

Abstract
Quorum sensing (QS), a bacterial cell-to-cell communication system mediated by small molecules and peptides, has received significant interest as a potential target to block infection. The common pathogen Pseudomonas aeruginosa uses QS to regulate many of its virulence phenotypes at high cell densities, and the LasR QS receptor plays a critical role in this process. Small molecule tools that inhibit LasR activity would serve to illuminate its role in P. aeruginosa virulence, but we currently lack highly potent and selective LasR antagonists, despite considerable research in this area. V-06–018, an abiotic small molecule discovered in a high-throughput screen, represents one of the most potent known LasR antagonists, but has seen little study since its initial report. Herein, we report a systematic study of the structure-activity relationships (SARs) that govern LasR antagonism by V-06–018. We synthesized a focused library of V-06–018 derivatives and evaluated the library for bioactivity using a variety of cell-based LasR reporter systems. The SAR trends revealed by these experiments allowed us to design probes with 10-fold greater potency than V-06–018 and 100-fold greater potency than other commonly used N-acyl l-homoserine lactone (AHL)-based LasR antagonists, along with high selectivities for LasR. Biochemical experiments to probe the mechanism of antagonism by V-06–018 and its analogs support these compounds interacting with the native ligand-binding site in LasR and, at least in part, stabilizing an inactive form of the protein. The compounds described herein are the most potent and efficacious antagonists of LasR known, and represent robust probes both for characterizing the mechanisms of LuxR-type QS and for chemical biology research in general in the growing QS field.
Keywords: N-acyl l-homoserine lactone, bacterial communication, intercellular signaling, LuxR-type receptor, small molecule probes, virulence
Graphical Abstract

Microbial resistance to antibiotics is emerging faster than new treatments are being developed, setting the stage for a public health crisis.1–2 As traditional antibiotics become less effective, interest has arisen in attenuating virulence via interference with nonessential pathways.3 Inhibition of quorum sensing (QS), a mode of bacterial communication dependent on the exchange of chemical signals, has been shown to reduce virulence phenotypes in multiple human pathogens without affecting cell viability.4–6 Accordingly, it has attracted significant interest as a potential anti-virulence strategy for combatting bacterial infections.7–8 Our laboratory9–11 and others12–15 are interested in the development of small molecule and peptide probes to dissect the mechanisms of QS and their roles in infection.
The prototypical QS circuit in Gram-negative bacteria is the LuxI/LuxR synthase/receptor pair, first discovered in the marine symbiont Vibrio fischeri.6 At low cell density, a LuxI-type enzyme synthesizes the QS signal, an N-acyl l-homoserine lactone (AHL), at a low basal rate. These low-molecular weight molecules can freely diffuse out of the cell, although in certain cases they are also actively exported.16 The concentration of AHL signal is largely proportional to cell density (and this correlation is highly dependent on the environment), but as a bacterial community grows, the level of AHL signal in the local environment likewise increases (Figure 1A). At high cell densities, the intracellular AHL concentration is sufficient for productive binding of the AHL to its cognate LuxR-type receptor, a transcription factor. The activated receptor:ligand complex then typically dimerizes and binds to DNA, which subsequently alters gene expression levels to promote group-beneficial behaviors. In pathogenic bacteria, these behaviors can include the production of toxic virulence factors and biofilm. Typically, once a “quorum” is achieved, expression of the LuxI-type synthase is also increased, amplifying AHL production in a positive “autoinduction” feedback loop.17
Figure 1:

(A) General schematic of LuxI/LuxR-type quorum sensing (QS) in Gram-negative bacteria. (B) Simplified view of QS in P. aeruginosa. LasI/R and RhlI/R are LuxI/R homologues. QscR is an “orphan” LuxR-type receptor and responds to OdDHL. PqsR is a LysR-type receptor that responds to the Pseudomonas quinolone signal (PQS). AHL synthases are omitted for clarity. (C) Structures of native agonist OdDHL (EC50 = 139 nM), non-AHL antagonist V-06–018 (IC50 = 5.2 μM), non-AHL agonist TP1-P (EC50 = 71 nM), and representative, synthetic AHL antagonist 4-bromo PHL (IC50 = 116 μM); potency values all obtained in the same P. aeruginosa LasR reporter (from ref. 30).20
Pseudomonas aeruginosa is an opportunistic pathogen that regulates many aspects of virulence using QS. This bacterium has a high rate of resistance to traditional antibiotics and causes infections that are especially dangerous for individuals with cystic fibrosis (CF), burn victims, and AIDS patients. The QS system in P. aeruginosa is relatively complex (Figure 1B),18 consisting of two LuxI/LuxR pairs (LasI/LasR and RhlI/RhlR) along with an orphan LuxR-type receptor (QscR), which lacks a related synthase and native AHL signal. LasI synthesizes N-(3-oxo-dodecanoyl)-l-homoserine lactone (OdDHL), which targets LasR but also strongly activates QscR. RhlI synthesizes N-butyryl-l-homoserine lactone (BHL), which targets RhlR. Additionally, P. aeruginosa has a LysR-type receptor, PqsR, which is unrelated to LuxR-type receptors and uses 2-heptyl-3-hydroxy-4-(1H)-quinolone (i.e., the Pseudomonas quinolone signal (PQS)) as its ligand. These four QS systems are intimately linked and control different aspects of P. aeruginosa virulence that are highly dependent on the environment (Figure 1B).9 LasR plays a central role in the QS hierarchy. For instance, LasR directly regulates the production of virulence factors such as elastase, alkaline protease, and exotoxin, and regulates rhamnolipid, HCN, and pyocyanin production via control of the rhl and pqs systems.18 Biofilm, a major virulence phenotype in P. aeruginosa, is also regulated by LasR via the rhl and pqs systems.19 In turn, LasR and RhlR are repressed by QscR, which again is strongly activated via LasR’s native signal, OdDHL.
The connection between QS and virulence in P. aeruginosa, and in other Gram-negative bacterial pathogens, has motivated the development of small molecules and macromolecules capable of inhibiting LuxI-type synthases,21 destroying or sequestering AHL signals,22 or blocking the binding of AHL signal to LuxR-type receptor.23 The latter competitive inhibition strategy has seen the most study to date, with significant contributions by the Spring,7 Bassler,24 Greenberg,25 and Meijler15 labs, as well as our lab.26 Due to its prominent position in the P. aeruginosa QS system (vide supra), much of the effort devoted to identifying small molecule modulators of QS in P. aeruginosa has focused on LasR. The majority of the known synthetic ligands that modulate LasR were identified by making systematic changes to the lactone “head group” and acyl “tail group” of OdDHL (e.g., 4-bromo PHL; Figure 1C).27–28 However, these past efforts have failed to yield compounds that antagonize LasR with both high efficacies and potencies.29 To our knowledge, none of these AHL analogs have lower than double-digit micromolar (μM) IC50 values in reporter gene assays of LasR activity in P. aeruginosa.30 These IC50 values contrast with the nanomolar (nM) EC50 value of LasR’s native ligand, OdDHL, and those of other non-native agonists (e.g., the triphenyl derivative TP-1; Figure 1C).25–26 The poor antagonism potencies for AHL analogs may be due, at least in part, to reliance on the AHL scaffold, which has several major liabilities for probe molecules. AHLs are susceptible to lactone hydrolysis, enzymatic degradation, and active efflux by P. aeruginosa.16, 31–32 These drawbacks make the development of non-AHL antagonists of LasR, and other LuxR-type receptors, highly desirable.30 That said, conversion of non-AHL scaffolds known to strongly agonize LasR (e.g., TP-1) into antagonists (i.e., “mode switching”) has also not provided sub-μM LasR antagonists so far,26 underscoring the challenges of this process.
High-throughput screens of small molecule libraries provide another pathway to identify non-AHL LasR antagonists.33 One such screen by Greenberg and coworkers in 2006 revealed the compound V-06–018, a β-keto amide with a phenyl head group and a nine carbon tail (Figure 1C).33 V-06–018 is a relatively potent LasR antagonist in both E. coli and P. aeruginosa LasR reporter strains (single digit micromolar IC50) and has been shown to inhibit genes and phenotypes related to virulence in P. aeruginosa.9, 33 The phenyl head group and aliphatic acyl tail of V-06–018 resemble that of the homoserine lactone head group and acyl tail of LasR’s native ligand, OdDHL (Figure 1C). However, as V-06–018 lacks a lactone moiety, it is not susceptible to hydrolysis or enzymatic cleavage by AHL lactonases.31–32 A prior study of ours also revealed that V-06–018 is not actively effluxed from P. aeruginosa by the promiscuous MexAB-OprM efflux pump, which is known to efflux both native and non-native AHLs with long acyl tails.16 Despite these desirable qualities, V-06–018 has seen practically no scrutiny from a structure–function perspective and no substantive use as a chemical probe since its initial report over a decade ago.30 We reasoned that the V-06–018 scaffold could provide entry into LasR antagonists with improved potencies along with robust physical properties, and in the current study we report our findings with regard to the first structure-function analysis of this scaffold. Our combined cell-based assays, synthesis, and iterative compound design revealed a set of new LasR antagonists based on V-06–018 with potencies, efficacies, and receptor selectivities in P. aeruginosa that, to our knowledge, surpass all known compounds reported to date. Follow on biochemical experiments on these compounds and V-06–018 support a mechanism of antagonism by which they interact with the OdDHL-binding site in LasR and, at least in part, stabilize an inactive form of the protein.
RESULTS AND DISCUSSION
V-06–018 is selective for LasR over RhlR and QscR in P. aeruginosa
We began our study by exploring the selectivity of V-06–018 for LasR over the other two LuxR-type receptors (RhlR and QscR) in P. aeruginosa, as other than its antagonistic activity in LasR,30 this profile was unknown. In view of the overlapping activities of these three receptors in P. aeruginosa (see Figure 1B), small molecule tools that are selective for LasR (or indeed any of these receptors) are of significant interest for use as mechanistic probes in this pathogen. We submitted V-06–018 to reporter gene assays in E. coli to examine its antagonistic activity (in competition with the receptors’ native or preferred ligand) and agonistic activity (alone) in LasR, RhlR, and QscR, using our previously reported methods (see Materials and Methods). In these reporter assays in a heterologous background (i.e., E. coli), each of the receptors was examined in isolation from the others, allowing for clearer selectivity profiles to be defined relative to using analogous P. aeruginosa reporter systems. Receptor activity was monitored via β-galactosidase production. These experiments revealed V-06–018 was only an antagonist of LasR, displayed no activity (as either an antagonist or agonist) in RhlR, and was only a very weak antagonist QscR at the highest concentrations tested (see Figure S1). This high receptor selectivity profile rendered the V-06–018 scaffold even more compelling for new LasR antagonist development in P. aeruginosa.
An efficient synthesis of V-06–018 and analogs
We next sought to devise a synthetic route to V-06–018 that was scalable and adaptable to analog synthesis. The only previously reported synthesis of V-06–018 gave the molecule in 5% yield, albeit in one step.24 That synthesis involved refluxing ethyl benzoyl acetate and nonylamine in ethanol. We reasoned the low yield for this reaction could be due to imine formation; therefore, we decided to protect the ketone in ethyl benzoyl acetate as a ketal (e.g., 2 → 3; Scheme 1), and then saponified the ester to access the carboxylic acid (4). Standard carbodiimide-mediated amide bond coupling (via EDC) of the acid with nonylamine proceeded smoothly to yield amide 5. Deprotection of the ketone furnished V-06–018 in 44% yield over four steps, in quantities typically greater than 100 mg. This synthetic route was advantageous as it could be easily modified to generate V-06–018 analogs with alternate tail groups (R’ in Scheme 1) through the coupling of different amines. In turn, alternate head groups could be incorporated by coupling different carboxylic acid building blocks (4), many of which are readily accessible from acylation reactions of substituted acetophenones using diethyl carbonate as an electrophile (e.g., 1 → 2; Scheme 1).34 We introduced both modifications in our subsequent synthesis of a focused library of V-06–018 analogs.
Scheme 1:
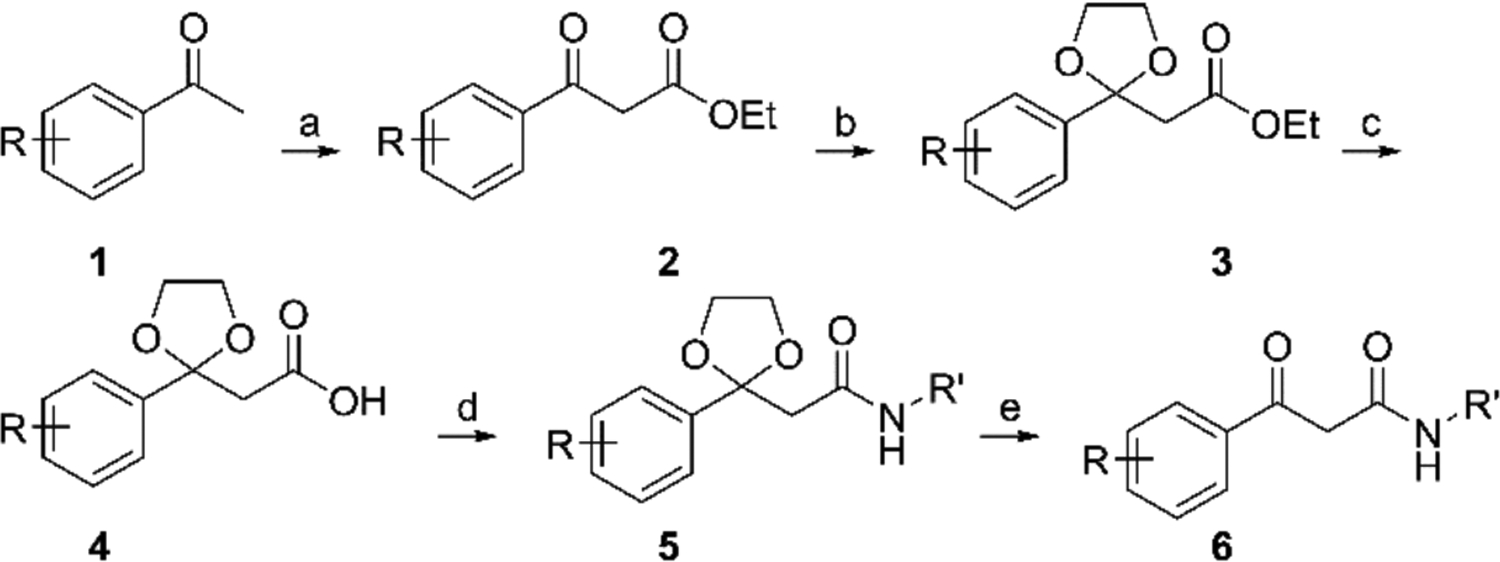
Synthesis of V-06–018 and related analogs. Reagents over arrows: a = NaH, (C2H5)2CO3, THF, Δ; b = C2H6O2, p-TsOH, benzene, Δ, Dean-Stark trap; c = 1:1 LiOH (1M, aq.), THF; d = EDC·HCl, DMAP,H2NR’, CH2Cl2; e = p-TsOH, acetone. See Materials and Methods and SI for additional details.
Structure-informed design of a V-06–018 analog library
We approached our design of V-06–018 analogs by first considering the binding mode of OdDHL to LasR (Figure 2). The reported X-ray structure of OdDHL bound to the LasR ligand-binding domain (LBD) indicates that the lactone, amide, and keto functionality in OdDHL can make several hydrogen bonds with residues in the LasR ligand-binding site (e.g., Tyr 56, Trp 60, Asp 73, and Ser 129).35 In view of their structural similarity (see Figure 1C), it is not unreasonable to assume that V-06–018 could target the same binding site on LasR as OdDHL. We therefore were interested in synthesizing analogs that could either gain or lose the ability to make the same hydrogen bonding contacts as OdDHL, to examine their effects on V-06–018 activity. As the phenyl head group of V-06–018 cannot engage in a hydrogen bond with LasR, we synthesized a series of analogs via Scheme 1 with alternate head groups (8, 12, 13, and 17–21; Figure 3) that either place a heteroatom in a position to potentially accept, or in the case of phenols 17 and 18, accept and/or donate a hydrogen bond.
Figure 2:
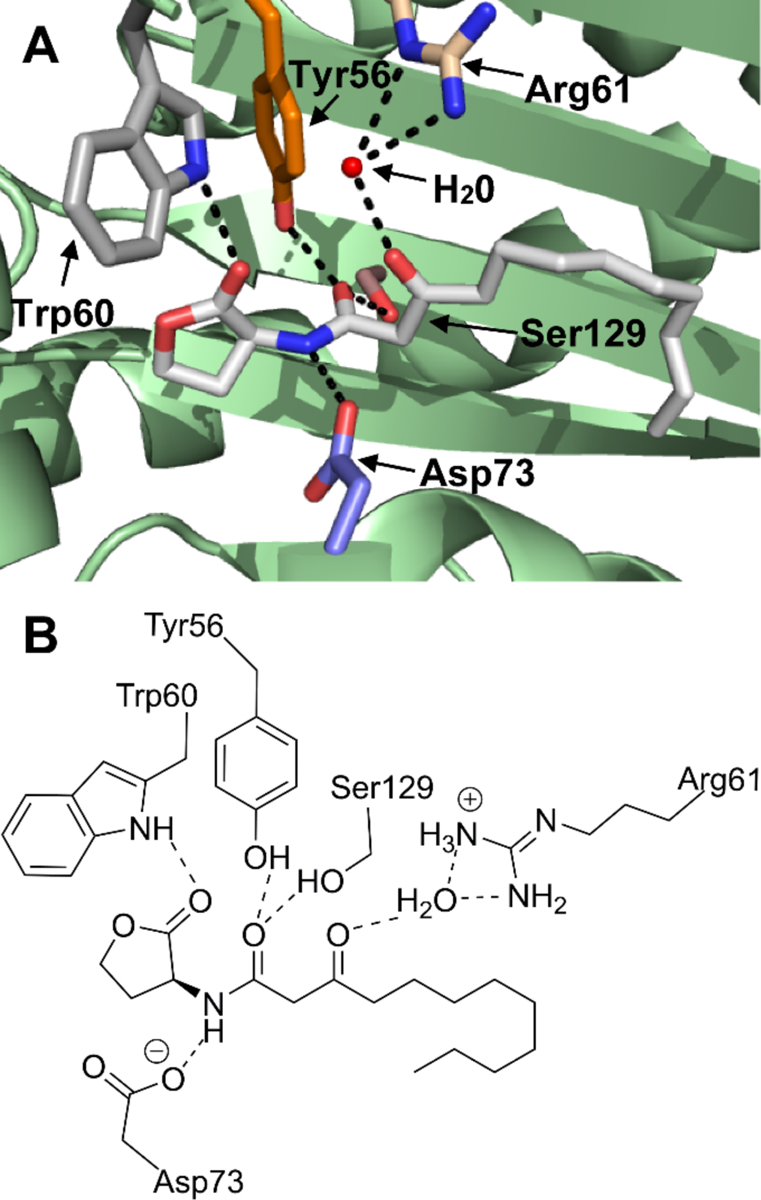
Three-dimensional (A) and two-dimensional (B) images of the OdDHL-binding site in the [LasR LBD:OdDHL]2 co-crystal structure (PDB ID: 2UV0).36 Dashed lines indicate putative hydrogen bonds between the labeled residues or water (shown as a red ball in part A) and OdDHL. OdDHL in part A is shown with carbon in grey, oxygen in red, and nitrogen in blue.
Figure 3:
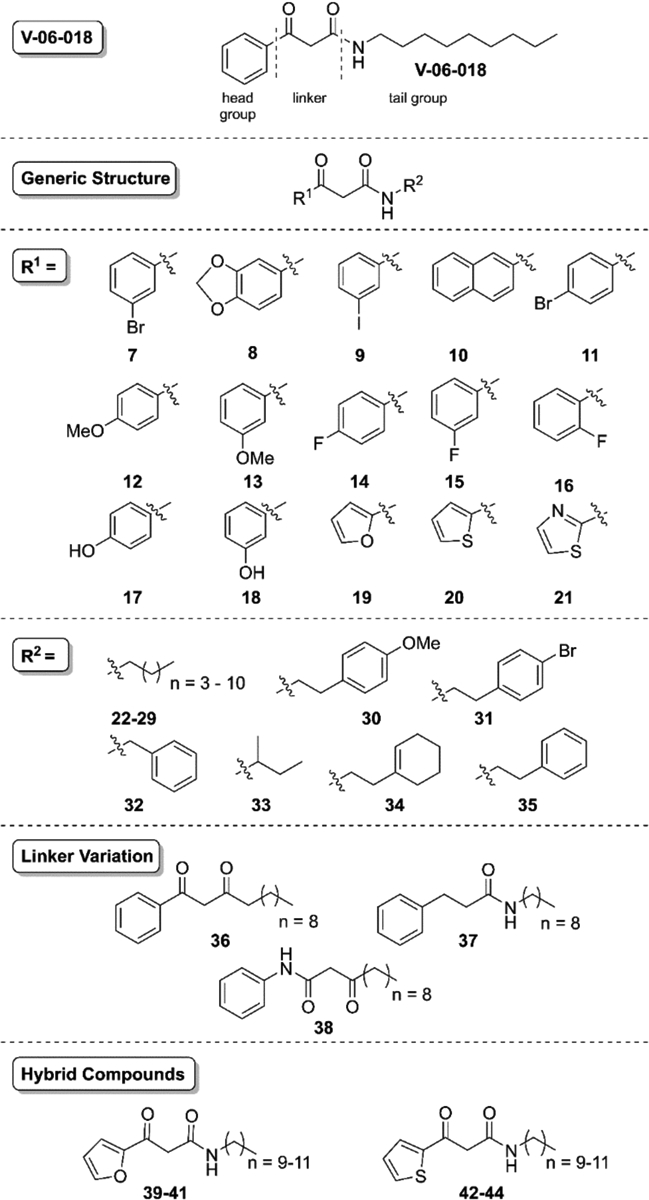
Library of V-06–018 analogs. Systematic changes were made to the head, tail, and linker regions of V-06–018 (see text). Compound 26 in this series, comprised of a phenyl head and nine carbon tail, is V-06–018.
To examine LasR’s tolerance for increased steric bulk on V-06–018’s headgroup, we synthesized napthyl derivative 10 (Figure 3). We also synthesized a variety of analogs with halogenated aryl headgroups (7, 9, 11, and 14-16) to explore electronic effects on activity. Within this set, compounds 9, 12, and 13 were also inspired by work reported by Spring and coworkers, who found that related molecules with these head groups were efficacious inhibitors of the production of QS-regulated virulence factors in P. aeruginosa.14 To alter the electronics and hydrogen-bonding ability of the V-06–018 headgroup without significantly increasing its size, we constructed a set of analogs with heterocyclic, aromatic headgroups (19-21).
Turning to the tail group of V-06–018, we again looked to OdDHL for guidance. The importance of hydrophobic contacts between ligands and the OdDHL acyl tail binding pocket in LasR has been noted (i.e., at residues Ala 127 and Leu 130),37–38 and AHL-based LasR agonists decrease in potency as their tails decrease from 12 carbons in length.39 To examine the importance of tail length for V-06–018’s antagonistic activity, we introduced five to twelve carbon tails via the amine coupling in Scheme 1, yielding compounds 22-29 (Figure 3; compound 26 is V-06–018). To mimic the molecular architecture of known AHL30 and TP-type26 antagonists of LasR, we included several derivatives with cyclic tail groups (30-32, 34 and 35). In addition, we examined an analog with a sec-butyl tail (33, racemic) to evaluate LasR’s tolerance for bulk at the position vicinal to the V-06–018 amide nitrogen. Lastly, to evaluate the importance of the heteroatoms in the “linker” region between the headgroup and tail, we synthesized diketone 36 and amide 37. Compound 38, a constitutional isomer of V-06–018, was reported previously by our lab;40 we included it here for comparison and to further expand our SAR analyses.
Evaluation of the V-06–018 library for LasR antagonism
We examined the activity of the V-06–018 library for LasR antagonism using a P. aeruginosa mutant strain (PAO-JP2, ΔlasIrhlI) that lacks the ability to synthesize OdDHL (or BHL) and contains a green fluorescent protein (GFP) reporter plasmid to examine LasR activity.16, 41 We used a P. aeruginosa LasR reporter as opposed to the E. coli LasR reporter introduced above, as we were most interested in the activity of the compounds (and their eventual use as probes) in the native organism. Further, as we previously showed that V-06–018 is not subject to active efflux by the MexAB-OprM pump in P. aeruginosa,16 we wanted to examine if these close analogs were also active in the presence of this pump. In this P. aeruginosa reporter system, compounds capable of LasR antagonism should reduce GFP production, and this loss can be quantitated by fluorescence (see Materials and Methods). To start, we screened the library for LasR antagonism at a concentration of 10 μM in competition against 150 nM OdDHL. Analogs with substituents on the head group were found to be generally less efficacious as LasR antagonists relative to V-06–018 (compounds 7–18, Figure 4A), suggestive that bulkier V-06–018 analogs may not be as well accommodated in the AHL binding site, regardless of their hydrogen bonding ability. Decreasing the size of the headgroup and including a polar atom was more fruitful. Two of the analogs based on five-membered heterocycles, furan 19 and thiophene 20, had equivalent efficacy to V-06–018 (~90% LasR antagonism). Not all hetero-cycles were effective as headgroups, however; thiazole 21 lost efficacy relative to V-06–018.
Figure 4:
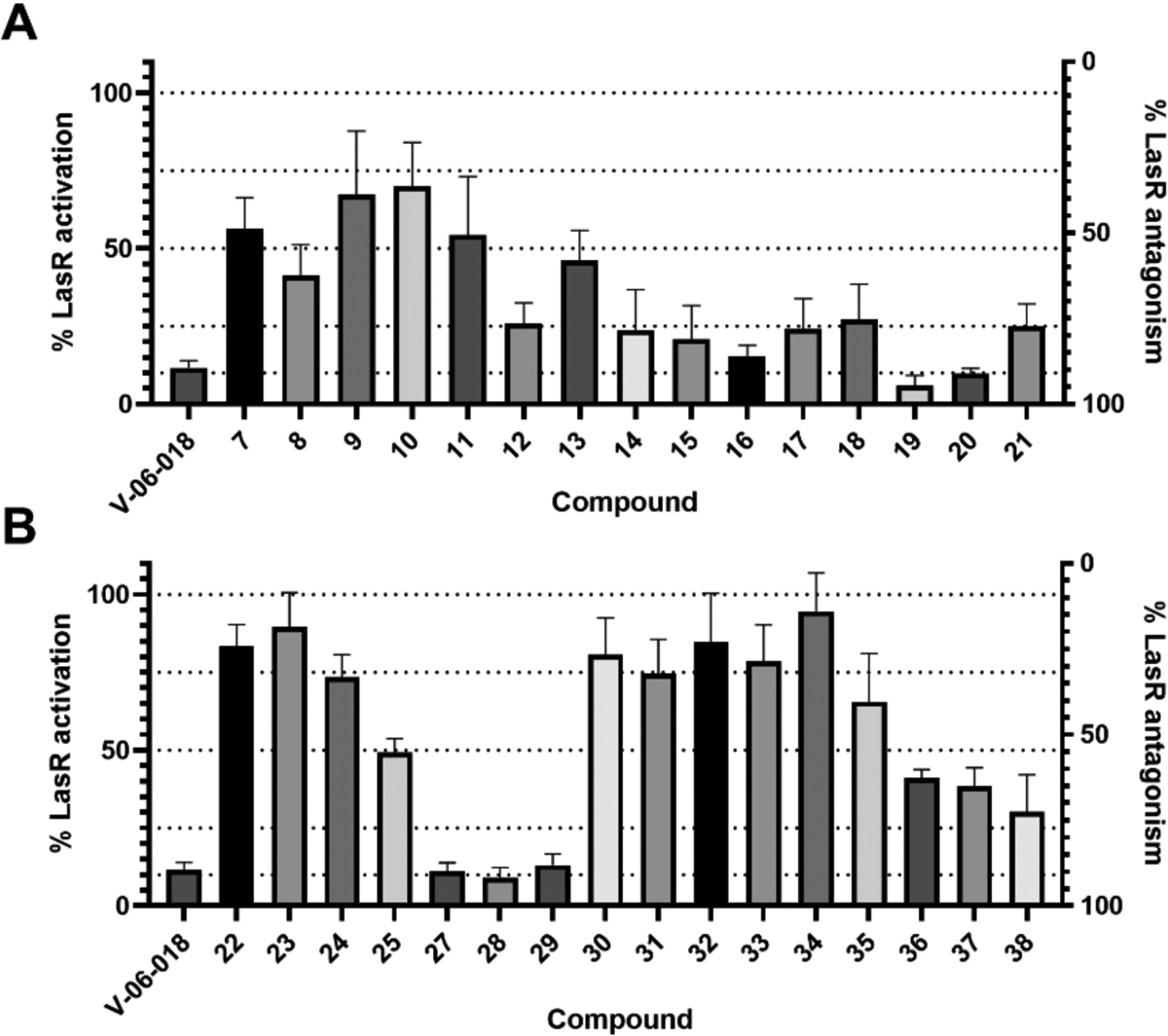
Primary LasR antagonism screening data in P. aeruginosa reporter PAO-JP2 for the (A) head group and (B) tail group and linker modified V-06–018 analogs. Compounds were screened at 10 μM in the presence of 150 nM OdDHL. Bacteria treated with 150 nM OdDHL only was defined as 100% LasR activity/0% LasR antagonism; conversely, bacteria treated with DMSO only (i.e., vehicle) was defined as 0% LasR activation/100% LasR antagonism. Error bars indicate SD of n ≥ 3 trials.
Turning to the tail group modified V-06–018 analogs, we found that only compounds with unbranched, acyclic alkyl tails were efficacious LasR antagonists (e.g., 27–29, Figure 4B). No compounds with cyclic moieties or branching (i.e., 30-35) in their tails were capable of antagonizing LasR by more than 50%. The length of the tail was also important; analogs 27-29, with 10- to 12-carbon tails, were equally as efficacious as V-06–018. The shorter tail analogs 22-25, however, antagonized LasR by less than 50%. These data suggest that binding interactions between LasR and these truncated V-06–018 analogs may have been reduced due to the lack of hydrophobic contacts (again, shown to be important for LasR:OdDHL binding).37–38 Modifications to the linker region also resulted in less active analogs. All three linker-modified compounds (36-38) lost efficacy relative to V-06–018, implicating the presence and position of the amide in V-06–018 as critical to LasR antagonism. Overall, these primary screening data indicated that only subtle alterations to the head and tail groups of V-06–018, and not the linker group, were tolerated for strong LasR antagonism.
Dose-response antagonism analysis of primary screening hits
To obtain a quantitative measure of compound potency, we performed dose-response analyses on the compounds that antagonized LasR ≥ 90% at 10 μM (19, 20, and 27-29) using the same P. aeruginosa LasR reporter strain and calculated their IC50 values (Table 1). We were excited to observe that each of these analogs was more potent than V-06–018. Increasing the length of the V-06–018 tail from 10 to 12 carbons (i.e., as in 27-29) led to a ~3–4-fold increase in potency. The heterocyclic analogs were also stronger LasR antagonists than V-06–018; furan 19 was approximately two-fold more potent than V-06–018, and thiophene 20 was closer to five-fold.
Table 1.
Potency and maximum LasR inhibition (efficacy) data for selected compounds in P. aeruginosa
| compound | IC50 (μM)a |
95% CI (μM)b |
Maximum Inhibition (%)c |
|---|---|---|---|
| V-06–018 (26) | 2.3 | (1.7 – 3.1) | 89 |
| 19 | 1.2 | (0.8 – 1.8) | 96 |
| 20 | 0.5 | (0.3 – 0.6) | 84 |
| 27 | 0.7 | (0.5 – 0.9) | 93 |
| 28 | 0.5 | (0.4 – 0.7) | 92 |
| 29 | 0.7 | (0.5 – 1.0) | 91 |
| 39 | 0.2 | (0.2 – 0.3) | 83 |
| 40 | 0.2 | (0.2 – 0.3) | 85 |
| 41 | 3.8 | (2.0 – 7.1) | 89 |
| 42 | 0.2d | (0.1 – 0.2) | 91 |
| 43 | 0.2d | (0.1 – 0.2) | 93 |
| 44 | 0.6 | (0.5 – 0.8) | 84 |
For details of PAO-JP2 reporter strain, see Materials and Methods. aAntagonism experiments performed by competing the compounds against OdDHL (1) at its approximate EC50 (150 nM for PAO-JP2) and inhibitory activity was measured relative to receptor activation at this EC50. IC50 values determined by testing compounds over a range of concentrations (0.64 nM – 50 μM). All assays performed in triplicate.
CI = confidence interval. 95% CIs calculated from the SEM of n ≥ 3 trials.
Denotes the best-fit value for the bottom of the computed dose-response curve.
Compound exhibited non-monotonic dose-response behavior. Reported IC50 corresponds to the antagonism portion of the curve. Full antagonism dose response curves are shown in Figure S2.
Second-generation V-06–108 analogs and LasR agonism profiles
Encouraged by the antagonistic activity profiles of our initial set of compounds, we designed and synthesized a set of “hybrid” second-generation V-06–018 analogs that combined features of the most active compounds. These compounds were comprised of a furan or thiophene head group united with 10, 11, or 12 carbon tails (compounds 39-44; see Figure 2), and were synthesized and evaluated for LasR antagonism in P. aeruginosa as described above. The second-generation compounds displayed a variety of activities in the LasR antagonism assay (listed in Table 1). Notably, furan derivatives 39 and 40, containing 10 or 11 carbon tails, respectively, were more potent than their parent compounds and were each 10-fold more potent than V-06–018. The 12-carbon furan analog 41, however, lost activity relative to its parent compounds.
We note that the thiophene analogs of 39 and 40, compounds 42 and 43, displayed non-monotonic partial agonism behavior in the LasR dose-response assays;29, 30 namely, at concentrations below 2 μM these compounds antagonized LasR, while at concentrations above 2 μM they agonized LasR. We have reported this activity profile for a series of ligands in reporter assays of LuxR-type proteins to date.27–28, 30 The antagonist portions of their dose-response curves indicated that 42 and 43 were each highly potent at lower concentrations, with IC50 values 10-fold lower than that of V-06–018. Interestingly, thiophene analog 44, differing by only one methylene unit than 43, lacked observable non-monotonic activity.
The discovery that two of the hybrid compounds were non-monotonic partial LasR agonists prompted us to measure dose-response agonism curves for all our most potent compounds (Figure S3). V-06–018 and compounds 27-29, comprised of phenyl headgroups, did not activate LasR. We also screened our first-generation library for LasR agonism at a single concentration (100 μM) and found that none of the analogs with phenyl headgroups activated LasR; however, thiophene 20 weakly agonized LasR (to 20%; Figure S4). We found that furans 39 and 40 could very weakly agonize LasR (7% and 4%, respectively) at the highest concentration screened (50 μM). Relative to 39 and 40, thiophenes 42 and 43 were stronger LasR agonists at 50 μM (30% and 22%, respectively), which matched their activity profile at this concentration in the dose-response antagonism analysis (as described above).
Activation in this cell-based reporter assay requires LasR to initiate transcription of gfp. This process requires LasR to adopt a conformation capable of homodimerization and productive DNA binding. Our results suggest that, at sufficiently high compound concentration, these furan and thiophene ligands can make contacts with LasR (either directly or indirectly via some other target) that promotes this process. However, contacts with just the head groups of 39, 40, 42, and 43 are presumably insufficient, as compounds 41 and 44, comprised of the same furan and thiophene head groups, respectively, yet linked to a twelve-carbon tail, failed to activate LasR even at high concentrations. These results suggest that contacts with the tail—specifically, a tail of 9–11 carbons—along with the head group are necessary for LasR agonism by this ligand class at high concentrations. Whether these ligands target the OdDHL binding site or another site on LasR, or another factor altogether, to promote LasR activation at these concentrations remains to be determined.
E. coli reporter assays indicate V-06–018 and analogs act directly via LasR
We next examined if our improved V-06–018 analogs elicit their antagonistic activity via acting directly on LasR using an E. coli LasR reporter system (see Materials and Methods).42–44 As high-lighted above, LasR is directly and indirectly regulated by other QS systems in P. aeruginosa, and thus activity profiles in the P. aeruginosa LasR reporter are a measure of this inter-regulated network. To address this question, we obtained dose-response curves for all of the compounds in Table 1 in an E. coli LasR reporter strain, and found that their relative efficacies and potencies largely tracked between the E. coli and P. aeruginosa reporters (Figure S5, Table S2). This alignment between the P. aeruginosa and E. coli reporter data suggests that these compounds elicit their effects via direct interactions with LasR. We note that all of our antagonists were less efficacious and potent against LasR in the E. coli reporter relative to P. aeruginosa. For example, the lead compound 40 was only four-fold more potent than V-06–018 in E. coli vs. being 10-fold more potent in P. aeruginosa. This reduction in potency also obscured the non-monotonic effects observed above for compounds 42 and 43. We postulate that this reduction in potency in E. coli is an artifact of differences in LasR expression levels between the two reporter systems (non-native level in E. coli vs. native level in P. aeruginosa).45 With more LasR present, higher concentrations of ligands are presumably required to inhibit LasR activity. Critically, the stronger efficacies and potencies of these V-06–018 derived antagonists in the native host background will increase their utility as probe molecules.
We were also curious to see if the new antagonists, like V-06–018, were selective for LasR over RhlR and QscR in P. aeruginosa. Screening representative compounds (39 and 40) in the E. coli RhlR and QscR reporter systems showed that 40 is highly LasR selective, with no observable activity in either RhlR or QscR (Figure S6). Compound 39 was found to be inactive in RhlR and, similar to V-06–018, only a weak QscR antagonist (~35% inhibition) at the very highest concentration tested. These results further underscore the receptor selectivity profile of the V-06–018 scaffold and the value of these compounds as chemical tools to study QS in P. aeruginosa.
P. aeruginosa reporter data support a competitive mechanism of LasR antagonism for V-06–018 and related compounds
We were interested to determine if V-06–018 and our new lead antagonists were acting as competitive LasR antagonists, and examined this question by testing them against OdDHL at varying concentrations in the P. aeruginosa LasR reporter assay. The observed potency of a competitive LasR antagonist should vary with OdDHL concentration, as both molecules are competing for space in the same ligand-binding site. We obtained antagonism dose response curves for V-06–018 and one of our lead compounds (40, which did not display non-monotonic behavior) in competition with OdDHL at 150 nM, 1 μM, and 10 μM (Figure 5). We observed an OdDHL-concentration-dependent decrease in the potency of both compounds. The relative potency trends for V-06–018 and 40 were also maintained, with compound 40 significantly more potent than V-06–018 at 150 nM and 1 μM. Unlike V-06–018, compound 40 was still capable of antagonizing LasR (to 55%) even in the presence of 10 μM OdDHL. These results are supportive of the ability of V-06–018 and its close analogs to act as competitive antagonists of LasR.
Figure 5:
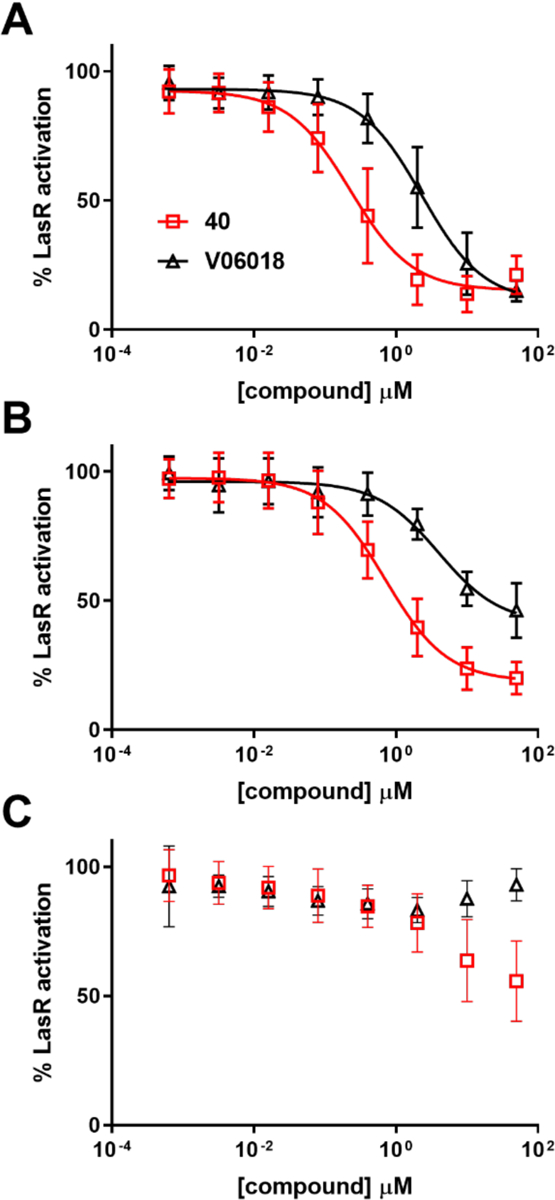
Dose-response LasR antagonism curves for V-06–018 and analog 40 in P. aeruginosa PAO-JP2. Dose-response curves of V-06–018 (black triangles) and 40 (red squares) in competition with (A) 150 nM, (B) 1 μM, and (C) 10 μM OdDHL. V-06–018 has IC50 values of 2.3 and 3.9 μM vs. 0.15 and 1 μM OdDHL, respectively; 40 has IC50 values of 0.2 and 0.7 μM vs. 0.15 and 1 μM OdDHL, respectively. IC50 values could not be calculated for these compounds in competition with 10 μM OdDHL (curves in part C).
Antagonists and non-classical partial agonist 42 solubilize LasR
We sought to further characterize the interactions between V-06–018 and related analogs with LasR, to understand how they engender receptor antagonism. Very little is known about the molecular mechanisms that lead to antagonism of LuxR-type receptors by small molecules, largely due to the instability of these proteins in vitro even in the presence of their native AHL ligand.46 LasR requires OdDHL throughout the production and purification process to be isolated, and has proven intractable to structural studies in full length form.36, 47–48 In principle, antagonists of LuxR-type proteins can operate by binding either in place of an AHL signal, or to a hypothetical, allosteric binding site. Once bound, antagonists can then cause antagonism by further destabilizing the protein (as has been shown for QscR and LasR)38, 47, 49 or by forming soluble complexes that are either incapable of dimerization or binding to DNA (as has been shown for CviR and LasR),50,51–52 or presumably combinations of these mechanisms (and potentially others). We were curious to investigate whether soluble LasR could be isolated when it was produced in the presence of V-06–018 or our new antagonists, or if it was destabilized in their presence relative to OdDHL. To test these questions, we produced LasR in E. coli grown in the presence of no compound (DMSO control) or 50 μM OdDHL, V-06–018, 40, or 42 (see Materials and Methods). After 16 h of protein production, we lysed the E. coli cells and separated the whole cell (WC) and soluble (S) lysate on an SDS-PAGE gel (Figure 6; quantitative analysis of the bands in the gel is provided in Table S3).
Figure 6.
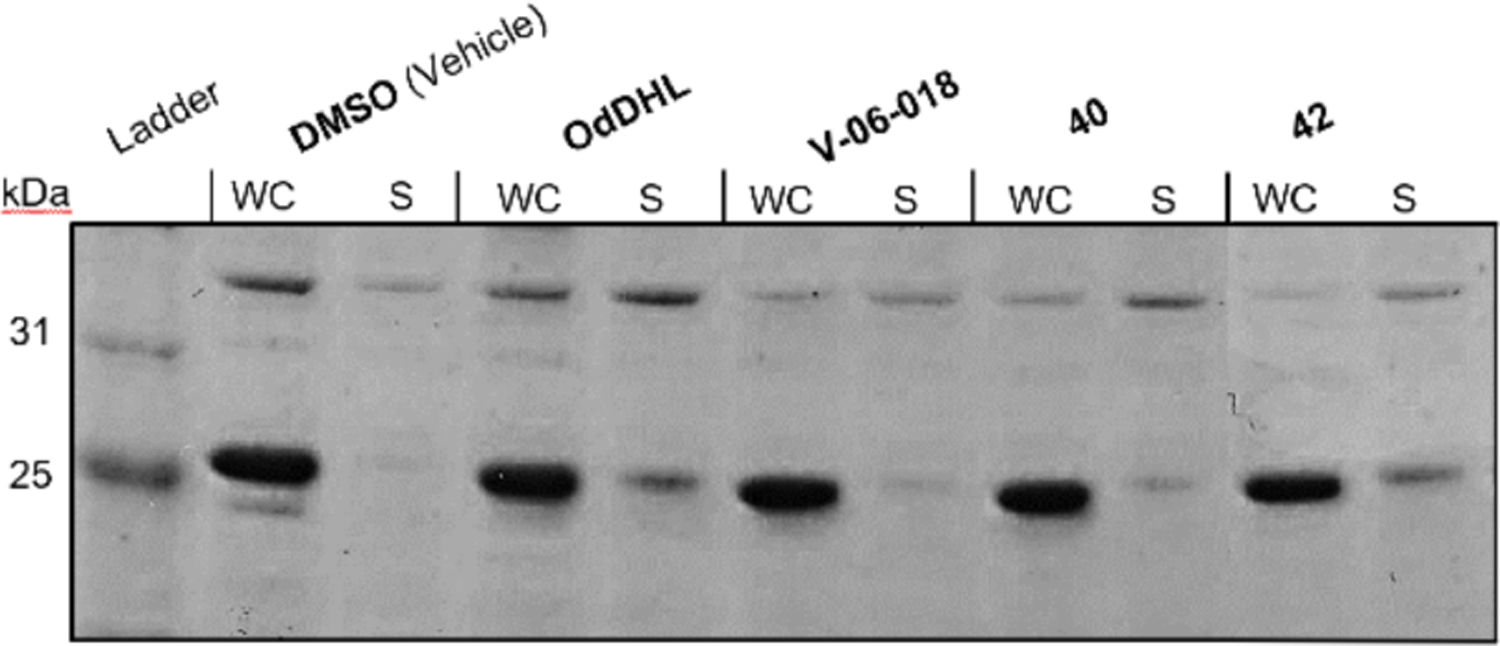
Characterization of LasR via SDS-PAGE gel in the presence of different ligands. Whole Cell (WC) and soluble (S) portions of E. coli cell lysates with LasR overexpressed in the presence of DMSO or 50 μM of OdDHL, V-06–018, 40, or 42. LasR has a mol. wt. of 27.9 kD.
As expected, we did not obtain any LasR in the soluble fraction of cells grown without exogenous compound (DMSO), while we obtained soluble LasR in the culture grown with exogenous OdDHL (S band ~30% as intense as WC band; Figure 6). These data recapitulate the finding that LasR requires a ligand to be soluble in vitro.53 We detected soluble bands for LasR produced in the presence of V-06–018 and furan 40. The bands were four-fold smaller than that of OdDHL (~7% as intense as WC band, vs. ~30% for OdDHL, Table S3), suggesting that these ligands do not solubilize LasR to the same extent as OdDHL. This result correlates with the previous report of Schneider and co-workers demonstrating that certain synthetic AHL-type antagonists (along with the close V-06–018 analog 38) form soluble complexes with LasR, albeit in less amounts than OdDHL.51 Schneider went on to show that these complexes were unable to bind to LasR’s target DNA using electrophoretic mobility shift assays (EMSAs), which allows for the interpretation that these ligands can stabilize an inactive LasR complex (e.g., incapable of dimerization or DNA binding). We also observed thiophene 42 solubilize LasR. The soluble band for 42 was more intense than those observed for V-06–018 and 40, and comparable to OdDHL (~30%). We note that 42 has a non-monotonic activity profile in the P. aeruginosa reporter assay and is capable of weak LasR agonism at higher concentrations; the larger quantity of LasR isolated in this experiment relative to V-06–018 and 40 (at 50 μM concentration) is therefore interesting and could arise due to this agonistic activity profile. Collectively, these SDS-PAGE data support the hypothesis that V-06–018 and related analogs act at LasR antagonists, at least in part, via inducing a soluble but inactive conformation of LasR. The reduced amount of protein in these soluble fractions relative to OdDHL suggests that V-06–018 and 40 may also cause antagonism by promoting LasR unfolding (i.e., destabilizing the receptor); thus, more than one mechanism of antagonism is likely operative. Further biochemical (e.g., EMSAs) and structural experiments are required to test these mechanistic hypotheses and are ongoing in our laboratory.
LasR mutants reveal residues critical for activation and inhibition by synthetic ligands
The results of the competitive LasR antagonism dose response assays, E. coli reporter assays, and protein production experiments outlined above suggest that V-06–018 and the lead analogs target LasR and interact with the OdDHL binding site to cause antagonism. In view of our original compound design, we were curious as to whether the residues in LasR that are known to govern LasR:OdDHL interactions (Figure 2) were also important to LasR antagonism by the V-06–018 ligand class, and applied a method utilized previously in our laboratory involving LasR mutants with modifications to the OdDHL binding site.35–36, 54 In this past work, a set of LasR single-point mutants were generated in which residues implicated in hydrogen bonding interactions with OdDHL were converted to residues incapable of hydrogen bonding but approximately the same steric size (e.g., Tyr → Phe). The mutant LasR proteins were then tested for activity using a LasR reporter plasmid in an E. coli host background (analogous to the E. coli LasR reporter assay system above). Compounds showing reduced activity in these mutants relative to wild-type LasR then can be postulated to make a contact with LasR that depends on the mutated residue. We tested V-06–018 and furan 39 at 100 μM in three LasR mutants with modifications to residues that make hydrogen-bonds to OdDHL (Tyr 56, Trp 60, and Ser 129; see Figure 3).54 Notably, all of these single-point LasR mutants (Y56F, W60F, and S129A) are still functional in the reporter assay, but are less active than wild-type LasR (as measured via reduced OdDHL potencies; Figure S7), reflective of the importance of these LasR:OdDHL interactions for activation. (As noted above, antagonists display reduced efficacy in general in this heterologous background relative to the native (P. aeruginosa) reporter system.)
V-06–018 was found to antagonize all three LasR mutants to a significantly lesser extent than wild-type LasR (Figure 7A). The same trend was true for furan 39. Tyr 56 and Ser 129 are believed to form hydrogen bonds with the amide carbonyl of OdDHL (Figure 2), and potentially could bind to one of the two linker carbonyl oxygens in V-06–018 and its analogs.36 Trp 60 hydrogen bonds with the lactone carbonyl oxygen of OdDHL, and it may be capable of hydrogen bonding with the furan oxygen of 39. An analogous hydrogen-bond to the head-group of V-06–018 is not possible, but the lower activity of V-06–018 in the W60F LasR mutant suggests that Trp 60 interacts in some other manner with V-06–018 to enforce antagonism. Further studies are necessary to pinpoint the specific molecular interactions that govern LasR antagonism by these two ligands. Nevertheless, these experiments with LasR mutants support V-06–018 and new antagonist 39 interacting with the OdDHL binding site in LasR.
Figure 7:
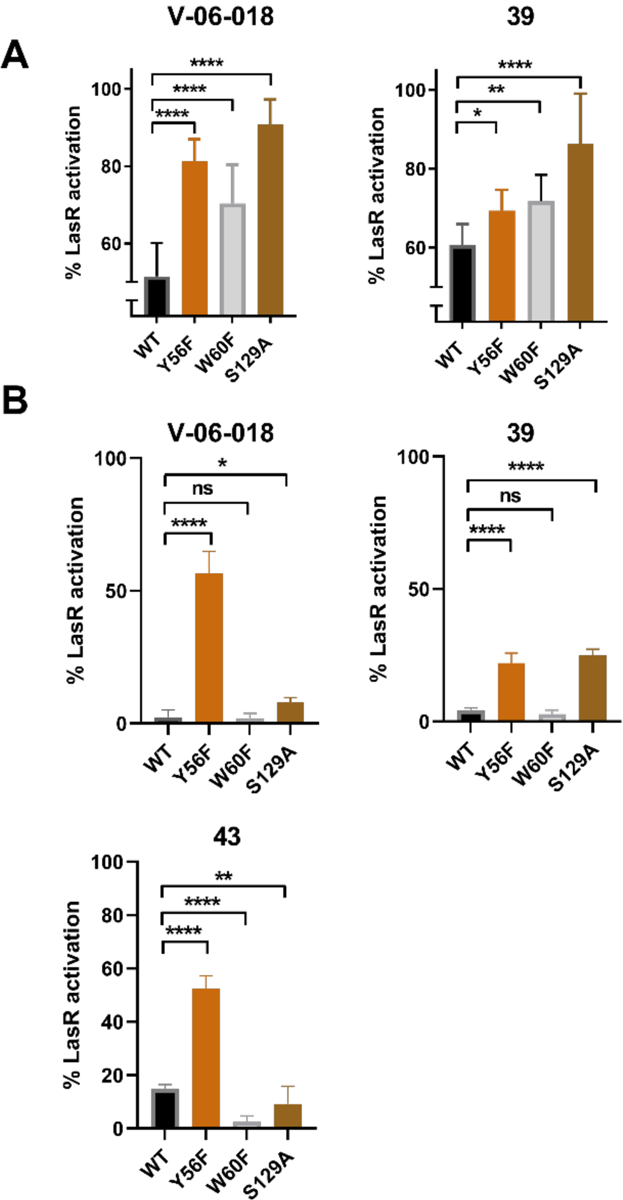
(A) LasR mutant antagonism data for V-06–018 and lead compound 39. Compounds tested at 100 μM against OdDHL at its approximate EC50 value in the specific E. coli LasR reporter strain (as indicated on the X-axis). (B) LasR mutant agonism data for V-06–018, 39, and 43. Compounds tested at 100 μM. For antagonism experiments, 100% is defined as the EC50 concentration of OdDHL in that specific LasR reporter strain (see Figure S6); for agonism experiments, 100% is defined as the activity of 100 μM OdDHL in that specific LasR reporter strain. Significance was assessed via a one-way ANOVA: **** = p < 0.0001; *** = p < 0.001; ** = p < 0.01; * = < 0.05. ns = no significant difference.
We also were curious to learn whether alternations to these LasR residues could impact the ability of our compounds to agonize LasR. Therefore, we examined the agonistic activities V-06–018, furan 39, and thiophene 43 in the three LasR mutant reporter strains at 100 μM; thiophene 43 was included in these agonism assays due to its non-monotonic agonism profile (see above). We were surprised to find that all three compounds agonized the LasR Y56F mutant to a significantly greater extent than wild-type LasR. For example, V-06–018, which does not agonize wild-type LasR, activated LasR Y56F to ~60% (relative to OdDHL) at 100 μM. In view of this unexpected result, we screened the remainder of our lead compounds in this LasR mutant reporter and found that they all were capable of activating the LasR Y56F mutant to some extent (from 9–56% at 100 μM; Figure S8). V-06–018 and 39 also agonized the LasR S129A mutant significantly more than wild-type LasR. These results suggest that removing the hydrogen bonds donated by Tyr 56 or Ser 129, or reducing sterics at these positions, may allow these V-06–018 type ligands more freedom to adjust their position in the LasR OdDHL binding pocket and adopt new contacts that engender LasR agonism as opposed to antagonism. None of our compounds were found to agonize the LasR W60F mutant; in fact, 43 lost agonistic activity in that mutant relative to wild-type LasR.
In our laboratory’s prior mutational studies of LasR, we observed compound 38 (Figure 3), a LasR antagonist and constitutional isomer of V-06–018, could agonize both the LasR Y56F and W60F mutants. We termed this transition from antagonist to agonist “Janus” behavior (after the two-faced Roman god).35 Here, we observed V-06–018 and compound 39 exhibit analogous “Janus” behavior in Y56F and S129A, but not in W60F (like 38). These results suggest that chemical modification of either the ligand (via chemical synthesis; i.e., V-06–018 → 38 or 39) or LasR (via mutagenesis of at least these three residues) is sufficient to alter contacts between the ligand and receptor to allow for either agonism or antagonism, or the degree thereof, and that these changes to molecular contacts are likely very subtle. The implications of these findings—specifically, that single point mutations can convert potent LasR antagonists into agonists—on the propensity for resistance to arise in P. aeruginosa to LasR antagonists did not escape our attention. We do note that the agonistic activity of these compounds is quite low (relative to OdDHL in wild type LasR). Additional experiments are required to explore the possibility of LasR mutants to arise naturally upon sustained treatment with V-06–018 or related analogs. However, our lab and others has shown previously that resistance to QS inhibitors, even if it was to develop, should be slow to spread through and not overtake a population of bacteria,55–56 supporting the continued search for such compounds. Moreover, the ability of V-06–018, 38, 39, and 43 to agonize the LasR mutants suggests that structural studies of these LasR mutant:ligand complexes could be particularly noteworthy, as they could illuminate the mechanisms by which these ligands both agonize LasR mutants and antagonize wild-type LasR. The heightened stability of LasR:agonist complexes relative to LasR:antagonist complexes could significantly enable such structural studies.
SUMMARY AND CONCLUSIONS
The work reported herein was motivated by the need for chemical probes of a key QS receptor, LasR, in the opportunistic pathogen P. aeruginosa. Despite considerable research to date, antagonists with sub-micromolar potencies, high efficacies, and selectivities for LasR over the other QS circuits in P. aeruginosa have been elusive. We performed the first structure-function analysis of the small molecule V-06–018, a promising yet unstudied LasR antagonist emerging from a high-throughput screen reported over 10 years ago.33 We developed a versatile and efficient synthetic route to V-06–018, produced a focused library of analogs using this route to explore the headgroup, linker, and tail portions of V-06–018, and evaluated the library for LasR modulatory ability using cell-based reporter systems. These screening data revealed stringent SARs for LasR antagonism by this ligand scaffold, including the requirement for a linear, alkyl tail group between nine to 12 carbons in length, an amide in the linker, an intolerance for substitution on the aryl head group, and a tolerance of certain 5-membered heterocyclic head groups. These SARs allowed us to design and synthesize second-generation LasR antagonists with nanomolar IC50 values in P. aeruginosa (e.g., 39 and 40). These compounds represent, to our knowledge, the most potent and efficacious synthetic antagonists of LasR to be reported, with IC50 values in P. aeruginosa 10-fold lower than V-06–018 and at least 100-fold lower than other AHL-based ligands.48 We note that we discovered these analogs after synthesizing fewer than 40 compounds; further development of the V-06–018 scaffold would likely yield even more potent compounds.
Our results indicate that the V-06–018 scaffold is quite selective for LasR over the other two LuxR-type receptors in P. aeruginosa, with 39, 40, and V-06–018 showing neither antagonistic nor agonistic activity in RhlR, 40 being inactive in QscR, and 39 and V-06–018 showing only modest antagonistic activity in QscR at the very highest concentrations tested. This activity profile is significant because the ability to selectively attenuate LasR activity in the midst of the highly inter-regulated QS system of this pathogen will facilitate mechanistic studies, and highlights the value of these V-06–018 analogs as chemical tools to study QS in P. aeruginosa.
We also report herein our investigations into the mechanism by which V-06–018 and related compounds modulate LasR activity. In the course of these studies, certain analogs were found to display interesting dual activity profiles—capable of strong LasR antagonism at nanomolar levels, yet LasR agonism at micromolar levels (i.e., non-monotonic partial agonists)—and we were intrigued by their mechanisms of action as well. Examination of the lead compounds against OdDHL at various concentrations and in an E. coli LasR reporter support a mechanism by which they bind competitively with OdDHL and interact directly with LasR. V-06–018 and furan antagonist 39 were found to be significantly less efficacious in LasR mutants that lack key residues in the ligand-binding site shown to make hydrogen-bonding contacts with OdDHL. This result is congruent with these compounds binding in the same site on LasR as or near to OdDHL. Protein production studies of LasR in the presence of V-06–018, furan-based antagonist 40, and thiophene-based antagonist 42 demonstrated that these compounds support folding of the protein into a soluble form, suggestive that they may stabilize an inactive form of the protein, analogous to the mechanism of CviR antagonism by the chlorolactone AHL analog (CL).50 V-06–018 and 40 also appear to reduce the amount of soluble LasR relative to 42 or the native agonist OdDHL, indicating that receptor destabilization could also contribute to the mechanism of inactivation by certain of these compounds. Finally, study of V-06–018 and furan-based antagonist 39 revealed that they were each capable of shifting from LasR antagonists to agonists in a LasR mutant lacking a single hydrogen-bonding motif in the ligand-binding site (e.g., Tyr 56→ Phe 56; removal of the Tyr hydroxyl). This finding indicates that subtle interactions of these ligands with LasR can have dramatic effects on receptor activity and suggests a novel route for exploring the mechanisms of this ligand class via structural studies of LasR mutant:ligand complexes. Overall, this study has provided a set of highly potent LasR antagonists that should find broad use as chemical probes of QS in P. aeruginosa, a robust chemical route to generate these compounds, and new insights into the mechanisms of LasR antagonism. These compounds and insights expand the understanding of LuxR-type QS in this important opportunistic pathogen.
MATERIALS AND METHODS
Chemistry
All chemicals were obtained from Sigma-Aldrich, Agros Organics, or TCI America. All reagents and solvents were used without further purification except for hexane, ethyl acetate, and dichloro-methane, which were distilled prior to use. Analytical thin-layer chromatography (TLC) was performed on 250 μm glass backed silica plates with F-254 fluorescent indicator from Silicycle. Visualization was performed using UV light and iodine. All new compounds were fully characterized for purity and identity; see SI for characterization data. Compound stock solutions were prepared in DMSO at appropriate concentrations and stored at −4 °C prior to use.
Representative procedures for the synthesis of V-06–018
Synthesis of ethyl 2-(2-phenyl-1,3-dioxolan-2-yl)acetate (3; R = H):
Ethyl benzoyl acetate (1.92 mL, 10 mmol, 1 equiv.), ethylene glycol (3.35 mL, 60 mmol, 6 equiv.), and p-toluene sulfonic acid (192 mg, 1 mmol, 0.1 equiv.) was added to a 250 mL round-bottom flask equipped with a Dean-Stark trap. The mixture was heated to reflux for approximately 24 h. The mixture was washed with saturated sodium bicarbonate (1 × 100 mL), water (1 ×100 mL), and saturated brine (1 × 100 mL). The organic portion was dried over magnesium sulfate and concentrated under reduced pressure. The crude material was purified by flash silica gel chromatography (20% ethyl acetate in hexane), and 3 was isolated as a colorless oil (1.87 g, 79% isolated yield).
Synthesis of 2-(2-phenyl-1,3-dioxolan-2-yl)acetic acid (4; R = H):
Compound 3 (287 mg, 1.2 mmol, 1 equiv.) was dissolved in THF (12 mL, 0.1 M) in a 100 mL round-bottom flask, after which aqueous 1M lithium hydroxide (12 mL, 12 mmol, 10 equiv.) was added. The reaction mixture was heated to 70 °C, and reaction progress was monitored by TLC. Upon consumption of the starting material, the organic layer was washed with saturated sodium bicarbonate (20 mL). The combined aqueous layers were extracted with ethyl acetate (20 mL). The pH of the combined aqueous layers was acidified with 10% aq. citric acid, and then extracted with ethyl acetate (3 × 20 mL). These organic portions were combined, dried over magnesium sulfate, and concentrated under reduced pressure to yield 4 as a colorless, crystalline solid that was >95% pure by 1H NMR and used without further purification (226 mg, 90% crude yield).
Synthesis of N-nonyl-2-(2-phenyl-1,3-dioxolan-2-yl)acetamide (5; R = H, R’= nonyl):
Acid 4 (226 mg, 1.08 mmol, 1 equiv.), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDCHCl; 207 mg, 1.62 mmol, 1.5 equiv.), 4-dimethylaminopyridine (DMAP; 20 mg, 0.162 mmol, 0.15 equiv.), and nonylamine (238 μL, 1.3 mmol, 1.2 equiv.) were dissolved in CH2Cl2 (10.8 mL, 0.1M), and the reaction mixture was stirred for ~15 h at room temperature. The reaction mixture was diluted into diethyl ether and washed with 1M HCl (2 × 30 mL), saturated sodium bicarbonate (2 × 30 mL), water (1 × 30 mL), and brine (1 × 30 mL). The organic portion was dried over magnesium sulfate and concentrated under reduced pressure to yield 5 as a colorless, crystalline solid that was >95% pure by 1H NMR and used without further purification (303 mg, 85% crude yield).
N-nonyl-3-oxo-3-phenylpropanamide (V-06–018, 26):
Compound 5 (303 mg, 0.92 mmol, 1 equiv.) and p-toluene sulfonic acid (175 mg, 0.92 mmol, 1 equiv.) were dissolved in acetone (9.2 mL, 0.1 M) in a 25 mL round-bottom flask. The reaction mixture was stirred at room temperature for 24 h. The mixture was diluted in diethyl ether (20 mL) and washed with saturated sodium bicarbonate (1 × 30 mL), water (1 × 30 mL), and brine (1 × 30 mL), then dried over magnesium sulfate and concentrated under reduced pressure. The resulting solid was purified by flash silica gel chromatography (20% ethyl acetate in hexanes) to give a V-06–018 (26) as a white solid (170 mg, 64% isolated yield).
Biology
A listing of all of the bacterial strains and plasmids used in this study is provided in Table S1. Bacteria were cultured in Luria-Bertani medium (LB) and grown at 37 °C. Growth was quantified by absorbance at 600 nm (OD600). Absorbance and fluorescence measurements were made on a Biotek Synergy 2 plate reader running Gen 5 software (version 1.05). Buffers used in biological experiments included: Z buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM H2O), phosphate buffer (60 mM Na2HPO4, 40 mM NaH2PO4), and phosphate buffered saline (137 mM NaCl, 2.68 mmol KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4). Dose-response curves were generated using GraphPad Prism software (version 8). Detailed descriptions of all biological experiments are provided in the SI.
P. aeruginosa reporter assay protocol
LasR reporter experiments in P. aeruginosa were performed as reported previously.30 Briefly, a single colony of P. aeruginosa PAO-JP241 was grown overnight in LB medium containing 300 μg/mL carbenicillin. Culture was diluted 1:100 in fresh LB medium without antibiotic. Subculture was grown to OD600 = 0.25–0.3. A 2-μL aliquot of compound stock solution (in DMSO) was added to the interior wells of black, clear-bottom 96-well plate. A 198-μL aliquot of bacterial culture was added to all compound containing wells. For antagonism experiments, at least three wells were filled with 198 μL of grown subculture (i.e., untreated subculture); the remainder of the subculture was treated with exogenous OdDHL (i.e., treated subculture) at various concentrations (150 nM, 1 μM, or 10 μM) prior to dispensing. Plates were incubated without shaking (static) for 6 h, after which GFP production was read for each well using a plate reader (excitation at 500 nM, emission at 540 nM) and normalized to cell growth. Activity was reported relative to cells containing only OdDHL.
E. coli reporter assay protocol
LasR, RhlR, and QscR assays in E. coli JLD271 (ΔsdiA) or DH5α utilized a β-galactosidase reporter and were conducted as previously reported.26 A representative protocol for the LasR assay is provided here. Briefly, a single colony of E. coli strain JLD271 bearing plasmids pJN105-L44 and pSC11-L42 was grown in LB medium. Overnight culture was diluted 1:10 in fresh LB medium with 100 μg/mL ampicillin and 10 μg/mL gentamicin and grown to an OD600 = 0.23–0.27. Once grown, arabinose was added to a final concentration of 4 mg/mL. A 2-μL aliquot of compound stock solution (in DMSO) or only DMSO (vehicle control) was added to the interior wells of a clear 96-well microtiter plate. For agonism assays, 198 μL aliquots of the subculture was dispensed into all internal wells. For antagonism assays, subculture was dispensed into at least three wells containing only DMSO; the remainder of the subculture was treated with the appropriate concentration of OdDHL and dispensed into all remaining interior wells. Plates were incubated at 37 °C with shaking at 200 rpm for 4 h.
To measure resulting β-galactosidase production, each interior well of a chemical-resistant 96-well plate (Costar 3879) was filled with 200 μL Z buffer, 8 μL CHCl3, and 4 μL 0.1% aqueous SDS. After the incubation period, the OD600 of each well of the bacteria-containing plate was measured. A 50-μL aliquot of each well of the bacteria-containing plate was transferred to the lysis-buffer containing chemical resistant plate, and the cells were lysed. A 100-μL aliquot from each well was transferred to a fresh clear-bottom 96-well plate. The Miller assay was started by adding 20 μL of the substrate ortho-nitrophenyl-β-galactoside (ONPG, 4 mg/mL in phosphate buffer) to each well. The plates were then incubated at 30 °C for 30 min and absorbances at 420 and 550 nm were read. Miller units were calculated for each well (see SI for detailed description). Activity was reported relative to wells containing only OdDHL.
LasR overexpression and SDS-PAGE protocols
E. coli BL21-DE3 harboring the pET17b (LasR) plasmid was grown overnight in LB medium from a single colony. The overnight culture was diluted 1:80 into fresh LB medium buffered with 100 mM MOPS, adjusted to pH 7, and grown to an OD600 = 0.5. Protein expression was induced by the addition of 0.4 M isopropyl β-d-1-thiogalactopyranoside (IPTG), and the culture was grown overnight at 17 °C. The next day, cells were pelleted by centrifugation. Whole cell and soluble portions of cell lysate were isolated and prepared via the Bacterial Protein Extraction Reagent (B-PER, ThermoFisher Scientific) according to package instructions. Cell lysates were run on a Biorad 10% SDS gel and stained with Coomassie. Band intensities were quantified using ImageJ (see Table S3).
Supplementary Material
ACKNOWLEDGEMENTS
Financial support for this work was provided by the NIH (R01 GM109403 and R35 GM131817). M.C.O. acknowledges support from the Arnold and Mabel Beckman Foundation through an Arnold O. Beckman Postdoctoral Fellowship. K.E.N. was supported in part by the UW-Madison NIH Chemistry-Biology Interface Training Program (T32 GM008505). NMR facilities in the UW-Madison Department of Chemistry were supported by the NSF (CHE-0342998) and a gift from Paul J. Bender. MS facilities in the UW-Madison Department of Chemistry were supported by the NSF (CHE-9974839).
ABBREVIATIONS
- QS
quorum sensing
- AHL
N-acyl-l-homoserine lactone
- SAR
structure-activity relationship
- OdDHL
N-(3-oxo-dodecanoyl)-l-homoserine lactone
Footnotes
The authors declare no competing financial interests.
REFERENCES
- 1.Brown ED; Wright GD, Antibacterial drug discovery in the resistance era. Nature 2016, 529 (7586), 336–43. [DOI] [PubMed] [Google Scholar]
- 2.Nathan C; Card O, Antibiotic Resistance - Problems, Progress, and Prospects. New Engl. J. of Med 2014, 371, 1761–1763. [DOI] [PubMed] [Google Scholar]
- 3.Allen RC; Popat R; Diggle SP; Brown SP, Targeting virulence: can we make evolution-proof drugs? Nat. Rev. Microbiol 2014, 12 (4), 300–8. [DOI] [PubMed] [Google Scholar]
- 4.Whiteley M; Diggle SP; Greenberg EP, Progress in and promise of bacterial quorum sensing research. Nature 2017, 551 (7680), 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rutherford ST; Bassler BL, Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb. Perspect. Med 2012, 2 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuqua C; Greenberg EP, Listening in on bacteria: acyl-homoserine lactone signalling. Nat. Rev. Mol. Cell. Biol 2002, 3 (9), 685–95. [DOI] [PubMed] [Google Scholar]
- 7.Galloway WR; Hodgkinson JT; Bowden SD; Welch M; Spring DR, Quorum Sensing in Gram-Negative Bacteria: Small-Molecule Modulation of AHL and AI-2 Quorum Sensing Pathways. Chem. Rev 2011, 111, 28–67. [DOI] [PubMed] [Google Scholar]
- 8.Sintim HO; Smith JA; Wang J; Nakayama S; Yan L, Paradigm shift in discovering next-generation anti-infective agents: targeting quorum sensing, c-di-GMP signaling and biofilm formation in bacteria with small molecules. Future Med. Chem 2010, 2 (6), 1005–1035. [DOI] [PubMed] [Google Scholar]
- 9.Welsh MA; Blackwell HE, Chemical Genetics Reveals Environment-Specific Roles for Quorum Sensing Circuits in Pseudomonas aeruginosa. Cell Chem. Biol 2016, 23 (3), 361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welsh MA; Eibergen NR; Moore JD; Blackwell HE, Small molecule disruption of quorum sensing cross-regulation in pseudomonas aeruginosa causes major and unexpected alterations to virulence phenotypes. J. Am. Chem. Soc 2015, 137 (4), 1510–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tal-Gan Y; Ivancic M; Cornilescu G; Yang T; Blackwell HE, Highly Stable, Amide-Bridged Autoinducing Peptide Analogues that Strongly Inhibit the AgrC Quorum Sensing Receptor in Staphylococcus aureus. Angew. Chem. Int. Ed. Engl 2016, 55 (31), 8913–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang B; Muir TW, Regulation of Virulence in Staphylococcus aureus: Molecular Mechanisms and Remaining Puzzles. Cell Chem. Biol 2016, 23 (2), 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Starkey M; Lepine F; Maura D; Bandyopadhaya A; Lesic B; He J; Kitao T; Righi V; Milot S; Tzika A; Rahme L, Identification of anti-virulence compounds that disrupt quorum-sensing regulated acute and persistent pathogenicity. PLoS Pathog. 2014, 10 (8), e1004321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodgkinson JT; Galloway WR; Wright M; Mati IK; Nicholson RL; Welch M; Spring DR, Design, synthesis and biological evaluation of non-natural modulators of quorum sensing in Pseudomonas aeruginosa. Org. Biomol. Chem 2012, 10 (30), 6032–44. [DOI] [PubMed] [Google Scholar]
- 15.Amara N; Mashiach R; Amar D; Krief P; Spieser SAH; Bottomley MJ; Aharoni A; Meijler MM, Covalent Inhibition of Bacterial Quorum Sensing. J. Am. Chem. Soc 2009. [DOI] [PubMed] [Google Scholar]
- 16.Moore JD; Gerdt JP; Eibergen NR; Blackwell HE, Active efflux influences the potency of quorum sensing inhibitors in Pseudomonas aeruginosa. Chembiochem 2014, 15 (3), 435–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner VE; Bushnell D; Passador L; Brooks AI; Iglewski BH, Microarray Analysis of Pseudomonas aeruginosa Quorum-Sensing Regulons: Effects of Growth Phase and Environment. J. Bacteriol 2003, 185 (7), 2080–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuster M; Greenberg EP, A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa. Int. J. Med. Microbiol 2006, 296 (2–3), 73–81. [DOI] [PubMed] [Google Scholar]
- 19.De Kievit TR; Gillis R; Marx S; Brown C; Iglewski BH, Quorum-sensing genes in Pseudomonas aeruginosa biofilms: their role and expression patterns. Appl. Environ. Microbiol 2001, 67 (4), 1865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The structure of TP1 was originally reported in 2006 (see ref. 25). In 2011, its structure was revised, from TP1-P (TP1- previous) to TP1-R (TP1- revised). Both isomers are bioactive and have similar activities in LasR. In the present study, we depict TP1-P and report a measurement of its EC50 previously made by our lab.; See: Zakhari JS; Kinoyama I; Struss AK; Pullanikat P; Lowery CA; Lardy M; Janda KD J. Am. Chem. Soc, 2011. 133 (11), 3840–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christensen QH; Grove TL; Booker SJ; Greenberg EP, A high-throughput screen for quorum-sensing inhibitors that target acyl-homoserine lactone synthases. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (34), 13815–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amara N; Krom BP; Kaufmann GF; Meijler MM, Macromolecular Inhibition of Quorum Sensing: Enzymes, Anbitodies, and Beyond. Chem. Rev 2011, 111, 195–208. [DOI] [PubMed] [Google Scholar]
- 23.Welsh MA; Blackwell HE, Chemical probes of quorum sensing: from compound development to biological discovery. FEMS Microbiol. Rev 2016, 40 (5), 774–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Loughlin CT; Miller LC; Siryaporn A; Drescher K; Semmelhack MF; Bassler BL, A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (44), 17981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muh U; Hare BJ; Duerkop BA; Schuster M; Hanzelka BL; Heim R; Olson ER; Greenberg EP, A structurally unrelated mimic of a Pseudomonas aeruginosa acyl-homoserine lactone quorum-sensing signal. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (45), 16948–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Reilly MC; Blackwell HE, Structure-Based Design and Biological Evaluation of Triphenyl Scaffold-Based Hybrid Compounds as Hydrolytically Stable Modulators of a LuxR-Type Quorum Sensing Receptor. ACS Infect. Dis 2016, 2 (1), 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geske GD; O’Neill JC; Miller DM; Wezeman RJ; Mattmann ME; Lin Q; Blackwell HE, Comparative analyses of N-acylated homoserine lactones reveal unique structural features that dictate their ability to activate or inhibit quorum sensing. Chembiochem 2008, 9 (3), 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geske GD; O’Neill JC; Miller DM; Mattman ME; Blackwell HE, Modulation of Bacterial Quorum Sensing with Synthetic Ligands: System Evaluation of N-Acylated Homoserine Lactones in Multiple Species and New Insights into Their Mechanisms of Action. J. Am. Chem. Soc 2007, 129, 13613–13625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.We note that Lipoxin A has been reported to inhibit LasR using an E. coli LasR reporter strain with a nanomolar IC50, but it only lowered LasR activity to 80% (relative to 100% activation by the native ligand). We are unaware of any other compound with a sub-micromolar IC50 in LasR and active in P. aeruginosa.; See: Wu B; Capilato JN; Pham MP; Walker J; Spur B; Rodriguez A; Perez LJ; Yin K FASEB J., 2016, 30 (6), 2400–2410. [DOI] [PubMed] [Google Scholar]
- 30.Moore JD; Rossi FM; Welsh MA; Nyffeler KE; Blackwell HE, A Comparative Analysis of Synthetic Quorum Sensing Modulators in Pseudomonas aeruginosa: New Insights into Mechanism, Active Efflux Susceptibility, Phenotypic Response, and Next-Generation Ligand Design. J. Am. Chem. Soc 2015, 137 (46), 14626–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yates EA; Philipp B; Buckley C; Atkinson S; Chhabra SR; Sockett RE; Goldner M; Dessaux Y; Camara M; Smith H; Williams P, N-Acylhomoserine Lactones Undergo Lactonolysis in a pH-, Temperature-, and Acyl Chain Length-Dependent Manner during Growth of Yersinia pseudotuberculosis and Pseudomonas aeruginosa. Infect. Immun 2002, 70 (10), 5635–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang F; Wang LH; Wang J; Dong YH; Hu JY; Zhang LH, Quorum quenching enzyme activity is widely conserved in the sera of mammalian species. FEBS Lett. 2005, 579 (17), 3713–7. [DOI] [PubMed] [Google Scholar]
- 33.Muh U; Schuster M; Heim R; Singh A; Olson ER; Greenberg EP, Novel Pseudomonas aeruginosa quorum-sensing inhibitors identified in an ultra-high-throughput screen. Antimicrob. Agents. Chemother 2006, 50 (11), 3674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang Y; Chen X; Zheng Y; Xue Z; Shu C; Yuan W; Zhang X, Highly diastereoselective and enantioselective synthesis of alpha-hydroxy beta-amino acid derivatives: Lewis base catalyzed hydrosilylation of alpha-acetoxy beta-enamino esters. Angew. Chem. Int. Ed. Engl 2011, 50 (32), 7304–7. [DOI] [PubMed] [Google Scholar]
- 35.Gerdt JP; McInnis CE; Schell TL; Rossi FM; Blackwell HE, Mutational analysis of the quorum-sensing receptor LasR reveals interactions that govern activation and inhibition by nonlactone ligands. Chem. Biol 2014, 21 (10), 1361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bottomley MJ; Muraglia E; Bazzo R; Carfi A, Molecular insights into quorum sensing in the human pathogen Pseudomonas aeruginosa from the structure of the virulence regulator LasR bound to its autoinducer. J. Biol. Chem 2007, 282 (18), 13592–600. [DOI] [PubMed] [Google Scholar]
- 37.Paczkowski JE; McCready AR; Cong JP; Li Z; Jeffrey PD; Smith CD; Henke BR; Hughson FM; Bassler BL, An Autoinducer Analogue Reveals an Alternative Mode of Ligand Binding for the LasR Quorum-Sensing Receptor. ACS Chem. Biol 2019, 14 (3), 378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCready AR; Paczkowski JE; Henke BR; Bassler BL, Structural determinants driving homoserine lactone ligand selection in the Pseudomonas aeruginosa LasR quorum-sensing receptor. Proc. Natl. Acad. Sci. U. S. A 2019, 116 (1), 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Passador L; Tucker KD; Guertin KR; Journet MP; Kende AS; Iglewski BH; Functional Analysis of the Pseudomonas aeruginosa Autoinducer PAI. J. Bacteriol 1996, 178 (20) 5995–6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McInnis CE; Blackwell HE, Design, synthesis, and biological evaluation of abiotic, nonlactone modulators of LuxR-type quorum sensing. Bioorg. Med. Chem 2011, 19 (16), 4812–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pearson JP; Pesci EC; Iglewski BH, Roles of Pseudomonas aeruginosa las and rhl Quorum-Sensing Systems in Control of Elastase and Rhamnolipid Biosynthesis Genes. J. Bacteriol 1997, 179 (18), 5756–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chugani SA; Whiteley M; Lee KM; D’Argenio D; Manoil C; Greenberg EP, QscR, a modulator of quorum-sensing signal synthesis and virulence in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (5), 2752–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindsay A; Ahmer BM, Effect of sdiA on biosensors of N-acylhomoserine lactones. J. Bacteriol 2005, 187 (14), 5054–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JH; Lequette Y; Greenberg EP, Activity of purified QscR, a Pseudomonas aeruginosa orphan quorum-sensing transcription factor. Mol. Microbiol 2006, 59 (2), 602–9. [DOI] [PubMed] [Google Scholar]
- 45.Wellington S; Greenberg EP, Quorum Sensing signal selectivity and the potential for interspecies cross talk. mBio 2019, 10 (2), e00146–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Churchill MEA; Chen L, Structural Basis of Acyl-homoserine Lactone-Dependent Signaling. Chem. Rev 2011, 111, 68–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Reilly MC; Dong SH; Rossi FM; Karlen KM; Kumar RS; Nair SK; Blackwell HE, Structural and Biochemical Studies of Non-native Agonists of the LasR Quorum-Sensing Receptor Reveal an L3 Loop “Out” Conformation for LasR. Cell Chem. Biol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zou Y; Nair SK, Molecular basis for the recognition of structurally distinct autoinducer mimics by the Pseudomonas aeruginosa LasR quorum-sensing signaling receptor. Chem. Biol 2009, 16 (9), 961–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wysoczynski-Horita CL; Boursier ME; Hill R; Hansen K; Blackwell HE; Churchill MEA, Mechanism of agonism and antagonism of the Pseudomonas aeruginosa quorum sensing regulator QscR with non-native ligands. Mol. Microbiol 2018, 108 (3), 240–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen G; Swem LR; Swem DL; Stauff DL; O’Loughlin CT; Jeffrey PD; Bassler BL; Hughson FM, A strategy for antagonizing quorum sensing. Mol. Cell 2011, 42 (2), 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suneby EG; Herndon LR; Schneider TL, Pseudomonas aeruginosa LasR.DNA Binding Is Directly Inhibited by Quorum Sensing Antagonists. ACS Infect. Dis 2017, 3 (3) 183–189. [DOI] [PubMed] [Google Scholar]
- 52.Morkunas B; Galloway WR; Wright M; Ibbeson BM; Hodgkinson JT; O’Connell KM; Bartolucci N; Della Valle M; Welch M; Spring DR, Inhibition of the production of the Pseudomonas aeruginosa virulence factor pyocyanin in wild-type cells by quorum sensing autoinducer-mimics. Org. Biomol. Chem 2012, 10 (42), 8452–64. [DOI] [PubMed] [Google Scholar]
- 53.Sappington KJ; Dandekar AA; Oinuma K; Greenberg EP, Reversible signal binding by the Pseudomonas aeruginosa quorum-sensing signal receptor LasR. MBio 2011, 2 (1), e00011–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerdt JP; McInnis CE; Schell TL; Blackwell HE, Unraveling the contributions of hydrogen-bonding interactions to the activity of native and non-native ligands in the quorum-sensing receptor LasR. Org. Biomol. Chem 2015, 13 (5), 1453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gerdt JP; Blackwell HE, Competition studies confirm two major barriers that can preclude the spread of resistance to quorum-sensing inhibitors in bacteria. ACS Chem. Biol 2014, 9 (10), 2291–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sully EK; Malachowa N; Elmore BO; Alexander SM; Femling JK; Gray BM; DeLeo FR; Otto M; Cheung AL; Edwards BS; Sklar LA; Horswill AR; Hall PR; Gresham HD, Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLOS Pathog. 2014, 10 (6), e1004174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.