

Abstract
Target vulnerability correlates the level of drug-target engagement required to generate a pharmacological response. High vulnerability targets are those that require only a relatively small fraction of occupancy to achieve the desired pharmacological outcome, whereas low vulnerability targets require high levels of engagement. Here we demonstrate that the slope of the correlation between drug-target residence time and the post-antibiotic effect (PAE) can be used to define the vulnerability of bacterial targets. For macrolides, a steep slope is observed between residence time on the E. coli ribosome and the PAE, indicating that the ribosome is a highly vulnerable drug target. Analysis of the residence time-PAE data for erythromycin, azithromycin, spiramycin, and telithromycin using a mechanistic pharmacokinetic-pharmacodynamic model that integrates drug-target kinetics into predictions of drug activity, lead to the successful prediction of the cellular PAE for tylosin which has the longest residence time (7.1 h) and PAE (5.8 h). Although the macrolide data support a connection between residence time, PAE and bactericidality, many bactericidal β-lactam antibiotics do not give a PAE illustrating the role of factors such as protein resynthesis in the expression of target vulnerability.
Keywords: Target vulnerability, ribosome, antibiotic, macrolide, residence time, post-antibiotic effect, β-lactam
Graphical Abstract

There is a continuing need to develop antibacterial agents with novel mechanisms of action to combat the emergence of resistance to existing antibiotics.1,2 However, despite this high level of need, and the ever more innovative approaches that are focused on the development of new drugs, the success rate of new drug approvals, both in antibacterial space and in other therapeutic areas, remains alarmingly low. One factor that may account for the high level of attrition in the drug discovery process is the heavy reliance on thermodynamic parameters for the selection and optimization of drug leads.3–7 Given that drug and target may not be at equilibrium in the human body, time-dependent target occupancy is a function of both the thermodynamics and kinetics of drug-target interactions.8 In particular, drugs that dissociate slowly from their targets may have extended activity after the drug has been eliminated, enabling dosing frequency to be reduced and leading to an increase in the therapeutic window if their rate of dissociation from off-target proteins is rapid. Since thermodynamic parameters such as IC50 values provide no information on the lifetime of the drug-target complex and can only inform on thermodynamic selectivity measured at constant drug concentration, the integration of drug-target kinetics into the discovery of new drugs is required if kinetic selectivity is also to be brought into play.3
Antibacterial activity depends on several important factors including the potency of the inhibitor that dictates the concentration of the drug that is required to produce inhibition (drug affinity),9 the kinetics of the drug-target interaction that controls how long the resulting inhibition lasts (time-dependent inhibition),10,11 and the level of target inhibition that results in bacterial death/stasis (target vulnerability).9 Significantly, although many targets may be essential for bacterial survival, those that must be continuously occupied at high levels (low vulnerability targets) are much more difficult to drug than those that only require low levels of occupancy to achieve the desired pharmacological outcome (high vulnerability targets).12–16 It follows that target vulnerability will influence whether kinetic selectivity can be employed since the pharmacological effect is more likely to persist after the drug has been eliminated for high vulnerability targets than for low vulnerability targets. An intimate relationship thus exists between target vulnerability, thermodynamic and/or kinetic selectivity, and drug pharmacokinetics (PK): in situations where a low vulnerability target requires continuous exposure to high levels of drug, selectivity will be driven primarily by thermodynamic considerations. In contrast, to take advantage of kinetic selectivity the initial drug concentration (Cmax) should be sufficient to engage the target but drug must then be eliminated rapidly.3,17,18 Target vulnerability will therefore directly impact dosing regimens since the drug effect may persist even at low drug concentrations if a highly vulnerable target is occupied by an inhibitor whose residence time is longer than the rate of drug elimination. This may be particularly important for antibacterial therapy where drugs are often dosed so that the drug concentration is continuously maintained above the MIC, and where presumably the target is 100% occupied, when in some cases drug efficacy may persist even when the concentration falls below the MIC and occupancy is less than 100%. In such circumstances it may be possible to reduce dosing frequency leading to an increase in the therapeutic window and improvements in safety. It follows that methods are needed to assess target vulnerability in order to guide medicinal chemistry efforts to optimize drug PK and aid in the establishment of appropriate dosing regimens.
Existing methods for evaluating target vulnerability include genetic approaches such as gene dosage experiments which have been applied to a number of systems including antibacterial targets.14,15 In addition, Wei and coworkers developed a strategy involving targeted protein degradation to assess the vulnerability of targets in Mycobacterium smegmatis.9 However, reducing the amount of active protein in a cell is fundamentally different from reducing protein activity through chemical inhibition, which necessarily results in a more stable but less active state of the protein. Thus, while changing the amount of protein may be a good way to assess vulnerability towards therapeutic approaches such as antisense RNA or targeted protein degradation,15,19 chemical inhibition is expected to provide a more realistic assessment of vulnerability for small molecule drug discovery programs. Previously, we demonstrated that the slope of the correlation between in vitro drug residence-time and the post-antibiotic effect (PAE), a metric for cellular time-dependent efficacy, provided insight into the relative vulnerability of two antibacterial targets, the LpxC enzyme from Pseudomonas aeruginosa (paLpxC) and the enoyl-ACP reductase FabI from Staphylococcus aureus (saFabI).11 A steeper slope was observed between residence time and PAE for paLpxC,10 compared to saFabI suggesting that the former target is more vulnerable.11 Vulnerability functions were then generated by fitting the kinetic and thermodynamic data to a mechanistic pharmacokinetic-pharmacodynamic (PK-PD) model that included the full kinetic scheme for target inhibition.10,11
A similar strategy is employed here to determine the relative vulnerability of the bacterial ribosome. The ribosome was chosen for two reasons: it is a central target for the action of antibacterial agents, and binding kinetic data exist for a series of macrolides that bind through a two-step induced-fit kinetic mechanism (Figure 1).20,21
Figure 1. The two-step induced-fit binding mechanism.

Ki is the inhibition constant for the first rapid equilibrium step and k5, k6 are the association and dissociation rate constants, respectively, for the second slow step.
Using a selection of macrolides with different residence times, we show that the correlation between residence time and PAE in E. coli has a steep slope indicating that the ribosome is a highly vulnerable target. The residence time-PAE data for four of the macrolides were analyzed using the mechanistic PK-PD model,10 leading to the successful prediction of the cellular PAE for tylosin. Comparison of the ribosomal vulnerability function with that of other targets, such as penicillin binding proteins (PBPs), illustrates the role of protein resynthesis in target vulnerability.
RESULTS AND DISCUSSION
Correlation between macrolide residence time and PAE in E. coli.
In the search for vulnerable drug targets, we speculated that evolutionary pressure may have led to the biosynthesis of compounds that engage highly vulnerable targets. In antibacterial space there are many classes of naturally produced molecules that inhibit bacterial growth and we chose to focus on a series of macrolide ribosome inhibitors for which binding kinetic data were available. Although macrolides are generally more active against Gram positive bacteria, the available binding kinetic data were for the E. coli ribosome, and thus microbiological experiments were also performed in this organism. We selected five macrolides for analysis that included erythromycin, azithromycin, spiramycin, telithromycin and tylosin, and that bind to the E. coli ribosome through a two-step mechanism with residence times varying from 17 min to 7 h (Figure 1).20,21
We first measured the MIC values for the macrolides against a wild-type strain of E. coli and an efflux pump mutant ΔacrAB (Table 1). The experimental MIC values were similar to those reported in the literature, ranging from 7–750 μM for the wild-type strain and 0.34–59 μM for the pump mutant.22–25 We then measured the PAE’s for both strains with erythromycin, azithromycin, spiramycin, and telithromycin, at concentrations ranging from 1x to 16xMIC. Because of the high MIC for spiramycin, the PAE was only determined for this compound in the pump mutant. The PAEs were calculated using a standard procedure in which the time for the bacteria to recover 1 log in growth after compound washout compared to bacteria treated with vehicle was determined by inspection (Figure S1). In general, the PAEs increased as a function of drug concentration and in keeping with our previous studies on LpxC and FabI,10 we used the PAEs obtained at 4xMIC for further analysis (Table 1).
Table 1.
Residence time and cellular PAE values of the macrolide ribosome inhibitors
| Inhibitor | Ki (nM) | MIC (μM) wild-type | MIC (μM) ΔacrAB | tR (h) | PAE (h)a wild-type | PAE (h)a ΔacrAB |
|---|---|---|---|---|---|---|
| Erythromycin | 393b | 89 | 0.34 | 0.28 ± 0.05b | 0.44 ± 0.28 | 0.2 ± 0.1 |
| Azithromycin | 48b | 6.7 | 0.67 | 1.11 ± 0.10b | 1.18 ± 0.1 | 1.3 ± 0.28 |
| Spiramycin | 2000d | 750 | 19.0 | 1.7 ± 0.57d | NDe | 1.7 ± 0.26 |
| Telithromycin | 500c | 7.4 | 2.46 | 2.03 ± 0.21c | 2.1 ± 0.46 | 2.74 ± 0.54 |
| Tylosin | 2,950 | 280 | 59 | 7.2 | NDe | 5.8 ± 0.8 |
PAE values are reported at 4xMIC. The wild-type E. coli strain is MG1655 and the pump mutant is MG1655 ΔacrAB.
Data at 25°C from Dinos, 2001.20
Data at 25°C from Kostopoulou, 2012.21
Data at 25°C from Brisson-Noel, 1988.26
ND, not determined. Due to the insolubility of spiramycin it was not possible to measure the PAE for this compound in the wild-type strain.
The PAEs ranged from 0.44 to 2.1 h for the wild-type strain and 0.2 to 2.7 h for the pump mutant. The similarity in PAE values between the wild-type and pump mutant strains for erythromycin, azithromycin and telithromycin suggests that the PAE is due to residence time and not rebinding since rebinding would be expected to lead to a longer PAE in the pump mutant. Because we could not determine PAE values for spiramycin in the wild-type strain due to the insolubility of this antibiotic at 2xMIC, the PAEs measured for the pump mutant at 4xMIC were compared with the reported residence times for the four antibiotics (Table 1). The data are plotted in (Figure 2) where it can be seen that an increase in residence time resulted in a prolongation of the PAE supporting a direct relationship between the lifetime of the drug-target complex and the delay in regrowth following compound washout.
Figure 2. Correlation between residence time and PAE.
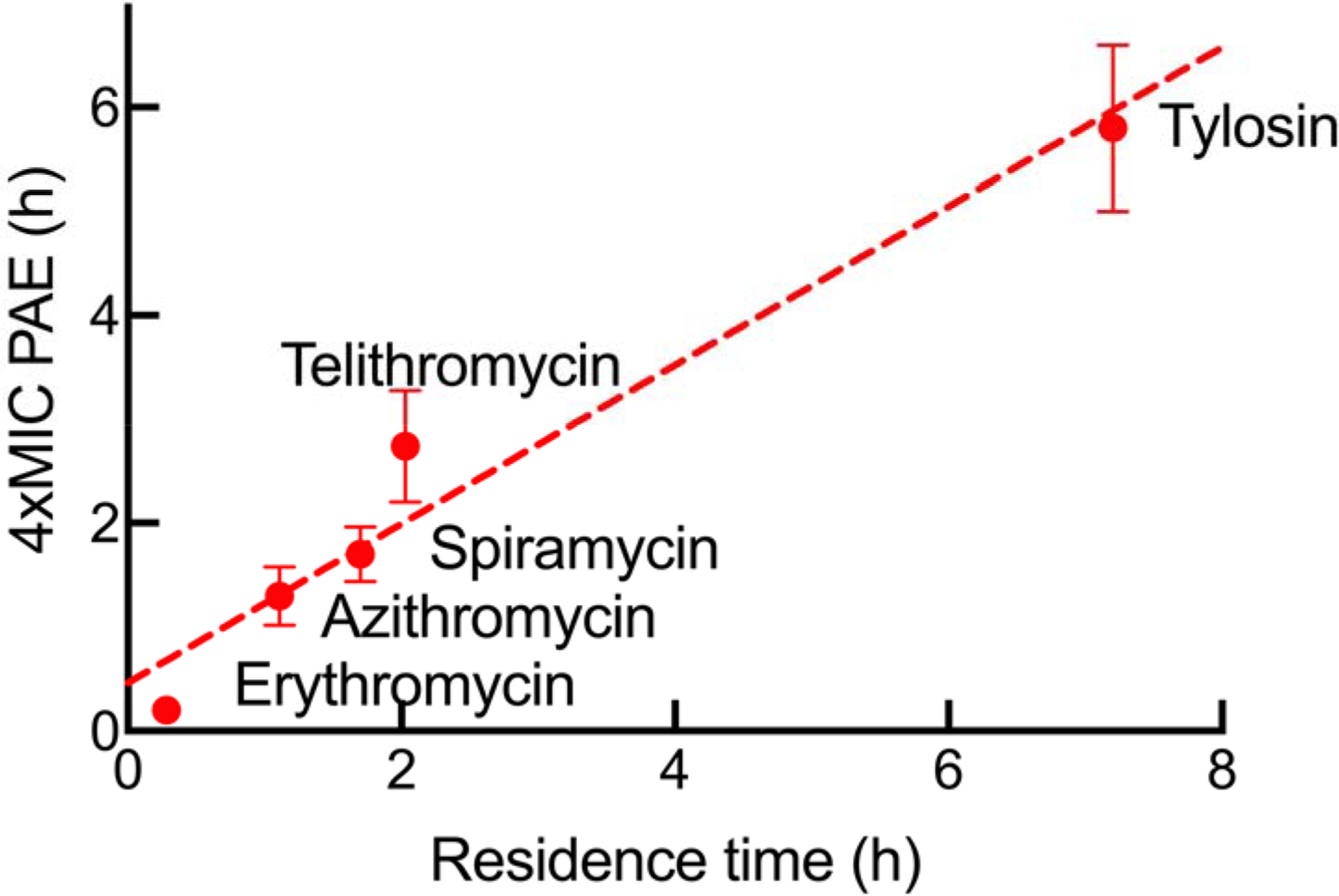
Experimental data are shown by solid symbols and the dashed-line is a linear fit to the data with a slope of 0.76 (R2 0.96). The reported residence-times were determined at 25°C while the PAEs were measured at 37°C against the E. coli ΔacrAB strain. Tylosin is included in the data fitting although the results of simulating and measuring the PAEs for tylosin are presented later in the results and discussion. The curve has not been constrained to pass through 0,0 as it is possible that a compound with no residence time could lead to an appreciable PAE as observed for inhibitors of LpxC.10
To further substantiate the role of residence time as a key driver in the PAE, we globally fit the pump mutant PAE data for the four macrolides to our kinetics-driven mathematical model (Equation 1). This model integrates the thermodynamics and kinetics of drug-target interactions into predictions of drug activity, in this case the PAE. Values for several parameters are needed for the modeling including Ki, k5 and k6 for the binding kinetics scheme shown in Figure 1, the rate of bacterial growth (kgrowth) and death (kkill), M, which is equal to Km/[S], and accounts for the impact of substrate concentration on compound binding, and the permeability parameter pm which takes into account the difference between the intracellular and extracellular drug concentration. The experimentally determined values of Ki, k5, k6 and kgrowth were used as input values for the modeling, and kkill was initially given the same value as kgrowth based on the presumption that macrolides are bacteriostatic (Table 2). An input value of 0.15 was used for M,27 and the input value for pm was estimated from the ratio of the MIC values for the efflux-pump and the wild-type strains. Since the ratio of pm over Ki is used in Equation 1, the input values for the composite parameter of pm/Ki are given in Table 2.
Table 2.
Input and output values of thermodynamic and kinetic parameters for the selected macrolides.
| Parameter | Erythromycin | Azithromycin | Spiramycin | Telithromycin | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Input value | Output value | Ratio | Input value | Output value | Ratio | Input value | Output value | Ratio | Input value | Output value | Ratio | |
| k5 (min−1)a | 0.1 | 0.1 | 1 | 0.1 | 0.2 | 2.3 | 2.2 | 0.8 | 0.4 | 0.5 | 0.4 | 0.8 |
| k6 (min−1)a | 0.06 | 0.05 | 0.8 | 0.02 | 0.004 | 0.3 | 0.007 | 0.006 | 0.9 | 0.008 | 0.003 | 0.4 |
| kgrowth (log10 h−1)b | 2.1 | 1.8 | 0.9 | 2.1 | 2.2 | 1.1 | 2.1 | 2.03 | 1 | 2.1 | 2 | 1 |
| kkill (log10 h−1)b | 2.1 | 2.8 | 1.3 | 2.1 | 2.5 | 1.2 | 2.1 | 2.4 | 1.1 | 2.1 | 2.6 | 1.2 |
| pm | 0.004 | 0.1 | 0.025 | 0.33 | ||||||||
| Ki (μM)a | 0.39 | 0.05 | 2.0 | 0.5 | ||||||||
| pm/Kic | 0.01 | 0.20 | 4.0 | 2 | 0.059 | 0.03 | 0.013 | 0.01 | 0.8 | 0.66 | 0.05 | 0.08 |
| M (Km/[S])d | 0.15 | 0.54 | 1.9 | 0.15 | 0.11 | 0.7 | 0.15 | 0.45 | 3 | 0.15 | 0.13 | 0.8 |
| R2,e | 0.98 | 0.99 | 0.99 | 0.98 | ||||||||
The PAE data obtained at different folds of MIC with the E. coli ΔacrAB pump mutant were fit to the kinetics driven mathematical PK/PD model (Equation 1). Fitting yielded the solid lines shown in Figure 3 and output values for each parameter required to generate the best fit to the data, and the ratio of the input and output value for each parameter are given.
Input values for the binding kinetic parameters determined at 25°C were taken from Dinos,2001,20 for erythromycin and azithromycin, from Brisson-Noel, 1988,26 for spiramycin, and from Kostopoulou, 2012,21 for telithromycin.
kgrowth, a seed value for the cell growth rate was obtained from control cultures grown without antibiotics and kkill was assumed to be the same as kgrowth as the compounds were reported to be bacteriostatic.
pm, the permeability factor, was estimated from the ratio of the MIC values for the wild-type and pump mutant strains. The pm values range from 0.004 to 0.33, giving pm/Ki values of 0.01–2.
M (Km/[S]) was seeded as 0.15, on the basis of the reported range for this parameter in cells.27
R2 is the goodness of fit.
Global fitting of the PAE data for the four antibiotics to Equation 1 using the input values generated the fitted curves in Figure 3 together with output values for each parameter (Table 2). The experimental data are well described by the model (R2 ≥ 0.98) and in general only a small variation (≤ 3-fold) was observed between the input and output values indicating that the parameters used in Equation 1 are sufficient to account for the PAE results. The exception is the value of pm/Ki which varied by as much as 30-fold. A similar discrepancy between the input and output values for pm/Ki was also observed for inhibitors of LpxC which we speculated was due to variation in compound permeation and the limitations of using the difference between the MIC values of the wild-type and pump mutant strains as the sole predictor of permeability (Table 2). The ability of the modeling to describe the experimental data again supports the hypothesis that the drug-target residence time of the ribosome inhibitors is responsible for the time-dependent activity of this series of antibiotics.
Figure 3. Fitting of the PAE data to the kinetics-driven mathematical model.

The experimental data are shown as squares and the lines represent the results of fitting to Equation 1. Input and output values for each parameter are given in Table 2. The decrease in CFUs at 1 h is due to a 1000-fold dilution of the bacterial culture into fresh media (see Figure S1).
Simulation of the PAE for tylosin using the kinetics-driven mathematical model.
Based on the assumption that the time-dependent activity of the ribosome inhibitors results from the residence-time of the drug-target complex, we used the mathematical model to predict the PAE of tylosin, a macrolide with a residence-time of 7.2 h. Input values for the binding kinetic parameters for tylosin were taken from Dinos, 2001,20 and a boundary factor of 2 was used which was estimated from the output values of the kinetic parameters obtained from fitting the PAE data of four ribosome-inhibitors to the model. In addition, a value of 0.31 with a boundary factor of 2.3 was used for M, which is an average within the standard deviation of the output values obtained from fitting the PAEs for the other macrolides to Equation 1. Values for kgrowth and kkill were 2 and 2.6 with the boundary factor of 1.1, respectively, and as the MIC values for tylosin are within 3-fold of those for spiramycin, the pm/Ki value for tylosin was assumed to be similar to or smaller than that of spiramycin (0.01). Parameter values were allowed to float between the boundary factors to give the best simulation of the experimental data by Equation 1. The simulated PAE growth curves at 1x, 2x and 4x MIC for tylosin are shown in Figure 4, and the PAE values are compared with the experimentally determined values in Table 3.
Figure 4. Simulation of the PAE for tylosin.

The experimental data are shown as squares and the lines represent the best simulation of the data by Equation 1 (R2 = 0.95) when the input values were allowed to float between the boundary factors. Input values for the parameters were as follows with the output values in parentheses: k5 1.36 (0.68) min−1, k6 0.0023 (0.0046) min−1, kgrowth 2.0 (1.9) log10 h−1, kkill 2.6 (2.4) log10 h−1, pm/Ki 0.01 (0.0027), and Km/[S] 0.31 (0.62). The decrease in CFUs at 1 h is due to a 1000-fold dilution of the bacterial culture into fresh media (see Figure S1).
Table 3.
In vitro and cellular kinetic and thermodynamic parameters for tylosin.
| Kia (μM) | k5a (min−1) | k6a (min−1) | tR (h) | MIC (μM) wild-type | MIC (μM) ΔacrAB | PAE (h) 4xMIC | PAE (h) 2xMIC | PAE (h) 1xMIC |
|---|---|---|---|---|---|---|---|---|
| 2.95 | 1.36 | 0.0023 | 7.2 | 280 | 59 | 5.8 ± 0.8 (4) | 2.9 ± 0.6 (2.9) | 2.3 ± 0.2 (1.6) |
Binding kinetic parameters were determined at 25°C,20 and experimental PAE data were collected at 37°C against the pump-mutant E. coli ΔacrAB strain. The experimental PAE values are the means ± standard deviation from duplicate experiments. The PAE values in parentheses are the predicted values obtained from the simulation.
In general, there was good agreement between the predicted and experimental PAE values for tylosin, supporting the ability of the model to integrate the kinetic parameters for drug binding into predictions of antibacterial activity. The data for tylosin are included in Figure 2 where a linear fit to the values for all 5 macrolides gives a slope of 0.76. Collectively, these data support a direct role of drug-target residence time in the post-antibiotic effect caused by the macrolides.
Target vulnerability: Slope of the residence time PAE correlation.
As previously discussed, the slope of the correlation between residence time and PAE provides insight into target vulnerability.11 For paLpxC a slope of 1.3 was observed compared to 0.27 for saFabI, suggesting that paLpxC is significantly more vulnerable than saFabI. In the present work we observed a slope of 0.76 for the macrolide inhibitors of the E. coli ribosome. However, the binding kinetic data used here were obtained at 25°C while the PAEs were measured at 37°C. In contrast, both the PAE and tR data for paLpxC and saFabI were obtained at 37°C. Since the kinetic rate constants for formation and breakdown of the drug-target complex are expected to be faster at 37°C,3 this would lead to a steeper slope. Even a two-fold increase in tR would increase the slope for the ribosome to a similar value to that for paLpxC, indicating that the ribosome is a highly vulnerable target.
The relationship between macrolide residence time and antibacterial activity has also been studied in other systems. In particular, Svetlov et al. studied the activity of erythromycin and solithromycin towards the Gram-positive organism Streptococcus pneumoniae.28 The measured residence time at 37°C for erythromycin was 10 min while biphasic dissociation of solithromycin was observed with residence time values of 2.3 and 49 h. While erythromycin was static, solithromycin was bactericidal, and the investigators proposed that the slow dissociation of the macrolide resulted in the change of mechanism from bacteriostatic to bactericidal. These observations are consistent with the work described here. In our studies the four macrolides with shorter residence times were bacteriostatic at every concentration, whereas tylosin was bactericidal (Figure S2). Nevertheless, even for the four bacteriostatic macrolides, an increase in residence time resulted in an increase in PAE, indicating that bactericidality was not essential for the translation of extended target occupancy to prolonged antibacterial activity.
β-Lactam Antibiotics.
The discussion above suggests that the extended residence time of bactericidal antibiotics might be more strongly coupled to the PAE than bacteriostatic compounds. The paLpxC inhibitors are bactericidal,29 whereas the saFabI inhibitors are bacteriostatic, and our previous work indicates that paLpxC is more vulnerable than saFabI.11 It follows then that β-lactam antibiotics, which are covalent inhibitors of penicillin binding proteins (PBPs), might be expected to give rise to very long PAEs given that these compounds are bactericidal. However, a survey of the literature revealed significant variation in the PAEs caused by different β-lactams such as penicillin G, ampicillin and cefamandole, which in some cases resulted in only very short or in no measurable PAEs in some species and strains.30,31 This variation in PAE may have several mechanistic explanations including the rate at which the covalent PBP-inhibitor complex deacylates and the rate of PBP resynthesis following exposure to β-lactams. For instance, E. coli PBPs inhibited by penicillin G were shown to have a half-lives of only 5–15 min,32–34 in contrast to the much slower deacylation of a PBP from the Gram positive bacterium Streptomyces R39 where the half-lives of the acyl-enzyme complex ranged from 9 to >100 h.35
While slower deacylation may account for the longer PAEs observed in some Gram positive organisms,30 the rate of protein synthesis is also a key factor in the translation of extended target engagement to prolonged antibacterial activity. In strains of Streptococcus pneumoniae and E. coli, normal growth resumed almost immediately following the addition of penicillinase to cultures treated with 50xMIC of penicillin. Using pulse labeling with tritiated phenylalanine, uridine and N-acetylglucosamine, it was shown that the rates of protein, RNA and cell wall synthesis quickly achieved pretreatment levels following removal of penicillin. In addition, using tritiated penicillin it was shown that normal growth resumed with rapidly synthesized new PBPs, rather than PBPs formed by deacylation of the existing penicillin-PBP acyl-enzyme complexes. Intriguingly, it was found that that newly synthesized PBPs required for growth resumption represented only a small fraction (<10%) of the pretreatment levels of PBPs. The lack of PAE in these systems is thus due to the low vulnerability of PBPs where only a small fraction of the total PBPs is required for cell wall synthesis and normal growth, and where the rate of PBP synthesis is rapid.36 Similarly, in Streptococcus pyogenes, exposure to 10xMIC of penicillin resulted in a PAE of 2.1 h which was attributed to the time to synthesize sufficient new PBPs to facilitate resumption of growth.37
Factors that modulate target vulnerability
Target vulnerability reflects the fraction of drug-target occupancy required to achieve the desired pharmacological outcome, and there are several molecular factors that control the translation of occupancy to effect. This includes the rate of target synthesis and the length of time the target must be occupied, which is related to the downstream events triggered by target occupancy. The PAE following compound washout integrates all these factors into a response. In the present case we show that the PAE correlates with the residence time of macrolides bound to the ribosome, indicating that the ribosome is a highly vulnerable target. The longest residence time macrolide examined here is bactericidal, supporting the link between residence time and macrolide cidality reported previously.28 However, examination of data for β-lactam antibiotics, which are bactericidal covalent inhibitors of PBPs, indicates that the translation of occupancy to effect in some strains and organisms is reduced either because the PBPs are intrinsically low vulnerability targets based on the fraction of PBP that must be inhibited to result in antibacterial activity and/or due to rapid target resynthesis Thus, targets that are resynthesized slowly are better targets for kinetic selectivity and for covalent inhibitor discovery. Inhibition of the ribosome will necessarily inhibit the synthesis of new ribosomes and it follows that other enzymes involved in transcription/translation might also be vulnerable targets. Taking this thought process one step further, combination therapy that includes an inhibitor of transcription/translation might increase the vulnerability of other targets by reducing the rate of protein synthesis, and anecdotally, we note reports where macrolides given in combination with β-lactams result in improved clinical outcome compared to the β-lactams alone.38–40
Moving forwards, it will be important to include measurements of protein turnover rates in decisions about whether kinetic selectivity can be employed as a mechanism to widen the therapeutic window. In addition, the rate of protein turnover should be also be explicitly included in antibacterial PK/PD models as it was for inhibitors of Bruton’s tyrosine kinase.41 In the present case we were able to model the macrolide PAEs without explicitly including the rate of ribosome synthesis in the PK/PD model presumably because the rate of synthesis is slow compared to other parameters that modulate the translation of occupancy to effect.
CONCLUSION
Target vulnerability is a key factor in drug development since it can directly impact the drug exposure required to achieve the desired pharmacological outcome. Assuming that continuous drug binding is required for efficacy, then high vulnerability targets will require lower levels of drug exposure than low vulnerability targets, assuming that other parameters such as drug-target binding kinetics, target turnover and drug permeability are similar. In particular, high vulnerability targets that turnover slowly will also be suitable for strategies that take advantage of kinetic selectivity using irreversible or slowly dissociating drugs, which can be dosed less frequently leading to improvements in the therapeutic window. Knowledge of target vulnerability thus provides useful guidelines and strategies for medicinal chemists since the PK profile required to inhibit high and low vulnerability targets may be very different. In particular, to take advantage of kinetic selectivity will require drugs that rapidly achieve a sufficient Cmax to engage the target but then which rapidly eliminate in order to minimize the exposure of off-target proteins from which the drug dissociates rapidly. It follows that strategies are needed to assess target vulnerability, and here we show that the slope of the correlation between residence time and PAE can inform on the vulnerability of antibacterial compounds. Similar strategies involving drug exposure and then removal can be envisaged in other disease areas and should prove useful in lead selection and optimization.
MATERIALS AND METHODS
Compounds.
All drugs were commercially available. Erythromycin was obtained from Sigma-Aldrich, azithromycin from TCI America, telithromycin from Gold biotechnology, spiramycin from Santa Cruz Biotech and tylosin from Alfa Aesar. Muller-Hinton broth and agar were purchased from Difco and all other chemicals were obtained from commercial vendors.
Bacterial strains.
Two strains of E. coli were used in this research. The wild-type strain of E. coli was MG1655 and the efflux pump mutant was E. coli MG1655 ΔacrAB which was a generous gift from Prof. Vincent Tam at the University of Houston.
Minimum Inhibitory Concentration (MIC).
Antibacterial susceptibility tests for aerobically growing bacteria were performed with the microbroth dilution assay according to the Clinical and Laboratory Standard Institute, using visual inspection of cells grown in transparent 96-well plates.42 Briefly, bacteria were grown to mid log phase (OD600 of 0.6–0.7) in cation-adjusted Mueller-Hinton (CAMH) media at 37 °C in an orbital shaker. A final inoculum concentration of 106 CFU/mL per well was added to media containing 2-fold dilutions of inhibitors to give final concentrations ranging from 0.03 to 160 μg/mL. The MIC was defined as the minimum concentration of inhibitor at which no visible growth could be detected after 24 h of incubation at 37 °C.
Post-antibiotic Effect (PAE).
Bacteria were grown to mid log phase (OD600 of 0.6–0.7) in CAMH media at 37 °C and then exposed to different concentrations of macrolides or vehicle (DMSO). After shaking for 1 h at 37°C, cultures were diluted 1000-fold into fresh CAMH media to remove unbound drug. Regrowth was monitored by taking 0.1 mL aliquots at 1 h time intervals and plating serial dilutions on Muller-Hinton agar plates. The CFUs were determined by counting colonies after overnight incubation at 37°C. The post-antibiotic effect (PAE) was calculated as the time required for the antibiotic-treated cell population to increase 1 log10 CFU minus the time needed for the control population to increase by 1 log10 CFU (Figure S1).
Cellular PAE modeling.
Mathematical analysis of the PAEs was performed using the method previously reported for paLpxC.10 Data were fit to Equation 1 assuming that the macrolides interact with the ribosome through a two-step binding mechanism.20,21
| Equation 1 |
In Equation 1, bacterial cell numbers (N, CFU/ml) depend on the balance between the rate of logarithmic cell growth (kgrowth) which is assumed to be due to the amount of available free enzyme (E and ES) and the rate of inhibitor-induced cell death (kkill) which is controlled by the amount of enzyme occupied by the inhibitor (EI and EI*). The amount of EI and EI* is calculated from the kinetics and thermodynamics of inhibitor-enzyme binding. Input values for the modeling are given in Tables 2 and 3.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant GM102864 to P.J.T.
Footnotes
Supporting Information
Post-antibiotic data for the macrolides.
CONFLICT OF INTEREST STATEMENT
The authors declare the following competing financial interest(s): P.J.T. is the cofounder of Chronus Pharmaceuticals Inc.
REFERENCES
- (1).Nathan C (2004) Antibiotics at the crossroads Nature, 431, 899–902. DOI: 10.1038/431899a [DOI] [PubMed] [Google Scholar]
- (2).von Nussbaum F; Brands M; Hinzen B; Weigand S; Habich D (2006) Antibacterial natural products in medicinal chemistry--exodus or revival? Angew Chem Int Ed Engl, 45, 5072–129. DOI: 10.1002/anie.200600350 [DOI] [PubMed] [Google Scholar]
- (3).Tonge PJ (2018) Drug-Target Kinetics in Drug Discovery ACS Chem Neurosci, 9, 29–39. DOI: 10.1021/acschemneuro.7b00185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Copeland RA; Pompliano DL; Meek TD (2006) Drug-target residence time and its implications for lead optimization Nat Rev Drug Discov, 5, 730–9. DOI: 10.1038/nrd2082 [DOI] [PubMed] [Google Scholar]
- (5).Copeland RA (2016) The drug-target residence time model: a 10-year retrospective Nat Rev Drug Discov, 15, 87–95. DOI: 10.1038/nrd.2015.18 [DOI] [PubMed] [Google Scholar]
- (6).Schuetz DA; de Witte WEA; Wong YC; Knasmueller B; Richter L; Kokh DB; Sadiq SK; Bosma R; Nederpelt I; Heitman LH; Segala E; Amaral M; Guo D; Andres D; Georgi V; Stoddart LA; Hill S; Cooke RM; De Graaf C; Leurs R; Frech M; Wade RC; de Lange ECM; A.P. I; Muller-Fahrnow A; Ecker GF (2017) Kinetics for Drug Discovery: an industry-driven effort to target drug residence time Drug Discov Today, 22, 896–911. DOI: 10.1016/j.drudis.2017.02.002 [DOI] [PubMed] [Google Scholar]
- (7).Zhang R; Monsma F (2009) The importance of drug-target residence time Curr Opin Drug Discov Devel, 12, 488–96. DOI: [PubMed] [Google Scholar]
- (8).Swinney DC (2009) The role of binding kinetics in therapeutically useful drug action Curr Opin Drug Discov Devel, 12, 31–9. DOI: [PubMed] [Google Scholar]
- (9).Wei JR; Krishnamoorthy V; Murphy K; Kim JH; Schnappinger D; Alber T; Sassetti CM; Rhee KY; Rubin EJ (2011) Depletion of antibiotic targets has widely varying effects on growth Proc Natl Acad Sci U S A, 108, 4176–81. DOI: 10.1073/pnas.1018301108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Walkup GK; You Z; Ross PL; Allen EK; Daryaee F; Hale MR; O’Donnell J; Ehmann DE; Schuck VJ; Buurman ET; Choy AL; Hajec L; Murphy-Benenato K; Marone V; Patey SA; Grosser LA; Johnstone M; Walker SG; Tonge PJ; Fisher SL (2015) Translating slow-binding inhibition kinetics into cellular and in vivo effects Nat Chem Biol, 11, 416–23. DOI: 10.1038/nchembio.1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Daryaee F; Chang A; Schiebel J; Lu Y; Zhang Z; Kapilashrami K; Walker SG; Kisker C; Sotriffer CA; Fisher SL; Tonge PJ (2016) Correlating Drug-Target Kinetics and In vivo Pharmacodynamics: Long Residence Time Inhibitors of the FabI Enoyl-ACP Reductase Chem Sci, 7, 5945–5954. DOI: 10.1039/C6SC01000H [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Datta S (1999) Understanding the Characteristics of a Good Anti-Infective Drug Target. J Parasit Dis, 23, 139–140. DOI: [Google Scholar]
- (13).Payne DJ; Gwynn MN; Holmes DJ; Pompliano DL (2007) Drugs for bad bugs: confronting the challenges of antibacterial discovery Nat Rev Drug Discov, 6, 29–40. DOI: 10.1038/nrd2201 [DOI] [PubMed] [Google Scholar]
- (14).Kaur P; Agarwal S; Datta S (2009) Delineating bacteriostatic and bactericidal targets in mycobacteria using IPTG inducible antisense expression PLoS ONE, 4, e5923 DOI: 10.1371/journal.pone.0005923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ramachandran V; Singh R; Yang X; Tunduguru R; Mohapatra S; Khandelwal S; Patel S; Datta S (2013) Genetic and chemical knockdown: a complementary strategy for evaluating an anti-infective target Adv Appl Bioinform Chem, 6, 1–13. DOI: 10.2147/AABC.S39198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Barry CE 3rd; Boshoff HI; Dartois V; Dick T; Ehrt S; Flynn J; Schnappinger D; Wilkinson RJ; Young D (2009) The spectrum of latent tuberculosis: rethinking the biology and intervention strategies Nat Rev Microbiol, 7, 845–55. DOI: 10.1038/nrmicro2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Vauquelin G; Charlton SJ (2010) Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action Br J Pharmacol, 161, 488–508. DOI: 10.1111/j.1476-5381.2010.00936.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Singh J; Petter RC; Baillie TA; Whitty A (2011) The resurgence of covalent drugs Nat Rev Drug Discov, 10, 307–17. DOI: 10.1038/nrd3410 [DOI] [PubMed] [Google Scholar]
- (19).Fisher SL; Phillips AJ (2018) Targeted protein degradation and the enzymology of degraders Curr Opin Chem Biol, 44, 47–55. DOI: 10.1016/j.cbpa.2018.05.004 [DOI] [PubMed] [Google Scholar]
- (20).Dinos GP; Michelinaki M; Kalpaxis DL (2001) Insights into the mechanism of azithromycin interaction with an Escherichia coli functional ribosomal complex Mol Pharmacol, 59, 1441–5. DOI: 10.1124/mol.59.6.1441 [DOI] [PubMed] [Google Scholar]
- (21).Kostopoulou ON; Petropoulos AD; Dinos GP; Choli-Papadopoulou T; Kalpaxis DL (2012) Investigating the entire course of telithromycin binding to Escherichia coli ribosomes Nucleic Acids Res, 40, 5078–87. DOI: 10.1093/nar/gks174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kouvela EC; Kalpaxis DL; Wilson DN; Dinos GP (2009) Distinct mode of interaction of a novel ketolide antibiotic that displays enhanced antimicrobial activity Antimicrob Agents Chemother, 53, 1411–9. DOI: 10.1128/AAC.01425-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Glassford I; Lee M; Wagh B; Velvadapu V; Paul T; Sandelin G; DeBrosse C; Klepacki D; Small MC; MacKerell AD Jr.; Andrade RB (2014) Desmethyl macrolides: synthesis and evaluation of 4-desmethyl telithromycin ACS Med Chem Lett, 5, 1021–6. DOI: 10.1021/ml5002097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Retsema J; Girard A; Schelkly W; Manousos M; Anderson M; Bright G; Borovoy R; Brennan L; Mason R (1987) Spectrum and mode of action of azithromycin (CP-62,993), a new 15-membered-ring macrolide with improved potency against gram-negative organisms Antimicrob Agents Chemother, 31, 1939–47. DOI: 10.1128/aac.31.12.1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Moore SD; Sauer RT (2008) Revisiting the mechanism of macrolide-antibiotic resistance mediated by ribosomal protein L22 Proc Natl Acad Sci U S A, 105, 18261–6. DOI: 10.1073/pnas.0810357105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Brisson-Noel A; Trieu-Cuot P; Courvalin P (1988) Mechanism of action of spiramycin and other macrolides J Antimicrob Chemother, 22 Suppl B, 13–23. DOI: 10.1093/jac/22.supplement_b.13 [DOI] [PubMed] [Google Scholar]
- (27).Bennett BD; Kimball EH; Gao M; Osterhout R; Van Dien SJ; Rabinowitz JD (2009) Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli Nat Chem Biol, 5, 593–9. DOI: 10.1038/nchembio.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Svetlov MS; Vazquez-Laslop N; Mankin AS (2017) Kinetics of drug-ribosome interactions defines the cidality of macrolide antibiotics Proc Natl Acad Sci U S A, 114, 13673–13678. DOI: 10.1073/pnas.1717168115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Barb AW; Jiang L; Raetz CR; Zhou P (2007) Structure of the deacetylase LpxC bound to the antibiotic CHIR-090: Time-dependent inhibition and specificity in ligand binding Proc Natl Acad Sci U S A, 104, 18433–8. DOI: 10.1073/pnas.0709412104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Bundtzen RW; Gerber AU; Cohn DL; Craig WA (1981) Postantibiotic suppression of bacterial growth Rev Infect Dis, 3, 28–37. DOI: [DOI] [PubMed] [Google Scholar]
- (31).Wilson DA; Rolinson GN (1979) The recovery period following exposure of bacteria to penicillins Chemotherapy, 25, 14–22. DOI: 10.1159/000237817 [DOI] [PubMed] [Google Scholar]
- (32).Tamura T; Imae Y; Strominger JL (1976) Purification to homogeneity and properties of two D-alanine carboxypeptidases I From Escherichia coli J Biol Chem, 251, 414–23. DOI: [PubMed] [Google Scholar]
- (33).Nicola G; Tomberg J; Pratt RF; Nicholas RA; Davies C (2010) Crystal structures of covalent complexes of beta-lactam antibiotics with Escherichia coli penicillin-binding protein 5: toward an understanding of antibiotic specificity Biochemistry, 49, 8094–104. DOI: 10.1021/bi100879m [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Georgopapadakou NH; Liu FY (1980) Penicillin-binding proteins in bacteria Antimicrob Agents Chemother, 18, 148–57. DOI: 10.1128/aac.18.1.148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Fuad N; Frere JM; Ghuysen JM; Duez C; Iwatsubo M (1976) Mode of interaction between beta-lactam antibiotics and the exocellular DD-carboxypeptidase--transpeptidase from Streptomyces R39 Biochem J, 155, 623–9. DOI: 10.1042/bj1550623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Tuomanen E (1986) Newly made enzymes determine ongoing cell wall synthesis and the antibacterial effects of cell wall synthesis inhibitors J Bacteriol, 167, 535–43. DOI: 10.1128/jb.167.2.535-543.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Yan S; Bohach GA; Stevens DL (1994) Persistent acylation of high-molecular-weight penicillin-binding proteins by penicillin induces the postantibiotic effect in Streptococcus pyogenes J Infect Dis, 170, 609–14. DOI: [DOI] [PubMed] [Google Scholar]
- (38).Lodise TP; Kwa A; Cosler L; Gupta R; Smith RP (2007) Comparison of beta-lactam and macrolide combination therapy versus fluoroquinolone monotherapy in hospitalized Veterans Affairs patients with community-acquired pneumonia Antimicrob Agents Chemother, 51, 3977–82. DOI: 10.1128/AAC.00006-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lee JH; Kim HJ; Kim YH (2017) Is beta-Lactam Plus Macrolide More Effective than beta-Lactam Plus Fluoroquinolone among Patients with Severe Community-Acquired Pneumonia?: a Systemic Review and Meta-Analysis J Korean Med Sci, 32, 77–84. DOI: 10.3346/jkms.2017.32.1.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ito A; Ishida T; Tachibana H; Nishiyama A; Furuta K; Tanaka M; Tokioka F; Yoshioka H; Arita M; Hashimoto T (2014) Beta lactam plus macrolide antibiotic combination therapy reduces the mortality of community acquired pneumococcal pneumonia more than beta lactam antibiotics alone Eur Respir J, 44. DOI: [Google Scholar]
- (41).Daryaee F; Zhang Z; Gogarty KR; Li Y; Merino J; Fisher SL; Tonge PJ (2017) A quantitative mechanistic PK/PD model directly connects Btk target engagement and in vivo efficacy Chem Sci, 8, 3434–3443. DOI: 10.1039/c6sc03306g [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).CLSI In Approved Standard M7-A5; 6 ed; Clinical and Laboratory Standards Institute: Wayne, PA, 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.