

Abstract
The focus on amyloid plaques and neurofibrillary tangles has yielded no Alzheimer’s disease (AD) modifying treatments in the past several decades, despite successful studies in preclinical mouse models. This inconsistency has caused a renewed focus on improving the fidelity and reliability of AD mouse models, with disparate views on how this can be accomplished. However, the interactive effects of the universal biological variables of AD that include age, APOE genotype, and sex are often overlooked. Age is the greatest risk factor for AD, while the ε4 allele of the human APOE gene, encoding apolipoprotein E, is the greatest genetic risk factor. Sex is the final universal biological variable of AD, as females develop AD at almost twice the rate of males and, importantly, female sex exacerbates the effects of APOE4 on AD risk and rate of cognitive decline. Therefore, this review evaluates the importance of context for understanding the role of APOE in preclinical mouse models. Specifically, we detail how human AD pathology is mirrored in current transgenic mouse models (“What”) and describe the critical need for introducing human APOE into these mouse models (“Who”). We next outline different methods for introducing human APOE into mice (“How”) and highlight efforts to develop temporally defined and location-specific human apoE expression models (“When” and “Where”). We conclude with the importance of choosing the human APOE mouse model relevant to the question being addressed, using the selection of transgenic models for testing apoE-targeted therapeutics as an example (“Why”).
Keywords: Alzheimer’s Disease (AD), familial AD transgenic mice (FAD-Tg), apolipoprotein E, sex and AD risk, APOE4 and AD risk, EFAD-Tg mouse model, APOE-TR mouse model, apoE as a therapeutic target
I. The “What” of APOE Transgenic AD Mouse Models: The Development of AD Pathology and the Effect of Universal Biological Variables
Alzheimer’s Disease (AD) is a complex, uniquely human condition that has eluded understanding and effective treatment for over a century. Nevertheless, dozens of transgenic (Tg) mouse models that recapitulate specific aspects of AD pathogenesis enable mechanistic interrogation and hypothesis testing impossible to achieve in human patients. In this first section, we describe the major pathological hallmarks of AD, review Tg mouse models that reproduce AD-like pathology, and introduce the universal biological variables of AD, specifically age, APOE and sex. These three variables are considered universal in that all people age, and all people have a biological sex and two APOE alleles, as compared to rare risk-enhancing mutations or to modifiable risk factors that impact only a certain proportion of the population.
Pathological Hallmarks of AD
Amyloid is a common quaternary protein structure consisting of parallel β-pleated sheets, providing the most condensed storage form for overabundant proteins (Chiti and Dobson, 2017). Unlike other quaternary structures, there is no amino acid sequence that defines amyloidogenic proteins; rather, it is the most condensed storage for any overproduced protein. In AD, these two overproduced proteins are amyloid-β (Aβ) peptide and microtubule associated protein tau (MAPT) that aggregate into amyloid structures termed amyloid plaques and neurofibrillary tangles (NFT). These two structures are the pathological hallmarks required for a definitive postmortem diagnosis of AD (Serrano-Pozo et al., 2011).
Because amyloid plaques are observed before NFT in humans, early hypotheses described amyloid plaques as precipitants to tangle formation, which then produce the progressive synaptic loss, neuronal atrophy, and cognitive decline that characterize the disease (Bloom, 2014, Selkoe and Hardy, 2016, Beyreuther and Masters, 1991). Evidence has largely disproven this “amyloid hypothesis,” with two major findings contradicting a direct connection between plaque load and cognitive deficits. First, dozens of trials with amyloid-targeting therapeutic agents have failed to produce any clinical benefit, despite pronounced reductions in amyloid pathology (recent conflicting Phase 3 results with aducanumab notwithstanding) (Liu et al., 2019, Mehta et al., 2017). Second, a significant subset of the elderly displays extensive amyloid plaque deposition yet age without signs of cognitive impairment (Bennett et al., 2006). Conversely, familial AD (FAD) is caused exclusively by mutations that enhance proteolytic processing of amyloid precursor protein (APP) to amyloid-β (Aβ), primarily the Aβ42 isoform, indicating a critical role for the Aβ peptide in the disease process (as reviewed by Van Cauwenberghe et al., 2016). The key research focus now resides on the soluble oligomeric Aβ (oAβ) species – the formation, toxicity, and persistence of which is influenced by numerous other pathologic factors beyond the scope of this review (e.g. inflammation, metabolic perturbation, and lipid homeostasis).
With NFT comprising the other pathological hallmark of AD, the role of tau has also been investigated extensively in preclinical studies. MAPT is a large, 134kb gene that may contain up to 16 exons in its mature RNA (Caillet-Boudin et al., 2015, Sergeant et al., 2005). Alternative splicing and other modifications result in the expression of 6 unique tau isoforms, which differ in that they possess either three or four copies of a C-terminus repeated region (3R or 4R) and anywhere from zero to two N-terminal inserts (N0, N1, N2) (Buee et al., 2000). These isoforms may be abnormally phosphorylated under various pathogenic conditions, thereby increasing the propensity of the protein to aggregate. Tau aggregates have numerous toxic effects on axonal transport, synaptic function, and neuronal survival, which may occur in both an amyloid-dependent and -independent manner (Ittner and Gotz, 2011, Chong et al., 2018).
Investigations into the aggregation cascades of Aβ and hyperphosphorylated tau have identified two distinct pathways (Figure 1) (Crespo et al., 2016, Powers and Powers, 2008, Necula et al., 2007, Ehrnhoefer et al., 2008, Perez et al., 2019, Uversky, 2010, Kjaergaard et al., 2018). In the “on pathway,” the given protein monomers aggregate to form oligomers, followed by continually larger structures (protofibrils, fibrils, and coiled coils for Aβ; paired helical filaments for tau), ultimately culminating in the stacking of β-pleated sheets in parallel to form the final amyloid structure (plaques or tangles). Although illustrated as discrete events, this pathway may more accurately be considered a series of transient intermediates. Alternatively, in the “off pathway,” the protein monomers combine to form soluble oligomers as the stable end-product. Ongoing research continues to debate the details (and existence) of these two pathways; however, this framework provides an explanation for amyloid-positive, cognitively normal individuals. The formation of persistent Aβ and tau oligomers through the “off” pathway – versus their transient, intermediate nature in the “on” pathway – also better explains the critical pathogenic role and marked toxicity of both oAβ and tau oligomers, which have been the subject of countless studies reviewed multiple times elsewhere (Cline et al., 2018, Sakono and Zako, 2010, Benilova et al., 2012, Lasagna-Reeves et al., 2011, Shafiei et al., 2017). While beyond the scope of this review, this framework has also been increasingly applied to other neurodegenerative conditions characterized by abnormal protein misfolding and accumulation, such as α-synuclein in Parkinson’s disease, in which the soluble oligomeric form may actually be the proximal neurotoxin (Bengoa-Vergniory et al., 2017).
Figure 1. Aβ and tau aggregation.
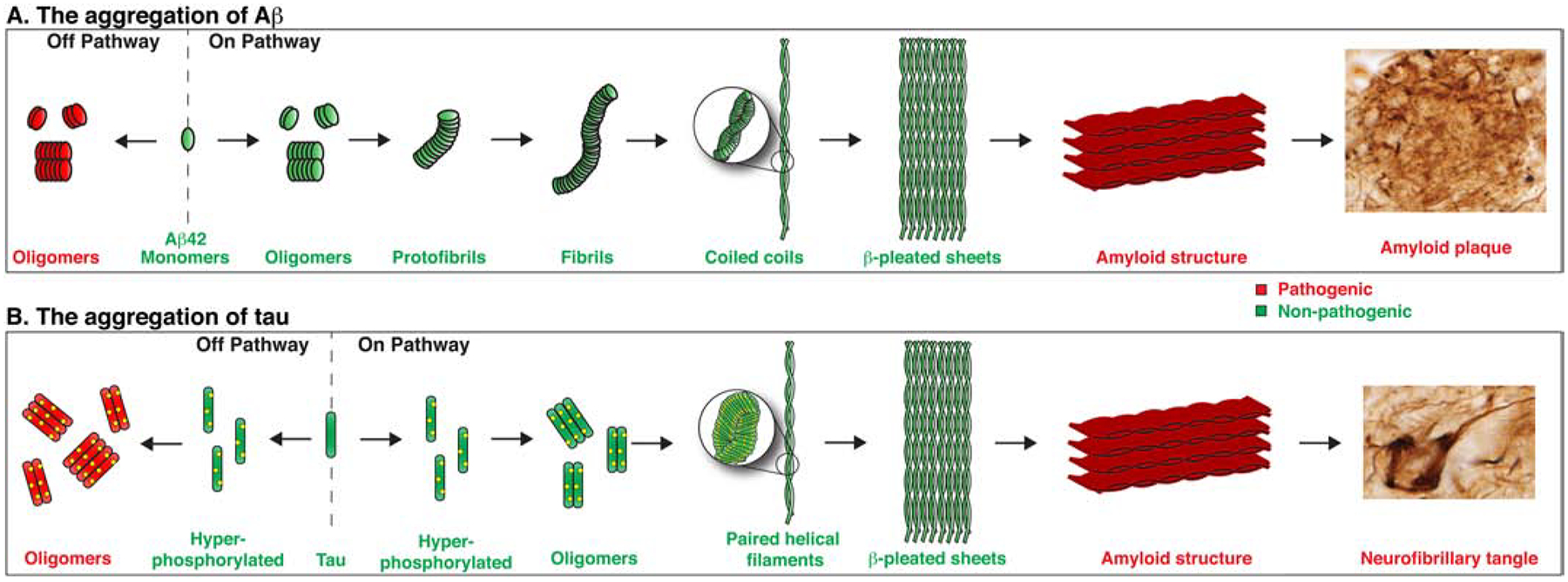
Two pathways for Aβ and tau monomer aggregation. In the on pathway, protein monomers sequentially aggregate to form larger structures culminating in an amyloid structure, either amyloid plaques for Aβ or NFT for tau. In the off pathway, protein monomers aggregate into oligomers – a persistent, stable, and pathogenic conformation. The upper panel was adapted from Pimplikar (2009); amyloid plaque and NFT staining adapted from Winblad and colleagues (2016).
Amyloid/Aβ Models
The critical role of Aβ in AD led to the development of Tg mice carrying FAD mutations (FAD-Tg) that increase the proteolytic processing of APP to Aβ42 or increase the Aβ42/Aβ40 ratio. Under neuron-specific promoters, mutations in human (h-) genes encoding APP, presenilin-1 or presenilin-2 genes (APP, PS1, and PS2, respectively) result in AD-like amyloid pathology in FAD-Tg mice (Oakley et al., 2006, Games et al., 1995, Duff et al., 1996, Holcomb et al., 1998, Hsiao et al., 1996, Radde et al., 2006, Mucke et al., 2000, Jankowsky et al., 2001). These mice are a powerful tool for the study of specific features of AD pathology and as preclinical AD models. A comparison of the regional development of Aβ pathology in brain of FAD-Tg mice and human AD patients is provided in Figure 2A. To aid in orientation, four particular regions (frontal cortex, hippocampus, cerebellum, and brain stem) common to both species, plus mouse olfactory bulb as an obvious difference, are labeled in the leftmost panel. Aβ deposition in human AD patients begins in frontal and temporal cortex before progressing to hippocampus and occipital cortex. In the final stages, amyloid deposition is saturated in the frontal and temporal cortices (Masters et al, 2015). Similarly, FAD-Tg mice exhibit plaque deposition beginning in the subiculum and deep layers of the frontal cortex, followed by a spread of deposits throughout the cortex, and into the hippocampus, and thalamus (Oakley et al., 2006, Tai et al., 2017, Youmans et al., 2012). Comparable to human AD, the most severe pathology is in the frontal cortex and subiculum. FAD-Tg models do sacrifice physiological relevance by overexpressing multiple mutant transgenes via heterologous promoters, resulting in Aβ accumulation occurring in early or mid-adulthood, compromising the impact of aging.
Figure 2. Progression of Aβ and tau pathology in humans and Tg mice.
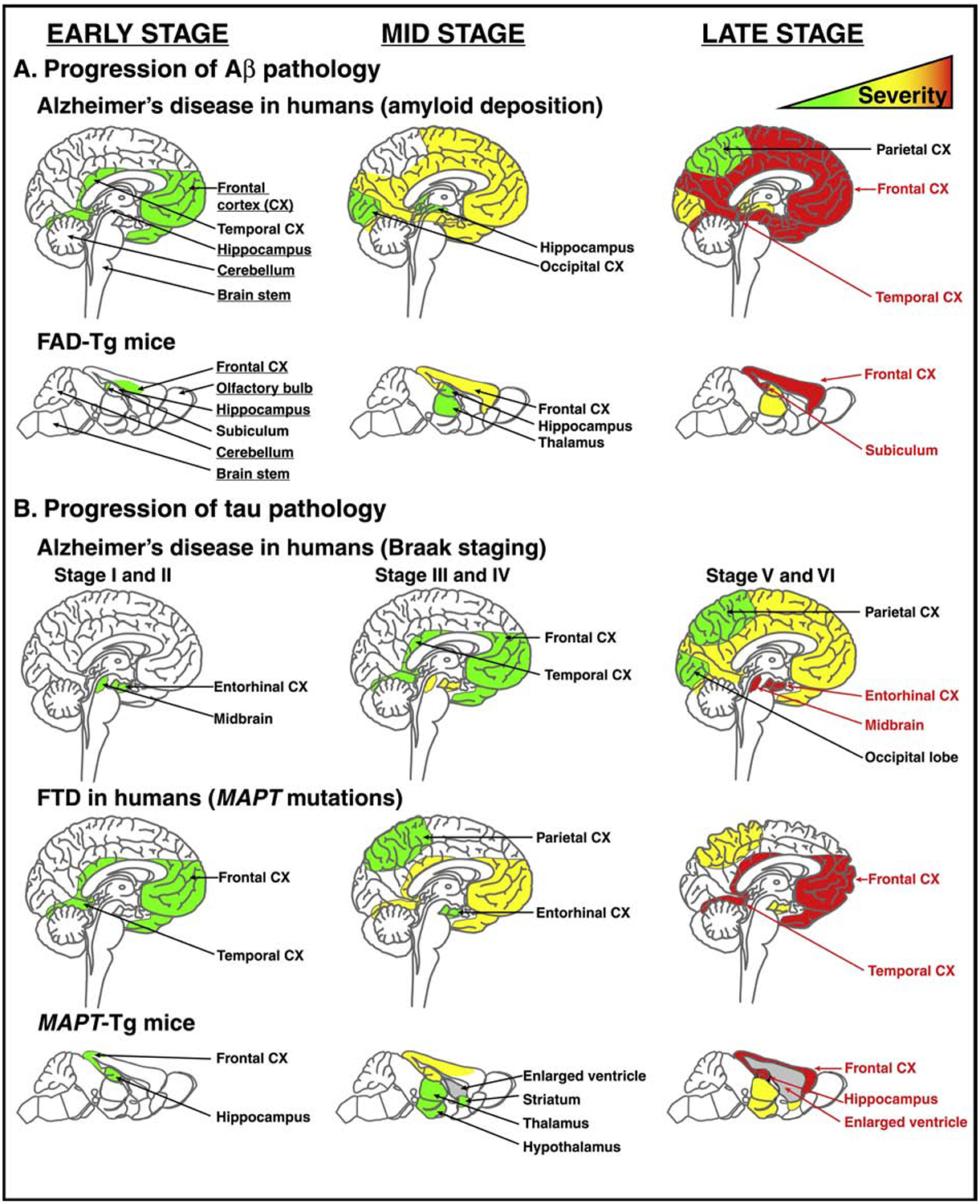
A) Comparison of Aβ pathology progression between AD human patients and FAD-Tg mouse models. B) Comparison of tau pathology progression among AD human patients (Braak staging), FTD human patients, and MAPT-Tg mouse models. The frontal cortex, hippocampus, cerebellum, and brainstem in both human and mouse brain are labeled in leftmost panel in A) to aid in orientation, with additional labels provided to indicate regions affected by pathological progression. Human Aβ and Braak staging panels were adapted from Masters and colleagues (2015). Human FTD was adapted from Wszolek and colleagues (2006). MAPT-Tg mouse pathology was adapted from Sahara and colleagues (2013).
More recent models have been engineered as humanized APP targeted replacement (TR) mice, in which the (mutated or normal) human Aβ coding domain is introduced and controlled by the mouse promoter and enhancer regions. In these mice, more physiologically relevant expression levels of APP mutants are still sufficient to induce AD-like pathology and cognitive deficits, with a modest worsening of pathology, but not cognition, with female sex (Saito et al., 2014, Masuda et al., 2016, Sakakibara et al., 2018, Pervolaraki et al., 2019). Given their relatively recent development, these APP-TR mice require further analysis to ensure they consistently recapitulate various features of amyloid pathology – such as age of onset, regional progression, and morphology – that have been extensively characterized in FAD-Tg mice.
Tau Models
Given the toxic effects of tau and following failures of Aβ-targeting therapies tested in FAD-Tg mouse models, mouse models of tau pathology have become more widespread as an alternative or complementary approach. Mouse (m-) tau is predominantly expressed as the three 4R isoforms and does not naturally aggregate into stable NFTs (Janke et al., 1999, Kampers et al., 1999). To account for these features, Tg mice that incorporate h-MAPT (MAPT-Tg) have been developed. Because h-tau expression is complex, MAPT-Tg mice have utilized different isoforms of h-tau with or without various MAPT mutations to induce tau protein aggregation and NFT formation, including P301S, P301L and ΔK280 (Roberson, 2012, Mocanu et al., 2008, Ramsden et al., 2005, Yoshiyama et al., 2007; as summarized in Koss et al., 2016, Jankowsky and Zheng, 2017). This extensive variation means that a diverse library of MAPT-Tg mouse models, which develop tau pathology to varying degrees over unique time courses, is available to study tau-mediated neurodegeneration. However, the complexity of h-tau makes standardization and comparison of MAPT-Tg mouse models challenging.
Importantly, while mutations in APP or PS1 cause FAD in humans, mutations in MAPT cause various forms of frontotemporal dementia (FTD) (Poorkaj et al., 1998, Spillantini and Goedert, 1998, Hutton et al., 1998, Irwin, 2016). Notably, FTD demonstrates a pattern of tauopathy distinct from the Braak staging characteristic of tauopathy in AD (Figure 2B). While tauopathy associated with AD in humans begins in the entorhinal cortex and midbrain (Braak and Braak, 1991, Hyman et al., 2012), FTD, as its name suggests, first impacts the frontal and temporal cortices with the entorhinal cortex only affected later in the disease (Irwin, 2016). As reviewed recently, MAPT-Tg mice more closely recapitulate a human FTD pattern than an AD pattern with pathology concentrated in the frontal cortex (Jankowsky and Zheng, 2017). Compared to even the most aggressive FAD-Tg models (e.g. 5xFAD), mutated h-MAPT drives massive neuronal loss in MAPT-Tg mice, resulting in significant enlargement of the ventricles and extensive atrophy of the frontal cortex (Ramsden et al., 2005, Yoshiyama et al., 2007, Jawhar et al., 2012). These unique features and limitations of MAPT-Tg mice should be considered in designing preclinical studies.
Amyloid/Aβ + Tau Models
Given the toxic and poorly understood interaction between tau and amyloid observed in AD, mouse models that combine both pathologies have also been studied (Oddo et al., 2003b, Oddo et al., 2003a, Jackson et al., 2016, Jankowsky and Zheng, 2017, Lippi et al., 2018, Saito et al., 2019). The most well-characterized of these models is the 3xTg mouse, which incorporates h-APP, PS1, and MAPT mutations (Billings et al., 2005, Oddo et al., 2003b, Oddo et al., 2003a). However, in these mice, cognitive deficits appear before any appreciable amyloid or tau pathology, which is the reverse of human AD. Numerous other mice that develop both tau and Aβ pathology via overexpressed FAD- or FTD-causing mutants have been reported (summarized in Lippi et al., 2018). An alternative strategy to insert non-mutated human APP and/or MAPT genes into mouse genome through targeted replacement “humanizes” the mice and provides a more physiologically relevant expression profile of APP or tau proteins, but these mice show mild pathology even at old age (Saito et al., 2019, Andorfer et al., 2003, Yetman et al., 2016). Moreover, despite important advances made in our ability to replicate the pathological hallmarks of AD with these mice, the models referenced in this section fail to adequately consider the universal biological variables of AD, which limits their translatability to human patients. Acting as a “decision guide” for choosing an APOE-Tg mouse model, the following sections of this review detail the progress made in incorporating the h-APOE gene into mouse models of AD and explore the enhanced physiological relevance of these models over their m-APOE-expressing counterparts (Table 1).
Table 1.
Decision guide for choosing APOE-Tg mouse models.
| The transgenic mouse models that mirror human AD pathology. | The introduction of human APOE into the transgenic mouse models. | The different methods of introducing human APOE into mouse models. | The development of mice with location- and time-specific apoE expression. | The importance of h-APOE-Tg mice for testing apoE-targeted therapeutics. |
|---|---|---|---|---|
|
|
|
|
|
Universal Biological Variables
The universal biological variables of AD are age, APOE genotype, and sex. Age is the greatest risk factor for AD, with APOE4 the greatest genetic risk factor. The human APOE gene encodes apolipoprotein E (apoE), with three isoforms: apoE2, apoE3 and apoE4. Sex is the other universal biological variable of AD, as females exhibit an almost two-fold greater lifetime AD risk and greater dementia burden compared to males (Barnes et al., 2005, Andersen et al., 1999, Oveisgharan et al., 2018, Vest and Pike, 2013). Underappreciated until relatively recently was the interaction between APOE4 and sex, with female APOE4 carriers possessing an increased AD risk compared to male APOE4 carriers (Ungar et al., 2014, Altmann et al., 2014, Fleisher et al., 2005, Breitner et al., 1999, Farrer et al., 1997, Martinez et al., 1998, Andersen et al., 1999, Bretsky et al., 1999, Molero et al., 2001, Corder et al., 2004). Few studies with FAD- and/or MAPT-Tg mice have stratified results by sex, but results are consistent with female sex exacerbating the development of AD-like pathology in these mice (Wang et al., 2003, Masuda et al., 2016, Gimenez-Llort et al., 2008, Bhattacharya et al., 2014). However, Tg mouse models that lack APOE4, or studies that use only male mice, produce results that cannot be fully interpreted either by sex, APOE, or the critical interaction found in female APOE4 carriers.
II. The “Who” of APOE Transgenic AD Mouse Models: The Role of Human APOE in AD Pathology
Proper consideration of APOE as a universal biological variable of AD in preclinical mouse studies requires recognition of the significant protein differences between m- and h-apoE. ApoE variation observed across humans is unique to our species, which makes modeling its significant impact on AD risk and pathogenesis challenging. In this section, we first discuss important distinctions between m- and h-apoE. Then, we review the various mouse models that incorporate h-APOE, either on its own in an otherwise normal mouse, or in combination with other Tg mutations or exogenous insults that produce AD-like pathology.
Notably, we do not describe in detail Tg mice that combine h-APOE expression with other late-onset AD risk-enhancing mutations. These models, such as the APOE4/TREM2*R47H mouse, are in early stages of development with limited data reported to date (Sasner, 2019). Moreover, these secondary mutations are quite rare, ranging from allele frequencies of 0.12–0.26% for TREM2*R47H in Caucasian populations to non-existent in Chinese or African-American populations (Guerreiro et al., 2013, Jonsson et al., 2013, Mehrjoo et al., 2015, Jay et al., 2017, Miyashita et al., 2014, Ma et al., 2014, Wang, 2018b, Guo et al., 2018, Jin et al., 2015). Thus, both the reliability and translatability of results gathered in these models are limited. Rather, in this section we discuss better characterized and more informative APOE-Tg mouse models that guide interpretation of AD-like pathology through the universal biological variables of AD.
APOE Transgenic Mice
Mouse vs. Human APOE
Differences between mouse and human apoE are numerous and significant. The human apoE protein exists in three main isoforms, while mice express only one form of apoE. These three human isoforms vary at only two of the 299 amino acids within the protein secondary structure (apoE2: Cys112/Cys158, apoE3: Cys112/Arg158, apoE4: Arg112/Arg158). Importantly, the homology between m- and h-apoE amino acid sequences is only 70% (Rajavashisth et al., 1985). Although m-apoE includes the equivalent of Arg112/Arg158 residues found in h-apoE4, the extent of the coding sequence variation between the two species produces substantial downstream consequences in the tertiary structure, as m-apoE functions as a single-domain protein, while h-apoE forms two distinct N- and C-terminus domains (Nguyen et al., 2014). Numerous studies comparing m- and h-apoE have identified significant differences, ranging from their basic function in lipid binding and transport to their effects on AD pathology (Weisgraber, 1994, Nguyen et al., 2014, Liao et al., 2015, Hudry et al., 2013, Fagan et al., 2002, Fagan et al., 1999). With regard to AD pathology, in spite of the negative consequences associated with the APOE4 allele, replacement of m-apoE with h-apoE critically delays AD pathology and decline in memory performance, while also introducing an isoform-specificity that mirrors human AD patients (APOE4 earlier and more severe; APOE2 later and more mild) in FAD-Tg mouse models (Balu et al., 2019, Bales et al., 1997, Bales et al., 2009, Youmans et al., 2012, Tai et al., 2011, Tai et al., 2017).
APOE Targeted Replacement
Mice expressing h-apoE isoforms have become a critical tool to study AD pathogenesis. Even without any additional FAD mutations, these mice exhibit isoform-specific differences in lipid physiology and synaptic function – both critical aspects of AD pathology. APOE-TR mice, in which the m-APOE coding sequence is replaced by that of a h-APOE allele, display alterations in synaptic number and structure, network connectivity, and behavior based on the specific apoE isoform expressed (Zhu et al., 2012, Sun et al., 2017, Neustadtl et al., 2017, Dumanis et al., 2013, Gillespie et al., 2016, Dumanis et al., 2009, Ji et al., 2003, Wang et al., 2005, Bour et al., 2008, Grootendorst et al., 2005, Koutseff et al., 2014). Some of these studies have also observed interaction between APOE genotype, sex, and aging, showing the ability of APOE-TR mice to reproduce the critical interaction of the universal biological variables of AD (Andrews-Zwilling et al., 2010, Leung et al., 2012). At a more fundamental level, the apoE4 protein is found at lower concentrations than apoE3 in these mice, and, in apoE3/E4 heterozygotes, the apoE4 isoform contributes to reduced apoE levels (Ramaswamy et al., 2005, Riddell et al., 2008a). This reduced presence of apoE4 may be due to its relative lack of structural stability and a greater propensity to undergo proteolytic degradation compared to apoE3 (Tamboli et al., 2014, Rohn, 2013). Functionally, apoE4 exhibits decreased affinity for lipids such as cholesterol compared to apoE3, with numerous studies demonstrating that apoE4-containing lipoproteins in the brain are smaller compared to apoE3-containing lipoproteins (Huang and Mahley, 2014, Weisgraber, 1994, Tai et al., 2014b, Hu et al., 2015, Heinsinger et al., 2016, Samieri et al., 2013). Reduced protein levels combined with hypolipidation of apoE4 contribute to the many pathological interactions described in the following sections and form the basis of several therapeutic strategies detailed at the end of this review. As a result, incorporating h-APOE into mouse models is critical for studying AD. However, one major limitation is that, although APOE2 is considered neuroprotective in humans, 100% of homozygous APOE2 mice have type III hyperlipoproteinemia, compared to only 10% of humans homozygous for APOE2 (Sullivan et al., 1998, Mahley et al., 1999, Koopal et al., 2017, Corsetti et al., 2018). Thus, APOE2-TR mice should be used with caution when compared with APOE3- or APOE4-TR mice, as the consequences of this condition, which is characterized by significantly increased plasma levels of chylomicron and VLDL remnants, for AD remain unknown.
Amyloid/Aβ Pathology and APOE
Given its wide-ranging toxic effects, reliable replication of oAβ pathophysiology is a necessary feature of preclinical AD models. This reproduction requires consideration of the differential effects of apoE isoforms on the formation and clearance of soluble oAβ. Across studies, Aβ deposition, soluble oAβ levels, and amyloid plaque pathology are consistently elevated in human AD brain samples in the presence of apoE4 compared to other isoforms (Tai et al., 2013, Hashimoto et al., 2012, Koffie et al., 2012, Hoglund et al., 2017). Also, oAβ efflux was diminished across engineered human vessels with apoE4 compared to apoE3 (Robert et al., 2017). The presence of any h-apoE isoform accelerates proteolytic degradation of soluble Aβ by microglia, but this effect is weaker with apoE4-containing particles compared to apoE3 and apoE2 (Jiang et al., 2008).
H-apoE-expressing FAD mouse models (FAD/APOE-Tg) reproduce this apoE isoform-dependent variation in Aβ pathology. In Tg2576, APP/PS1, PDAPP, and 5xFAD mice, genetic replacement of m-APOE with one of the three human alleles introduced a delayed isoform-specific effect on oAβ levels, Aβ deposition, and amyloid plaque pathology in the brain, with levels highest in apoE4-expressing mice (Bales et al., 2009, Pankiewicz et al., 2014, Youmans et al., 2012, Tai et al., 2013, Castellano et al., 2011, Pankiewicz et al., 2017, Kim et al., 2011, Fryer et al., 2005, Holtzman et al., 2000, Tai et al., 2017). Viral-mediated expression of human apoE in PDAPP, APP/PS1, and Tg2576 mice also induced an isoform-dependent effect on Aβ pathology, again highest in apoE4-expressing mice (Hudry et al., 2013, Dodart et al., 2005). Importantly, studies in EFAD mice, which overexpress Aβ via 5xFAD mutations and express h-apoE3 (E3FAD) or h-apoE4 (E4FAD), reproduce the interactive effects of APOE4 and female sex on AD pathology. These interactive effects include increased Aβ pathology, greater cognitive deficits, decreased microglial interaction with plaques and lower microglial TREM2 expression, and increased cerebrovascular pathology in female APOE4 carriers compared to male APOE4 carriers (Tai et al., 2017, Stephen et al., 2019, Thomas et al., 2016, Balu et al., 2019). Moreover, with EFAD mice, the interactive effects of age, APOE genotype, and sex on survival rates were examined with female E4FADs exhibiting the shortest lifespan and male E3FADs the longest, with male E4FADs and female E3FADs in between (Balu et al., 2019). These findings demonstrate the importance of FAD/APOETg mice in modeling the apoE/Aβ interaction, which cannot be achieved with mice expressing their natural apoE protein.
Tau Pathology and APOE
Compared to the APOE4/Aβ interaction, the correlation between tau pathology and APOE4 is relatively poorly established in human patients. One recent study showed that apoE4 reduced the age of onset for FTD patients, independent of Aβ pathology (Koriath et al., 2019), while data from Rush University’s longitudinal human cohorts suggest that apoE isoforms only modulate tau pathology in the presence of Aβ pathology (Farfel et al., 2016). Neither of these studies stratified participants by sex; thus, these conflicting results may be the product of our limited understanding of the effects of the universal biological variables of AD on tau pathology.
In attempts to further elucidate the relationship between APOE genotype and tau pathology, MAPT-Tg mouse models that express h-APOE (MAPT/APOE-Tg) have been developed. In a key study, the Holtzman group demonstrated that, compared to P301STg mice crossed with APOE3- or APOE2-TR mice, P301S/APOE4-TR mice demonstrated increased tau levels, inflammation, and brain atrophy (Shi et al., 2017). However, a follow-up report from the Bu group employing viral-mediated P301L MAPT expression in APOE-TR mice found no exacerbation of tau pathology, inflammation, or cognitive deficits with apoE4 (Zhao et al., 2018). In fact, this second study observed exacerbation of this pathology in the presence of apoE2. However, the complete penetrance of Type III hyperlipoproteinemia in APOE2-TR mice as detailed above represents an important caveat for interpreting results from this model. Moreover, these two studies utilized different MAPT mutants (P301S vs P301L) and different expression methods (Tg vs. viral). Further study of MAPT/APOE-Tg mice is certainly warranted in order to better define the relationship between h-apoE isoforms and tau pathology, especially given the observations that late-stage AD may become increasingly amyloid-independent in spite of continued accumulation of pathology (Hyman, 2011). As noted in Section I, investigators must pay particular attention to whether the MAPT-Tg mutation employed more closely models human AD or FTD, although the clinical data described at the beginning of this subsection (Koriath et al., 2019, Farfel et al., 2016) suggest that the relationship of both pathologies with APOE status is worthy of further investigation.
Amyloid/Aβ + Tau Pathology and APOE
Recent work suggests that the presence of amyloid plaques and phosphorylated tau synergistically promote neurodegeneration in AD patients (Pascoal et al., 2017a, Pascoal et al., 2017b, Fortea et al., 2014, Desikan et al., 2012). However, information on the interaction between the two proteins is severely limited in the context of APOE genotype and sex. Although human cohorts are in majority APOE4 carriers and females, many studies do not stratify their human populations by APOE genotype and sex. Therefore, models expressing h-apoE in combination with Aβ and tau pathologies provide greater insight into their interactions.
To study the effects of h-apoE on the interaction between Aβ and tau pathology, a cross between APOE4-TR and 3xTg mice was developed (3xTg/APOE-TR) (Oddo et al., 2009). This initial study noted that h-apoE4 delayed both Aβ and tau pathology compared to m-apoE. Further studies observed that female 3xTg/APOE4-TR exhibited greater Aβ pathology compared to male 3xTg/APOE4-TR and female 3xTg, demonstrating a synergistic interaction between APOE4 genotype and female sex (Hou et al., 2015). While analysis of sex differences was an important inclusion, the comparison of h-apoE4 to m-apoE (rather than to other h-apoE isoforms) was a major limitation of these studies. When a full h-apoE isoform comparison was performed, greater tau pathology was observed in 3xTg/APOE4-TR compared to 3xTg/APOE3-TR or 3xTg/APOE2-TR mice (Bennett et al., 2013). However, because the majority of that study focused on acute effects of traumatic brain injury, a longitudinal analysis of 3xTg/APOE-TR mice on a wider range of AD pathological markers is still needed to further characterize this model.
Modifiable Risk Factors and APOE
Amyloid plaques and NFTs together constitute the classic AD pathology, but the vast majority of patients with dementia, including those with a clinical AD diagnosis, do not develop these pathological hallmarks on an otherwise healthy background. This so-called “mixed pathology” is actually the most commonly observed phenotype in demented patients, and, in particular, cerebrovascular disease has been identified as a major contributor to dementia (Bennett, 2017, Matthews et al., 2009). Another study estimated that roughly 30% of the AD burden could be attributed to modifiable risk factors, including hypertension, diabetes mellitus, smoking, obesity, and physical inactivity (Norton et al., 2014). APOE4 allele increases the risk for cardiovascular disease and diabetes and correlates with increased plasma low-density lipoprotein cholesterol levels (Bennet et al., 2007, Xu et al., 2016, El-Lebedy et al., 2016). Synergistic effects have also been observed between APOE4 and these modifiable factors in AD risk (Peila et al., 2002). Therefore, apoE4 could be considered to act as a “first hit” primer for the central nervous system (CNS) to be further compromised by a “second hit” (e.g. traumatic brain injury (TBI), diabetes, vascular disease, etc.), such that apoE4 exacerbates the effects of the modifiable risk factor on brain pathology (Mahley et al., 2006).
Associations between modifiable risk factors and APOE genotype are reproduced in APOE-TR mice. APOE4-TR mice that are genetically obese or placed on a high-fat diet (HFD) – two models of diabetes/dyslipidemia – show greater disturbances in glucose and lipid metabolism (Arbones-Mainar et al., 2016, Johnson et al., 2017, Arbones-Mainar et al., 2008). APOE4-TR mice on HFD performed worse in Morris water maze testing and had less cerebral blood flow than APOE3-TR mice. Moreover, EFAD mice on HFD exhibited greater signs of AD pathology and glial activation in apoE4- than apoE3-expressing mice (Johnson et al., 2017, Johnson et al., 2019, Moser and Pike, 2017, Nam et al., 2018). In a separate study of EFAD mice, apoE4 exacerbated cerebrovascular pathology and cognitive deficits following exposure to the bacterial endotoxin lipopolysaccharide (LPS), a model of chronic, low-grade peripheral inflammation characteristic of comorbid conditions such as diabetes (Marottoli et al., 2017). APOE4 carriage also exacerbated pathology following TBI in multiple studies in APOE-TR, FAD/APOE-Tg, and 3xTg/APOE-TR mouse models (Laskowitz et al., 2010, Bennett et al., 2013, Cao et al., 2017). These findings that connect apoE4 to exacerbated AD and vascular pathology following metabolic and/or inflammatory insults underscore the importance of incorporating h-APOE in modeling modifiable risk factors for AD. The contribution of these modifiable risk factors to AD burden also suggests that future studies should incorporate measures of metabolism, inflammation, and vascular function as efficacy readouts along with Aβ and tau pathology to enhance translatability to human disease.
III. The “How” of APOE Transgenic AD Mouse Models: Transgenic Strategies for Incorporating Human APOE
After the APOE4 allele was identified as the greatest genetic risk factor for AD, the demand for mouse models expressing h-APOE grew rapidly. As research technologies have advanced, several different strategies to introduce h-APOE into mice have emerged. In this portion of the review, we provide an overview of these strategies and comment on their contributions to AD research over the past three decades.
Multi-Gene Cluster
An initial method of introducing h-APOE consisted of recombining the entire ~58 kilobase locus of human chromosome 19 that encodes APOE and two members of the apolipoprotein C family (APOC1 and APOC2) into a large artificial chromosome, such as the P1 bacteriophage-derived or yeast artificial chromosome (PAC or YAC, respectively) (Simonet et al., 1993, Allan et al., 1995, Loring et al., 1996). Transfer of the entire multi-gene cluster provided valuable information on many distal regulatory elements. However, transfecting these large strands of DNA resulted in the overexpression of apoE in irrelevant tissues, with no expression in the brain, reducing their relevance to AD (Simonet et al., 1993, Allan et al., 1995).
Heterologous Promoters
A common method of introducing APOE is with heterologous promoter constructs, ensuring expression in CNS cell types. As reviewed previously by our group, heterologous promoters, including Thy1, GFAP, PGK, PDGF, transferrin, and NSE, drive the expression of apoE in either neurons or astrocytes in various brain regions (Tai et al., 2011, Bowman et al., 1995, Smith et al., 1998, Sun et al., 1998, Buttini et al., 1999, Tesseur et al., 2000b, Tesseur et al., 2000a, Huber et al., 2000). With these models, cell-and region-specific effects of h-apoE isoforms were elucidated (discussed in greater detail in Section IV below). However, the use of heterologous promoters introduced two major challenges. First, as these mice were developed in an APOE−/− background, only promoter-specific cells produced apoE. Second, the cells that produced apoE generally expressed it at higher than physiological levels and outside of the endogenous regulatory mechanisms. Thus, while useful for studying certain aspects of apoE biology, heterologous promoters are insufficient to accurately model h-APOE expression in mice.
Endogenous Promoters
The APOE-TR model is the predominant method to express h-APOE in mice. These mice were engineered through homologous recombination to contain the h-APOE coding sequences in place of the m-APOE coding sequences, but with intact mouse regulatory sequences (Sullivan et al., 1997). As described in the preceding sections of this review, APOE-TR mice exhibit isoform-specific phenotypes that mirror human patients, ranging from reduced apoE4 protein levels in the brain (compared to apoE3 or apoE2) to memory/cognitive deficits in APOE4 carriers, particularly females (Koutseff et al., 2014, Zhu et al., 2012, Dumanis et al., 2009, Bour et al., 2008, Riddell et al., 2008a). APOE-TR mice provide a more patient-relevant baseline to explore the cognitive and pathologic effects of various insults that are modulated by apoE isoforms (e.g. TBI, high-fat diet, etc.), and the crosses with other Tg models of AD detailed above in Section II are critical for modeling the interaction of h-apoE with amyloid and tau pathology.
IV. The “When” and “Where” of APOE Transgenic AD Mouse Models: Location- and Time-specific Effects of ApoE on AD Pathology
Development of pathology and cognitive decline in AD patients occurs over the course of decades and involves numerous interactions between the brain and peripheral tissues. In the next section of this review, we will discuss recent advances in the temporal-and regional-specificity of apoE expression. These studies represent preliminary efforts to better understand how the isoform-specific impact of apoE varies over the lifespan or with the location of apoE expression.
Temporal Specificity
Having described numerous interactions between apoE and AD pathology in the above sections of this review, we must also recognize that AD pathogenesis is characterized by a long prodromal period before onset of clinical symptoms. Aβ aggregation, tau accumulation, synaptic loss, and even neuronal death occur years to decades before any appreciable cognitive deficit in patients (Sperling et al., 2011, Jack et al., 2010, Gomez-Isla et al., 1996). Because each copy of the APOE4 allele accelerates age of onset by approximately eight years, apoE4 likely produces detrimental effects at a young age (Liu et al., 2013). Determining the precise time course of apoE isoform-specific effects on AD pathogenesis will eventually facilitate optimal treatment strategies for patients.
Two recent studies controlled temporal expression of h-apoE in mice to further define the timeline of apoE4-related AD pathology. In APP/PS1 mice engineered to express h-apoE in astrocytes by an inducible promoter, the critical window for the effect of apoE4 in promoting Aβ aggregation was early in life (<6M) (Liu et al., 2017a). In that study, apoE4 had no effect during the rapid phase of insoluble amyloid (e.g. fibrils and plaques) deposition later in life (>6M). Conversely, induction of apoE3 at any age did not exacerbate amyloid pathology and, in fact, reduced Aβ-associated gliosis and increased PSD-95, suggesting that both the temporal window and the specific apoE isoform are critical for modulating AD pathology. In a complementary study, anti-sense oligonucleotides (ASOs) were used to knock down h-apoE expression in APP/PS1 mice at various time points. When apoE (either apoE3 or apoE4) was reduced at birth, Aβ aggregation and deposition were also significantly reduced, but when apoE was not knocked down until 6 weeks of age (after Aβ aggregation had begun), no change in overall amyloid burden was noted at 4M (Huynh et al., 2017). Both of these studies demonstrated increased pathology with apoE4 vs. apoE3 and indicated that apoE isoform effects are restricted to the initial seeding and oligomerization of Aβ42. However, a key limitation of these studies was that they did not stratify for sex and, for the ASO study in particular, only analyzed amyloid aggregation, without evaluating the other AD pathological factors that, as mentioned above, become more prominent later in disease progression. Utilizing these time-limited APOE expression models in broader studies will enhance our understanding of the time course over which apoE isoforms differentially modulate AD pathology development.
Cell and Region Specificity
In addition to the varying effects of apoE on AD pathogenesis with time, the location of apoE expression also plays an important role in disease progression. Regional analysis of APOE-TR mice reveals that apoE protein levels are highest in cerebellum and lowest in hippocampus and frontal cortex (Sullivan et al., 2004). In the EFAD mice, apoE levels, regardless of isoform, are cerebellum > cortex > hippocampus (Youmans et al., 2012). This regional variation may provide some explanation as to why AD pathology preferentially affects the hippocampal regions first, as lower apoE levels could provide less protection against early Aβ aggregation. Importantly, we must note that certain observations, such as delayed amyloid pathology in mice expressing any h-apoE isoform compared to APOE knockout (KO) mice described in Section II, support the hypothesis that apoE4 is a loss of positive function; conversely, other observations, such as reduced amyloid pathology following early reductions in apoE reviewed here in Section IV, might conversely suggest a gain of toxic function. Two explanations may help resolve this apparent conflict. First, the specific apoE isoform exerts a critical impact on Aβ pathology, such that apoE3 provides greater protection than apoE4. Moreover, study results depend on the particular form of amyloid being quantified. As shown in Figure 1, the “on” vs. “off” pathways of Aβ aggregation allow for increased oAβ to occur simultaneously with decreased plaques, and vice versa. Thus, particular attention should be paid in future studies into the apoE/Aβ interaction to sensitively and specifically detecting these various aggregated species. The ability of APOE-TR and FAD/APOE-Tg mice to recapitulate human regional brain expression patterns in the context of the three apoE isoforms make them a useful tool for these studies and for other mechanistic investigations into the influence of apoE on other AD-relevant pathology.
In contrast to the natural expression patterns produced in APOE-TR mice, heterologous promoter constructs (introduced in Section III above) drive h-APOE expression toward specific cell types or regions within the brain. Using Thy1 and PDGF promoters resulted in apoE expression in hippocampal and cortical neurons, while the PGK promoter directed neuronal expression throughout the entire mouse brain (Tesseur et al., 2000b). Use of these promoter constructs demonstrated that neuronal expression of h-apoE4 contributed to tau hyperphosphorylation, which, in follow-up experiments, caused significant axonal dysfunction and degeneration (Tesseur et al., 2000a). Subsequent studies using either Thy1 or NSE promoter constructs combined neuron-specific apoE expression with additional insults, such as crossing with h-APP mice or administering the excitotoxin kainic acid, to reveal increased Aβ pathology and neurodegeneration, respectively, in the presence of apoE4 compared to apoE3 (Buttini et al., 1999, Brecht et al., 2004, Van Dooren et al., 2006). NSE promoter-driven apoE expression also revealed apoE4-mediated deficits in cognitive behavioral tasks that was worse in female mice, which may be explained by particular toxicity of apoE4 fragments to GABAergic interneuron populations (Raber et al., 1998, Andrews-Zwilling et al., 2010, Knoferle et al., 2014). The other major cell-specific apoE expression model utilizes the GFAP promoter to restrict h-apoE expression to astrocytes, which are the major cell type that produces apoE protein in the brain. Mice transfected with GFAP-apoE demonstrate isoform-specific worsening in neuronal morphology, microvascular pathology, motor function, and cognitive performance (Smith et al., 1998, Sun et al., 1998, Tang et al., 2009, Meng et al., 2015, Chaudhari et al., 2016, Ji et al., 2003, Hartman et al., 2001, van Meer et al., 2007).
Because the heterologous promoter strategy results in overexpression of apoE significantly above normal protein levels, these mice have not received widespread use. Instead, APOE-TR mice, which express h-apoE at normal physiological levels, are more prevalent given their greater accuracy and translatability. However, models of neuron-and astrocyte-specific expression still fill an important niche for mechanistic investigation of the isoform-specific effects of apoE.
Central vs. Peripheral APOE Expression
In recent years, appreciation of the interactions between peripheral and CNS apoE on AD pathology has grown. The apoE protein does not cross the blood-brain barrier, meaning that peripheral apoE (expressed primarily by hepatocytes) and CNS apoE (expressed primarily by astrocytes) form two distinct “pools” (reviewed recently in (Chernick et al., 2019)). While the contributions of peripheral apoE to AD modifiable risk factors and comorbid pathologies (high cholesterol, type 2 diabetes, cardiovascular disease) are described above, the development of new APOE-Tg models that spatially restrict the expression of apoE has illuminated the direct role of peripheral versus CNS apoE on AD pathology as well. Knocking out m-APOE in the brain did not result in cognitive deficits, whereas full APOE-KO mice did exhibit cognitive deficits (Lane-Donovan and Herz, 2016). However, this study utilized m-APOE, which limits translatability to h-APOE and AD.
To build on those findings, researchers from the Bu and Nielsen groups recently presented data from mice expressing h-apoE isoforms specifically in either vascular mural cells or hepatocytes, with apoE4 worsening vascular and neuropathology as compared to apoE3 or apoE2 in both cases (Yamazaki et al., 2018, Patra et al., 2018). When crossed with APP/PS1 mice, liver-specific apoE4 expression worsened Aβ pathology while liver-derived apoE3 was protective (Alzforum, 2018). Notably, in these studies, region-specific apoE isoform expression occurred on an APOE-KO background, which complicates the interpretation of their findings. In a separate article, the Holtzman group recently reported that specific reduction of h-apoE expression in the livers of APP/PS1/APOE-TR mice did not affect brain Aβ plaque deposition (Huynh et al., 2019). As only the Holtzman results have been peer-reviewed and published, further study on the role of peripheral apoE in modulating brain pathology is necessary. Moreover, because these studies used the APP/PS1 model, analysis naturally focused on amyloid pathology, but the effect of central vs. peripheral apoE expression on other pathological factors is also worthy of exploration. These cell-type or region-specific inducible models will be critical tools for the next phase of AD research, particularly with the heightened focus recently on the brain-periphery connection.
V. The “Why” of APOE Transgenic AD Mouse Models: Preclinical Testing of Therapeutic Strategies Targeting ApoE
The above-reviewed APOE-Tg preclinical mouse models highlight the critical role of h-apoE isoforms in modulating AD pathology and the importance of APOE4 in unmasking the increased risk in females. Another consequence of the pleiotropic effects of apoE in the brain is that a drug acting through improving positive functions or reducing toxic gain of functions of apoE at a foundational level could provide wide-ranging therapeutic benefit. AD mouse models can be used to refine a hypothesis, dissect a signaling mechanism or, as in our example, test a therapeutic targeting apoE. The choice of a mouse model requires not simply choosing a mouse that develops the relevant pathology, but fully understanding the strengths and limitations of the model. In this final section, we evaluate six major categories of therapeutic compounds in development that target apoE and discuss the importance of choosing the specific APOE-Tg mouse model relevant to the hypothesized mechanism of action of each candidate. Based on the universal biological variables of AD, we assume the importance of h-APOE and the use of male and female cohorts. In addition, while a particular model of pathology may be indicated as the primary model, examination in models that mimic other pathological mechanisms is also suggested as the role of APOE in AD pathology is multifaceted.
ApoE2 Overexpression (Figure 3.1)
Figure 3. Therapeutic strategies to target apoE.
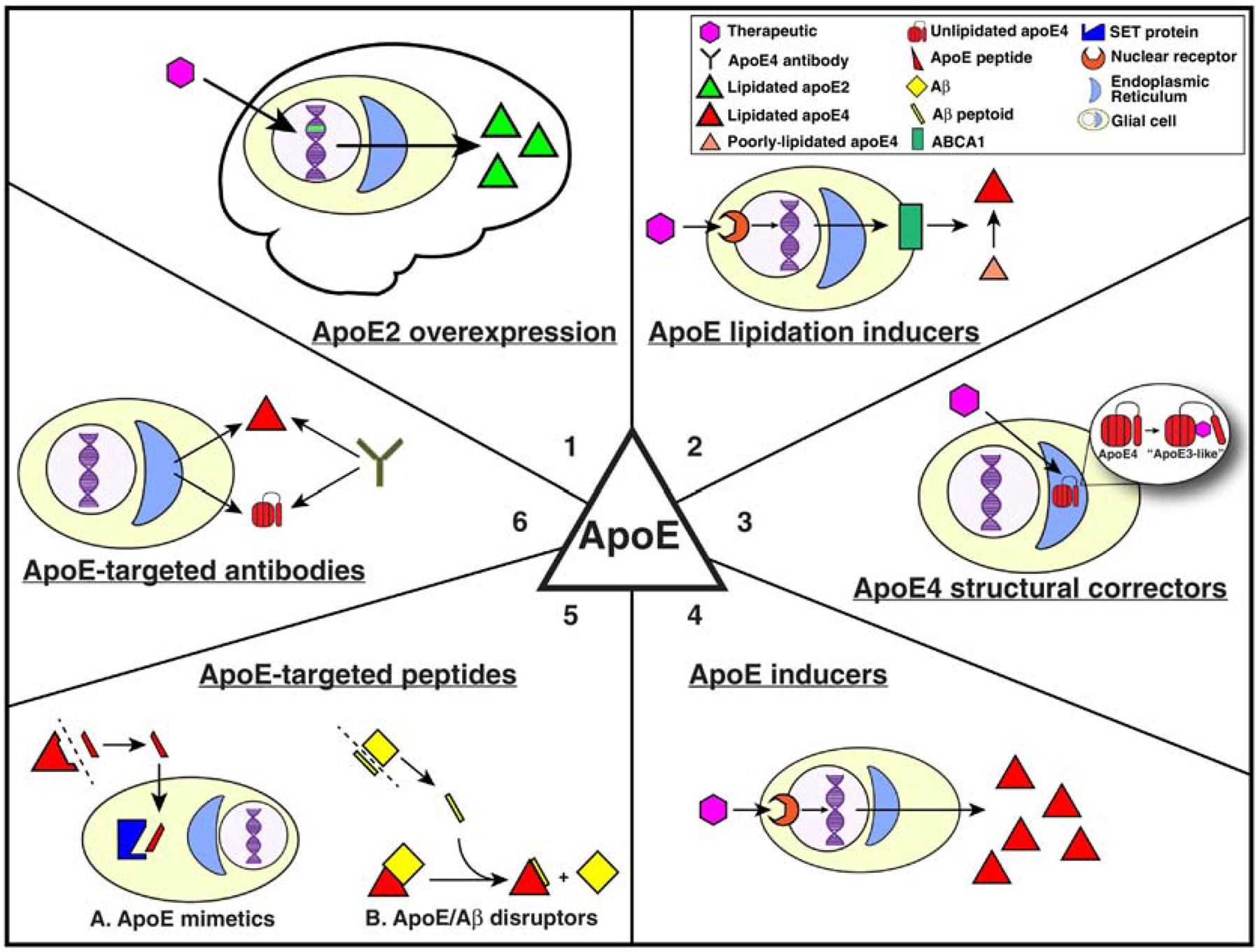
Six major categories of apoE-targeted therapeutic candidates in development are ideal for preclinical testing in APOE-Tg mouse models. These include apoE2 overexpression, apoE4 structural correctors, apoE lipidation promoters, apoE inducers, apoE-directed peptides, and antibodies that target various forms of apoE.
While APOE4 constitutes the most important genetic risk factor for AD, the APOE2 allele induces a significant protective effect, lowering AD risk two- to four-fold for one or two copies, respectively, compared to APOE3 carriers (Bertram et al., 2007, Corder et al., 1994). Thus, overexpression of apoE2 has been postulated as a potential therapeutic strategy to correct apoE4-associated deficits. Indeed, intracerebroventricular injection of lentiviral APOE2 reduced hippocampal Aβ load in PDAPP mice (Dodart et al., 2005). More recently, a single thalamic injection of an adeno-associated virus (AAV)rh.10h vector engineered to contain the APOE2 coding sequence significantly decreased brain Aβ levels in APP/PS1/APOE4 mice (Zhao et al., 2016). An open label Phase 1 clinical trial (NCT03634007) to determine maximum tolerated dose and assess brain apoE2 expression levels with this vector in 15 APOE4 homozygous MCI/AD patients was scheduled to begin in October 2019.
Despite positive results, numerous challenges remain in the development of this virus-based therapeutic. Along with ensuring intracisternal injection safety in human patients, potential adverse effects of apoE2 overexpression and detrimental interactions of the new apoE2 protein with endogenous apoE4 must both be identified. Additionally, the use of an APOE-Tg model to test this strategy should include both Aβ and tau pathology (only Aβ has been utilized to date), with careful attention paid to readouts that assess inflammation as well, given the diverse effects of apoE on AD pathology.
More generally, we lack a full understanding of the APOE2 protective mechanism. ApoE2 may be in a functional continuum with apoE3 and apoE4 (Table 2), based on relative apoE protein levels and lipid binding affinities observed in both APOE-TR mice and human cortical samples (Conejero-Goldberg et al., 2014, Riddell et al., 2008b, Tai et al., 2014a). Other observations, including significantly reduced binding to low-density lipoprotein receptors (LDLRs), suggest apoE2 may have a unique function compared to apoE3 and apoE4 (Mahley et al., 2006). Further complicating an apoE isoform functional continuum is the recent identification of the APOE3 Christchurch (R136S) mutation, which delayed symptom onset in a familial AD patient for several decades (Arboleda-Velasquez et al., 2019). However, the APOE3 Christchurch mutation is an exceedingly rare mutation that requires homozygosity to delay FAD. Moreover, case reports describing carriers of this mutation suggest it confers an even greater risk for type III hyperlipoproteinemia than the apoE2 allele (Wardell et al., 1987). Additional study is critical before comparison can be made between the APOE3 Christchurch mutation and common APOE genotypes.
Table 2.
Functional and pathologic differences among apoE isoforms in the periphery and central nervous system.
| Readouts in Human Periphery | APOE2 | APOE3 | APOE4 |
|---|---|---|---|
| Cholesterol | Low | Intermediate | High |
| Plasma apoE levels | High | Intermediate | Low |
| VLDL affinity | Low | Similar to APOE2 | High |
| LDL affinity | High | Similar to APOE4 | Low |
| HDL affinity | High | Similar to APOE2 | Low |
| Plasma LDL levels | Low | Intermediate LDL | High |
|
|
||
|
|
||
| Cardiovascular risk | Low | Intermediate | High |
| Type III hyperlipoproteinemia risk | Develops in < 10% of APOE2/2 (requires second genetic or environmental factor). | No risk | No risk |
| Readouts in Human CNS | APOE2 | APOE3 | APOE4 |
| ApoE levels | Similar to ApoE3 | High | Low |
| AD risk | Low | Intermediate | High |
With the known limitation of type III hyperlipoproteinemia in APOE2-TR mice, the AAV delivery strategy offers an alternative for studying these allelic differences and, more generally, the effects of apoE2 in vivo. Virus-mediated expression of apoE2 on an APOE3- or APOE4-TR background will facilitate identification of physiologic differences and help probe continual vs. discrete functions of the isoforms. Future research efforts such as these will provide valuable insight into the protective mechanism of apoE2 and, potentially, illuminate new routes to harness this protection for a therapeutic purpose.
ApoE Lipidation Inducers (Figure 3.2)
Reduced lipidation of apoE4, observed both in vitro and in vivo, is a structural deficit that has been implicated in several pathological deficits, and, thus, is a hypothesized therapeutic target for AD (Wahrle et al., 2004, Fitz et al., 2012, Wahrle et al., 2008, Rebeck, 2017, Wahrle et al., 2005). Nascent apoE particles secreted by glial cells are lipidated primarily by cholesterol transport proteins ABCA1 and ABCG1, leading to the hypothesis that increasing expression of these transporters will reverse apoE4 lipidation deficits. The primary molecular target for increasing ABCA1/G1 is the liver X receptor (LXR), a nuclear receptor that serves as the primary transcription factor controlling ABCA1/G1 expression (Qiu et al., 2001, Costet et al., 2000). LXR agonists TO901317 and GW3965 decrease Aβ pathological burden and improve memory performance in various FAD-Tg mouse models (Riddell et al., 2007, Fitz et al., 2010, Cui et al., 2011, Koldamova et al., 2005, Vanmierlo et al., 2011, Burns et al., 2006, Terwel et al., 2011, Donkin et al., 2010). However, LXRα-stimulated production of triglycerides in the liver leads to adverse effects, precluding clinical development of these agents (Schultz et al., 2000, Quinet et al., 2006). Newer, selective LXRβ agonists have been synthesized and characterized (reviewed in El-Gendy et al., 2018), with compounds from both Bristol-Myers Squibb and Wyeth entering Phase 1 trials. However, BMS-852927 (NCT01651273) decreased circulating neutrophil levels, while LXR-623 (Wyeth, NCT00366522) produced CNS-related adverse effects (Katz et al., 2009, Kirchgessner et al., 2016). From these two trials, it is difficult to determine whether the side effects were due to LXR agonism or were unique to the individual drug candidates being tested.
Despite challenges in achieving selectivity, preclinical development of LXR agonists continues, and strategies to bypass LXRα-mediated side effects have been reported (Fan et al., 2018). Because ABCA1 deficiency has been linked to both amyloid-dependent and -independent AD-related pathology (Karasinska et al., 2009, Karasinska et al., 2013, Corona et al., 2015, Fitz et al., 2014, Castranio et al., 2018), a diverse set of preclinical APOE-TR models will be appropriate for future in vivo studies with these compounds. Specifically, APOE-TR mice provide a platform for evaluating target engagement with assays ranging from native gel electrophoresis for apoE-containing particle size to size exclusion chromatography techniques that evaluate shifts in lipid and lipoprotein profiles (LaDu et al., 1994, LaDu et al., 2012). FAD/APOE-Tg models would enable testing of amyloid-dependent effects, and combining APOE-TR with a second hit, such as HFD-, TBI- or LPS-induced inflammation, could be used to analyze amyloid-independent effects. Moreover, peripheral ABCA1 modulates vascular cholesterol deposition (Oram, 2002, Stukas et al., 2014), permitting a comparison between CNS vs. peripheral apoE-expressing mice, provided these Tg models become better characterized in the next several years. These testing strategies using APOE-Tg mice enhance translational impact that has been limited to date by the use of FAD-Tg mice expressing m-apoE (Moutinho and Landreth, 2017).
ApoE4 Structural Correctors (Figure 3.3)
Small-molecule structure correctors of apoE4 remain in an early stage of development. ApoE4 structural correctors were initially identified in 2005 from a library of small molecules that bind to apoE4 to prevent the N- to C-terminal domain interaction, “converting” it to an apoE3-like structure (Ye et al., 2005). While beneficial functional effects have been reported in N2a neuroblastoma cells and iPSC-derived neurons (Brodbeck et al., 2011, Wang et al., 2018a, Chen et al., 2012, Mahley and Huang, 2012), the lack of any reported animal studies limits the therapeutic potential of these compounds. Although structure correcting drugs have been developed for other conditions (e.g. Ivacaftor for cystic fibrosis), evidence of bioavailability, tolerability, and efficacy in animal models is necessary to establish structure correctors as therapeutic candidates for AD. In particular, APOE-TR mice serve as a basic model to test the hypothesis that expression levels and lipidation of apoE4 mirror those of apoE3 following treatment. As in vitro studies have described a reversal of apoE isoform-dependent tau pathology in neurons (Wang et al., 2018a), efficacy studies could utilize a MAPT/APOE-Tg model to evaluate the translation of these results from cells to animals, although such a model must be better established. A more fundamental issue is the use of neuronal cells for the production of apoE as it is widely accepted that glial cells, particularly astrocytes, are the primary apoE-expressing cell type in the brain.
ApoE Inducers (Figure 3.4)
In both human and APOE-TR mouse brains, apoE4 levels are lower than those of apoE3 (Riddell et al., 2008a, Martinez-Morillo et al., 2014). To address the reduced levels of apoE4 in the brain, compounds that increase apoE expression, particularly in APOE4 carriers, have been proposed as therapeutic agents for AD. The most prominent such compound is bexarotene (Bex), a retinoid X receptor (RXR) agonist that is an FDA-approved chemotherapeutic. However, because RXR is a nuclear receptor in the same family as LXR, it is critical to keep in mind that while Bex may induce the expression of APOE, it also targets ABCA1/G1. Thus, Bex could also have been classified as a potential therapeutic to induce apoE lipidation (Figure 3.2). In a high-profile study, Cramer and colleagues demonstrated rapid clearance of Aβ and reversal of numerous functional deficits with Bex administration, though results from follow-up experiments were mixed (Veeraraghavalu et al., 2013, Tesseur et al., 2013, Price et al., 2013, Fitz et al., 2013, Cramer et al., 2012). A Phase 2 human clinical trial (NCT01782742) found no change in amyloid burden, with adverse increases in serum triglycerides (Cummings et al., 2016). Another proposed apoE-inducing therapeutic for AD is probucol, which reduces blood-brain barrier (BBB) dysfunction, cognitive deficits, and synaptic impairment in wild-type mice when paired with a diabetogenic diet or following intracerebroventricular Aβ peptide injections (Mamo et al., 2019, Santos et al., 2012). A Phase 1b/2a clinical trial with probucol is listed as complete nearly two years ago, though results have yet to be reported (NCT02707458)
Novel analogs of Bex, along with other LXR/RXR agonists, have been developed in the past year, and new screens for apoE inducers have been performed as well (Yuan et al., 2019, Ren et al., 2019, Pollinger et al., 2019, Fan et al., 2016, Finan et al., 2016). Most of these newly-described compounds have limited (or no) in vivo data supporting their efficacy, thus moving into animal models represents an important next phase of their development. Given that reduced apoE4 or apoE3 levels have been found to correlate with increased brain Aβ deposition in mice and humans (DeMattos, 2004, Lambert et al., 2005, Bales et al., 2009, Gupta et al., 2011), an FAD/APOE-Tg mouse (such as EFAD, or APP/PS1/APOE-TR) will be a useful model to study whether apoE inducers can reverse the development of Aβ pathology. Importantly, a study by our group with Bex revealed reduction in soluble Aβ levels in E4FAD mice, but not E3FAD mice, highlighting the clarity of analysis provided by a model that incorporates APOE genotype (Tai et al., 2014a, Koster et al., 2017). Studies with h-APOE inducible mice discussed in Section IV also emphasize a critical period of AD progression during which apoE inducers would provide the highest therapeutic impact.
ApoE-targeted Peptides (Figure 3.5)
Several peptides that target various aspects of apoE-mediated pathology via diverse mechanisms have been developed. The first peptide, CS-6253, consists of 26 amino acids from the C-terminus of the apoE protein that form an amphipathic helical structure. The peptide acts to stabilize ABCA1 in the cell membrane, thus facilitating cholesterol efflux in vitro and improving apoE4 lipidation in APOE4-TR mice (Hafiane et al., 2015, Boehm-Cagan et al., 2016). Another peptide with similar function, named 4F for its four phenylalanine residues, has been studied extensively for cardiovascular disease before it was observed to increase apoE4 lipidation in vitro and reduce Aβ pathology and cognitive deficits in APP/PS1 mice (Handattu et al., 2009, Chernick et al., 2018). Additionally, two peptides based on the receptor-binding domain of apoE (COG133, consisting of residues 133–149 of apoE, and COG1410, derived from residues 138–149) have been described as anti-inflammatory and neuroprotective following testing in FAD mice with a knockout of the nitric oxide synthase 2 gene; another similar peptide reversed pathology and cognitive deficits in 3xTg and 5xFAD mice (Lynch et al., 2005, Laskowitz et al., 2007, Christensen et al., 2011, Vitek et al., 2012, Sawmiller et al., 2019). Notably, none of the aforementioned peptides (Figure 3.5.A) has been studied in an AD model that incorporates h-APOE expression, a critical limitation for a drug with proposed mechanism of action mimicking or enhancing h-apoE lipidation, as the h-apoE isoforms are known to have functional and pathological differences.
Finally, a peptoid based on residues 12–28 of the Aβ42 sequence has been reported to block the apoE/Aβ42 interaction (Figure 3.5.B), with beneficial reductions in amyloid pathology and cognition in APP/PS1 and, with an earlier peptide version, APP/PS1/APOE-TR mice (Sadowski et al., 2004, Liu et al., 2017b, Liu et al., 2014). Whether blocking this interaction will ultimately prove beneficial or harmful in human AD patients remains to be determined, as the authors aim to specifically prevent the apoE/Aβ42 interaction. The drug instead likely inhibits all interaction regardless of apoE isoform, potentially ameliorating any protective actions of apoE3 or apoE2 (Pankiewicz et al., 2014). Further, the possibility exists that this peptoid simply disrupts Aβ aggregation independent of apoE; testing in a FAD/APOE-KO mouse alongside the FAD/APOE-Tg models expressing various isoforms would rule out this possibility and show the importance of apoE – or the apoE4 isoform in particular – in the mechanism of action of this drug candidate.
ApoE-targeted Antibodies (Figure 3.6)
Monoclonal antibodies have become an attractive therapeutic option for many diseases due to their high specificity and minimal off-target effects compared to small molecules. However, poor results with Aβ-directed antibodies in late-stage trials may temper excitement for therapeutic antibodies targeting other AD-related proteins. Efficacy of several apoE-targeting antibodies has been reported, with one recently described that preferentially binds non-lipidated and aggregated forms of apoE3 and apoE4 to reduce Aβ pathology in APP/PS1/APOE4-TR mice (Kim et al., 2012, Liao et al., 2014, Liao et al., 2018). Continued research into apoE-directed antibodies as AD therapeutics will require a broader assessment of their effects. While published studies noted a decrease in Aβ plaque burden, no other changes in CNS pathological markers of AD have been reported. Moreover, no changes in apoE levels were observed, and any changes in apoE structure or function (e.g. lipidation state) were not described.
Over the next several years, it is likely that antibodies will be developed that preferentially target single apoE isoforms, bind to functional lipidated apoE rather than aggregated forms, and/or alter brain levels of apoE (either by targeting apoE for degradation to decrease apoE or by stabilizing lipoproteins to increase apoE). With regard to preclinical testing of these antibodies, the failure to clinically translate reduced plaques to improved cognition with Aβ-directed monoclonal antibodies suggests that we should explore other potential mechanisms of action aside from amyloid plaque reduction. Thus, despite positive observations with one anti-apoE antibody in APP/PS1/APOE4-TR mice, these antibodies could also be examined for their effects on apoE isoform-dependent tau pathology (i.e. in an MAPT/APOE-Tg model) or on apoE-dependent inflammation (i.e. APOE-TR mice injected with LPS). A more diverse preclinical profiling of apoE-targeted antibodies will answer critical questions about their desired therapeutic profile and better position them for clinical development as AD treatments.
VI. Conclusion: Choosing the Right Mouse Model
Because mouse APP and MAPT genes do not readily produce toxic Aβ species or NFT, modeling AD pathology in mice remains a challenge. New models are continually generated, with a recent AlzForum search yielding 179 different AD mouse models, each of which is often characterized for only a single feature of interest. Given the complexity of the disease, hundreds of additional models could be generated without ever producing one that fully recapitulates human AD or is characterized beyond a gene of interest to provide context and allow for informed conclusions. Thus, the focus of AD transgenics should be on maximizing the reliability and validity of the mouse model to answer a given research question. In order to accomplish this goal, the universal biological variables of AD must be considered. FAD- and MAPT-Tg mice, along with newer “humanized” APPTR and MAPT-TR mice, recapitulate aspects of AD-like (or FTD-like) pathology but do not permit interpretation of the effects of APOE or sex as universal biological variables of AD. Incorporation of h-APOE is critical for accurate preclinical modeling of the disease and for exposing the increased risk to female APOE4 carriers, and, in this review, we have described numerous contexts for understanding apoE isoform-dependent impact in mice. APOE4 exacerbates amyloid pathology in FAD-Tg mouse models, may affect tau pathology in MAPT-Tg mouse models, and aggravates the effects of a second insult that reproduces modifiable risk factors. An even more nuanced interrogation has occurred with Tg mice that have been engineered to express h-APOE only in specific cell types, in certain brain regions, in CNS vs. peripheral compartments, or at certain times throughout the lifespan. Thus, diverse AD mouse models that express h-APOE are available for future mechanistic studies, though it is critical that the correct models be chosen to test the hypothesis at hand. To demonstrate this point, we concluded by reviewing apoE-targeted therapeutics and, in particular, an analysis of their results in appropriate APOE-Tg mouse models.
HIGHLIGHTS.
What: AD is uniquely human, but molecular mechanisms can be measured in Tg-mice.
Who: A universal biological variable of AD, APOE is critical for preclinical models.
How: There are several strategies for developing an APOE-Tg mice.
When/Where: Developmental and region-specific models probe APOE-expression patterns.
Why: Therapeutic strategies targeting apoE can exploit the relevant mouse model(s).
Acknowledgements
The authors thank Deebika Balu and Ana Carolina Valencia-Olvera for their review of the manuscript and figures.
Funding
The LaDu lab is funded by NIH (NIA) R01 AG056472, R01 AG057008, UH2/3 NS10012, R56 AG058655, 1R44 AG060826, UIC-DPI Phase 2 POC Award, and generous philanthropic support. C.T.L. received support from NIH (NIA) T32AG57468, the American Heart Association (20PRE35150022), and the University of Illinois at Chicago Center for Clinical and Translational Science (NIH UL1TR002003).
ABBREVIATIONS
- 3xTg
triple-transgenic (mutated human APP, PS1, and MAPT) mouse
- 3xTg/APOE-TR
triple-transgenic (mutated human APP, PS1, and MAPT) mouse expressing human APOE
- 5xFAD
transgenic mouse harboring five familial AD mutations (three in APP, 2 in PS1)
- Aβ
amyloid-β
- AAV
adeno-associated virus
- ABCA1
ATP-binding cassette protein A1
- AD
Alzheimer’s disease
- ASOs
anti-sense oligonucleotides
- APOC
apolipoprotein C gene
- APOE
apolipoprotein E gene
- ApoE
apolipoprotein E
- APP
amyloid precursor protein
- APP
amyloid precursor protein gene
- APP/PS1
transgenic mouse expressing mutant human APP and PS1
- APP/PS1/APOE-TR
APP/PS1 transgenic mouse expressing a human apoE isoform
- BBB
blood-brain barrier
- Bex
Bexarotene
- CNS
central nervous system
- EFAD
5xFAD transgenic mouse expressing a human apoE isoform
- FAD
familial Alzheimer’s disease
- FAD/APOE-Tg
FAD transgenic mice expressing human APOE
- FAD/APOE-KO
APOE-null FAD mice
- FAD-Tg
FAD transgenic mice
- FTD
frontotemporal dementia
- FTDP-17
frontotemporal dementia and parkinsonism linked to chromosome 17
- GFAP
glial fibrillary acidic protein
- h-
human
- HFD
high-fat diet
- iPSC
induced pluripotent stem cells
- KO
knock-out
- LDLR
low-density lipoprotein receptor
- LPS
lipopolysaccharides
- LXR
liver X receptor
- MAPT
microtubule-associated protein tau
- MAPT-Tg
transgenic mouse with human tau
- MAPT/APOE-Tg
transgenic mouse with human tau and human APOE
- MCI
mild cognitive impairment
- M
months
- m-
mouse
- NFT
neurofibrillary tangles
- NSE
neuron-specific enolase
- oAβ
oligomeric Aβ
- PAC
phage P1-derived artificial chromosome
- PDAPP
PDGF promoter-driven APP expression transgenic mouse
- PDGF
platelet-derived growth factor
- PGK
phosphoglycerate kinase
- PP2A
protein phosphatase 2A
- PS1
presenilin-1
- PS1
presenilin-1 gene
- RXR
retinoid X receptor
- TR
targeted replacement
- Tg
transgenic
- Thy1
thymocyte-differentiation antigen 1
- TREM2
gene encoding the triggering receptor expressed on myeloid cells 2
- TBI
traumatic brain injury
- YAC
yeast artificial chromosome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
The authors have no conflicting interests to disclose.
References
- Alzforum, 2018. ApoE has hand in Alzheimer’s beyond Aβ, beyond the brain [Online]. alzforum.org: FBRI LLC. Available: https://www.alzforum.org/news/research-news/apoe-has-hand-alzheimers-beyond-av-beyond-brain [Accessed October 27 2019].
- Allan CM, Walker D & Taylor JM 1995. Evolutionary duplication of a hepatic control region in the human apolipoprotein E gene locus. Identification of a second region that confers high level and liver-specific expression of the human apolipoprotein E gene in transgenic mice. J Biol Chem, 270, 26278–81. [DOI] [PubMed] [Google Scholar]
- Altmann A, Tian L, Henderson VW, Greicius MD & Alzheimer’s Disease Neuroimaging Initiative, I. 2014. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann Neurol, 75, 563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen K, Launer LJ, Dewey ME, Letenneur L, Ott A, Copeland JR, Dartigues JF, Kragh-Sorensen P, Baldereschi M, Brayne C, Lobo A, Martinez- Lage JM, Stijnen T & Hofman A 1999. Gender differences in the incidence of AD and vascular dementia: The EURODEM Studies. EURODEM Incidence Research Group. Neurology, 53, 1992–7. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, De Silva R, Tucker KL, Barde YA, Duff K & Davies P 2003. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem, 86, 582–90. [DOI] [PubMed] [Google Scholar]
- Andrews-Zwilling Y, Bien-Ly N, Xu Q, Li G, Bernardo A, Yoon SY, Zwilling D, Yan TX, Chen L & Huang Y 2010. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci, 30, 13707–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arboleda-Velasquez JF, Lopera F, O’hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, Leyton-Cifuentes D, Chen K, Baena A, Aguillon D, Rios-Romenets S, Giraldo M, Guzman-Velez E, Norton DJ, Pardilla-Delgado E, Artola A, Sanchez JS, Acosta-Uribe J, Lalli M, Kosik KS, Huentelman MJ, Zetterberg H, Blennow K, Reiman RA, Luo J, Chen Y, Thiyyagura P, Su Y, Jun GR, Naymik M, Gai X, Bootwalla M, Ji J, Shen L, Miller JB, Kim LA, Tariot PN, Johnson KA, Reiman EM & Quiroz YT 2019. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat Med, 25, 1680–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbones-Mainar JM, Johnson LA, Altenburg MK & Maeda N 2008. Differential modulation of diet-induced obesity and adipocyte functionality by human apolipoprotein E3 and E4 in mice. Int J Obes (Lond), 32, 1595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbones-Mainar JM, Johnson LA, Torres-Perez E, Garcia AE, Perez-Diaz S, Raber J & Maeda N 2016. Metabolic shifts toward fatty-acid usage and increased thermogenesis are associated with impaired adipogenesis in mice expressing human APOE4. Int J Obes (Lond), 40, 1574–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Liu F, Wu S, Lin S, Koger D, Delong C, Hansen JC, Sullivan PM & Paul SM 2009. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J Neurosci, 29, 6771–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B & Paul SM 1997. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet, 17, 263–4. [DOI] [PubMed] [Google Scholar]
- Balu D, Karstens AJ, Loukenas E, Maldonado Weng J, York JM, Valencia-Olvera AC & Ladu MJ 2019. The role of APOE in transgenic mouse models of AD. Neurosci Lett, 707, 134285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes LL, Wilson RS, Bienias JL, Schneider JA, Evans DA & Bennett DA 2005. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry, 62, 685–91. [DOI] [PubMed] [Google Scholar]
- Bengoa-Vergniory N, Roberts RF, Wade-Martins R & Alegre-Abarrategui J 2017. Alpha-synuclein oligomers: a new hope. Acta Neuropathol, 134, 819–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I, Karran E & De Strooper B 2012. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci, 15, 349–57. [DOI] [PubMed] [Google Scholar]
- Bennet AM, Di Angelantonio E, Ye Z, Wensley F, Dahlin A, Ahlbom A, Keavney B, Collins R, Wiman B, De Faire U & Danesh J 2007. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA, 298, 1300–11. [DOI] [PubMed] [Google Scholar]
- Bennett DA 2017. Mixed pathologies and neural reserve: Implications of complexity for Alzheimer disease drug discovery. PLoS Med, 14, e1002256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC & Wilson RS 2006. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology, 66, 1837–44. [DOI] [PubMed] [Google Scholar]
- Bennett RE, Esparza TJ, Lewis HA, Kim E, Mac Donald CL, Sullivan PM & Brody DL 2013. Human apolipoprotein E4 worsens acute axonal pathology but not amyloid-beta immunoreactivity after traumatic brain injury in 3xTG-AD mice. J Neuropathol Exp Neurol, 72, 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Mcqueen MB, Mullin K, Blacker D & Tanzi RE 2007. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet, 39, 17–23. [DOI] [PubMed] [Google Scholar]
- Beyreuther K & Masters CL 1991. Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer’s disease: precursor-product relationships in the derangement of neuronal function. Brain Pathol, 1, 241–51. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Haertel C, Maelicke A & Montag D 2014. Galantamine slows down plaque formation and behavioral decline in the 5XFAD mouse model of Alzheimer’s disease. PLoS One, 9, e89454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, Mcgaugh JL & Laferla FM 2005. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron, 45, 675–88. [DOI] [PubMed] [Google Scholar]
- Bloom GS 2014. Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol, 71, 505–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm-Cagan A, Bar R, Liraz O, Bielicki JK, Johansson JO & Michaelson DM 2016. ABCA1 Agonist Reverses the ApoE4-Driven Cognitive and Brain Pathologies. J Alzheimers Dis, 54, 1219–1233. [DOI] [PubMed] [Google Scholar]
- Bour A, Grootendorst J, Vogel E, Kelche C, Dodart JC, Bales K, Moreau PH, Sullivan PM & Mathis C 2008. Middle-aged human apoE4 targeted-replacement mice show retention deficits on a wide range of spatial memory tasks. Behav Brain Res, 193, 174–82. [DOI] [PubMed] [Google Scholar]
- Bowman BH, Jansen L, Yang F, Adrian GS, Zhao M, Atherton SS, Buchanan JM, Greene R, Walter C, Herbert DC, Weaker FJ, Chiodo LK, Kagan-Hallet K & Hixson JE 1995. Discovery of a brain promoter from the human transferrin gene and its utilization for development of transgenic mice that express human apolipoprotein E alleles. Proc. Natl. Acad. Sci. USA, 92, 12115–12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H & Braak E 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol, 82, 239–59. [DOI] [PubMed] [Google Scholar]
- Brecht WJ, Harris FM, Chang S, Tesseur I, Yu GQ, Xu Q, Dee Fish J, Wyss-Coray T, Buttini M, Mucke L, Mahley RW & Huang Y 2004. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci, 24, 2527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitner JC, Wyse BW, Anthony JC, Welsh-Bohmer KA, Steffens DC, Norton MC, Tschanz JT, Plassman BL, Meyer MR, Skoog I & Khachaturian A 1999. APOE-epsilon4 count predicts age when prevalence of AD increases, then declines: the Cache County Study. Neurology, 53, 321–31. [DOI] [PubMed] [Google Scholar]
- Bretsky PM, Buckwalter JG, Seeman TE, Miller CA, Poirier J, Schellenberg GD, Finch CE & Henderson VW 1999. Evidence for an interaction between apolipoprotein E genotype, gender, and Alzheimer disease. Alzheimer Dis Assoc Disord, 13, 216–21. [DOI] [PubMed] [Google Scholar]
- Brodbeck J, Mcguire J, Liu Z, Meyer-Franke A, Balestra ME, Jeong DE, Pleiss M, Mccomas C, Hess F, Witter D, Peterson S, Childers M, Goulet M, Liverton N, Hargreaves R, Freedman S, Weisgraber KH, Mahley RW & Huang Y 2011. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J Biol Chem, 286, 17217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buee L, Bussiere T, Buee-Scherrer V, Delacourte A & Hof PR 2000. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev, 33, 95–130. [DOI] [PubMed] [Google Scholar]
- Burns MP, Vardanian L, Pajoohesh-Ganji A, Wang L, Cooper M, Harris DC, Duff K & Rebeck GW 2006. The effects of ABCA1 on cholesterol efflux and Abeta levels in vitro and in vivo. J Neurochem. [DOI] [PubMed] [Google Scholar]
- Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L & Mahley RW 1999. Expression of human apolipoprotein E3 or E4 in the brains of Apoe−/− mice: isoform-specific effects on neurodegeneration. J Neurosci, 19, 4867–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillet-Boudin ML, Buee L, Sergeant N & Lefebvre B 2015. Regulation of human MAPT gene expression. Mol Neurodegener, 10, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Gaamouch FE, Meabon JS, Meeker KD, Zhu L, Zhong MB, Bendik J, Elder G, Jing P, Xia J, Luo W, Cook DG & Cai D 2017. ApoE4-associated phospholipid dysregulation contributes to development of Tau hyper-phosphorylation after traumatic brain injury. Sci Rep, 7, 11372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, Demattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ & Holtzman DM 2011. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med, 3, 89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castranio EL, Wolfe CM, Nam KN, Letronne F, Fitz NF, Lefterov I & Koldamova R 2018. ABCA1 haplodeficiency affects the brain transcriptome following traumatic brain injury in mice expressing human APOE isoforms. Acta Neuropathol Commun, 6, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari K, Wong JM, Vann PH & Sumien N 2016. Exercise, but not antioxidants, reversed ApoE4-associated motor impairments in adult GFAP-ApoE mice. Behav Brain Res, 305, 37–45. [DOI] [PubMed] [Google Scholar]
- Chen HK, Liu Z, Meyer-Franke A, Brodbeck J, Miranda RD, Mcguire JG, Pleiss MA, Ji ZS, Balestra ME, Walker DW, Xu Q, Jeong DE, Budamagunta MS, Voss JC, Freedman SB, Weisgraber KH, Huang Y & Mahley RW 2012. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J Biol Chem, 287, 5253–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernick D, Ortiz-Valle S, Jeong A, Swaminathan SK, Kandimalla KK, Rebeck GW & Li L 2018. High-density lipoprotein mimetic peptide 4F mitigates amyloid-beta-induced inhibition of apolipoprotein E secretion and lipidation in primary astrocytes and microglia. J Neurochem, 147, 647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernick D, Ortiz-Valle S, Jeong A, Qu W & Li L 2019. Peripheral versus central nervous system APOE in Alzheimer’s disease: Interplay across the blood-brain barrier. Neurosci Lett, 708, 134306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F & Dobson CM 2017. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu Rev Biochem, 86, 27–68. [DOI] [PubMed] [Google Scholar]
- Chong FP, Ng KY, Koh RY & Chye SM 2018. Tau Proteins and Tauopathies in Alzheimer’s Disease. Cell Mol Neurobiol, 38, 965–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen DJ, Ohkubo N, Oddo J, Van Kanegan MJ, Neil J, Li F, Colton CA & Vitek MP 2011. Apolipoprotein E and peptide mimetics modulate inflammation by binding the SET protein and activating protein phosphatase 2A. J Immunol, 186, 2535–42. [DOI] [PubMed] [Google Scholar]
- Cline EN, Bicca MA, Viola KL & Klein WL 2018. The Amyloid-beta Oligomer Hypothesis: Beginning of the Third Decade. J Alzheimers Dis, 64, S567–S610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conejero-Goldberg C, Gomar JJ, Bobes-Bascaran T, Hyde TM, Kleinman JE, Herman MM, Chen S, Davies P & Goldberg TE 2014. APOE2 enhances neuroprotection against Alzheimer’s disease through multiple molecular mechanisms. Mol Psychiatry, 19, 1243–50. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC Jr., Rimmler JB, Locke PA, Conneally PM, Schmader KE & et al. 1994. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet, 7, 180–4. [DOI] [PubMed] [Google Scholar]
- Corder EH, Ghebremedhin E, Taylor MG, Thal DR, Ohm TG & Braak H 2004. The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex, and APOE polymorphism. Ann N Y Acad Sci, 1019, 24–8. [DOI] [PubMed] [Google Scholar]
- Corona AW, Kodoma N, Casali BT & Landreth GE 2015. ABCA1 is Necessary for Bexarotene-Mediated Clearance of Soluble Amyloid Beta from the Hippocampus of APP/PS1 Mice. J Neuroimmune Pharmacol, 11, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsetti JP, Sparks CE, Bakker SJL, Gruppen EG & Dullaart RPF 2018. Roles of high apolipoprotein E blood levels and HDL in development of familial dysbetalipoproteinemia in epsilon2epsilon2 subjects. Clin Biochem, 52, 67–72. [DOI] [PubMed] [Google Scholar]
- Costet P, Luo Y, Wang N & Tall AR 2000. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J Biol Chem, 275, 28240–5. [DOI] [PubMed] [Google Scholar]
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA & Landreth GE 2012. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science, 335, 1503–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo R, Villar-Alvarez E, Taboada P, Rocha FA, Damas AM & Martins PM 2016. What Can the Kinetics of Amyloid Fibril Formation Tell about Off-pathway Aggregation? J Biol Chem, 291, 2018–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W, Sun Y, Wang Z, Xu C, Xu L, Wang F, Chen Z, Peng Y & Li R 2011. Activation of liver X receptor decreases BACE1 expression and activity by reducing membrane cholesterol levels. Neurochem Res, 36, 1910–21. [DOI] [PubMed] [Google Scholar]
- Cummings JL, Zhong K, Kinney JW, Heaney C, Moll-Tudla J, Joshi A, Pontecorvo M, Devous M, Tang A & Bena J 2016. Double-blind, placebo-controlled, proof-of-concept trial of bexarotene in moderate Alzheimer’s disease. Alzheimer’s Research & Therapy, 8, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demattos RB 2004. Apolipoprotein E Dose-Dependent Modulation of beta-Amyloid Deposition in a Transgenic Mouse Model of Alzheimer’s Disease. J Mol Neurosci, 23, 255–62. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Mcevoy LK, Thompson WK, Holland D, Brewer JB, Aisen PS, Sperling RA, Dale AM & Alzheimer’s Disease Neuroimaging, I. 2012. Amyloid-beta--associated clinical decline occurs only in the presence of elevated P-tau. Arch Neurol, 69, 709–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Marr RA, Koistinaho M, Gregersen BM, Malkani S, Verma IM & Paul SM 2005. Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA, 102, 1211–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, May S, Chan J, Fan J, Collins J & Wellington CL 2010. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem, 285, 34144–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-Tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J & Younkin S 1996. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature, 383, 710–3. [DOI] [PubMed] [Google Scholar]
- Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, Weeber EJ, Turner RS, Xu B, Rebeck GW & Hoe HS 2009. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci, 29, 15317–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumanis SB, Dibattista AM, Miessau M, Moussa CE & Rebeck GW 2013. APOE genotype affects the pre-synaptic compartment of glutamatergic nerve terminals. J Neurochem, 124, 4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A & Wanker EE 2008. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol, 15, 558–66. [DOI] [PubMed] [Google Scholar]
- El-Gendy BEM, Goher SS, Hegazy LS, Arief MMH & Burris TP 2018. Recent Advances in the Medicinal Chemistry of Liver X Receptors. J Med Chem, 61, 10935–10956. [DOI] [PubMed] [Google Scholar]
- El-Lebedy D, Raslan HM & Mohammed AM 2016. Apolipoprotein E gene polymorphism and risk of type 2 diabetes and cardiovascular disease. Cardiovasc Diabetol, 15, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Holtzman DM, Munson G, Mathur T, Schneider D, Chang LK, Getz GS, Reardon CA, Lukens J, Shah JA & Ladu MJ 1999. Unique lipoproteins secreted by primary astrocytes from wild type, apoE (−/−), and human apoE transgenic mice. J Biol Chem, 274, 30001–7. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM & Holtzman DM 2002. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol Dis, 9, 305–18. [DOI] [PubMed] [Google Scholar]
- Fan J, Zareyan S, Zhao W, Shimizu Y, Pfeifer TA, Tak JH, Isman MB, Van Den Hoven B, Duggan ME, Wood MW, Wellington CL & Kulic I 2016. Identification of a Chrysanthemic Ester as an Apolipoprotein E Inducer in Astrocytes. PLoS One, 11, e0162384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Zhao RQ, Parro C, Zhao W, Chou HY, Robert J, Deeb TZ, Raynoschek C, Barichievy S, Engkvist O, Maresca M, Hicks R, Meuller J, Moss SJ, Brandon NJ, Wood MW, Kulic I & Wellington CL 2018. Small molecule inducers of ABCA1 and apoE that act through indirect activation of the LXR pathway. J Lipid Res, 59, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfel JM, Yu L, De Jager PL, Schneider JA & Bennett DA 2016. Association of APOE with tau-tangle pathology with and without beta-amyloid. Neurobiol Aging, 37, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N & Van Duijn CM 1997. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA, 278, 1349–56. [PubMed] [Google Scholar]
- Finan GM, Realubit R, Chung S, Lutjohann D, Wang N, Cirrito JR, Karan C & Kim TW 2016. Bioactive Compound Screen for Pharmacological Enhancers of Apolipoprotein E in Primary Human Astrocytes. Cell Chem Biol, 23, 1526–1538. [DOI] [PubMed] [Google Scholar]
- Fitz NF, Cronican A, Pham T, Fogg A, Fauq AH, Chapman R, Lefterov I & Koldamova R 2010. Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J Neurosci, 30, 6862–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, Cronican AA, Saleem M, Fauq AH, Chapman R, Lefterov I & Koldamova R 2012. Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J Neurosci, 32, 13125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, Cronican AA, Lefterov I & Koldamova R 2013. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science, 340, 924–c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, Castranio EL, Carter AY, Kodali R, Lefterov I & Koldamova R 2014. Improvement of memory deficits and amyloid-beta clearance in aged APP23 mice treated with a combination of anti-amyloid-beta antibody and LXR agonist. J Alzheimers Dis, 41, 535–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher A, Grundman M, Jack CR Jr., Petersen RC, Taylor C, Kim HT, Schiller DH, Bagwell V, Sencakova D, Weiner MF, Decarli C, Dekosky ST, Van Dyck CH, Thal LJ & Alzheimer’s Disease Cooperative, S. 2005. Sex, apolipoprotein E epsilon 4 status, and hippocampal volume in mild cognitive impairment. Arch Neurol, 62, 953–7. [DOI] [PubMed] [Google Scholar]
- Fortea J, Vilaplana E, Alcolea D, Carmona-Iragui M, Sanchez-Saudinos MB, Sala I, Anton-Aguirre S, Gonzalez S, Medrano S, Pegueroles J, Morenas E, Clarimon J, Blesa R, Lleo A & Alzheimer’s Disease Neuroimaging I 2014. Cerebrospinal fluid beta-amyloid and phospho-tau biomarker interactions affecting brain structure in preclinical Alzheimer disease. Ann Neurol, 76, 223–30. [DOI] [PubMed] [Google Scholar]
- Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM & Holtzman DM 2005. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci, 25, 2803–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F & et al. 1995. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature, 373, 523–7. [DOI] [PubMed] [Google Scholar]
- Gillespie AK, Jones EA, Lin YH, Karlsson MP, Kay K, Yoon SY, Tong LM, Nova P, Carr JS, Frank LM & Huang Y 2016. Apolipoprotein E4 Causes Age-Dependent Disruption of Slow Gamma Oscillations during Hippocampal Sharp-Wave Ripples. Neuron, 90, 740–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez-Llort L, Arranz L, Mate I & De La Fuente M 2008. Gender-specific neuroimmunoendocrine aging in a triple-transgenic 3xTg-AD mouse model for Alzheimer’s disease and its relation with longevity. Neuroimmunomodulation, 15, 331–43. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Price JL, Mckeel DW Jr., Morris JC, Growdon JH & Hyman BT 1996. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci, 16, 4491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grootendorst J, Bour A, Vogel E, Kelche C, Sullivan PM, Dodart JC, Bales K & Mathis C 2005. Human apoE targeted replacement mouse lines: h-apoE4 and h-apoE3 mice differ on spatial memory performance and avoidance behavior. Behav Brain Res, 159, 1–14. [DOI] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J & Alzheimer Genetic Analysis, G. 2013. TREM2 variants in Alzheimer’s disease. N Engl J Med, 368, 117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta VB, Laws SM, Villemagne VL, Ames D, Bush AI, Ellis KA, Lui JK, Masters C, Rowe CC, Szoeke C, Taddei K & Martins RN 2011. Plasma apolipoprotein E and Alzheimer disease risk: The AIBL study of aging. Neurology, 76, 1091–1098. [DOI] [PubMed] [Google Scholar]
- Hafiane A, Bielicki JK, Johansson JO & Genest J 2015. Novel Apo E-Derived ABCA1 Agonist Peptide (CS-6253) Promotes Reverse Cholesterol Transport and Induces Formation of prebeta-1 HDL In Vitro. PLoS One, 10, e0131997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handattu SP, Garber DW, Monroe CE, Van Groen T, Kadish I, Nayyar G, Cao D, Palgunachari MN, Li L & Anantharamaiah GM 2009. Oral apolipoprotein A I mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer’s disease. Neurobiol Dis, 34, 525–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman RE, Wozniak DF, Nardi A, Olney JW, Sartorius L & Holtzman DM 2001. Behavioral phenotyping of GFAP-apoE3 and -apoE4 transgenic mice: apoE4 mice show profound working memory impairments in the absence of Alzheimer’s-like neuropathology. Exp Neurol, 170, 326–44. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Serrano-Pozo A, Hori Y, Adams KW, Takeda S, Banerji AO, Mitani A, Joyner D, Thyssen DH, Bacskai BJ, Frosch MP, Spires-Jones TL, Finn MB, Holtzman DM & Hyman BT 2012. Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide. J Neurosci, 32, 15181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinsinger NM, Gachechiladze MA & Rebeck GW 2016. Apolipoprotein E Genotype Affects Size of ApoE Complexes in Cerebrospinal Fluid. J Neuropathol Exp Neurol, 75, 918–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglund K, Kern S, Zettergren A, Borjesson-Hansson A, Zetterberg H, Skoog I & Blennow K 2017. Preclinical amyloid pathology biomarker positivity: effects on tau pathology and neurodegeneration. Transl Psychiatry, 7, e995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, Mcgowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K & Duff K 1998. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med, 4, 97–100. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, Mckeel D, Wozniak D & Paul SM 2000. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA, 97, 2892–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X, Adeosun SO, Zhang Q, Barlow B, Brents M, Zheng B & Wang J 2015. Differential contributions of ApoE4 and female sex to BACE1 activity and expression mediate Abeta deposition and learning and memory in mouse models of Alzheimer’s disease. Front Aging Neurosci, 7, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F & Cole G 1996. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science, 274, 99–102. [DOI] [PubMed] [Google Scholar]
- Hu J, Liu CC, Chen XF, Zhang YW, Xu HX & Bu GJ 2015. Opposing effects of viral mediated brain expression of apolipoprotein E2 (apoE2) and apoE4 on apoE lipidation and A beta metabolism in apoE4-targeted replacement mice. Mol Neurodegener, 10, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y & Mahley RW 2014. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis, 72 Pt A, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber G, Marz W, Martin JR, Malherbe P, Richards JG, Sueoka N, Ohm T & Hoffmann MM 2000. Characterization of transgenic mice expressing apolipoprotein E4(C112R) and apolipoprotein E4(L28P; C112R). Neuroscience, 101, 211–8. [DOI] [PubMed] [Google Scholar]
- Hudry E, Dashkoff J, Roe AD, Takeda S, Koffie RM, Hashimoto T, Scheel M, Spires-Jones T, Arbel-Ornath M, Betensky R, Davidson BL & Hyman BT 2013. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med, 5, 212ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Perez-Tur J & Hardy J 1998. Genetics of Alzheimer’s disease. Essays in Biochemistry, 33, 117–31. [DOI] [PubMed] [Google Scholar]
- Huynh TV, Liao F, Francis CM, Robinson GO, Serrano JR, Jiang H, Roh J, Finn MB, Sullivan PM, Esparza TJ, Stewart FR, Mahan TE, Ulrich JD, Cole T & Holtzman DM 2017. Age-Dependent Effects of apoE Reduction Using Antisense Oligonucleotides in a Model of beta-amyloidosis. Neuron, 96, 1013–1023 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh TV, Wang C, Tran AC, Tabor GT, Mahan TE, Francis CM, Finn MB, Spellman R, Manis M, Tanzi RE, Ulrich JD & Holtzman DM 2019. Lack of hepatic apoE does not influence early Abeta deposition: observations from a new APOE knock-in model. Mol Neurodegener, 14, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT 2011. Amyloid-dependent and amyloid-independent stages of Alzheimer disease. Arch Neurol, 68, 1062–4. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV & Montine TJ 2012. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement, 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ 2016. Tauopathies as clinicopathological entities. Parkinsonism Relat Disord, 22 Suppl 1, S29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM & Gotz J 2011. Amyloid-beta and tau--a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci, 12, 65–72. [DOI] [PubMed] [Google Scholar]
- Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC & Trojanowski JQ 2010. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurology, 9, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Rudinskiy N, Herrmann AG, Croft S, Kim JM, Petrova V, Ramos-Rodriguez JJ, Pitstick R, Wegmann S, Garcia-Alloza M, Carlson GA, Hyman BT & Spires-Jones TL 2016. Human tau increases amyloid beta plaque size but not amyloid beta-mediated synapse loss in a novel mouse model of Alzheimer’s disease. Eur J Neurosci, 44, 3056–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Beck M, Stahl T, Holzer M, Brauer K, Bigl V & Arendt T 1999. Phylogenetic diversity of the expression of the microtubule-associated protein tau: implications for neurodegenerative disorders. Brain Res Mol Brain Res, 68, 119–28. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG & Borchelt DR 2001. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng, 17, 157–65. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL & Zheng H 2017. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol Neurodegener, 12, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawhar S, Trawicka A, Jenneckens C, Bayer TA & Wirths O 2012. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol Aging, 33, 196 e29–40. [DOI] [PubMed] [Google Scholar]
- Jay TR, Von Saucken VE & Landreth GE 2017. TREM2 in Neurodegenerative Diseases. Mol Neurodegener, 12, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Gong Y, Gan W, Beach T, Holtzman DM & Wisniewski T 2003. Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer’s disease patients. Neuroscience, 122, 305–15. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P & Landreth GE 2008. ApoE promotes the proteolytic degradation of Abeta. Neuron, 58, 681–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SC, Carrasquillo MM, Benitez BA, Skorupa T, Carrell D, Patel D, Lincoln S, Krishnan S, Kachadoorian M, Reitz C, Mayeux R, Wingo TS, Lah JJ, Levey AI, Murrell J, Hendrie H, Foroud T, Graff-Radford NR, Goate AM, Cruchaga C & Ertekin-Taner N 2015. TREM2 is associated with increased risk for Alzheimer’s disease in African Americans. Mol Neurodegener, 10, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Torres ER, Impey S, Stevens JF & Raber J 2017. Apolipoprotein E4 and Insulin Resistance Interact to Impair Cognition and Alter the Epigenome and Metabolome. Sci Rep, 7, 43701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Torres ER, Weber Boutros S, Patel E, Akinyeke T, Alkayed NJ & Raber J 2019. Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J Cereb Blood Flow Metab, 39, 770–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, Van Duijn CM, Thorsteinsdottir U, Kong A & Stefansson K 2013. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med, 368, 107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampers T, Pangalos M, Geerts H, Wiech H & Mandelkow E 1999. Assembly of paired helical filaments from mouse tau: implications for the neurofibrillary pathology in transgenic mouse models for Alzheimer’s disease. FEBS Lett, 451, 39–44. [DOI] [PubMed] [Google Scholar]
- Karasinska JM, Rinninger F, Lutjohann D, Ruddle P, Franciosi S, Kruit JK, Singaraja RR, Hirsch-Reinshagen V, Fan J, Brunham LR, Bissada N, Ramakrishnan R, Wellington CL, Parks JS & Hayden MR 2009. Specific loss of brain ABCA1 increases brain cholesterol uptake and influences neuronal structure and function. J Neurosci, 29, 3579–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasinska JM, De Haan W, Franciosi S, Ruddle P, Fan J, Kruit JK, Stukas S, Lutjohann D, Gutmann DH, Wellington CL & Hayden MR 2013. ABCA1 influences neuroinflammation and neuronal death. Neurobiol Dis, 54, 445–55. [DOI] [PubMed] [Google Scholar]
- Katz A, Udata C, Ott E, Hickey L, Burczynski ME, Burghart P, Vesterqvist O & Meng X 2009. Safety, pharmacokinetics, and pharmacodynamics of single doses of LXR-623, a novel liver X-receptor agonist, in healthy participants. J Clin Pharmacol, 49, 643–9. [DOI] [PubMed] [Google Scholar]
- Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, Basak JM, Finn MB & Holtzman DM 2011. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J Neurosci, 31, 18007–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Eltorai AE, Jiang H, Liao F, Verghese PB, Kim J, Stewart FR, Basak JM & Holtzman DM 2012. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J Exp Med, 209, 2149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchgessner TG, Sleph P, Ostrowski J, Lupisella J, Ryan CS, Liu X, Fernando G, Grimm D, Shipkova P, Zhang R, Garcia R, Zhu J, He A, Malone H, Martin R, Behnia K, Wang Z, Barrett YC, Garmise RJ, Yuan L, Zhang J, Gandhi MD, Wastall P, Li T, Du S, Salvador L, Mohan R, Cantor GH, Kick E, Lee J & Frost RJ 2016. Beneficial and Adverse Effects of an LXR Agonist on Human Lipid and Lipoprotein Metabolism and Circulating Neutrophils. Cell Metab, 24, 223–33. [DOI] [PubMed] [Google Scholar]
- Kjaergaard M, Dear AJ, Kundel F, Qamar S, Meisl G, Knowles TPJ & Klenerman D 2018. Oligomer Diversity during the Aggregation of the Repeat Region of Tau. ACS Chem Neurosci, 9, 3060–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoferle J, Yoon SY, Walker D, Leung L, Gillespie AK, Tong LM, Bien-Ly N & Huang YD 2014. Apolipoprotein E4 Produced in GABAergic Interneurons Causes Learning and Memory Deficits in Mice. Journal of Neuroscience, 34, 14069–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Hashimoto T, Tai HC, Kay KR, Serrano-Pozo A, Joyner D, Hou S, Kopeikina KJ, Frosch MP, Lee VM, Holtzman DM, Hyman BT & Spires-Jones TL 2012. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-beta. Brain, 135, 2155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova RP, Lefterov IM, Staufenbiel M, Wolfe D, Huang S, Glorioso JC, Walter M, Roth MG & Lazo JS 2005. The Liver X Receptor Ligand T0901317 Decreases Amyloid-β Production in Vitro and in a Mouse Model of Alzheimer’s Disease. J Biol Chem, 280, 4079–88. [DOI] [PubMed] [Google Scholar]
- Koopal C, Marais AD, Westerink J & Visseren FL 2017. Autosomal dominant familial dysbetalipoproteinemia: A pathophysiological framework and practical approach to diagnosis and therapy. J Clin Lipidol, 11, 12–23 e1. [DOI] [PubMed] [Google Scholar]
- Koriath C, Lashley T, Taylor W, Druyeh R, Dimitriadis A, Denning N, Williams J, Warren JD, Fox NC, Schott JM, Rowe JB, Collinge J, Rohrer JD & Mead S 2019. ApoE4 lowers age at onset in patients with frontotemporal dementia and tauopathy independent of amyloid-beta copathology. Alzheimers Dement (Amst), 11, 277–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss DJ, Robinson L, Drever BD, Plucinska K, Stoppelkamp S, Veselcic P, Riedel G & Platt B 2016. Mutant Tau knock-in mice display frontotemporal dementia relevant behaviour and histopathology. Neurobiol Dis, 91, 105–23. [DOI] [PubMed] [Google Scholar]
- Koster KP, Smith C, Valencia-Olvera AC, Thatcher GR, Tai LM & Ladu MJ 2017. Rexinoids as Therapeutics for Alzheimer’s Disease: Role of APOE. Curr Top Med Chem, 17, 708–20. [DOI] [PubMed] [Google Scholar]
- Koutseff A, Mittelhaeuser C, Essabri K, Auwerx J & Meziane H 2014. Impact of the apolipoprotein E polymorphism, age and sex on neurogenesis in mice: pathophysiological relevance for Alzheimer’s disease? Brain Res, 1542, 32–40. [DOI] [PubMed] [Google Scholar]
- Ladu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS & Frail DE 1994. Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem, 269, 23403–6. [PubMed] [Google Scholar]
- Ladu MJ, Munson GW, Jungbauer L, Getz GS, Reardon CA, Tai LM & Yu C 2012. Preferential interactions between ApoE-containing lipoproteins and Abeta revealed by a detection method that combines size exclusion chromatography with non-reducing gel-shift. Biochim Biophys Acta, 1821, 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Mann D, Richard F, Tian J, Shi J, Thaker U, Merrot S, Harris J, Frigard B, Iwatsubo T, Lendon C & Amouyel P 2005. Is there a relation between APOE expression and brain amyloid load in Alzheimer’s disease? J Neurol Neurosurg Psychiatry, 76, 928–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane-Donovan C & Herz J 2016. High-Fat Diet Changes Hippocampal Apolipoprotein E (ApoE) in a Genotype- and Carbohydrate-Dependent Manner in Mice. PLoS One, 11, e0148099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Jackson GR & Kayed R 2011. Tau oligomers as potential targets for immunotherapy for Alzheimer’s disease and tauopathies. Curr Alzheimer Res, 8, 659–65. [DOI] [PubMed] [Google Scholar]
- Laskowitz DT, Mckenna SE, Song P, Wang H, Durham L, Yeung N, Christensen D & Vitek MP 2007. COG1410, a novel apolipoprotein E-based peptide, improves functional recovery in a murine model of traumatic brain injury. J Neurotrauma, 24, 1093–107. [DOI] [PubMed] [Google Scholar]
- Laskowitz DT, Song P, Wang H, Mace B, Sullivan PM, Vitek MP & Dawson HN 2010. Traumatic brain injury exacerbates neurodegenerative pathology: improvement with an apolipoprotein E-based therapeutic. J Neurotrauma, 27, 1983–95. [DOI] [PubMed] [Google Scholar]
- Leung L, Andrews-Zwilling Y, Yoon SY, Jain S, Ring K, Dai J, Wang MM, Tong L, Walker D & Huang Y 2012. Apolipoprotein E4 causes age- and sex-dependent impairments of hilar GABAergic interneurons and learning and memory deficits in mice. PLoS One, 7, e53569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Hori Y, Hudry E, Bauer AQ, Jiang H, Mahan TE, Lefton KB, Zhang TJ, Dearborn JT, Kim J, Culver JP, Betensky R, Wozniak DF, Hyman BT & Holtzman DM 2014. Anti-ApoE antibody given after plaque onset decreases Abeta accumulation and improves brain function in a mouse model of Abeta amyloidosis. J Neurosci, 34, 7281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Zhang TJ, Jiang H, Lefton KB, Robinson GO, Vassar R, Sullivan PM & Holtzman DM 2015. Murine versus human apolipoprotein E4: differential facilitation of and co-localization in cerebral amyloid angiopathy and amyloid plaques in APP transgenic mouse models. Acta Neuropathol Commun, 3, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Li A, Xiong M, Bien-Ly N, Jiang H, Zhang Y, Finn MB, Hoyle R, Keyser J, Lefton KB, Robinson GO, Serrano JR, Silverman AP, Guo JL, Getz J, Henne K, Leyns CE, Gallardo G, Ulrich JD, Sullivan PM, Lerner EP, Hudry E, Sweeney ZK, Dennis MS, Hyman BT, Watts RJ & Holtzman DM 2018. Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Invest, 128, 2144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippi SLP, Smith ML & Flinn JM 2018. A Novel hAPP/htau Mouse Model of Alzheimer’s Disease: Inclusion of APP With Tau Exacerbates Behavioral Deficits and Zinc Administration Heightens Tangle Pathology. Front Aging Neurosci, 10, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Liu CC, Kanekiyo T, Xu H & Bu G 2013. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol, 9, 106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Zhao N, Fu Y, Wang N, Linares C, Tsai CW & Bu G 2017a. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron, 96, 1024–32 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PP, Xie Y, Meng XY & Kang JS 2019. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct Target Ther, 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Breitbart A, Sun Y, Mehta PD, Boutajangout A, Scholtzova H & Wisniewski T 2014. Blocking the apolipoprotein E/amyloid beta interaction in triple transgenic mice ameliorates Alzheimer’s disease related amyloid beta and tau pathology. J Neurochem, 128, 577–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Park S, Allington G, Prelli F, Sun Y, Marta-Ariza M, Scholtzova H, Biswas G, Brown B, Verghese PB, Mehta PD, Kwon YU & Wisniewski T 2017b. Targeting Apolipoprotein E/Amyloid beta Binding by Peptoid CPO_Abeta17–21 P Ameliorates Alzheimer’s Disease Related Pathology and Cognitive Decline. Sci Rep, 7, 8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loring JF, Paszty C, Rose A, Mcintosh TK, Murai H, Pierce JE, Schramm SR, Wymore K, Lee VM, Trojanowski JQ & Peterson KR 1996. Rational design of an animal model for Alzheimer’s disease: introduction of multiple human genomic transgenes to reproduce AD pathology in a rodent. Neurobiol Aging, 17, 173–82. [DOI] [PubMed] [Google Scholar]
- Lynch JR, Wang H, Mace B, Leinenweber S, Warner DS, Bennett ER, Vitek MP, Mckenna S & Laskowitz DT 2005. A novel therapeutic derived from apolipoprotein E reduces brain inflammation and improves outcome after closed head injury. Exp Neurol, 192, 109–16. [DOI] [PubMed] [Google Scholar]
- Ma J, Zhou Y, Xu J, Liu X, Wang Y, Deng Y, Wang G, Xu W, Ren R, Liu X, Zhang Y, Wang C, Tang H & Chen S 2014. Association study of TREM2 polymorphism rs75932628 with late-onset Alzheimer’s disease in Chinese Han population. Neurol Res, 36, 894–6. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Huang Y & Rall SC Jr. 1999. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J Lipid Res, 40, 1933–49. [PubMed] [Google Scholar]
- Mahley RW, Weisgraber KH & Huang Y 2006. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A, 103, 5644–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW & Huang Y 2012. Small-molecule structure correctors target abnormal protein structure and function: structure corrector rescue of apolipoprotein e4-associated neuropathology. J Med Chem, 55, 8997–9008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamo JCL, Lam V, Brook E, Mooranian A, Al-Salami H, Fimognari N, Nesbit M & Takechi R 2019. Probucol prevents blood-brain barrier dysfunction and cognitive decline in mice maintained on pro-diabetic diet. Diab Vasc Dis Res, 16, 87–97. [DOI] [PubMed] [Google Scholar]
- Marottoli FM, Katsumata Y, Koster KP, Thomas R, Fardo DW & Tai LM 2017. Peripheral Inflammation, Apolipoprotein E4, and Amyloid-beta Interact to Induce Cognitive and Cerebrovascular Dysfunction. ASN Neuro, 9, 1759091417719201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez M, Campion D, Brice A, Hannequin D, Dubois B, Didierjean O, Michon A, Thomas-Anterion C, Puel M, Frebourg T, Agid Y & Clerget-Darpoux F 1998. Apolipoprotein E epsilon4 allele and familial aggregation of Alzheimer disease. Arch Neurol, 55, 810–6. [DOI] [PubMed] [Google Scholar]
- Martinez-Morillo E, Hansson O, Atagi Y, Bu G, Minthon L, Diamandis EP & Nielsen HM 2014. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol, 127, 633–43. [DOI] [PubMed] [Google Scholar]
- Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA & Cummings JL 2015. Alzheimer’s Disease. Nat Rev Dis Primers, 1:15056. [DOI] [PubMed] [Google Scholar]
- Masuda A, Kobayashi Y, Kogo N, Saito T, Saido TC & Itohara S 2016. Cognitive deficits in single App knock-in mouse models. Neurobiol Learn Mem, 135, 73–82. [DOI] [PubMed] [Google Scholar]
- Matthews FE, Brayne C, Lowe J, Mckeith I, Wharton SB & Ince P 2009. Epidemiological pathology of dementia: attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study. PLoS Med, 6, e1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrjoo Z, Najmabadi A, Abedini SS, Mohseni M, Kamali K, Najmabadi H & Khorram Khorshid HR 2015. Association Study of the TREM2 Gene and Identification of a Novel Variant in Exon 2 in Iranian Patients with Late-Onset Alzheimer’s Disease. Med Princ Pract, 24, 351–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Jackson R, Paul G, Shi J & Sabbagh M 2017. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin Investig Drugs, 26, 735–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng FT, Zhao J, Fang H & Liu YJ 2015. The influence of chronic stress on anxiety-like behavior and cognitive function in different human GFAP-ApoE transgenic adult male mice. Stress, 18, 419–26. [DOI] [PubMed] [Google Scholar]
- Miyashita A, Wen Y, Kitamura N, Matsubara E, Kawarabayashi T, Shoji M, Tomita N, Furukawa K, Arai H, Asada T, Harigaya Y, Ikeda M, Amari M, Hanyu H, Higuchi S, Nishizawa M, Suga M, Kawase Y, Akatsu H, Imagawa M, Hamaguchi T, Yamada M, Morihara T, Takeda M, Takao T, Nakata K, Sasaki K, Watanabe K, Nakashima K, Urakami K, Ooya T, Takahashi M, Yuzuriha T, Serikawa K, Yoshimoto S, Nakagawa R, Saito Y, Hatsuta H, Murayama S, Kakita A, Takahashi H, Yamaguchi H, Akazawa K, Kanazawa I, Ihara Y, Ikeuchi T & Kuwano R 2014. Lack of genetic association between TREM2 and late-onset Alzheimer’s disease in a Japanese population. J Alzheimers Dis, 41, 1031–8. [DOI] [PubMed] [Google Scholar]
- Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, Petrova O, Schonig K, Bujard H, Mandelkow E, Zhou L, Rune G & Mandelkow EM 2008. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci, 28, 737–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molero AE, Pino-Ramirez G & Maestre GE 2001. Modulation by age and gender of risk for Alzheimer’s disease and vascular dementia associated with the apolipoprotein E-epsilon4 allele in Latin Americans: findings from the Maracaibo Aging Study. Neurosci Lett, 307, 5–8. [DOI] [PubMed] [Google Scholar]
- Moser VA & Pike CJ 2017. Obesity Accelerates Alzheimer-Related Pathology in APOE4 but not APOE3 Mice. eNeuro, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutinho M & Landreth GE 2017. Therapeutic potential of nuclear receptor agonists in Alzheimer’s disease. J Lipid Res, 58, 1937–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K & Mcconlogue L 2000. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci, 20, 4050–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam KN, Wolfe CM, Fitz NF, Letronne F, Castranio EL, Mounier A, Schug J, Lefterov I & Koldamova R 2018. Integrated approach reveals diet, APOE genotype and sex affect immune response in APP mice. Biochim Biophys Acta Mol Basis Dis, 1864, 152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necula M, Kayed R, Milton S & Glabe CG 2007. Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J Biol Chem, 282, 10311–24. [DOI] [PubMed] [Google Scholar]
- Neustadtl AL, Winston CN, Parsadanian M, Main BS, Villapol S & Burns MP 2017. Reduced cortical excitatory synapse number in APOE4 mice is associated with increased calcineurin activity. Neuroreport, 28, 618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, Dhanasekaran P, Nickel M, Mizuguchi C, Watanabe M, Saito H, Phillips MC & Lund-Katz S 2014. Influence of domain stability on the properties of human apolipoprotein E3 and E4 and mouse apolipoprotein E. Biochemistry, 53, 4025–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton S, Matthews FE, Barnes DE, Yaffe K & Brayne C 2014. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol, 13, 788–94. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R & Vassar R 2006. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci, 26, 10129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP & Laferla FM 2003a. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging, 24, 1063–70. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y & Laferla FM 2003b. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron, 39, 409–21. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Cheng D & Laferla FM 2009. Genetically altering Abeta distribution from the brain to the vasculature ameliorates tau pathology. Brain Pathol, 19, 421–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oram JF 2002. The cholesterol mobilizing transporter ABCA1 as a new therapeutic target for cardiovascular disease. Trends Cardiovasc Med, 12, 170–5. [DOI] [PubMed] [Google Scholar]
- Oveisgharan S, Arvanitakis Z, Yu L, Farfel J, Schneider JA & Bennett DA 2018. Sex differences in Alzheimer’s disease and common neuropathologies of aging. Acta Neuropathologica, 136, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiewicz JE, Guridi M, Kim J, Asuni AA, Sanchez S, Sullivan PM, Holtzman DM & Sadowski MJ 2014. Blocking the apoE/Abeta interaction ameliorates Abeta-related pathology in APOE epsilon2 and epsilon4 targeted replacement Alzheimer model mice. Acta Neuropathol Commun, 2, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiewicz JE, Baquero-Buitrago J, Sanchez S, Lopez-Contreras J, Kim J, Sullivan PM, Holtzman DM & Sadowski MJ 2017. APOE Genotype Differentially Modulates Effects of Anti-Abeta, Passive Immunization in APP Transgenic Mice. Mol Neurodegener, 12, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoal TA, Mathotaarachchi S, Mohades S, Benedet AL, Chung CO, Shin M, Wang S, Beaudry T, Kang MS, Soucy JP, Labbe A, Gauthier S & Rosa-Neto P 2017a. Amyloid-beta and hyperphosphorylated tau synergy drives metabolic decline in preclinical Alzheimer’s disease. Mol Psychiatry, 22, 306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoal TA, Mathotaarachchi S, Shin M, Benedet AL, Mohades S, Wang S, Beaudry T, Kang MS, Soucy JP, Labbe A, Gauthier S, Rosa-Neto P & Alzheimer’s Disease Neuroimaging I 2017b. Synergistic interaction between amyloid and tau predicts the progression to dementia. Alzheimers Dement, 13, 644–53. [DOI] [PubMed] [Google Scholar]
- Patra K, Giannisis A & Nielsen H 2018. Neuropathological findings driven by an APOEε4 liver phenotype. Alzheimer’s and Dementia, 14, P652. [Google Scholar]
- Peila R, Rodriguez BL, Launer LJ & Honolulu-Asia Aging S 2002. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes, 51, 1256–62. [DOI] [PubMed] [Google Scholar]
- Perez C, Miti T, Hasecke F, Meisl G, Hoyer W, Muschol M & Ullah G 2019. Mechanism of Fibril and Soluble Oligomer Formation in Amyloid Beta and Hen Egg White Lysozyme Proteins. J Phys Chem B, 123, 5678–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervolaraki E, Hall SP, Foresteire D, Saito T, Saido TC, Whittington MA, Lever C & Dachtler J 2019. Insoluble Abeta overexpression in an App knock-in mouse model alters microstructure and gamma oscillations in the prefrontal cortex, affecting anxiety-related behaviours. Dis Model Mech, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimplikar SW Reassessing the amyloid cascade hypothesis of Alzheimer’s disease. Int J Biochem Cell Biol, 41, 1261–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollinger J, Gellrich L, Schierle S, Kilu W, Schmidt J, Kalinowsky L, Ohrndorf J, Kaiser A, Heering J, Proschak E & Merk D 2019. Tuning Nuclear Receptor Selectivity of Wy14,643 towards Selective Retinoid X Receptor Modulation. J Med Chem, 62, 2112–26. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M & Schellenberg GD 1998. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol, 43, 815–25. [DOI] [PubMed] [Google Scholar]
- Powers ET & Powers DL 2008. Mechanisms of protein fibril formation: nucleated polymerization with competing off-pathway aggregation. Biophys J, 94, 379–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AR, Xu G, Siemienski ZB, Smithson LA, Borchelt DR, Golde TE & Felsenstein KM 2013. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science, 340, 924–d. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Cavelier L, Chiu S, Yang X, Rubin E & Cheng JF 2001. Human and mouse ABCA1 comparative sequencing and transgenesis studies revealing novel regulatory sequences. Genomics, 73, 66–76. [DOI] [PubMed] [Google Scholar]
- Quinet EM, Savio DA, Halpern AR, Chen L, Schuster GU, Gustafsson JA, Basso MD & Nambi P 2006. Liver X receptor (LXR)-beta regulation in LXRalpha-deficient mice: implications for therapeutic targeting. Mol Pharmacol, 70, 1340–9. [DOI] [PubMed] [Google Scholar]
- Raber J, Wong D, Buttini M, Orth M, Bellosta S, Pitas RE, Mahley RW & Mucke L 1998. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc Natl Acad Sci USA, 95, 10914–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaggi F, Wolburg H, Gengler S, Haass C, Ghetti B, Czech C, Holscher C, Mathews PM & Jucker M 2006. Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep, 7, 940–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajavashisth TB, Kaptein JS, Reue KL & Lusis AJ 1985. Evolution of apolipoprotein E: mouse sequence and evidence for an 11-nucleotide ancestral unit. Proc Natl Acad Sci USA, 82, 8085–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy G, Xu Q, Huang YD & Weisgraber KH 2005. Effect of domain interaction on apolipoprotein E levels in mouse brain. J Neurosci, 25, 10658–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden M, Kotilinek L, Forster C, Paulson J, Mcgowan E, Santacruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M & Ashe KH 2005. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci, 25, 10637–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeck GW 2017. The role of APOE on lipid homeostasis and inflammation in normal brains. J Lipid Res, 58, 1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G, Bao W, Zeng Z, Zhang W, Shang C, Wang M, Su Y, Zhang XK & Zhou H 2019. Retinoid X Receptor Alpha Nitro-ligand Z-10 and Its Optimized Derivative Z-36 Reduce beta-Amyloid Plaques in Alzheimer’s Disease Mouse Model. Mol Pharm, 16, 480–488. [DOI] [PubMed] [Google Scholar]
- Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, Ring RH, Kirksey Y, Aschmies S, Xu J, Kubek K, Hirst WD, Gonzales C, Chen Y, Murphy E, Leonard S, Vasylyev D, Oganesian A, Martone RL, Pangalos MN, Reinhart PH & Jacobsen JS 2007. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol Cell Neurosci, 34, 621–8. [DOI] [PubMed] [Google Scholar]
- Riddell DR, Zhou H, Atchison K, Warwick HK, Atkinson PJ, Jefferson J, Xu L, Aschmies S, Kirksey Y, Hu Y, Wagner E, Parratt A, Xu J, Li Z, Zaleska MM, Jacobsen JS, Pangalos MN & Reinhart PH 2008a. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci, 28, 11445–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell DR, Zhou H, Atchison K, Warwick HK, Atkinson PJ, Jefferson J, Xu L, Aschmies S, Kirksey Y, Hu Y, Wagner E, Parratt A, Xu J, Li ZT, Zaleska MM, Jacobsen JS, Pangalos MN & Reinhart PH 2008b. Impact of Apolipoprotein E (ApoE) Polymorphism on Brain ApoE Levels. J Neurosci, 28, 11445–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED 2012. Mouse models of frontotemporal dementia. Ann Neurol, 72, 837–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert J, Button EB, Yuen B, Gilmour M, Kang K, Bahrabadi A, Stukas S, Zhao W, Kulic I & Wellington CL 2017. Clearance of beta-amyloid is facilitated by apolipoprotein E and circulating high-density lipoproteins in bioengineered human vessels. Elife, 6, e29595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT 2013. Proteolytic cleavage of apolipoprotein E4 as the keystone for the heightened risk associated with Alzheimer’s disease. Int J Mol Sci, 14, 14908–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski M, Pankiewicz J, Scholtzova H, Ripellino JA, Li Y, Schmidt SD, Mathews PM, Fryer JD, Holtzman DM, Sigurdsson EM & Wisniewski T 2004. A synthetic peptide blocking the apolipoprotein E/beta-amyloid binding mitigates beta-amyloid toxicity and fibril formation in vitro and reduces beta-amyloid plaques in transgenic mice. Am J Pathol, 165, 937–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara N, Perez PD, Lin WL, Dickson DW, Ren Y, Zeng H, Lewis J & Febo M 2013. Age-related decline in white matter integrity in a mouse model of tauopathy: An in vivo diffusion tensor magnetic resonance imaging study. Neurobiol Aging, 35, 1364–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N & Saido TC 2014. Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci, 17, 661–3. [DOI] [PubMed] [Google Scholar]
- Saito T, Mihira N, Matsuba Y, Sasaguri H, Hashimoto S, Narasimhan S, Zhang B, Murayama S, Higuchi M, Lee VMY, Trojanowski JQ & Saido TC 2019. Humanization of the entire murine Mapt gene provides a murine model of pathological human tau propagation. J Biol Chem, 294, 12754–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara Y, Sekiya M, Saito T, Saido TC & Iijima KM 2018. Cognitive and emotional alterations in App knock-in mouse models of Abeta amyloidosis. BMC Neurosci, 19, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakono M & Zako T 2010. Amyloid oligomers: formation and toxicity of Abeta oligomers. FEBS J, 277, 1348–58. [DOI] [PubMed] [Google Scholar]
- Samieri C, Lorrain S, Buaud B, Vaysse C, Berr C, Peuchant E, Cunnane SC & Barberger-Gateau P 2013. Relationship between diet and plasma long-chain n-3 PUFAs in older people: impact of apolipoprotein E genotype. J Lipid Res, 54, 2559–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos DB, Peres KC, Ribeiro RP, Colle D, Dos Santos AA, Moreira ELG, Souza DOG, Figueiredo CP & Farina M 2012. Probucol, a lipid-lowering drug, prevents cognitive and hippocampal synaptic impairments induced by amyloid beta peptide in mice. Exp Neurol, 233, 767–75. [DOI] [PubMed] [Google Scholar]
- Sasner M 2019. B6(SJL)-Apoetm1.1(APOE*4)Adiuj Trem2em1Adiuj/J [Online]. Jax.org: The Jackson Laboratory. Available: https://www.jax.org/strain/028709 [Accessed December 30 2019].
- Sawmiller D, Habib A, Hou HY, Mori T, Fan AR, Tian J, Zeng J, Giunta B, Sanberg PR, Mattson MP & Tan J 2019. A Novel Apolipoprotein E Antagonist Functionally Blocks Apolipoprotein E Interaction With N-terminal Amyloid Precursor Protein, Reduces beta-Amyloid-Associated Pathology, and Improves Cognition. Biol Psychiatry, 86, 208–20. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD & Shan B 2000. Role of LXRs in control of lipogenesis. Genes Dev, 14, 2831–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ & Hardy J 2016. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med, 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant N, Delacourte A & Buee L 2005. Tau protein as a differential biomarker of tauopathies. Biochim Biophys Acta, 1739, 179–97. [DOI] [PubMed] [Google Scholar]
- Serrano-Pozo A, Frosch MP, Masliah E & Hyman BT 2011. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med, 1, a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafiei SS, Guerrero-Munoz MJ & Castillo-Carranza DL 2017. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front Aging Neurosci, 9, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, Mccue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C, Alzheimer’s Disease Neuroimaging I, Fagan AM, Miller BL, Boxer AL, Seeley WW, Butovsky O, Barres BA, Paul SM & Holtzman DM 2017. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature, 549, 523–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonet WS, Bucay N, Lauer SJ & Taylor JM 1993. A far-downstream hepatocyte-specific control region directs expression of the linked human apolipoprotein E and C-I genes in transgenic mice. J Biol Chem, 268, 8221–9. [PubMed] [Google Scholar]
- Smith JD, Sikes J & Levin JA 1998. Human apolipoprotein E allele-specific brain expressing transgenic mice. Neurobiol Aging, 19, 407–13. [DOI] [PubMed] [Google Scholar]
- Sperling RA, Jack CR Jr. & Aisen PS 2011. Testing the right target and right drug at the right stage. Sci Transl Med, 3, 111cm33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG & Goedert M 1998. Tau protein pathology in neurodegenerative diseases. Trends Neurosci, 21, 428–33. [DOI] [PubMed] [Google Scholar]
- Stephen TL, Cacciottolo M, Balu D, Morgan TE, Ladu MJ, Finch CE & Pike CJ 2019. APOE genotype and sex affect microglial interactions with plaques in Alzheimer’s disease mice. Acta Neuropathol Commun, 7, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukas S, Robert J & Wellington CL 2014. High-density lipoproteins and cerebrovascular integrity in Alzheimer’s disease. Cell Metab, 19, 574–91. [DOI] [PubMed] [Google Scholar]
- Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH & Maeda N 1997. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem, 272, 17972–80. [DOI] [PubMed] [Google Scholar]
- Sullivan PM, Mezdour H, Quarfordt SH & Maeda N 1998. Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe*2. J Clin Invest, 102, 130–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PM, Mace BE, Maeda N & Schmechel DE 2004. Marked regional differences of brain human apolipoprotein E expression in targeted replacement mice. Neuroscience, 124, 725–33. [DOI] [PubMed] [Google Scholar]
- Sun GZ, He YC, Ma XK, Li ST, Chen DJ, Gao M, Qiu SF, Yin JX, Shi J & Wu J 2017. Hippocampal synaptic and neural network deficits in young mice carrying the human APOE4 gene. CNS Neurosci Ther, 23, 748–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Wu S, Bu G, Onifade MK, Patel SN, Ladu MJ, Fagan AM & Holtzman DM 1998. Glial fibrillary acidic protein-apolipoprotein E (apoE) transgenic mice: astrocyte-specific expression and differing biological effects of astrocyte-secreted apoE3 and apoE4 lipoproteins. J Neurosci, 18, 3261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai LM, Youmans KL, Jungbauer L, Yu C & Ladu MJ 2011. Introducing Human APOE into Abeta Transgenic Mouse Models. Int J Alzheimers Dis, 2011, 810981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai LM, Bilousova T, Jungbauer L, Roeske SK, Youmans KL, Yu C, Poon WW, Cornwell LB, Miller CA, Vinters HV, Van Eldik LJ, Fardo DW, Estus S, Bu G, Gylys KH & Ladu MJ 2013. Levels of soluble apolipoprotein E/amyloid-beta (Abeta) complex are reduced and oligomeric Abeta increased with APOE4 and Alzheimer disease in a transgenic mouse model and human samples. J Biol Chem, 288, 5914–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai LM, Koster KP, Luo J, Lee SH, Wang YT, Collins NC, Ben Aissa M, Thatcher GR & Ladu MJ 2014a. Amyloid-beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J Biol Chem, 289, 30538–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai LM, Mehra S, Shete V, Estus S, Rebeck GW, Bu GJ & Ladu MJ 2014b. Soluble apoE/A beta complex: mechanism and therapeutic target for APOE4-induced AD risk. Molecular Neurodegeneration, 9, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai LM, Balu D, Avila-Munoz E, Abdullah L, Thomas R, Collins N, Valencia-Olvera AC & Ladu MJ 2017. EFAD Transgenic Mice as a Human APOE Relevant Preclinical Model of Alzheimer’s Disease. J Lipid Res, 58, 1733–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamboli IY, Heo D & Rebeck GW 2014. Extracellular proteolysis of apolipoprotein E (apoE) by secreted serine neuronal protease. PLoS One, 9, e93120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang KF, Cai L & Zhou JN 2009. Observation of the density and size of cells in hippocampus and vascular lesion in thalamus of GFAP-apoE transgenic mice. Neurosci Bull, 25, 167–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwel D, Steffensen KR, Verghese PB, Kummer MP, Gustafsson JA, Holtzman DM & Heneka MT 2011. Critical role of astroglial apolipoprotein E and liver X receptor-alpha expression for microglial Abeta phagocytosis. J Neurosci, 31, 7049–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, Van Dorpe J, Bruynseels K, Bronfman F, Sciot R, Van Lommel A & Van Leuven F 2000a. Prominent axonopathy and disruption of axonal transport in transgenic mice expressing human apolipoprotein E4 in neurons of brain and spinal cord. Am J Pathol, 157, 1495–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, Van Dorpe J, Spittaels K, Van Den Haute C, Moechars D & Van Leuven F 2000b. Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am J Pathol, 156, 951–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, Lo AC, Roberfroid A, Dietvorst S, Van Broeck B, Borgers M, Gijsen H, Moechars D, Mercken M, Kemp J, D’hooge R & De Strooper B 2013. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science, 340, 924–e. [DOI] [PubMed] [Google Scholar]
- Thomas R, Zuchowska P, Morris AW, Marottoli FM, Sunny S, Deaton R, Gann PH & Tai LM 2016. Epidermal growth factor prevents APOE4 and amyloid-beta-induced cognitive and cerebrovascular deficits in female mice. Acta Neuropathol Commun, 4, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungar L, Altmann A & Greicius MD 2014. Apolipoprotein E, gender, and Alzheimer’s disease: an overlooked, but potent and promising interaction. Brain Imaging Behav, 8, 262–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN 2010. Mysterious oligomerization of the amyloidogenic proteins. FEBS J, 277, 2940–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cauwenberghe C, Van Broeckhoven C & Sleegers K 2016. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med, 18, 421–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dooren T, Muyllaert D, Borghgraef P, Cresens A, Devijver H, Van Der Auwera I, Wera S, Dewachter I & Van Leuven F 2006. Neuronal or glial expression of human apolipoprotein e4 affects parenchymal and vascular amyloid pathology differentially in different brain regions of double- and triple-transgenic mice. Am J Pathol, 168, 245–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meer P, Acevedo S & Raber J 2007. Impairments in spatial memory retention of GFAP-apoE4 female mice. Behav Brain Res, 176, 372–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanmierlo T, Rutten K, Dederen J, Bloks VW, Van Vark-Van Der Zee LC, Kuipers F, Kiliaan A, Blokland A, Sijbrands EJ, Steinbusch H, Prickaerts J, Lutjohann D & Mulder M 2011. Liver X receptor activation restores memory in aged AD mice without reducing amyloid. Neurobiol Aging, 32, 1262–72. [DOI] [PubMed] [Google Scholar]
- Veeraraghavalu K, Zhang C, Miller S, Hefendehl JK, Rajapaksha TW, Ulrich J, Jucker M, Holtzman DM, Tanzi RE, Vassar R & Sisodia SS 2013. Comment on “ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models”. Science, 340, 924–f. [DOI] [PubMed] [Google Scholar]
- Vest RS & Pike CJ 2013. Gender, sex steroid hormones, and Alzheimer’s disease. Horm Behav, 63, 301–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitek MP, Christensen DJ, Wilcock D, Davis J, Van Nostrand WE, Li FQ & Colton CA 2012. APOE-mimetic peptides reduce behavioral deficits, plaques and tangles in Alzheimer’s disease transgenics. Neurodegener Dis, 10, 122–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, Kowalewski T & Holtzman DM 2004. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem, 279, 40987–93. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM & Holtzman DM 2005. Deletion of Abca1 Increases Aβ Deposition in the PDAPP Transgenic Mouse Model of Alzheimer Disease. J Biol Chem, 280, 43236–42. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, Jain S, Hirsch-Reinshagen V, Wellington CL, Bales KR, Paul SM & Holtzman DM 2008. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest, 118, 671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wilson WA, Moore SD, Mace BE, Maeda N, Schmechel DE & Sullivan PM 2005. Human apoE4-targeted replacement mice display synaptic deficits in the absence of neuropathology. Neurobiol Dis, 18, 390–8. [DOI] [PubMed] [Google Scholar]
- Wang C, Najm R, Xu Q, Jeong DE, Walker D, Balestra ME, Yoon SY, Yuan H, Li G, Miller ZA, Miller BL, Malloy MJ & Huang Y 2018a. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med, 24, 647–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Tanila H, Puolivali J, Kadish I & Van Groen T 2003. Gender differences in the amount and deposition of amyloidbeta in APPswe and PS1 double transgenic mice. Neurobiol Dis, 14, 318–27. [DOI] [PubMed] [Google Scholar]
- Wang P, Guo Q, Zhou Y, Chen K, Xu Y, Ding D, Hong Z & Zhao Q 2018b. Lack of association between triggering receptor expressed on myeloid cells 2 polymorphism rs75932628 and late-onset Alzheimer’s disease in a Chinese Han population. Psychiatr Genet, 28, 16–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardell MR, Brennan SO, Janus ED, Fraser R & Carrell RW 1987. Apolipoprotein E2-Christchurch (136 Arg----Ser). New variant of human apolipoprotein E in a patient with type III hyperlipoproteinemia. J Clin Invest, 80, 483–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisgraber KH 1994. Apolipoprotein E: structure-function relationships. Adv Protein Chem, 45, 249–302. [DOI] [PubMed] [Google Scholar]
- Winblad B, Amouyel P, Andrieu S, Ballard C, Brayne C, Brodaty H, Cedazo-Minquez A, Dubois B, Edvardsson D, Feldman H & et al. 2016. Defeating Alzheimer’s disease and other dementias: a priority for European science and society. Lancet Neurol. 15, 455–532. [DOI] [PubMed] [Google Scholar]
- Wszolek ZK, Tsuboi Y, Ghetti B et al. 2006. Frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Orphanet J Rare Dis, 1, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Zhao J, Zhang Y, Ma X, Dai Q, Zhi H, Wang B & Wang L 2016. Apolipoprotein E Gene Variants and Risk of Coronary Heart Disease: A Meta-Analysis. Biomed Res Int, 2016, 3912175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Liu CC, Oue H, Kurti A, Yamazaki A, Fryer J, Kanekiyo T & Bu G 2018. ApoE4 conditionally expressed in cerebrovasculature impairs endothelial functions and induces cognitive deficits. Alzheimer’s and Dementia, 14, P1457–P1458. [Google Scholar]
- Ye S, Huang Y, Mullendorff K, Dong L, Giedt G, Meng EC, Cohen FE, Kuntz ID, Weisgraber KH & Mahley RW 2005. Apolipoprotein (apo) E4 enhances amyloid {beta} peptide production in cultured neuronal cells: ApoE structure as a potential therapeutic target. Proc Natl Acad Sci USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yetman MJ, Fowler SW & Jankowsky JL 2016. Humanized Tau Mice with Regionalized Amyloid Exhibit Behavioral Deficits but No Pathological Interaction. PLoS One, 11, e0153724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ & Lee VM 2007. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron, 53, 337–51. [DOI] [PubMed] [Google Scholar]
- Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, Kim J, Eimer WA, Estus S, Rebeck GW, Weeber EJ, Bu G, Yu C & Ladu MJ 2012. APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem, 287, 41774–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan C, Guo X, Zhou Q, Du F, Jiang W, Zhou X, Liu P, Chi T, Ji X, Gao J, Chen C, Lang H, Xu J, Liu D, Yang Y, Qiu S, Tang X, Chen G & Zou L 2019. OAB-14, a bexarotene derivative, improves Alzheimer’s disease-related pathologies and cognitive impairments by increasing beta-amyloid clearance in APP/PS1 mice. Biochim Biophys Acta Mol Basis Dis, 1865, 161–180. [DOI] [PubMed] [Google Scholar]
- Zhao L, Gottesdiener AJ, Parmar M, Li M, Kaminsky SM, Chiuchiolo MJ, Sondhi D, Sullivan PM, Holtzman DM, Crystal RG & Paul SM 2016. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer’s disease mouse models. Neurobiol Aging, 44, 159–172. [DOI] [PubMed] [Google Scholar]
- Zhao N, Liu CC, Van Ingelgom AJ, Linares C, Kurti A, Knight JA, Heckman MG, Diehl NN, Shinohara M, Martens YA, Attrebi ON, Petrucelli L, Fryer JD, Wszolek ZK, Graff-Radford NR, Caselli RJ, Sanchez-Contreras MY, Rademakers R, Murray ME, Koga S, Dickson DW, Ross OA & Bu G 2018. APOE epsilon2 is associated with increased tau pathology in primary tauopathy. Nat Commun, 9, 4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Nwabuisi-Heath E, Dumanis SB, Tai LM, Yu C, Rebeck GW & Ladu MJ 2012. APOE genotype alters glial activation and loss of synaptic markers in mice. Glia, 60, 559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]