

Abstract
Neuropsychiatric symptoms, such as avolition, apathy, and anhedonia, precede the onset of debilitating motor symptoms in Huntington’s disease (HD), and their development may give insight into early disease progression and treatment. However, the neuronal and circuit mechanisms of premanifest HD pathophysiology are not well-understood. Here, using a transgenic rat model expressing the full-length human mutant HD gene, we find early and profound deficits in reward motivation in the absence of gross motor abnormalities. These deficits are accompanied by significant and progressive dysfunction in corticostriatal processing and communication among brain areas critical for reward-driven behavior. Together, our results define early corticostriatal dysfunction as a possible pathogenic contributor to psychiatric disturbances and may help identify potential pharmacotherapeutic targets for the treatment of HD.
Keywords: corticostriatal, Huntington’s disease, motivation, nucleus accumbens, prefrontal cortex
Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disease that typically manifests in adulthood with debilitating motor symptoms (Harper 1992; Vonsattel and DiFiglia 1998). Characteristically, HD patients suffer from involuntary choreic or “dance-like” movements, progressing to brady- and hypokinesia at advanced stages. However, cognitive and emotional abnormalities are also highly prevalent and usually appear early during a stage that precedes motor symptoms by a decade or more (Paulsen et al. 2008; Epping et al. 2016; Martinez-Horta et al. 2016). In particular, lack of motivation to pursue reward is an early manifestation of HD dysfunction and represents a core neuropsychiatric feature of the disease (Paulsen et al. 2001; Naarding et al. 2009; Tabrizi et al. 2013; Van Duijn et al., 2014; Martinez-Horta et al. 2016; Sousa et al. 2018). This motivational impairment potentiates with disease progression (Van Duijn et al., 2014) and is highly difficult to treat (Krishnamoorthy and Craufurd 2011; Berman et al. 2012), severely impacting the quality of life of HD patients and caregivers (Hamilton et al. 2003; Papoutsi et al. 2014).
Progressive neural dysfunction and degeneration in motor-related areas of the brain, particularly the primary motor cortex and the striatum, are linked to the development of the pathognomonic motor symptoms (Vonsattel and DiFiglia 1998; Cepeda et al. 2007; Bunner and Rebec 2016). However, the neural substrates of motivational disturbances in HD have been far less studied. Evidence from human imaging studies in non-HD patients identifies a prominent role for brain reward regions, such as the medial prefrontal cortex (PFC) and the nucleus accumbens (NAc), in apathetic or avolitional symptoms. Specifically, reduced volume of the PFC (Huey et al. 2015) and the NAc (Paul et al. 2005; Carriere et al. 2014) as well as altered PFC–NAc functional connectivity (Alexopoulos et al. 2013) are positively associated with apathy severity ratings. Interestingly, significant atrophy of the NAc is already evident in premanifest HD patients (Majid et al. 2011; van den Bogaard et al. 2011a; van den Bogaard et al. 2011b), and in early manifest HD, widespread reduction in task-specific functional coupling is measured between various prefrontal cortical regions (Thiruvady et al. 2007; Unschuld et al. 2013). However, despite these findings, it is unknown how dysfunctional corticoaccumbal circuit dynamics evolve over disease progression and whether they are associated with impaired motivation.
Here, we use the BACHD-Tg5 transgenic rat model of HD to delineate the development of age-dependent motivational deficits and associated corticoaccumbal network abnormalities. The Tg5 rat model uses a human bacterial artificial chromosome (BAC) containing the full-length HTT genomic sequence with 97 CAG/CAA repeats and all regulatory elements (Yu-Taeger et al. 2012; Manfré et al. 2018). Prior work has established that the Tg5 model recapitulates many of the genetic changes (Yu-Taeger et al. 2017) and mutant Htt (mHtt) protein aggregation patterns found in HD patients (Yu-Taeger et al. 2012). In the present set of experiments, the progressive ratio (PR) task was used to assess reward-motivated behavior, and in vivo optogenetic stimulation and electrophysiological recordings in the PFC and NAc were used to examine functional neural network activity across ages. Tg5 rats demonstrated early and sustained deficits in motivation for reward, and session-wide and event-related field potential recordings revealed dynamic phenotypic alterations in the coordination of corticoaccumbal activity that arose during maladaptive behavior. These findings will undoubtedly advance our knowledge of the etiology of motivational deficits in HD and give crucial insight into avenues for treatment.
Materials and Methods
Subjects
Thirty-two male rats per age group (3, 6, and 12 mo. old) per genotype (WT and BACHD-Tg5) were provided by CHDI (Los Angeles, CA) from the University of Tübingen (Tübingen, Germany). Prior to surgery, rats were group-housed in plastic cages with ad libitum access to food and water. Following surgery, they were singly-housed and food-restricted to 85–90% free-feeding body weight to facilitate training and testing of food-maintained operant behavior. All rodent holding rooms were maintained at 24 °C and 40–50% humidity under a 12-h light/dark cycle with lights on at 07:00 h. Research facilities were certified by the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC), and experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at University of Maryland School of Medicine and in accordance with the Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011).
Behavior
Progressive Ratio (PR) Task
Rats were tested in operant conditioning chambers (MedAssociates, Inc., St. Albans, VT) as previously described (Hernandez and Cheer 2012), and data collection and programming were conducted using PC computers with a Med-PC interface (MedAssociates, Inc.). Rats were first trained on a fixed ratio 1 (FR1) schedule of reinforcement for chocolate-flavored food pellets (#F0299, Bio-Serv, Flemington, NJ). Once at least 80 pellets were obtained within 30 min, rats were switched to a fixed ratio 5 (FR5) schedule of reinforcement until they reached the same criteria in 2 consecutive sessions. Next, an FR5ITI10 schedule was introduced in which an inter-trial interval (ITI) of 10 s was presented after each pellet delivery. Finally, rats were tested on a progressive ratio (PR) schedule of reinforcement with a 10-s ITI to quantify the effort they were willing to expend for food pellets (Hodos 1961; Arnold and Roberts 1997). The number of lever presses required to earn each pellet, or response ratio, increased exponentially on successive trials (response ratio = [5 × e(0.2×reward number) – 5]), yielding ratios of 1, 2, 4, 6, 9, 12, 15, 20, 25, 32, 40, 50, 62, 77, 95, 118, etc. The final ratio attained (i.e., breakpoint) reflects reinforcer efficacy and inherent motivation to obtain the reinforcer. Each session came to an end whenever 20 min elapsed without a pellet delivery.
Free Food Pellet Consumption
To assess food pellet consumption under free access conditions, rats were given 1-h ad libitum access to chocolate pellets once/day for 3 days while having ad libitum access to chow in their home cages.
Locomotor Activity
Rats were placed in individual home cages equipped with horizontal infrared sensors (Accuscan Instruments, Inc., Columbus, OH) measuring spontaneous locomotor activity by number of beam breaks. After one 20-min habituation session, two 40-min test sessions were administered on consecutive days.
Saccharin Preference Test
A reduction in saccharin preference is a validated measure of anhedonia (Harkin et al. 2002; Shimamoto et al. 2015; Spierling et al. 2017), and under ad libitum feeding conditions, rats were given a series of 2-bottle choice tests between saccharin and water. To obtain a concentration-response function, 6 saccharin concentrations (0%, 0.009%, 0.055%, 0.15%, 0.55%, and 0.9% w/v) were presented concurrently with water. Each concentration was presented for 2 h/day for at least 2 consecutive days, and average intake of saccharin was compared to intake of water to obtain percent saccharin preference [(saccharin intake)/(saccharin + water intake) × 100] at each concentration.
In Vivo Optogenetic Stimulation of mPFC with Simultaneous NAc Recordings
Optogenetic Stimulation in Anesthetized Animals
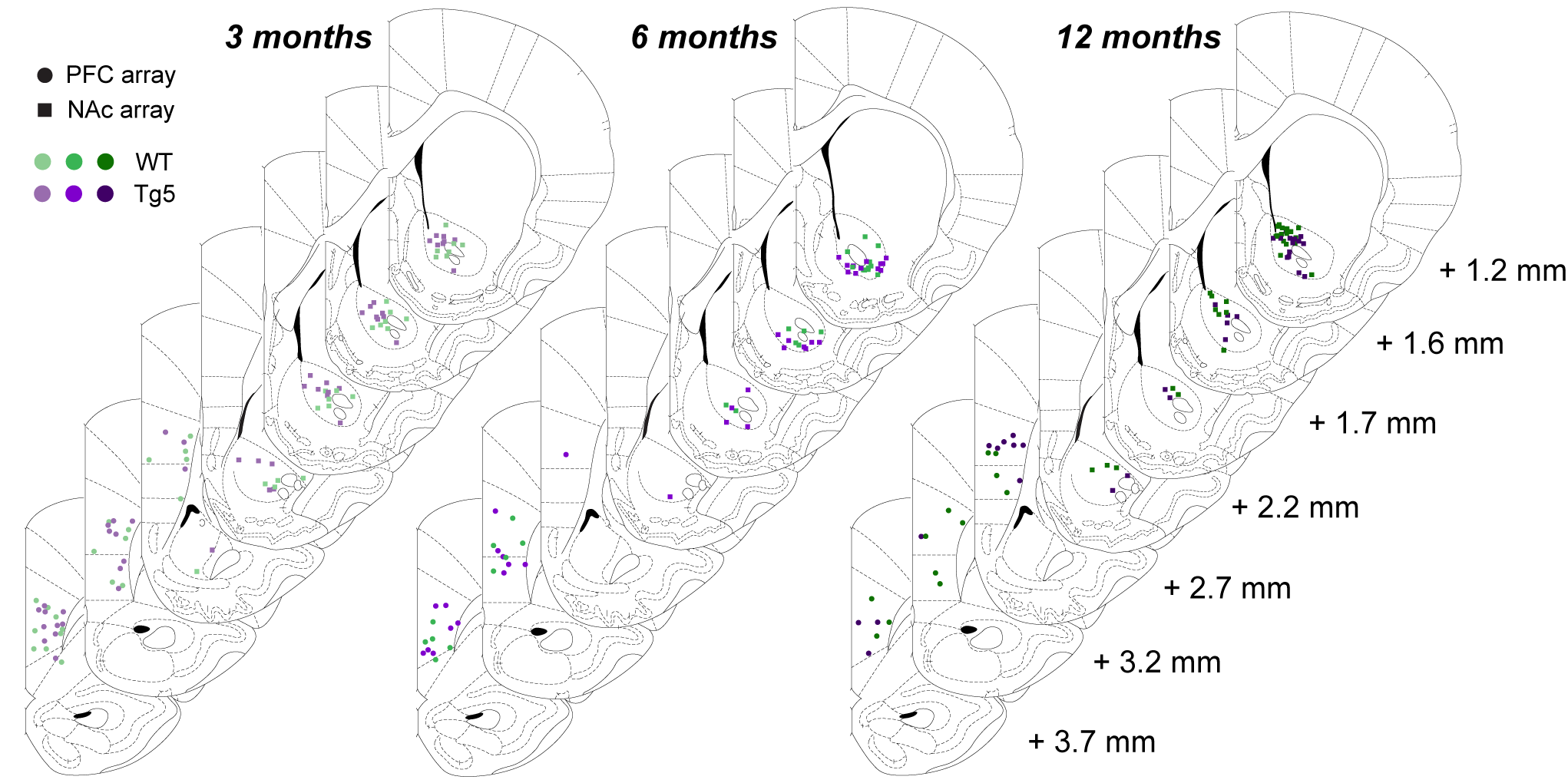
Following established methods (Britt et al. 2012; Ji and Neugebauer 2012; Nakamura et al. 2013; Suska et al. 2013; Liang et al. 2015), animals were transduced by injecting 500 μl of AAV.CaMKII.hChR2(H134R).EYFP (University of North Carolina Vector Core, Chapel Hill, NC) in the prelimbic (PrL) cortex (AP + 3.0, ML ± 0.5, DV –3.2 mm from Bregma, Paxinos and Watson 2006). Four weeks after injection, rats were anesthetized using urethane (1.5 mg/kg, ip). An optic fiber (200 nm diameter; ThorLabs, Inc., Newton, NJ) was inserted into the PrL cortex, and a custom-made 16-channel tetrode (Nguyen et al. 2009) was lowered into the striatum so that the lower 4-channel bundle was in the NAc (AP + 1.4, ML ± 1.2, DV –7.0). Neural activity was recorded as described below. Optogenetic stimulation was delivered through an optical patch cable (200 nm diameter) coupled to a 473-nm laser (BL473T3; Shanghai Laser and Optics Century Co., Ltd., Shanghai, China). Laser power at the fiber tip was adjusted to 15 mW, and laser stimulation was administered at 5, 10, 15, 20, or 30 Hz (5-ms pulse width) for a total of 2 s, repeating every 60 s (Master 9, AMPI, Jerusalem, Israel). Each recording included 10 stimulation trains at each frequency, and an additional recording was collected where no light stimulation was delivered (i.e., no laser stimulation). Transfections and positioning of optical fibers and electrode were verified histologically (Fig. S1).
In Vivo Recordings During Operant Behavior
Surgery
Rats were implanted with 2 fixed microwire arrays with electrodes made of Teflon-insulated stainless steel (0.25 mm inter-electrode space, 0.5 mm inter-row space; Microprobes, Gaithersburg, MD). Each rat had an 8-channel array aimed at the PrL (AP + 3.2, ML ± 0.5, DV –3.2 mm from Bregma) and a 16-channel array aimed at the NAc (AP + 1.2, ML ± 1.2, DV –6.5). Electrodes were fixed to the skull with acrylic cement secured with stainless steel bone screws. A stainless steel wire from each array served as a ground and was inserted caudal to the arrays in the midbrain/cerebellum interface. Rats were administered topical antibiotic and analgesic ointment as well as injectable analgesics for 48 h following surgery and were allowed at least 7 days of recovery.
Recordings
Neural activity was recorded using commercially available hardware and software, including headstages and programmable amplifiers, filters, and multichannel spike-sorting software (Plexon Inc., Dallas, TX). Signals were routed to a differential preamplifier and relayed to a Multichannel Acquisition Processor (Plexon Inc.), which allows for computer-controlled, channel-specific signal amplification (gain steps 1–30, total gain 1000–32 000×), filtering (second-order 500 Hz low cut, 5 kHz high cut), and analog to digital conversion (48 simultaneous sampling 12-bit converters, 40 kHz). Raw local field potential (LFP) voltage traces and whole-session power spectral densities (PSDs) were visually inspected to confirm recording quality before further analysis. Analysis of LFPs was performed using NeuroExplorer software (NeuroExplorer Inc., Nex Technologies, Madison, AL), the FieldTrip analysis package (Oostenveld et al. 2011), and custom-written Matlab (MathWorks, Natick, MA) scripts using inbuilt functions wherever possible to increase reproducibility. Correct electrode positioning was verified histologically (Fig. S2).
Statistical Analyses
Behavior
The primary dependent measures were mean responses, breakpoints, pellets earned, locomotor activity counts, and pellet consumption and were analyzed with 2-factor ANOVA (Geno. × Age). Time to complete each ratio and saccharin preference were analyzed with separate 2-factor mixed ANOVA for each age (Geno. × Trials or Saccharin Concentration) or 2-factor mixed ANOVA for each genotype (Age × Trials or Saccharin Concentration). Since WT and Tg5 rats achieved different breakpoints on the PR schedule, their time to complete each ratio could only be assessed on trials that both groups completed. Where appropriate, planned contrasts to assess phenotypic differences at each age were performed with the Bonferroni procedure, and results were considered significant if P < 0.05. All statistical analyses were performed using Prism (GraphPad Software, La Jolla, CA).
Laser Stimulation-Evoked LFPs
For each stimulation frequency, raw power was measured by fast Fourier transform (FFT) calculations in a 4 Hz band centered on that frequency and averaged over 10 stimulations. The LFP power ratio was defined as Plight/Pbaseline, where Plight is the power in a frequency band in the presence of light stimulation and Pbaseline is the power in the same band during periods of no stimulation (Cardin et al. 2009). Power ratios across frequency (Geno. × Freq. and Age × Freq.) and percent change in evoked power ratio in Tg5 compared to WT rats (Age × Freq.) were analyzed with 2-factor mixed ANOVA.
Session-wide and Event-related LFPs
LFPs were first filtered via a combination of a discrete wavelet transform and then empirical mode decomposition, followed by a butterworth notch filter (Akwei-Sekyere 2015). This resulted in a reduction of powerline noise that kept the rest of the frequency content unaltered. Session-wide power spectra were calculated using Welch’s FFT-based method (1024 frequencies between 1 and 200 Hz, smoothed with a Gaussian kernel with half-width = 3 points) and normalized as log power in dB (Berke 2009). Coherence magnitude values were determined with the same parameters except with 512 frequencies. LFP power and coherence were calculated as area under the curve (AUC) and integrated across frequency bandwidths specified in previous reports (Hernandez and Cheer 2012). AUCs were tested for normality before further analysis with 2-factor ANOVA (Geno. × Age). Event-related oscillatory activity was examined in a 10-s window centered on reward delivery. Perievent wavelet power and coherence were derived from an FFT-based continuous wavelet transform algorithm. The resulting power and coherence magnitude values were averaged separately over groups and then scaled to the same colormap to allow for comparisons between groups. Power and coherence within each frequency band were normalized via z-score to the first second of the 10-s window and then examined with 2-way mixed ANOVA (Geno. × Time Bin and Age × Time Bin). Peak reward-evoked PFC–NAc wavelet coherence across various frequency bandwidths was correlated with earned reinforcers under the PR schedule using linear regression.
Results
Early HD Deficits in Reward-motivated Behavior Persist Through Advanced Ages
To examine motivation for reward at early and advanced ages in the HD model, 3, 6, and 12 mo. old rats Tg5 and WT rats were trained to lever-press for sweetened food pellets initially under a fixed-ratio (FR1 and FR5) and then under a progressive ratio (PR) schedule of reinforcement, where the response ratio on each trial escalates exponentially until responding ceases altogether (Fig. 1a). This is known as the “breakpoint” and is considered an index of the animal’s motivation or willingness to work for the reinforcer as well as the subjective value of the reinforcer. Although there were no differences in performance among the Tg5 and WT rats under the FR1 and FR5 schedules at any age (Fig. S3A–C), Tg5 rats demonstrated a pronounced deficit in motivation under the PR schedule as early as 3 mo. (Fig. 1a,b). Similar to previous work (Jansson et al. 2014; Clemensson, Clemensson, Fabry, et al. 2017), Tg5 rats reached lower breakpoints (Fig. 1b, **P < 0.01, ***P < 0.001) [Genotype (Geno.): F(1,83) = 45.34, P < 0.0001; Age: F(2,83) = 5.778, P < 0.01; Geno. × Age: F(2,83) = 3.647, P < 0.05] and earned fewer pellets (Fig. S3D, **P < 0.01, ****P < 0.0001) [Geno.: F(1,83) = 68.24, P < 0.0001; Age: F(2,83) = 7.338, P < 0.01; Geno. × Age: F(2,83) = 1.875, ns] than WT counterparts at all ages. In contrast, when the same food pellets were made freely available, both genotypes consumed similar amounts (Fig. 1d) [Geno.: F(1.44) = 1.065, ns; Age: F(2,44) = 13.21, P < 0.0001; Geno. × Age: F(2,44) = 1.599, ns]. Importantly, at all ages examined, gross locomotor activity measured by beam crossings in the home cage was not compromised in the Tg5 groups (Fig. 1c) [Geno.: F(1,38) = 3.068, ns; Age: F(2,38) = 34.65, P < 0.0001; Geno. × Age: F(2,38) = 2.057, ns], and at 3 mo. of age, rate of reward-driven operant responding was maintained at WT levels, with Tg5 rats exhibiting similar latencies to complete each response ratio on matched trials (Fig. 1e) [Geno.: F(1,28) = 0.3279, ns; Trial: F(5 140) = 9.592, P < 0.0001; Geno. × Trial: F(5 140) = 1.590, ns; also see Supplemental Results], before declining in performance at 6 (Fig. 1F *P < 0.05) [Geno.: F(1,11) = 5.213, P < 0.05; Trial: F(5,55) = 17.90, P < 0.0001; Geno. × Trial: F(5,55) = 1.553, ns] and 12 (Fig. 1g, *P < 0.05, ***P < 0.001) [Geno.: F(1,27) = 5.772, P < 0.05; Trial: F(5 135) = 11.45, P < 0.0001; Geno. × Trial: F(5 135) = 3.402, P < 0.01] mo. of age. These data suggest that attenuated breakpoints under the PR schedule are not explained simply by emerging HD-mediated impairment in motor function. Indeed, in a standard assessment of anhedonia (Harkin et al. 2002; Shimamoto et al. 2015; Spierling et al. 2017), Tg5 rats showed lower preference for saccharin solution in a two-bottle choice test at the peak (3 mo. and 12 mo.) and highest (3, 6, 12 mo.) doses of the concentration-response function compared to WTs (Fig. 1h–j, *P < 0.05, **P < 0.01, ***P < 0.001) [3 mo. – Geno.: F(1,14) = 8.267, P < 0.05; Concentration (Conc.): F(4,56) = 53.65, P < 0.0001; Geno. × Conc.: F(4,56) = 10.20, P < 0.0001; 6 mo. – Geno.: F(1,14) = 4.792, P < 0.05; Conc.: F(4,56) = 11.66, P < 0.0001; Geno. × Conc.: F(4,56) = 1.988, ns; 12 mo. – Geno.: F(1,16) = 8.623, P < 0.01; Conc.: F(4,64) = 92.91, P < 0.0001; Geno. × Conc.: F(4,64) = 3.247, P < 0.05; also see Supplemental Results, Fig. S4]. This downward shift in the saccharin preference function indicates a reduction in reward efficacy in the Tg5 vs. WT rats and is consistent with early onset of anhedonia in the BACHD mouse model of HD (Hult Lundh et al. 2013). Together with diminished PR performance, this provides evidence that the Tg5 rats demonstrate early anhedonia and motivational dysfunction at 3 mo. that is sustained through advanced ages.
Figure 1.

Early and sustained motivational and hedonic deficits in the absence of gross motor abnormalities in HD. (a) Representative cumulative response records from 3 mo. WT and Tg5 rats performing the PR task. (b) Tg5 rats show reduced breakpoints under a PR schedule across all ages (n = 89). (c) Significant age-related decline in locomotor activity counts in both genotypes (n = 44). (d) Similar consumption of highly-palatable food pellets under free-access conditions across all ages in Tg5 vs. WT rats (n = 50). At 6 mo., WT rats consume significantly more pellets than at 12 mo. (e–g) On matched PR trials, Tg5 rats show similar latency as WT rats to complete each response ratio at 3 mo. of age before declining in performance at 6 and 12 mo. of age (n = 73). (h–j) Tg5 rats demonstrate lower preference for saccharin solution than WT rats at all ages (n = 50). Unless otherwise stated, data are represented as mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001 vs. Tg5; # P < 0.05 vs. other ages.
Compromised Corticostriatal Communication in HD
Given evidence of HD-mediated impairment in motivation for reward (Fig. 1), we next examined the integrity of communication among areas of the brain participating in reward processing across early and advanced ages. Efficacy of PFC–NAc neural transmission was investigated with anesthetized in vivo optical stimulation and electrophysiological recordings (Fig. 2). Tg5 and WT rats were transduced with an adeno-associated viral vector to express calcium calmodulin kinas II (CaMKII)-driven channelrhodopsin 2 (ChR2) in projection neurons of the PFC (Fig. 2a); subsequently, cortical pyramidal neurons were optically stimulated over a range of frequencies while concurrent field potential recordings were performed in the NAc (Fig. 2b,c). Faithful transmission of stimulation was expressed as frequency-specific increases in power of the accumbal LFP (Fig. 2e,f) and normalized to each animal’s baseline power with the LFP power ratio (Fig. 2d,g–I; Methods). PFC stimulation evoked notably lower increases in the accumbal LFP power ratio in Tg5 vs. WT rats at 3 mo. (Fig. 2g, *P < 0.05) [Geno.: F(1,13) = 3.807, ns; Frequency (Freq.): F (5,65) = 3.576, P < 0.01; Geno. × Freq.: F(5,65) = 2.659, P < 0.05; also see Supplemental Results, Fig. S5] and 6 mo. (Fig. 2h, *P < 0.05) [Geno.: F(1,11) = 2.292, ns; Freq.: F(5,55) = 3.300, P < 0.01; Geno. × Freq.: F(5,55) = 1.621, ns]. Across all frequencies examined, Tg5 rats exhibited very little change in NAc LFP power ratio; and at the peak of the frequency-response curve, Tg5 rats demonstrated significantly less evoked power, indicating compromised information transfer between the PFC and NAc in HD animals. Similar results were revealed when the Tg5 NAc power ratio was expressed as a percent change from WT [Age: F(2,18) = 1.437, ns; Freq.: F(5,90) = 13.02, P < 0.0001; Age × Freq.: F(10,90) = 1.870, ns] (Fig. 2d). However, by 12 mo., no significant evoked power differences were observed between genotypes (Fig. 2d,i) [Geno.: F(1,14) = 0.8153, ns; Freq.: F(5,70) = 10.09, P < 0.0001; Geno. × Freq.: F(5,70) = 0.5114, ns]. From these findings alone, it is not clear whether compromised PFC–NAc functional connectivity is due to alterations in the PFC, NAc or both. However, early impairments in PFC–NAc stimulation efficiency followed by apparent normalization at advanced ages may reflect progressive decline in neuronal function followed by compensatory modifications. Specifically, antecedent HD-mediated deficits in PFC–NAc communication at 3 and 6 mo. may lead to adaptions in the corticoaccumbal circuit that render it hypersensitive to exogenous stimulation of PFC afferents at 12 mo., resulting in normalization of evoked accumbal LFP power. Taken together, these recordings nevertheless reveal early-onset of substantial dysfunction in communication among brain regions critical for reward-driven behavior specifically in Tg5 rats.
Figure 2.

Optogenetic stimulation reveals compromised integrity of corticostriatal communication in HD. (a) ChR2 transduction and optogenetic stimulation site in the PFC and extracellular tetrode recording site in the NAc. (b,c) Upper: Representative unfiltered and filtered NAc LFP recordings during 20 Hz optogenetic stimulation of the PFC in 3 mo. rats. Lower: Representative FFTs of NAc power at baseline and during 20 Hz PFC stimulation. (d) Evoked NAc power across frequency ranges as percent change from WT. Tg5 rats had much smaller changes in NAc power following optogenetic PFC stimulation compared to WT rats at 3 and 6 mo. but not 12 mo. (n = 21, *P < 0.05, **P < 0.01, ***P < 0.001 vs. NL). (e–g) Power ratio from NAc LFP recordings across PFC stimulation frequencies. Evoked LFP power is greatly attenuated in Tg5 compared to WT rats at 3 mo. and 6 mo. but not 12 mo. (n = 44, *P < 0.05 vs. Tg5; # P < 0.05, ## P < 0.01, ### P < 0.001 vs. NL). Unless otherwise stated, data are represented as mean ± SEM.
Early and Progressive Dysfunction of the Corticostriatal Circuit in HD
In order to assess longitudinal changes in endogenous neural processing in corticostriatal areas associated with reward seeking, in vivo local LFP recordings were collected simultaneously in the PFC and NAc during performance on the PR task in separate groups of 3, 6, and 12 mo. old rats (Fig. 3a,b). LFPs reflect synaptic input potentials (EPSPs and IPSPs) as well as slower membrane voltage changes (Buzsaki 2006) averaged over a large population of neurons. Systematic, coordinated activity results in rhythmic oscillations in the LFP that are often related to external stimuli. In particular, accumbal gamma frequency oscillations (30.1–120 Hz; Fig. 3c) are frequently associated with reward-motivated behavior (van der Meer and Redish 2009; Kalenscher et al. 2010) as well as reward receipt (Berke 2009; van der Meer and Redish 2009; Kalenscher et al. 2010). Session-wide estimates of the LFP frequency content revealed significant alterations in signal strength within the PFC and the NAc in Tg5 vs. WT rats (Fig. 3d–i). Tg5 rats exhibited normal spectral densities of the PFC LFP at 3 (Fig. 3d) and 6 (Fig. 3e) mo., but power of high frequency gamma oscillations was attenuated compared to WT rats by 12 mo. (Fig. 3F, *P < 0.05) [High Gamma – Geno.: F(1,41) = 8.028, P < 0.01; Age: F(2,41) = 0.3597, ns; Geno. × Age: F(2,41) = 0.7741, ns]. Within the NAc, however, power of both low and high gamma frequency oscillations was reduced in Tg5 animals as early as 3 mo. (Fig. 3g, *P < 0.05) [Low Gamma – Geno.: F(1,42) = 8.128, P < 0.01; Age: F(2,42) = 1.540, ns; Geno. × Age: F(2,42) = 8.389, P < 0.001; High Gamma – Geno.: F(1,42) = 10.94, P < 0.01; Age: F(2,42) = 0.5498, ns; Geno. × Age: F(2,42) = 4.763, P < 0.05]. By 6 mo., there was a normalization of accumbal LFP power compared to WT counterparts (Fig. 3h), but genotype differences in high gamma power re-emerged by 12 mo. (Fig. 3i, *P < 0.05). Given prior evidence of poor PFC–NAc neural transmission at 6 mo. (Fig. 2d,h), the normalized accumbal LFP power in 6 mo. old Tg5 rats measured here may reveal dynamic compensatory adaptation in the ventral striatal network, which weakens again and perhaps definitively by 12 mo.
Figure 3.

Progressive corticostriatal network dysfunction in HD. (a) Multi-electrode extracellular recording sites in the PFC and NAc. (b) Representative simultaneous LFP recordings collected during PR task performance. (c) Representative power spectrum illustrating functional frequency bandwidths [Δ: 0–3 Hz, θ: 3.1–7 Hz, α: 7.1–12 Hz, β: 12.1–30 Hz, low γ: 30.1–55 Hz, high γ: 55.1–120 Hz]. Inset: Magnification of 0–12 Hz range. (d–f) Intra-PFC power spectra demonstrate attenuated high gamma power at 12 mo. in Tg5 vs. WT rats. Insets: AUC of frequency bandwidth power (n = 47). (g–i) Intra-NAc power spectra reveal genotype differences in gamma frequency power across ages. Compared to WT rats, Tg5 rats display lower power in both low and high gamma frequency ranges at 3 mo., normalization of power at 6 mo., and then reemergence of abnormally lower power in the high gamma range at 12 mo. Insets: AUC of frequency bandwidth power (n = 48). (j–l) LFP–LFP coherence between the PFC and NAc is diminished in Tg5 rats at 6 and 12 mo., but not 3 mo. Insets: AUC of frequency bandwidth coherence (n = 47). Unless otherwise stated, data are represented as mean or mean ± SEM. *P < 0.05 vs. Tg5.
These regional deficits in field potential strength were accompanied by inter-regional disruptions in neural synchrony. Correlation and coherence among LFPs are measures of functional coupling among distributed neuronal populations and are critical to a variety of cognitive functions (Uhlhaas and Singer 2006). Consistent with prior work (Berke 2009), session-wide LFP–LFP coherence between the PFC and NAc was robust in both the theta and high gamma frequency ranges (Fig. 3j–l), indicating that these 2 regions couple strongly in these bandwidths. At 3 mo., there were no apparent differences in PFC–NAc coherence among Tg5 and WT rats across any frequency bands (Fig. 3j). However, by 6 mo., high gamma frequency coherence was diminished in the Tg5 compared to WT animals (Fig. 3k, *P < 0.05) [High Gamma – Geno.: F(1,41) = 13.86, P < 0.001; Age: F(2,41) = 0.6787, ns; Geno. × Age: F(2,41) = 1.417, ns], and this significant reduction in gamma coherence persisted through 12 mo. (Fig. 3l, *P < 0.05). These findings illustrate a progressive frequency-specific disconnection in the corticoaccumbal circuit. All together, these results from simultaneous PFC and NAc recordings in awake animals reveal dynamic changes in both local and corticostriatal networks that appear as early as 3 mo. and continue to develop over time, resulting in substantial reduction of circuit-wide neural connectivity by 12 mo. of age in Tg5 rats.
Aberrant Reward-evoked Corticostriatal Network Activity in HD
Simultaneous recordings in the PFC and NAc allowed monitoring of ongoing corticostriatal network dynamics during reward-motivated behavior. In particular, coherence of oscillatory activity among PFC and NAc networks was examined at the time of reinforcement. Although there were no robust genotype differences in evoked power of the PFC (Fig. S7) or NAc (Fig. S8) LFP upon reward delivery, there were prominent differential reward-induced changes in corticoaccumbal network synchrony among Tg5 and WT animals (Fig. 4). At 3 mo. (Fig. 4a–c), significant evoked coherence was present in low frequency bandwidths (e.g., delta, alpha, theta) in WT rats († P < 0.05) and select low (e.g., theta) and high (e.g., beta, low gamma, high gamma) frequency bandwidths in Tg5 rats (& P < 0.05), but overall, there were only minor differences between the genotypes (*P < 0.05) [3 mo. Delta – Geno.: F(1,10) = 1.344, ns; Block: F(9,90) = 7.315, P < 0.0001; Geno. × Block: F(9,90) = 1.797, ns; Theta – Geno.: F(1,8) = 0.2952, ns; Block: F(9,72) = 19.01, P < 0.0001; Geno. × Block: F(9,72) = 0.6968, ns; Alpha – Geno.: F(1,9) = 1.198, ns; Block: F(9,81) = 10.82, P < 0.0001; Geno. × Block: F(9,91) = 1.299, ns; Beta – Geno.: F(1,11) = 3.301, ns; Block: F(9,99) = 8.050, P < 0.0001; Geno. × Block: F(9,99) = 1.457, ns; Low Gamma – Geno.: F(1,10) = 2.729, ns; Block: F(9,90) = 17.25, P < 0.0001; Geno. × Block: F(9,90) = 0.8772, ns; High Gamma – Geno.: F(1,11) = 1.135, ns; Block: F(9,99) = 9.933, P < 0.0001; Geno. × Block: F(9,99) = 0.6781, ns; also see Supplemental Results, Fig. S6]. However, at 6 mo. (Fig. 4d–f), reward delivery was followed by few significant elevations in PFC–NAc coherence in WT rats (e.g., beta; † P < 0.05) but robust elevations across most frequency ranges in Tg5 rats (e.g., delta, theta, alpha, low gamma, high gamma; & P < 0.05) [6 mo. Delta – Geno.: F(1,11) = 2.412, ns; Block: F(9,99) = 7.973, P < 0.0001; Geno. × Block: F(9,99) = 3.100, P < 0.01; Theta – Geno.: F(1,10) = 3.708, ns; Block: F(9,90) = 10.91, P < 0.0001; Geno. × Block: F(9,90) = 2.380, P < 0.05; Alpha – Geno.: F(1,12) = 2.642, ns; Block: F(9 108) = 6.464, P < 0.0001; Geno. × Block: F(9 108) = 3.252, P < 0.01; Beta – Geno.: F(1,12) = 0.113, ns; Block: F(9 108) = 6.065, P < 0.0001; Geno. × Block: F(9 108) = 0.5311, ns; Low Gamma – Geno.: F(1,12) = 6.816, P < 0.05; Block: F(9 108) = 12.47, P < 0.0001; Geno. × Block: F(9 108) = 6.019, P < 0.0001; High Gamma – Geno.: F(1,13) = 3.387, ns; Block: F(9 117) = 7.431, P < 0.0001; Geno. × Block: F(9 117) = 4.777, P < 0.0001]. This resulted in notable genotype differences in coherence of PFC–NAc network oscillations over nearly all frequency bandwidths (*P < 0.05, **P < 0.01, ****P < 0.0001), indicating widespread potentiation of oscillatory coupling across the frequency spectrum in the Tg5 rats at this age. By 12 mo. (Fig. 4g–i), WT rats demonstrated significant event-related coherence only in the delta-range frequencies († P < 0.05), while Tg5 rats showed significant event-related coherence restricted to the theta-range frequencies (& P < 0.05) [12 mo. Delta – Geno.: F(1,7) = 3.415, ns; Block: F(9,63) = 4.456, P < 0.001; Geno. × Block: F(9,63) = 2.083, P < 0.05; Theta – Geno.: F(1,8) = 8.584, P < 0.05; Block: F(9,72) = 26.62, P < 0.0001; Geno. × Block: F(9,72) = 6.810, P < 0.0001; Alpha – Geno.: F(1,10) = 0.3904, ns; Block: F(9,90) = 2.687, P < 0.01; Geno. × Block: F(9,90) = 1.872, ns; Beta – Geno.: F(1,9) = 0.3079, ns; Block: F(9,81) = 5.581, P < 0.0001; Geno. × Block: F(9,81) = 2.611, P < 0.05; Low Gamma – Geno.: F(1,9) = 2.850, ns; Block: F(9,81) = 4.489, P < 0.0001; Geno. × Block: F(9,81) = 4.592, P < 0.0001; High Gamma – Geno.: F(1,8) = 0.1547, ns; Block: F(9,72) = 2.526, P < 0.05; Geno. × Block: F(9,72) = 3.548, P < 0.01]. Prior to reward delivery, Tg5 rats exhibited notably lower coherence in the low gamma bandwidth compared to their WT counterparts (*P < 0.05, **P < 0.01). Similar to results from 6 mo., 12 mo. old Tg5 rats demonstrated greater increases in theta-range coherence at reward delivery when compared to WTs (**P < 0.001). Across all ages, reward delivery consistently elicited increases in theta frequency coherence. Notably, stronger evoked PFC–NAc theta coherence was associated with better performance on the PR task in WT (Fig. 5; R2 = 0.4188, P < 0.01) but not Tg5 rats [R2 = 0.01913, ns; WT vs. Tg5 slopes: F(1,31) = 4.74082, P < 0.05]. Overall, these findings suggest compromised synchrony and coordination among neural activity in the corticoaccumbal network upon reward delivery. Whereas WT rats generally display brief increases in PFC–NAc coherence, Tg5 rats demonstrate prolonged event-related potentiation of PFC–NAc coherence, suggesting considerable temporally-patterned dysfunction in network-wide reward processing with potential for disruption of reward-directed behavior.
Figure 4.

Abnormal reward-evoked changes in corticostriatal network dynamics in HD. Left: Comparison of WT and Tg5 average wavelet spectrograms demonstrates dysregulation of LFP–LFP coherence among the PFC and NAc regions upon reward delivery (0 s, dashed line). Right: Frequency-specific PFC–NAc coherence within the 10-s window centered around reward delivery and normalized to the first second of the window. (a–c) At 3 mo., there were robust elevations in PFC–NAc coherence in response to reward delivery in both WT and Tg5 but few differences in coherence among the genotypes (n = 12). (d–f) By 6 mo., increases in LFP–LFP coherence at reward delivery were present, and Tg5 vs. WT rats showed prominent potentiation of network coherence upon reward delivery across most frequency bandwidths, including delta, theta, alpha, low gamma, and high gamma (n = 13). (g–i) At 12 mo., abnormally elevated PFC–NAc coherence in the theta frequency range persisted in Tg5 compared to WT rats. However, there was overall attenuation of evoked coherence with normalization of coherence magnitude in the delta, alpha, low gamma, and high gamma bandwidths and with significantly lower coherence in the low gamma frequency range pre-reward delivery in the Tg5 vs. WT rats (n = 10). Unless otherwise stated, data are represented as mean or mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. Tg5; WT: † P < 0.05 vs. Bin 1; Tg5: & P < 0.05 vs. Bin 1.
Figure 5.

Reward-evoked corticostriatal theta synchrony predicts motivational state in WT but not HD rats. Among all WT animals, higher peak reward-evoked PFC–NAc coherence in the theta frequency range (3.1–7 Hz) was correlated with a greater number of reinforcers earned on the PR task (n = 17; R2 = 0.4188, P < 0.01). Consistent with abnormally elevated reward-evoked theta coherence in the Tg5 animals across all ages, no strong relationship among theta coherence and PR task performance was measured in the Tg5 rats (n = 18; R2 = 0.01913, ns).
Discussion
Here, we demonstrate early onset of profound motivational and hedonic deficits that are sustained through advanced ages in a transgenic rat model of HD. These deficits are accompanied by significant and progressive dysfunction in the coordination of distributed neural activity between and within brain regions critical for reward-driven behavior (Table 1). During the early period at 3 mo., Tg5 rats already exhibited diminished motivation for reward as well as notable corticoaccumbal and NAc-specific circuit dysfunction. Onset of goal-directed motor impairments and the persistence of avolition over later months were associated with further development of intra-PFC and corticoaccumbal network-wide abnormalities. Together, these findings offer new and fundamental insight into the longitudinal progression of HD-mediated affective disturbances and associated dysfunction in the neural substrates of reward-motivated behavior. Furthermore, they define early corticoaccumbal dysfunction as a possible key pathogenic contributor to a prominent motivational endophenotype of HD and may help identify potential pharmacotherapeutic targets for its successful treatment.
Table 1.
Summary of behavioral and neurophysiological impairments in Tg5 vs. WT rats across ages
| Age | |||
|---|---|---|---|
| 3 mo. | 6 mo. | 12 mo. | |
| Behavior | |||
| Motor (gross locomotor) | – | – | – |
| Motor (operant response rate) | – | ↓ | ↓ |
| Progressive ratio (PR) breakpoints | ↓ | ↓ | ↓ |
| Saccharin preference | ↓ | ↓ | ↓ |
| Neural Dynamics | |||
| PFC synchrony | – | – | ↓ |
| NAc synchrony | ↓ | – | ↓ |
| PFC–NAc functional connectivity (anesthetized, stimulated) | ↓ | ↓ | – |
| PFC–NAc functional connectivity (awake, session-wide) | – | ↓ | ↓ |
| PFC–NAc functional connectivity (awake, perievent) | – | ↑ | ↑↓ |
↓Decrease in this measure compared to WT; ↑Increase in this measure compared to WT.
In human patients and animal models of HD, an outstanding question is whether HD-mediated neural dysfunction originates in the striatum or in brain regions that interact with it (Laforet et al. 2001; Petersén et al. 2005; Cepeda et al. 2007; Raymond et al. 2011). In the present study, a reduction in reward-motivated behavior (Table 1, Fig. 1b,h, Fig. S3D–E) was accompanied by intact intra-PFC synchrony (Fig. 3d) but compromised optically-evoked PFC–NAc communication (Fig. 2d,g) and profound deficits in intra-NAc synchrony (Fig. 3g) already at 3 mo. Reduced NAc synchronization could be a consequence of PFC–NAc disconnection, but it could also be the cause of impaired coupling between these regions as synchronization of neural responses is necessary for propagation across sparsely connected networks (Abeles 1991). From these data, it is difficult to differentiate between the source of dysfunction in the coordination of PFC and NAc neural activity. Quite likely, HD-mediated neural dysfunction may be concurrently evolving in both brain regions already at 3 mo., contributing to the marked reduction in the pursuit of reward.
By 6 mo., progressive alterations in the corticoaccumbal network resulted in significant impairment in optically-evoked (Fig. 2d,h) and endogenous (Fig. 3k) PFC–NAc functional connectivity in Tg5 vs. WT rats (Table 1). At the same time, intra-PFC oscillations remained unaffected (Fig. 3e), while intra-NAc oscillations normalized to WT levels (Fig. 3h). The potentiation of accumbal LFP power from 3 to 6 mo. of age likely reflects ongoing neural compensation for abnormal processes within the NAc or within areas that interact with it. This is also consistent with elevations in reward-evoked PFC–NAc coherence in Tg5 rats during the PR task (Fig. 4d–f). Specifically, Tg5 rats exhibited significantly lower session-wide PFC–NAc coherence in the high gamma bandwidth compared to their WT counterparts (Fig. 3k), but they demonstrated irregular, high coherence time-locked to reward delivery across nearly all frequency bandwidths (Fig. 4d–f). While it seems counter-intuitive that lower session-wide coherence between the PFC and NAc would be accompanied by elevated coherence post-reward, these findings are consistent with work from HD patients and other animal models of HD that reveal corticostriatal disconnection often accompanied by heightened or dysregulated activity in the striatum (Cepeda et al. 2007; Raymond et al. 2011; Veldman and Yang 2018). Adaptive behavior requires critical integration and segregation of neural activity with controlled spatiotemporal dynamics (Uhlhaas and Singer 2006). Our data reveal low coordination among distributed neural assemblies outside of salient task events, which indicates a deficit in corticostriatal communication at baseline. However, in response to a stimulus that may drive PFC–NAc communication (e.g., reward delivery), the abnormally robust and sustained increase in coherence observed may reflect heightened striatal receptivity to input. This maladaptive long-lasting coordination at reinforcement may indeed be one of the principal contributors to compromised reward-seeking behavior in the Tg5 rats.
Of significant interest is how compromised PFC–NAc network integrity at 6 mo. may contribute to the emergence of reward-driven motor deficits in the Tg5 rats (Fig. 1F). The onset of longer latencies during reward seeking at this age may reflect the development of the HD motor phenotype. Although we did not observe pathological locomotor behavior across any age, others have reported impaired motor function in the Tg5 rats starting between 1 (i.e., rotarod) (Yu-Taeger et al. 2012) and 4 mo. (i.e., open field) (Abada et al. 2013). However, motivated behavioral deficits could also be a manifestation of corticostriatal and accumbal impairments in neural processing. Medium spiny neurons of the NAc receive and integrate convergent glutamatergic inputs from cortical and limbic regions that are important for generating and regulating motivated behaviors (Mogenson et al. 1980; Meredith et al. 2008; Sesack and Grace 2010). In turn, they project to motor-related regions that enable the execution of these behaviors. Therefore, advancing dysfunction in the NAc and in the larger corticoaccumbal network may critically interfere with the translation of motivation into action, likely contributing to the significant avolition and anhedonia observed in this study.
By 12 mo. of age, Tg5 rats experience cognitive and emotional deficits (Jansson et al. 2014; Clemensson, Clemensson, Riess, et al. 2017; Manfré et al. 2018) and advanced neuropathology (Yu-Taeger et al. 2012; Teo et al. 2016), involving the formation of mHtt aggregates in both the cortex and NAc (Yu-Taeger et al. 2012). Consistent with these findings, we report widespread neural dysfunction in the corticoaccumbal circuit, including attenuation of the PFC (Fig. 3f) and accumbal (Fig. 3i) networks in addition to impaired PFC–NAc functional connectivity (Fig. 3l, Fig. 4g–i). Interestingly, at 12 mo., optical stimulation of the corticoaccumbal pathway evoked a NAc response that was potentiated over earlier ages (Fig. S5B) and comparable to WT levels (Fig. 2d,i). These findings, nevertheless, align with prior work, mentioned above, that reveals that corticostriatal disconnection at advanced ages is often accompanied by potentiated excitability and dysregulation of the striatum (Cepeda et al. 2007; Raymond et al. 2011; Veldman and Yang 2018). For instance, ensemble recordings of the PFC (Walker et al. 2008) and dorsal striatum (Miller et al. 2008, 2010) demonstrated a loss of coordinated single-unit spiking activity within each region in fully symptomatic animals. Additional studies identified reduced glutamatergic input to striatal MSNs (Cepeda et al. 2003; Singaraja et al. 2011), enhanced MSN firing rates (Rebec et al. 2006; Miller et al. 2008), and overall more highly excitable MSNs (Cepeda et al. 2003; Joshi et al. 2009; André et al. 2011). Our translational corticostriatal field potential measures complement these previous findings and extend observations across early and advanced ages, helping to identify and characterize onset of HD-mediated dysfunction.
Overall, the present study extends earlier work focused on the motor cortex and dorsal striatum to the investigation of corticostriatal regions critically involved in motivated behavior. For the first time, we demonstrate significant and progressive deficits in the recruitment and coordination of the PFC and NAc during impaired reward-seeking behavior in HD animals. These findings offer previously unseen insight to the pathological neural processes that may underlie emergent motivational dysfunction in the HD phenotype and may help to develop successful biomarkers and treatments for the disease.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Notes
This work was supported by the CHDI Foundation (award to J.F.C.) and the National Institutes of Health (F32 DA043967 to N.E.Z.). Conflict of Interest: None declared.
References
- Abada Y-SK, Nguyen HP, Schreiber R, Ellenbroek B. 2013. Assessment of motor function, sensory motor gating and recognition memory in a novel BACHD transgenic rat model for huntington disease. PLoS One. 8:e68584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeles M. 1991. Corticonics: Neural Circuits of the Cerebral Cortex [WWW Document]. Camb Core. URL /core/books/corticonics/7BF149062695412A32FFC1255C98B410
- Akwei-Sekyere S. 2015. Powerline noise elimination in biomedical signals via blind source separation and wavelet analysis. PeerJ. 3:e1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulos GS, Hoptman MJ, Yuen G, Kanellopoulos D, Seirup JK, Lim KO, Gunning FM. 2013. Functional connectivity in apathy of late-life depression: a preliminary study. J Affect Disord. 149:398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- André VM, Cepeda C, Fisher YE, Huynh M, Bardakjian N, Singh S, Yang XW, Levine MS. 2011. Differential electrophysiological changes in striatal output neurons in Huntington’s disease. J Neurosci Off J Soc Neurosci. 31:1170–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold JM, Roberts DC. 1997. A critique of fixed and progressive ratio schedules used to examine the neural substrates of drug reinforcement. Pharmacol Biochem Behav. 57:441–447. [DOI] [PubMed] [Google Scholar]
- Berke JD. 2009. Fast oscillations in cortical-striatal networks switch frequency following rewarding events and stimulant drugs. Eur J Neurosci. 30:848–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman K, Brodaty H, Withall A, Seeher K. 2012. Pharmacologic treatment of apathy in dementia. Am J Geriatr Psychiatry Off J Am Assoc Geriatr Psychiatry. 20:104–122. [DOI] [PubMed] [Google Scholar]
- Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, Bonci A. 2012. Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron. 76:790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunner KD, Rebec GV. 2016. Corticostriatal dysfunction in Huntington’s disease: the basics. Front Hum Neurosci. 10:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G. 2006. Rhythms of the brain. New York, NY: Oxford University Press. [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai L-H, Moore CI. 2009. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 459:663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriere N, Besson P, Dujardin K, Duhamel A, Defebvre L, Delmaire C, Devos D. 2014. Apathy in Parkinson’s disease is associated with nucleus accumbens atrophy: a magnetic resonance imaging shape analysis. Mov Disord Off J Mov Disord Soc. 29:897–903. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Calvert CR, Hernández-Echeagaray E, Nguyen OK, Jocoy E, Christian LJ, Ariano MA, Levine MS. 2003. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington’s disease. J Neurosci Off J Soc Neurosci. 23:961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Wu N, André VM, Cummings DM, Levine MS. 2007. The corticostriatal pathway in Huntington’s disease. Prog Neurobiol. 81:253–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemensson EKH, Clemensson LE, Fabry B, Riess O, Nguyen HP. 2017. a. Further investigation of phenotypes and confounding factors of progressive ratio performance and feeding behavior in the BACHD rat model of Huntington disease. PLoS One. 12:e0173232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemensson EKH, Clemensson LE, Riess O, Nguyen HP. 2017. b. The BACHD Rat Model of Huntington disease shows signs of fronto-striatal dysfunction in two operant conditioning tests of short-term memory. PLoS One. 12:e0169051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epping EA, Kim J-I, Craufurd D, Brashers-Krug TM, Anderson KE, McCusker E, Luther J, Long JD, Paulsen JS, PREDICT-HD Investigators and Coordinators of the Huntington Study Group . 2016. Longitudinal psychiatric symptoms in prodromal Huntington’s disease: a decade of data. Am J Psychiatry. 173:184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JM, Salmon DP, Corey-Bloom J, Gamst A, Paulsen JS, Jerkins S, Jacobson MW, Peavy G. 2003. Behavioural abnormalities contribute to functional decline in Huntington’s disease. J Neurol Neurosurg Psychiatry. 74:120–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkin A, Houlihan DD, Kelly JP. 2002. Reduction in preference for saccharin by repeated unpredictable stress in mice and its prevention by imipramine. J Psychopharmacol Oxf Engl. 16:115–123. [DOI] [PubMed] [Google Scholar]
- Harper PS. 1992. The epidemiology of Huntington’s disease. Hum Genet. 89:365–376. [DOI] [PubMed] [Google Scholar]
- Hernandez G, Cheer JF. 2012. Effect of CB1 receptor blockade on food-reinforced responding and associated nucleus accumbens neuronal activity in rats. J Neurosci. 32:11467–11477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodos W. 1961. Progressive ratio as a measure of reward strength. Science. 134:943–944. [DOI] [PubMed] [Google Scholar]
- Huey ED, Lee S, Brickman AM, Manoochehri M, Griffith E, Devanand DP, Stern Y, Grafman J. 2015. Neuropsychiatric effects of neurodegeneration of the medial versus lateral ventral prefrontal cortex in humans. Cortex J Devoted Study Nerv Syst Behav. 73:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hult Lundh S, Nilsson N, Soylu R, Kirik D, Petersén Å. 2013. Hypothalamic expression of mutant huntingtin contributes to the development of depressive-like behavior in the BAC transgenic mouse model of Huntington’s disease. Hum Mol Genet. 22:3485–3497. [DOI] [PubMed] [Google Scholar]
- Jansson EKH, Clemens LE, Riess O, Nguyen HP. 2014. Reduced motivation in the BACHD rat model of Huntington disease is dependent on the choice of food deprivation strategy. PLoS One. 9:e105662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. 2012. Modulation of medial prefrontal cortical activity using in vivo recordings and optogenetics. Mol Brain. 5:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi PR, Wu N-P, André VM, Cummings DM, Cepeda C, Joyce JA, Carroll JB, Leavitt BR, Hayden MR, Levine MS, et al. 2009. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J Neurosci Off J Soc Neurosci. 29:2414–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalenscher T, Lansink CS, Lankelma JV, Pennartz CMA. 2010. Reward-associated gamma oscillations in ventral striatum are regionally differentiated and modulate local firing activity. J Neurophysiol. 103:1658–1672. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy A, Craufurd D. 2011. Treatment of apathy in Huntington’s disease and other movement disorders. Curr Treat Options Neurol. 13:508–519. [DOI] [PubMed] [Google Scholar]
- Laforet GA, Sapp E, Chase K, McIntyre C, Boyce FM, Campbell M, Cadigan BA, Warzecki L, Tagle DA, Reddy PH, et al. 2001. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington’s disease. J Neurosci Off J Soc Neurosci. 21:9112–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Watson GDR, Alloway KD, Lee G, Neuberger T, Zhang N. 2015. Mapping the functional network of medial prefrontal cortex by combining optogenetics and fMRI in awake rats. Neuroimage. 117:114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majid DSA, Aron AR, Thompson W, Sheldon S, Hamza S, Stoffers D, Holland D, Goldstein J, Corey-Bloom J, Dale AM. 2011. Basal ganglia atrophy in prodromal Huntington’s disease is detectable over one year using automated segmentation. Mov Disord Off J Mov Disord Soc. 26:2544–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfré G, Novati A, Faccini I, Rossetti AC, Bosch K, Molteni R, Riva MA, Van der Harst JE, Nguyen HP, Homberg JR. 2018. BACHD rats expressing full-length mutant huntingtin exhibit differences in social behavior compared to wild-type littermates. PLoS One. 13:e0192289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Horta S, Perez-Perez J, van Duijn E, Fernandez-Bobadilla R, Carceller M, Pagonabarraga J, Pascual-Sedano B, Campolongo A, Ruiz-Idiago J, Sampedro F, et al. , Spanish REGISTRY investigators of the European Huntington’s Disease Network . 2016. Neuropsychiatric symptoms are very common in premanifest and early stage Huntington’s Disease. Parkinsonism Relat Disord. 25:58–64. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Baldo BA, Andrezjewski ME, Kelley AE. 2008. The structural basis for mapping behavior onto the ventral striatum and its subdivisions. Brain Struct Funct. 213:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Walker AG, Fowler SC, von Hörsten S, Riess O, Johnson MA, Rebec GV. 2010. Dysregulation of coordinated neuronal firing patterns in striatum of freely behaving transgenic rats that model Huntington’s disease. Neurobiol Dis. 37:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Walker AG, Shah AS, Barton SJ, Rebec GV. 2008. Dysregulated information processing by medium spiny neurons in striatum of freely behaving mouse models of Huntington’s disease. J Neurophysiol. 100:2205–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogenson GJ, Jones DL, Yim CY. 1980. From motivation to action: functional interface between the limbic system and the motor system. Prog Neurobiol. 14:69–97. [DOI] [PubMed] [Google Scholar]
- Naarding P, Janzing JGE, Eling P, van der Werf S, Kremer B. 2009. Apathy is not depression in Huntington’s disease. J Neuropsychiatry Clin Neurosci. 21:266–270. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Baratta MV, Cooper DC. 2013. A method for high fidelity optogenetic control of individual pyramidal neurons in vivo. J Vis Exp JoVE. 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DP, Layton SP, Hale G, Gomperts SN, Davidson TJ, Kloosterman F, Wilson MA. 2009. Micro-drive array for chronic in vivo recording: tetrode assembly. J Vis Exp JoVE. 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostenveld R, Fries P, Maris E, Schoffelen J-M. 2011. FieldTrip: open source software for advanced analysis of MEG, EEG, and invasive electrophysiological data. Comput Intell Neurosci. 2011:156869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papoutsi M, Labuschagne I, Tabrizi SJ, Stout JC. 2014. The cognitive burden in Huntington’s disease: pathology, phenotype, and mechanisms of compensation. Mov Disord Off J Mov Disord Soc. 29:673–683. [DOI] [PubMed] [Google Scholar]
- Paul RH, Brickman AM, Navia B, Hinkin C, Malloy PF, Jefferson AL, Cohen RA, Tate DF, Flanigan TP. 2005. Apathy is associated with volume of the nucleus accumbens in patients infected with HIV. J Neuropsychiatry Clin Neurosci. 17:167–171.15939969 [Google Scholar]
- Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, Guttman M, Johnson S, MacDonald M, Beglinger LJ, et al. , Predict-HD Investigators and Coordinators of the Huntington Study Group . 2008. Detection of Huntington’s disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 79:874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS, Ready RE, Hamilton JM, Mega MS, Cummings JL. 2001. Neuropsychiatric aspects of Huntington’s disease. J Neurol Neurosurg Psychiatry. 71:310–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. 2006. The rat brain in stereotaxic coordinates: Hard Cover Edition. Elsevier. [Google Scholar]
- Petersén A, Gil J, Maat-Schieman MLC, Björkqvist M, Tanila H, Araújo IM, Smith R, Popovic N, Wierup N, Norlén P, et al. 2005. Orexin loss in Huntington’s disease. Hum Mol Genet. 14:39–47. [DOI] [PubMed] [Google Scholar]
- Raymond LA, André VM, Cepeda C, Gladding CM, Milnerwood AJ, Levine MS. 2011. Pathophysiology of Huntington’s disease: time-dependent alterations in synaptic and receptor function. Neuroscience. 198:252–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebec GV, Conroy SK, Barton SJ. 2006. Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: variations with behavioral state and repeated ascorbate treatment. Neuroscience. 137:327–336. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Grace AA. 2010. Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 35:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto A, Holly EN, Boyson CO, DeBold JF, Miczek KA. 2015. Individual differences in anhedonic and accumbal dopamine responses to chronic social stress and their link to cocaine self-administration in female rats. Psychopharmacology (Berl). 232:825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singaraja RR, Huang K, Sanders SS, Milnerwood AJ, Hines R, Lerch JP, Franciosi S, Drisdel RC, Vaid K, Young FB, et al. 2011. Altered palmitoylation and neuropathological deficits in mice lacking HIP14. Hum Mol Genet. 20:3899–3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa M, Moreira F, Jesus-Ribeiro J, Marques I, Cunha F, Canário N, Freire A, Januário C. 2018. Apathy profile in Parkinson’s and Huntington’s Disease: a comparative cross-sectional study. Eur Neurol. 79:13–20. [DOI] [PubMed] [Google Scholar]
- Spierling SR, Mattock M, Zorrilla EP. 2017. Modeling hypohedonia following repeated social defeat: Individual vulnerability and dopaminergic involvement. Physiol Behav. 177:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suska A, Lee BR, Huang YH, Dong Y, Schlüter OM. 2013. Selective presynaptic enhancement of the prefrontal cortex to nucleus accumbens pathway by cocaine. Proc Natl Acad Sci U S A. 110:713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, Borowsky B, Landwehrmeyer B, Frost C, Johnson H, et al. , TRACK-HD Investigators . 2013. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 12:637–649. [DOI] [PubMed] [Google Scholar]
- Teo RTY, Hong X, Yu-Taeger L, Huang Y, Tan LJ, Xie Y, To XV, Guo L, Rajendran R, Novati A, Calaminus C, et al. 2016. Structural and molecular myelination deficits occur prior to neuronal loss in the YAC128 and BACHD models of Huntington disease. Hum Mol Genet. 25:2621–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiruvady DR, Georgiou-Karistianis N, Egan GF, Ray S, Sritharan A, Farrow M, Churchyard A, Chua P, Bradshaw JL, Brawn T-L, et al. 2007. Functional connectivity of the prefrontal cortex in Huntington’s disease. J Neurol Neurosurg Psychiatry. 78:127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W. 2006. Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron. 52:155–168. [DOI] [PubMed] [Google Scholar]
- Unschuld PG, Liu X, Shanahan M, Margolis RL, Bassett SS, Brandt J, Schretlen DJ, Redgrave GW, Hua J, Hock C, et al. 2013. Prefrontal executive function associated coupling relates to Huntington’s disease stage. Cortex J Devoted Study Nerv Syst Behav. 49:2661–2673. [DOI] [PubMed] [Google Scholar]
- Van den Bogaard SJA, Dumas EM, Acharya TP, Johnson H, Langbehn DR, Scahill RI, Tabrizi SJ, van Buchem MA, van der Grond J, Roos RAC, TRACK-HD Investigator Group . 2011. a. Early atrophy of pallidum and accumbens nucleus in Huntington’s disease. J Neurol. 258:412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bogaard SJA, Dumas EM, Ferrarini L, Milles J, van Buchem MA, van der Grond J, Roos RAC. 2011. b. Shape analysis of subcortical nuclei in Huntington’s disease, global versus local atrophy – results from the TRACK-HD study. J Neurol Sci. 307:60–68. [DOI] [PubMed] [Google Scholar]
- Van der Meer MAA, Redish AD. 2009. Low and high gamma oscillations in rat ventral striatum have distinct relationships to behavior, reward, and spiking activity on a learned spatial decision task. Front Integr Neurosci. 3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duijn E, Craufurd D, Hubers AAM, Giltay EJ, Bonelli R, Rickards H, Anderson KE, van Walsem MR, van der Mast RC, Orth M, Landwehrmeyer GB, European Huntington’s Disease Network Behavioural Phenotype Working Group . 2014. Neuropsychiatric symptoms in a European Huntington’s disease cohort (REGISTRY). J Neurol Neurosurg Psychiatry. 85:1411–1418. [DOI] [PubMed] [Google Scholar]
- Veldman MB, Yang XW. 2018. Molecular insights into cortico-striatal miscommunications in Huntington’s disease. Curr Opin Neurobiol. 48:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. 1998. Huntington disease. J Neuropathol Exp Neurol. 57:369–384. [DOI] [PubMed] [Google Scholar]
- Walker AG, Miller BR, Fritsch JN, Barton SJ, Rebec GV. 2008. Altered information processing in the prefrontal cortex of Huntington’s disease mouse models. J Neurosci Off J Soc Neurosci. 28:8973–8982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu-Taeger L, Bonin M, Stricker-Shaver J, Riess O, Nguyen HHP. 2017. Dysregulation of gene expression in the striatum of BACHD rats expressing full-length mutant huntingtin and associated abnormalities on molecular and protein levels. Neuropharmacology. 117:260–272. [DOI] [PubMed] [Google Scholar]
- Yu-Taeger L, Petrasch-Parwez E, Osmand AP, Redensek A, Metzger S, Clemens LE, Park L, Howland D, Calaminus C, Gu X, et al. 2012. A novel BACHD transgenic rat exhibits characteristic neuropathological features of Huntington disease. J Neurosci Off J Soc Neurosci. 32:15426–15438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.