

ABSTRACT
Saccharomyces cerevisiae is the most extensively studied yeast and, over the last century, provided insights on the physiology, genetics, cellular biology and molecular mechanisms of eukaryotes. More recently, the increase in the discovery of wild strains, species and hybrids of the genus Saccharomyces has shifted the attention towards studies on genome evolution, ecology and biogeography, with the yeast becoming a model system for population genomic studies. The genus currently comprises eight species, some of clear industrial importance, while others are confined to natural environments, such as wild forests devoid from human domestication activities. To date, numerous studies showed that some Saccharomyces species form genetically diverged populations that are structured by geography, ecology or domestication activity and that the yeast species can also hybridize readily both in natural and domesticated environments. Much emphasis is now placed on the evolutionary process that drives phenotypic diversity between species, hybrids and populations to allow adaptation to different niches. Here, we provide an update of the biodiversity, ecology and population structure of the Saccharomyces species, and recapitulate the current knowledge on the natural history of Saccharomyces genus.
Keywords: Saccharomyes genus, yeast ecology, yeast hybrids, biodiversity
The increase in the discovery of wild strains, species and hybrids of the genus Saccharomyces has shifted the attention towards studies on genome evolution, ecology and biogeography. Here, we provide an update of the biodiversity and population genomics of the Saccharomyces species and recapitulate the current knowledge on the natural history of Saccharomyces genus.
FOREWORDS
The genus Saccharomyces includes S. cerevisiae a well-known organism in industrial baking and fermentation processes as well as in bioenergy and biomedical fields (Mager and Winderickx 2005; Fukuda, Kondo and Tamalampudi 2009; Sicard and Legras 2011). Since the release of the full genome sequence of S. cerevisiae in 1996, extensive functional annotations has started making it the most well-known eukaryotic system to date (Goffeau et al. 1996). The availability of a reliable reference genome and the development of high-throughput sequencing subsequently facilitated the whole-genome sequencing and the robust annotation of a large number of Saccharomyces species (Cliften et al. 2003; Kellis et al. 2003; Nakao et al. 2009; Scannell et al. 2011; Liti et al. 2013; Hewitt et al. 2014; Baker et al. 2015; Naseeb et al. 2018). With the increase of ecological surveys of Saccharomyces species in nature, these species become models for studies on population genomics (Liti et al. 2009; Wang et al. 2012; Duan et al. 2018; Peter et al. 2018). Moreover, robust whole genome sequencing, led to large-scale genomic studies of a variety of strains of Saccharomyces species, providing insight into their evolution and natural variation (Warringer et al. 2011; Bergstrom et al. 2014; Gallone et al. 2016; Peter et al. 2018).
Research on ecological diversity, population genomics and phenotypic variation for industrial application for both wild and domesticated Saccharomyces species have been excelling throughout the last decade. However, the biodiversity and true niche and abundance of the different species remain ambiguous. In this review, we present an overview on the genus Saccharomyces focusing on the species biodiversity, ecological niches and population genomics.
SPECIES OF THE GENUS SACCHAROMYCES
The name Saccharomyces was proposed by J. Meyen in 1838, with S. cerevisiae being the first described species. In 1870, M. Reess presented a description of the genus and species that included the yeasts associated with fermentation (Rainieri, Zambonelli and Kaneko 2003). The Saccharomyces sensu stricto group was initially described in 1970 in the second edition of ‘The Yeast: A Taxonomic Study’, (Lodder 1970) and originally comprised of 21 species (Teresa Fernandez-Espinar, Barrio and Querol 2003). Over the years, the Saccharomyces genus has evolved through taxonomic rearrangements, in which several taxa have been removed and placed in the sister group Saccharomyces sensu lato (Fig. 1). In the past, conventional taxonomic methods were employed which had limitations such as the differentiation of strains within a species based solely on morphological and a few physiological characteristics. These limitations have encouraged the integration of molecular methods, such as DNA re-association, chromosomal karyotyping, restriction fragment length polymorphism (RFLP) and sequencing of multiple loci for the classification of Saccharomyces species (Vaughan-Martini, Martini and Cardinali 1993; Guillamon et al. 1994; Naumov et al. 2000b; Kurtzman and Piškur 2006). In 2003, Kurtzman and Barnette established Saccharomyces complex as a monophyletic group phylogenetically distinct from Saccharomyces sensu lato species. Species of the ‘sensu lato’ group were then reclassified into new species, thus resulting in the termination of the phrases ‘sensu stricto’ and ‘sensu lato’ (Fig. 1A and B, Kurtzman and Robnett 2003). Saccharomyces species propagate asexually via budding but are also capable of mating followed by meiosis when the nutrients in the environment become scarce. The presence of sexual reproduction in the yeasts enabled taxonomists to differentiate between the species using the biological species concept (BSC), where only hybridization events within the same species will produce fertile hybrids. Therefore, the production of sterile offspring indicates that the parents belong to two different species (Naumov 1996). The use of BSC has been the method of choice for the taxonomy of budding yeasts given in support of molecular methods. Currently, the advances in DNA sequencing technology allowed quick acquisition of a large amount of genomic data for several species. This enabled a solid resolution of the yeast taxonomy to the strain level and prompted another revision of the classification of the Saccharomyces species based on phylogenetic analysis (Fig. 1C).
Figure 1.

The genus Saccharomyces taxonomic rearrangements. The panels show the main changes in the Saccharomyces species taxonomy within the sensu stricto group over the years. A) In 1998, 14 species were included in the ‘sensu stricto’ group (Vaughan-Martini and Martini 1998). B) In 2003, several species were reclassified and removed abolishing the group names ‘sensu stricto’ and ‘sensu lato’ (Kurtzman and Robnett 2003). Wild species previously isolated were confirmed as distinct Saccharomyces species using molecular and genetic hybridization methods, adding S. mikatae, S. kudriavzevii and S. cariocanus to the group (Naumov et al. 2000b). C) From the year 2003 to 2011 further novel species were discovered from nature and other species were reclassified (Naumov 2000a; Wang and Bai 2008; Libkind et al. 2011). D) Now, the Saccharomyces genus consists of eight species and two natural hybrids (Boynton and Greig 2014; Naseeb et al. 2017). Previous taxonomical names of the species are in parenthesis.
The genus Saccharomyces is now consisting of eight species, namely; S. cerevisiae, S. paradoxus, S. mikatae, S. jurei, S. kudriavzevii, S. arboricola, S. eubayanus and S. uvarum. Some of these species are parents of natural hybrids that either formed spontaneously in the wild without the involvement of humans or in habitats created by humans e.g. industrial environments (Fig. 1D). All the initially described Saccharomyces species were linked to domestication and S. paradoxus, the closest relative to S. cerevisiae, was the first wild Saccharomyces species to be isolated from oak and birch sap in Russia and Ukraine. Based on DNA re-association and genetic hybridization analyses, species that were previously described as S. cerevisiae var. tetrasporus and S. cerevisiae var. terrestris are now known as synonyms of S. paradoxus (Martini 1989; Naumov 1996). Subsequently, two Saccharomyces species were isolated from decayed leaves and soil in Japan and one from the Drosophila species in Brazil that were reproductively isolated, with distinct chromosomal profiles, (Naumov, Naumova and Louis 1995a; Naumov et al. 1995b). The species isolated from Brazil was described as S. cariocanus (now reclassified as S. paradoxus based on the low sequence divergence between the species), while the two species from Japan were described as S. kudriavzevii and S. mikatae (Naumov et al. 2000b). In 2008, S. arboricola was isolated from the bark of broadleaf trees in China (Wang and Bai 2008).
The classification of S. uvarum and S. bayanus was controversial and went through several revisions (Naumov 1996; Nguyen and Gaillardin 1997; Nguyen, Lepingle and Gaillardin 2000). S. bayanus has been recognized as a complex cryotolerant species separated into two varieties; the heterogenous strains belonging to S. bayanus var. bayanus and the homogenous strains S. bayanus var. uvarum (Vaughan-Martini and Martini 2011). S. bayanus var. uvarum consist of a pure lineage strain with no genomic contribution from other Saccharomyces species, thus is now known as a distinct species named S. uvarum (Fig. 1C, Rainieri et al. 1999; Nguyen, Lepingle and Gaillardin 2000; Pulvirenti et al. 2000). Recently, the separation of S. bayanus into two varieties based on the BSC is considered taxonomically invalid and S. uvarum stands as s real species and not a variety of S. bayanus (Nguyen and Boekhout 2017). However, S. bayanus var. bayanus is now recognized as a natural hybrid rather than a true species. The isolation of S. eubayanus from a southern beech (Nothofagus spp.) tree in Patagonia, Argentina resolved the taxonomic classification of S. bayanus. A comparative genomic analysis revealed that the S. bayanus CBS 380T genome is composed of 67% S. uvarum and 33% S. eubayanus sequences with introgressions from S. cerevisiae, making S. bayanus a hybrid between these three species (Fig. 1D, Libkind et al. 2011). The latest addition to the Saccharomyces genus is S. jurei, which was isolated from oak bark and the surrounding soil in the pre-Alps near Saint-Aubin, France. This species is reproductively isolated and phylogenetically distinct from all members of the Saccharomyces species. S. jurei is genealogically closely related to S. mikatae, S. paradoxus and S. cerevisiae based on sequences of the internal transcribed region (ITS1–5.8S-ITS2) and the D1/D2 domains of the 26S rRNA (Naseeb et al. 2017). Whole-genome sequencing and phylogenetic analyses of a concatenation of 101 universally distributed orthologs placed S. jurei and S. mikatae in a monophyletic group. In addition, the S. jurei species possesses two chromosomal translocations, one of which is shared with the two S. mikatae strains IFO1815 and IFO1816, suggesting a common evolutionary history (Naseeb et al. 2018).
BIOGEOGRAPHY OF SACCHAROMYCES SPECIES
Saccharomyces cerevisiae
The phylogenetic analysis of wild and domesticated S. cerevisiae strains has revealed a complex population structure (Fay and Benavides 2005; Liti et al. 2009). The first population genomic studies used genome wide single nucleotide polymorphisms (SNPs) analysis to cluster S. cerevisiae strains into five delineated populations that correlated with isolation regions and fermentation types: North American, Malaysian, West African, sake and wine/European (Table 1). However, some strains (primarily human-related) were not assigned to a specific lineage and were labelled as mosaic due to the polymorphic nature of their genome (Liti et al. 2009). A further study surveying New Zealand habitats found seven distinct S. cerevisiae subpopulations isolated from soil, bark, flowers and spontaneous ferment (Goddard et al. 2010). Interestingly, the New Zealand strains are phylogenetically closely related to the European population, as shown by the number of shared alleles (Cromie et al. 2013). Another large-scale field survey of primeval forests in China resulted in the isolation of 99 wild S. cerevisiae strains belonging to eight distinct lineages that were partially reproductively isolated (10.2% to 89.1% spore viability) (Wang et al. 2012). More recently, genome-wide SNPs analyses of over 200 wild and domesticated Chinese strains revealed two new wild lineages increasing the number of the Chinese populations to twelve (Duan et al. 2018). Phylogenetic analysis of the Chinese strains and S. cerevisiae of worldwide origins revealed one of the Chinese populations to be the most ancient, forming the basal lineage of the phylogenetic tree. The high number of genetically diverged lineages present in this region indicated that the species had an Asian origin (Duan et al. 2018). Such view has been recently supported by Peter and co-workers in their analysis of SNPs in 1011 S. cerevisiae strains of domesticated, human and wild origins using statistical dimension reduction tools, which supported the hypothesis of an origin of this species outside China (Peter et al. 2018).
Table 1.
Common niches and global distribution of the wild Saccharomyces populations.
| Species | Ecology | Populations |
|---|---|---|
| S. cerevisiae | Broadly associated with bark and soil Fagales order trees | Asian, European, North American and South American |
| S. paradoxus | Broadly associated with bark and soil of Qurecus spp. | Asian, European, North American (America A/Europe, America B and America C) |
| S. eubayanus | Broadly associated with Nothofagus spp. | Patagonian A, Patagonian B/Holarctic (North America and Tibet strains) and West Chinese |
| S. uvarum | Broadly associated with Nothofagus spp. and other Fagales spp. | South American A/Holarctic, South American B and Australasian |
| S. kudriavzevii | Decayed leaf, soil, bark of mainly Quercus spp. | European (Portugal, Spain & France) and Asian (Taiwan and Japan) |
| S. arboricola | The bark of Quercus fabri, Castanopsis orthacantha, soil and seeds, Fruiting body of Auricularia polytricha | Asian (China and Taiwan) and Australasian (New Zealand) |
| S. mikatae | Soil and decayed leaf | Asian (Japan) |
| S. jurei | Bark and soil of Quercus robur | European (France) |
A distinct monophyletic lineage of a wild population of S. cerevisiae associated with Mediterranean oak (MO) was only detected in southern Europe (Almeida et al. 2015). The MO population is closely related to the wine population based on genome wide analysis. Strains of the wild MO population were shown to be the source of the ancestor domesticated strains (wine strains) based on population demographics analysis (Almeida et al. 2015). However, Duan et al. (2018) proposed that the wine strains originated in Asia as proven by clustering a few wild Chinese isolates with the wine lineage and sharing horizontally transferred genes between strains of the two populations.
A novel South American population was isolated from Brazil and grouped into a single clade that is clearly separated from the other previously known populations. Some of these strains displayed a mosaic genome, and 54% of the Brazilian strains had only a small amount of introgression from the wine population strains, suggesting a previous domestication in the history of S. cerevisiae (Barbosa et al. 2016). More sampling in a systematic way which will encourage the exploration of undescribed Saccharomyces populations.
Saccharomyces paradoxus
In contrast to S. cerevisiae, S. paradoxus has been almost completely limited to wild environments, and forms well-structured populations that are related to a geographic origin and that are less phenotypically diverse than S. cerevisiae (Liti et al. 2009, Warringer et al. 2011). S. paradoxus strains were originally designated into three geographically-structured populations: Far Eastern, European and North American, with a less defined Hawaiian population represented by a single strain (Liti, Barton and Louis 2006; Liti, Barton and Louis 2009) (Table 1). These populations are partially reproductively isolated and are diverged by 1.5% to 4.6% (Liti, Barton and Louis 2006). The North American population is further divided into three lineages: America A/Europe, America B and America C, (Table 1). The America A/Europe lineage includes European strains that are thought to have recently migrated to North America. The American populations shows about 2.0% to 3% inter-lineage nucleotide divergence based on the genes POP2 and RPB2 (Leducq et al. 2014). These lineages co-exist in partial sympatry in North America, showing secondary contact of original populations that diverged allopatrically (Kuehne et al. 2007; Leducq et al. 2014). The secondary introduction of a diverged population also expanded the geographical distribution of the European population. S. paradoxus strains that are highly similar to the European ones have also been detected in New Zealand; it has been proposed that the European strains were introduced to the region through the shipment of oak acorns from Australia or the United Kingdom (Zhang et al. 2010). In addition to the cases of occupancy overlap with the America A and B lineages, the American lineages are generally broadly separated along a north-south gradient in North America. The lineages show phenotypic divergence reflecting the differences in their ability to adapt to local temperature that influenced their distribution (Leducq et al. 2014). Partial post-zygotic isolation has been demonstrated within and between the genetically and phenotypically diverged North American populations that were associated with chromosomal rearrangements, indicating the early stages of speciation (Charron, Leducq and Landry 2014a).
Saccharomyces eubayanus
S. eubayanus strains were initially isolated in Patagonia (Argentina) and are clustered into two lineages: Patagonia A and Patagonia B. A few strains that were later isolated from North America (Wisconsin) were identified as being a mixture of the two lineages (Table 1). The Patagonia B lineage is diverged from the Patagonia A lineage, revealing a divergence of 0.93%, based on the sequences of nine nuclear genes and a mitochondrial gene (Peris et al. 2014). A single S. eubayanus strain that was isolated from New Zealand was clustered with the Patagonia B lineage, according to the phylogenetic analyses of six loci (Gayevskiy and Goddard 2016). The distribution of S. eubayanus has extended to Far East Asia, where three lineages have been discovered in different regions of China: West China, Sichuan and Tibet/Lager. The genetic diversity within the Asian population is up to 7.57% (multilocus analysis may overestimate sequence divergence between species in comparison to genome-wide analysis), which was higher than what has been recorded between the Patagonia A and B lineages (Bing et al. 2014). Multilocus phylogenetic analyses of previously known strains and of strains from North America (Washington, North Carolina and Canada) have identified a new clade that includes strains with a Holarctic distribution genetically closely related to the Patagonia B population (0.56% genetic distance based on the complete genome). Based on the latest molecular analyses, the three main S. eubayanus populations have been recognized as Patagonia A, Patagonia B/Holarctic including strains from North America and Tibet and West Chinese (Peris et al. 2016a). Extensive sampling of Nothofagus sp. trees in South America revealed a uniquely high isolation frequency of S. eubayanus strains and genome-wide sequencing added depth to the phylogeny of the specie populations (Eizaguirre et al. 2018; Langdon et al. 2019; Nespolo et al. 2019). Adding to the complexity of the S. eubayanus populations, six sub-populations are now recognized (PA1, PA2, PB1, PB2, PB3 and Holarctic) in addition to admixture populations (Langdon et al. 2019).
Saccharomyces uvarum
The whole-genome data of the S. uvarum strains that are associated with wild and domesticated environments in North and South America, Eurasia and Australasia have been phylogenetically analysed and grouped into three clades: South American A/Holarctic, South America B and Australasia (Table 1) (Almeida et al. 2014). The South American A/Holarctic clade primarily includes strains that have been isolated from Holarctic regions, along with a few South American strains, while the B clade only contains South American strains. The Australasian lineage is distinctly separated from the other populations, with 4.4% genome divergence, and is partially reproductively isolated from the other S. uvarum strains. The highest level of species diversity has been found in the Southern Hemisphere, where two populations have diverged by 1%. This high level of diversity was demonstrated by the pairwise nucleotide diversity of the South American isolates compared to the Holarctic and Australasian isolates (0.689 vs 0.141 and 0.162, respectively). The low diversity of the Holarctic isolates and the phylogenetic grouping of the strains within the South American A lineage suggests that the Holarctic population is derived from the South American A population and only recently migrated into the Northern Hemisphere (Almeida et al. 2014).
Saccharomyces kudriavzevii
The S. kudriavzevii species is currently represented by Asian strains that have been isolated from Japan and Taiwan and European strains that have been isolated from Portugal , Spain and France (Table 1) (Naumov et al. 2000b; Sampaio and Goncalves 2008; Lopes, Barrio and Querol 2010; Erny et al. 2012; Naumov, Lee and Naumova 2013). Multilocus sequence analyses of the S. kudriavzevii strains that have been isolated from Europe (Spain and Portugal) have revealed that the strains are closely related, with a nucleotide diversity of 0.21%. These strains are diverged by 0.51% from the Japanese type strain (IFO 1802T); consequently, they were assigned to an Iberian/European population (Peris et al. 2016b). Based on genome-wide sequencing analysis, a single Japanese S. kudriavzevii strain (IFO 1803) was shown to be diverged from the other known strains by ∼4% (Hittinger et al. 2010). Recently, a large number of S. kudriavzevii strains were isolated from the Italian Carnic Alps that showed phenotypic variation (Alsammar 2018). These strains are closely related to the European strains (CA111 and ZP629) based on multiloci analysis, but form a distinct sub-population based on whole genome SNPs analysis (Alsammar 2018). A feature that distinctly differentiates the European strains from the Asian strains is the ability to utilize galactose of the former. The Japanese strains have retained pseudogenes of the seven GAL pathway genes, but they are heavily mutated, rendering them non-functional (Hittinger, Rokas and Carroll 2004; Hittinger et al. 2010). The previous population genomics study of S. kudriavzevii did not include the Taiwanese strains, however, phylogenetic analyses of the D1/D2 and ITS1 sequences clustered most of the Taiwanese strains with the Japanese IFO 1803 strain, while others were grouped with the Portuguese strains and the Japanese type strain IFO 1802T (Naumov, Lee and Naumova 2013). Interestingly, the distribution of S. kudriavzevii seems to be restricted to Europe and Asia, since it has not been isolated from other regions that are densely populated with well-structured populations of Saccharomyces species, such as North or South America. A comprehensive population genomics study for this species that includes all the strains that have been isolated from the different regions has not yet been conducted, however it seems clear that S. kudriavzevii strains are grouped into an Asian population (that includes the Japanese and Taiwanese strains) and a European population composed of the strains that have been isolated from Portugal, Spain and France (Table 1).
Saccharomyces arboricola
To date, the distribution of S. arboricola has been limited to Far East Asia (China and Taiwan) and Australasia (New Zealand, Table 1) (Wang and Bai 2008; Naumov, Lee and Naumova 2013; Gayevskiy and Goddard 2016). The Chinese strains closely resemble the Taiwanese strains, as the type strain exhibits ITS and D1/D2 sequences that are identical to the Taiwanese strains (Wang and Bai 2008; Naumov, Lee and Naumova 2013). Nine S. arboricola strains that were isolated from soil in New Zealand possess a genome divergence of 2.6% from a Chinese reference strain (Gayevskiy and Goddard 2016).
Saccharomyces mikatae and Saccharomyces jurei
S. mikatae has only been isolated in Japan, and it encompasses two strains, IFO 1815T and IFO 1816 (Table 1) (Naumov et al. 2000b). Similarly, S. jurei has been found only in Europe, with two strains, NCYC 3947T and NCYC 3962, isolated from oak bark and soil, respectively, in the French pre-Alps (Naseeb et al. 2017).
SACCHAROMYCES INTERSPECIFIC HYBRIDS
Species of Saccharomyces readily hybridize due to the absence of significant prezygotic barriers, and produce hybrids that are sterile primarily due to sequence divergence among the species (Morales and Dujon 2012). Hybrids among Saccharomyces species are common in industrial fermentation environments involved in brewing and wine making process (Fig. 2) (Sicard and Legras 2011), however, they are scarcely reported in wild (Barbosa et al. 2016) and medical samples (Peris et al. 2018). Hybridization is advantageous in Saccharomyces evolution, since it introduces high genetic variation leading to novel lineage conferring hybrid vigour and wider adaptation potential (Gonzalez et al. 2007; Belloch et al. 2008; Piatkowska et al. 2013).
Figure 2.

Common Saccharomyces hybrids and the source of their isolation. Saccharomyces may hybridize forming double or triple hybrids that are of industrial significance. Most of the known hybrids are associated with domestication activities and a few strains isolated from non-fermentation environments.
The most well-known industrial hybrid is S. pastorianus, resulting from the cross between S. cerevisiae and S. eubayanus (Fig. 1D, syn. S. carlsbergensis). This hybrid has been used for centuries in brewing and is responsible for lager production, which is conducted at low temperatures (5–14°C), in contrast to ale brewing which occurs at higher temperatures (15–24°C) and is carried out by S. cerevisiae (Sicard and Legras 2011). The cold-tolerant nature of S. pastorianus allows the species to ferment at low temperatures, a trait inherited from the S. eubayanus parent; meanwhile, the S. cerevisiae sub-genome contributes to the hybrid's ability to ferment maltotriose (Hebly et al. 2015).
Array comparative genomic hybridization analysis of several S. pastorianus strains identified two distinct lineages, based on differences in chromosome content, chromosome structure and ploidy, namely; Saaz-type (group 1) and Frohberg (group 2), named after the region of initial isolation and the region of brewing, respectively (Dunn and Sherlock 2008). The origin of the S. eubayanus lager yeast parent was thought to be South American, due to the high abundance of this species in that region, introduced to European brewing after early trans-Atlantic trade (Libkind et al. 2011). However, brewing originated in Bavaria during medieval time and rapidly expanded in the 1400s, long before the beginning of the trans-Atlantic trade in the 1500s. Following the S. eubayanus discovery in Patagonia, Asian populations of the species were isolated from various regions in China, and the genome of a Tibetan strain was shown to be 99.82% similar to the S. eubayanus subgenome of the lager yeast making it the more likely parent of the lager yeasts, with S. cerevisiae being the other parent. This discovery led scientists to hypothesize that S. eubayanus was introduced to Europe through the silk road (Bing et al. 2014). However, genome-wide pairwise nucleotide sequence divergence analysis revealed regions in the Tibetan strains that are more similar to North Carolina strains than to S. pastorianus, which was also supported by phylogenetic analysis (Peris et al. 2016a). Based on these findings Peris et al. (2016a) concluded that none of the known S. eubayanus is with certainty the nearest parent of S. pastorianus.S. eubayanus has still not been isolated in Europe, although DNA signals of the species were detected in soil of Italian mountain regions (Alsammar et al. 2019).
The genetic differences between group 1 and group 2 lager yeasts was explained by independent hybridization of group 1 and group 2 lager hybrids (Monerawela et al. 2015). However, the presence of conserved chromosomal translocation events in strains of both groups suggest a common ancestor (Walther, Hesselbart and Wendland 2014; Okuno et al. 2016). The latest SNPs analysis by Okuno et al. (2016) sheds light on the evolution of the lager yeasts, which suggests at least a single common hybridization event between the groups. The authors proposed two possible theories to explain the hybridization origin of the lager yeasts (Fig. 3): 1. A common ancestor originates from the hybridization of a diploid ale-type S. cerevisiae and a diploid S. eubayanus resulting in group 2 (4n) strains. Chromosomal deletions in S. cerevisiae genome of the 4n hybrid gave rise to to group 1 (3n) strains (Fig. 3A). 2. An initial hybridization of a haploid ale-type S. cerevisiae with a diploid S. eubayanus producing the ancestral group 1 (3n) yeasts, followed by a second hybridization with haploid S. cerevisiae strain resulting in the ancestor of group 2 (4n) yeasts (Fig. 3B).
Figure 3.
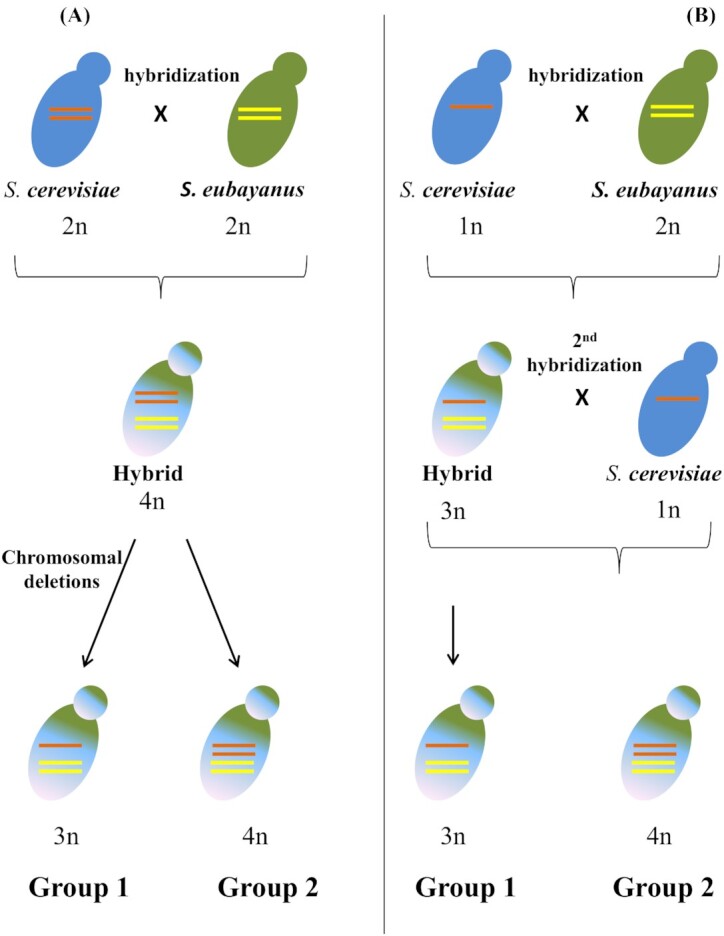
Origin of S. pastorianus group 1 and group 2 strains based on two theories. (A) hybridization between diploid S. cerevisiae and S. eubayanus followed by chromosomal deletions in the S. cerevisiae sub- genome of group 1 strains. (B) The hybridization of a haploid S. cerevisiae and a diploid S. eubayanus lead to a triploid hybrids (3n) followed by a second hybridization event in group 2 strains (4n) (Figure adapted from Okuno et al. (2016).
Genetic analysis of beer, wine and cider Saccharomyces strains lead to the discovery of other natural double interspecific hybrids (Fig. 2), S. cerevisiae x S. uvarum and S. cerevisiae x S. kudriavzevii, including triple hybrids, S. cerevisiae x S. kudriavzevii x S. uvarum (Masneuf et al. 1998; Bradbury et al. 2006; Gonzalez et al. 2006; Lopandic et al. 2007; Gonzalez, Barrio and Querol 2008; Peris et al. 2012a). S. cerevisiae x S. kudriavzevii hybrids were also isolated from clinical samples and dietry supplement (Peris et al. 2012a).
Phylogenetic analysis of the European S. kudriavzevii strains showed that they are more closely related to the natural S. cerevisiae x S. kudriavzevii hybrids (associated with fermentation in central Europe) than the Asian strains, thus indicating an hybridization of European origin (Sampaio and Goncalves 2008; Lopes, Barrio and Querol 2010). Unlike S. cerevisiae, S. kudriavzevii has not been found in fermentative environments, suggesting that the hybridization event between S. cerevisiae and S. kudriavzevii may have taken place in the wild before the hybrids expanded to domesticated settings (Belloch et al. 2009).
The proportion of S. kudriavzevii genome relative to S. cerevisiae genome in the hybrids differs between strains (Belloch et al. 2009; Erny et al. 2012; Peris et al. 2012b; Borneman et al. 2016). The hybrids with a higher proportion of S. cerevisiae sub-genome are better adapted to fermentation stresses, while the hybrids with higher amount of S. kudriavzevii sub-genome are more efficient at fermentation at low temperature (Belloch et al. 2008; Peris et al. 2012b).
Other hybrids have also been isolated in the wild such as those between S. cerevisiae x S. paradoxus (Barbosa et al. 2016). The clear introgressions in S. cerevisiae coming from S. paradoxus genome support the occurrence of hybridization of these two species in the wild (Barbosa et al. 2016). A considerable number of open reading frames (ORFs) belonging to S. paradoxus were recorded to be introgressed in the genomes of S. cerevisiae analysed by Peter et al 2018. Recently, S. cerevisiae x S. paradoxus hybrids were isolated from processed olives and olive products (Fig. 2). These hybrids in addition to other S. cerevisiae x S. paradoxus previously isolated from the similar substrates formed a distinct lineage named the ‘olives clad’(Pontes et al. 2019).
Genetic analysis of the North American S. paradoxus lineages that are partially sympatric revealed hybridization events within the natural lineages suggesting the occurance of hybridization in nature. The genome of the hybrid species (SpC*) is a mosaic of the North American lineage SpB and SpC genotypes due to the secondary contact between the parental lineages.The phenotypic growth response of the hybrid lineage is unique, corresponding to conditions of the contact region between the hybrid's parents (Leducq et al. 2016). Recently, novel intraspecific hybrids (SpD) generated between backcrossing of the hybrid species SpC* and its parental lineage SpB were isolated from natural environments. SpD hybrids revealed partial reproduction isolation with the North American lineages and a distinct growth and transcriptome profiles, thus leading to the increasing chance of hybrid formation and persistence in nature (Eberlein et al. 2019).
ECOLOGY OF SACCHAROMYCES SPECIES
The fermentation processes of domesticated Saccharomyces species have been thoroughly studied, leading early ecological studies to investigate fermentation-related environments, such as breweries and vineyards, as the typical habitats of Saccharomyces species (Sampaio and Gonçalves 2017). However, most Saccharomyces species are now recognized as being wild species that are isolated from environments not related to human activity (Naumov, Naumova and Sniegowski 1998; Naumov 2000a; Wang and Bai 2008; Libkind et al. 2011; Naseeb et al. 2017). Some species are present in both wild habitats and domesticated environments (Almeida et al. 2014; Peter et al. 2018). The ecology of S. cerevisiae extends to human guts and may be correlated to disorders such as irritable bowel syndrome (Nash et al. 2017; Sokol et al. 2017). The differentiation between wild and domesticated Saccharomyces populations reflects distinct genomic evolutions history shown by differences in chromosomal cores and subtelomeres (Yue et al. 2017).
The hybrids S. pastorianus and S. bayanus have not been isolated from natural environments and are strictly associated with brewing environments (Rainieri et al. 2006). Subsequently, they were maintained due to brewing-related selection pressures (Dunn and Sherlock 2008; Libkind et al. 2011). S. paradoxus, S. mikatae, S. jurei, S. kudriavzevii, S. arboricola and S. eubayanus are purely wild species, while S. cerevisiae and S. uvarum encompass domesticated and wild strains . The wild species are commonly associated with tree substrates, such as bark, soil, leaves, exudates and litter. The frequent isolation of Saccharomyces species, especially S. paradoxus and S. cerevisiae, from Quercus spp. (oak) led to the hypothesis that this particular tree is the yeasts’ natural habitat (Naumov, Naumova and Sniegowski 1998; Sniegowski, Dombrowski and Fingerman 2002; Johnson et al. 2004; Sampaio and Goncalves 2008; Hyma and Fay 2013; Charron et al. 2014b). However, Saccharomyces species have also been isolated from several other tree species (Table 1), extending the their habitat to the order Fagales (Sampaio and Goncalves 2008; Libkind et al. 2011; Alsammar 2018). In fact, the absence of Quercus spp. from South America encouraged the exploration of native tree species, such as Nothofagus sp. (Southern beech, a member of the order Fagales), as well as the sugar-rich fruiting stromata of Cyttaria hariotii (a tree parasite) which resulted in the isolation of S. eubayanus and S. uvarum (Libkind et al. 2011). These species have also been isolated from Araucaria araucana, a native South American tree (Rodriguez et al. 2014). The presence of these species in the Southern Hemisphere is correlated with the native tree species, suggesting that the species are well-established in this region (Rodriguez et al. 2014). In contrast, S. uvarum has been isolated in at low frequency, primarily from Quercus spp. in Europe and was also isolated from the Nothofagus that are present in New Zealand and Tasmania (Almeida et al. 2014). S. eubayanus have been detected in North America and China, primarily associated with Quercus spp. (Bing et al. 2014, Peris et al. 2016a), while the isolation of a single S. eubayanus in New Zealand was from sampling fruits, bark and soil of trees that were native to the region (Gayevskiy and Goddard 2016).
Oak trees are the most common host for Saccharomyces species in the Northern hemisphere. S. paradoxus specifically is frequently isolated from oak bark, soil and exudates; in some cases, this species has been isolated in sympatry with S. cerevisiae (Naumov, Naumova and Sniegowski 1998; Sniegowski, Dombrowski and Fingerman 2002; Sampaio and Goncalves 2008; Sampaio and Gonçalves 2017). Large ecological surveys of Saccharomyces species have demonstrated the specificity of S. paradoxus to oak trees. Tha majority of trees sampled from different regions in Canada harboured a 3-fold higher percentage of S. paradoxus compared to other tree species (Charron et al. 2014b). Sampling of various trees in the United States has also revealed a significant association of S. paradoxus with oak trees (Sylvester et al. 2015). However, neither of these studies successfully isolated S. cerevisiae, whose presence may have been restricted by the northern limit of the sampling regions. Both species differ in their thermal growth profiles, with S. cerevisiae having a higher optimum temperature than S. paradoxus. Therefore, the absence of S. cerevisiae may have been affected by lower temperatures of the sampling areas (Sweeney et al. 2004; Salvado et al. 2011). A possible explanation for the general frequent isolation of Saccharomyces species from trees bark is that the species might seek refuge in the tree bark during seasonal changes (Goddard and Greig 2015). Despite the common association of Saccharomyces species with oak tree bark, Kowallik and Greig (2016) showed that samples of leaf litter surrounding oak trees yielded a higher abundance of S. paradoxus than from bark suggesting that the yeasts may be dispersed from tree bark to litter by rainwater or insects (Kowallik and Greig 2016). The tree bark niche for the Saccharomyces species is not fully understood, as the sugar content of this habitat is too low to support the growth of Crabtree-positive yeast species (Boynton and Greig 2014). The presence of Saccharomyces species on bark has been correlated with the presence of hexoses sugars, which may explain the species’ occurrence (Sampaio and Goncalves 2008). Analysis of the human gut microbiome revealed the abundance of S. cerevisiae found in 92.2% of the sampled volunteers, indicating that the species is a common resident of the gut (Nash et al. 2017). A shift in the abundance of S. cerevisiae in the human gut was shown to be associated with inflammatory bowel disease microbiota dysbiosis (Sokol et al. 2017).
S. uvarum is associated with wine and cider fermentation, however, it is not considered to be fully domesticated, as strains have been isolated from several natural environments (Sampaio and Goncalves 2008; Libkind et al. 2011). Although the numbers of S. uvarum isolates are generally low in comparison to other species, they have a global distribution, with the Southern Hemisphere harbouring a high abundance of the species (Almeida et al. 2014). Similarly, S. cerevisiae has a global distribution being isolated from natural environments in North America, China and Europe, as well as domesticated ones such as vineyards, fruits and insects (Sniegowski, Dombrowski and Fingerman 2002; Stefanini et al. 2012; Wang et al. 2012; Hyma and Fay 2013; Almeida et al. 2015). Phylogenetic analysis of S. cerevisiae species has revealed that the wild strains have the oldest lineages and are located at the root of the phylogenetic tree; moreover, wild strains have a higher genetic diversity than most domesticated strains, suggesting that the domesticated strains are derived from the natural populations (Fay and Benavides 2005; Wang et al. 2012; Almeida et al. 2015). The association of S. cerevisiae with Drosophila spp., bees and wasps, especially in regions that are populated with fruits, represents a source of the yeast's dispersal that maintains genetic diversity and protection during unfavourable seasonal climates (Goddard et al. 2010; Stefanini et al. 2012; Buser et al. 2014).
Despite the enrichment culture's sensitivity for the isolation of the Saccharomyces species from environmental samples (Kowallik, Miller and Greig 2015), the method may introduce biases toward the isolation of one or a few species that can outcompete others in the selection media. If the Saccharomyces species are outgrown by other species in the sample, the actual species distribution may be underestimated (Boynton and Greig 2014; Goddard and Greig 2015). Moreover, the enrichment culture method will not reveal the actual abundance of the species in a natural environment, as a single cell might propagate, forming cell clones and lead to an overestimation of the species’ existence. For example, Kowallik, Miller and Greig (2015) reported that S. paradoxus was rare on oak bark, as demonstrated when the bark samples were inoculated in a malt extract medium that had been supplemented with lactic acid and was outcompeted by surrounding microbial species.
Differences in growth temperatures of the Saccharomyces species influence their ecological interactions in nature. Wild species with different temperature growth profiles have been reported to occupy the same habitat (Sweeney et al. 2004; Sampaio and Goncalves 2008; Paget, Schwartz and Delner 2014) such as the coexistence of S. paradoxus and S. cerevisiae on oak bark from a single sampling site in North American (Sniegowski, Dombrowski and Fingerman 2002). Moreover, the incubating tree bark at high (30°C) and low temperatures (10°C) resulted in the isolation of S. cerevisiae coupled with S. kudriavzevii and S. paradoxus and with S. uvarum (Sampaio and Goncalves 2008). The thermo-niche adaptation is due to differences in optimal growth temperatures and circadian temperature changes that allows the alternating growth of the species, thus preventing the abundance of one species over the other.
DNA SIGNALS OF SACCHAROMYCES SPECIES IN NATURE
To avoid culturing biases and to determine the actual abundance of the Saccharomyces species in their natural habitat a high-throughput sequencing of environmental DNA (eDNA) extracted from bark, soil and vineyard samples was employed by several research groups (Taylor et al. 2014; Kowallik, Miller and Greig 2015; Dashko et al. 2016; Alsammar et al. 2019). Pyrosequencing of bark samples and bark infusions did not result in the detection of any of the Saccharomyces species (Kowallik, Miller and Greig 2015). High-throughput sequencing of grapes collected from vineyards of different regions in New Zealand yielded only S. cerevisiae at an abundance of 1:20 000 (Taylor et al. 2014). DNA signatures of S. cerevisiae, S. paradoxus, S. mikatae and S. pastorianus were detected in oak bark and soil of vineyard trees and wine must samples in Slovenia. The Saccharomyces were rare in the bark and soil samples, however, S. cerevisiae and S. paradoxus were the dominant species in must samples (Dashko et al. 2016). Targeting Saccharomyces eDNA based on the size of the ITS region extracted from soil surrounding different tree species at varying altitudes succeeded in the detection of most species of the Saccharomyces species in low abundance in comparison to other fungi (Alsammar et al. 2019). Although S. mikatae was not isolated outside Asia, metagenomic signature of the species has been detected in grape must in Europe (Dashko et al. 2016) and in soil surrounding oak, spruce and beech trees (Alsammar et al. 2019), suggesting a wider distribution of the species. Also, S. jurei has not yet been isolated from areas other than its original isolation region. However, eDNA of this species was detected in soil surrounding different tree species in Italy which encourages further sampling in the mountain regions across Europe (Alsammar et al. 2019). These findings indicate that these substrates may not be the natural niche of the Saccharomyces species, a theory that contradicts the adaptation model, which postulates that for an organism to be adapted to a niche, it must be abundant in that niche (Goddard and Greig 2015). Given the low abundance and habitat diversity of S. cerevisiae, it has been proposed that is it a nomad, that is not adapted to a specific niche. Although the nomad model was applied to S. cerevisiae, the criteria of this model, such as the presence of species in low abundance, could also be applied to the other species of the Saccharomyces genus (Goddard and Greig 2015). Extensive sampling of various habitats is needed to confirm the nomad nature of the wild Saccharomyces species.
CONCLUSIONS AND PERSPECTIVES
Domestication processes have contributed greatly to the evolution of genome Saccharomyces species (Gallone et al. 2016; Dujon and Louis 2017). In the last few decades, researchers started to discover a large biodiversity of Saccharomyces species in the natural environment, prompting to focus their studies on the ecology and distribution of wild species (Sniegowski, Dombrowski and Fingerman 2002; Sampaio and Goncalves 2008; Charron et al. 2014b; Kowallik, Miller and Greig 2015; Sylvester et al. 2015; Kowallik and Greig 2016; Alsammar et al. 2019), genome evolution of the Saccharomyces species and their hybrids (Dunn and Sherlock 2008; Morales and Dujon 2012; Piatkowska et al. 2013; Hewitt et al. 2014; Dujon and Louis 2017; Peris et al. 2018), population genomics (Liti et al. 2009; Schacherer et al. 2009; Louis 2011; Peter et al. 2018) and phenotype variation (Warringer et al. 2011; Naseeb et al. 2016). The feasibility of whole-genome sequencing allowed the redefinition of the Saccharomyces species taxonomy based on the phylogeny rather than the concept of reproductive isolation and helped the identification of diverged populations of the yeast's species and strains according to their geography, environmental niche and human domestication (Peter et al. 2018).
Species belonging to the Saccharomyces genus are now known to be residing in soil, bark, decaying leaves, insect guts and in healthy and diseased human guts. The optimization of isolation techniques allowed the detection of new species and targeted metagenomic approaches were able to assess the degree of Saccharomyces species biodiversity present in the wild. For further insights on the natural history and evolution of Saccharomyces species more sampling of novel niches in different regions of the world would be desirable.
FUNDING
This work was supported by BBSRC (BB/L021471/1) and the EU Framwork Programme for Research H2020-MSCA-ITN-2017 (764364). HA was funded by the Kuwait government through Kuwait University.
ACKNOWLEDGMENT
The authors wish to thank BBSRC and the EU Commission for the funding.
Conflict of interest . None declared.
REFERENCES
- Almeida P, Barbosa R, Zalar Pet al.. A population genomics insight into the Mediterranean origins of wine yeast domestication. Mol Ecol. 2015;24:5412–27. [DOI] [PubMed] [Google Scholar]
- Almeida P, Goncalves C, Teixeira Set al.. A Gondwanan imprint on global diversity and domestication of wine and cider yeast Saccharomyces uvarum. Nat Commun. 2014;5:4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsammar HF, Naseeb S, Brancia LBet al.. Targeted metagenomics approach to capture the biodiversity of Saccharomyces genus in wild environments. Env Microbiol Rep. 2019;11:206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsammar HF. Environmnetal Genomics of the Saccharomyces sensu stricto Group. Thesis, The University of Manchester, UK, Manchester, 2018.
- Baker E, Wang B, Bellora Net al.. The genome sequence of Saccharomyces eubayanus and the domestication of lager-brewing yeasts. Mol Biol Evol. 2015;32:2818–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa R, Almeida P, Safar SVet al.. Evidence of natural hybridization in Brazilian wild lineages of Saccharomyces cerevisiae. Genome Biol Evol. 2016;8:317–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloch C, Orlic S, Barrio Eet al.. Fermentative stress adaptation of hybrids within the Saccharomyces sensu stricto complex. Int J Food Microbiol. 2008;122:188–95. [DOI] [PubMed] [Google Scholar]
- Belloch C, Perez-Torrado R, Gonzalez SSet al.. Chimeric genomes of natural hybrids of Saccharomyces cerevisiae and Saccharomyces kudriavzevii. Appl Environ Microbiol. 2009;75:2534–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom A, Simpson JT, Salinas Fet al.. A high-definition view of functional genetic variation from natural yeast genomes. Mol Biol Evol. 2014;31:872–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing J, Han PJ, Liu WQet al.. Evidence for a Far East Asian origin of lager beer yeast. Curr Biol. 2014;24:R380–1. [DOI] [PubMed] [Google Scholar]
- Borneman AR, Forgan AH, Kolouchova Ret al.. Whole genome comparison reveals high levels of inbreeding and strain redundancy across the spectrum of commercial wine strains of saccharomyces cerevisiae. G3. 2016;6:957–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boynton PJ, Greig D. The ecology and evolution of non-domesticated Saccharomyces species. Yeast. 2014;31:449–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury JE, Richards KD, Niederer HAet al.. A homozygous diploid subset of commercial wine yeast strains. Antonie Van Leeuwenhoek. 2006;89:27–37. [DOI] [PubMed] [Google Scholar]
- Buser CC, Newcomb RD, Gaskett ACet al.. Niche construction initiates the evolution of mutualistic interactions. Ecol Lett. 2014;17:1257–64. [DOI] [PubMed] [Google Scholar]
- Charron G, Leducq JB, Bertin Cet al.. Exploring the northern limit of the distribution of Saccharomyces cerevisiae and Saccharomyces paradoxus in North America. FEMS Yeast Res. 2014b;14:281–8. [DOI] [PubMed] [Google Scholar]
- Charron G, Leducq JB, Landry CR. Chromosomal variation segregates within incipient species and correlates with reproductive isolation. Mol Ecol. 2014a;23:4362–72. [DOI] [PubMed] [Google Scholar]
- Cliften P, Sudarsanam P, Desikan Aet al.. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301:71–6. [DOI] [PubMed] [Google Scholar]
- Cromie GA, Hyma KE, Ludlow CLet al.. Genomic sequence diversity and population structure of Saccharomyces cerevisiae assessed by RAD-seq. G3. 2013;3:2163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashko S, Liu P, Volk Het al.. Changes in the relative abundance of two Saccharomyces species from oak forests towwine fermentations. Front Microbiol. 2016;7:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S-F, Han P-J, Wang Q-Met al.. The origin and adaptive evolution of domesticated populations of yeast from Far East Asia. 2018;9:2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon BA, Louis EJ. Genome diversity and evolution in the budding yeasts (Saccharomycotina). Genetics. 2017;206:717–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn B, Sherlock G. Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res. 2008;18:1610–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberlein C, Hénault M, Fijarczyk Aet al.. Hybridization is a recurrent evolutionary stimulus in wild yeast speciation. Nat Commun. 2019;10:923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizaguirre JI, Peris D, Rodriguez MEet al.. Phylogeography of the wildl lager-brewing ancestor (Saccharomyces eubayanus) in Patagonia. Environ Microbiol. 2018;20:3732–43. [DOI] [PubMed] [Google Scholar]
- Erny C, Raoult P, Alais Aet al.. Ecological success of a group of Saccharomyces cerevisiae x Saccharomyces kudriavzevii hybrids in the Northern European wine-making environment. Appl Environ Microbiol. 2012;78:3256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Benavides JA. Evidence for domesticated and wild populations of Saccharomyces cerevisiae. PLos Genet. 2005;1:66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda H, Kondo A, Tamalampudi S. Bioenergy: sustainable fuels from biomass by yeast and fungal whole-cell biocatalysts. Biochem Eng J. 2009;44:2–12. [Google Scholar]
- Gallone B, Steensels J, Prahl Tet al.. Domestication and divergence of Saccharomyces cerevisiae beer yeasts. Cell. 2016;166:1397–410. e1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayevskiy V, Goddard MR. Saccharomyces eubayanus and Saccharomyces arboricola reside in North Island native New Zealand forests. Environ Microbiol. 2016;18:1137–47. [DOI] [PubMed] [Google Scholar]
- Goddard MR, Anfang N, Tang Ret al.. A distinct population of Saccharomyces cerevisiae in New Zealand: evidence for local dispersal by insects and human-aided global dispersal in oak barrels. Environ Microbiol. 2010;12:63–73. [DOI] [PubMed] [Google Scholar]
- Goddard MR, Greig D. Saccharomyces cerevisiae: a nomadic yeast with no niche? FEMS Yeast Res. 2015;15:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey Het al.. Life with 6000 genes. Science. 1996;274:563–47. [DOI] [PubMed] [Google Scholar]
- Gonzalez SS, Barrio E, Gafner Jet al.. Natural hybrids from Saccharomyces cerevisiae, Saccharomyces bayanus and Saccharomyces kudriavzevii in wine fermentations. FEMS Yeast Res. 2006;6:1221–34. [DOI] [PubMed] [Google Scholar]
- Gonzalez SS, Barrio E, Querol A. Molecular characterization of new natural hybrids of Saccharomyces cerevisiae and S. kudriavzevii in brewing. Appl Environ Microbiol. 2008;74:2314–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez SS, Gallo L, Climent MAet al.. Enological characterization of natural hybrids from Saccharomyces cerevisiae and S. kudriavzevii. Int J Food Microbiol. 2007;116:11–18. [DOI] [PubMed] [Google Scholar]
- Guillamon JM, Barrio E, Huerta Tet al.. Rapid characterization of four species of the Saccharomyces sensu stricto complex according to mitochondrial DNA patterns. Int J Syst Bacteriol. 1994;44:708–14. [DOI] [PubMed] [Google Scholar]
- Hebly M, Brickwedde A, Bolat Iet al.. S. cerevisiae x S. eubayanus interspecific hybrid, the best of both worlds and beyond. FEMS Yeast Res. 2015;15:1–14. [DOI] [PubMed] [Google Scholar]
- Hewitt SK, Donaldson IJ, Lovell SCet al.. Sequencing and characterisation of rearrangements in three S. pastorianus strains reveals the presence of chimeric genes and gives evidence of breakpoint reuse. PLoS One. 2014;9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT, Goncalves P, Sampaio JPet al.. Remarkably ancient balanced polymorphisms in a multi-locus gene network. Nature. 2010;464:54–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT, Rokas A, Carroll SB. Parallel inactivation of multiple GAL pathway genes and ecological diversification in yeasts. Proc Natl Acad Sci USA. 2004;101:14144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyma KE, Fay JC. Mixing of vineyard and oak-tree ecotypes of Saccharomyces cerevisiae in North American vineyards. Mol Ecol. 2013;22:2917–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LJ, Koufopanou V, Goddard MRet al.. Population genetics of the wild yeast Saccharomyces paradoxus. Genetics. 2004;166:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi Met al.. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–54. [DOI] [PubMed] [Google Scholar]
- Kowallik V, Greig D. A systematic forest survey showing an association of Saccharomyces paradoxus with oak leaf litter. Env Microbiol Rep. 2016;8:833–41. [DOI] [PubMed] [Google Scholar]
- Kowallik V, Miller E, Greig D. The interaction of Saccharomyces paradoxus with its natural competitors on oak bark. Mol Ecol. 2015;24:1596–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehne HA, Murphy HA, Francis CAet al.. Allopatric divergence, secondary contact, and genetic isolation in wild yeast populations. Curr Biol. 2007;17:407–11. [DOI] [PubMed] [Google Scholar]
- Kurtzman CP, Piškur J. Taxonomy and phylogenetic diversity among the yeasts. (Sunnerhagen P, Piskur J, eds.) Comparative Genomics: Using Fungi as Models. Berlin Heidelberg, Berlin, Heidelberg: Springer, 2006, 29–46. [Google Scholar]
- Kurtzman CP, Robnett CJ. Phylogenetic relationships among yeasts of the ‘Saccharomyces complex’ determined from multigene sequence analyses. FEMS Yeast Res. 2003;3:417–32. [DOI] [PubMed] [Google Scholar]
- Langdon QK, Peris D, Eizaguirre JIet al.. Genomic diversity and global distribution of Saccharomyces eubayanus, the wild ancestor of hybrid lager-brewing yeasts, bioRxiv. 2019:709535, DOI:10.1101/709535.
- Leducq JB, Charron G, Samani Pet al.. Local climatic adaptation in a widespread microorganism. Proc Biol Sci. 2014;281:20132472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leducq JB, Nielly-Thibault L, Charron Get al.. Speciation driven by hybridization and chromosomal plasticity in a wild yeast. Nat Microbiol. 2016;1:15003. [DOI] [PubMed] [Google Scholar]
- Libkind D, Hittinger CT, Valerio Eet al.. Microbe domestication and the identification of the wild genetic stock of lager-brewing yeast. Proc Natl Acad Sci USA. 2011;108:14539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Barton DB, Louis EJ. Sequence diversity, reproductive isolation and species concepts in Saccharomyces. Genetics. 2006;174:839–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Carter DM, Moses AMet al.. Population genomics of domestic and wild yeasts. Nature. 2009;458:337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Nguyen Ba AN, Blythe Met al.. High quality de novo sequencing and assembly of the Saccharomyces arboricolus genome. BMC Genomics. 2013;14:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodder JL. The Yeast, A Taxonomic Study. North-Holland: Amsterdam: Elsevier, 1970. [Google Scholar]
- Lopandic K, Gangl H, Wallner Eet al.. Genetically different wine yeasts isolated from Austrian vine-growing regions influence wine aroma differently and contain putative hybrids between Saccharomyces cerevisiae and Saccharomyces kudriavzevii. FEMS Yeast Res. 2007;7:953–65. [DOI] [PubMed] [Google Scholar]
- Lopes CA, Barrio E, Querol A. Natural hybrids of S. cerevisiae x S. kudriavzevii share alleles with European wild populations of Saccharomyces kudriavzevii. FEMS Yeast Res. 2010;10:412–21. [DOI] [PubMed] [Google Scholar]
- Louis EJ. Population genomics and speciation in yeasts. Fungal Biol Rev. 2011;25:136–42. [Google Scholar]
- Mager WH, Winderickx J. Yeast as a model for medical and medicinal research. Trends Pharmacol Sci. 2005;26:265–73. [DOI] [PubMed] [Google Scholar]
- Martini AV. Saccharomyces paradoxus comb. nov., a newly separated species of the Saccharomyces sensu stricto complex based upon nDNA/nDNA homologies. Syst Appl Microbiol. 1989;12:179–82. [Google Scholar]
- Masneuf I, Hansen J, Groth Cet al.. New hybrids between Saccharomyces sensu stricto yeast species found among wine and cider production strains. Appl Environ Microbiol. 1998;64:3887–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monerawela C, James TC, Wolfe KHet al.. Loss of lager specific genes and subtelomeric regions define two different Saccharomyces cerevisiae lineages for Saccharomyces pastorianus Group I and II strains. FEMS Yeast Res. 2015;15:1–11. [DOI] [PubMed] [Google Scholar]
- Morales L, Dujon B. Evolutionary role of interspecies hybridization and genetic exchanges in yeasts. Microbiol Mol Biol Rev. 2012;76:721–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao Y, Kanamori T, Itoh Tet al.. Genome sequence of the lager brewing yeast, an interspecies hybrid. DNA Res. 2009;16:115–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naseeb S, Alsammar H, Burgis Tet al.. Whole genome sequencing, de Novo assembly and phenotypic profiling for the new budding yeast species Saccharomyces jurei. G3. 2018;8:2967–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naseeb S, Carter Z, Minnis Det al.. Widespread impact of chromosomal inversions on gene expression uncovers robustness via phenotypic buffering. Mol Biol Evol. 2016;33:1679–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naseeb S, James SA, Alsammar Het al.. Saccharomyces jurei sp. nov., isolation and genetic identification of a novel yeast species from Quercus robur. Int J Syst Evol Microbiol. 2017;67:2046–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash AK, Auchtung TA, Wong MCet al.. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome. 2017;5:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumov G. Genetic identification of biological species in the Saccharomyces sensu stricto complex. J Ind Microbiol. 1996;17:295–302. [Google Scholar]
- Naumov GI, James SA, Naumova ESet al.. Three new species in the Saccharomyces sensu stricto complex: Saccharomyces cariocanus, Saccharomyces kudriavzevii and Saccharomyces mikatae. Int J Syst Evol Microbiol. 2000b;50 Pt 5:1931–42. [DOI] [PubMed] [Google Scholar]
- Naumov GI, Lee CF, Naumova ES. Molecular genetic diversity of the Saccharomyces yeasts in Taiwan: Saccharomyces arboricola, Saccharomyces cerevisiae and Saccharomyces kudriavzevii. Antonie Van Leeuwenhoek. 2013;103:217–28. [DOI] [PubMed] [Google Scholar]
- Naumov GI, Naumova ES, Hagler ANet al.. A new genetically isolated population of the Saccharomyces sensu stricto complex from Brazil. Antonie Van Leeuwenhoek. 1995b;67:351–5. [DOI] [PubMed] [Google Scholar]
- Naumov GI, Naumova ES, Louis EJ. Two new genetically isolated populations of the Saccharomyces sensu stricto complex from japan. J Gen Appl Microbiol. 1995a;41:499–505. [DOI] [PubMed] [Google Scholar]
- Naumov GI, Naumova ES, Sniegowski PD. Saccharomyces paradoxus and Saccharomyces cerevisiae are associated with exudates of North American oaks. Can J Microbiol. 1998;44:1045–50. [PubMed] [Google Scholar]
- Naumov GI. Saccharomyces bayanus var. uvarum comb.nov., a new variety established by genetic analysis. Mikrobiologiia. 2000a;69:410–4. [PubMed] [Google Scholar]
- Nespolo RF, Villarroel CA, Oporto CIet al.. An Out-of-Patagonia dispersal explains most of the worldwide genetic distribution in Saccharomyces eubayanus, bioRxiv. 2019:709253, DOI:10.1101/709253.
- Nguyen H-V, Gaillardin C. Two subgroups within the Saccharomyces bayanus species evidenced by PCR amplification and restriction polymorphism of the non-transcribed spacer 2 in the ribosomal DNA unit. Syst Appl Microbiol. 1997;20:286–94. [Google Scholar]
- Nguyen HV, Boekhout T. Characterization of Saccharomyces uvarum (Beijerinck, 1898) and related hybrids: assessment of molecular markers that predict the parent and hybrid genomes and a proposal to name yeast hybrids. FEMS Yeast Res. 2017;17:1–19. [DOI] [PubMed] [Google Scholar]
- Nguyen HV, Lepingle A, Gaillardin CA. Molecular typing demonstrates homogeneity of Saccharomyces uvarum strains and reveals the existence of hybrids between S. uvarum and S. cerevisiae, including the S. bayanus type strain CBS 380. Syst Appl Microbiol. 2000;23:71–85. [DOI] [PubMed] [Google Scholar]
- Okuno M, Kajitani R, Ryusui Ret al.. Next-generation sequencing analysis of lager brewing yeast strains reveals the evolutionary history of interspecies hybridization. DNA Res. 2016;23:67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paget CM, Schwartz J-M, Delneri D. Environmental systems biology of cold-tolerant phenotype in Saccharomyces species adapted to grow at different temperatures. 2014;23:5241–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris D, Belloch C, Lopandic Ket al.. The molecular characterization of new types of Saccharomyces cerevisiae x S. kudriavzevii hybrid yeasts unveils a high genetic diversity. Yeast. 2012a;29:81–91. [DOI] [PubMed] [Google Scholar]
- Peris D, Langdon QK, Moriarty RVet al.. Complex ancestries of lager-brewing hybrids were shaped by standing variation in the wild yeast Saccharomyces eubayanus. PLos Genet. 2016a;12:e1006155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris D, Lopes CA, Belloch Cet al.. Comparative genomics among Saccharomyces cerevisiae x Saccharomyces kudriavzevii natural hybrid strains isolated from wine and beer reveals different origins. BMC Genomics. 2012b;13:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris D, Perez-Torrado R, Hittinger CTet al.. On the origins and industrial applications of Saccharomyces cerevisiae x Saccharomyces kudriavzevii hybrids. Yeast. 2018;35:51–69. [DOI] [PubMed] [Google Scholar]
- Peris D, Perez-Traves L, Belloch Cet al.. Enological characterization of Spanish Saccharomyces kudriavzevii strains, one of the closest relatives to parental strains of winemaking and brewing Saccharomyces cerevisiae x S. kudriavzevii hybrids. Food Microbiol. 2016b;53:31–40. [DOI] [PubMed] [Google Scholar]
- Peris D, Sylvester K, Libkind Det al.. Population structure and reticulate evolution of Saccharomyces eubayanus and its lager-brewing hybrids. Mol Ecol. 2014;23:2031–45. [DOI] [PubMed] [Google Scholar]
- Peter J, De Chiara M, Friedrich Aet al.. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature. 2018;556:339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatkowska EM, Naseeb S, Knight Det al.. Chimeric protein complexes in hybrid species generate novel phenotypes. PLos Genet. 2013;9:e1003836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontes A, Čadež N, Gonçalves Pet al.. A quasi-domesticate relic hybrid population of Saccharomyces cerevisiae × S. paradoxus adapted to Olive Brine. Front Genet. 2019;10:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulvirenti A, Nguyen H, Caggia Cet al.. Saccharomyces uvarum, a proper species within Saccharomyces sensu stricto. FEMS Microbiol Lett, 2000;192:191–6. [DOI] [PubMed] [Google Scholar]
- Rainieri S, Kodama Y, Kaneko Yet al.. Pure and mixed genetic lines of Saccharomyces bayanus and Saccharomyces pastorianus and their contribution to the lager brewing strain genome. Appl Environ Microbiol. 2006;72:3968–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainieri S, Zambonelli C, Hallsworth JEet al.. Saccharomyces uvarum, a distinct group within Saccharomyces sensu stricto. FEMS Microbiol Lett. 1999;177:177–85. [DOI] [PubMed] [Google Scholar]
- Rainieri S, Zambonelli C, Kaneko Y. Saccharomyces sensu stricto: systematics, genetic diversity and evolution. J Biosci Bioeng. 2003;96:1–9. [PubMed] [Google Scholar]
- Rodriguez ME, Perez-Traves L, Sangorrin MPet al.. Saccharomyces eubayanus and Saccharomyces uvarum associated with the fermentation of Araucaria araucana seeds in Patagonia. FEMS Yeast Res. 2014;14:948–65. [DOI] [PubMed] [Google Scholar]
- Salvado Z, Arroyo-Lopez FN, Guillamon JMet al.. Temperature adaptation markedly determines evolution within the genus Saccharomyces. Appl Environ Microbiol. 2011;77:2292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio JP, Goncalves P. Natural populations of Saccharomyces kudriavzevii in Portugal are associated with oak bark and are sympatric with S. cerevisiae and S. paradoxus. Appl Environ Microbiol. 2008;74:2144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampaio JP, Gonçalves P. Biogeography and ecology of the genus Saccharomyces. (Buzzini P, Lachance M-A, Yurkov A, eds.), Yeasts in Natural Ecosystems: Ecology, Cha: Springer International Publishing, 2017, 131–53. [Google Scholar]
- Scannell DR, Zill OA, Rokas Aet al.. The awesome power of yeast evolutionary genetics: new genome sequences and strain resources for the Saccharomyces sensu stricto genus. G3. 2011;1:11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacherer J, Shapiro JA, Ruderfer DMet al.. Comprehensive polymorphism survey elucidates population structure of Saccharomyces cerevisiae. Nature. 2009;458:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicard D, Legras JL. Bread, beer and wine: yeast domestication in the Saccharomyces sensu stricto complex. C R Biol. 2011;334:229–36. [DOI] [PubMed] [Google Scholar]
- Sniegowski PD, Dombrowski PG, Fingerman E. Saccharomyces cerevisiae and Saccharomyces paradoxus coexist in a natural woodland site in North America and display different levels of reproductive isolation from European conspecifics. FEMS Yeast Res. 2002;1:299–306. [DOI] [PubMed] [Google Scholar]
- Sokol H, Leducq V, Aschard Het al.. Fungal microbiota dysbiosis in IBD. Gut. 2017;66:1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanini I, Dapporto L, Legras JLet al.. Role of social wasps in Saccharomyces cerevisiae ecology and evolution. Proc Natl Acad Sci USA. 2012;109:13398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney JY, Kuehne HA, Sniegowski PD. Sympatric natural Saccharomyces cerevisiae and S. paradoxus populations have different thermal growth profiles. FEMS Yeast Res. 2004;4:521–5. [DOI] [PubMed] [Google Scholar]
- Sylvester K, Wang QM, James Bet al.. Temperature and host preferences drive the diversification of Saccharomyces and other yeasts: a survey and the discovery of eight new yeast species. FEMS Yeast Res. 2015;15. [DOI] [PubMed] [Google Scholar]
- Taylor MW, Tsai P, Anfang Net al.. Pyrosequencing reveals regional differences in fruit-associated fungal communities. Environ Microbiol. 2014;16:2848–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teresa Fernandez-Espinar M, Barrio E, Querol A. Analysis of the genetic variability in the species of the Saccharomyces sensu stricto complex. Yeast. 2003;20:1213–26. [DOI] [PubMed] [Google Scholar]
- Vaughan-Martini A, Martini A, Cardinali G. Electrophoretic karyotyping as a taxonomic tool in the genus Saccharomyces. Antonie Van Leeuwenhoek. 1993;63:145–56. [DOI] [PubMed] [Google Scholar]
- Vaughan-Martini A, Martini A. 44 - Saccharomyces Meyen ex Reess. (Kurtzman CP, FellJW,eds.). The Yeasts (Fourth Edition). Amsterdam: Elsevier; 1998, 358–71. [Google Scholar]
- Vaughan-Martini A, Martini A. Chapter 61 - Saccharomyces Meyen ex Reess (1870) A2 - Kurtzman, Cletus P. (Fell JW, BoekhoutT,eds.) The Yeasts (Fifth Edition). London: Elsevier; 2011, 733–46. [Google Scholar]
- Walther A, Hesselbart A, Wendland J. Genome sequence of Saccharomyces carlsbergensis, the world's first pure culture lager yeast. G3. 2014;4:783–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QM, Liu WQ, Liti Get al.. Surprisingly diverged populations of Saccharomyces cerevisiae in natural environments remote from human activity. Mol Ecol. 2012;21:5404–17. [DOI] [PubMed] [Google Scholar]
- Wang SA, Bai FY. Saccharomyces arboricolus sp. nov., a yeast species from tree bark. Int J Syst Evol Microbiol. 2008;58:510–4. [DOI] [PubMed] [Google Scholar]
- Warringer J, Zorgo E, Cubillos FAet al.. Trait variation in yeast is defined by population history. PLos Genet. 2011;7:e1002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J-X, Li J, Aigrain Let al.. Contrasting evolutionary genome dynamics between domesticated and wild yeasts. Nat Genet. 2017;49:913–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Skelton A, Gardner RCet al.. Saccharomyces paradoxus and Saccharomyces cerevisiae reside on oak trees in New Zealand: evidence for migration from Europe and interspecies hybrids. FEMS Yeast Res. 2010;10:941–7. [DOI] [PubMed] [Google Scholar]