

Case Study.
After studying the material in this chapter, the reader should be able to respond to the following case study:
A 25-year-old female was seen by her primary care physician complaining of fatigue. Physical exam revealed pale conjunctiva. The remainder of the exam was unremarkable. The patient admitted to experiencing heavier than normal bleeding during menses. CBC results:
| WBC | 8.1 × 109/L |
| Hemoglobin | 8.6 g/dL |
| MCV | 65 fL |
| MCHC | 29 % |
| Platelets | 172 × 109/L |
Because of the low MCV, a blood film review was performed. There appeared to be immature granulocytes present, (eosinophils and basophils were normal) which required performing a manual differential:
| Cell | % |
| Segmented neutrophils | 9 |
| Band neutrophils | 21 |
| Metamyelocytes | 16 |
| Myelocytes | 14 |
| Lymphocytes | 30 |
| Monocytes | 7 |
| Eosinophils | 2 |
| Basophils | 1 |
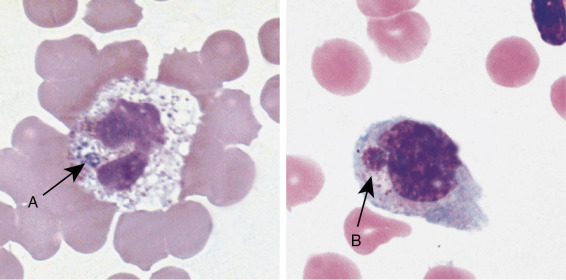
Figure 26.1.
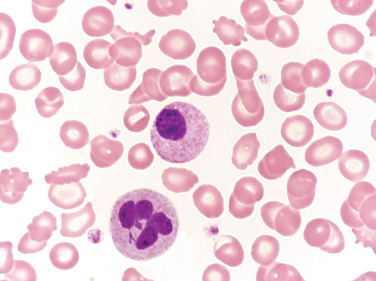
Peripheral Blood Film for the Patient in the Case Study.
The cell on the top was classified as a myelocyte. The other immature neutrophils reported in the differential (not shown) appeared similar in morphology. The cell on the bottom was classified as a neutrophil. (Wright-Giemsa stain, X1000.)
(Courtesy Nicholas Brehl, Indiana University, Indianapolis, IN.)
-
1.
Do you agree with the cell on top being classified as a myelocyte? If not, provide an explanation in morphologic terms.
-
2.
The patient was subsequently diagnosed with iron deficiency anemia. If cells identified as band neutrophils and metamyelocytes appeared very much like the cell depicted, except for some nuclear indentations, what other hematologic diagnosis is suspected?
-
3.
Provide an approach to correcting the results of the WBC differential that does not require repeating the test.
-
4.
What are clinical implications of reporting the manual differential results above if a left shift does not actually exist?
This chapter concentrates on leukocyte disorders that are not caused by clonal or neoplastic changes in hematopoietic precursor cells. The causes can be genetic or acquired and involve one or more lineages: neutrophil, lymphocyte, monocyte, eosinophil, and basophil, affecting the number of circulating cells, morphology, or both. Many of these disorders are associated with significant clinical manifestations, although some are benign in nature.
Congenital defects of leukocyte number and function
There are more than 350 primary immunodeficiency disorders, each caused by genetic abnormalities that disrupt normal function of innate or adaptive immune systems.1 Cellular functions, number of cells or both are affected, and the resulting clinical manifestations are often severe and widespread. This chapter highlights some of the more common and well-known diseases with significant peripheral blood film abnormalities.
Severe combined immune deficiency (SCID) is a group of genetic immunodeficiencies affecting both cellular and humoral immunity. Almost all patients with SCID have a marked decrease in circulating T cells, poorly functioning B cells, hypogammaglobulinemia, and profound clinical manifestations. Left untreated, most patients die within the first two years of life. Two examples of SCID are γc deficiency and adenosine deaminase (ADA) deficiency.
Severe combined immune deficiency
Gamma chain deficiency, or X-linked SCID, is the most common form of SCID and is caused by mutations in the IL2RG gene located at Xq13.1.2 IL2RG normally codes for the common γ chain in leukocyte receptors that bind with interleukins 2, 4, 7, 9, 15 and 21. These interleukins normally provide growth, differentiation and survival signals for B, T and NK cells. These signals are disrupted becasue of altered leukocyte receptors in γc deficiency. Patients become symptomatic between 3 to 6 months of age as protective maternal immunoglobulins are depleted, presenting without tonsils or lymph nodes along with severe life-threatening recurring infections. Circulating T and natural killer (NK) lymphocytes are nearly absent. B cells are adequate in number but are dysfunctional. Children with γc deficiency fail to thrive and death usually occurs before age 2 unless treatment with hematopoietic stem cell transplant is successful.3 Gene therapy has been effective in clinical trials; however, there is a significant risk of leukemic transformation.
Autosomal recessive adenosine deaminase ADA deficiency represents 10% to 20% of SCID cases and is caused by one of many mutations in the ADA gene located at chromosome 20q13.12.4 Adenosine deaminase is a key component of the metabolic breakdown of adenosine triphosphate (ATP) and RNA. ADA deficiency results in an intra- and extracellular accumulation of adenosine, which is lymphotoxic, leading to profound decreases in T, B, and NK cells. Patients experience a range of recurring, life-threatening bacterial, viral, and fungal infections beginning early in life. In addition, there are skeletal abnormalities, neurologic deficits, and skin rashes. As with X-linked SCID, stem cell transplant and gene therapy (experimental) are used to reconstitute the failed immune system.
Wiskott-Aldrich syndrome
Wiskott-Aldrich syndrome (WAS), is classified as a combined immunodeficiency.1 It is a rare X-linked disease caused by one of more than 400 mutations in the WAS gene, which results in decreased levels of WASp protein.5 WASp is important in cytoskeletal remodeling and nuclear transcription in hematopoietic cells. T cells are decreased; B cells, T cells and NK cells, neutrophils and monocytes are dysfunctional which leads to bacterial, viral and fungal infections. There is a risk of bleeding due to thrombocytopenia and small abnormal platelets. Therapies using eltrombopag and romiplostim have been somewhat successful in increasing the platelet count in WAS hematopoietic stem cell transplant is potentially curative; however, up to 55% of transplanted patients develop significant autoimmune cytopenias.5 Gene therapy has been successful in clinical trials, although there is a substantial risk for the development of acute leukemia.6
22q11 syndromes
22q11 deletion syndromes, also classified as combined immunodeficiency, include DiGeorge syndrome, autosomal dominant Opitz GBBB, Sedlackova syndrome, Caylor cardiofacial syndrome, Shprintzen syndrome, and conotruncal anomaly face syndrome.1 , 7 All the disorders within the 22q11 deletion syndrome have variable degrees of immunodeficiency because of the absence or decreased size of the thymus and low numbers of T lymphocytes. The underlying genetic abnormality is a microdeletion in chromosome band 22q11.2, most likely involving TBX1 and occurs in approximately 1 in 3000 to 6000 births.8 The 22q11 deletion is associated with a broad range of problems such as cardiac defects, palatal abnormalities, distinctive facial features, developmental delays, psychiatric disorders, short stature, kidney disease, and hypocalcemia. Hematologic issues include thrombocytopenia and large platelets, autoimmune cytopenias, and increased risk of malignancy. Patients are often treated with thymic tissue transplantation or fully matched peripheral blood T cell transplantation, however, the death rate is high and many succumb to the disease before 1 year of age.
Bruton tyrosine kinase deficiency
Classified as an antibody deficiency, Bruton tyrosine kinase (BTK) deficiency (X-linked agammaglobulinemia) is a primary immunodeficiency disease characterized by reductions in all serum immunoglobulin isotypes and profoundly decreased or absent B cells.1 BTK deficiency is caused by a mutation in the gene encoding Bruton tyrosine kinase, resulting in decreased production of BTK, which is important for B cell development, differentiation, and signaling.1 , 9 Without BTK, lymphocytes fail to fully mature, leading to severe hypogammaglobulinemia and an inability to produce specific antibodies. Infants with BTK deficiency display symptoms between 4 and 6 months, once maternal antibodies have cleared. Recurring life-threatening bacterial infections ensue. Risk of fungal and viral (except enterovirus) infection is low because of normal T cell function. Treatment consists of immunoglobulin replacement therapy.
Chédiak-Higashi syndrome
Chédiak-Higashi syndrome is a rare autosomal recessive disease of immune dysregulation. Only 500 cases had been reported worldwide as of 2008.10 Chédiak-Higashi syndrome is associated with a mutation in the CHS1 LYST gene on chromosome 1q42.1-2 that encodes for a protein that regulates the morphology and function of lysosome-related organelles.10 , 11 Many types of cells in the body are affected and exhibit abnormally large lysosomes, which contain fused dysfunctional granules. Clinical manifestations begin in infancy with partial albinism and severe recurrent life-threatening bacterial infections. Hematologic findings in Chédiak-Higashi syndrome include giant lysosomal granules in granulocytes, monocytes, and lymphocytes (Figure 26.2 ). These fused granules result in leukocyte dysfunction. Patients often have bleeding issues as a result of abnormal dense granules in platelets. Death occurs before the age of 10 years.
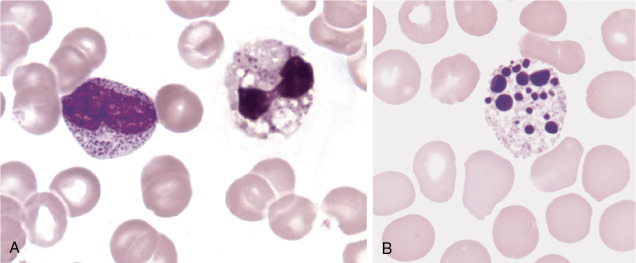
Figure 26.2.
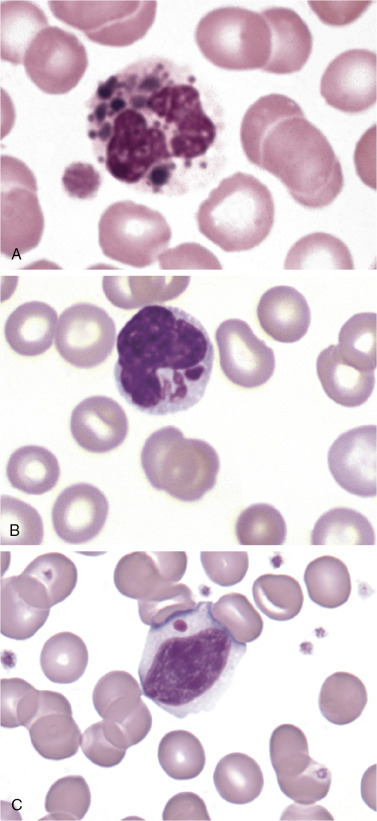
Cells from a Patient with Chédiak-Higashi Syndrome.
(A), Neutrophil with large dark lysosomal granules. (B), Monocyte with large azure granules. (C), Lymphocyte with one large azure granule. (Peripheral blood, Wright-Giemsa stain, ×1000.)
Pseudo-Chédiak-Higashi granules are cytoplasmic inclusions that resemble the fused lysosomal granules in Chédiak-Higashi syndrome. Pseudo-Chédiak-Higashi granules have been reported in patients with acute myeloid leukemia, chronic myeloid leukemia, and myelodysplastic syndrome (MDS).12., 13., 14.
Congenital defects of phagocytes
The congenital neutropenias (CNs) are a rare group of genetic diseases characterized by low neutrophil count, increased risk of infection, organ dysfunction, and a high rate of leukemic transformation.15 , 16 CNs usually present in the first year of life as recurrent fevers caused by bacterial and fungal infections, which are often life threatening. Improvements in treatment, including antibiotic prophylaxis and use of granulocyte colony-stimulating factor (G-CSF), have decreased morbidity and mortality rates; however, higher doses of G-CSF have been associated with increased risk of malignant transformation.17 Twenty-four genes have been identified as causing CN.15 Some more common and better known CNs are described in Table 26.1
TABLE 26.1.
Congenital Neutropenias
| Name | Gene | Gene Location | Inheritance | Hematologic/Clinical Features |
|---|---|---|---|---|
| Elastase deficiency | ELANE | 19p13.3 | Dominant | Severe/permanent neutropenia, bone marrow maturation arrest. Recurring infections. Risk for MDS/leukemia |
| Cyclic neutropenia (SCN1) | ELANE | 19p13.3 | Dominant | Neutropenia cycles every 21 days along with mouth ulcers, fevers, and bacterial infections |
| Kostmann disease (SCN3) | HAX1 | 1q21.3 | Recessive | Cognitive and neurologic defects, recurring infections, seizures, risk for MDS/leukemia |
| X-linked neutropenia | WAS | Xp11.4-p11.21 | X-linked | Severe/permanent neutropenia, maturation arrest, recurring infections, monocytopenia |
| Glycogen storage type Ib | SLC37A4 | 11q23.3 | Recessive | Hypoglycemia, lactic acidosis, hyperlipidemia, hepatomegaly, increased risk of infection |
MDS, Myelodysplastic syndrome.
Leukocyte adhesion disorders (defects of motility)
As leukocytes leave circulation to move to a site of infection, they adhere to endothelial cells and the tissue matrix and roll along vessel walls. Selectins and other adhesion molecules expressed on endothelial cells and leukocytes mediate this process, interacting with ligands on the surface of leukocytes to slow the speed of the leukocytes in circulation.18 Ligand binding induces high-affinity attachment of integrins with endothelial cell receptors. Leukocyte adhesion disorders (LADs) are rare autosomal recessive inherited conditions resulting in the inability of neutrophils and monocytes to move from circulation to the site of inflammation (called extravasation).Consequences of these disorders are recurrent severe bacterial and fungal infections. Hematopoietic stem cell transplant is the only curative treatment.
LAD I is caused by a mutation in ITGB2, the gene encoding the CD18 subunit of β2 integrins, resulting in either a decreased or truncated form of the β2 integrin, which is necessary for adhesion to endothelial cells, recognition of bacteria, and outside-in signaling.19 , 20 These activities are compromised in individuals with LAD 1. Shortly after birth, patients suffer from recurrent infections, often affecting skin and mucosal infections. Lymphadenopathy, splenomegaly, and neutrophilia are common findings. The clinical severity, including number of infections and survival, depends on the amount of β2 integrins produced.20 Infant mortality rate is high in LAD I.
LAD II presents in a similar manner as LAD 1, however leukocytes have normal β2 integrins. There are molecular defects in SLC35C1, which codes for a fucose transporter that moves fucose from the endoplasmic reticulum to the Golgi region. Fucose is needed for posttranslational fucosylation of glycoconjugates, which are required for synthesis of selectin ligands.21 In LAD II, selectin synthesis is compromised. Clinically, LAD II patients have recurring infections, neutrophilia, growth retardation, a coarse face, and other physical deformities. In LAD II the defective fucose transporter leads to an inability to produce functional selectins and defective leukocyte recruitment, which leads to recurring infections. Other clinical findings related to defective fucose transport are absence of blood group H antigen, growth retardation, and neurologic defects.22
LAD III is caused by mutations in Kindlin-3.23 Kindlin-3 protein along with talin are required for activation of β integrin and leukocyte rolling. In LAD III, leukocytes and platelets have normal expression of integrins; however, there is failure in response to external signals that normally results in leukocyte activation. Clinically, LAD III patients experience a mild LAD I-like immunodeficiency with recurrent infections. Additionally, there is decreased platelet integrin GPIIbβ3 (glycoprotein IIb/IIIa), resulting in bleeding similar to that seen in Glanzmann thrombasthenia.
Like LAD, Shwachman-Diamond syndrome (SDS) is a defect in leukocyte motility. SDS is a rare autosomal recessive disease caused by mutations in the SBDS gene located at 7q11.22.24 This affects the SBDS protein product which has an important role in ribosomal maturation, cell proliferation and bone marrow microenvironment.25 Patients are usually diagnosed in infancy with exocrine pancreatic insufficiency associated malabsorption, malnutrition, chronic steatorrhea, and failure to thrive. There is bone marrow failure, including cytopenias, myelodysplasia, and an increased risk of acute leukemia. Patients also suffer from recurring infections, skeletal abnormalities, skin and dental problems, and cognitive issues. Hematopoietic stem cell transplant is an option for patients with bone marrow failure, myelodysplastic syndrome (MDS), and acute leukemia.
Defects of respiratory burst
Chronic granulomatous disease (CGD) is a rare condition caused by the decreased ability of neutrophils to undergo a respiratory burst after phagocytosis of foreign organisms. Approximately 60% of cases are X-linked recessive and 40% are autosomal recessive.26 X-linked recessive patients experience a more severe disease course and have shorter lifespans than autosomal recessive patients. CGD is caused by mutations in genes responsible for proteins that make up the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase. Under normal conditions, phagocytosis of foreign organism leads to phosphorylation and binding of cytosolic p47phos and p67phos.17 Primary granules containing antibacterial neutrophil elastase and cathepsin G and secondary granules containing the cytochrome complex gp91phox and gp22phox migrate to the phagolysosome. NADPH oxidase forms when p47phos and p67phos along with p40phox and RAC2 combine with the cytochrome complex. Superoxide is generated in the phagolysosome when an electron from NADPH is added to oxygen. NADPH has additional regulatory functions in the generation of other antimicrobial agents. Most cases of CGD are due to mutations in gp91phox or p47. The genes implicated in CGD and protein products are listed in Table 26.2 .
TABLE 26.2.
Genes Involved in Chronic Granulomatous Disease
| Inheritance | Gene | Location | Protein Product |
|---|---|---|---|
| XLR | CYBB | Xp21.1 | gp91phºx |
| AR | CYBA | 16q24 | p22phºx |
| AR | NCF1 | 7q11.23 | P47phºx |
| AR | NCF2 | 1q25 | p67phºx |
| AR | NCF4 | 22q13.1 | P40phºx |
AR, Autosomal recessive; XLR, sex-linked recessive.
CGD patients experience life-threatening catalase-positive bacterial and fungal infections. Advancements in care, including the routine use of prophylactic antibiotics and azole antifungals, have improved the clinical course of the disease and increased survival rates.27 , 28 CGD is diagnosed through a test that uses a fluorescent probe dihydrorhodamine to measure intracellular production of reactive oxygen species.26
Whim syndrome
WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis syndrome) is classified as a defect in intrinsic and innate immunity. WHIM results from mutations in the CXCR4 gene located at 2q22.29 Normal CXCR4 protein regulates movement of white blood cells between the bone marrow and peripheral blood. Neutrophils accumulate in the bone marrow (myelokathexis), which results in low numbers of circulating neutrophils. The retained neutrophils in the marrow exhibit degenerative, pyknotic, morphologic changes. In addition to neutropenia, lymphopenia, monocytopenia, and hypogammaglobulinemia are present. As a result, patients experience recurrent bacterial infections and are highly susceptible to human papillomavirus (HPV) infection, which leads to warts, which can be widespread and resistant to treatment. Treatment consists of antibiotic prophylaxis, immunoglobulin replacement therapy, and G-CSF.30 CXCR4 receptor antagonist is a targeted treatment proving to be effective at increasing neutrophil and lymphocyte counts.
Morphologic abnormalities of leukocytes without associated immunodeficiency
Pelger-Huët anomaly
Pelger-Huët anomaly (PHA), also known as true or congenital PHA, is an autosomal dominant disorder characterized by decreased nuclear segmentation and distinctive coarse chromatin clumping pattern. PHA potentially affects all leukocytes, although morphologic changes are most obvious in mature neutrophils.31 The prevalence of PHA is approximately 1 in 4785 in the United States.31 The disorder is a result of a mutation in the lamin β-receptor gene. 2 The lamin β receptor is an inner nuclear membrane protein that combines β-type lamins and heterochromatin and plays a major role in leukocyte nuclear shape changes that occur during normal maturation. Mutations in the lamin β-receptor gene result in the morphologic changes characteristic of PHA. Pelger-Huët (PH) nuclei may appear round, ovoid, or peanut shaped. Bilobed forms—the characteristic spectacle-like (“pince-nez”) morphology with the nuclei attached by a thin filament can also be seen (Figure 26.3 ). Reports of patients with homozygous PHA are rare. Virtually all identified cases are heterozygotes, in which greater than 68% of neutrophils exhibit PH morphology.32 Neutrophils in PHA appear to function normally.33 In heterozygous PHS, individuals are clinically normal, while in homozygous PHS, cognitive impairment, heart defects, and skeletal abnormalities may occur.31 , 24
Figure 26.3.
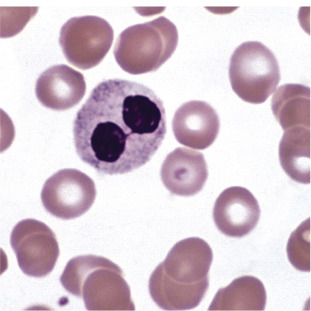
Pelger-Huët Cell.
Pince-nez form with two rounded segments connected by a filament. Notice the dense chromatin pattern. (Peripheral blood, Wright-Giemsa stain, ×1000.)
Pseudo- or Acquired Pelger-Huët anomaly
Neutrophils with similar morphology to PHA can be seen in patients with MDS, acute myeloid leukemia, and myeloproliferative neoplasm. Acquired or pseudo-PHA neutrophils are also associated with severe bacterial infections, HIV, tuberculosis, and mycoplasma pneumonia.33 Drugs known to induce PHA morphology include immunosuppressants, chemotherapies, valproate, sulfisoxazole, fluconazole, ganciclovir, hematopoietic growth factors, and ibuprofen.3 , 35
Because nuclei of PHA neutrophils are round, oval, or peanut shaped, cells may be misclassified as myelocytes, metamyelocytes, or band neutrophils in the white blood cell (WBC) differential. This can lead to unnecessary tests, misdiagnosis, and inappropriate treatment. Because PHA neutrophils maintain normal function and their presence is not associated with inflammation, infection, or other causes of a neutrophilic left shift, it is recommended that PHA neutrophils be classified as segmented neutrophils with an appropriate interpretive comment.36 There are morphologic differences between PHA neutrophils and normal neutrophils that can aid in differentiating between the two. Cell size is smaller, the nucleus/cytoplasm (N/C) ratio of PHA neutrophils is lower, and chromatin is darker, more coarse, and more densely clumped than band neutrophils, metamyelocytes, and myelocytes. Metamyelocytes and myelocytes generally show some degree of cytoplasmic basophilia, whereas PHA neutrophils exhibit nearly colorless cytoplasm except for that imparted by normal cytoplasmic granulation. Pseudo-PHA cells can exhibit hypogranularity in MDS.
Differentiating between true and pseudo-PHA can be challenging. It is important to consider the number of cells exhibiting characteristic morphology. In true PHA the number of affected cells is higher than in pseudo-PHA (>68% vs. <35%, respectively).32 , 37 In true PHA, all WBC lineages can be affected in terms of nuclear shape and chromatin structure. In pseudo-PHA the phenomenon is restricted to neutrophils, except in MDS where monocytes, eosinophils, and basophils may be affected. Furthermore, if true PHA is suspected, careful examination of peripheral blood films of family members may reveal characteristic morphology. Clinical correlation can also help to differentiate true and pseudo-PHA.
Neutrophil hypersegmentation
Normal neutrophils contain three to five lobes that are separated by filaments. Hypersegmented neutrophils have more than five lobes and are most often associated with megaloblastic anemia, in which hypersegmented neutrophils are usually larger than normal (Figure 26.4 ). Hypersegmented neutrophils can also be seen in MDS where they represent a form of dysplasia. Less often hypersegmented neutrophils can be found in hereditary neutrophil hypersegmentation. In this disorder individuals are asymptomatic and have no signs of megaloblastic anemia.
Figure 26.4.
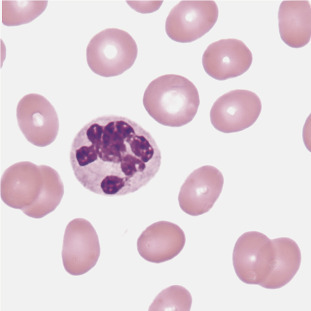
Hypersegmented Neutrophil.
(Peripheral blood, Wright-Giemsa stain, ×1000.)
(From Rodak, B. F., & Carr, J. H. [2017]. Clinical Hematology Atlas. [5th ed.]. St. Louis: Saunders.)
Alder-Reilly anomaly
Alder-Reilly anomaly (AR) is a rare inherited disorder characterized by granulocytes (monocytes and lymphocytes less often) with large, darkly staining metachromatic cytoplasmic granules (Figure 26.5 ). AR anomaly was initially reported in patients with gargoylism; however, it can be seen in otherwise healthy individuals.38 The characteristic granulation, called Reilly bodies, is also found in the mucopolysaccharidoses (MPSs). The cytoplasmic granules contain partially digested mucopolysaccharides. Leukocyte function is not affected in AR. AR bodies in neutrophils may resemble heavy toxic granulation, however, Reilly bodies can also be present in monocytes and lymphocytes, whereas toxic granulation occurs only in neutrophils. Also, neutrophilia with left shift, Döhle bodies, or both often accompany toxic granulation. These are not associated with AR.
Figure 26.5.
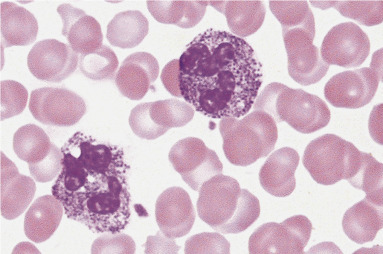
Neutrophils from a Patient with Alder-Reilly Anomaly.
Note the dark granules present in both cells. Such granules may also be seen in eosinophils and basophils. (Peripheral blood, Wright-Giemsa stain, ×1000.)
(Courtesy Dennis R. O’Malley, US Labs, Irvine, CA.)
May-Hegglin anomaly
May-Hegglin anomaly is a rare, autosomal dominant disorder characterized by variable thrombocytopenia, giant platelets, and large Döhle body-like inclusions in neutrophils, eosinophils, basophils, and monocytes (Figure 26.6 ). May-Hegglin anomaly is caused by a mutation in the MYH9 gene on chromosome 22q12-13.39 There is disordered production of myosin heavy chain type IIA, which affects megakaryocyte maturation and platelet fragmentation when shedding from megakaryocytes
Figure 26.6.
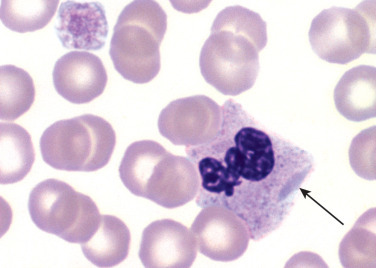
Neutrophil and Giant Platelet from a Patient with May-Hegglin Anomaly.
Note the large, elongated, bluish inclusion in the neutrophil cytoplasm. (Peripheral blood, Wright-Giemsa stain, ×1000.)
The basophilic Döhle body-like leukocyte inclusions in May-Hegglin anomaly are composed of precipitated myosin heavy chains. True Döhle bodies consist of lamellar rows of rough endoplasmic reticulum. Most individuals with May-Hegglin anomaly are asymptomatic, but a few have mild bleeding tendencies related to the degree of thrombocytopenia.
Lysosomal storage diseases
Lysosomal storage disorders (LSDs) are a group of more than 50 inherited enzyme deficiencies resulting from mutations in genes that code for the production of lysosomal enzymes.40 The result is flawed degradation of phagocytized material and buildup of undigested substrates within lysosomes. This causes cell dysfunction, cell death, and a range of clinical symptoms. All cells containing lysosomes can be affected. LSDs are classified according to the underdegraded macromolecule that accumulates in the cell. The categories include sphingolipidoses, oligosaccharidoses, mucolipidoses, mucopolysaccharidoses (MPS), lipoprotein storage disorders, and lysosomal transport defects. More common LSDs are presented.
Mucopolysaccharidoses
The MPS are a family of inherited disorders of mucopolysaccharide or glycoaminoglycan (GAG) degradation. Each MPS is caused by deficient activity of an enzyme necessary for the degradation of dermatan sulfate, heparan sulfate, keratan sulfate, and/or chondroitin sulfate. The partially degraded material builds up in lysosomes and results in serious physical and cognitive problems and shortened survival. Table 26.3 provides genetic and clinical information about MPS subtypes along with the deficient enzymes and accumulated substrates.
TABLE 26.3.
Mucopolysaccharidoses Disorders
| Name | Subtype | Enzyme Deficiency | AS | Genetics | Clinical Features |
|---|---|---|---|---|---|
| MPS I | Hurler syndrome | α-l-iduronidase | DS + HS | IDUA 4p16.3 | Coarse facies, short stature, corneal clouding, joint stiffening, umbilical hernia, dysostosis multiplex, hepatosplenomegaly, frequent upper respiratory infections, cognitive impairment; death before 10 y |
| MPS I | Scheie syndrome | α-l-iduronidase | DS + HS | IDUA 4p16.3 | Hepatomegaly, joint contractures, cardiac valve abnormalities, corneal clouding; prolonged survival but with disability |
| MPS II | Hunter syndrome | Iduronate sulfatase | DS + HS | IDS Xq28 | Coarse facial features, short stature, skeletal deformities, joint stiffness, mental retardation |
| MPS III A | Sanfilippo syndrome | Heparan N-sulfatase | HS | SGSH 17q25.3 | Hirsutism, coarse facial features, behavioral problems, aggressive behavior, speech delay, hepatomegaly; most die before 20 y |
| MPS III B | Sanfilippo syndrome | α-N-acetylglucosaminidase | HS | NAGLU 17q21.2 | Cardiomegaly, coarse facial features, progressive dementia, convulsions, survive into 20s, 30s |
| MPS III C | Sanfilippo syndrome | Heparan acetyl-CoA: α-glucosaminidase N-acetyltransferase | HS | HGSNAT 8p11.21 | Delayed psychomotor development, behavioral problems, sleeping/hearing problems, coarse facial features, recurrent infection, diarrhea, epilepsy, and retinitis pigmentosa |
| MPS IV A | Morquio syndrome | Galactose-6-sulfatase | KS + CS | GALNS 16q24.3 | Musculoskeletal abnormalities, short stature, pulmonary and cardiac dysfunction, hearing loss corneal clouding; most die before 30 y |
| MPS IV B | Morquio syndrome | β-Galactosidase | KS | GLB1 3p22.3 | Musculoskeletal abnormalities, short stature, pulmonary and cardiac dysfunction, hearing loss corneal clouding; most die before 30 y |
AS, accumulated substrate; CS, Chondroitin-6-sulfate; DS, dermatan sulfate; HS, heparin sulfate; KS, keratan sulfate.
Adapted from Wraith, J. E., & Jones, S. (2014). Mucopolysaccharidosis type I. Pediatr Endocrinol Rev, 12(1), 102–106; Francisca, M., Coutinho, L., & Alves, S. (2015). From bedside to cell biology: A century of history on lysosomal dysfunction. Gene, 555(1), 50–58; Meyer, A., Kossow, K., Gal, A., et al. (2007). Scoring evaluation of the natural course of mucopolysaccharidosis type IIIA (Sanfilippo syndrome type A). Pediatrics, 120(5), 1255–1261; and Morrone, A., Caciotti, A., Atwood, R., et al. (2014). Morquio A syndrome-associated mutations: A review of alterations in the GALNS gene and a new locus-specific database. Human Mutation, 35(11), 1271–1279.
The peripheral blood of a patient with MPS may show metachromatic Reilly bodies in neutrophils, monocytes, and lymphocytes. Macrophages in the bone marrow can also demonstrate cytoplasmic metachromatic material. Diagnosis relies on assays that measure the specific enzymes involved. MPS treatment consists of enzyme replacement therapy or hematopoietic stem cell transplantation (Hurler patients only). Emerging therapies include additional enzyme replacement therapies, improved antiinflammatories, gene therapy, nanoparticles, and substrate reduction therapy.41
The sphingolipidoses are inherited disorders in which lipid catabolism is defective (Figure 26.7 ). Two of these disorders, Gaucher and Neimann-Pick diseases are characterized by macrophages with distinctive morphology.
Figure 26.7.
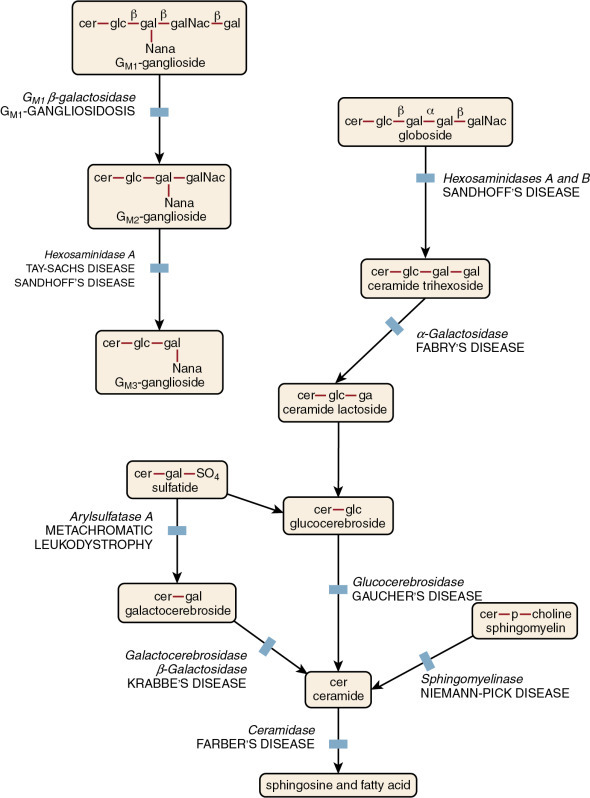
Pathways and Diseases of Sphingolipid Metabolism.
(From Orkin, S. H., Fisher, D. E., & Look, A. T., et al. [2009]. Nathan and Oski’s hematology of infancy and childhood. [7th ed.]. Philadelphia: Saunders.)
Gaucher disease
Gaucher disease is the most common of the lysosomal lipid storage diseases. It is an autosomal recessive disorder caused by a defect or deficiency in the catabolic enzyme β-glucocerebrosidase (gene located at 1q21-q22), which is necessary for glycolipid metabolism. As a result, there is an accumulation of unmetabolized substrate sphingolipid glucocerebroside in macrophages throughout the body, including osteoclasts in bone and microglia in the brain. At least 1 in 17 Ashkenazi Jews are carriers.42 More than 400 genetic mutations have been reported, and although some correlations have been found with specific mutations and disease severity and course, most cases (phenotypes) cannot be predicted by genotype.43
There are three types of Gaucher disease and type I is by far the most common. Neurologic symptoms are key factors to differentiate the subtypes. The phenotype is quite heterogeneous, with some patients being asymptomatic, whereas others experience a multitude of clinical problems. Demographic and clinical information associated with the three types of Gaucher disease are provided in Table 26.4 .
TABLE 26.4.
Clinical Subtypes of Gaucher Disease
| Type I: Non-Neuronopathic | Type II: Acute Neuronopathic | Type III: Subacute Neuronopathic | |
|---|---|---|---|
| Age at presentation | Childhood/adulthood | Infancy | Childhood/adulthood |
| Hepatosplenomegaly | + → +++ | + | + → +++ |
| Skeletal abnormality | – → +++ | – | ++ → +++ |
| CNS disease | – | +++ | + → +++ |
| Life span | 6–80 + years | <2 years | 2–60 years |
| Ethnicity | Ashkenazi Jews | Panethnic | Panethnic, Swedes |
CNS, Central nervous system.
Note: Absence and severity of features are indicated by – to +++.
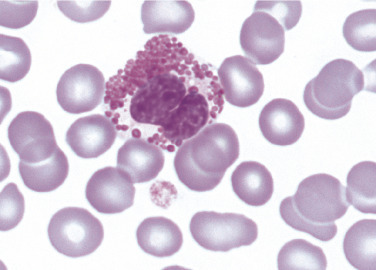
Bone marrow replacement by Gaucher cells contribute to anemia and thrombocytopenia, which are common findings in these patients. Gaucher cells are distinctive macrophages, single or in clusters, exhibiting abundant fibrillar blue-gray cytoplasm with a striated or wrinkled appearance (sometimes described as onion skin-like) (Figure 26.8 ). Gaucher cells stain positive with trichrome, aldehyde fuchsin, periodic acid-Schiff (PAS) and acid phosphatase.40 A commercially available test for β-glucosidase (glucocerebrosidase) is available to confirm diagnosis. Genetic testing is sometimes used in Ashkenazi Jews to screen for the most common mutations, including N370S, 84GG, IVS2 + 1G> A, and L444P. 40 In all three forms of Gaucher disease, there is a fifteenfold increase for developing hematologic malignancies such as plasma cell neoplasm, chronic lymphocytic leukemia, non-Hodgkin lymphoma, and acute leukemia.44
Figure 26.8.
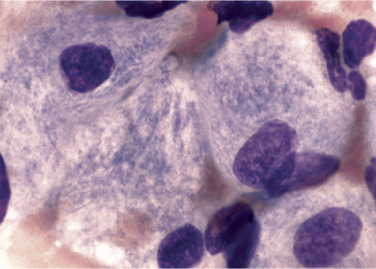
Characteristic Macrophages in Patient with Gaucher Disease.
Note cytoplasmic striations in the cytoplasm. (Bone marrow, hematoxylin and eosin stain, ×1000.)
(From Rodak, B. F., & Carr, J. H. [2013]. Clinical Hematology Atlas. [4th ed.]. St. Louis: Saunders.)
Treatment of Gaucher disease includes enzyme replacement therapy with recombinant glucocerebrosidase. Agents are also available to reduce glucocerebroside. Allogeneic stem cell transplantation offers the potential for cure; however, treatment-related mortality rate is high and no studies have determined the safety or efficacy of transplant compared with standard of care therapies.45 Pseudo-Gaucher cells can be found in bone marrow of some patients with thalassemia,46 chronic myeloid leukemia,47 acute lymphoblastic leukemia,48 non-Hodgkin lymphoma,49 and plasma cell neoplasms.50 , 51 In these diseases, pseudo-Gaucher cells most likely form as a result of excessive cell turnover, which overwhelms the glucocerebrosidase enzyme, rather than a true decrease in the enzyme. Electron microscopy shows that pseudo-Gaucher cells do not contain the tubular inclusions described in Gaucher cells.
Niemann-Pick disease
Niemann-Pick disease (NP) is characterized by an accumulation of fat in cellular lysosomes of vital organs, which impairs their function, leading to a range of clinical findings. NP has three subtypes. Types A and B are caused by recessive mutations in the SMPD1 gene located within the chromosomal region 11p15.4. This results in a deficiency of lysosomal hydrolase enzyme acid sphingomyelinase (ASM) and a subsequent buildup of the substrate sphingomyelin in the liver, spleen, and lungs.40 More than 180 mutations in SMPD1 have been reported.40 Foam cells and sea-blue histiocytes can be seen in the bone marrow. Foam cells are macrophages with cytoplasm packed with lipid-filled lysosomes that appear as small vacuoles (foam) after staining (Figure 26.9 ). Sea blue histiocytes are macrophages with lipofuscin-, glycophospholipid-, and sphingomyelin contained in -cytoplasmic granules, 1 to 3 μ in diameter, that appear blue with Wright stain. Type A (acute neuronopathic form) NP mostly affects Eastern European Jews. Type A presents in infancy and is associated with failure to thrive, lymphadenopathy, hepatosplenomegaly, vision problems, and rapid neurodegenerative decline that results in death, usually by 4 years of age. In type A, there is less than 5% of normal sphingomyelinase activity. In type B (the non-neuronopathic form) there is approximately 10% to 20% normal enzyme activity.52 Type B NP is more common in individuals of Northern African descent and presents in the first decade to adulthood with a variable clinical course. Although there is no neurocognitive impairment, patients experience massive hepatosplenomegaly, heart disease, and pulmonary insufficiency.53 Diagnosis of types A and B NP is based on enzymatic quantitation of sphingomyelinase activity in cell or tissue extracts. Genetic testing screens for three genes responsible for more than 90% of cases in the Ashkenazi Jewish population and one gene in approximately 90% of type B patients from North Africa.40, 53 Treatment primarily consists of enzyme replacement therapy, although it is ineffective at slowing the progression of neurologic disease in type A patients.
Figure 26.9.
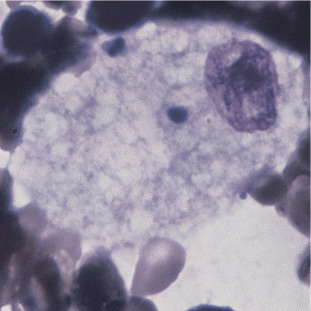
Niemann-Pick Cell. Note eccentric nucleus and bubble-like pattern of storage deposit in the cytoplasm. (Bone marrow, hematoxylin and eosin stain, ×1000.)
(From Rodak, B. F., & Carr, J. H. [2017]. Clinical Hematology Atlas. [5th ed.]. St. Louis: Saunders.)
Type C NP is an autosomal recessive lipidosis in which mutations in NPC1 or NP2 gene (95% and 5% of cases, respectively) causes impaired cellular trafficking and homeostasis of cholesterol.54 The result is buildup of unesterified cholesterol in lysosomes. The clinical presentation in type C NP is heterogeneous with regard to age of onset and type and severity of neurologic and psychiatric symptoms, as well as visceral involvement.40 , 54 Early diagnosis is important because substrate reduction therapy can slow the progression of neurologic damage55; however, because of its heterogeneous presentation, diagnosis is often challenging. A suspicion index tool for type C disease has been found to be highly sensitive and specific for arriving at a presumptive diagnosis.56 Confirmation is obtained through genetic testing or biochemical staining of cultured fibroblasts with filipin to demonstrate unesterified cholesterol in cellular vesicles. Therapy focuses on symptom relief. The prognosis in type C NP is poor, with most patients dying before the age of 25 years.57
Quantitative abnormalities of leukocytes
Neutrophils
Normal relative neutrophil count in adults, bands plus segmented forms, is approximately 50% to 70%. Normal absolute neutrophil count (ANC) is approximately 2 to 7.7 × 109/L. In patients with a neutrophilic left shift (presence of immature cells in the myelocytic cell line), some clinicians prefer to include metamyelocytes and myelocytes in addition to band and segmented forms when calculating the ANC.
Neutrophilia
An absolute increase in neutrophils greater than 7.0 × 109/L in adults or 8.5 × 109/L in children is called neutrophilia. Neutrophilia can occur as a result of catecholamine-induced shift in neutrophils from the marginal pool (cells normally adhering to vessel walls) to the circulating pool. More often, neutrophilia occurs when there is an increase in bone marrow production of the neutrophil series or there is a transfer of neutrophils from the bone marrow storage pool to the circulating pool. Neutrophilia is often accompanied by a left shift. Box 26.1 lists conditions and drugs associated with neutrophilia.
BOX 26.1. Causes of Neutrophilia.
-
•
Emotional stress
-
•
Strenuous exercise
-
•
Trauma/injury
-
•
Pregnancy: labor and delivery
-
•
Eclampsia
-
•
Surgery
-
•
Infections: bacterial, some viral
-
•
Burns
-
•
Myocardial infarction
-
•
Pancreatitis
-
•
Vasculitis
-
•
Colitis
-
•
Autoimmune disease
-
•
Acute hemorrhage
-
•
Chronic blood loss
-
•
Hemolysis
-
•
Smoking
-
•
Chronic blood loss
-
•
Metabolic ketoacidosis
-
•
Uremia
-
•
Leukocyte adhesion deficiency
-
•
Familial cold urticaria
-
•
Hereditary neutrophilia
-
•
Leukemia
-
•
Chronic myeloproliferative neoplasm
-
•
Steroids
-
•
Lithium
-
•
Colony-stimulating factors (granulocyte CSF)
-
•
Cortisol
-
•
Epinephrine
Adapted from Dale, D. C., & Welte, K. (2016). Neutropenia and neutrophilia. In: Kaushansky, K., Lichtman, M. A., Prchal, J. T., et al. (Eds.), Williams Hematology. (9th ed., pp. 991–1004). New York: McGraw-Hill; and Reichard, K. (2010). Non-neoplastic granulocytic and monocytic disorders, excluding neutropenia. In Foucar, K., Reichard, K., & Czuchlewski, D. (Eds.), Bone Marrow Pathology. (3rd ed., Vol 1, pp. 180–205). Chicago: American Society for Clinical Pathology Press.
The term leukemoid reaction refers to a reactive neutrophilic leukocytosis greater than 50 × 109/L with a shift to the left. Leukemoid reactions are usually caused by acute and chronic infections, metabolic disease, or inflammation or occur as part of an inflammatory response to malignancy. When there is a suspected leukemoid reaction, chronic myelogenous (myeloid)leukemia should be ruled out. Distinguishing features between the two are listed in Table 26.5 .
TABLE 26.5.
Distinguishing Features between Leukemoid Reaction and Chronic Myeloid Leukemia
| Leukemoid Reaction | Chronic Myeloid Leukemia | |
|---|---|---|
| Eosinophils & basophils | Normal count and morphology | Often elevated; may show mixed granulation |
| Neutrophil morphology | Toxic granulation and Döhle bodies often | May see pseudo-Pelger-Huët forms |
| Platelet count | Normal | Elevated early; decreased late |
| Platelet morphology | Normal | Possible giant, hypogranular, and/or bizarre forms |
| Hemoglobin | Normal | Often anemic |
| LAP score | Elevated | Decreased |
| Genetics | Normal | Positive for t(9;22); BCR-ABL |
LAP, Leukocyte alkaline phosphatase.
The term leukoerythroblastic reaction refers to the simultaneous presence of immature neutrophils, nucleated red blood cells, and teardrop red blood cells (RBCs). A leukoerythroblastic reaction is sometimes accompanied by neutrophilia. A leukoerythroblastic reaction suggests either (1) a space-occupying lesion in the bone marrow such as metastatic tumor, fibrosis, lymphoma, or leukemia, or a marked increase in one of the normal marrow cells (e.g., erythroid hyperplasia seen in hemolytic anemia); or (2) primary myelofibrosis.
Neutropenia
Neutropenia is defined as a decrease in the ANC to less than 2.0 × 109/L in white adults or 1.3 × 109/L in black adults. The risk of infection increases as the ANC falls to less than 1.0 × 109/L. Severe neutropenia (<0.5 × 109/L) further increases the risk. Agranulocytosis refers to a neutrophil count of less than 0.1 × 109/L. Some causes of neutropenia are (1) increased rate of removal or destruction of peripheral blood neutrophils ; (2) fewer neutrophils released from the bone marrow to the blood because of decreased production or ineffective hematopoiesis, where neutrophils are present in the bone marrow but not released into circulation because they are defective; (3) decreased ratio of circulating versus marginal pool of neutrophils; or (4) a combination of these.
Acquired neutropenia
Acquired forms of neutropenia include are much more common than congenital syndromes. Box 26.2 provides a list of acquired causes of neutropenia.
BOX 26.2. Causes of Acquired Neutropenia.
Drugs
-
•
Anticancer
-
•
Analgesics/antiinflammatories: acetaminophen, aspirin, diclofenac, ibuprofen, indomethacin, linezolid, naproxen, sulfasalazine
-
•
Antibiotics: β-lactams, cephalosporins, chloramphenicol, clindamycin, gentamicin, penicillin, sulfonamides, trimethoprim-sulfamethoxazole, tetracycline, vancomycin
-
•
Antivirals: acyclovir, ganciclovir, abacavir, zidovudine
-
•
Antifungals: terbinafine, Amphotericin B
-
•
Anticonvulsants: carbamazepine, phenytoin, valproate
-
•
Antihistamines: brompheniramine, chlorphenamine, cimetidine, famotidine, ranitidine
-
•
Antimalarials: chloroquine, dapsone, quinine
-
•
Antithyroids: propylthiouracil, methimazole, carbimazole
-
•
Cardiovascular: amiodarone, captopril, clopidogrel, flecainide, furosemide, methyldopa, procainamide, propranolol, quinidine, spironolactone, thiazide diuretics, ticlopidine
-
•
Antianxiety/hypnotics: benzodiazepines, meprobamate
-
•
Psychotropics: amitriptyline, amoxapine, clozapine, doxepin, olanzapine, phenothiazines, risperidone
-
•
Hypoglycemics: chlorpropamide, tolbutamide
-
•
Phenothiazines: chlorpromazine, phenothiazines
Other
-
•
Levamisole (adulterated cocaine)
-
•
Immunologic
-
•
Radiation
-
•
Toxins
-
•
Overwhelming infections
-
•
Splenomegaly
-
•
Hemodialysis
-
•
Aplastic anemia
-
•
Copper deficiency
-
•
Alcoholism
-
•
Tumor metastasis to bone marrow/space-occupying lesion
-
•Hematologic malignancy
-
•Myelodysplastic syndrome
-
•Large granular lymphocyte syndrome
-
•Leukemia
-
•Plasma cell neoplasm
-
•
Adapted from Andres, E., & Maloisel, F. (2008). Idiosyncratic drug-induced agranulocytosis or acute neutropenia. Curr Opin Hematol, 15(1), 15–21; Andrès, E., & Mourot-Cottet, R. (2017). Non-chemotherapy drug-induced neutropenia—an update. Exp Opin Drug Saf, 16(11), 1235–1242; Gibson, C., & Berliner, N. (2014). How we evaluate and treat neutropenia in adults. Blood, 124(8), 1251–1258; and Pick, A. M., & Nystrom, K. K. (2014). Nonchemotherapy drug-induced neutropenia and agranulocytosis: could medications be the culprit? J Pharm Pract, 27(5), 447–452.
Drug-induced neutropenia.
Medications are the most common causes of acquired neutropenia. Neutropenia has been associated with almost all classes of drugs and is a result of myeloid suppression or immunologic response. Many agents used to treat cancer are known to induce neutropenia, including traditional myelosuppressive chemotherapeutics, targeted therapies, and immunotherapies. Nonchemotherapy drug-induced neutropenia and agranulocytosis most often present as idiosyncratic reactions. The annual rate of occurrence of drug-induced agranulocytosis is 2.3 to 15.4 cases per million in the United States.58 The mortality rate is 10% to 16%.58
Immune neutropenia.
In neonatal alloimmune neutropenia (NAN), maternal immunoglobulin G (IgG) crosses the placenta and binds to paternal human neutrophil antigens (HNA) found on fetal leukocytes. Antibody-coated neutrophils are removed from circulation by the reticuloendothelial system, resulting in an ANC of less than 0.5 × 109/L. Of the nine HNA located on Fcγ-receptor IIIb (FcγRIIIb), HNA-1a, -1b, -1c, -1d, and -2 are most often implicated in NAN.59 , 60 The incidence of NAN is not well defined; it varies from .02% to 0.8% of live births.61., 62., 63. Resulting infections are most often not life threatening, and neutropenia resolves within 6 months after clearance of material IgG.
Autoimmune neutropenia (AIN) is associated with IgG autoantibodies against one or more HNA. Primary AIN usually presents around 7 to 9 months of age.64 The disease tends to be self-limiting, and 90% of patients spontaneously recover by 2 years of age.64 , 65 Secondary AIN is more common in adults and is associated with connective tissue disorders, Felty syndrome, hematologic neoplasms, solid tumors, primary immunodeficiencies, bacterial and viral infections, and posttransplantation complications and as idiosyncratic reactions to a number of medications.64 , 66 Immunologic mechanisms involved in drug-induced neutropenia include formation of immune complexes, haptens, drug-induced formation of neutrophil autoantibodies, and T-lymphocyte toxicity.66
Eosinophils
Several factors influence the number of eosinophils in circulation: bone marrow proliferation rate and release into the bloodstream, movement from the blood into extravascular tissues, and cell survival and destruction after eosinophils have moved into the tissues.
Eosinophilia
Eosinophilia is defined as an absolute eosinophil count greater than 0.4 × 109/L. A major function of eosinophils is degranulation, where substances are released that damage an offending organism (i.e., parasites) or target cell. Nonmalignant causes of eosinophilia are generally a result of cytokine stimulation, especially from interleukin-3 and interleukin-5 (IL-3 and IL-5).67 , 68 Eosinophilia is associated with parasitic infections, especially helminths. It is also associated with allergic reactions, including asthma, rhinitis, urticaria, and atopic dermatitis; scabies infestation, scarlet fever, HIV, primary biliary cirrhosis, hepatitis, autoimmune disorders, drug reactions, and some hematologic neoplasms.68., 69., 70.
Eosinopenia is defined as an absolute eosinophil count of less than 0.09 × 109/L and can be difficult to detect because the reference interval is low. Eosinopenia often accompanies other cytopenias in conditions that result in marrow hypoplasia, specifically involving leukocytes. Eosinopenia has been reported in autoimmune disorders, steroid therapy, stress, sepsis, and acute inflammatory states.71 , 72
Basophils
Basophilia is defined as an absolute basophil count greater than 0.15 × 109/L and is associated with chronic myeloid leukemia, allergic rhinitis, hypersensitivity to drugs or food, chronic infections, hypothyroidism, chronic inflammatory conditions, radiation therapy, and bee stings.73 , 74
Monocytes
Monocytosis is defined as an absolute monocyte count greater than 1.0 × 109/L in adults and greater than 3.5 × 109/L in neonates. Monocytosis is associated with numerous conditions because of their role in acute and chronic inflammation and infections, immunologic conditions, hypersensitivity reactions, and tissue repair. Monocytosis is often the first sign of recovery after myelosuppression. It is also seen in congenital cyclic neutropenia, where monocytosis occurs during periods of neutropenia in the 21-day cycle. A list of conditions associated with monocytosis is provided in Box 26.3 .
BOX 26.3. Conditions Associated With Monocytosis.
-
•Infection
-
•Tuberculosis
-
•Brucellosis
-
•Leishmaniasis
-
•Fungal
-
•Subacute bacterial endocarditis
-
•Malaria
-
•
-
•
Recovery from acute infection
-
•
Recovery from myelosuppression
-
•Immunologic/autoimmune
-
•Systemic lupus erythematosus
-
•Rheumatoid arthritis
-
•Autoimmune neutropenia
-
•Inflammatory bowel disease
-
•Myositis
-
•Sarcoidosis
-
•
-
•
Cyclic neutropenia
-
•
Postsplenectomy
-
•Drugs
-
•Colony-stimulating factors
-
•Olanzapine
-
•
-
•Malignancy
-
•Hodgkin disease
-
•Myelodysplastic/myeloproliferative neoplasms
-
•Myelodysplastic syndromes
-
•Acute myeloid leukemia involving the monocyte lineage
-
•
Adapted from Lynch, D. T., & Foucar, K. F. (2016). Ask the hematopathologists: diagnostic approach to monocytosis. Hematologist, 13(4), 4; Robinson, R. L., Burk, M. S., & Raman, S. (2003). Fever, delirium, autonomic instability, and monocytosis associated with olanzapine. J Postgrad Med, 49(1), 96; and Tsolia, M., Drakonaki, S., Messaritaki, A., et al. (2002). Clinical features, complications and treatment outcome of childhood brucellosis in central Greece. J Infect, 44(4), 257–262.
Monocytopenia, defined as an absolute monocyte count of less than 0.2 × 109/L, is very rare in conditions that do not also involve cytopenias of other lineages, such as aplastic anemia or chemotherapy-induced cytopenias. Monocytopenia has been found in patients receiving steroid therapy75 or hemodialysis and in sepsis.76 Viral infections, especially those caused by the Epstein-Barr virus (EBV), can also cause monocytopenia.77 Profound monocytopenia is associated with hairy cell leukemia.78
Lymphocytes
Identification of lymphocytosis varies with the age of the individual. Children between 2 weeks and 8 to 10 years of age have higher absolute lymphocyte counts than adults. In adults, lymphocytes represent 20% to 40% of circulating leukocytes. Lymphocytosis in children is defined as an absolute lymphocyte count greater than 10.0 × 109/L, whereas in adults it is defined as a count greater than 5.0 × 109/L. Newborns have lymphocyte counts similar to those of adults.
Lymphocytosis is usually accompanied by changes in lymphocyte morphology. See Table 26.6 for a list of disorders associated with reactive lymphocytosis.
TABLE 26.6.
Causes of Nonmalignant Lymphocytosis and Lymphocytopenia
| Lymphocytosis | Lymphocytopenia |
|---|---|
| Reactive Morphology | Congenital Immunodeficiencies
Infections
|
Infections
Nonreactive Morphology
|
Adapted from Vasu, S., & Caligiuri, M. A. (2016). Lymphocytosis and lymphocytopenia. In: Kaushansky, K., Lichtman, M. A., Prchal, J. T., et al. (Eds.), Williams Hematology. (9th ed., pp. 1199–1210). New York: McGraw-Hill; and Foucar, K. (2010). Non-neoplastic disorders of lymphoid cells. In Foucar, K., Reichard, K., & Czuchlewski, D. (Eds.), Bone Marrow Pathology. (3rd ed., Vol 2, pp. 448–473). Chicago: American Society for Clinical Pathology Press.
Lymphocytopenia in children is defined as an absolute lymphocyte count less than 2.0 × 109/L, whereas in adults it is defined as a count less than 1.0 × 109/L. Nonmalignant causes of lymphocytopenia can be subdivided into inherited and acquired and are listed in Table 26.6.
Secondary morphological changes
Neutrophils
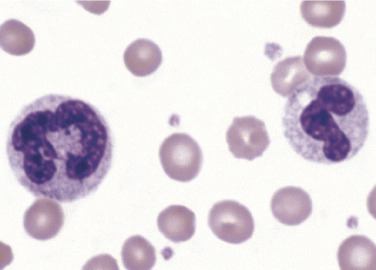
Physiologic response to infection, inflammation, stress, or administration of recombinant colony-stimulating factor (CSF)results in neutrophilia, often with a shift to the left and occasionally with the presence of blasts. The severity of infection, inflammation, or dose of CSF influences ANC and level of immaturity of neutrophils released into circulation. Additionally, neutrophils may exhibit changes in morphology such as toxic granulation, Döhle bodies, and cytoplasmic vacuoles. Toxic granulation appears as dark, blue-black granules in the cytoplasm of neutrophils, usually in segmented and band forms. Toxic granules are peroxidase positive and reflect an increase in acid mucosubstance within primary, azurophilic granules that may enhance bactericidal activity.79 There is a positive correlation between level of C-reactive protein (an acute phase protein) and the number of neutrophils with toxic granulation; therefore toxic granulation is suggestive of inflammation.7 , 80 In addition, toxic granulation can be seen in various infections as well as in patients who have received G-CSF.81 Toxic granulation can mimic granulation found in Alder-Reilly anomaly. One helpful defining characteristic of toxic granulation is that in most cases, not all neutrophils are equally affected (Figure 26.10 ). Table 26.7 highlights reactive neutrophil morphologic changes and associated conditions.
Figure 26.10.
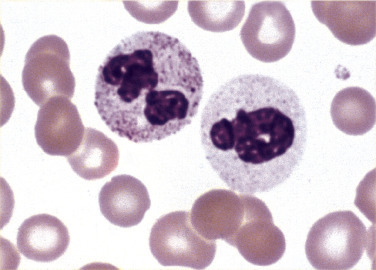
Toxic Granulation.
Note that one neutrophil contains toxic granulation and the other does not. Also note that the toxic granules are clustered in some areas of the cytoplasm. Both of these findings help in distinguishing toxic granulation from poor staining or from the dark granules seen in Alder-Reilly anomaly. (Peripheral blood, Wright-Giemsa stain, ×1000.)
TABLE 26.7.
Reactive Morphologic Changes in Neutrophils
| Reactive Change | Appearance | Associated with |
|---|---|---|
| Toxic granulation | Dark, blue-black cytoplasmic granules | Inflammation, infection, G-CSF |
| Döhle bodies | Intracytoplasmic pale blue round or elongated bodies between 1 and 5 μm in diameter, usually adjacent to cellular membranes. | Infection, G-CSF, pregnancy, burns |
| Cytoplasmic vacuoles | Small to large circular clear areas in cytoplasm, rarely may contain organism | Bacterial infection, autophagocytosis secondary to drug ingestion, acute alcoholism, or excess storage of sample before making blood film |
G-CSF, Granulocyte colony-stimulating factor.
Döhle bodies are cytoplasmic inclusions consisting of remnants of ribosomal ribonucleic acid (RNA) arranged in parallel rows.82 Döhle bodies are typically found in band and segmented neutrophils (Figure 26.11 ) and can appear together with toxic granulation. They are intracytoplasmic, pale blue round or elongated inclusions between 1 and 5 μm in diameter. Döhle bodies are usually located close to cellular membranes. A delay in preparing the blood film after collection may affect Döhle body appearance in that they are more grey than blue or in some cases may not be visible. Döhle bodies are relatively nonspecific. Their presence has been associated with a wide range of conditions, including bacterial infections, sepsis, and pregnancy.82 , 83 Döhle bodies are similar in appearance to the inclusions found in May-Hegglin anomaly. However, in May-Hegglin anomaly, Döhle body-like inclusions can also be seen in eosinophils, basophils, and monocytes.
Figure 26.11.
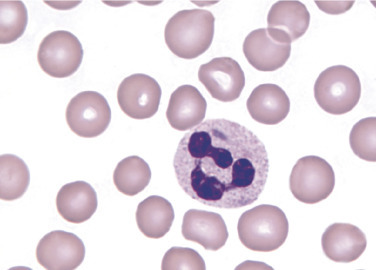
Neutrophil Containing a Döhle Body.
(Peripheral blood, Wright-Giemsa stain, ×1000.)
Cytoplasmic vacuolation of neutrophils is encountered less often than toxic granules and Döhle bodies. Vacuoles generally reflect phagocytosis, either of self (autophagocytosis) or of extracellular material (Figure 26.12 ). Autophagocytic vacuoles tend to be small (approximately 2 μm) and distributed throughout the cytoplasm. Autophagocytosis can be induced by specimen storage in ethylene diamintetraacetic (EDTA) for more than 2 hours, autoantibodies, acute alcoholism, and exposure to high doses of radiation.84., 85., 86. Phagocytic vacuoles induced by either bacteria or fungi are suggestive of sepsis. When phagocytic vacuoles are seen, a careful examination sometimes reveals organisms within the vacuoles. Phagocytic vacuoles tend to be large (up to 6 μm) and often accompanied by toxic granulation.
Figure 26.12.
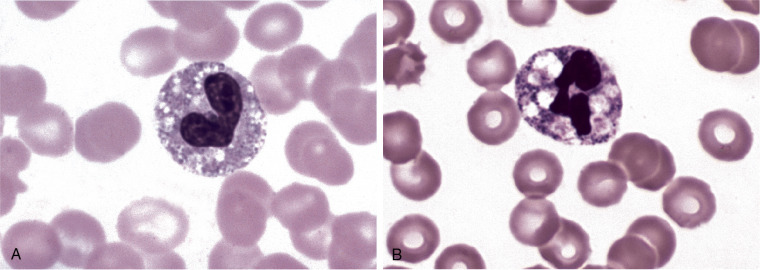
Cytoplasmic vacuoles.
(A), Band neutrophil with autophagocytic vacuoles. Note their small size. (B), Neutrophil with phagocytic vacuoles. Note their larger size. Other evidence of toxicity in this cell is the pyknotic nucleus. (Peripheral blood, Wright-Giemsa stain, ×1000.)
Cases of ehrlichiosis and anaplasmosis have been increasing in the United States over the past decade.85 Ehrlichia and Anaplasma are small, obligate, intracellular bacteria transmitted by ticks to humans and other vertebrate hosts. These organisms grow as a cluster (morulae) in neutrophils (Anaplasma phagocytophilum and rarely in Ehrlichia ewingii) (Figure 26.13 A) and in monocytes (Ehrlichia chaffeensis) (Figure 26.13B). Leukopenia, thrombocytopenia, and elevated liver enzymes are common laboratory findings, and anemia occurs in about half the cases ofehrlichiosis.88 Intracellular aggregates of purple colored particles (morulae) in neutrophils or monocytes may occasionally be detected in the first week of infection on a Wright-Giemsa stained peripheral blood film or a buffy coat preparation. Morulae can be mistaken for Döhle bodies in neutrophils. Polymerase chain reaction testing is often required to confirm diagnosis.87 Early detection is essential because antibiotic treatment with doxycycline is effective and can prevent serious complications.
Figure 26.13.
Intracellular Organisms.
(A),Anaplasma phagocytophilum in a neutrophil. (B),Ehrlichia chaffeensis in a monocytic cell. (Peripheral blood, Wright-Giemsa stain, ×1000.)
(Courtesy J. Stephen Dumler, The Johns Hopkins Medical Institution, Baltimore, MD.)
Pyknotic nuclei in neutrophils generally indicate imminent cell death. In a pyknotic nucleus, water has been lost and the chromatin becomes dense and dark; however, chromatin or filaments can still be seen between nuclear lobes (depending on whether the cell is a band or segmented form).89 , 90
Necrotic nuclei are found in dead neutrophils. They are rounded nuclear fragments with no filaments and no chromatin pattern (Figure 26.14 ). Increased numbers of pyknotic or necrotic cells suggest that an extended amount of time has elapsed between blood collection and blood film preparation.
Figure 26.14.
Pyknotic and Necrotic Cells.
(A), Upper cell is a neutrophil whose nucleus is dehydrated, which makes it very dark and dense. Note that there is still a filament between the segments. This is referred to as a pyknotic cell. The cell is also vacuolated. (B), Neutrophil that has died. Note that the nucleus has disintegrated into numerous rounded spheres of DNA with no filaments. This is referred to as a necrotic or necrobiotic cell. (Peripheral blood, Wright-Giemsa stain, ×1000.)
(B from Carr, J. H., & Rodak, B. F. [2009]. Clinical Hematology Atlas. [3rd ed.]. St. Louis: Saunders.)
Cytoplasmic swelling of neutrophils is a result of osmotic swelling of the cytoplasm or by increased adhesion to the glass slide in stimulated neutrophils. Regardless of the cause, the result is a variation in neutrophil size or neutrophil anisocytosis (Figure 26.15 ).
Figure 26.15.
Neutrophil Anisocytosis.
The neutrophil to the left is larger than the other neutrophil. This is often caused by cytoplasmic swelling. (Peripheral blood, Wright-Giemsa stain, ×1000.)
Eosinophils and basophils
Eosinophils can sometimes appear hypogranular (Figure 26.16 ). Hypogranular eosinophils have been associated with acute lymphoblastic leukemia and hypereosinophilic syndrome.91 , 92 In vitro disruption of the cellular membrane may occur during the process of making the blood film as eosinophils are fragile. To promote accuracy, it is recommended that fractured eosinophils be counted as eosinophils in the manual WBC differential.35
Figure 26.16.
Partially Degranulated Eosinophil.
This cell was found on the blood film for a patient with trichinosis. (Peripheral blood, Wright-Giemsa stain, ×1000.)
Basophil granules are water soluble, and it is not uncommon for them to be partially or completely washed away during staining.93 Sometimes few or no granules remain; only a pinkish tinge in or around the cell is seen. This can hamper proper identification of basophils when performing manual WBC differentials.
Monocytes
Reactive changes in monocytes are an uncommon finding. Reactive monocytes exhibit thin, band-like, or segmenting nuclei (Figure 26.17 ). Reactive changes also include increased cytoplasmic volume, irregular cytoplasmic borders, and changes in granule size and coloration. Reactive monocytes have been associated with infection, recovery from myelosuppression, and administration of granulocyte-macrophage CSF.
Figure 26.17.
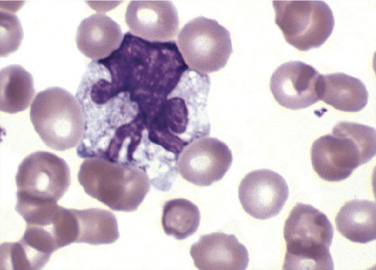
Reactive Monocyte.
Note the contorted nucleus. Other evidence of toxicity is the several vacuoles in the cytoplasm. (Peripheral blood, Wright-Giemsa stain, ×1000.)
Lymphocytes
Lymphocytes that exhibit reactive morphology have been classified using various terms, including reactive, variant, atypical, transformed, effector, plasmacytoid, Turk cells, Downey, and immunoblasts. The International Council for Standardization in Hematology (ICSH) recommends using reactive when the lymphocyte exhibits morphology consistent with a benign etiology and abnormal when lymphocyte morphology suggests a malignant or clonal etiology.36
Reactive changes in lymphocyte morphology occur as lymphocytes are stimulated when interacting with antigens in peripheral lymphoid organs (Figures. 26.18 and 26.19 ). B and T lymphocyte activation results in the transformation of small, resting lymphocytes into proliferating larger cells. These reactive lymphocytes spill into peripheral circulation. Reactive lymphocytes often present as a heterogeneous population of various shapes and sizes. There is variation in the nuclear to cytoplasmic ratio, nuclear shape, and chromatin pattern, which is clumped, but some cells may contain less condensed chromatin. Nucleoli may be visible. An increase in basophilic cytoplasm that varies in intensity within and between cells is a common finding. The cytoplasm may be indented by surrounding RBCs; however, other cells, including blasts, may also show similar indentation. A plasmacytoid lymphocyte is a type of reactive lymphocyte that has some morphologic features of plasma cells (Figure 26.20 ). If such cells are numerous, lymphoma or Waldenstrom’s macroglobulinemia may be suspected. True plasma cells are not commonly encountered on blood film review.
Figure 26.18.
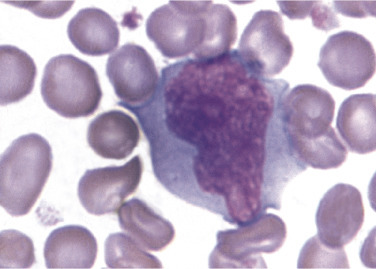
Reactive (variant) Lymphocyte.
(Peripheral blood, Wright-Giemsa stain, ×1000.)
Figure 26.19.
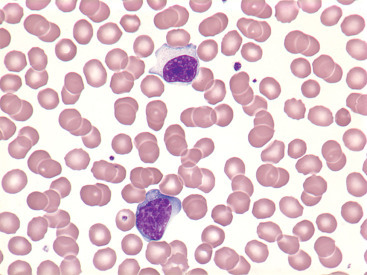
Reactive (Variant) Lymphocytes.
Cells from a patient with infectious mononucleosis. (Peripheral blood, Wright-Giemsa stain, ×1000.)
Figure 26.20.
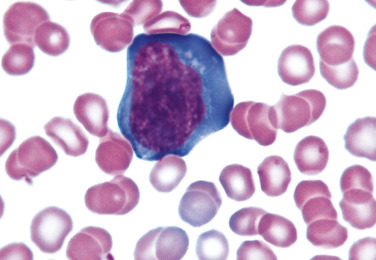
Reactive (Variant) lymphocyte (Plasmacytoid).
(Peripheral blood, Wright-Giemsa stain, ×1000.)
Infectious mononucleosis
Most humans are subclinically infected with Epstein-Barr virus. EBV has been associated with several benign and malignant diseases but has been proven to be the causative agent in only a few, including infectious mononucleosis (IM). When primary infection with EBV occurs in childhood, it often goes unnoticed. Early childhood infection with EBV is not usually identified as the symptoms are generally mild and overlap with other viral syndromes. The incubation period of IM is approximately 3 to 7 weeks, and during this time EBV preferentially infects B lymphocytes through attachment of viral envelope glycoprotein 350/220 to CD21 (C3d complement receptors).94 The oropharynx epithelial cells are also infected, but the mechanism is unclear because these cells do not express CD21. The cellular response in IM is important in the control of the infection and is characterized by proliferation and activation of NK lymphocytes, CD4+ T cells, and CD8+ memory cytotoxic T cells (EBV-CTLs) in response to B cell infection. Most of the circulating reactive lymphocytes represent activated T cells.
Common clinical manifestations of IM include sore throat, dysphagia, fever, chills, cervical lymphadenopathy, fatigue, and headache. An absolute lymphocytosis is present with up to 50% or more reactive forms. Complications are generally mild and include hepatosplenomegaly (and elevated transaminases), hemolytic anemia, and moderate thrombocytopenia. In rare cases patients develop aplastic anemia, disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, Guillain-Barré syndrome, or other neurologic complication.95 The incidence of clinically apparent IM in the United States is 500 cases per 100,000 annually. The highest frequency of clinically overt IM is in young adults aged 15 to 24 years, although IM has been reported in patients 3 months to 70 years of age.96
Heterophile antibodies stimulated by EBV that cross-react with antigens found on sheep and horse RBCs. Rapid screening procedures detect heterophile antibodies. However, not everyone with IM will produce heterophile antibodies, especially children. Definitive testing (if needed) for EBV infection includes a panel of antigen and antibody tests for viral capsid antigen (VCA), Epstein-Barr nuclear antigen (EBNA), and IgG/IgM antibodies against VCA and EBNA. Cytomegalovirus can cause a mononucleosis syndrome with similar clinical features. Clinical and laboratory findings associated with IM are summarized in Box 26.4 .
BOX 26.4. Infectious Mononucleosis: Clinical and Laboratory Findings.
| Clinical Manifestations | Laboratory Test Results |
|---|---|
Common
Less Common
|
|
EBV, Epstein-Barr virus; WBC, white blood cell
Summary
-
•
The primary immunodeficiency disorders are a group of inherited diseases of the innate or adaptive immune system that often result in severe clinical manifestations.
-
•
Both γc deficiency and adenosine deaminase (ADA) deficiency are examples of severe combined immune deficiency (SCID), characterized by defects in cellular and humoral immunity, with a wide range of severe clinical symptoms and death within the first few years of life.
-
•
Wiskott-Aldrich syndrome is a combined immunodeficiency disease with numerous hematologic and clinical findings and increased risk for leukemic transformation.
-
•
DiGeorge syndrome is a disease characterized by a severe decrease in T cells, physical abnormalities, neurologic/psychiatric issues, and hematologic manifestations.
-
•
Bruton tyrosine kinase (BTK) deficiency (X-linked agammaglobulinemia) results in a profound decrease in B cells, hypogammaglobulinemia, and severe, life-threatening infections.
-
•
Chédiak-Higashi syndrome is an extremely rare condition affecting various cells in the body where granules fuse within lysosomes, disrupting normal function, leading to a wide range of serious clinical symptoms and early death.
-
•
The congenital neutropenias manifest early in life as recurrent life-threatening bacterial and fungal infections with increased risk for leukemic transformation.
-
•
Leukocyte adhesion disorders arise as a result of impaired ability of neutrophils and monocytes to move to sites of infection. Clinical findings include recurring severe infections, organomegaly, and neurologic defects.
-
•
Shwachman-Diamond syndrome is characterized by exocrine pancreatic insufficiency, digestive issues, infections, physical anomalies, neurologic defects, bone marrow failure, and increased risk of leukemia.
-
•
Chronic granulomatous disease results from a failure in the neutrophil respiratory burst after ingestion of organisms. Patients suffer from severe recurrent infections; however, advances in the use of antimicrobial agents have been effective at improving the clinical condition.
-
•
WHIM (warts, hypogammaglobulinemia, infections, and myelokathexis syndrome) is a disorder of white blood cell (WBC) movement from the bone marrow to blood resulting in leukopenia, hypogammaglobulinemia, bacterial infections, human papillomavirus infection, and warts.
-
•
Pelger-Huët anomaly is a genetic disorder resulting in hypolobulated mature leukocytes. These cells can be confused with immature neutrophils. An acquired form of hyposegmentation called pseudo-Pelger-Huët is associated with hematologic neoplasms and other disorders.
-
•
Alder-Reilly anomaly is a manifestation of mucopolysaccharidosis characterized by metachromatic granules in leukocytes, which can be confused with toxic granulation.
-
•
May-Hegglin anomaly is characterized by thrombocytopenia, giant platelets, and Döhle body-like inclusions in leukocytes. Most cases are asymptomatic.
-
•
Lysosomal storage disorders are congenital deficiencies of lysosomal enzymes and impaired digestion of macromolecules, which accumulate and impair cellular functions.
-
•
The mucopolysaccharidoses are associated with a specific defect in an enzyme necessary for the degradation of glycoaminoglycan (GAG). Partially digested GAGs build up and disrupt cellular functions leading to serious physical and neurologic problems.
-
•
Gaucher disease is characterized by a deficiency in β-glucocerebrosidase and accumulation of cell sphingolipids leading to a range of clinical manifestations and increased risk for malignant transformation
-
•
Niemann-Pick disease is caused by a deficiency in sphingomyelinase and buildup of fats in various organs in the body, resulting in neurologic defects, organomegaly, failure to thrive, and early death.
-
•
Reactive changes in neutrophils include a left shift, Döhle bodies, toxic granulation, and vacuoles.
-
•
Reactive morphology in monocytes includes segmenting nuclei, changes in granule color and size, and irregular cytoplasmic borders.
-
•
Reactive changes in lymphocytes include increased size, increased basophilic cytoplasm, and morphologic heterogeneity.
Now that you have completed this chapter, go back and read again the case study at the beginning and respond to the questions presented.
Review questions
Answers can be found in the Appendix.
-
1.Which of the following inherited leukocyte disorders is caused by a mutation in the lamin B receptor?
-
a.Pelger-Huët anomaly
-
b.Chédiak-Higashi disease
-
c.Alder-Reilly anomaly
-
d.May-Hegglin anomaly
-
a.
-
2.Which of the following inherited leukocyte disorders involves mutations in nonmuscle myosin heavy-chain IIA?
-
a.Pelger-Huët anomaly
-
b.Chédiak-Higashi disease
-
c.Alder-Reilly anomaly
-
d.May-Hegglin anomaly
-
a.
-
3.Which of the following inherited leukocyte disorders might be seen in Hurler syndrome?
-
a.Pelger-Huët anomaly
-
b.Chédiak-Higashi disease
-
c.Alder-Reilly anomaly
-
d.May-Hegglin anomaly
-
a.
-
4.Which of the following lysosomal storage diseases is characterized by macrophages with striated cytoplasm and storage of glucocerebroside?
-
a.Sanfilippo syndrome
-
b.Gaucher disease
-
c.Fabry disease
-
d.Niemann-Pick disease
-
a.
-
5.The neutrophils in chronic granulomatous disease are incapable of producing:
-
a.Hydrogen peroxide
-
b.Hypochlorite
-
c.Superoxide
-
d.All of the above
-
a.
-
6.Individuals with X-linked SCID have a mutation that affects their ability to synthesize:
-
a.Deaminase
-
b.Oxidase
-
c.IL-2 receptor
-
d.IL-8 receptor
-
a.
-
7.An absolute lymphocytosis with reactive lymphocytes suggests which of the following conditions?
-
a.DiGeorge syndrome
-
b.Bacterial infection
-
c.Parasitic infection
-
d.Viral infection
-
a.
-
8.What leukocyte cytoplasmic inclusion is composed of ribosomal RNA?
-
a.Primary granules
-
b.Toxic granules
-
c.Döhle bodies
-
d.Howell-Jolly bodies
-
a.
-
9.The expected complete blood cell count (CBC) results for women in active labor would include:
-
a.High total white blood cell (WBC) count with increased lymphocytes
-
b.High total WBC count with a slight shift to the left in neutrophils
-
c.Normal WBC count with increased eosinophils
-
d.Low WBC count with increased monocytes
-
a.
-
10.Which of the following is true of an absolute increase in lymphocytes with reactive morphology?
-
a.The population of lymphocytes appears morphologically homogeneous.
-
b.They are usually effector B cells.
-
c.The reactive lymphocytes have increased cytoplasm with variable basophilia.
-
d.They are most commonly seen in bacterial infections.
-
a.
References
- 1.Picard C., Bobby Gaspar H., Al-Herz W. International Union of Immunological Societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. 2018;38:96–128. doi: 10.1007/s10875-017-0464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Puck J.M., Deschenes S.M., Porter J.C. The interleukin-2 receptor g chain maps to Xq13.1 and is mutated in X-linked severe combined immunodeficiency, SCIDX1. Hum Mol Genet. 1993;2(8):1099–1104. doi: 10.1093/hmg/2.8.1099. [DOI] [PubMed] [Google Scholar]
- 3.Allenspach E., Rawlings D.J., & Scharenberg A.M. X-linked severe combined immunodeficiency. In: Adam M.P., Ardinger H.H., Pagon R.A., editors. GeneReviews® [Internet] University of Washington, Seattle; Seattle (WA): 2016. https://www.ncbi.nlm.nih.gov/books/NBK1410/pp. 1993R–2016. Accessed July 17, 2018. [Google Scholar]
- 4.Bradford K.L., Moretti F.A., Carbonaro-Sarracino D.A. Adenosine deaminase (ADA)-deficient severe combined immune deficiency (SCID): molecular pathogenesis and clinical manifestations. J Clin Immunol. 2017;37:626–637. doi: 10.1007/s10875-017-0433-3. [DOI] [PubMed] [Google Scholar]
- 5.Candotti F.J. Clinical manifestations and pathophysiological mechanisms of the Wiskott-Aldrich syndrome. Clin Immunol. 2018;38:13–27. doi: 10.1007/s10875-017-0453-z. [DOI] [PubMed] [Google Scholar]
- 6.Braun C.J., Witzel M., Paruzynski A. Gene therapy for Wiskott-Aldrich syndrome—long-term reconstitution and clinical benefits, but increased risk for leukemogenesis. Rare Diseases. 2014;2(1):e947749. doi: 10.4161/21675511.2014.947749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morsheimer M., Brown Whitehorn T.F., Heimall J. The immune deficiency of chromosome 22q11.2 deletion syndrome. Am J Med Genet Part A. 2017;173:2366–2372. doi: 10.1002/ajmg.a.38319. [DOI] [PubMed] [Google Scholar]
- 8.Lambert M.P., Arulselvan A., Schott A. The 22q11.2 deletion syndrome: cancer predisposition, platelet abnormalities and cytopenias. Am J Med Genet. 2017:1–7. doi: 10.1002/ajmg.a.38474. [DOI] [PubMed] [Google Scholar]
- 9.Shillitoe B., & Gennery A. X-linked agammaglobulinemia: outcomes in the modern era. Clin Immunol. 2017;183:54–62. doi: 10.1016/j.clim.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan J., De Domenico I., & Ward D.M. Chediak-Higashi syndrome. Curr Opin Hematol. 2008;15(1):22–29. doi: 10.1097/MOH.0b013e3282f2bcce. [DOI] [PubMed] [Google Scholar]
- 11.Introne W., Boissy R.E., & Gahl W.A. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab. 1999;68(2):283–303. doi: 10.1006/mgme.1999.2927. [DOI] [PubMed] [Google Scholar]
- 12.Daneshbod Y., & Medeiros L.J. Pseudo Chediak-Higashi anomaly in acute monoblastic leukemia. Blood. 2016;128(21):2583. doi: 10.1182/blood-2016-08-732339. [DOI] [PubMed] [Google Scholar]
- 13.La Gioia A., Bombara M., Fiorini F. Pseudo-Chédiak-Higashi granules and other unusual cytoplasmic inclusions in refractory anaemia with excess blasts-2. Br J Haematol. 2017;176:156. doi: 10.1111/bjh.14405. [DOI] [PubMed] [Google Scholar]
- 14.Tsai I.M., Tsai C.C., & Ladd D.J. Pseudo-Chediak-higashi anomaly in chronic myelogenous leukemia with myelofibrosis. Am J Clin Pathol. 1977;67(6):608–609. doi: 10.1093/ajcp/67.6.608. [DOI] [PubMed] [Google Scholar]
- 15.Donadieu J., Beaupain B., Fenneteau O. Congenital neutropenia in the era of genomics: classification, diagnosis, and natural history. Br J Haematol. 2017;179:557–574. doi: 10.1111/bjh.14887. [DOI] [PubMed] [Google Scholar]
- 16.Hauck, F, & Klein, C. Pathogenic mechanisms and clinical implications of congenital neutropenia syndromes. Curr Opin Allergy Clin Immunol. 2013;13(6):596–606. doi: 10.1097/ACI.0000000000000014. [DOI] [PubMed] [Google Scholar]
- 17.Rosenberg P.S., Alter B.P., Bolyard A.A. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. 2006;107(12):4628–4635. doi: 10.1182/blood-2005-11-4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawrence M.B., Kansas G.S., Kunkel E.J. Threshold levels of fluid shear promote leukocyte adhesion through selectins (CD62L,P,E) J Cell Biol. 1997;136(3):717–727. doi: 10.1083/jcb.136.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris E.S., McIntyre T.M., Prescott S.M. The leukocyte integrins. J Biol Chem. 2000;275(31):23409–23412. doi: 10.1074/jbc.R000004200. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt S., Moser M., & Sperandio M. The molecular basis of leukocyte recruitment and its deficiencies. Mol Immunol. 2013;55(1):49–58. doi: 10.1016/j.molimm.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 21.Sperandio M., Gleissner C.A., & Ley K. Glycosylation in immune cell trafficking. Immunol Rev. 2009;230(1):97–113. doi: 10.1111/j.1600-065X.2009.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gazit Y., Mory A., Etzioni A. Leukocyte adhesion deficiency type II: long-term follow-up and review of the literature. J Clin Immunol. 2010;30(2):308–313. doi: 10.1007/s10875-009-9354-0. [DOI] [PubMed] [Google Scholar]
- 23.Moser M., Nieswandt B., Ussar S. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14(3):325–330. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
- 24.Myers K. Shwachman-Diamond syndrome. In: Adam M.P., Ardinger H.H., Pagon R.A., editors. GeneReviews® [Internet] University of Washington, Seattle; Seattle (WA): 2014. https://www.ncbi.nlm.nih.gov/books/NBK1756/pp. 1993–2017. Accessed July 17, 2018. [Google Scholar]
- 25.Nelson, A. & Myers, K. Diagnosis, treatment and molecular pathology of Shwachman-Diamond syndrome. Hematol Oncol Clin North Am. 2018;32(4):687–700. doi: 10.1016/j.hoc.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 26.Chiriaco M., Salfa I., Di Matteo G. Chronic granulomatous disease: clinical, molecular, and therapeutic aspects. Pediatr Allergy Immunol. 2016;27(3):242–253. doi: 10.1111/pai.12527. [DOI] [PubMed] [Google Scholar]
- 27.Arnold D.E., & Heimall J.R. A review of chronic granulomatous disease. Adv Ther. 2017;34(12):2543–2557. doi: 10.1007/s12325-017-0636-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dinauer M.C. Primary immune deficiencies with defects in neutrophil function. Hematology Am Soc Hematol Educ Program. 2016;2016(1):43–50. doi: 10.1182/asheducation-2016.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dotta L., Tassone L., & Badolato R. Clinical and genetic features of warts, hypogammaglobulinemia, infections and myelokathexis (WHIM) syndrome. Curr Mol Med. 2011;11(4):317–325. doi: 10.2174/156652411795677963. [DOI] [PubMed] [Google Scholar]
- 30.Badolato R., Donadieu J., & the WHIM Research Group How I treat warts, hypogammaglobulinemia, infections, and myelokathexis syndrome. Blood. 2017;130(23):2491–2498. doi: 10.1182/blood-2017-02-708552. [DOI] [PubMed] [Google Scholar]
- 31.Colella R., & Hollensead S.C. Understanding and recognizing the Pelger-Huët anomaly. Am J Clin Pathol. 2012;137(3):358–366. doi: 10.1309/AJCP3G8MDUXYSCID. [DOI] [PubMed] [Google Scholar]
- 32.Skendzel L.P., & Hoffman G.C. The Pelger anomaly of leukocytes: forty-one cases in seven families. Am J Clin Pathol. 1962;37:294–301. doi: 10.1093/ajcp/37.3.294. [DOI] [PubMed] [Google Scholar]
- 33.Johnson C.A., Bass D.A., Trillo A.A. Functional and metabolic studies of polymorphonuclear leukocytes in the congenital Pelger-Huët anomaly. Blood. 1980;55(3):466–469. [PubMed] [Google Scholar]
- 34.Cunningham J.M., Patnaik M.M., Hammerschmidt D.E. Historical perspective and clinical implications of the Pelger-Huët cell. Am J Hematol. 2009;84(2):116–119. doi: 10.1002/ajh.21320. [DOI] [PubMed] [Google Scholar]
- 35.Wang E. Pseudo-Pelger Huet anomoly induced by medications. Am J Clin Pathol. 2011;135:291–303. doi: 10.1309/AJCPVFY95MAOBKRS. [DOI] [PubMed] [Google Scholar]
- 36.Palmer L., Briggs C., McFadden S. ICSH recommendations for the standardization of nomenclature and grading of peripheral blood cell morphological features. Int J Lab Hematol. 2015;37:287–303. doi: 10.1111/ijlh.12327. [DOI] [PubMed] [Google Scholar]
- 37.Shetty V.T., Mundle S.D., & Raza A. Pseudo Pelger-Huët anomaly in myelodysplastic syndrome: hyposegmented or apoptotic neutrophil? Blood. 2001;98(4):1273–1275. doi: 10.1182/blood.v98.4.1273. [DOI] [PubMed] [Google Scholar]
- 38.Brunning R.D. Morphologic alterations in nucleated blood and marrow cells in genetic disorders. Hum Pathol. 1970;1:99–124. [Google Scholar]
- 39.Kunishima S., Kojima T., Tanaka T. Mapping of a gene for May-Hegglin anomaly to chromosome 22q. Hum Genet. 1999;105(5):379–383. doi: 10.1007/s004390051119. [DOI] [PubMed] [Google Scholar]
- 40.Ferreira C.R., & Gahl W.A. Lysosomal storage diseases. Transl Sci Rare Dis. 2017;2:1–71. doi: 10.3233/TRD-160005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giugliani R., Federhen A., Vairo F. Emerging drugs for the treatment of mucopolysaccharidoses. Expert Opin Emerg Drugs. 2016;21(1):9–26. doi: 10.1517/14728214.2016.1123690. [DOI] [PubMed] [Google Scholar]
- 42.Beutler E., Nguyen N.J., Henneberger M.W. Gaucher disease: gene frequencies in the Ashkenazi Jewish population. Am J Hum Genet. 1993;52(1):85–88. [PMC free article] [PubMed] [Google Scholar]
- 43.Stenson P.D., Mort M., Ball E.V. The human gene mutation database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133(1):1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosenbloom B.E., Weinreb N.J., Zimran A. Gaucher disease and cancer incidence: a study from the Gaucher Registry. Blood, 2005;105(12):4569–4572. doi: 10.1182/blood-2004-12-4672. [DOI] [PubMed] [Google Scholar]
- 45.Somaraju U.R., & Tadepalli K. Hematopoietic stem cell transplantation for Gaucher disease. Cochrane Database Syst Rev. 2017;10:CD006974. doi: 10.1002/14651858.CD006974.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma P., Khurana N., & Singh T. Pseudo-Gaucher cells in Hb E disease and thalassemia intermedia. Hematology. 2007;12(5):457–459. doi: 10.1080/10245330701393675. [DOI] [PubMed] [Google Scholar]
- 47.Helbig G., Janikowska A., & Kyrcz-Krzemien S. Aggregates of pseudo-Gaucher cells after treatment of chronic myeloid leukemia in blastic phase. Int J Hematol. 2015;101(1):3–4. doi: 10.1007/s12185-014-1693-9. [DOI] [PubMed] [Google Scholar]
- 48.Carrington P.A., Stevens R.F., & Lendon M. Pseudo-Gaucher cells. J Clin Pathol. 1992;45(4):360. doi: 10.1136/jcp.45.4.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cozzolino L., Picardi M., Pagliuca S. B-cell non-Hodgkin lymphoma and pseudo-Gaucher cells in a lymph node fine needle aspiration. Cytopathology. 2016;27(2):134–136. doi: 10.1111/cyt.12254. [DOI] [PubMed] [Google Scholar]
- 50.Shenjere P., B, & Banerjee S.B. Pseudo-Gaucher cells in multiple myeloma. Int J Surg Pathol. 2008;16(2):176–179. doi: 10.1177/1066896907311120. [DOI] [PubMed] [Google Scholar]
- 51.Dubois-Galopin F., & Berger M.G. Waldenström macroglobulinemia with pseudo-Gaucher cells. Blood. 2010;116(18):3388. doi: 10.1182/blood-2009-12-258574. [DOI] [PubMed] [Google Scholar]
- 52.Schuchman E.H. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. Int J Clin Pharmacol Ther. 2009;47(Suppl. 1):S48–S57. doi: 10.5414/cpp47048. [DOI] [PubMed] [Google Scholar]
- 53.Schuchman E.H., & Desnick R.J. Types A and B Niemann-Pick disease. Mol Genet Metab. 2017;120:27–33. doi: 10.1016/j.ymgme.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McKay Bounford K., & Gissen P. Genetic and laboratory diagnostic approach in Niemann Pick disease type C. J Neurol. 2014;261(Suppl. 2):569–575. doi: 10.1007/s00415-014-7386-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mengel E., Klünemann H.H., Lourenço C.M. Niemann-Pick disease type C symptomatology: an expert-based clinical description. Orphanet J Rare Dis. 2013;8:166. doi: 10.1186/1750-1172-8-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wijburg F.A., Sedel F., Pineda M. Development of a suspicion index to aid diagnosis of Niemann-Pick disease type C. Neurology. 2012;78(20):1560–1567. doi: 10.1212/WNL.0b013e3182563b82. [DOI] [PubMed] [Google Scholar]
- 57.Evans W.R.H., Hendriksz C.J. Niemann-Pick type C disease – the tip of the iceberg? A review of neuropsychiatric presentation, diagnosis and treatment. BJPsych Bull. 2017;41(2):109–114. doi: 10.1192/pb.bp.116.054072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andrès E., Mourot-Cottet R. Non-chemotherapy drug-induced neutropenia - an update. Expert Opin Drug Saf. 2017;16(11):1235–1242. doi: 10.1080/14740338.2017.1376645. [DOI] [PubMed] [Google Scholar]
- 59.Reil A., Sachs U.J., Siahanidou T. HNA-1d: a new human neutrophil antigen located on Fcg receptor IIIb associated with neonatal immune neutropenia. Transfusion. 2013;53:2145–2151. doi: 10.1111/trf.12086. [DOI] [PubMed] [Google Scholar]
- 60.van den Tooren-de Groot R., Ottink M., Huiskes E. Management and outcome of 35 cases with foetal/neonatal alloimmune neutropenia. Acta Paediatr. 2014;103:e467–e474. doi: 10.1111/apa.12741. [DOI] [PubMed] [Google Scholar]
- 61.BUX J., Jung K.D., Kauth T. Serological and clinical aspects of granulocyte antibodies leading to alloimmune neonatal neutropenia. Transfus Med. 1992;2:143–149. doi: 10.1111/j.1365-3148.1992.tb00148.x. [DOI] [PubMed] [Google Scholar]
- 62.Z˙upa´nska B., Uhrynowska M., Guz K. The risk of antibody formation against HNA1a and HNA1b granulocyte antigens during pregnancy and its relation to neonatal neutropenia. Transfus Med. 2001;11:377–382. doi: 10.1046/j.1365-3148.2001.00325.x. [DOI] [PubMed] [Google Scholar]
- 63.Williams B.A., & Fung Y. Alloimmune neonatal neutropenia: can we afford the consequences of a missed diagnosis? J Paediatr Child Health. 2006;42:59–61. doi: 10.1111/j.1440-1754.2006.00774.x. [DOI] [PubMed] [Google Scholar]
- 64.Afzal W., Owlia M.B., Hasni. Autoimmune neutropenia updates: etiology, pathology, and treatment. South Med J. 2017;110(4):300–307. doi: 10.14423/SMJ.0000000000000637. [DOI] [PubMed] [Google Scholar]
- 65.Dale D.C. How I manage children with neutropenia. Br J Haematol. 2017;178:351–363. doi: 10.1111/bjh.14677. [DOI] [PubMed] [Google Scholar]
- 66.Autrel-Moignet A., & Lamy T. Autoimmune neutropenia. Presse Med. 2014;43(4):e105–e118. doi: 10.1016/j.lpm.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 67.Korenaga M., Hitoshi Y., Yamaguchi N. The role of interleukin-5 in protective immunity to Strongyloides venezuelensis infection in mice. Immunology. 1991;72(4):502–507. [PMC free article] [PubMed] [Google Scholar]
- 68.Simon D., & Simon H.U. Eosinophilic disorders. J Allergy Clin Immunol. 2007;119(6):1291–1300. doi: 10.1016/j.jaci.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 69.Simon D., Braathen L.R., & Simon H.U. Eosinophils and atopic dermatitis. Allergy. 2004;59(6):561–570. doi: 10.1111/j.1398-9995.2004.00476.x. [DOI] [PubMed] [Google Scholar]
- 70.Filley W.V., Holley K.E., Kephart G.M. Identification by immunofluorescence of eosinophil granule major basic protein in lung tissues of patients with bronchial asthma. Lancet. 1982;2(8288):11–16. doi: 10.1016/s0140-6736(82)91152-7. [DOI] [PubMed] [Google Scholar]
- 71.Abidi K., Khoudri I., Belayachi J. Eosinopenia is a reliable marker of sepsis on admission to medical intensive care units. Crit Care. 2008;12(2):R59. doi: 10.1186/cc6883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Abidi K., Belayachi J., Derras Y. Eosinopenia, an early marker of increased mortality in critically ill medical patients. Intensive Care Med. 2011;37(7):1136–1142. doi: 10.1007/s00134-011-2170-z. [DOI] [PubMed] [Google Scholar]
- 73.Jimenez C., Gasalla R. Incidence and clinical significance of peripheral and bone marrow basophilia. J Med. 1987;18(5–6):293–303. [PubMed] [Google Scholar]
- 74.May M.E., & Waddell C.C. Basophils in peripheral blood and bone marrow. A retrospective review. Am J Med. 1984;76(3):509–511. doi: 10.1016/0002-9343(84)90671-5. [DOI] [PubMed] [Google Scholar]
- 75.Chakraborty A., Blum R.A., Cutler D.L. Pharmacoimmunodynamic interactions of interleukin-10 and prednisone in healthy volunteers. Clin Pharmacol Ther. 1999;65(3):304–318. doi: 10.1016/S0009-9236(99)70110-4. [DOI] [PubMed] [Google Scholar]
- 76.Nockher W.A., Wiemer J., & Scherberich J.E. Haemodialysis monocytopenia: differential sequestration kinetics of CD141CD161 and CD1411 blood monocyte subsets. Clin Exp Immunol. 2001;123(1):49–55. doi: 10.1046/j.1365-2249.2001.01436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Savard M., & Gosselin J. Epstein-Barr virus immunossuppression of innate immunity mediated by phagocytes. Virus Res. 2006;119(2):134–145. doi: 10.1016/j.virusres.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 78.Bethel K. Pathology of hairy-cell leukaemia. Best Pract Res Clin Haematol. 2003;16(1):15–31. doi: 10.1016/s1521-6926(02)00087-7. [DOI] [PubMed] [Google Scholar]
- 79.van de Vyver A., Delport E.F., Esterhuizen M. The correlation between C-reactive protein and toxic granulation of neutrophils in the peripheral blood. S Afr Med J. 2010;100(7):442–444. doi: 10.7196/samj.3912. [DOI] [PubMed] [Google Scholar]
- 80.Kabutomori O., Iwatani Y., & Kanakura Y. Toxic granulation neutrophils and C-reactive protein. Arch Intern Med, 2000;160(21):3326–3327. doi: 10.1001/archinte.160.21.3326-a. [DOI] [PubMed] [Google Scholar]
- 81.Kabutomori O., Kanakura Y., & Watani Y. Induction of toxic granulation in neutrophils by granulocyte colony-stimulating factor. Eur J Haematol. 2002;69(3):187–188. doi: 10.1034/j.1600-0609.2002.02767.x. [DOI] [PubMed] [Google Scholar]
- 82.Prokocimer M., & Potasman I. The added value of peripheral blood cell morphology in the diagnosis and management of infectious diseases—part 1: basic concepts. Postgrad Med J. 2008;84:579–585. doi: 10.1136/pgmj.2008.069609. [DOI] [PubMed] [Google Scholar]
- 83.Abernathy M.R. Döhle bodies associated with uncomplicated pregnancy. Blood. 1966;27(3):380–385. [PubMed] [Google Scholar]
- 84.Boxer L.A., & Stossel T.P. Effects of anti-human neutrophil antibodies in vitro. quantitative studies. J Clin Invest. 1974;53(6):1534–1545. doi: 10.1172/JCI107704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Davidson R.J., & McPhie J.L. Cytoplasmic vacuolation of peripheral blood cells in acute alcoholism. J Clin Pathol. 1980;33(12):1193–1196. doi: 10.1136/jcp.33.12.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Holley T.R., Van Epps D.E., Harvey R.L. Effect of high doses of radiation on human neutrophil chemotaxis, phagocytosis and morphology. Am J Pathol. 1974;75(1):61–72. [PMC free article] [PubMed] [Google Scholar]
- 87.Dahlgren F.S., Mandel E.J., Krebs J.W. Increasing incidence of Ehrlichia chaffeensis and Anaplasma phagocytophilum in the United States, 2000-2007. Am J Trop Med Hyg. 2011;85(1):124–131. doi: 10.4269/ajtmh.2011.10-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ismail N., Bloch K.C., & McBride J.W. Human ehrlichiosis and anaplasmosis. Clin Lab Med. 2010;30(1):261–292. doi: 10.1016/j.cll.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Taneja R., Parodo J., Jia S.H. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit Care Med. 2004;32(7):1460–1469. doi: 10.1097/01.ccm.0000129975.26905.77. [DOI] [PubMed] [Google Scholar]
- 90.Son Y.O., Jang Y.S., Heo J.S. Apoptosis-inducing factor plays a critical role in caspase-independent, pyknotic cell death in hydrogen peroxide-exposed cells. Apoptosis. 2009;14(6):796–808. doi: 10.1007/s10495-009-0353-7. [DOI] [PubMed] [Google Scholar]
- 91.Catovsky D., Bernasconi C., Verdonck P.J. The association of eosinophilia with lymphoblastic leukaemia or lymphoma: a study of seven patients. Br J Haematol. 1980;45(4):1365–2141. doi: 10.1111/j.1365-2141.1980.tb07174.x. [DOI] [PubMed] [Google Scholar]
- 92.Kim H., Lee Y., Lee D. A case of idiopathic hypereosinophilic syndrome with hypersegmented and hypogranular eosinophils. Clin Lab Haematol. 1999;21(6):427–430. [PubMed] [Google Scholar]
- 93.Scordino T. Basophil with washed out granules. 2016. https://imagebank.hematology.org/image/60505/basophil-with-washed-out-granules?type = atlas Accessed January 23, 2019. ASH Image Bank.
- 94.Fingeroth J.D., Weis J.J., Tedder T.F. Epstein-Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc Natl Acad Sci U S A. 1984;81(14):4510–4514. doi: 10.1073/pnas.81.14.4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Luzuriaga K., & Sullivan J.L. Infectious mononucleosis. N Engl J Med, 2010;362(21):1993–2000. doi: 10.1056/NEJMcp1001116. [DOI] [PubMed] [Google Scholar]
- 96.Crawford D.H., Macsween K.F., Higgins C.D. A cohort study among university students: identification of risk factors for Epstein-Barr virus seroconversion and infectious mononucleosis. Clin Infect Dis. 2006;43(3):276–282. doi: 10.1086/505400. [DOI] [PubMed] [Google Scholar]