immature and dividing enterocytes present in the intestinal crypts. Regardless of their site of predilection, these infections all destroy enterocytes in the gastrointestinal mucosa and so reduce its absorptive surface, leading to malabsorption diarrhea with attendant loss of both fluid and electrolytes. The pathogenesis of enteric virus infections can be even more complex than simple virus-mediated destruction of enterocytes; for example, rotaviruses produce a protein (nsp4) that itself causes secretion of fluid into the bowel (intestinal hypersecretion), even in the absence of substantial virus-mediated damage. In suckling neonates, undigested lactose from ingested milk passes through the small bowel to the large bowel, where it exerts an osmotic effect that further exacerbates fluid loss. Animals with severe diarrhea can rapidly develop pronounced dehydration, hemoconcentration, acidosis that inhibits critical enzymes and metabolic pathways, hypoglycemia, and systemic electrolyte disturbances (typically, decreased sodium and increased potassium), and diarrhea can be quickly fatal in very young or otherwise compromised animals.
Enteric virus infections generally begin in the stomach or proximal small intestine, and they then spread caudally as a “wave” that sequentially affects the jejunum, ileum, and large bowel. As the infection progresses through the bowel, absorptive cells destroyed by the infecting virus are quickly replaced by immature enterocytes from the intestinal crypts. The presence of increased numbers of these immature enterocytes contributes to malabsorption and intestinal hypersecretion (fluid and electrolyte loss). Similarly, adaptive immune responses lead to mucosal IgA and systemic IgG production in animals that survive, conferring resistance to reinfection. Enteric virus infections in neonates are frequently associated with infections by other enteric pathogens, including bacteria (e.g., enterotoxigenic or enteropathogenic Escherichia coli) and protozoa such as Cryptosporidium spp., probably because of the common factors (crowding, poor sanitation) that predispose to these infections.
Viral Infection of the Skin
In addition to being a site of initial infection, the skin may be invaded secondarily via the blood stream. Thus skin lesions that accompany viral infections can be either localized, such as papillomas, or disseminated. In animals, erythema (reddening) of the skin as a consequence of systemic viral infections is most obvious on exposed, hairless, non-pigmented areas such as the snout, ears, paws, scrotum, and udder. In addition to papillomas (warts), virus-induced lesions that commonly affect the skin of virus-infected animals are variously described as macules, papules, vesicles, and pustules. Viruses of particular families tend to produce characteristic cutaneous lesions, frequently in association with similar lesions in the oral and nasal mucosa, the teats and genitalia, and at the junction of the hooves and skin of ungulates. Vesicles are especially important cutaneous lesions, because they are characteristic of foot-and-mouth disease and other viral diseases that can mimic it, although vesicles clearly can occur in diseases that are not caused by viruses. Vesicles are essentially discrete “blisters” that result from accumulation of edema fluid within the affected epidermis, or separation of the epidermis from the underlying dermis (or mucosal epithelium from the submucosa). Vesicles rupture quickly to leave focal ulcers.
Papules are either localized (e.g., orf) or disseminated (e.g., lumpy skin disease) epithelial proliferations that are characteristic of poxvirus infections. These proliferative and raised lesions frequently become extensively encrusted with inflammatory exudate.
Virus infections that result in widespread endothelial injury in blood vessels throughout the body, including those of the subcutaneous tissues, can produce subcutaneous edema and erythema or hemorrhages in the skin and elsewhere (including the oral cavity and internal organs).
Viral Infection of the Central Nervous System
The central nervous system (brain and spinal cord) is exquisitely susceptible to serious, often fatal injury by certain viral infections. Viruses can spread from distal sites to the brain via nerves (as previously described), or via the blood. To spread from the blood, viruses first must overcome the obstacle of the blood–brain barrier formed by the endothelial lining and mesenchymal wall of blood vessels within the brain and spinal cord. It remains somewhat enigmatic as to how most viruses cross this barrier to assess the parenchyma of the central nervous system, whether by passage in virus-infected leukocytes or by active or passive transport through the vascular wall (Figure 3.13 ).
Figure 3.13.
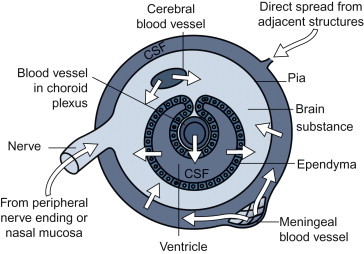
Routes of viral invasion of the central nervous system. CSF, cerebrospinal fluid.
[From Medical Microbiology, C. A. Mims, J. H. Playfair, I. M. Roitt, D. Wakelin, R. Williams. Mosby, St. Louis, MO (1993), with permission.]
Once present within the central nervous system, a number of viruses can quickly spread to cause progressive infection of neurons and/or glial cells (astrocytes, microglia, and oligodendrocytes). Lytic infections of neurons, whether caused by togaviruses, flaviviruses, herpesviruses, or other viruses, leads to encephalitis or encephalomyelitis characterized by neuronal necrosis, phagocytosis of neurons (neuronophagia), and perivascular infiltrations of inflammatory cells (perivascular cuffing). In contrast, virulent rabies virus infection of neurons is non-cytocidal and evokes little inflammatory reaction, but it is uniformly lethal for most mammalian species.
Other characteristic pathologic changes are produced by various viruses, and by prions that cause slowly progressive diseases of the central nervous system. In bovine spongiform encephalopathy in cattle and scrapie in sheep, for example, there is slowly progressive neuronal degeneration and vacuolization. In contrast, infection of glial cells in dogs with canine distemper leads to progressive demyelination.
In most cases, central nervous system infection seems to be a dead end in the natural history of viruses—shedding and transmission of most neurotropic viruses do not depend on pathogenetic events in the nervous system. There are important exceptions, however. Rabies virus infection causes behavioral changes in the host that favor transmission of the virus to other hosts. The alphaherpesviruses depend on the delivery of virus from cranial and spinal sensory ganglia to epithelial sites. Epithelial shedding, which follows virus emergence from ganglia during recrudescence, is important because it offers the opportunity for transmission long after primary lesions have resolved. The prion agent of bovine spongiform encephalopathy is iatrogenically spread by the inclusion of ruminant central nervous system tissue in meat and bone meal fed to cattle. All in all, it seems anomalous that neurotropism should be the outstanding characteristic of so many of the most notorious pathogens of animals and zoonotic pathogens of humans, and yet be the pathogenetic characteristic least related to virus perpetuation in nature, emphasizing perhaps that the irreparable damage that is of such grave consequence to the host is of such little consequence to the virus.
Viral Infection of the Hemopoietic System and Immune Effects
The hemopoietic system includes: (1) the myeloid tissues, specifically the bone marrow and cells derived from it—erythrocytes, platelets, monocytes, and granulocytes, and (2) the lymphoid tissues, which include the thymus, lymph nodes, spleen, mucosal-associated lymphoid tissues and, in birds, the cloacal bursa. As cells that populate the myeloid and lymphoid systems, including lymphocytes, dendritic cells, and cells of the mononuclear phagocytic system (monocytes and macrophages) are all derived from bone marrow (or equivalent hemopoietic tissue) precursors, it is convenient to group them together under the heading of the hemopoietic system and to dispense with obsolete terminology such as “lymphoreticular” or “reticuloendothelial” systems. Importantly, lymphocytes and mononuclear phagocytes (blood monocytes, tissue macrophages, dendritic cells) are responsible for adaptive immunity (see Chapter 4), thus viral infections of these cells can have profound effects on immunity.
Infection and damage to mononuclear phagocytes can protect an invading virus from phagocytic removal, and suppress or inhibit both the innate and adaptive immune response to it. Some of the most destructive and lethal viruses known exhibit this tropism: filoviruses, arenaviruses, hantaviruses, orbiviruses such as African horse sickness and bluetongue viruses, certain bunyaviruses such as Rift Valley fever virus, alphaviruses such as Venezuelan equine encephalitis virus, and flaviviruses such as yellow fever virus. After initial invasion, infection with these viruses begins with their uptake by dendritic cells and/or macrophages in lymphoid tissues (lymph nodes, thymus, bone marrow, Peyer’s patches, and the white pulp of the spleen). Viral infection can then spread in these tissues, frequently leading to cytolysis of adjacent lymphocytes and immune dysfunction.
Viral infections can result in either specific acquired immunodeficiency or generalized immunosuppression. A relevant example of this phenomenon is provided by infection of the cloacal bursa (bursa of Fabricius) in chickens (the site of B cell differentiation in birds) with infectious bursal disease virus, which leads to atrophy of the bursa and a severe deficiency of B lymphocytes, equivalent to bursectomy. The result is an inability of severely affected birds to develop antibody-mediated immune responses to other infectious agents, which in turn leads to an increase in susceptibility to bacterial infections such as those caused by Salmonella spp. and E. coli, and other viruses. Since the discovery of acquired immunodeficiency syndrome (AIDS) in humans and its etiologic agent, human immunodeficiency virus (HIV), similar viruses have been discovered in monkeys (simian immunodeficiency viruses), cattle (bovine immunodeficiency virus), and cats (feline immunodeficiency virus). In susceptible animals, these viruses individually can infect and destroy specific but different cells of the immune system, thereby causing immunosuppression of different types and severity.
Many other viruses (e.g., classical swine fever virus, bovine viral diarrhea virus, canine distemper virus, feline and canine parvoviruses) that cause systemic infections, especially those that infect mononuclear phagocytes and/or lymphocytes, may temporarily but globally suppress adaptive immune responses, both humoral and cell-mediated. Affected animals are predisposed to diseases caused by other infectious agents during the period of virus-induced immunosuppression, a phenomenon that can also occur following vaccination with certain live-attenuated vaccines. The immune response to unrelated antigens may be reduced or abrogated in animals undergoing such infections.
Virus-induced immunosuppression may in turn lead to enhanced virus replication, such as the reactivation of latent herpesvirus, adenovirus, or polyomavirus infections. Similarly, immunosuppression associated with administration of cytotoxic drugs or irradiation for chemotherapy or organ transplantation can predispose to recrudescence of herpesviruses and, potentially, others.
Viral Infection of the Fetus
Most viral infections of the dam have no harmful effect on the fetus, although severe infections of the dam can sometimes lead to fetal death and expulsion (abortion) in the absence of fetal infection. However, some viruses can cross the placenta to infect the fetus (Table 3.3 ). Such infections occur most commonly in young dams (such as first-calf heifers) that are exposed during pregnancy to pathogenic viruses to which they have no immunity, as a consequence of lack of either appropriate vaccination or natural infection. The outcome of fetal viral infection is dependent upon the properties (virulence and tropism) of the infecting virus, as well as the gestational age of the fetus at infection. Severe cytolytic infections of the fetus, especially in early gestation, are likely to cause fetal death and resorption or abortion, which also is dependent on the species of animal affected—abortion is especially common in those species in which pregnancy is sustained by fetal production of progesterone (such as sheep), whereas pregnancy is less likely to be terminated prematurely in multiparous species in which pregnancy is maintained by maternally derived progesterone (such as swine).
Table 3.3.
Viral Infections of the Fetus or Embryo
| Animal | Family/Genus | Virus | Syndrome |
|---|---|---|---|
| Cattle | Herpesviridae/Varicellovirus | Infectious bovine rhinotracheitis virus | Fetal death, abortion |
| Retroviridae/Deltaretrovirus | Bovine leukemia virus | Inapparent infection, leukemia | |
| Reoviridae/Orbivirus | Bluetongue virus | Fetal death, abortion, congenital defects | |
| Bunyaviridae/Bunyavirus | Akabane virus | Fetal death, abortion, stillbirth, congenital defects | |
| Flaviviridae/Pestivirus | Bovine viral diarrhea virus | Fetal death, abortion, congenital defects, inapparent infection with life-long carrier status and shedding | |
| Horses | Herpesviridae/Varicellovirus | Equine herpesvirus 1 | Fetal death, abortion, neonatal disease |
| Arteriviridae/Arterivirus | Equine arteritis virus | Fetal death, abortion | |
| Swine | Herpesviridae/Varicellovirus | Pseudorabies virus | Fetal death, abortion |
| Parvoviridae/Parvovirus | Swine parvovirus | Fetal death, abortion, mummification, stillbirth, infertility | |
| Flaviviridae/Flavivirus | Japanese encephalitis virus | Fetal death, abortion | |
| Flaviviridae/Pestivirus | Classical swine fever (hog cholera) virus | Fetal death, abortion, congenital defects, inapparent infection with life-long carrier status and shedding | |
| Sheep | Reoviridae/Orbivirus | Bluetongue virus | Fetal death, abortion, congenital defects |
| Bunyaviridae/Phlebovirus | Rift Valley fever virus | Fetal death, abortion | |
| Bunyaviridae/Nairovirus | Nairobi sheep disease virus | Fetal death, abortion | |
| Flaviridae/Pestivirus | Border disease virus | Congenital defects | |
| Dogs | Herpesviridae/Varicellovirus | Canine herpesvirus | Perinatal death |
| Cats | Parvoviridae/Parvovirus | Feline panleukopenia virus | Cerebellar hypoplasia |
| Retroviridae/Gammaretrovirus | Feline leukemia virus | Inapparent infection, leukemia, fetal death | |
| Mice | Parvoviridae/Parvovirus | Rat virus | Fetal death |
| Arenaviridae/Arenavirus | Lymphocytic choriomeningitis virus | Inapparent infection, with life-long carrier status and shedding | |
| Chicken | Picornaviridae/Enterovirus | Avian encephalomyelitis virus | Congenital defects, fetal death |
| Retroviridae/Alpharetrovirus | Avian leukosis/sarcoma viruses | Inapparent infection, leukemia, other diseases |
Teratogenic viruses are those that can cause developmental defects after in-utero infection. The outcome of infections of pregnant animals with teratogenic viruses is influenced to a great extent by gestational age. Thus, viral infections that occur during critical stages of organogenesis in the developing fetus can have devastating consequences from virus-mediated infection and destruction of progenitor cells before they can populate organs such as the brain. For example, Akabane virus, Cache Valley virus, bovine viral diarrhea virus, and bluetongue virus can all cause teratogenic brain defects in congenitally infected ruminants.
Although immune competence generally is developed by mid-gestation, viral infections before this time can lead to a weak and ineffectual immune response that leads to persistent postnatal infection, such as persistent bovine viral diarrhea virus infection in cattle and congenital lymphocytic choriomeningitis virus infection in mice.
Viral Infection of Other Organs
Almost any organ may be infected with one or another kind of virus via the blood stream, but most viruses have well-defined organ and tissue tropisms that reflect the factors described earlier (presence of receptors, intracellular and other physiological or physical co-factors, etc.). The clinical importance of infection of various organs and tissues depends, in part, on their role in the physiologic well-being of the animal. In addition to the organs and tissues already described (respiratory tract, gastrointestinal tract, skin, brain and spinal cord, hemopoietic tissues), viral infections of the heart and liver can also have especially devastating consequences. The liver is the target of relatively few viral infections of animals, in marked contrast to the numerous hepatitis viruses (hepatitis A, B, and C viruses in particular) and other viruses (e.g., yellow fever virus) that are important causes of severe liver disease in humans. In animals, Rift Valley fever virus, mouse hepatitis virus, and infectious canine hepatitis virus characteristically affect the liver, as do several abortigenic herpesviruses after fetal infections (e.g., infectious bovine rhinotracheitis virus, equine herpesvirus 1, pseudorabies virus). Virus-mediated cardiac injury is relatively uncommon in animals, but is characteristic of bluetongue and some other endotheliotrophic viral infections, and alphavirus infections of Atlantic salmon and rainbow trout.
Non-specific Pathophysiological Changes in Viral Diseases
Some of the adverse consequences of viral infections cannot be attributed to direct cell destruction by the virus, to immunopathology, or to the effects of increased concentrations of endogenous adrenal glucocorticoids in response to the stress of the infection. Viral diseases are accompanied frequently by a number of vague general clinical signs, such as fever, malaise, anorexia, and lassitude. Cytokines (interleukin-1 in particular) produced in the course of innate immune responses to infection may be responsible for some of these signs, which collectively can significantly reduce the animal’s performance and impede recovery. Less characterized are the potential neuropsychiatric effects of persistent viral infection of particular neuronal tracts, such as that caused by Borna disease virus. Borna disease virus infection is not lytic in neurons, but induces bizarre changes in the behavior of rats, cats, and horses.
Viruses that cause widespread vascular injury can result in disseminated hemorrhages and/or edema as a result of increased vascular permeability. Vascular injury in these so-called hemorrhagic viral fevers, which include Dengue hemorrhagic fever, yellow fever, Ebola, and different hantavirus infections in humans, and bluetongue in ruminants, can result either from viral infection of endothelial cells or the systemic release from other infected cells of vasoactive and inflammatory mediators such as tissue necrosis factor. Widespread endothelial injury leads to coagulation and thrombosis that may precipitate disseminated intravascular coagulation, which is the common pathway that leads to death of animals and humans infected with a variety of viruses that directly or indirectly cause vascular injury.
Virus-induced Immunopathology
Viruses typically cause direct damage to the host cells they infect cells by subverting their metabolic machinery. Inflammatory responses that accompany most viral infections also can potentially contribute to disease pathogenesis, as described earlier. However, in certain instances, it is the host’s immune response triggered by viral infection that mediates tissue injury and disease, particularly in those viruses that cause persistent, non-cytocidal infections. Thus the immune response can exert a two-edged role in the pathogenesis of viral diseases. Infiltration of virus-infected tissues by lymphocytes and macrophages, release of cytokines and other mediators, and the resultant inflammation are all typical of viral infections. These responses are critical to the initial control of infection, as well as to eventual virus clearance and induction of protective immunity (see Chapter 4). However, there is a delicate balance between the protective and destructive effects of host antiviral immune responses, and between the ability of the virus to replicate and spread in the face of the host’s protective response. Indeed, there are viral infections in which such manifestations of the immune response are the cardinal factors in the onset and progression of the associated disease.
Immune-mediated tissue injury caused by individual viruses involves any one or more of the four types (I–IV) of immunopathologic (hypersensitivity) reactions. The distinction between these various mechanisms is increasingly less defined, but this classification system is useful for mechanistic understanding. Most viruses that cause diseases with a defined immune-mediated component involve type IV hypersensitivity reactions, and a few involve type III reactions. Type I reactions are anaphylactic-type reactions mediated by antigen-specific IgE and mast-cell-derived mediators such as histamine and heparin, and the activation of serotonin and plasma kinins; with the exception of its potential role in inflammation, this mechanism is probably unimportant in the pathogenesis of most viral infections. Similarly, type II hypersensitivity reactions that involve antibody-mediated lysis of cells, either directly through complement activation or via cells that bind to the Fc portion of bound antibodies, are of uncertain significance in the pathogenesis of viral diseases in animals.
Type III hypersensitivity reactions are caused by complexes of antigen and antibody (immune complexes) that initiate inflammation and tissue damage. Immune complexes circulate in blood in the course of most viral infections. The fate of the immune complexes depends on the ratio of antibody to antigen. In infections in which there is a large excess of antibody as compared with circulating virus, or even if there are equivalent amounts of antibody and virus, the virus is typically cleared by tissue macrophages. However, in some persistent infections, viral proteins (antigens) and/or virions are released continuously into the blood but the antibody response is weak and antibodies are of low avidity. In these instances, immune complexes are deposited in small blood vessels that function as filters, especially those of the renal glomeruli. Immune complexes continue to be deposited in glomeruli over periods of weeks, months, or even years, leading to their accumulation and subsequent immune-complex mediated glomerulonephritis.
Lymphocytic choriomeningitis virus infection in mice infected in utero or as neonates provides a classic example of immune complex disease associated with a persistent viral infection. Viral antigens are constantly present in the blood and, although there is specific immune dysfunction (“tolerance”), small amounts of non-neutralizing antibody are formed as mice age, leading to the formation of immune complexes that are deposited progressively within the walls of the glomerular capillaries. Depending on the strain of mouse, the end result may be glomerulonephritis leading to death from renal failure. Circulating immune complexes may also be deposited in the walls of the small blood vessels in the skin, joints, and choroid plexus, where they also cause tissue injury. A similar disease pathogenesis can occur in other persistent viral infections of animals, such as Aleutian mink disease (parvovirus infection), feline leukemia, and equine infectious anemia.
Unlike the other hypersensitivity reactions, type IV reactions, also called delayed hypersensitivity reactions, are mediated by T lymphocytes and macrophages. Cytotoxic T lymphocytes are critical components of the adaptive immune response that leads to clearance of virus-infected cells. Specifically, cytotoxic T lymphocytes recognize viral antigens expressed along with major histocompatibility (MHC) class I molecules on the surface of infected cells, which they then bind and lyse (see Chapter 4). This mechanism, however, can lead to ongoing destruction of host cells during certain viral infections, including those persistent infections caused by non-cytocidal viruses. Examples include neurological diseases induced by Borna disease virus. The respiratory tract is especially vulnerable to this type of immune-mediated disease, including infection with influenza and parainfluenza viruses. For example, Sendai virus is a non-cytolytic respiratory pathogen in rodents. Disease is minimal following Sendai virus infection of T-cell-deficient animals, whereas disease is severe in immunocompetent animals. T cells induce severe necrotizing bronchiolitis and interstitial pneumonia, with destruction of type II pneumocytes, rendering the alveoli incapable of repair.
Experimental lymphocytic choriomeningitis virus infection of adult mice has been extensively studied as a type IV immune-mediated disease accompanying a non-cytolytic viral infection (Figure 3.14 ). After intracerebral inoculation, the virus replicates harmlessly in the meninges, ependyma, and choroid plexus epithelium until about the seventh day, when (CD)8+ class I MHC-restricted cytotoxic T cells invade the central nervous tissues to lyse the infected cells, which in turn results in extensive inflammation producing meningitis, cerebral edema, neurological signs such as convulsions, and death. Likewise, intraperitoneal inoculation of virus results in immune-mediated hepatitis and severe lymphoid depletion in the spleen. The death of infected mice can be prevented by chemical immunosuppression, by X-irradiation, or by prior treatment with antilymphocyte serum.
Figure 3.14.
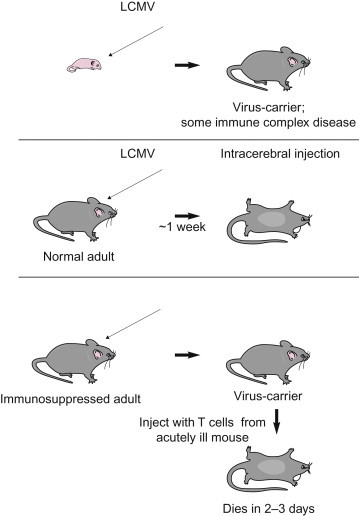
Injection of lymphocytic choriomeningitis virus (LCMV) into a newborn mouse produces a persistent infection with only minor pathological changes (top). Intracerebral injection of a normal adult mouse produces a fulminating disease that quickly kills the animal (middle). However, a T-deficient mouse (e.g. neonatally thymectomized or treated with anti-thymocyte serum) tolerates an intracerebral injection of LCMV (bottom). This carrier state can be broken by an injection of T cells (but not serum) from a mouse acutely ill with LCM.
(Adapted from Introduction to Immunology. J. W. Kimball, p. 462. Copyright 1983 MacMillan Publishing, with permission.)
Viruses and Autoimmune Disease
It repeatedly has been proposed, with little definitive evidence, that subtle (subclinical or asymptomatic) viral infections are responsible for autoimmune diseases in animals and humans. Proposed mechanisms for this largely hypothetical phenomenon focus on either unregulated or misdirected immune responses precipitated by a viral infection, or the presence of shared or equivalent antigens on infectious agents and host cells (molecular mimicry). Molecular mimicry clearly is responsible for immune-mediated diseases initiated by microbial infection, as classically illustrated by rheumatic heart disease in humans that is initiated by group A Streptococcus infection. In viruses, individual epitopes have been identified in several viruses that are also present in animal tissue, such as muscle or nervous tissue (e.g., myelin basic protein). These antibodies to these epitopes potentially might contribute to immune-mediated tissue during the course of viral infection, but their pathogenic role, if any, in initiating and potentiating autoimmune disease remains uncertain.
Persistent Infection and Chronic Damage to Tissues and Organs
Persistent infections of one type or another are produced by a wide range of viruses, and are common in veterinary medicine. Apart from enteric and respiratory viruses that cause transient infections that remain localized to their respective target organs, most other categories of viral infections include examples of chronic infection. Foot-and-mouth disease, for example, usually is an acute, self-limiting infection, but a carrier state of uncertain epidemiological relevance occurs in which virus persists in the oropharynx of low numbers of convalescent animals. In other instances, such as those associated with immunodeficiency viral infections, persistent viral infections lead to chronic diseases, even when the acute manifestations of infection have been trivial or subclinical. Finally, persistent infections can lead to continuing tissue injury, often with an immune-mediated basis.
Persistent viral infections are important for several reasons. For example, they may be reactivated and cause recrudescent episodes of disease in the individual host, or they may lead to immunopathologic disease or to neoplasia. Persistent infection may allow survival of a particular virus in individual animals and herds, even after vaccination. Similarly, persistent infections may be of epidemiologic importance—the source of contagion in long-distance virus transport and in reintroduction after elimination of virus from a given herd, flock, region, or country. For convenience, persistent viral infections may be subdivided into several categories:
Persistent infections, per se, in which infectious virus is demonstrable continuously, whether or not there is ongoing disease. Disease may develop late, often with an immunopathologic or neoplastic basis. In other instances, disease is not manifest in persistently infected animals; for example, in the deer mouse (Peromyscus maniculatus), the reservoir rodent host of Sin Nombre virus, and the etiologic agent of hantavirus pulmonary syndrome in humans, virus is shed in urine, saliva, and feces probably for the life of the animal, even in the face of neutralizing antibody.
A striking proportion of persistent infections involve the central nervous system. The brain is somewhat sequestered from systemic immune activity by the blood–brain barrier and, further, neurons express very little MHC antigen on their surface, thereby conferring some protection against destruction by cytotoxic T lymphocytes.
Latent infections, in which infectious virus is not demonstrable except when reactivation occurs. For example, in infectious pustular vulvovaginitis, the sexually transmitted disease caused in cattle by bovine herpesvirus 1, virus usually cannot be isolated from the latently infected carrier cow except when there are recrudescent lesions. Viral latency may be maintained by restricted expression of genes that have the capacity to kill the cell. During latency, herpesviruses express only a few genes that are necessary in the maintenance of latency, notably so-called latency-associated transcripts. During reactivation, which is often stimulated by immunosuppression and/or by the action of a cytokine or hormone, the whole viral genome is transcribed again. This strategy protects the virus during its latent state from all host immune actions that would normally result in virus clearance.
Slow infections, in which quantities of infectious virus gradually increase during a very long preclinical phase that eventually leads to a slowly progressive disease (e.g., ovine progressive pneumonia).
Acute infections with late clinical manifestations, in which continuing replication of the causative virus is not involved in the progression of the disease. For example, in the cerebellar atrophy syndrome that occurs in young cats as a result of fetal infection with feline panleukopenia virus, virus cannot be isolated at the time neurologic damage is diagnosed. In fact, because of this, the cerebellar syndrome was for many years considered to be an inherited malformation.
It may be noted that these categories are defined primarily in terms of the extent and continuity of virus replication during the long period of persistence. The presence or absence of shedding and disease are secondary issues as far as this categorization is concerned. Further, some persistent infections possess features of more than one of these categories. For example, all retrovirus infections are persistent and most exhibit features of latency, but the diseases they cause may be delayed following infection or only manifest as slowly progressive diseases. The variety of patterns of persistent viral infections is shown diagrammatically in Figure 3.15 .
Figure 3.15.
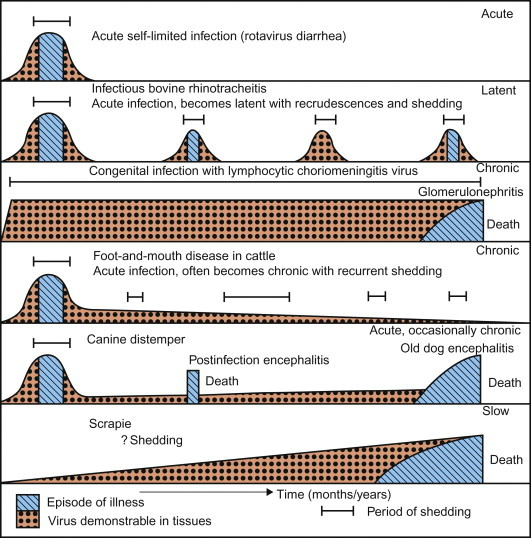
The shedding of virus and the occurrence of clinical signs in acute self-limited infections and various kinds of persistent infection, as exemplified by the diseases indicated. The time scale is notional and the duration of various events approximate.
Individual viruses employ a remarkable variety of strategies for successful evasion of host immune and inflammatory responses in vivo. These mechanisms include non-cytocidal infections without expression of immunogenic proteins, replication in cells of the immune system or subversion of host innate and adaptive immunity (see Chapter 4), and infection of non-permissive, resting, or undifferentiated cells. Some viruses have evolved strategies for evading neutralization by the antibody they elicit. Ebola virus, for example, uses an “immune decoy” to evade neutralizing antibody—specifically, a secreted viral protein that binds circulating antibody. The surface glycoproteins of filoviruses, arenaviruses, bunyaviruses (e.g., Rift Valley fever virus) and some arteriviruses (e.g., porcine reproductive and respiratory syndrome virus and lactate dehydrogenase-elevating virus) are heavily glycosylated, which may serve to mask the neutralizing epitopes contained in these proteins. Antigenic drift is especially characteristic of persistent RNA viral infection, particularly persistent RNA virus infections such as those associated with lentiviruses (e.g., equine infectious anemia virus). During persistent infection, sequential antigenic variants are produced, with each successive variant sufficiently different to evade the immune response raised against the preceding variant. In equine infectious anemia, clinical signs occur in periodic cycles, with each cycle being initiated by the emergence of a new viral variant. In addition to providing a mechanism for escape from immune elimination, each new variant may be more virulent than its predecessor, and this may directly affect the severity and progression of the disease.
The integration of retroviral proviral DNA into the genome of the host germ-line cells assures indefinite maintenance from one generation to the next; such proviral DNA can also lead to induction of tumors (oncogenesis).
Virus-Induced Neoplasia
The revolution in molecular cell biology has provided remarkable insights into the mechanisms of regulation of cell growth and differentiation, and these insights have, in turn, advanced understanding of the mechanisms underpinning failures of regulatory processes that are expressed as neoplasia. The genetic changes that are ultimately responsible for neoplasia may be caused by chemical or physical agents or viruses, but all involve certain common cellular pathways. Interestingly, while there are a substantial number of both RNA and DNA viruses that are oncogenic in animals, only a relatively few viruses have been definitively linked to human cancer.
The discoveries of the viral etiology of avian leukemia by Ellerman and Bang and of avian sarcoma by Rous, in 1908 and 1911 respectively, were long regarded as curiosities unlikely to be of any fundamental significance. However, study of these avian viruses and related retroviruses of mice has increased our overall understanding of neoplasia greatly, and since the 1950s there has been a steady stream of discoveries clearly incriminating other viruses in a variety of benign and malignant neoplasms of numerous species of mammals, birds, amphibians, reptiles, and fish. Many avian retroviruses are major pathogens of poultry, and other retroviruses produce neoplasms in domestic animals. Similarly, several different DNA viruses have been determined to be responsible for cancers in humans and animals.
Any discussion of virus-induced neoplasia requires that a few commonly used terms are defined: a neoplasm is a new growth (syn. tumor); neoplasia is the process that leads to the formation of neoplasms (syn. carcinogenesis); oncology is the study of neoplasia and neoplasms; a benign neoplasm is a growth produced by abnormal cell proliferation that remains localized and does not invade adjacent tissue; in contrast, a malignant neoplasm (syn. cancer) is locally invasive and may also be spread to other parts of the body (metastasis). Carcinomas are cancers of epithelial cell origin, whereas sarcomas are cancers that arise from cells of mesenchymal origin. Solid neoplasms of lymphocytes are designated lymphosarcoma or malignant lymphoma (syn. lymphoma), whereas leukemias are cancers of hemopoietic origin characterized by circulation of cancerous cells.
Neoplasms arise as a consequence of the dysregulated growth of cells derived from a single, genetically altered progenitor cell. Thus, although neoplasms are often composed of several cell types, they are considered to originate from a monoclonal outgrowth of a single cell. It recently has been proposed that neoplasms arise from cells with properties and function similar to those of the stem cells that are present in normal tissue. Specifically, many normal tissues contain a small population of resident, long-lived stem cells that are tissue progenitors; these cells can divide to produce either terminally differentiated, relatively short-lived cells with limited replicative ability, or additional long-lived stem cells. As cancers are immortal and have unlimited ability to replicate, it is assumed that they, too, must contain stem cells that arise either from normal tissue stem cells or from differentiated cells that assume stem cell-like properties.
The Cellular Basis of Neoplasia
Neoplasia is the result of non-lethal genetic injury, as may be acquired by chemical or physical damage, or from viral infections. Some cancers, however, arise randomly through the accumulation of spontaneous genetic mutations. A neoplasm results from the clonal expansion of a single cell that has suffered genetic damage, typically in one of four types of normal regulatory genes: (1) proto-oncogenes, which are cellular genes that regulate growth and differentiation; (2) tumor suppressor genes that inhibit growth, typically by regulating the cell cycle; (3) genes that regulate apoptosis (programmed cell death; see Chapter 4); (4) genes that mediate DNA repair. Carcinogenesis involves a multi-step progression resulting from the cumulative effects of multiple mutations.
Once developed, neoplasms are: (1) self-sufficient, in that they have the capacity to proliferate without external stimuli; for example, as the result of unregulated oncogene activation; (2) insensitive to normal regulatory signal that would limit their growth, such as transforming growth factor-β and the cyclin-dependent kinases that normally regulate orderly progression of cells through the various phases of the cell cycle; (3) resistant to apoptosis because of either the activation of anti-apoptotic molecules or the inhibition of mediators of apoptosis such as p53; (4) limitless potential for replication. Cancers also may have the ability to invade and spread to distant tissues (metastasis), and neoplasms typically promote the proliferation of new blood vessels that support their growth.
Neoplasia, regardless of cause, is the result of unregulated cellular proliferation. In the normal sequence of events during cellular proliferation, a growth factor binds to its specific cellular receptor, leading to signal transduction that ultimately results in nuclear transcription, which in turn leads to the cell entering and progressing through the cell cycle until it divides. Proto-oncogenes are normal cellular genes that encode proteins that function in normal cellular growth and differentiation; they include (1) growth factors; (2) growth factor receptors; (3) intracellular signal transducers; (4) nuclear transcription factors; (5) cell-cycle control proteins. Oncogenes are derived by mutation of their normal cellular proto-oncogene counterparts, and the expression of oncogenes results in production of oncoproteins that mediate autonomous (unregulated) growth of neoplastic cells.
The development of cancer (malignant neoplasia) is a protracted, multi-step process that reflects the accumulation of multiple mutations. A potentially neoplastic clone of cells must bypass apoptosis (programmed death), circumvent the need for growth signals from other cells, escape from immunologic surveillance, organize its own blood supply, and possibly metastasize. Thus, tumors other than those induced by rapidly transforming retroviruses like Rous sarcoma virus generally do not arise as the result of a single event, but by a series of steps leading to progressively greater loss of regulation of cell division.
Oncogenic DNA and RNA viruses have been identified in both animals and humans, including retroviruses, papillomaviruses, herpesviruses, and several other DNA viruses. Cells transformed by non-defective retroviruses also express the full range of viral proteins, and new virions bud from their membranes. In contrast, transformation by DNA viruses usually occurs in cells undergoing non-productive infection in which viral DNA is integrated into the cellular DNA of the transformed cells or, in the case of papillomaviruses and herpesviruses, in which the viral DNA remains episomal. Certain virus-specific antigens are demonstrable in transformed cells. Some tumor-associated antigens are expressed on the plasma membrane where, in vivo, they constitute potential targets for immunologic attack.
Oncogenic RNA Viruses
Retrovirus Pathogenesis
Retroviruses are a significant cause of neoplasia in many species of animals, including cattle, cats, non-human primates, mice, and chickens, among others. Their pathogenesis is linked to their propensity to integrate randomly within the genome of host cells, thereby being infectious mutagens. The consequences of such integration are largely innocuous and clinically silent, and only seldom result in oncogenesis. As described in Chapter 14, retroviruses can be biologically divided into exogenous (horizontally transmissible) agents, or endogenous, in which case they are integrated within the host genome. Retroviruses can be either replication-competent or replication-defective.
Rarely, a replication-competent retrovirus will integrate into the genome of host germ cells. A complete DNA copy of the viral genome (known as the provirus) may thereafter be transmitted in the germ line DNA from parent to progeny (i.e., via ova or sperm) and, over the course of evolution, may be perpetuated in every individual of an animal species. Such retroviruses are said to be endogenous. As long as endogenous retroviruses remain replication-competent, they may also be horizontally transmissible like their exogenous relatives. Over the course of time, multiple endogenous retroviruses become integrated throughout the genome, either through new exposures, or more commonly when provirus genomes are replicated during cell division, they can then be integrated elsewhere in the genome as retrotransposons. As millennia pass, many of these “retro-elements” become replication-defective but their DNA continues to have the potential to reintegrate as retrotransposons, and their partial genes may continue to encode proteins. These reintegration events, when involving functioning host genes, may result in spontaneous mutations within the germ line of the species. This process is known as insertional mutagenesis. It is not necessarily in the best interest of the host to carry potentially deleterious viral mutagens within its genome, so the host evolves to lack somatic cell receptors for its endogenous viruses, or the host may mutate, truncate, methylate, or even evict proviral sequences over time. In essence, endogenous retroviruses and their hosts are in a constant state of co-evolution. Some anciently acquired endogenous retroviruses have actually become vital to host physiology. During pregnancy of mammals (except monotremes), endogenous retro-elements are expressed to high levels during embryo implantation and placental development, inducing immunosuppressive and cell fusion (syncytium formation) effects that are vital to mammalian placental and fetal development. Syncytin-1 is one such gene product that is an endogenous retro-element env-derived fusigenic glycoprotein that is critical in syncytial trophoblast formation. It is a highly conserved and essential gene among placental animals.
Endogenous proviruses, like other host genes, are expressed differentially in different tissues, at different ages, and under the control of various stimuli, including hormones and immune states. When a dividing cell is co-expressing two or more proviruses, proviral genomes may recombine to form new retroviral variants with novel ability to infect somatic cells through alternate receptors. This has been illustrated in lymphoma-prone inbred mouse strains, with each mouse strain having different constellations of pro-viral integrations that recombine to become replication-competent infectious viruses that can target vulnerable tissues through novel receptor–ligand interactions. Although this has been extensively studied, the consequences of proviral recombination and induction of neoplasia by viral recombinants is largely an artificial phenomenon that is unique to the inbred mouse.
Retrovirus-Induced Neoplasia
Oncogenic retroviruses are classified as chronic transforming or acute transforming retroviruses. These two major types of transforming retroviruses induce neoplasia in significantly different ways.
Chronic Transforming Retroviruses
Chronic transforming retroviruses induce neoplasia through random integrations into the genome of somatic cells. They exert their effect as “cis-activating” retroviruses that transform cells by becoming integrated in the host-cell DNA close to a cell growth regulating gene, and thus usurping normal cellular regulation of this gene. These cell growth regulating host genes are termed “oncogenes,” or cellular oncogenes (c-onc). Despite the terminology implying that they are oncogenic, c-onc genes are host genes that encode important cell signaling products that regulate normal cell proliferation and quiescence. The presence of an integrated provirus, with its strong promoter and enhancer elements, upstream from a c-onc gene may amplify the expression of the c-onc gene greatly. This is the likely mechanism whereby the weakly oncogenic endogenous avian leukosis viruses produce neoplasia. When avian leukosis viruses cause malignant neoplasia, the viral genome has generally been integrated at a particular location, immediately upstream from a host c-onc gene. Integrated avian leukosis provirus increases the synthesis of the normal c-myc oncogene product 30- to 100-fold. Experimentally, only the viral long-terminal repeats (LTR) need be integrated to cause this effect; furthermore, by this mechanism c-myc may also be expressed in cells in which it is not normally expressed or is normally expressed at much lower levels.
Not all chronic transforming retroviruses require insertional mutagenesis in regions of c-onc genes to be oncogenic. Both exogenous and endogenous mouse mammary tumor viruses carry an extra viral gene sequence that encodes a super-antigen (Sag) that stimulates proliferation of lymphocytes. Expression of Sag stimulates massive B cell proliferation and mouse mammary tumor virus replication in the dividing B cells, with subsequent homing of virus-expressing lymphocytes to mammary tissue. Both lymphomas and mammary tumors may ensue, but oncogenesis does not require alteration of host oncogenes. The exogenous ovine retroviruses that cause nasal carcinomas and pulmonary adenocarcinomas (Jaagsiekte) infect epithelial target cells, and transformation is related to expression of the viral env gene. Bovine leukemia virus is an exogenous retrovirus that causes chronic leukosis and B cell lymphoma. The virus encodes tax, rex, R3, and G4 genes in the 3′ end of its viral genome. The tax gene functions as a transactivator of host genes. Bovine leukemia virus is closely related to human T lymphotrophic virus 1 (HTLV-1), which has a similar viral genome. In contrast to cis-activating retroviruses, these viruses are examples of “trans-activating” retroviruses.
Acute Transforming Retroviruses
Acute transforming retroviruses are directly oncogenic by carrying an additional viral oncogene, v-onc, and are classified as “transducing” retroviruses. The retroviral v-onc originates from a host c-onc gene, and the transforming activity of the v-onc is accentuated by mutation. Given the high error rate of reverse transcription, v-onc gene homologs of c-onc genes will always carry mutations and the strongly promoted production of the viral oncoprotein will readily exceed that of the normal cellular oncoprotein. The result can be uncontrolled cell growth. Because c-onc genes are the precursors of v-onc genes, c-onc genes are also called “proto-oncogenes.” Wherever acute transforming retroviruses integrate in the host genome, it is the v-onc that is directly responsible for the rapid malignant change that occurs in cells infected with these viruses. Over 60 different v-onc genes have been identified, and retroviruses have been instrumental in identifying their cellular homologues.
The v-onc is usually incorporated into the viral RNA in place of part of one or more normal viral genes. Because such viruses have lost some of their viral genetic sequences, they are usually incapable of replication, and are therefore termed “defective” retroviruses. Defective retroviruses circumvent their defective replicative ability by utilizing non-defective “helper” retroviruses for formation of infectious virions. An exception is Rous sarcoma virus, in that its genome contains a viral oncogene (v-src) in addition to its full complement of functioning viral genes (gag, pol, and env); thus Rous sarcoma virus is both replication-competent and an acute transforming virus. Rous sarcoma virus is one of the most rapidly acting carcinogens known, transforming cultured cells in a day or so and causing neoplasia and death in chickens in as little as 2 weeks after infection.
Although retrovirus v-onc genes often preclude virus replication, v-onc genes have been acquired over time by retroviruses, most likely because they cause cellular proliferation. As most retroviruses replicate during cell division, this favors virus growth and perpetuation in nature. Defective retroviruses carrying a v-onc gene are always found in the company of a replication-competent helper virus that supplies missing functions, such as an environmentally stable envelope. The advantage to both viruses is presumably that when they are together they can infect more cells and produce more progeny of both viruses.
The various v-onc genes and the proteins they encode are assigned to major classes: growth factors (such as v-sis); growth factor receptors and hormone receptors (such as v-erbB); intracellular signal transducers (such as v-ras); and nuclear transcription factors (such as v-jun). The oncoprotein products of the various retroviral v-onc genes act in many different ways to affect cell growth, division, differentiation, and homeostasis:
-
•
v-onc genes usually contain only that part of their corresponding c-onc gene that is transcribed into messenger RNA—in most instances they lack the introns that are so characteristic of eukaryotic genes
-
•
v-onc genes are separated from the cellular context that normally controls gene expression, including the normal promoters and other sequences that regulate c-onc gene expression
-
•
v-onc genes are under the control of the viral LTRs, which not only are strong promoters but also are influenced by cellular regulatory factors. For some retrovirus v-onc genes, such as myc and mos, the presence of viral LTRs is all that is needed for tumor induction
-
•
v-onc genes may undergo mutations (deletions and rearrangements) that alter the structure of their protein products; such changes can interfere with normal protein–protein interactions, leading to escape from normal regulation
-
•
v-onc genes may be joined to other viral genes in such a way that their functions are modified. For example, in Abelson murine leukemia virus the v-abl gene is expressed as a fusion protein with a gag protein; this arrangement directs the fusion protein to the plasma membrane where the Abl protein functions. In feline leukemia virus, the v-onc gene fms is also expressed as a fusion protein with a gag protein, thus allowing the insertion of the Fms oncoprotein in the plasma membrane.
Many acute transforming retroviruses induce solid tumors in addition to hemopoietic tumors. These viruses are termed “sarcoma” viruses. In addition to many avian leukosis virus-derived sarcoma viruses that have incorporated various v-onc genes, several acute transforming defective sarcoma viruses have been isolated from sarcomas of cats naturally infected with exogenous feline leukemia virus, a woolly monkey infected with a simian retrovirus, and several sarcoma viruses have been isolated from laboratory rodents infected with both exogenous and endogenous retroviruses. Acute transforming defective retroviruses are significant as oncogens in individual animals, but are not naturally transmissible agents.
Oncogenic DNA Viruses
Although retroviruses are the most important oncogenic viruses in animals, certain DNA viruses are also significant, including papillomaviruses, polyomaviruses, herpesviruses, and potentially others (Table 3.4 ). DNA tumor viruses interact with cells in one of two ways: (1) productive infection, in which the virus completes its replication cycle, resulting in cell lysis, or (2) non-productive infection, in which the virus transforms the cell without completing its replication cycle. During such non-productive infection, the viral genome or a truncated version of it is integrated into the cellular DNA; alternatively, the complete genome persists as an autonomously replicating plasmid (episome). The genome continues to express early gene functions. The molecular basis of oncogenesis by DNA viruses is best understood for polyomaviruses, papillomaviruses, and adenoviruses, all of which contain genes that behave as oncogenes, including tumor suppressor genes. These oncogenes appear to act by mechanisms similar to those described for retrovirus oncogenes: they act primarily in the nucleus, where they alter patterns of gene expression and regulation of cell growth. In every case the relevant genes encode early proteins having a dual role in virus replication and cell transformation. With a few possible exceptions, the oncogenes of DNA viruses have no homologue or direct ancestors (c-onc genes) among cellular genes of the host.
Table 3.4.
Viruses that can Induce Tumors in Domestic or Laboratory Animals or Humans
| Family/Genus | Virus | Kind of Tumor |
|---|---|---|
| DNA Viruses | ||
| Poxviridaea/Leporipoxvirus | Rabbit fibroma virus and squirrel fibroma virus | Fibromas and myxomas in rabbits and squirrels (hyperplasia rather than neoplasia) |
| Poxviridaea/Yatapoxvirus | Yaba monkey tumor virus | Histiocytoma in monkeys |
| Herpesviridae/Alphaherpesvirinae/Mardivirus | Marek’s disease virus | T cell lymphoma in fowl |
| Herpesviridae/Gammaherpesvirinae/Rhadinovirus | Ateline herpesvirus 2 and saimirine herpesvirus 2 | Nil in natural hosts, lymphomas and leukemia in certain other monkeys |
| Herpesviridae/Gammaherpesvirinae/Lymphocryptovirus | Epstein–Barr virus | Burkitt’s lymphoma, nasopharyngeal carcinoma, and B cell lymphomas in humans and monkeys |
| Baboon herpesvirus | Lymphoma in baboons | |
| Herpesviridae/Gammaherpesvirinae/Rhadinovirus | Cottontail rabbit herpesvirus | Lymphoma in rabbits |
| Alloherpesviridae/ranid herpesvirus | Lucké frog herpesvirus | Renal adenocarcinoma in frogs and tadpoles |
| Adenoviridae/Mastadenovirus | Many adenoviruses | Tumors in newborn rodents, no tumors in natural hosts |
| Papillomaviridae/multiple genera | Cottontail rabbit papillomavirus | Papillomas, skin cancers in rabbits |
| Bovine papillomavirus 4 | Papillomas, carcinoma of intestine, bladder | |
| Bovine papillomavirus 7 | Papillomas, carcinoma of eye | |
| Human papillomaviruses 5, 8 | Squamous cell carcinoma | |
| Human papillomaviruses 16, 18 | Genital carcinomas | |
| Polyomaviridae/Polyomavirus | Murine polyomavirus and simian virus 40 | Tumors in newborn rodents |
| Reverse Transcribing Viruses | ||
| Hepadnaviridae/Orthohepadnavirus | Human, woodchuck hepatitis viruses | Hepatocellular carcinomas in humans and woodchucks |
| Hepadnaviridae/Avihepadnavirus | Duck hepatitis virus | Hepatocellular carcinomas in ducks |
| Retroviridae/Alpharetrovirus | Avian leukosis viruses | Leukosis (lymphoma, leukemia), osteopetrosis, nephroblastoma in fowl |
| Rous sarcoma virus | Sarcoma in fowl | |
| Avian myeloblastosis virus | Myeloblastosis in fowl | |
| Retroviridae/Betaretrovirus | Mouse mammary tumor virus | Mammary carcinoma in mice |
| Mason–Pfizer monkey virus | Sarcoma and immunodeficiency disease in monkeys | |
| Ovine pulmonary adenocarcinoma virus (Jaagsiekte virus) | Pulmonary adenocarcinoma in sheep | |
| Retroviridae/Gammaretrovirus | Feline leukemia virus | Leukemia in cats |
| Feline sarcoma virus | Sarcoma in cats | |
| Murine leukemia and sarcoma viruses | Leukemia, lymphoma, and sarcoma in mice | |
| Avian reticuloendotheliosis virus | Reticuloendotheliosis in fowl | |
| Retroviridae/Deltaretrovirus | Bovine leukemia virus | Leukemia (B cell lymphoma) in cattle |
| HTLV 1 and 2 viruses and simian HTLV viruses | Adult T cell leukemia and hairy cell leukemia in humans, leukemia in monkeys | |
| RNA Viruses | ||
| Flaviviridae/Hepacivirus | Hepatitis C virus | Hepatocellular carcinoma in humans |
Not true oncogenic viruses. They differ from all other viruses listed in that poxviruses replicate in cytoplasm and do not affect the cellular genome.
The protein products of DNA virus oncogenes are multifunctional, with particular functions that mimic functions of normal cellular proteins related to particular domains of the folded protein molecule. They interact with host-cell proteins at the plasma membrane or within the cytoplasm or nucleus.
Oncogenic Polyomaviruses and Adenoviruses
During the 1960s and 1970s, two members of the family Polyomaviridae, murine polyomavirus and simian virus 40 (SV40), as well as certain human adenoviruses (types 12, 18, and 31) were shown to induce malignant neoplasms following their inoculation into baby hamsters and other rodents. With the exception of murine polyomavirus, none of these viruses induces cancer under natural conditions in its natural host, rather they transform cultured cells of certain other species and provide experimental models for analysis of the molecular events in cell transformation.
Polyomavirus- or adenovirus-transformed cells do not produce virus. Viral DNA is integrated at several sites in the chromosomes of the cell. Most of the integrated viral genomes are complete in the case of the polyomaviruses, but defective in the case of the adenoviruses. Only certain early viral genes are transcribed, albeit at an unusually high rate. By analogy with retrovirus genes, they are now called oncogenes. Their products, demonstrable by immunofluorescence, used to be known as tumor (T) antigens. A great deal is now known about the role of these proteins in transformation. Virus can be rescued from polyomavirus-transformed cells—that is, virus can be induced to replicate, by irradiation, treatment with certain mutagenic chemicals, or co-cultivation with certain types of permissive cells. This cannot be done with adenovirus-transformed cells, as the integrated adenovirus DNA contains substantial deletions.
It should be stressed that the integration of viral DNA does not necessarily lead to transformation. Many or most episodes of integration of polyomavirus or adenovirus DNA have no recognized biological consequence. Transformation by these viruses in experimental systems is a rare event, requiring that the viral transforming genes be integrated in the location and orientation needed for their expression. Even then, many transformed cells revert (abortive transformation). Furthermore, cells displaying the characteristics of transformation do not necessarily produce neoplasms.
Oncogenic Papillomaviruses
Papillomaviruses produce papillomas (warts) on the skin and mucous membranes of most animal species (see Chapter 11). These benign neoplasms are hyperplastic epithelial outgrowths that generally regress spontaneously. Occasionally, however, they may progress to malignancy, which in part is a property of specific virus strains. Virus-induced papillomas occur in many species, and papillomaviruses are also the cause of sarcoids in horses, some human oropharyngeal carcinomas, and cervical carcinoma in women, and are associated with some squamous cell carcinomas in cats and dogs.
In benign warts, the papillomavirus DNA is episomal, meaning it is not integrated into the host-cell DNA and persists as an autonomously replicating episome, whereas in papillomavirus-induced cancers the viral DNA is integrated into that of the host. Thus, integration probably is necessary for malignant transformation, as the pattern of integration is clonal within cancers: each cancer cell carries at least one, and often many incomplete copies of the viral genome. The site of virus integration is random, and there is no consistent association with cellular proto-oncogenes. For some papillomaviruses, integration disrupts one of the early genes, E2, which is a viral repressor. Other viral genes may also be deleted, but the viral oncogenes (e.g., E6 and E7) remain intact, are expressed efficiently, and cause the malignant transformation. The proteins expressed by the viral oncogenes interact with cellular growth regulating proteins produced by proto-oncogenes and tumor suppressors such as p53 to block apoptosis and promote cellular proliferation. Another relevant example is bovine papillomavirus type 1 E5 oncoprotein, which alters the activity of cell membrane proteins involved in regulating cellular proliferation—for example, platelet derived growth factor receptor.
Oncogenic Hepadnaviruses
Mammalian, but not avian, hepadnaviruses are associated strongly with naturally occurring hepatocellular carcinomas in their natural hosts. Woodchucks that are chronically infected with woodchuck hepatitis virus almost inevitably develop hepatocellular carcinoma, even in the absence of other carcinogenic factors. Oncogenesis induced by mammalian hepadnaviruses is a multifactorial process, and there are differences in the cellular mechanisms responsible for carcinogenesis associated with different viruses. Whereas ground squirrel and woodchuck hepatitis viruses activate cellular oncogenes, the mode of action of human hepatitis B virus is uncertain, as it apparently has no consistent site of integration or oncogene association. The hepatocellular regeneration accompanying cirrhosis of the liver also promotes the development of neoplasia in hepatitis virus-infected humans, but there is no cirrhosis in the animal models. The likelihood of hepadnavirus-associated carcinoma is greatest in animals (and humans) infected at birth.
Oncogenic Herpesviruses
Oncogenic Alphaherpesviruses
Marek’s disease virus of chickens transforms T lymphocytes, causing them to proliferate to produce a generalized polyclonal T lymphocyte neoplasm. The disease is preventable by vaccination with live-attenuated virus vaccines that lack the retrovirus v-onc genes that are present in Marek’s disease virus.
Oncogenic Gammaherpesviruses
Herpesviruses of the subfamily Gammaherpesvirinae are lymphotropic and the etiologic agents of lymphomas and carcinomas in hosts ranging from amphibians to primates, including humans. Epstein–Barr virus (human herpesvirus 4) in otherwise healthy young human adults causes infectious mononucleosis (glandular fever), in which there is B lymphocyte proliferation that resolves. The mechanism by which the virus goes on to produce malignancy in some individuals has been best studied in Burkitt’s lymphoma, a malignant B cell lymphoma that occurs in children in East Africa and less frequently in children in other parts of the world. The Epstein–Barr viral genomic DNA is present in multiple copies of episomal DNA in each cell of most African Burkitt’s lymphomas. Lymphoma cells express viral nuclear antigen, but do not produce virus. These cells also contain a characteristic 8:14 chromosomal translocation. Burkitt’s lymphoma may develop as a consequence of c-myc deregulation resulting from this translocation, which in turn causes an arrest of normal cellular maturation and differentiation processes. Some Burkitt’s lymphomas also have mutations in the cellular tumor suppressor gene, p53.
The subfamily Gammaherpesvirinae includes several other viruses that cause lymphomas in heterologous primate hosts (e.g., herpesvirus siamiri), human herpesvirus 8, associated with Kaposi sarcoma mostly in humans with AIDS, and bovine malignant catarrhal fever virus, an acute fatal lymphoproliferative disease of cattle and certain wild ruminants (see Chapter 9). These viruses are lymphotropic and contain numerous unspliced genes that appear to have been captured from the host during virus replication, over considerable evolutionary time. These captured genes typically encode proteins that (1) regulate cell growth, (2) are immunosuppressive, or (3) are enzymes involved in nucleic acid metabolism—they include genes encoding cytokine or cytokine receptor homologues, regulatory proteins such as cyclins that control the cell cycle, and proteins such as bcl2 that can block apoptosis. The function of these various virus-encoded proteins is consistent with the lymphotropic and transforming properties of these viruses. Thus these herpesviruses seem to have evolved/acquired different strategies to overcome cell cycle arrest, apoptosis, and activation of cellular immunity, all to favor virus replication and survival, all also causing lymphocyte proliferation and transformation.
Certain members of the family Alloherpesviridae are associated with neoplasia in their respective hosts, including renal adenocarcinomas of frogs and epithelial tumors of salmonid fish.
Oncogenic Poxviruses
Although some poxviruses are regularly associated with the development of benign tumor-like lesions (see Chapter 7), there is no evidence that these ever become malignant, nor is there evidence that poxvirus DNA is ever integrated into cellular DNA. A very early viral protein produced in poxvirus-infected cells displays homology with epidermal growth factor and is probably responsible for the epithelial hyperplasia characteristic of many poxvirus infections. For some poxviruses (e.g., fowlpox, orf, and rabbit fibroma viruses), epithelial hyperplasia is a dominant clinical manifestation and may be a consequence of a more potent form of the poxvirus epidermal growth factor homologue.