

Abstract
Mutations in the human ATP13A2 gene are associated with an early-onset form of Parkinson’s disease (PD) known as Kufor Rakeb Syndrome (KRS). Patients with KRS show increased iron deposition in the basal ganglia, suggesting iron toxicity-induced neurodegeneration as a potential pathogenesis associated with the ATP13A2 mutation. Previously we demonstrated that functional losses of ATP13A2 disrupt the lysosome’s ability to store excess iron, leading to reduce survival of dopaminergic neuronal cells. To understand the possible mechanisms involved, we studied a Caenorhabditis elegans mutant defective in catp-6 function, an ortholog of human ATP13A2 gene. Here we show that catp-6 mutant worms have defective autophagy and lysosomal function, demonstrate characteristic PD phenotypes including reduced motor function and dysregulated iron metabolism. Additionally, these mutants have defective mitochondrial health, which is rescuable via iron chelation or mitophagy induction.
Keywords: ATP13A2; catp-6; lysosomes; iron metabolism; mitochondrial function; urolithin A; iron chelation; TFEB, Parkinson’s disease; C. elegans
Introduction
Parkinson’s disease is a progressive neurodegenerative disorder for which etiology and pathogenesis is still incompletely understood. Over the last several years, many genes associated with autosomal recessive forms of Parkinsonism including parkin (PARK2), DJ-1 (PARK7), PINK1 (PARK6) and ATP13A2 (PARK9) have been identified (Kitada et al. 1998; Bonifati et al. 2003; Valente et al. 2004; Ramirez et al. 2006). Genetic changes associated with homozygous or compound heterozygous mutations in the ATP13A2 gene have specifically been shown to result in a rare juvenile form of PD, Kufor Rakeb Syndrome (KRS)(Behrens et al. 2010). The disease is partially responsive to L-DOPA therapy and is characterized by features including pyramidal signs, supranuclear gaze palsy, dystonia and dementia (Najim al-Din et al. 1994; Hampshire et al. 2001; Williams et al. 2005).
The ATP13A2 gene encodes a lysosomal transmembrane type 5 ATPase pump that maintains lysosomal function (Ramirez et al. 2006; Dehay et al. 2012; Usenovic et al. 2012). In lysosomes, ATP13A2 has been suggested to function as an ATP-dependent cation transporter, although this has not been directly demonstrated (Ramirez et al. 2006). Deletion of the yeast ortholog of ATP13A2, ypk9 has been shown to increase sensitivity to heavy metals including cadmium, manganese, selenium and nickel, suggesting a critical role for ATP13A2 in the transport and regulation of these metals (Gitler et al. 2009; Schmidt et al. 2009). Previous work from our own laboratory demonstrated that functional losses in ATP13A2 disrupt the lysosome’s ability to store excess iron, eventually leading to reduce survival of dopaminergic neuronal cells (Rajagopalan et al. 2016).
Since neurons are heavily dependent on mitochondria for their function, we asked whether dysregulated iron homeostasis resulting from ATP13A2 loss affects mitochondrial function. We studied this in a C.elegans catp-6 mutant, which lacks an ortholog of the human ATP13A2 gene. We show that similar to human ATP13A2, catp-6 loss of function affects lysosomal function, and plays an important role in the maintenance of both iron homeostasis and mitochondrial function. Therapeutically, we show that iron chelation or compounds that induce mitophagy can rescue mitochondrial pathologies associated with ATP13A2 loss of function.
Materials and Methods
Caenorhabditis elegans strains and maintenance
C. elegans strains used in the study are: N2 (Bristol), RB2510 W08D2.5(ok3473) IV and NL5901 (pkIs2386 [unc-54p::alphasynuclein::YFP + unc-119(+)]). The strains were obtained from the Caenorhabditis Genetics Center (CGC), University of Minnesota, MN, USA. The allele ok3473 is a knockout for W08D2.5/catp-6 gene with an estimated deletion size of 900 bp as reported by CGC. The deletion results in a significant loss in catp-6 mRNA and protein levels (Figure 1A,B and C).
Figure 1: Loss of catp-6 affects autophagy and lysosomal function in C. elegans.
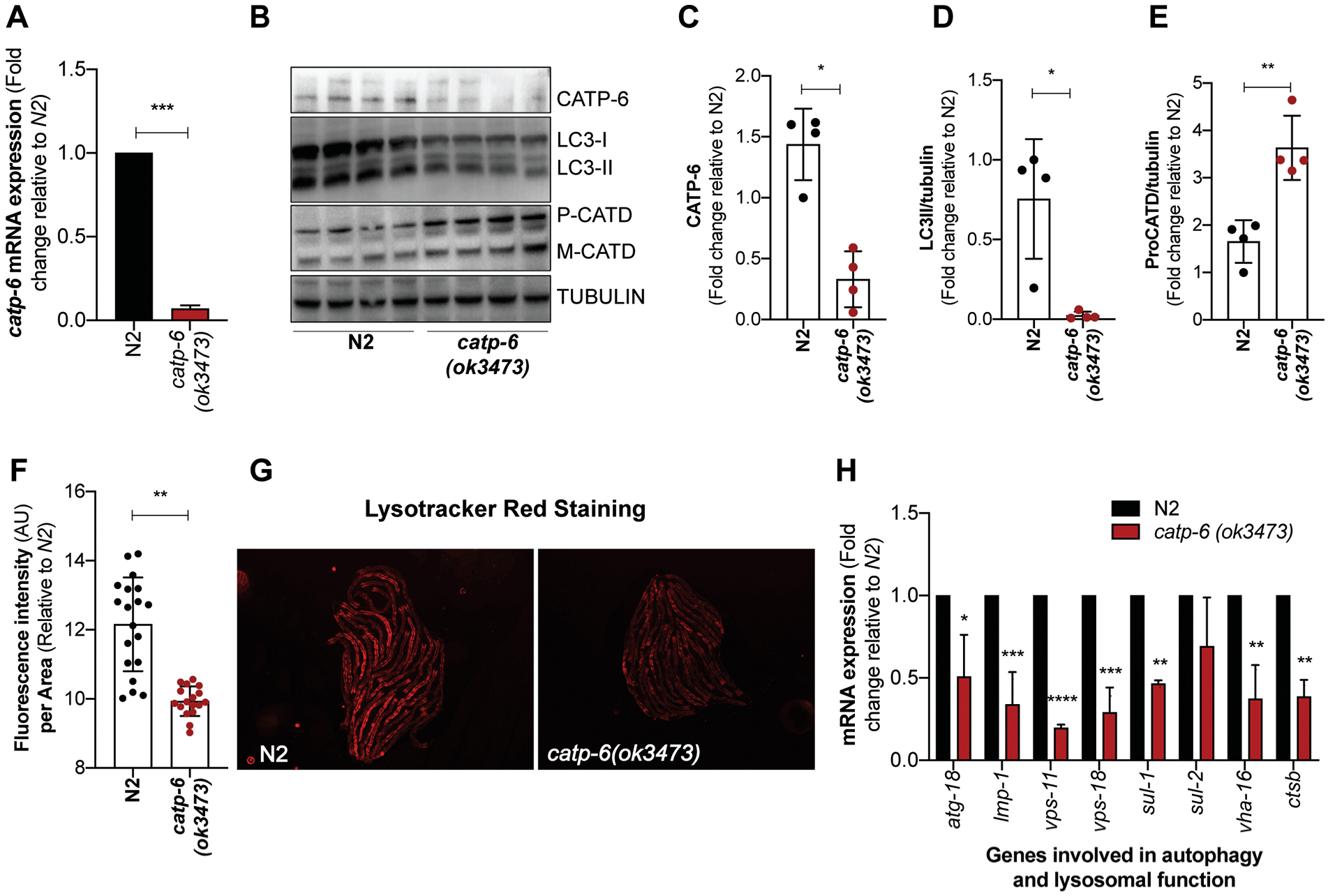
(A) Graph represents fold change ± SD in the mRNA expression of catp-6 in catp-6(ok3473) mutant relative to wild type N2 worms (n=2, p-values calculated using unpaired Student’s t-test). (B) Immunoblot showing the level of CATP-6, autophagosome specific cleaved LGG-1/LC3-II protein and lysosome specific aspartyl protease ASP-3/cathepsin D (CATD) which exists as procathepsin D (P) and mature (M) cathepsin D and loading control γ-tubulin. (C-E) Graph represents the densitometric analysis of the bands, showing average band intensity ± SD of CATP-6, cleaved LGG-1/LC3-II and P-CATD normalized to loading control γ-tubulin (n=4, p-values calculated using unpaired Student’s t-test) (F) Graph represents the mean fluorescence intensity ± SD of LysoTracker Red stain normalized to the body size for wild type N2 and catp-6(ok3473) worms. Quantification was performed on the whole body of an individual worm using NIH image J software (n=2, p-values calculated using unpaired Student’s t-test). (G) Representative images of the N2 and catp-6(ok3473) worms showing LysoTracker red fluorescence. (H) Graph represents fold change ± SD in the mRNA expression of autophagy and lysosomal specific genes in catp-6(ok3473) mutant worms relative to wild type N2 worms (n=2, p-values calculated using ordinary two-way ANOVA by Multiple t-tests with correction for multiple comparisons using the Holm-Sidak method). All the p-values are represented as *p < 0.05, **p<0.01 ***p<0.001 and ****p<0.0001.
C. elegans strains were maintained at 20°C under standard laboratory conditions as described previously (Stiernagle 2006). Worm populations were maintained in 60 mm NGM agar plates (3 g/L NaCl, 17 g/L agar; 2.5 g/L peptone; 1 mM CaCl2, 5 mg/L cholesterol, 1 mM MgSO4, 25 mM KPO4) seeded with OP50 Escherichia coli. During experiments synchronized population of worms were obtained by a 2-hour egg-lay from day-1 adult hermaphrodites on E. coli OP50 seeded 60 mm NGM agar plates. Post 2-hour adults were removed and the eggs were left to develop into adults at 20°C.
Compound preparation and treatment
A 100 mM stock of Urolithin A (UA) (sc-475514) and TFEB enhancer was prepared in sterile DMSO (Sigma) and stored in small aliquots at −20°C. From the stock solution, 130 μL of the working solution (50 μM UA or TFEB enhancer) was prepared by mixing 1.5 μL of stock solution (or DMSO only for control plates, 0.05% DMSO) with 128.5 μL of sterile water, and was added to the top of the 35 mm NGM plates (3 mL NGM agar) already seeded with a bacterial OP50 lawn. Stock solution of Calcium Disodium Ethylenediaminetetraacetic acid (CaEDTA) (Sigma) was prepared at 75 mM in sterile water and stored at 4°C. The 35 mm NGM plates (3 mL NGM agar) already seeded with a bacterial OP50 lawn was spotted with 100 μL of stock solution at a final concentration of 2.5 mM CaEDTA. Control plates for CaEDTA were treated with 100 μL of sterile water. Compound solution was distributed over the entire plate surface and allowed to dry in a sterile hood with lid open for at least 45 minutes in order to ensure complete drying of the agar/bacterial surface. We have seen that not doing so keeps the agar/bacterial surface moist, increasing the chances of worm loss from the moist edges of the plate. We chose to add compounds on top of the seeded plates and not in the agar plate to ensure maximum bioavailability and ensure stability of the compounds. Plates were then allowed to sit at 20°C for 24 hours before use or before moving into 4°C. Plates were stored for no longer than two weeks.
During experiments day-1 hermaphrodites were allowed to lay eggs for 2 hours on the non-compound treated plate. Equal number of synchronized eggs (35–40 per plate) were then picked with platinum pick and moved to a compound-treated plate and allowed to develop into adults at 20°C. Young day-1 adult worms were then used for the mitochondrial toxicity assay.
Mitochondrial toxicity assay
Mitochondrial toxin rotenone (complex I inhibitor) was used to assess the mitochondrial health. A 100 mM stock of rotenone was prepared in the sterile DMSO and stored in small aliquots at −20°C. From the stock solution, final working solution of rotenone (25 and 50 μM) was prepared in S-basal solution (5.85 g/L NaCl, 1g/L K2HPO4, 6 g/L KH2PO4, H2O to 1 L) and dispensed in a 96-well flat bottom plate (40 μL per well). All the wells including control wells were matched to consist 0.05%. Synchronized populations of day-1 adult worms (grown on control, CaEDTA, TFEB enhancer and UA) were picked and transferred to individual wells (10–15 worms per well). Three hours post-rotenone exposure, worms were scored dead or alive under the microscope. Worms that failed to display touch-provoked movement were scored as dead. Values represent percent survival ± SD.
Mounting worms and imaging
Microscopic slides containing pads of 2% agarose were prepared 30 minutes before mounting worms. Around 2–5 μL of 2 mM levamisole was pipetted out on the center of the agarose pads. Worms were transferred to the levamisole drop, and a cover slip was placed on top before imaging.
Adult lifespan assays
Plate preparation:
A 450 mM stock solution of ferric ammonium citrate (FAC) (Sigma) was prepared in a sterile water. The 35 mm FuDR (Sigma) (10 μg/ml or 40.6 μM) containing NGM plates (3 mL NGM agar) already seeded with a bacterial OP50 lawn was spotted with FAC to make a final concentration of (15–50 mM) in sterile water. Control plates were treated with (9–30%) ammonium citrate (Sigma) as negative iron control. For zinc treated plates, 1 M stock solution of ZnSo4 was prepared in sterile water and spotted at a final concentration of 500 μM. Plates were stored at 20°C overnight and then used for worms transfer or stored at 4°C for no longer than two weeks.
Age-synchronized day-1 worms (n=35 per plate) were then picked and transferred to FAC, zinc or control FuDR plates. Worms were transferred to fresh-compound treated plates every alternate day for 1st and 2nd week and then once a week. Worms were scored dead or alive every alternated day. Worms that failed to display touch-provoked movement were scored as dead. Worms that died from causes other than aging, such as sticking to the plate walls, internal hatching of eggs (‘bagging’), or gonadal extrusion were censored. All lifespan experiments were performed at 20°C. Survival graphs were plotted using GraphPad Prism 8 (GraphPad Software, Inc., La Jolla, CA).
Thrashing assay for motor function
Egg-lay synchronized populations of N2, catp-6(ok3473) and (pkIs2386 [unc-54p::alphasynuclein::YFP + unc-119(+)]) worms were grown at 20°C. Thrashing rate was measured when worms reached reproductive adult stage. Thrashing was measured by recording a 30 second video of day-1 worms transferred to a sterile glass slide in a drop of S basal buffer. The worms were allowed to acclimate to the environment for 30 second and then video recorded under Leica stereomicroscope using corresponding software (30 second, n=30 in triplicate). For analysis, the video was slowed down and movement of the worm’s head and/or tail to the same side was counted as one thrash. Worms which curled up or did not show any movement were not included in the analysis. Thrashing rate for the individual worm was plotted and average thrashing rate was determined.
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR) of iron metabolism genes
RNA was isolated from 400–500 day-1 adult N2 and catp-6(ok3473) worms grown at 20°C. The worms were washed with 1X M9 buffer (g/L Na2HPO4, 3 g/L KH2PO4, 5 g/L NaOH, 1 mL/L of 1M MgSO4 solution) to get rid of any attached bacteria and finally collected in 300 μL of RNA lysis buffer supplied with a Zymogen RNA isolation kit. RNA was isolated according to the supplier protocol. The RNA concentration was quantified using NanoDrop 2000 Spectrophotometer. A total of 2000 ng RNA was subsequently reverse-transcribed to cDNA using iScript cDNA Synthesis Kit. Real time PCR was then performed using SYBR green master mix in a Light cycler 480II system. The relative gene expression was calculated by 2−ΔΔCt method. The sequence of the qRT primers used for the analysis is provided in a Table 1 and were obtained from the previously published study (Lapierre et al. 2013; van den Ecker et al. 2015).
Table 1:
Sequence of the primers used in study
| Gene | Primer | Sequence(5'−3') |
|---|---|---|
| catp-6 | Forward | AAGTCTAACGCCACAATTCCCACCGA |
| Reverse | CCAAAAGTGATCGACAAGGAAT | |
| atg-18 | Forward | AAATGGACATCGGCTCTTTG |
| Reverse | TGATAGCATCGAACCATCCA | |
| lmp-1 | Forward | ATCCGCCACCGCTTCGCATT |
| Reverse | TCGAGCTCCCACTCTTTGGCG | |
| vps-11 | Forward | TCCGCTTGTCGTCCTGGAGC |
| Reverse | TCACACGCCGAGCACTTGGT | |
| vps-18 | Forward | CGAGCCGGCGCCAGTTGTAA |
| Reverse | TCCATCCGGCGAAAGCCACG | |
| sul-1 | Forward | CAGGATGGGATGAGTGGCACG |
| Reverse | GGGTCTTCTGGTCCATGCGGC | |
| sul-2 | Forward | ATGGCAGCAGAAGGCACCCG |
| Reverse | GCCATTTTCCAACCATGCCAGTTGC | |
| vha-16 | Forward | AGGCGCTGACTCGCGGACTT |
| Reverse | TGGTCTCTGGTGAAGAGTTCCGGTG | |
| ctsb/cpr-1 | Forward | CGCCAAGGACAAGCACTTCGGA |
| Reverse | ACCTTGGCCTTTCCGGCGAC | |
| fpn-1 | Forward | GGACT CAAAGGCGTCAAGTT |
| Reverse | TAGGATGCGGCGAGGTTAT | |
| aco-1 | Forward | GTCGTAGATCTTGCCGCTATG |
| Reverse | GGCTTCAAGGTTTCCGTAGT | |
| aco-2 | Forward | TCACTCCTCTTCGTCCATCT |
| Reverse | GGACGTTTCAATCTGTCTTTCAC | |
| lpd-8 | Forward | GCTTTGTACAAGCAGCTCGAT |
| Reverse | CGCTGAGCTGAAATCGTATGTT | |
| smf-3 | Forward | CTCGTTTGGTACTGTGGATGTT |
| Reverse | CAGAGAGGGATTACTCCGTTAGA | |
| abtm-1 | Forward | TCAGATCGCAGGCAGTTTAC |
| Reverse | GTGAAAGCATGTCCTTGTGAATAC | |
| mfn-1 | Forward | CTTGCAGGCTAGGTATCGTTAT |
| Reverse | TTGCAGGAACCTGGAATATGA | |
| ftn-2 | Forward | GTTAACAAGCAGATCAACATT |
| Reverse | ACGCTCCTCATCCGATT | |
| Genotype primers | ||
| catp-6 | outer primer-Forward | CTTAAAATTTCGCGGCTGAG |
| catp-6 | outer primer-Reverse | TAAGGCCTTCCAAAAGAGCA |
| catp-6 | Inner primer-Forward | TTTTCGGCTTAGAAAACAGCA |
| catp-6 | Inner primer-Reverse | TGGTGAGCTCGATGAATACG |
Mitochondrial membrane potential quantification using TMRM (tetramethylrhodamine)
TMRM plate preparation:
From a stock solution of 100 μM TMRM (Thermo, T668) prepared in DMSO a final concentration of 150 nM TMRM (diluted in water) was added to the top of the 35 mm NGM plates (3 mL NGM agar) already seeded with a bacterial OP50 lawn. TMRM solution was distributed over the entire plate surface and allowed to dry in a sterile hood with lid open for at least 45 minutes in order to ensure complete drying of the agar/bacterial surface. Since TMRM is light-sensitive, plates were covered with aluminum foil and were allowed to sit at 20°C for 24 hours before use.
Egg-lay synchronized populations of N2 and catp-6(ok3473) worms were grown at 20°C. Late L3 larval stage worms were picked and transferred onto TMRM plates for 24 hours at 20°C and then transferred to non-TMRM plates for 1 hour to remove excess stain. As a positive control we transferred 20–30 N2 worms from the 24 hours TMRM treated N2 plate onto FCCP supplemented non-TMRM plate for 1 hour. About 20–30 worms per strain were picked and mounted on agarose pad as mentioned above for visualization of TMRM fluorescence under Zeiss Imager Z1 fluorescence microscope using rhodamine filters. Quantification of images was performed using NIH ImageJ software. We quantified the fluorescence intensity of the TMRM stain in the whole body for an individual worm and normalized it to the body size of the worm. Graph represents the average fluorescence intensity ± SD intensity of all the worms shown in the image.
LysoTracker Red staining
LysoTracker Red stain plate preparation:
From a stock solution of 1 mM LysoTracker Red DND-99 (Molecular Probes) prepared in DMSO a final concentration of 100 nM stain (diluted in water) was added to the top of the 35 mm NGM plates (3 mL NGM agar) already seeded with a bacterial OP50 lawn. LysoTracker Red stain solution was distributed over the entire plate surface and allowed to dry in a sterile hood with lid open for at least 45 minutes in order to ensure complete drying of the agar/bacterial surface.
Egg-lay synchronized populations of N2 and catp-6(ok3473) worms were grown at 20°C. Larval L4 stage N2 and catp-6(ok3473) worms were picked and transferred onto LysoTracker Red stained plates for 24 hours at 20°C. Post-24 hours, worms were picked and washed in a fresh drop of 1X M9 buffer and mounted on 2% agarose pad slides for microscopic visualization as mentioned above. Worm images were taken in a Zeiss Imager Z1 fluorescence microscope using rhodamine filters. Image analysis was performed using Image J (NIH) software by measuring mean LysoTracker fluorescence of each worm normalized to it’s body size. Graph represents mean fluorescence intensity ± SD of all the worms shown in the image.
Basal and maximum oxygen consumption rate (OCR)
Basal and maximum OCR was measured using Seahorse XF96 equipment (Seahorse Bioscience Inc., North Billerica, MA, USA). Egg-lay synchronized day-5 adult N2 and catp-6(ok3473) worms (~200 per strain, n=20 worms per well) were used for measuring the basal and maximum OCR according to previously described protocol (NG AND GRUBER 2019). On the day of the assay we washed animals three times with 1X M9 buffer in a 15 mL conical tube to get rid of any attached bacteria. We transferred ~20 animals to each well of a Seahorse XF24 Cell Culture Microplate with 200 μL of 1X M9 buffer. We first measured the basal respiration rate in every well, followed by injection of Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (Cayman) in the wells to uncouple mitochondria.
We injected sodium azide at the end of the assay in every well to completely block mitochondrial respiration. These compounds were prepared in the final solution of 1X M9 buffer and were injected from the reagent ports automatically to the wells at the time indicated. All respiration parameters were normalized to the number of animals per well. Bar graph represents the average basal and maximum OCR ± SD.
Worm protein extraction, SDS-PAGE and Immunoblotting
Synchronized day-1 adult N2 and catp-6 mutant worms were grown at 20°C. 400 worms per sample were collected in 1X M9 buffer. Worms were washed twice and suspended in RIPA lysis buffer supplemented with complete EDTA-free protease inhibitor cocktail (ROCHE). Worm pellets were sonicated for 15 cycles at maximum intensity in Bioruptor sonicator (Diagenode). Lysate was centrifuged at 1000 × g for 1 minute and clear protein lysate was transferred to fresh eppendorf. After protein estimation equal amount of protein was used for immunoblotting. Polyacrylamide gels (10%−15%) were used for the separation of proteins. 35 μg of total proteins were transferred to 0.2 μm nitrocellulose membranes using the BioRad blotting system. All membranes were blocked with blocking buffer. Primary antibodies γ-Tubulin (T1450), ATP13A2 (LS-B11104), cathepsin D Antibody (C-20) (sc-6486), LC3B Antibody (CST #2775) were used for immunoblotting of total proteins. Goat anti-rabbit-IgG-HRP (CST, 7074), horse anti-mouse IgG-HRP (CST, 7076) were used as secondary antibody in all immunoblotting experiments. Chemiluminescence intensities were detected with ChemiDoc imaging system (BioRad) and quantified using Image J software (NIH). γ-tubulin levels were used to normalize the intensities of bands.
Statistical analysis
Survival graphs were plotted using GraphPad Prism 8 (GraphPad Software, Inc., La Jolla, CA). All the statistical analysis of survival curve P-values was performed using the Log-rank (Mantel-Cox) test through the online software OASIS (http://sbi.postech.ac.kr/oasis). The p-values for the quantification of the immunoblot band intesnity, LysosTracker Red stain assay were determined using unpaired Student’s test. The p-values for the quantification of relative fold mRNA difference in Figure 1D and 2C were calculated using ordinary two-way ANOVA by Multiple t-tests with correction for multiple comparisons using the Holm-Sidak method. The p- value for thrashing rate and TRMRM staining assay were determined using ordinary one-way ANOVA and Tukey’s multiple comparisons test. The The p-values for OCR assay in Figure 3E was determined using ordinary two-way ANOVA and Sidak’s multiple comparison test. The p-values for rotenone toxicity assay were determined using ordinary two-way ANOVA and Tukey’s multiple comparisons test. All the experiments were repeated at least twice. p-values were designated as: *P < 0.05, **P<0.01 and ***P<0.001, ****P<0.0001, ns-not significant
Figure 2: Reduced motor function and dysregulated iron metabolism in catp-6 mutants.
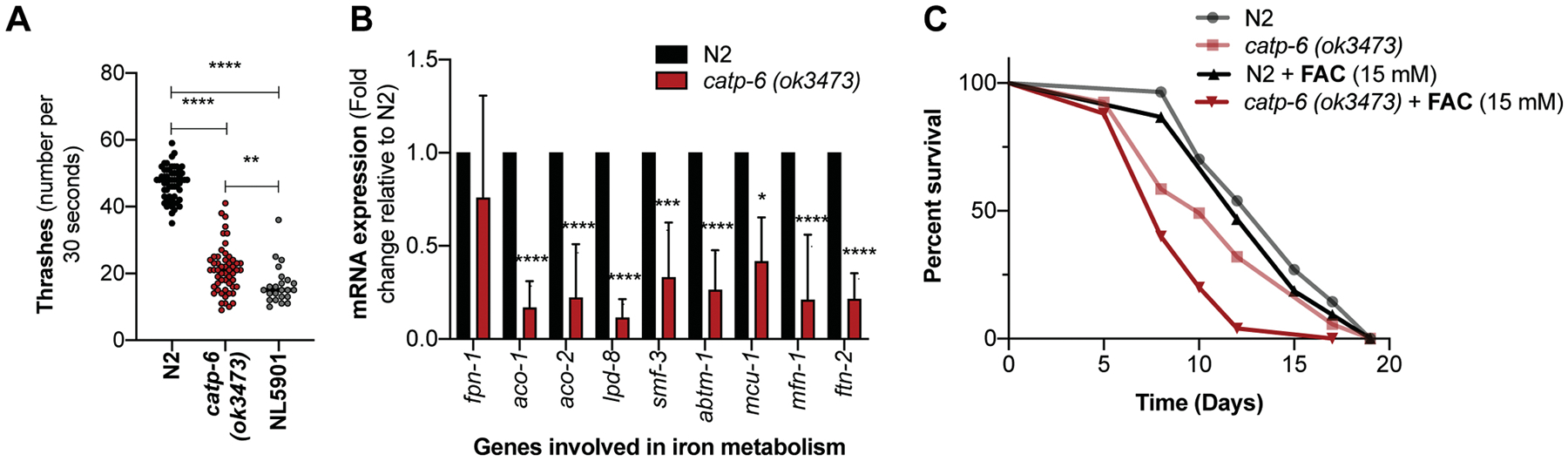
(A) Quantification of the number of thrashes in N2 versus catp-6(ok3473) mutant worms. Graph represents average number of thrashes ± SD over the period of 30 seconds, where Individual dot represents single worm. NL5901 (pkIs2386 [unc- 54p::alphasynuclein::YFP + unc-119(+)] strain expressing human α-synuclein in muscles shows motor defects and served as a positive control for the assay. (n=3, p-values calculated using ordinary one-way ANOVA and Tukey’s multiple comparisons test). (B) Graph represents fold change ± SD in the mRNA expression of iron-metabolizing genes in catp-6(ok3473) mutant worms relative to wild type N2 worms (n=5, p-values calculated using two-way ANOVA analysis). (C) The effect of ferric ammonium citrate (FAC) or ammonium citrate exposure as a negative control on survival of N2 and catp-6(ok3473) mutant worms. N2 control [13.78 ± 0.44] (median-14), N2 FAC (15 mM) [13.43 ± 0.58] (median-13), catp-6(ok3473) [11.64 ± 0.59] (median-11), and catp-6(ok3473) FAC (15 mM) [9.04 ± 0.36] (median-8). [Mean average lifespan ± SEM], (n=2, p-value calculated using Mantel-Cox Log rank test). All the p-values are represented as *p < 0.05, **p<0.01 and ***p<0.001, ****p<0.0001.
Figure 3: Defective mitochondrial function in catp-6 mutants.
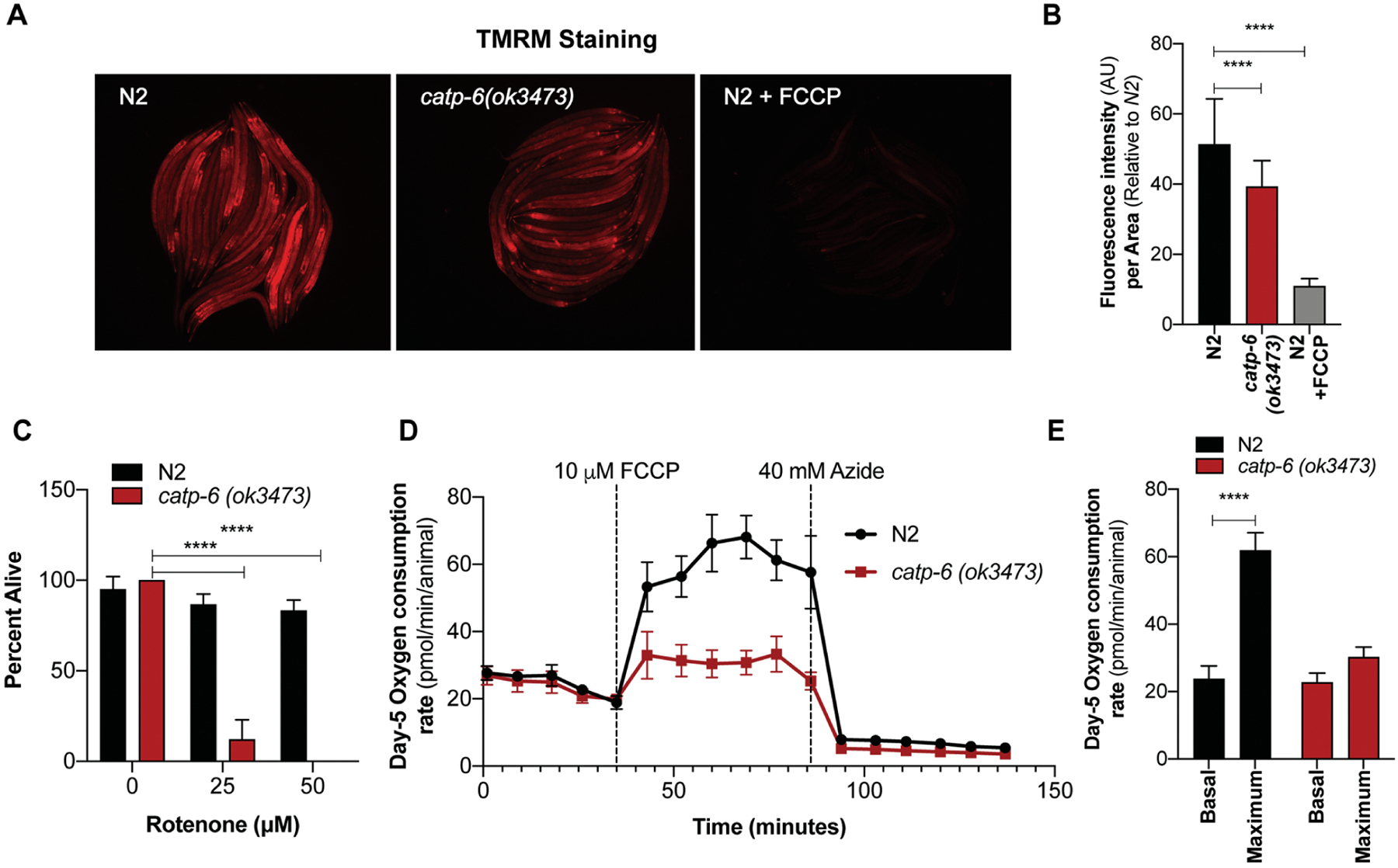
(A) Representative images of TMRM-stained N2, catp-6(ok3473) and TMRM-N2 worms exposed to 10 μM FCCP for 1 hour (positive control). (B) Graph represents the average mean fluorescence intensity ± SD of TMRM stain normalized to the body size. Quantification was performed on the whole body of an individual worm using NIH image J software (n=2, p-value calculated using ordinary one-way ANOVA and Tukey’s multiple comparisons test). (C) Enhanced sensitivity of catp-6(ok3473) mutant worms to the mitochondrial toxin rotenone measured as percent survival ± SD (n=2, p-value calculated using ordinary two-way ANOVA and Tukey’s multiple comparisons test). (D-E) Mitochondrial function in N2 and catp-6(ok3473) mutant worms as quantitated by oxygen consumption rate using the Seahorse. Bar graph represents average OCR consumption ± SD of day-5 N2 and catp-6(ok3473) worms (n=2, p-value calculated using ordinary two-way ANOVA and Sidak’s multiple comparison test). All the p-values are represented as *p < 0.05, **p<0.01 and ***p<0.001, ****p<0.0001.
Results
Loss of catp-6 affects autophagy and lysosomal function in C. elegans
The human ATP13A2 gene shares close homology with the C. elegans CATP genes, with CATP-6 being the closest homolog (Figure S1A). Given the important role of ATP13A2 in regulating lysosomal function, we asked whether loss of catp-6 in C. elegans also affects lysosomal function or not. We used catp-6(ok3473) mutant and genotype confirmed an estimated deletion of 900 bp as reported by the CGC. Deletion results in a significant loss of catp-6 mRNA (Figure 1A) and protein levels (Figure 1B, 1C). We assessed autophagy and lysosomal function in these mutants by quantifying protein levels of autophagosome (cleaved LGG-1/LC3-II) and lysosome specific aspartyl protease ASP-3/cathepsin D (CATD), respectively. We see protein level of cleaved LGG-1/LC3-II is significantly reduced in catp-6 mutant, suggesting decreased autophagosomes in catp-6 mutant (Figure 1B, 1D). This decrease in cleaved LGG-1/LC3-II level seems not to result from enhanced clearance of autophagosomes by lysosomes, but rather due to defect in early autophagosome formation, as lysosomal function is defective in this mutant. We tested lysosomal function by monitoring the level of premature (P-CATD) and mature (M-CATD) form of pH dependent lysosomal enzyme aspartyl protease ASP-3/CATD. We see that levels of P-CATD are significantly high in the catp-6 mutants compared to wild type N2 worms, suggesting reduced conversion of P-CATD to M-CATD, which is directly dependent on the acidity of the lysosomes. In support, we see lysosomal pH of catp-6 mutant worms is less acidic, as seen by significantly low LysosTracker Red staining when compared to wild type N2 worms (Figure 1F, 1G). LysosTracker Red is a pH dependent stain that stains lower in worms with inability to maintain acidic lysosome. Finally, we also see that catp-6 mutant worms show significantly low mRNA expression of several key genes required for autophagy and lysosomal function (Figure 1H) (Lapierre et al. 2013). All together these results show that loss in catp-6 affects autophagy and lysosomal function.
Reduced motor function and dysregulated iron metabolism in catp-6 mutants
Mutations in the ATP13A2 gene in humans results in parkinsonism-like phenotypes characterized by a loss of motor function, hence we first investigated whether catp-6 mutant displayed any observable motor function defect. Indeed, we found that the thrashing rate (body bends) of catp-6 mutant worms was significantly reduced compared to wild type N2 worms (catp-6 vs N2, 21 vs 47 average thrashes per 30 seconds) (Figure 2A). As a control for thrashing assay we used NL5901 transgenic worms, which expresses human α-synuclein under control of muscle specific unc-54 promoter cells and how motor function defect. As expected we see a significant motor function defects in NL5901 worms as their thrashing rate was significantly low (NL5901 vs N2, 16 vs 47 average thrashes per 30 seconds) (Figure 2A). We also observed a significant delay in the rate of development in catp-6 mutants when compared to wild type N2 worms (Figure S1B).
We next asked whether catp-6 mutant worms showed any signs of dysregulated iron metabolism. We observed several of the core genes required for metabolizing iron were significantly down-regulated in catp-6 mutants compared to wild type N2 worms (Figure 2B). Interestingly, we also observed that catp-6 mutant worms showed enhanced sensitivity towards externally supplied iron, suggesting an inability to regulate iron. The magnitude of suppression of mean average lifespan upon exposure to 15 mM ferric ammonium citrate (FAC) was much higher in catp-6 mutant worms 22% (p=0.0008), (catp-6(ok3473) FAC (15 mM) [9.04 ± 0.36] vs catp-6(ok3473) [11.64 ± 0.59], Mean average lifespan ± SEM) with no significant changes in mean average lifespan of wild type N2 worms at similar dose (N2 FAC (15 mM) [13.43 ± 0.58] vs N2 control [13.78 ± 0.44], Mean average lifespan ± SEM) (Figure 2C, S1C). We also tested the effects of zinc supplementation on catp-6 mutant, which was recently shown to have altered zinc homeostasis (BAESLER et al. 2019). We see magnitude of mean average lifespan suppression upon exposure to zinc was significantly high in catp-6 mutants (21.4% p=0.0004, zinc treated catp-6 vs untreated catp-6, 10.17 ± 0.46 vs 12.94 ± 0.45, mean average lifespan ± SEM) compared to wild type N2 worms (9.0% p=0.02, zinc treated N2 vs untreated N2, 13.42 ± 0.43 vs 14.75 ± 0.59, mean average lifespan ± SEM) (Figure S1D).
Defective mitochondrial function in catp-6 mutants
Defects in mitochondrial function underlie many neurodegenerative diseases, including PD. The catp-6 mutants have previously been shown to have an enhanced neuronal loss upon expression of just a single copy α-synuclein, a protein associated with mitochondrial dysfunction (Cooper et al. 2018). The fact that mutation in catp-6 gene is enough to enhance susceptibility towards neuronal loss suggest defect in key process or organelle effecting α-synuclein pathology. We, therefore, asked whether mutation in catp-6 alone results in enhanced mitochondrial defects. A characteristic feature of defective mitochondria is that they are unable to maintain membrane potential. Using the membrane potential based fluorescent dye tetramethylrhodamine (TMRM), which fluoresces only in healthy mitochondria, we demonstrated that catp-6 mutants show significantly lower TMRM fluorescence compared to wild type N2 worms (Figure 3A–B). As a positive control for the assay we exposed TMRM-stained N2 worms to the mitochondrial uncoupler FCCP (10 μM for 1 hour) known to reduce mitochondrial membrane potential. In addition, catp-6 mutants displayed increased sensitivity to the mitochondrial toxin, rotenone (Figure 3C). Moreover, catp-6 mutant worms displayed deficits in their maximum respiration capacity compared to wild type worms (Figure 3D–E). Taken together, these results suggest that catp-6 mutant worms have multiple defects in mitochondrial function.
Iron chelation and mitophagy induction rescue mitochondrial defects in catp-6 mutants
Next, we asked whether the observed defects in mitochondrial function could result from CATP-6 mediated dysregulation in iron metabolism. We used a known iron chelator Calcium Disodium Ethylenediaminetetraacetic acid (CaEDTA), which has been previously shown by our group to reduce iron levels and to increase lifespan in worms (Klang et al. 2014). We found that catp-6 mutant worms treated with 2.5 mM CaEDTA showed significant protection against rotenone toxicity at lower concentration (25 μM) and showed a protective trend to a higher concentration of rotenone (50 μM), which was however not significant (Figure 4A). Significant protection against rotenone toxicity was also observed in catp-6 mutants following treatment with a 50 μM dose of novel Transcription factor EB (TFEB) activator (recently discovered by our laboratory-not published) (Chamoli et al. 2017) and urolithin A (Ryu et al. 2016) both of which induce mitophagy in C. elegans (Figure 4B).
Figure 4: Iron chelation and mitophagy induction rescue sensitivity of catp-6 to rotenone exposure.

Sensitivity of N2 and catp-6(ok3473) mutant worms to the mitochondrial toxin, rotenone, measured as percent survival ± SD in worms exposed to (A) the 2.5 mM iron chelator calcium-EDTA (Ca-EDTA) or (B) the 50 μM mitophagy-inducing agents (TFEB enhancer and urolithin A) throughout development (n=2, p-value calculated using ordinary two-way ANOVA and Tukey’s multiple comparisons test). All the p-values are represented as *p < 0.05, **p<0.01 and ***p<0.001, ****p<0.0001.
Discussion
ATP13A2 is a proton-pumping lysosomal ATPase whose mutation is associated with a rare juvenile onset form of PD known as KRS. ATP13A2 is highly expressed within dopaminergic Substantia Nigra pars compacta (DAergic SNpc) neurons and its expression has been reported to be decreased within DAergic SNpc neurons in post-mortem PD tissues and in the brains of patients with Lewy body disorder (LBD) (murphy et al. 2013). The physiological function of ATP13A2, and hence the role of its dysfunction in KRS and other related conditions, however remains elusive.
Lysosomes are principle reservoirs for chelatable iron (Kurz et al. 2004; KURZ et al. 2006; Kurz et al. 2008; Kurz et al. 2011; Terman AND Kurz 2013). Inhibition of vacuolar proton-pumping ATPase activity and subsequent increases in lysosomal pH have been reported to result in the release of chelatable ferrous iron from the lysosomes into the cytosol. Lysosomal pH has been reported to be compromised in ATP13A2 mutant fibroblasts and in DAergic cell lines in which levels have been genetically reduced (Dehay et al. 2012). Interestingly, cytosolic iron levels have been reported to be elevated in the midbrain DAergic neurons of KRS patients.
The ATP13A2 transcript has been shown to contain a hypoxia response element (HRE) within its 5’ end, suggesting that its expression may be regulated by HIF1α. Transcription factor HIF1α is part of a highly conserved complex which serves to coordinately regulate a number of neuroprotective genes involved in cellular stress responses, including the maintenance of iron homeostasis and energy metabolism (Siddiq et al. 2005; Nakayama 2009; Greer et al. 2012). Previous work from our laboratory demonstrated expression of ATP13A2 within vulnerable midbrain DAergic neurons in vivo to be regulated by HIF1α signaling and knockdown of ATP13A2 to inhibit HIF1α-mediated abrogation of cytosolic iron elevation and neurotoxicity in cultured dopaminergic DAergic cells under conditions of mitochondrial stress (Rajagopalan et al. 2016). These data suggest that ATP13A2 likely plays an important role in the maintenance of both cellular iron and mitochondrial homeostasis within these neurons.
Simialr to ATP13A2, loss in its C. elegans homolog catp-6 has been previously identified as the modifier of human α-synuclein in a RNAi based screen of PD worm models (Hamamichi et al. 2008). Also, recently it was shown that C. elegans catp-6 mutation predisposes worms towards α-synuclein induced sensitivity to various stresses including mitohcondrial stressor paraquat (Cooper et al. 2018). In order to explore the potential mechanistic link between losses in ATP13A2, iron and mitochondrial dysfunction, we studied C. elegans catp-6 mutant. Our results show that, in conjunction with reductions in motility, catp-6 mutant worms exhibit a reduction in iron regulation as indicated by alterated expression of several iron-metabolizing genes and increased sensitivity to exogenous iron exposure. Data showing catp-6 mutants have defective lysosmal function suggest that alteration in iron homeostais in these mutant may result due to loss of chelatable iron from the lysosmes. Additionally, we also show defective mitochondrial function in these mutants as demonstrated by reduced mitochondrial membrane potential and maximal respiration rate as well as increased sensitivity to the mitochondrial toxin, rotenone. The latter was found to be rescued via either iron chelation or induction of mitophagy (Figure 4). The ability of iron chelation to prevent rotenone sensitivity is of particular importance as it suggests that mitochondrial dysfunction in the catp-6 mutants is causally linked to iron elevation. We hypothesize (Figure 5) that defective mitochondrial function in catp-6/ATP13A2 mutants may be due to direct uptake of iron into mitochondria by the mitochondrial calcium uniporter (MCU-1), as has previously been reported in cultured hepatocytes (Uchiyama et al. 2008). Alternatively, role of catp-6 in maintaining lysosmal function may effect the sorting nexin 3(Snx3)-retromer-mediated recycling of iron transporters and thus the iron homeostasis. This function of Snx3-retromer-mediated recycling of iron transporters is inhibited in dopamine neurons expressing α-synuclein which leads to neurodegeneration (Patel et al. 2018). This is in part why iron chelators has also been found effective in preventing α-synculein induced neurodegenration (Patel et al. 2018). The mitophagy inducer urolithin A is safe for humans and was reported to elicit improved mitochondrial health in a recent Phase-I clinical trial (Andreux et al. 2019). Interestingly, urolithin A is also a human gut metabolite which results from the transformation of ellagic acid (high in pomegranates) by the gut bacteria (Landete 2011). This raises the exciting possibility of the use of dietary interventions to treat ATP13A2 loss-of-function in patients. Our findings, together with recent reports documenting a role for catp-6 in maintaining Ca2+ (Narayanaswamy et al. 2019) and Zn2+ (Baesler et al. 2019) homeostasis also highlights the potential of using catp-6 mutants as a platform for drug discovery and the identification of new genes regulating metal-induced toxicity.
Figure 5: Schematic overview of the results.
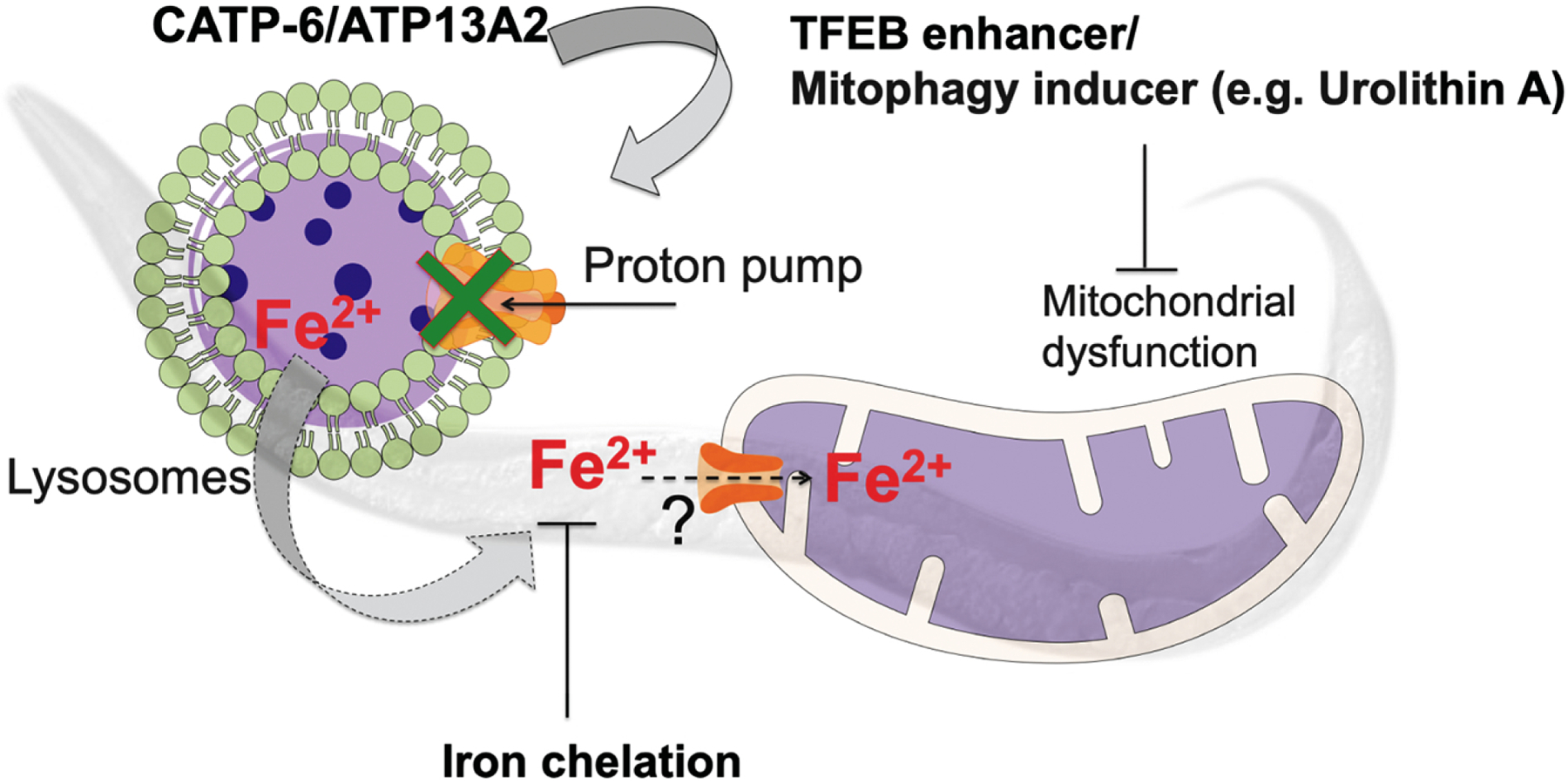
The catp-6/ATP13A2 gene encodes a lysosomal transmembrane type 5 ATPase pump that helps to maintain autophagy and lysosomal function. Loss in CATP-6 function results in dysregulated iron metabolism effecting mitochondrial function and rendering it sensitive to mitochondrial stress. Iron chelation, TFEB enhancement or mitophagy inductions all separately rescue sensitivity to the mitochondrial toxin, rotenone.
Supplementary Material
Supp. Figure 1: (A) CATP-6 shares closest homology with human ATP13A2 protein as identified by STRING database (Szklarczyk et al. 2015). (B) Representative image of egg-lay synchronized populations of N2 and catp-6(ok3473) worms grown at 20°C. Images were taken under stereomicroscope post-48 hours of egg lay. (C) External supplementation of iron is toxic to worms. Survival plot of N2 worms exposed to different doses of ferric ammonium citrate (FAC) or control ammonium citrate. N2 control [13.78 ± 0.44] (median-15), N2 FAC (15 mM) [13.43 ± 0.58] (median-10) ns, N2 FAC (30 mM) [10.94 ± 0.58] (median-12) p<0.0001, N2 FAC (50 mM) [9.65 ± 0.36] (median-9) p<0.0001. [Mean average lifespan ± SEM], n=2, p-value calculated using Mantel-Cox Log rank test. (D) External supplementation of zinc is toxic to worms. Survival plot of N2 worms exposed to 500 μM ZnSO4 or water control. N2 control [14.75 ± 0.59], N2 ZnSO4 (500 μM) [13.42 ± 0.43], catp-6(ok3473) control [12.94 ± 0.45], catp-6(ok3473) ZnSO4 (500 μM) [10.17 ± 0.46]. [Mean average lifespan ± SEM]. n=2, p-value calculated using Mantel-Cox Log rank test.
Highlights.
Loss in C. elegans catp-6 disrupts autophagy and lysosmal function
CATP-6 mediates iron homesotais and mitochondrial function
Iron chelation and mitophagy induction as potential therapeutic approach to resuce mitochondrial defects in catp-6/ATP13A2 mutants
Acknowledgments
This project was funded by NIH R21 grant NS0957 awarded to JKA. MC is supported by a postdoctoral fellowship from the Larry L. Hillblom Foundation. We thank Anand Rane and Dr. Dipa Bhaumik for help with laboratory resources. Strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andreux PA, Blanco-Bose W, Ryu D, Burdet F, Ibberson M et al. , 2019. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nature Metabolism 1: 595–603. [DOI] [PubMed] [Google Scholar]
- Baesler J, Kopp JF, Pohl G, Aschner M, Haase H et al. , 2019. Zn homeostasis in genetic models of Parkinson’s disease in Caenorhabditis elegans. J Trace Elem Med Biol 55: 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens MI, Bruggemann N, Chana P, Venegas P, Kagi M et al. , 2010. Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov Disord 25: 1929–1937. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ et al. , 2003. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299: 256–259. [DOI] [PubMed] [Google Scholar]
- Chamoli M, Chinta S, Schmidt M, Lithgow GJ and Andersen J, 2017. ROLE OF PHARMACOLOGICALLY INDUCED-TFEB IN AGING AND AGE-RELATED NEURODEGENERATION. Innovation in Aging 1: 1224–1224. [Google Scholar]
- Cooper JF, Spielbauer KK, Senchuk MM, Nadarajan S, Colaiacovo MP et al. , 2018. alpha-synuclein expression from a single copy transgene increases sensitivity to stress and accelerates neuronal loss in genetic models of Parkinson’s disease. Exp Neurol 310: 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH et al. , 2012. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 109: 9611–9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S et al. , 2009. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet 41: 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer SN, Metcalf JL, Wang Y and Ohh M, 2012. The updated biology of hypoxia-inducible factor. EMBO J 31: 2448–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamichi S, Rivas RN, Knight AL, Cao S, Caldwell KA et al. , 2008. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc Natl Acad Sci U S A 105: 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampshire DJ, Roberts E, Crow Y, Bond J, Mubaidin A et al. , 2001. Kufor-Rakeb syndrome, pallido-pyramidal degeneration with supranuclear upgaze paresis and dementia, maps to 1p36. J Med Genet 38: 680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y et al. , 1998. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608. [DOI] [PubMed] [Google Scholar]
- Klang IM, Schilling B, Sorensen DJ, Sahu AK, Kapahi P et al. , 2014. Iron promotes protein insolubility and aging in C. elegans. Aging (Albany NY) 6: 975–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz T, Eaton JW and Brunk UT, 2011. The role of lysosomes in iron metabolism and recycling. Int J Biochem Cell Biol 43: 1686–1697. [DOI] [PubMed] [Google Scholar]
- Kurz T, Gustafsson B and Brunk UT, 2006. Intralysosomal iron chelation protects against oxidative stress-induced cellular damage. FEBS J 273: 3106–3117. [DOI] [PubMed] [Google Scholar]
- Kurz T, Leake A, Von Zglinicki T and Brunk UT, 2004. Relocalized redox-active lysosomal iron is an important mediator of oxidative-stress-induced DNA damage. Biochem J 378: 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz T, Terman A, Gustafsson B and Brunk UT, 2008. Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell Biol 129: 389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landete JM, 2011. Ellagitannins, ellagic acid and their derived metabolites: A review about source, metabolism, functions and health. Food research international 2011 v.44 no.5: pp. 1150–1160. [Google Scholar]
- Lapierre LR, De Magalhaes Filho CD, McQuary PR, Chu CC, Visvikis O et al. , 2013. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun 4: 2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KE, Cottle L, Gysbers AM, Cooper AA and Halliday GM, 2013. ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol Commun 1: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najim al-Din AS, Wriekat A, Mubaidin A, Dasouki M and Hiari M, 1994. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol Scand 89: 347–352. [DOI] [PubMed] [Google Scholar]
- Nakayama K, 2009. Cellular signal transduction of the hypoxia response. J Biochem 146: 757–765. [DOI] [PubMed] [Google Scholar]
- Narayanaswamy N, Chakraborty K, Saminathan A, Zeichner E, Leung K et al. , 2019. A pH-correctable, DNA-based fluorescent reporter for organellar calcium. Nat Methods 16: 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng LF, and Gruber J, 2019. Measurement of Respiration Rate in Live Caenorhabditis elegans. Bio-protocol 9: e3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D, Xu C, Nagarajan S, Liu Z, Hemphill WO et al. , 2018. Alpha-synuclein inhibits Snx3-retromer-mediated retrograde recycling of iron transporters in S. cerevisiae and C. elegans models of Parkinson’s disease. Hum Mol Genet 27: 1514–1532. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Rane A, Chinta SJ and Andersen JK, 2016. Regulation of ATP13A2 via PHD2-HIF1alpha Signaling Is Critical for Cellular Iron Homeostasis: Implications for Parkinson’s Disease. J Neurosci 36: 1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D et al. , 2006. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38: 1184–1191. [DOI] [PubMed] [Google Scholar]
- Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N et al. , 2016. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med 22: 879–888. [DOI] [PubMed] [Google Scholar]
- Schmidt K, Wolfe DM, Stiller B and Pearce DA, 2009. Cd2+, Mn2+, Ni2+ and Se2+ toxicity to Saccharomyces cerevisiae lacking YPK9p the orthologue of human ATP13A2. Biochem Biophys Res Commun 383: 198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S et al. , 2005. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem 280: 41732–41743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T, 2006. Maintenance of C. elegans. WormBook: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D et al. , 2015. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43: D447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman A, and Kurz T, 2013. Lysosomal iron, iron chelation, and cell death. Antioxid Redox Signal 18: 888–898. [DOI] [PubMed] [Google Scholar]
- Uchiyama A, Kim JS, Kon K, Jaeschke H, Ikejima K et al. , 2008. Translocation of iron from lysosomes into mitochondria is a key event during oxidative stress-induced hepatocellular injury. Hepatology 48: 1644–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usenovic M, Tresse E, Mazzulli JR, Taylor JP and Krainc D, 2012. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci 32: 4240–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K et al. , 2004. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304: 1158–1160. [DOI] [PubMed] [Google Scholar]
- van den Ecker D, Hoffmann M, Muting G, Maglioni S, Herebian D et al. , 2015. Caenorhabditis elegans ATAD-3 modulates mitochondrial iron and heme homeostasis. Biochem Biophys Res Commun 467: 389–394. [DOI] [PubMed] [Google Scholar]
- Williams DR, Hadeed A, al-Din AS, Wreikat AL and Lees AJ, 2005. Kufor Rakeb disease: autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov Disord 20: 1264–1271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp. Figure 1: (A) CATP-6 shares closest homology with human ATP13A2 protein as identified by STRING database (Szklarczyk et al. 2015). (B) Representative image of egg-lay synchronized populations of N2 and catp-6(ok3473) worms grown at 20°C. Images were taken under stereomicroscope post-48 hours of egg lay. (C) External supplementation of iron is toxic to worms. Survival plot of N2 worms exposed to different doses of ferric ammonium citrate (FAC) or control ammonium citrate. N2 control [13.78 ± 0.44] (median-15), N2 FAC (15 mM) [13.43 ± 0.58] (median-10) ns, N2 FAC (30 mM) [10.94 ± 0.58] (median-12) p<0.0001, N2 FAC (50 mM) [9.65 ± 0.36] (median-9) p<0.0001. [Mean average lifespan ± SEM], n=2, p-value calculated using Mantel-Cox Log rank test. (D) External supplementation of zinc is toxic to worms. Survival plot of N2 worms exposed to 500 μM ZnSO4 or water control. N2 control [14.75 ± 0.59], N2 ZnSO4 (500 μM) [13.42 ± 0.43], catp-6(ok3473) control [12.94 ± 0.45], catp-6(ok3473) ZnSO4 (500 μM) [10.17 ± 0.46]. [Mean average lifespan ± SEM]. n=2, p-value calculated using Mantel-Cox Log rank test.