

Abstract
Acute disseminated encephalomyelitis (ADEM) is an acute multifocal demyelinating disease of the central nervous system that typically follows an infectious illness. Its clinical course in most cases is monophasic; however, relapsing ADEM is rarely seen, which poses a diagnostic challenge for distinguishing this disease from multiple sclerosis (MS). Although typically encountered in children, it also occurs in adults with disease characteristics slightly different from the pediatric cases. Formerly, ADEM occurred particularly often in children with measles. However, the illness most often follows a non-descript viral or even bacterial infectious illness. ADEM occurs throughout the world, and may even be more common in less-developed countries, where MS is rare, than in developed ones, where MS is common. Children seldom get MS as opposed to adults, indicating that ADEM constitutes a distinct entity from MS. The prognosis of ADEM is generally good, but severe neurologic sequelae after ADEM are occasionally seen. In this chapter, the etiology, clinical/laboratory/radiologic characteristics, treatment options, and prognosis of ADEM are discussed.
Keywords: ADEM, pathogenesis, histology, MRI, prognosis, treatment, variants, MS, NMO, EAE
Introduction
Acute disseminated encephalomyelitis (ADEM) is an acute multifocal demyelinating disease of the central nervous system (CNS) that typically follows an infectious illness. Its clinical course in most cases is monophasic; however, relapsing ADEM occurs occasionally and this may pose a diagnostic challenge for distinguishing this disease from multiple sclerosis (MS). Although typically encountered in children, it also occurs in adults with disease characteristics different from the pediatric cases. Formerly, ADEM occurred particularly often in children with measles. However, the disease most often follows a non-descript viral or even bacterial infectious illness. ADEM occurs throughout the world, and may even be more common in less-developed countries where MS is rare than in developed ones where MS is common. Children seldom get MS as opposed to adults, indicating that ADEM constitutes a distinct entity from MS. The prognosis of ADEM is generally good, but severe neurologic sequelae after ADEM are occasionally seen. In this chapter, the etiology, clinical/laboratory/radiologic characteristics, treatment options, and prognosis of ADEM are discussed.
Etiology
ADEM is an autoimmune disease resulting from para- or postinfectious complications. The prototypical disease was first reported in 1790, occurring in a 23-year-old-woman who presented with lower-extremity weakness and bladder retention 1 week after a measles rash (Croft, 1969). Later, it became apparent that several different viruses, mainly exanthamatous, could produce febrile encephalopathy shortly after the infection. Hence, the illnesses were named after the disease-causing viruses, such as varicella encephalomyelitis, rubella encephalomyelitis, and rabies encephalomyelitis, also known as neuroparalytic accident. Table 35.1 lists many of the known infections associated with ADEM. Although there is a temporal relationship between the febrile illness and onset of neurologic symptoms, the neurologic illness is probably not due to direct invasion of the infectious agent into the CNS. This is because: (1) no infectious agent has been consistently found in the CNS of affected cases whether by cerebrospinal fluid (CSF) analysis, brain biopsy, or postmortem; (2) the pathology of acute viral encephalitis differs from ADEM; and (3) vaccination with non-viable viruses can produce the same clinical and pathologic illness. It is likely that the infectious agents trigger an autoimmune response against the CNS antigens, which in turn produces the CNS pathology.
Table 35.1.
Infectious agents associated with acute disseminated encephalomyelitis
| Viruses | Vaccinations | Bacterial |
|---|---|---|
| Measles | Measles | Streptococcus |
| Mumps | Mumps | Borrelia burgdorferi |
| Rubella | Rubella | Legionella |
| Coxsackie | Rabies (Semple-type) | Mycoplasma pneumoniae |
| Coronavirus | Tetanus | Salmonella |
| Herpes (HSV, HHV-6, VZV, EBV, CMV) | Oral polio | Rickettsia rickettsii |
| Influenza A and B | Influenza | Chlamydia |
| Hepatitis A and B | Pertussis | Campylobacter |
| HTLV-1 | Hepatitis B | Leptospira |
| HIV | Japanese encephalitis virus | |
| Dengue virus | Tick-borne encephalitis | |
| Smallpox | Yellow fever |
HSV, herpes simplex virus; HHV, human herpesvirus; VZV, varicella-zoster virus; EBV, Epstein–Barr virus; CMV, cytomegalovirus; HTLV-1, human T-lymphotropic virus-1; HIV, human immunodeficiency virus.
The term ADEM should not be extended to cases of acute encephalitis, which are caused by direct invasion of the viruses into the CNS, even though the latter can result in demyelination as well. Some of these viruses, such as Epstein–Barr virus, can produce ADEM and acute encephalitis (Domachowske et al., 1996, Fujimoto et al., 2003, Doja et al., 2006). The distinguishing feature between two illnesses is usually a rather abrupt and fulminant course with acute encephalitis as opposed to ADEM, which usually has a subacute onset and greater white-matter involvement (Hartung and Grossman, 2001). Human immunodeficiency virus (HIV) encephalopathy is associated with both gray- and white-matter involvement (Letendre et al., 2009, Kuper et al., 2011). Typical HIV leukoencephalopathy is seen in advanced disease stages and the pathologic features in most cases are not demyelinating. Although histologic lesions show myelin pallor, active demyelination of axons is not seen (Power et al., 1993). The immunostaining for myelin basic protein (MBP) from the affected areas is normal and lipid-laden macrophages are not seen (Power et al., 1993). However, rare cases of HIV encephalopathy presenting as an ADEM-like illness, with demyelination and relative sparing of axons, have been reported (Jones et al., 1988, Gray et al., 1991, von Giesen et al., 1994). HTLV-I infection causes slowly progressive myelopathy and is associated with demyelination, but through a different mechanism than ADEM. HTLV-I does not infect oligodendrocytes or neurons but preferentially infects CD4 + and CD8 + T cells, which, when activated by the virus, could lead to demyelination (Hollsberg, 1997, Kannian et al., 2012). It may also infect microglia, which may become activated and release cytokines toxic to myelin (Hollsberg, 1997).
Other forms of encephalitis that may also cause demyelination but distinct from ADEM include subacute sclerosing panencephalitis, a chronic progressive measles infection of the brain, rubella panencephalitis, varicella-zoster virus (VZV) encephalitis, and human herpesvirus-6 (HHV-6). In panencephalitis caused by rubella, prominent white-matter changes with axonal fragmentation are seen (Townsend et al., 1975, Townsend et al., 1976). VZV encephalitis in AIDS patients can produce demyelination. In addition to inflammatory demyelination, VZV may also directly infect oligodendrocytes, since viral inclusion bodies can be seen in these cells (Gray et al., 1991, Gray et al., 1994, Amlie-Lefond et al., 1995). HHV-6 can produce demyelination also by directly infecting oligodendrocytes (De Bolle et al., 2005). HHV-6 is also one of several hypothesized etiologic agents for MS (Challoner et al., 1995).
Another form of demyelinating disease seen in HIV is progressive multifocal leukoencephalopathy (PML), which is caused by infection of oligodendrocytes by the JC virus, a polyomavirus. PML is seen under conditions that impair immunity, such as acquired immunodeficiency syndrome (AIDS), posttransplant, lymphoma, and autoimmune diseases receiving treatment with chemotherapy or immunomodulators such as natalizumab, rituximab, or efalizumab (Carson et al., 2009). PML lesions usually occur in the deep white matter and are typically solitary, but they can rarely be multifocal (Astrom et al., 1958). Also, cortical demyelination from PML has been described (Sweeney et al., 1994, Shintaku et al., 2000, Moll et al., 2008). The frontal and parieto-occipital regions are usually affected, but deep gray matter, brainstem, cerebellum, and spinal cord can be involved (von Einsiedel et al., 1993, Bienfait et al., 1998, Kastrup et al., 2002, Bernal-Cano et al., 2007). The demyelinating lesions seen in PML lack inflammatory reaction and necrosis, but have accompanying reactive astrocytes and oligodendroglial nuclear inclusions (Astrom and Stoner, 1994, Aksamit, 2006). However, upon immune reconstitution, as may occur in AIDS patients after highly active antiretroviral therapy or in autoimmune diseases once immunosuppression is stopped, PML lesions once containing little inflammation now develop intense inflammatory reaction with edema and necrosis (Tan et al., 2009). This immune reconstitution inflammatory syndrome can produce diffuse white-matter changes, as seen on magnetic resonance imaging (MRI), even in areas initially devoid of any pathology, for example in MS after cessation of natalizumab therapy (Tan et al., 2011, Gheuens et al., 2012).
Pathogenesis
The pathogenesis of ADEM has most resemblance to the animal model experimental autoimmune encephalomyelitis (EAE), which is an acute demyelinating disease induced by immunization of animals with myelin protein products (Rivers et al., 1933). After immunization with CNS antigens emulsified in Freund's complete adjuvant, animal recipients present with a monophasic illness causing tetraparesis and incontinence. Histologic analysis shows inflammatory demyelinating lesions typically in the spinal cord and in some animal strains in the brain. Analogous to EAE, in humans ADEM cases have been observed after immunization with Semple rabies vaccine, a live attenuated vaccine that in the past was contaminated with rabbit or goat CNS tissue (Hemachudha et al., 1987). However, newer vaccines are developed in cultures of human diploid cells and hence do not have the same risk for producing ADEM. Since EAE is a T-cell-mediated disease, as demonstrated by adoptive transfer of the disease through T cells and not through serum factors to recipient animals, it is likely that ADEM is also a T-cell-mediated disease by its close resemblance to the EAE model (Paterson, 1960). Furthermore, in vitro studies also show increased T-cell reactivity to MBP in blood and CSF samples taken from ADEM patients (Lisak and Zweiman, 1977). In addition to MBP, other myelin proteins may be the target of autoimmune responses, such as proteolipid protein and myelin-oligodendrocyte glycoprotein, since both of these can also produce EAE (Kuerten et al., 2006).
Several theories have been postulated to explain how infections or vaccinations prime T-cell responses against myelin antigens in the CNS. It is likely that more than one immune mechanism is involved in triggering ADEM in susceptible patients. One of the more popular mechanisms is “molecular mimicry,” whereby an infectious agent contains several epitopes that are similar in structure to the endogenous myelin epitopes (Fujinami and Oldstone, 1985). During an immune response against the invading organism, some of the T cells directed against the infectious epitopes become capable of cross-reacting with self myelin peptides, hence leading to an autoimmune response (Fujinami and Oldstone, 1985). These cross-reactive T cells proliferate in response to self-antigenic stimulation and release chemokines that further recruit more lymphocytes and macrophages to the site of activation, further enhancing demyelination, neuronal, and axonal injury. The well-known examples of molecular mimicry include cross-reactivity between Campylobacter jejuni and GQ1b ganglioside in the Fisher-Miller variant of Guillain–Barré syndrome and between GM1 ganglioside in the acute motor axonal neuropathy variant of Guillain–Barré syndrome (Jacobs et al., 1995, Oomes et al., 1995, Rees et al., 1995).
Another mechanism whereby myelin-reactive T cells could be generated is through non-specific activation of naturally occurring autoreactive T cells by virus or bacterial “superantigens” (Jorens et al., 2000). Such foreign antigens can activate a wide variety of CD4 + T cells, some of which may have some reactivity against myelin epitopes. Upon activation, these T cells may proliferate and help generate an inflammatory response to self antigens. For example, in poststreptococcal related ADEM cases, superantigens contained in Streptococcus pyogenes cell wall may non-selectively activate T cells by simultaneously binding to the major histocompatibiltity complex class II molecule and T-cell receptor (Burns et al., 1992, Jorens et al., 2000). An alternative hypothesis may be that some viruses inhibit certain types of T cells, such as suppressor T lymphocytes (Sakaguchi et al., 1995, Grant et al., 2008). These cells have an important role in inhibiting potentially autoreactive T cells (Sakaguchi et al., 1995, Grant et al., 2008). If suppressor T cells are inhibited by certain viruses, then removal of their suppressor function may lead to activation of myelin-specific T cells.
ADEM may also result from activation of previously primed immune cells through reinfection by the same organism. Astrocytes and microglia in the CNS can function as antigen presenting cells. Direct infection of these cells by certain viruses may result in activation of a subset of T cells that were primed during the initial infection with the same organism. These previously primed T cells may mount an inflammatory response against the CNS epitopes, including myelin. This hypothesis has been observed in murine CNS disease caused by lymphocytic choriomeningitis virus (Merkler et al., 2006).
Epidemiology
ADEM is an uncommon illness and hence epidemiologic studies are complicated by small case series from limited centers. ADEM can occur at any age, with higher frequency in children than adults. Predominant age of occurrence in children is 5–8 years (Hynson et al., 2001, Tenembaum et al., 2002, Anlar et al., 2003) and adults between 19 and 61 years (Schwarz et al., 2001). In children, there appears to be no gender predominance (Dale et al., 2000, Leake et al., 2004), although some studies have reported a slight male incidence (Murthy et al., 2002, Tenembaum et al., 2002). The diagnosis of ADEM is usually made in the setting of a viral illness or vaccination. Hence, the incidence of disease varies with the triggering infectious agent. A higher occurrence of ADEM has been described in the winter and spring months in small studies, but the illness can occur throughout the whole year (Murthy et al., 2002, Leake et al., 2004). One pediatric study from San Diego County, CA, reported the estimated incidence of ADEM to be about 0.4/100 000/year (Leake et al., 2004). Most patients (93%) in this study had signs of infection 21 days prior to the neurologic symptoms, and 5% had received vaccination in the preceding month (Leake et al., 2004). Worldwide distribution of ADEM is unknown in part because regional cases of ADEM are often linked to specific vaccinations. Incidence of ADEM after measles is 1:1000, vaccinia 1:63 to 1:300,000, varicella 1:10,000, and rubella 1:20 000 (Gibbons et al., 1956, Spillane and Wells, 1964). Cases after mumps and scarlet fever have also been described (Gibbons et al., 1956).
Clinical presentation of ADEM and variants
Clinical manifestations of ADEM can be pleotropic. Symptoms can range from non-specific, such as malaise and fatigue, to fulminant, progressing rapidly to obtundation and coma. The clinical course of ADEM is classically monophasic, with most patients (70–77%) reporting an antecedent infection or vaccination (Amit et al., 1986, Hynson et al., 2001, Tenembaum et al., 2002). In children, a prodromal phase precedes the neurologic symptoms and consists of malaise, fever, headache, nausea, and vomiting, progressing within days to maximal neurologic symptoms. The presenting neurologic manifestation varies according to the initial CNS site affected by the inflammation. As the disease progresses, multiple CNS sites become involved, with resulting deficits including hemiplegia (76%), ataxia (18–65%), cranial nerve deficits (22–45%), optic neuritis (7–23%), myelitis (24%), speech impairment (5–21%), hemiparesthesias (2–3%), and seizures (13–35%) (Hynson et al., 2001, Murthy et al., 2002, Tenembaum et al., 2002, Tenembaum et al., 2007, Anlar et al., 2003, Leake et al., 2004).
Although ADEM typically has a monophasic course, recurrences after a delay are not uncommon. The incidence of these relapses has been variably reported from 5.5% to as high as 21% (Hung et al., 2001, Hynson et al., 2001, Murthy et al., 2002, Tenembaum et al., 2002, Leake et al., 2004). Several terms have been used to describe relapsing forms of ADEM, such as recurrent, relapsing, pseudorelapsing, and multiphasic ADEM, all differing in time between recrudescences, clinical presentation, and distribution of MRI lesions. In order to avoid misdiagnosis and to have uniform criteria for classification, the International Pediatric MS Study Group has proposed three types of variations in ADEM recrudescences (Tenembaum et al., 2007). Using the Study Group criteria, ADEM is defined as an acute or subacute neurologic event associated with multiple CNS lesions, and, more importantly, no prior similar neurologic event and no prior evidence of CNS demyelination. If a subsequent neurologic event takes place within 4 weeks of tapering steroid therapy or within 3 months from the initial event, then this event is not considered a “true” relapse but still temporally related to the initial event. Such a relapse has also been dubbed “pseudorelapsing ADEM” or “steroid-dependent ADEM.” A true recurrent ADEM is defined as a new clinical event occurring at least 3 months after the initial ADEM presentation and at least 4 weeks after the completion of steroid treatment. Importantly, the clinical presentation and distribution of demyelinating lesions on MRI should be very similar to the initial ADEM event. Multiphasic ADEM is defined as future ADEM episodes, but involving new clinical deficits and MRI lesions different from the initial ADEM event. Recrudescences occur at least 3 months after the initial ADEM episode and greater than 4 weeks after the completion of steroid treatment.
A hyperacute variant of ADEM is known as hemorrhagic leukoencephalopathy (AHL), also variably called AHEM or acute necrotizing hemorrhagic leukoencephalitis (ANHLE). It was first described by Hurst in 1941 and is part of the same spectrum of disease as ADEM, except that it follows a rapid, fulminant clinical course. Its clinical course is monophasic, and it is more rare than ADEM, observed in 2% of children with ADEM (Tenembaum et al., 2002). AHL occurs within days to a few weeks after an infection, usually upper respiratory, or vaccination. Clinical presentation includes fever, headache, confusion, seizures, and weakness, progressing rapidly to stupor and coma. Computed tomography or MRI scans show large lesions associated with edema and mass effect, tissue displacement, and petechial or frank hemorrhages. Death can occur within days from the onset of encephalopathy due to herniation caused by cerebral edema and hemorrhage. Most patients do not survive this illness and few who endure are left with permanent deficits. With rapid treatment with steroids, plasma exchange (PLEX), or other immunosuppressive agents, more favorable outcomes have been noted (Seales and Greer, 1991, Rosman et al., 1997).
Besides ADEM and AHL, there are certain isolated syndromes, such as optic neuritis, acute cerebellar ataxia (Bickerstaff's encephalitis), and transverse myelitis, that may also follow an infection or vaccination. In contrast to the typical ADEM, prodromal symptoms in these restricted syndromes usually do not occur and the clinical presentation is only limited to the relevant CNS site of injury. Imaging studies confirm CNS involvement relevant to the clinical manifestation and diffusely distributed lesions are not seen, as in ADEM. These restricted syndromes are more frequently seen in adults than in children. It is uncertain whether these isolated syndromes represent a restricted form of ADEM or entirely different forms of inflammatory demyelinating diseases. However, if such syndromes do occur subsequent to an infectious illness and follow a monophasic course, then it is more likely that they represent a restricted form of ADEM. But, if an antecedent infection is not observed and these syndromes have a relapsing course, then other diseases need to be considered, such as MS, neuromyelitis optica (NMO), and neurologic complication of connective tissue diseases.
Adult ADEM
In adults, clinical manifestations of ADEM are slightly different from the pediatric population. In contrast to children, fever (15%), meningism (15%), loss of consciousness (19%), and seizures (4%) are uncommon (Schwarz et al., 2001). Other symptoms and their frequency of occurrence include motor (77%), sensory (65%), brainstem (62%), ataxia (38%), spinal (15%), and aphasia (8%). The median age of onset is 33 (range 19–61), with slight female predominance (65%) (Schwarz et al., 2001). Antecedent infection is found in only 46% of adult patients, which is lower than children. In a case series of adult patients initially diagnosed with ADEM, 35% developed clinically definite MS over a period of 38 months (Schwarz et al., 2001). Also, adult patients with the final diagnosis of ADEM at the end of follow-up period were older than those with MS (median age 33 versus 26), more often had an antecedent infection, had infratentorial lesions, a higher CSF albumin fraction, and lower incidence of oligoclonal bands (OCBs) (Schwarz et al., 2001). However, no single clinical, radiologic, or laboratory feature clearly predicted the final diagnosis of ADEM versus MS in this study (Schwarz et al., 2001).
Radiologic characteristics
MRI is the imaging of choice for evaluating patients for possible ADEM. Since ADEM evolves over several days, initial MRI may be normal if done at the onset of prodromal symptoms (Bennetto and Scolding, 2004, Menge et al., 2007). During the progressive course of the disease, T2 MRI sequences show several lesions scattered throughout the brain involving both the white- and gray-matter structures, such as cerebral cortices, thalamus, basal ganglia, brainstem, and cerebellum (Fig. 35.1) (Schwarz et al., 2001, Menge et al., 2007). Although most lesions show contrast enhancement, implying that they developed relatively simultaneously, a minority of lesions do not enhance. This is because as these lesions evolve over several weeks, some of those having the earliest onset no longer demonstrate enhancement. In ADEM, contrast-enhancing lesions have been observed in 30–100% of patients (Khong et al., 2002, Tenembaum et al., 2002, Lim et al., 2003). The pattern of enhancement can be variable, from complete enhancement of smaller lesions to incomplete enhancement of larger lesions, such as ring-shaped, open ring, gyral, and nodular patterns (Caldemeyer et al., 1994, van der Meyden et al., 1994). Rarely, large solitary lesions can be seen in the brain, often resembling neoplasm. The correct diagnosis in these cases can only be confirmed by biopsy. In addition to the brain, spinal cord can also be affected in ADEM, occurring in about 11–28% of cases (Dale et al., 2000, Hynson et al., 2001, Tenembaum et al., 2002, Anlar et al., 2003). Spinal cord lesions are typically large and appear edematous on MRI. Often there is a predilection for the thoracic cord, but lesions can occur at any level.
Fig. 35.1.

(A) Fluid-attenatuated inversion recovery (FLAIR) T2 magnetic resonance imaging (MRI) demonstrating juxtacortical and subcortical lesions dispersed throughout the brain (arrows). (B) Postcontrast T1 MRI showing slight enhancement of some of the lesions (arrows). The diminutive contrast enhancement implies more chronic lesions.
Serial MRI after the initial episode plays a vital role in establishing the diagnosis of ADEM. In monophasic ADEM, MRI obtained at least 6 months after the initial episode should show resolving lesions and, more importantly, no evidence of any new lesions. Complete resolution of MRI lesions can occur in 37–75% and partial resolution in 25–53% (Amit et al., 1986, Kesselring et al., 1990, Dale et al., 2000, Khong et al., 2002, Tenembaum et al., 2002). Further specificity for the diagnosis of ADEM can be obtained by continual MRI follow-up over 2–3 years. If there is accrual of additional T2 lesions over time, then patients need to be evaluated for possible MS.
Histopathology
The hallmark of ADEM is multifocal perivascular inflammation, with infiltration of lymphocytes and macrophages into the parenchyma (Fig. 35.2 ). Adjacent to the areas of inflammation, myelin loss occurs with relative axonal sparing. There is proliferation of endothelial cells and fibrin deposits are seen within the vascular lumens. Plasma cells and granulocytes are only rarely seen in ADEM. Most lesions are seen in the white matter but gray matter is also involved in many cases. Viral inclusion bodies are not seen on hematoxylin and eosin-stained sections. Considering the extent of perivascular inflammation in ADEM, some have suggested that ADEM is a form of vasculopathy with secondary myelin destruction. This view is buttressed by the similarities between ADEM histopathology and that of EAE in primates (Behan et al., 1972, Behan et al., 1973).
Fig. 35.2.
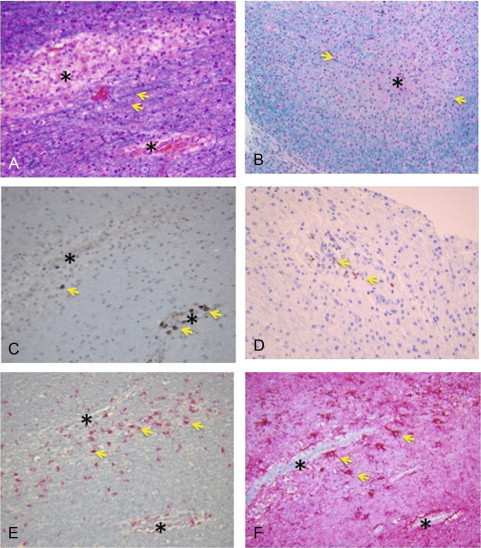
Acute disseminated encephalomyelitis. Photomicrographs showing a multiple sclerosis lesion with surrounding inflammatory response and loss of myelin. (A) Hematoxylin & eosin-luxol fast blue (H&E-LFB) stain showing perivascular loss of myelin (arrows showing myelin stained with LFB). (B) High magnification showing loss of myelin in the lesion center (asterisk) surrounded by intact myelin stained with LFB (arrows). (C and D) Stain for CD3 + T cells showing accumulation of T cells around the lesions (arrows). (E) CD68 stain showing macrophages surrounding blood vessels. (F) Glial fibrillary acidic protein (GFAP) stain showing reactive astrocytes (gliosis) surrounding blood vessels (arrows).
(Courtesy of Peter Pytel, MD, Department of Pathology, University of Chicago.)
The histopathology of AHL is more severe than ADEM. Grossly, the brain appears swollen and punctate hemorrhages are seen. Perivascular infiltrates contain neutrophils and eosinophils, in contrast to the “routine” ADEM. Fibrinoid necrosis is commonly seen in the blood vessels in AHL. Hemorrhages within blood vessels along with venous thrombosis are also observed. The pathology of AHL in humans is almost identical to the hyperacute form of EAE in animals (Levine, 1974, Hart and Earle, 1975).
In the MS type of demyelinating disease, lymphocyte phenotype mediating myelin and axonal damage has been extensively studied. In MS, direct damage to the tissue is caused by CD8 + T cells, macrophages and microglia (release of nitric oxide and reactive oxygen species), with CD4 + Th1 and TH17 cells releasing proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-6, IL-12, and IL-17 (Miller, 2012). However, the pathologic role of similar immune cells and cytokines has not been carefully elucidated in ADEM. Histologic studies from a few cases have shown the presence of TNF-α and IL-1β in lesions of ADEM (Kadhim et al., 2003). Studies using CSF analysis are confounded by timing of CSF collection and treatment influence, but they have shown elevated levels of TNF-α, IL-1b, and, inconsistently, IL-6 (Ichiyama et al., 2002, Dale and Morovat, 2003).
Differential diagnosis
ADEM usually presents with subacute encephalopathy, which has a broad differential (Table 35.2 ). No specific criteria exist for diagnosing ADEM, but the antecedent history, temporal course of the illness, neuroimaging, CSF analysis, and probably repeat imaging during remission are most important in arriving at the diagnosis and excluding other causes of encephalopathy. In the differential diagnosis, conditions to consider and rule out relevant to treatment and prognosis are MS and NMO. Table 35.3 provides key clinical, radiologic, and laboratory features distinguishing ADEM from MS and NMO. The key points to consider in diagnosing typical ADEM are antecedent infectious history, subacute encephalopathy, presence of multifocal lesions on brain MRI in the white and gray matter, most of which are contrast-enhancing, and CSF analysis showing mild to moderate pleocytosis, normal to slightly elevated protein, normal glucose, and absence of OCBs. The diagnosis can be further reassured if follow-up MRI after 6 months or more continues to show lesion resolution and absence of new lesions. Features that herald a MS course include relatively benign clinical presentation, MRI lesions concentrated more or less around the periventricular and pericallosal areas, MRI evidence of chronic lesions (T1 lesions), absence of gray-matter lesions, and positive OCBs. OCBs have been reported anywhere from 0 to 29% of ADEM cases and 64–92% of pediatric MS cases (Dale et al., 2000, Hynson et al., 2001, Tenembaum et al., 2002, Pohl et al., 2004). In one study, pediatric patients with MS were more likely to have OCBs on presentation than ADEM (64% versus 29%), although this difference was not found to be statistically significant (Dale et al., 2000). Hence, presence of OCBs is somewhat helpful for prognostic purposes, but cannot be used by itself in differentiating MS from ADEM. Clinical and MRI follow-up over time is probably the best method for determining whether patients initially presenting with demyelinating disease go on to develop MS in the future. Features that portend NMO diagnosis include longitudinally extensive transverse myelitis, absence of lesions in the brain on MRI, and severe unilateral or bilateral optic neuritis. Pediatric patients with NMO may also have prodromal symptoms but no clear evidence of antecedent infection in most cases.
Table 35.2.
Differential diagnosis of acute disseminated encephalomyelitis (ADEM)
| Vascular | Strokes, CADASIL, amyloid angiopathy, PRES, eclampsia |
| Infectious | Viral or bacterial encephalitis, HIV encephalopathy, PML, abscess |
| Toxic | Inhaled heroin, carbon monoxide |
| Autoimmune | Multiple sclerosis, neurosarcoidosis, Behçet's disease, primary CNS angiitis, vasculitis due to connective tissue diseases such as lupus and Sjögren's disease |
| Metabolic | Mitochondrial diseases (MELAS, Leber's hereditary optic neuropathy), adrenoleukodystrophy, central and extrapontine myelinolysis |
| Iatrogenic | Methotrexate, tacrolimus, cyclosporine |
| Neoplastic | Neoplasms, metastasis, and paraneoplastic syndromes |
CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; PRES, posterior reversible encephalopathy syndrome; HIV, human immunodeficiency virus; PML, progressive multifocal leukoencephalopathy; CNS, central nervous system; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes.
Table 35.3.
Features of the major central nervous system demyelinating diseases
| NMO | Multiple sclerosis | ADEM | |
|---|---|---|---|
| Demographics | |||
| Age of onset (median) in years | 40 | 30 | 6.5 (pediatric); 33 (adults)† |
| Sex F:M | 8:1 (relapsing) | 2:1 (relapsing) | 1:1 |
| Ethnicity | ↑ Incidence in non-white | Usually white | All populations at risk |
| Clinical course | |||
| Monophasic | 20% | NA | > 95% |
| Relapsing | 80% | 85% initially | < 5% |
| Primary progressive | NA | 15% | NA |
| MRI features | |||
| Brain white-matter lesions | Typically absent at onset | Oval, usually perpendicular to ventricles | Large, disseminated; rarely periventricular |
| Spinal cord lesions | Longitudinally extensive | Punctate | Punctate to rarely large longitudinal |
| Optic nerve lesions | Unilateral or bilateral, with poor recovery | Usually unilateral, with good recovery | Unilateral or bilateral with good recovery |
| CSF findings | |||
| WBC count | ↑↑↑ Lymphocytes, neutrophils | ↑ Lymphocytes | ↑↑ Lymphocytes, occasionally neutrophils |
| Total protein | Elevated | Typically normal | Normal to elevated |
| Oligoclonal bands (OCBs) | Typically absent* | > 90% present | Variable (0–29%)‡ |
| Pathology | |||
| Gray matter | Demyelination with necrosis | Demyelination | Demyelination (AHL : hemorrhage) |
| White matter | Demyelination with necrosis | Demyelination | Demyelination (AHL : hemorrhage) |
| Lesion immunopathology | |||
| Macrophages | +++ | +++ | +++ |
| T cells | +/– | +++ | +++ |
| Eosinophils | ++ | None | Rare |
| Neutrophils | +++ | None | Occasional |
| Antibodies | +++ | Variable | Variable |
| Complement | ++ | Variable | Variable |
| Coexisting autoimmune diseases | 30–50% | Infrequent | Not seen |
| Disability progression | Relapse-related | Independent of relapses | Relapse-related in recurrent/multifocal |
NMO, neuromyelitis optica; ADEM, acute disseminated encephalomyelitis; MRI, magnetic resonance imaging; CSF, cerebrospinal fluid; WBC, white blood cell; AHL, acute hemorrhagic leukoencephalopathy.
OCBs may be seen during active disease, but disappear during remission. In ADEM, persistence of OCBs during disease remission raises suspicion for later risk of developing multiple sclerosis.
The CSF findings in NMO include moderate pleocytosis, elevated protein, and absence of OCBs. A serum marker for NMO is available commercially, NMO IgG antibody. This NMO-IgG binds selectively to aquaporin-4 water channel, which is found in high amounts in astrocytic foot processes at the blood–brain barrier (Lennon et al., 2004). The NMO-IgG antibody test is useful in identifying NMO from MS, with sensitivity of 73% and specificity of 91% (Lennon et al., 2004). Specific radiologic and laboratory criteria have been proposed to differentiate NMO from MS: (1) a spinal cord MRI lesion extending contiguously three or more vertebral segments; (2) a brain MRI that fails to meet diagnostic criteria for MS; and (3) a positive NMO-IgG antibody titer (Wingerchuk et al., 2006). Two of these three supporting criteria need to be met for the diagnosis of NMO.
Other mimics to exclude based on imaging are Schilder's disease and tumefactive MS, which are seen as large tumor-like lesions associated with edema and mass effect; Marburg variant of MS, which also presents with encephalopathy and multiple, large lesions on MRI; brain abscess, which is usually a solitary, complete ring-enhancing lesion with hypointense center on MRI. Symmetric bithalamic lesions can be seen in cerebral venous thrombosis, hypernatremia, acute necrotizing encephalopathy, and extrapontine myelinolysis (Cusmai et al., 1994, Mizuguchi et al., 1995, Ruggieri et al., 1998, Hartfield et al., 1999, Suwa et al., 1999). Basal ganglia lesions can be seen in organic aciduria and infantile bilateral striatal necrosis (Brismar and Ozand, 1994, Fujita et al., 1994).
Treatment
Due to rarity of ADEM, acute presentation, and urgency of treatment, no clinical trials of therapeutic agents have been systematically studied. Hence, reports of therapeutic efficacy are based only on small case series and anecdotal experience. Symptomatic and supportive care involves admission of patients usually to a step-down unit or intensive care unit due to moderate to severe encephalopathy and the presence of cerebral edema. However, in rare cases, clinical presentation is benign and spontaneous improvement has been observed (Kimura et al., 1996).
Treatment of choice is high-dose glucocorticoid administered systemically for several days (3–5), followed by an extended oral steroid taper over 6–8 weeks. Recrudescences are fewer when this regimen is employed compared to a short steroid taper, strongly suggesting a benefit from glucocorticoid treatment, as might be expected for a T-cell-mediated process. A good response to therapy has been observed in most studies (Wang et al., 1996, Apak et al., 1999, Schwarz et al., 2001, Tenembaum et al., 2002). In a large, non-randomized study of 121 patients with ADEM, 43 were treated with steroids and others did not receive such treatment. The onset of symptom improvement was sooner in patients receiving steroids than those who did not (5.46 versus 15.6 days) (Rust et al., 1997). However, the time to maximal recovery was similar in the two groups (Rust et al., 1997).
There are cases of ADEM or even fulminant presentation such as AHL where steroids alone are not sufficient for suppressing inflammation and improving clinical findings. PLEX, intravenous immunoglobulin (IVIG), and immunosuppressive drugs have been used with some success (Seales and Greer, 1991, Stricker et al., 1992, Rodriguez et al., 1993, Markus et al., 1997, Pradhan et al., 1999, Sahlas et al., 2000). Plasma exchange or IVIG is usually the second step in recalcitrant cases. PLEX is administered every other day for a total of five sessions, with removal of 1–1.5 L plasma volume. Maintenance doses at 4-week intervals may be needed for the next 3–4 months. In a variety of severe demyelinating CNS diseases (MS, NMO, ADEM), PLEX use has been associated with moderate to marked improvement in 40% of patients (Keegan et al., 2002). Also, in several case reports, PLEX use early in the disease course has been reported to be very successful, at times associated with fairly complete recovery (Stricker et al., 1992, Miyazawa et al., 2001, RamachandranNair et al., 2005). IVIG is given at a dose of 400 mg/kg/day for 5 consecutive days. Also, maintenance doses at 4-week intervals may be needed for the next 3–4 months. Several case reports have documented the successful use of IVIG either alone or in combination with steroids in ADEM (Kleiman and Brunquell, 1995, Nishikawa et al., 1999, Straussberg et al., 2001). Of the immunosuppressive agents, cyclophosphamide has been more often used in ADEM after steroid failure. Its use is rather reserved, often as a third-line agent after inadequate response from PLEX or IVIG.
Prognosis
The prognosis of ADEM has significantly improved from former times. This is probably due to a drastic decrease in wild-type measles infections and the use of safer and more efficient vaccinations. Also, there is a widespread use of steroids once ADEM is diagnosed, which has been found to be beneficial in limiting clinical symptoms. Full recovery occurs in many patients and improvement with minor residual symptoms in up to 70–90% (Shahar et al., 2002, Gupte et al., 2003, Menge et al., 2005). However, mortality from ADEM may still be as high as 5%. This may particularly be the case in patients with more ominous clinical presentation, such as those with infratentorial lesions and large spinal cord lesions. In the case of AHL, the mortality can be as high as 50% (Borlot et al., 2011). A greater percentage of pediatric patients recover completely from ADEM compared to adults (60–80% versus 46%), implying greater recovery potential of the developing CNS in children (Schwarz et al., 2001).
Limited observational studies have reported the natural course of ADEM without any treatment. Case studies from India, Japan, and Russia have suggested that clinical symptoms of ADEM in children gradually improve over several weeks, with up to 50–70% of patients achieving full recovery without treatment (Kimura et al., 1996, Murthy et al., 2002, Idrissova et al., 2003). In one of these studies, children with more extensive brain lesions had partial recovery compared to those with fewer lesions (Murthy et al., 2002). Antecedent infection was not found to correlate with the clinical outcome (Murthy et al., 2002). However, another study suggested variable clinical outcome depending on the type of antecedent infection, with favorable outcome in 70% of patients without prior infection, 54% post-varicella, and 43% post-rubella infections (Idrissova et al., 2003). Nonetheless, these studies, when taken together, do suggest a favorable outcome in most patients without any specific treatment. However, it is generally agreed that ADEM needs to be promptly treated upon diagnosis because of the beneficial effects of limiting clinical symptoms and possibly preventing recurrences.
In addition to the physical disability, long-term cognitive deficits have been reported in ADEM. In one study, children aged 6–15 had neuropsychologic testing done 3.5 years after ADEM presentation. Mild cognitive deficits in attention, executive function, and behavior were observed in these children, even in those with complete resolution of MRI lesions (Hahn et al., 2003). In another study with a mean follow-up of 3.9 years, children with ADEM before the age of 5 had lower IQ scores and educational achievement as well as behavioral problems when compared to age- and sex-matched healthy controls (Jacobs et al., 2004). Patients with onset of ADEM at an older age also had slower verbal processing when assessed after a mean of 2.2 years (Jacobs et al., 2004). Based on these studies, it is likely that subtle cognitive disability persists after a severe bout of CNS demyelination despite the lack of any clear physical or radiologic findings. Perhaps the neuroanatomic basis for these cognitive deficits is subtle damage in the cortical regions of the brain, an area that is difficult to visualize on routine MRI scans. Non-conventional MRI techniques such as diffusion tensor, magnetization transfer ratio, MR spectroscopy, and high-resolution cortical imaging may be more sensitive in assessing damage in the cortical gray matter than the conventional MRI measures.
Future directions
There are several issues in this disease that continue to be enigmatic. Much attention has been paid to the antecedent infections or vaccinations associated with ADEM. However, little is known about the host factors such as genetic background that may predispose to ADEM. Also, it is not clear why most cases of ADEM are monophasic and only some follow a multiphasic course. There are no clear prognostic biomarkers, especially with risk to subsequently developing MS. Without any controlled trials, the ideal treatment protocol for ADEM remains to be determined. For example, steroids seem to be the first-line therapy, but would the outcome be better if PLEX or IVIG is combined with steroids? Pathologic mechanisms in ADEM are still not well understood, despite its similarities to EAE. It appears that ADEM is a T-cell-mediated disease, yet therapies that modify B-cell responses, such as PLEX and IVIG, are successful in treating this illness. The interaction of T and B cells in ADEM needs to be further explored, especially as this may be relevant to therapy. Finally, cases of ADEM are probably underreported. More comprehensive studies need to be carried out worldwide in order to understand fully its epidemiology, as it may be relevant to shedding light on triggering factors, pathogenesis of the disease, and possibly preventive measures.
References
- Aksamit A.J. Review of progressive multifocal leukoencephalopathy and natalizumab. Neurologist. 2006;12:293–298. doi: 10.1097/01.nrl.0000250948.04681.96. [DOI] [PubMed] [Google Scholar]
- Amit R., Shapira Y., Blank A. Acute, severe, central and peripheral nervous system combined demyelination. Pediatr Neurol. 1986;2:47–50. doi: 10.1016/0887-8994(86)90040-8. [DOI] [PubMed] [Google Scholar]
- Amlie-Lefond C., Kleinschmidt-DeMasters B.K., Mahalingam R. The vasculopathy of varicella-zoster virus encephalitis. Ann Neurol. 1995;37:784–790. doi: 10.1002/ana.410370612. [DOI] [PubMed] [Google Scholar]
- Anlar B., Basaran C., Kose G. Acute disseminated encephalomyelitis in children: outcome and prognosis. Neuropediatrics. 2003;34:194–199. doi: 10.1055/s-2003-42208. [DOI] [PubMed] [Google Scholar]
- Apak R.A., Kose G., Anlar B. Acute disseminated encephalomyelitis in childhood: report of 10 cases. J Child Neurol. 1999;14:198–201. doi: 10.1177/088307389901400312. [DOI] [PubMed] [Google Scholar]
- Astrom K.E., Stoner G.L. Early pathological changes in progressive multifocal leukoencephalopathy: a report of two asymptomatic cases occurring prior to the AIDS epidemic. Acta Neuropathol. 1994;88:93–105. doi: 10.1007/BF00294365. [DOI] [PubMed] [Google Scholar]
- Astrom K.E., Mancall E.L., Richardson E.P., Jr. Progressive multifocal leuko-encephalopathy; a hitherto unrecognized complication of chronic lymphatic leukaemia and Hodgkin's disease. Brain. 1958;81:93–111. doi: 10.1093/brain/81.1.93. [DOI] [PubMed] [Google Scholar]
- Behan P.O., Behan W.M., Feldman R.G. Cell-mediated hypersensitivity to neural antigens. Occurrence in human patients and nonhuman primates with neurological diseases. Arch Neurol. 1972;27:145–152. doi: 10.1001/archneur.1972.00490140049008. [DOI] [PubMed] [Google Scholar]
- Behan P.O., Kies M.W., Lisak R.P. Immunologic mechanisms in experimental encephalomyelitis in nonhuman primates. Arch Neurol. 1973;29:4–9. doi: 10.1001/archneur.1973.00490250022002. [DOI] [PubMed] [Google Scholar]
- Bennetto L., Scolding N. Inflammatory/post-infectious encephalomyelitis. J Neurol Neurosurg Psychiatry. 2004;75(Suppl 1):i22–i28. doi: 10.1136/jnnp.2003.034256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal-Cano F., Joseph J.T., Koralnik I.J. Spinal cord lesions of progressive multifocal leukoencephalopathy in an acquired immunodeficiency syndrome patient. J Neurovirol. 2007;13:474–476. doi: 10.1080/13550280701469178. [DOI] [PubMed] [Google Scholar]
- Bienfait H.P., Louwerse E.S., Portegies P. Progressive multifocal leukoencephalopathy presenting as a solitary gray matter lesion. J Neurol. 1998;245:557–558. doi: 10.1007/s004150050243. [DOI] [PubMed] [Google Scholar]
- Borlot F., da Paz J.A., Casella E.B. Acute hemorrhagic encephalomyelitis in childhood: case report and literature review. J Pediatr Neurosci. 2011;6:48–51. doi: 10.4103/1817-1745.84408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brismar J., Ozand P.T. CT and MR of the brain in the diagnosis of organic acidemias. Experiences from 107 patients. Brain Dev. 1994;16(Suppl):104–124. doi: 10.1016/0387-7604(94)90103-1. [DOI] [PubMed] [Google Scholar]
- Burns J., Littlefield K., Gill J. Bacterial toxin superantigens activate human T lymphocytes reactive with myelin autoantigens. Ann Neurol. 1992;32:352–357. doi: 10.1002/ana.410320308. [DOI] [PubMed] [Google Scholar]
- Caldemeyer K.S., Smith R.R., Harris T.M. MRI in acute disseminated encephalomyelitis. Neuroradiology. 1994;36:216–220. doi: 10.1007/BF00588134. [DOI] [PubMed] [Google Scholar]
- Carson K.R., Focosi D., Major E.O. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a Review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol. 2009;10:816–824. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- Challoner P.B., Smith K.T., Parker J.D. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci U S A. 1995;92:7440–7444. doi: 10.1073/pnas.92.16.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft P.B. Para-infectious and post-vaccinal encephalomyelitis. Postgrad Med J. 1969;45:392–400. doi: 10.1136/pgmj.45.524.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusmai R., Bertini E., Di Capua M. Bilateral, reversible, selective thalamic involvement demonstrated by brain MR and acute severe neurological dysfunction with favorable outcome. Neuropediatrics. 1994;25:44–47. doi: 10.1055/s-2008-1071582. [DOI] [PubMed] [Google Scholar]
- Dale R.C., Morovat A. Interleukin-6 and oligoclonal IgG synthesis in children with acute disseminated encephalomyelitis. Neuropediatrics. 2003;34:141–145. doi: 10.1055/s-2003-41281. [DOI] [PubMed] [Google Scholar]
- Dale R.C., de Sousa C., Chong W.K. Acute disseminated encephalomyelitis, multiphasic disseminated encephalomyelitis and multiple sclerosis in children. Brain. 2000;123:2407–2422. doi: 10.1093/brain/123.12.2407. [DOI] [PubMed] [Google Scholar]
- De Bolle L., Van Loon J., De Clercq E. Quantitative analysis of human herpesvirus 6 cell tropism. J Med Virol. 2005;75:76–85. doi: 10.1002/jmv.20240. [DOI] [PubMed] [Google Scholar]
- Doja A., Bitnun A., Jones E.L. Pediatric Epstein–Barr virus-associated encephalitis: 10-year review. J Child Neurol. 2006;21:385–391. doi: 10.1177/08830738060210051101. [DOI] [PubMed] [Google Scholar]
- Domachowske J.B., Cunningham C.K., Cummings D.L. Acute manifestations and neurologic sequelae of Epstein–Barr virus encephalitis in children. Pediatr Infect Dis J. 1996;15:871–875. doi: 10.1097/00006454-199610000-00008. [DOI] [PubMed] [Google Scholar]
- Fujimoto H., Asaoka K., Imaizumi T. Epstein–Barr virus infections of the central nervous system. Intern Med. 2003;42:33–40. doi: 10.2169/internalmedicine.42.33. [DOI] [PubMed] [Google Scholar]
- Fujinami R.S., Oldstone M.B. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230:1043–1045. doi: 10.1126/science.2414848. [DOI] [PubMed] [Google Scholar]
- Fujita K., Takeuchi Y., Nishimura A. Serial MRI in infantile bilateral striatal necrosis. Pediatr Neurol. 1994;10:157–160. doi: 10.1016/0887-8994(94)90050-7. [DOI] [PubMed] [Google Scholar]
- Garg R.K. Acute disseminated encephalomyelitis. Postgrad Med J. 2003;79:11–17. doi: 10.1136/pmj.79.927.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheuens S., Smith D.R., Wang X. Simultaneous PML-IRIS after discontinuation of natalizumab in a patient with MS. Neurology. 2012;78:1390–1393. doi: 10.1212/WNL.0b013e318253d61e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons J.L., Miller H.G., Stanton J.B. Para-infectious encephalomyelitis and related syndromes; a critical review of the neurological complications of certain specific fevers. Q J Med. 1956;25:427–505. [PubMed] [Google Scholar]
- Grant C., Oh U., Yao K. Dysregulation of TGF-beta signaling and regulatory and effector T-cell function in virus-induced neuroinflammatory disease. Blood. 2008;111:5601–5609. doi: 10.1182/blood-2007-11-123430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray F., Chimelli L., Mohr M. Fulminating multiple sclerosis-like leukoencephalopathy revealing human immunodeficiency virus infection. Neurology. 1991;41:105–109. doi: 10.1212/wnl.41.1.105. [DOI] [PubMed] [Google Scholar]
- Gray F., Belec L., Lescs M.C. Varicella-zoster virus infection of the central nervous system in the acquired immune deficiency syndrome. Brain. 1994;117:987–999. doi: 10.1093/brain/117.5.987. [DOI] [PubMed] [Google Scholar]
- Gupte G., Stonehouse M., Wassmer E. Acute disseminated encephalomyelitis: a review of 18 cases in childhood. J Paediatr Child Health. 2003;39:336–342. doi: 10.1046/j.1440-1754.2003.00154.x. [DOI] [PubMed] [Google Scholar]
- Hahn C.D., Miles B.S., MacGregor D.L. Neurocognitive outcome after acute disseminated encephalomyelitis. Pediatr Neurol. 2003;29:117–123. doi: 10.1016/s0887-8994(03)00143-7. [DOI] [PubMed] [Google Scholar]
- Hart M.N., Earle K.M. Haemorrhagic and perivenous encephalitis: a clinical-pathological review of 38 cases. J Neurol Neurosurg Psychiatry. 1975;38:585–591. doi: 10.1136/jnnp.38.6.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartfield D.S., Loewy J.A., Yager J.Y. Transient thalamic changes on MRI in a child with hypernatremia. Pediatr Neurol. 1999;20:60–62. doi: 10.1016/s0887-8994(98)00092-7. [DOI] [PubMed] [Google Scholar]
- Hartung H.P., Grossman R.I. ADEM: distinct disease or part of the MS spectrum? Neurology. 2001;56:1257–1260. doi: 10.1212/wnl.56.10.1257. [DOI] [PubMed] [Google Scholar]
- Hemachudha T., Griffin D.E., Giffels J.J. Myelin basic protein as an encephalitogen in encephalomyelitis and polyneuritis following rabies vaccination. N Engl J Med. 1987;316:369–374. doi: 10.1056/NEJM198702123160703. [DOI] [PubMed] [Google Scholar]
- Hollsberg P. Pathogenesis of chronic progressive myelopathy associated with human T-cell lymphotropic virus type I. Acta Neurol Scand Suppl. 1997;169:86–93. doi: 10.1111/j.1600-0404.1997.tb08156.x. [DOI] [PubMed] [Google Scholar]
- Hung K.L., Liao H.T., Tsai M.L. The spectrum of postinfectious encephalomyelitis. Brain Dev. 2001;23:42–45. doi: 10.1016/s0387-7604(00)00197-2. [DOI] [PubMed] [Google Scholar]
- Hurst E.W. Acute haemorrhagic leucoencephalitis: a previously undefined entity. Med J Aust. 1941;2:1–6. [Google Scholar]
- Hynson J.L., Kornberg A.J., Coleman L.T. Clinical and neuroradiologic features of acute disseminated encephalomyelitis in children. Neurology. 2001;56:1308–1312. doi: 10.1212/wnl.56.10.1308. [DOI] [PubMed] [Google Scholar]
- Ichiyama T., Shoji H., Kato M. Cerebrospinal fluid levels of cytokines and soluble tumour necrosis factor receptor in acute disseminated encephalomyelitis. Eur J Pediatr. 2002;161:133–137. doi: 10.1007/s00431-001-0888-2. [DOI] [PubMed] [Google Scholar]
- Idrissova Zh.R., Boldyreva M.N., Dekonenko E.P. Acute disseminated encephalomyelitis in children: clinical features and HLA-DR linkage. Eur J Neurol. 2003;10:537–546. doi: 10.1046/j.1468-1331.2003.00639.x. [DOI] [PubMed] [Google Scholar]
- Jacobs B.C., Endtz H., van der Meche F.G. Serum anti-GQ1b IgG antibodies recognize surface epitopes on Campylobacter jejuni from patients with Miller Fisher syndrome. Ann Neurol. 1995;37:260–264. doi: 10.1002/ana.410370218. [DOI] [PubMed] [Google Scholar]
- Jacobs R.K., Anderson V.A., Neale J.L. Neuropsychological outcome after acute disseminated encephalomyelitis: impact of age at illness onset. Pediatr Neurol. 2004;31:191–197. doi: 10.1016/j.pediatrneurol.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Jones H.R., Jr., Ho D.D., Forgacs P. Acute fulminating fatal leukoencephalopathy as the only manifestation of human immunodeficiency virus infection. Ann Neurol. 1988;23:519–522. doi: 10.1002/ana.410230515. [DOI] [PubMed] [Google Scholar]
- Jorens P.G., VanderBorght A., Ceulemans B. Encephalomyelitis-associated antimyelin autoreactivity induced by streptococcal exotoxins. Neurology. 2000;54:1433–1441. doi: 10.1212/wnl.54.7.1433. [DOI] [PubMed] [Google Scholar]
- Kadhim H., De Prez C., Gazagnes M.D. In situ cytokine immune responses in acute disseminated encephalomyelitis: insights into pathophysiologic mechanisms. Hum Pathol. 2003;34:293–297. doi: 10.1053/hupa.2003.34. [DOI] [PubMed] [Google Scholar]
- Kannian P., Yin H., Doueiri R. Distinct transformation tropism exhibited by human T lymphotropic virus type 1 (HTLV-1) and HTLV-2 is the result of postinfection T cell clonal expansion. J Virol. 2012;86:3757–3766. doi: 10.1128/JVI.06900-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastrup O., Maschke M., Diener H.C. Progressive multifocal leukoencephalopathy limited to the brain stem. Neuroradiology. 2002;44:227–229. doi: 10.1007/s00234-001-0714-6. [DOI] [PubMed] [Google Scholar]
- Keegan M., Pineda A.A., McClelland R.L. Plasma exchange for severe attacks of CNS demyelination: predictors of response. Neurology. 2002;58:143–146. doi: 10.1212/wnl.58.1.143. [DOI] [PubMed] [Google Scholar]
- Kesselring J., Miller D.H., Robb S.A. Acute disseminated encephalomyelitis. MRI findings and the distinction from multiple sclerosis. Brain. 1990;113:291–302. doi: 10.1093/brain/113.2.291. [DOI] [PubMed] [Google Scholar]
- Khong P.L., Ho H.K., Cheng P.W. Childhood acute disseminated encephalomyelitis: the role of brain and spinal cord MRI. Pediatr Radiol. 2002;32:59–66. doi: 10.1007/s00247-001-0582-6. [DOI] [PubMed] [Google Scholar]
- Kimura S., Nezu A., Ohtsuki N. Serial magnetic resonance imaging in children with postinfectious encephalitis. Brain Dev. 1996;18:461–465. doi: 10.1016/s0387-7604(96)00046-0. [DOI] [PubMed] [Google Scholar]
- Kleiman M., Brunquell P. Acute disseminated encephalomyelitis: response to intravenous immunoglobulin. J Child Neurol. 1995;10:481–483. doi: 10.1177/088307389501000612. [DOI] [PubMed] [Google Scholar]
- Kuerten S., Lichtenegger F.S., Faas S. MBP-PLP fusion protein-induced EAE in C57BL/6 mice. J Neuroimmunol. 2006;177:99–111. doi: 10.1016/j.jneuroim.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Kuper M., Rabe K., Esser S. Structural gray and white matter changes in patients with HIV. J Neurol. 2011;258:1066–1075. doi: 10.1007/s00415-010-5883-y. [DOI] [PubMed] [Google Scholar]
- Leake J.A., Albani S., Kao A.S. Acute disseminated encephalomyelitis in childhood: epidemiologic, clinical and laboratory features. Pediatr Infect Dis J. 2004;23:756–764. doi: 10.1097/01.inf.0000133048.75452.dd. [DOI] [PubMed] [Google Scholar]
- Lennon V.A., Wingerchuk D.M., Kryzer T.J. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- Letendre S.L., Ellis R.J., Everall I. Neurologic complications of HIV disease and their treatment. Top HIV Med. 2009;17:46–56. [PMC free article] [PubMed] [Google Scholar]
- Levine S. Hyperacute, neutrophilic, and localized forms of experimental allergic encephalomyelitis: a review. Acta Neuropathol. 1974;28:179–189. doi: 10.1007/BF00719023. [DOI] [PubMed] [Google Scholar]
- Lim K.E., Hsu Y.Y., Hsu W.C. Multiple complete ring-shaped enhanced MRI lesions in acute disseminated encephalomyelitis. Clin Imaging. 2003;27:281–284. doi: 10.1016/s0899-7071(02)00552-1. [DOI] [PubMed] [Google Scholar]
- Lisak R.P., Zweiman B. In vitro cell-mediated immunity of cerebrospinal-fluid lymphocytes to myelin basic protein in primary demyelinating diseases. N Engl J Med. 1977;297:850–853. doi: 10.1056/NEJM197710202971602. [DOI] [PubMed] [Google Scholar]
- Markus R., Brew B.J., Turner J. Successful outcome with aggressive treatment of acute haemorrhagic leukoencephalitis. J Neurol Neurosurg Psychiatry. 1997;63:551. doi: 10.1136/jnnp.63.4.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menge T., Hemmer B., Nessler S. Acute disseminated encephalomyelitis: an update. Arch Neurol. 2005;62:1673–1680. doi: 10.1001/archneur.62.11.1673. [DOI] [PubMed] [Google Scholar]
- Menge T., Kieseier B.C., Nessler S. Acute disseminated encephalomyelitis: an acute hit against the brain. Curr Opin Neurol. 2007;20:247–254. doi: 10.1097/WCO.0b013e3280f31b45. [DOI] [PubMed] [Google Scholar]
- Merkler D., Horvath E., Bruck W. “Viral deja vu” elicits organ-specific immune disease independent of reactivity to self. J Clin Invest. 2006;116:1254–1263. doi: 10.1172/JCI27372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E. Multiple sclerosis. Adv Exp Med Biol. 2012;724:222–238. doi: 10.1007/978-1-4614-0653-2_17. [DOI] [PubMed] [Google Scholar]
- Miyazawa R., Hikima A., Takano Y. Plasmapheresis in fulminant acute disseminated encephalomyelitis. Brain Dev. 2001;23:424–426. doi: 10.1016/s0387-7604(01)00256-x. [DOI] [PubMed] [Google Scholar]
- Mizuguchi M., Abe J., Mikkaichi K. Acute necrotising encephalopathy of childhood: a new syndrome presenting with multifocal, symmetric brain lesions. J Neurol Neurosurg Psychiatry. 1995;58:555–561. doi: 10.1136/jnnp.58.5.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll N.M., Rietsch A.M., Ransohoff A.J. Cortical demyelination in PML and MS: similarities and differences. Neurology. 2008;70:336–343. doi: 10.1212/01.WNL.0000284601.54436.e4. [DOI] [PubMed] [Google Scholar]
- Murthy S.N., Faden H.S., Cohen M.E. Acute disseminated encephalomyelitis in children. Pediatrics. 2002;110:e21. doi: 10.1542/peds.110.2.e21. [DOI] [PubMed] [Google Scholar]
- Nishikawa M., Ichiyama T., Hayashi T. Intravenous immunoglobulin therapy in acute disseminated encephalomyelitis. Pediatr Neurol. 1999;21:583–586. doi: 10.1016/s0887-8994(99)00042-9. [DOI] [PubMed] [Google Scholar]
- Oomes P.G., Jacobs B.C., Hazenberg M.P. Anti-GM1 IgG antibodies and Campylobacter bacteria in Guillain–Barré syndrome: evidence of molecular mimicry. Ann Neurol. 1995;38:170–175. doi: 10.1002/ana.410380208. [DOI] [PubMed] [Google Scholar]
- Paterson P.Y. Transfer of allergic encephalomyelitis in rats by means of lymph node cells. J Exp Med. 1960;111:119–136. doi: 10.1084/jem.111.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl D., Rostasy K., Reiber H. CSF characteristics in early-onset multiple sclerosis. Neurology. 2004;63:1966–1967. doi: 10.1212/01.wnl.0000144352.67102.bc. [DOI] [PubMed] [Google Scholar]
- Power C., Kong P.A., Crawford T.O. Cerebral white matter changes in acquired immunodeficiency syndrome dementia: alterations of the blood–brain barrier. Ann Neurol. 1993;34:339–350. doi: 10.1002/ana.410340307. [DOI] [PubMed] [Google Scholar]
- Pradhan S., Gupta R.P., Shashank S. Intravenous immunoglobulin therapy in acute disseminated encephalomyelitis. J Neurol Sci. 1999;165:56–61. doi: 10.1016/s0022-510x(99)00072-6. [DOI] [PubMed] [Google Scholar]
- RamachandranNair R., Rafeequ M., Girija A.S. Plasmapheresis in childhood acute disseminated encephalomyelitis. Indian Pediatr. 2005;42:479–482. [PubMed] [Google Scholar]
- Rees J.H., Gregson N.A., Hughes R.A. Anti-ganglioside GM1 antibodies in Guillain–Barré syndrome and their relationship to Campylobacter jejuni infection. Ann Neurol. 1995;38:809–816. doi: 10.1002/ana.410380516. [DOI] [PubMed] [Google Scholar]
- Rivers T.M., Sprunt D.H., Berry G.P. Observations on attempts to produce acute disseminated encephalomyelitis in monkeys. J Exp Med. 1933;58:39–53. doi: 10.1084/jem.58.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M., Karnes W.E., Bartleson J.D. Plasmapheresis in acute episodes of fulminant CNS inflammatory demyelination. Neurology. 1993;43:1100–1104. doi: 10.1212/wnl.43.6.1100. [DOI] [PubMed] [Google Scholar]
- Rosman N.P., Gottlieb S.M., Bernstein C.A. Acute hemorrhagic leukoencephalitis: recovery and reversal of magnetic resonance imaging findings in a child. J Child Neurol. 1997;12:448–454. doi: 10.1177/088307389701200707. [DOI] [PubMed] [Google Scholar]
- Ruggieri M., Polizzi A., Pavone L. Thalamic syndrome in children with measles infection and selective, reversible thalamic involvement. Pediatrics. 1998;101:112–119. doi: 10.1542/peds.101.1.112. [DOI] [PubMed] [Google Scholar]
- Rust R.S., Dodson W., Prensky A.L. Classification and outcome of acute disseminated encephalomyelitis. Ann Neurol. 1997:491. [Google Scholar]
- Sahlas D.J., Miller S.P., Guerin M. Treatment of acute disseminated encephalomyelitis with intravenous immunoglobulin. Neurology. 2000;54:1370–1372. doi: 10.1212/wnl.54.6.1370. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S., Sakaguchi N., Asano M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- Schwarz S., Mohr A., Knauth M. Acute disseminated encephalomyelitis: a follow-up study of 40 adult patients. Neurology. 2001;56:1313–1318. doi: 10.1212/wnl.56.10.1313. [DOI] [PubMed] [Google Scholar]
- Seales D., Greer M. Acute hemorrhagic leukoencephalitis. A successful recovery. Arch Neurol. 1991;48:1086–1088. doi: 10.1001/archneur.1991.00530220108029. [DOI] [PubMed] [Google Scholar]
- Shahar E., Andraus J., Savitzki D. Outcome of severe encephalomyelitis in children: effect of high-dose methylprednisolone and immunoglobulins. J Child Neurol. 2002;17:810–814. doi: 10.1177/08830738020170111001. [DOI] [PubMed] [Google Scholar]
- Shintaku M., Matsumoto R., Sawa H. Infection with JC virus and possible dysplastic ganglion-like transformation of the cerebral cortical neurons in a case of progressive multifocal leukoencephalopathy. J Neuropathol Exp Neurol. 2000;59:921–929. doi: 10.1093/jnen/59.10.921. [DOI] [PubMed] [Google Scholar]
- Spillane J.D., Wells C.E. The neurology of Jennerian vaccination. A clinical account of the neurological complications which occurred during the smallpox epidemic in south wales in 1962. Brain. 1964;87:1–44. doi: 10.1093/brain/87.1.1. [DOI] [PubMed] [Google Scholar]
- Straussberg R., Schonfeld T., Weitz R. Improvement of atypical acute disseminated encephalomyelitis with steroids and intravenous immunoglobulins. Pediatr Neurol. 2001;24:139–143. doi: 10.1016/s0887-8994(00)00229-0. [DOI] [PubMed] [Google Scholar]
- Stricker R.B., Miller R.G., Kiprov D.D. Role of plasmapheresis in acute disseminated (postinfectious) encephalomyelitis. J Clin Apher. 1992;7:173–179. doi: 10.1002/jca.2920070403. [DOI] [PubMed] [Google Scholar]
- Suwa K., Yamagata T., Momoi M.Y. Acute relapsing encephalopathy mimicking acute necrotizing encephalopathy in a 4-year-old boy. Brain Dev. 1999;21:554–558. doi: 10.1016/s0387-7604(99)00078-9. [DOI] [PubMed] [Google Scholar]
- Sweeney B.J., Manji H., Miller R.F. Cortical and subcortical JC virus infection: two unusual cases of AIDS associated progressive multifocal leukoencephalopathy. J Neurol Neurosurg Psychiatry. 1994;57:994–997. doi: 10.1136/jnnp.57.8.994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan K., Roda R., Ostrow L. PML-IRIS in patients with HIV infection: clinical manifestations and treatment with steroids. Neurology. 2009;72:1458–1464. doi: 10.1212/01.wnl.0000343510.08643.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan I.L., McArthur J.C., Clifford D.B. Immune reconstitution inflammatory syndrome in natalizumab-associated PML. Neurology. 2011;77:1061–1067. doi: 10.1212/WNL.0b013e31822e55e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenembaum S., Chamoles N., Fejerman N. Acute disseminated encephalomyelitis: a long-term follow-up study of 84 pediatric patients. Neurology. 2002;59:1224–1231. doi: 10.1212/wnl.59.8.1224. [DOI] [PubMed] [Google Scholar]
- Tenembaum S., Chitnis T., Ness J. Acute disseminated encephalomyelitis. Neurology. 2007;68(Suppl 2):S23–S36. doi: 10.1212/01.wnl.0000259404.51352.7f. [DOI] [PubMed] [Google Scholar]
- Townsend J.J., Baringer J.R., Wolinsky J.S. Progressive rubella panencephalitis. Late onset after congenital rubella. N Engl J Med. 1975;292:990–993. doi: 10.1056/NEJM197505082921902. [DOI] [PubMed] [Google Scholar]
- Townsend J.J., Wolinsky J.S., Baringer J.R. The neuropathology of progressive rubella panencephalitis of late onset. Brain. 1976;99:81–90. doi: 10.1093/brain/99.1.81. [DOI] [PubMed] [Google Scholar]
- van der Meyden C.H., de Villiers J.F., Middlecote B.D. Gadolinium ring enhancement and mass effect in acute disseminated encephalomyelitis. Neuroradiology. 1994;36:221–223. doi: 10.1007/BF00588135. [DOI] [PubMed] [Google Scholar]
- von Einsiedel R.W., Fife T.D., Aksamit A.J. Progressive multifocal leukoencephalopathy in AIDS: a clinicopathologic study and review of the literature. J Neurol. 1993;240:391–406. doi: 10.1007/BF00867351. [DOI] [PubMed] [Google Scholar]
- von Giesen H.J., Arendt G., Neuen-Jacob E. A pathologically distinct new form of HIV associated encephalopathy. J Neurol Sci. 1994;121:215–221. doi: 10.1016/0022-510x(94)90355-7. [DOI] [PubMed] [Google Scholar]
- Wang P.N., Fuh J.L., Liu H.C. Acute disseminated encephalomyelitis in middle-aged or elderly patients. Eur Neurol. 1996;36:219–223. doi: 10.1159/000117253. [DOI] [PubMed] [Google Scholar]
- Wingerchuk D.M., Lennon V.A., Pittock S.J. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–1489. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]