

MULTIPLE SCLEROSIS AS AN AUTOIMMUNE DISEASE
multiple sclerosis is the most important inflammatory disease of the central nervous system (CNS). In contrast to other encephalitides, the inflammatory changes underlying multiple sclerosis do not seem to be direct responses to microbial infection. Instead, they are caused and sustained by autoimmune responses. The central hypothesis is that T lymphocytes with receptors for central nervous system myelin components enter the brain, respond locally to their target antigen, and (indirectly) attack local cells. These autoaggressive T cells trigger an inflammatory cascade that is responsible for all the neurological deficits seen in affected individuals.
This concept takes issue with several time-honoured dogmas of neuroimmunology. How can immune cells mount an attack against one of the body's own tissues? What are the origin and nature of these misguided immune cells? It has been maintained traditionally that the central nervous system is exempt from physiological immune mechanisms. How then can we explain pathological autoimmune responses occurring within the central nervous system? Both sets of questions must be answered convincingly in order to understand the pathogenesis of multiple sclerosis.
First, we explain the general organization of the immune system, then discuss mechanisms that result in autoimmune attack against self, and see which of these mechanisms contribute to the pathogenesis of multiple sclerosis. We consider the central nervous system tissues as an environment for immune reactivity. We examine the global conditions that allow or prevent immune responses in central nervous system tissues, and then specifically describe the changed central nervous system arrangements in the lesions of multiple sclerosis.
The immune system is the body's main defence force. It must protect against the myriad of environmental microbial organisms and also against potentially cancerous cells arising within tissues of high cellular turnover. The immune system works as a formidable killing machine. It is designed to spot, encircle and neutralize any suspicious structure appearing in the body and threatening its well-being. In this context, suspicious means any material deviating from healthy self tissues. If immune cells lose their ability to distinguish suspicious foreign and intact self components, they may attack and damage normal tissues – and thus cause autoimmune disease. These are not rare and exotic afflictions, but count among the most important problems in clinical medicine. ‘Autoimmune diseases together constitute the third-greatest clinical burden, after cardiovascular diseases and cancer’ (Nossal 2001). Autoimmunity may affect diverse organs, causing diseases ranging from rheumatoid arthritis, insulin-dependent diabetes mellitus, ulcerative colitis and systemic lupus erythematosus to multiple sclerosis. It should be stressed, however, that the autoimmune concept in multiple sclerosis lacks formal proof. However, it rests on several diverse lines of evidence, which, taken together strongly suggest that immunopathological events, which we presume to be of an autoimmune nature, are responsible for the occurrence and development of the disease.
One of the strongest arguments in favour of an immune pathogenesis for multiple sclerosis comes from the morphology of its lesions. As detailed in Chapter 12, the lesion in multiple sclerosis is characterized by perivascular round cell infiltration, and these accumulating lymphocytes spill into the surrounding parenchyma. Active lesions in multiple sclerosis are almost indistinguishable from areas of inflammation seen in diseases of proven autoimmune pathogenesis, especially models of experimentally induced autoimmune encephalomyelitis (EAE; see below). The second line of evidence supporting the autoimmune hypothesis is genetic and rests on the association of disease susceptibility with polymorphic genes that are potentially involved in autoimmune responses (see Chapter 3). The best-documented example is the major histocompatibility complex (MHC), whose class II products are required for the presentation of autoantigen to T lymphocytes and are, in addition, crucially involved in the development of the immune repertoire. Finally, the autoimmune pathogenesis of multiple sclerosis is endorsed by the relative success of therapies that suppress or modulate the immune response (see Chapter 18). For example, β-interferon (IFN-β) has a favourable effect in reducing relapse rates and decreasing lesion load as assessed by magnetic resonance imaging (MRI). The same is true for Copaxone (copolymer-1), which diverts myelin-reactive T cells from the pathogenic Th1 (T helper 1) profile to the regulatory Th2 (T helper 2) pattern. Finally, some antibodies that blindfold or eliminate activated T cells were found to be beneficial in clinical trials (von Andrian and Engelhardt 2003).
IMMUNE RESPONSES: INNATE AND ADAPTIVE
Evolution has provided two protective immune systems, the innate and the adaptive. Both share the ability to identify a potentially harmful external (or internal) structure, and then mount a response designed to neutralize this threat. At the same time, either type of response must be selective, meaning that the destructive potential must be directed exclusively to the suspected target, while the body's own tissues are completely spared. The two immune systems fulfil their tasks admirably well, although they use radically different principles.
The innate immune response is phylogenetically old. It acts in worms and insects as well as mammals. It is fully preformed in the healthy organism. Its elements are present in the tissues irrespective of microbial threat. In stark contrast, the more modern adaptive immune system formed much later in evolution. It is only found in vertebrates. Adaptive immune responses only offer protection following a first encounter with a microbial target. This contact leads to a maximal response concentrated on the particular microbe, with the generation of killer cells or molecules that neutralize and eliminate the target with maximal efficiency.
Both responses have their advantages and drawbacks. The preformed, innate immune response can act more or less instantaneously against an enemy, but its weapons are quite blunt. Due to good but not perfect self–nonself discrimination, the reaction may create some collateral damage to healthy tissues. In contrast, adaptive immune responses need time to develop fully but, once established, are extremely specific for the pathogen and maximally efficient. To mount an almost ideal defence, both immune systems join forces, forming one coherent two-tiered system of protection that combines and correlates their independent mechanisms. It follows that their regulatory signalling mechanisms are tightly interconnected through bridging cells and soluble molecules.
Innate immunity
Each living organism, from amoeba to human, lives in a sea of microbes that threaten to invade and decompose the organism. This happens after death. The healthy living body, however, is protected by efficient mechanisms that hold back most microorganisms, and quickly inactivate those that have intruded into the organism. Protection comes from robust outer membranes that act as almost impermeable antimicrobial barriers, plus mechanisms of innate immunity that form the first line of defence against those microbes that manage to breach the barriers (see Table 11.1 ).
Table 11.1.
Comparison of innate immune signalling proteins in Drosophila and mammals
| Drosophila | Mammals | |
|---|---|---|
| Pattern recognition receptors | ||
| Non-signalling | GNBP (3) | CD14, MD-2 |
| LPS | ? | TLR4 |
| PG, LP, zymosan | ? | TLR2 |
| Flagellin | ? | TLR5 |
| Bacterial DNA | ? | TLR9 |
| Orphan receptors | 18 wheeler, dTLR3–9 | Other TLRs |
| Cytokines and receptors | ||
| TNF-α/TNFR | TNF-like/TNFR-likea | TNF-α/TNFR1&2 |
| Spätzle/Toll | Spätzle/Toll | ? |
| IL-1β/IL-1R-related | None | IL-1β/IL-1R (8) |
| Intracellular signalling components | ||
| Adapters | Tube, dMyD88 | MyD88, Tollip |
| Pelle-like kinases | Pelle | IRAK (4) |
| TRAF | DTRAF1–3 | TRAF1–6 |
| MAP3K | dTAK1, dMEKK | TAK1, TAB1 & 2, NIK, MEKK1–3 |
| Atypical PKC | DaPKC | PKC-ζ and λ, p62 |
| RIP-like proteins | IMD | RIP |
| NF-κB components | ||
| IBs | Cactus | IκBα, β, γ, ɛ, |
| IκB kinases: | ||
| IKKα/β | DMIKKβ (IRD5) | IKKα, β |
| IKKγ | DmIKKγ (KENNY) | IKKγ/NEMO/IKKAP |
| IKKɛ-like | DmIKKɛ/dIK2 | IKKɛ/i, TBK/NAK/T2K |
| NF-κB precursors | Relish | p105, p100 |
| and products | Relish N & C | p50, p52 |
| NK-κB subunits | Dorsal, Dif | RelA/p65, RelB, c-Rel |
Both known and putative innate immune signalling proteins are categorized into pattern recognition receptors, cytokines and their receptors, intracellular signalling components, and NF-κB-related kinases and transcription factors. The number of orthologs per genome is indicated in parentheses.
The Drosophila TNFR-like predicted gene contains a TNFR-like cysteine-rich extracellular domain but does not include a TNFR-related intracellular domain.
Reproduced with permission fromSilverman and Maniatis (2001).
© 2006
In insects, a particular gene, Toll, encodes receptor structures that specifically discriminate microbial structures. Toll receptor-mediated recognition stimulates particular migratory cells in the fly haemolymph, ‘haemocytes’, to swallow small microbes, or encapsulate larger microbes and fungi. Soon after this discovery, Toll-like genes and receptors were found throughout phylogeny, including within the human genome. Indeed, Toll-like receptors are now recognized as the key antimicrobial structures. We now know of an ever-expanding family of Toll-like receptors that recognize different components of microbial organisms (Hoffmann et al 1999).
Toll-like receptors bind bacterial membrane components (endotoxin), polysaccharides of bacterial capsule structures, viral RNA and bacterial DNA. They are present in many cell types – but mainly professional antigen-presenting cells (dendritic cells), macrophages, polymorphonuclear leucocytes and all components of the phagocytic system (Akira et al 2001). As in insects, activation of Toll-like receptors on vertebrate cells leads to activation, which, in the case of phagocytes, may stimulate phagocytosis and secretion of soluble antimicrobial effector proteins – most prominently defensins and proteases. Both act by perforating the microbial membrane and thus destabilizing the organism.
Members of the Toll-like receptor family are involved in pattern recognition. Globally, they distinguish molecules found on bacteria and viruses but rarely, if ever, on vertebrate cells. Thus, they discriminate microbial structures from self but do not sharply identify the exact nature of a microbial product. They share this recognition strategy with other innate receptors, for example mannoside receptor or C-reactive protein, and thus differ from immune receptors (T- and B-cell receptors), which recognize small circumscripted molecular determinants or epitopes.
If the innate immune receptors act quite bluntly, this is also true for innate effector molecules. Defensins, for example, kill bacteria by inserting holes into their membranes, but at the same time quite often also affect the surrounding host tissue. In addition, the molecules provide chemotactic signals that attract dendritic cells to sites of fresh infection (Yang et al 2002).
In general, the relative disadvantage of limited self–nonself discrimination is outweighed by the speed of innate immune reactivity. Responses occur rapidly, almost immediately after bacterial infection, and reach any location of the affected organism. As will be shown later, innate immune responsiveness is almost ideally complemented by adaptive immunity. This is superior in terms of specificity. It is important to note that the rules that operate in immune responses against foreign antigens also pertain to pathological autoimmune responses. Responses of the innate immune system can profoundly influence the course and intensity of autoimmune responses delivered by the adaptive immune system (Bachmann and Kopf 2001).
Adaptive immunity: immune repertoire and immune surveillance
The adaptive immune system is much more powerful and elaborate than its innate counterpart. Its enormous efficiency depends on two qualities – precision of the actual response and immunological memory. Immune responses are highly specific. If, for example, an infectious agent enters the body, the ensuing response focuses exclusively on this microbe, ignoring other potential antigens for the moment. In this way, the immune response can be maximally efficient whilst exerting minimum effort. Morever, typical immune responses leave a specific imprint on the immune system. Thus, they imprint immunological memory. A microbe entering the body for a second time will trigger a much more vigorous immune reaction than on first encounter. The immune system remembers the old microbial acquaintance and responds with a more intense and quicker set of reactions.
The structural basis for immune specificity and memory resides in the clonal diversity of preformed lymphocytes. The immune system is composed of lymphocyte families – clones – each of which is characterized by diverse surface receptors for antigen. Ideally, each clonal receptor can bind and recognize just one antigenic structure. Thus, a foreign antigen intruding into the tissue binds and selects those lymphocyte receptors with the best fit. These cells are activated to multiply and differentiate to effector cells responsible for neutralizing the antigen.
How can a specific immune cell spot its antigen in the body? How can antigen-specific immune cells fulfil their protective mission? The answer is by immune surveillance. At any time there are millions of immune cells, especially of the antigen experienced memory type, roaming through the body's organs, scanning tissues for intruded microbes or for newly arisen tumour cells. However, immune surveillance relies not just on random migration. Patrolling immune cells have antennae for chemical signals that attract them to suspicious areas in a tissue. The attracting signals are chemokines, small molecular proteins secreted by dendritic cells when activated by a microbial structure. Dendritic cells play a pivotal role in linking innate and adaptive immune responses. Activation by microbes happens by mechanisms of innate immunity, while attraction of memory T cells leads to adaptive immunity.
However, dendritic cells are much more than just sensors. They take up bacteria and digest their antigenic structures to make them recognizable by T lymphocytes. Dendritic cells are professional antigen-presenting cells. They offer freshly digested antigen to attracted memory T cells. In addition, having picked up antigen, dendritic cells leave the peripheral tissue and migrate through lymphatic vessels to the nearest immune organ – often a lymph node – there they import and present the antigen to freshly generated naive T cells.
T LYMPHOCYTES
The adaptive immune system relies on two main protagonists, T cells and B cells. Both are practically indistinguishable by morphological criteria, and both lineages also develop from common progenitors residing in the bone marrow. The two differ radically, however, in their function. The main role of B lymphocytes is the production of humoral antibodies, but they play an additional role in presenting antigen to T lymphocytes. T lymphocytes are the main regulatory cells in the immune system, helping B lymphocytes to mount an optimal antibody response and downregulate ongoing immune responses; also, T cells are effector cells in the responses of delayed-type hypersensitivity.
T-cell receptors
There are two classes of T-cell antigen receptors. The majority of T cells use the αβ receptor (see also Chapter 12). These include most CD4+ and CD8+ T cells, which recognize peptide antigens in the molecular context of MHC class I and class II, respectively. A minority of T cells, whose recognition properties are much less well known, use instead a pair of γδ T-cell receptors. Both classes of T cell are consistently found in the infiltrates of demyelinating diseases, but whilst lymphocytes using αβ receptors have been characterized as definitely pathogenic effectors, the role of γδ T cells remains enigmatic (see below).
Although T- and B-cell (immunoglobulin) receptors share some elementary principles, they differ radically in other respects. Both receptor types are composed of two identical heavy (H) and two identical light (L) chains. Like immunoglobulin genes, T-cell receptors are encoded by C, V and J genes, arranged in a large cluster located on human chromosome 7 (Figure 11.1 ). The antigen-recognizing surfaces of the αβ and γδ T cells are formed by individual combinations of these V, J and D genes, rearranged and spliced by the recombinase machinery. Further diversification of the T-cell receptor is achieved by inclusion of N region nucleotides. In contrast to B-cellular immunoglobulins, T-cell receptors remain unchanged throughout an immune response. They do not sharpen their affinity by somatic hypermutation upon contact with antigen, and there is no intraclonal class switching of T-cell receptor.
Figure 11.1.
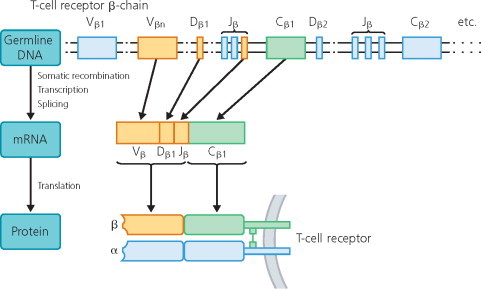
Rearrangement of germline genes in formation of the T-cell receptor. The germline DNA encoding the T-cell receptor β chain genes contains 65 (46 functional) variable (Vβ), 13 joining (Jβ; not all are shown), two diversity (Dβ) and two constant (Cβ) segments (Rowen et al 1996). The V domain of the T-cell receptor β chain is encoded by three gene segments, Vβ, Dβ and Jβ. Rearrangement of these gene segments generates a functional exon that is transcribed and spliced to join VDJ to C. The resulting mRNA is translated to yield the T-cell receptor β chain protein. The mRNA coding the T-cell receptor α chain is constructed by similar mechanisms (not shown).
Adapted from Hohlfeld (1997).
© 2006
T-cell receptors bind their antigen complex by a surface formed from the complementarity-forming region 3 and a bound antigenic peptide (Figure 11.2 ). Antigen binding causes a structural change in the T-cell receptor molecule, which triggers a cascade of signals that ultimately arrives in the nucleus and results in T-cell activation – expressed as transcription of activation-related genes. The T-cell receptor chains are anchored in the cell membrane, but they do not have cytoplasmic tails sufficient to import the activation signal into the cell. This is done by a cluster of molecules of the CD3 class, sticking around the T-cell receptor chains and extending long protein domains into the cytoplasm. The CD3 cytoplasmic domains contain a sequence motif termed ITAM (immunoreceptor tyrosine-based activation motif), originally discovered in B cells but also present in T lymphocytes and mast cells (Reth 1989). In response to antigen binding, protein phosphorylation of ITAM and conformational changes of the CD3 cytoplasmic domains lead to the binding and activation of a relay of signal proteins that transport the information into the nucleus (M.M. Davis 2002). Why is T-cell receptor signal transmission so complicated, and why does it involve so many individual components? The answer is that intermolecular cooperation facilitates fine tuning of the signal strength. As will be seen later, it is the strength of the antigenic signal plus additional stimuli that determines the functional character of a newly triggered immune response.
Figure 11.2.
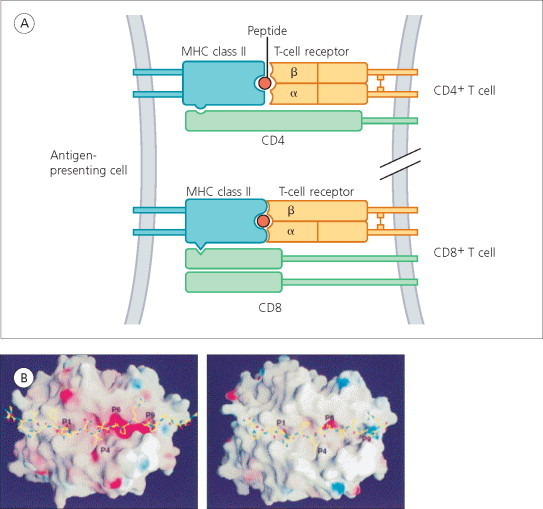
(A) Antigen recognition by CD4+ T cells (top) and CD8+ cells (bottom). The T-cell receptor of CD4+ cells recognizes an antigen peptide bound to an MHC class II molecule (such as HLA-DR, -DP or -DQ) on the surface of an antigen-presenting cell. The receptor of CD8+ T cells recognizes an antigen peptide bound to MHC class I molecule (such as HLA-A, -B or -C). CD4 and CD8 act as coreceptors. (B) Presentation of major encephalitogenic peptides by HLA-DR antigens. Crystallography of HLA-DR2a–myelin basic protein and HLA-DR2b–myelin basic protein complexes. Left is a top view of the HLA-DR2a–myelin basic protein 86–105 complex. Right is a top view of the HLA-DR2b–myelin basic protein 85–99 complex.
(A) from Y. Li et al (2000). © 2000, with permission from Elsevier. Adapted from Hohlfeld (1997).
Antigen recognition by T lymphocytes is very different from their B-cell relatives. A mature B lymphocyte can recognize a correctly folded antigen by direct ligation of its membrane immunoglobulin receptor, be it surface bound or in solution. Antigen recognition by the T cell is altogether different. It requires antigen first to be taken up, processed and presented by an antigen-presenting cell. These cells internalize the antigenic protein and split it into small peptide fragments, whose length may vary between eight and several dozen amino acids. Within the cytoplasm of the antigen-presenting cell, some peptides are bound to molecules encoded by the MHC. Principally, all classic MHC molecules comprise two protein chains, which form a deep and polymorphous cleft. Its molecular shape is determined by the amino acid sequence of each MHC molecule, which is highly polymorphic (see also Chapter 3). During processing, the emerging antigenic peptides compete for binding to the MHC molecule cleft, and the peptide with the best fit wins. The newly formed complex of peptide and histocompatibility antigen complex is then transported to the surface of the antigen-presenting cell for the attention of specific T cells. To be activated, T cells must recognize determinants on the lips of the MHC molecule along with peptide side chains that point out of the groove.
There are, however, two sets of MHC molecules with differing structural details – both in terms of peptide binding characteristics and the sets of T cells to which they present antigen. MHC class I proteins are formed by one polymorphic H chain and one monomorphic L chain (β2-microglobulin). Class I MHC molecules preferentially bind relatively short peptides within the endoplasmic reticulum. Many are derived from endogenous protein components. Class I embedded peptides are presented to CD8+ T cells (Figure 11.2A, bottom), the major T-cell subset containing most of the cytotoxic killer lymphocytes. In contrast, MHC class II proteins can bind longer peptide fragments and do so in a special vesicular compartment distinct from classic lysosomes. Most of the class II bound peptides come from exogenous proteins, which have been taken up by the antigen-presenting cell and degraded intracellularly. Class II restricted peptides are presented to CD4+ T lymphocytes (Figure 11.2A, top, and 11.2B), the cell class comprising helper T cells, along with effector cells involved in cellular hypersensitivity responses (Germain 1994).
In addition to activation by peptide binding, T-cell receptors can be stimulated by ‘superantigens’. These microbial products bind to the outside of MHC molecules and the T-cell receptor, rather than binding to its groove and antigen-binding complementarity-determining region 3 (CDR3) surface. Thus, individual superantigens do not distinguish single T-cell clones, which are defined by their peptide–MHC specificity, but bind to V gene-specific structures outside the CDR3. Different superantigens bind distinct panels of Vβ proteins, thus implying that a given superantigen activates all T-cell clones using this segment (Herman et al 1991). Activation of T cells by microbial superantigen is of considerable clinical interest because this mechanism may contribute to the pathogenic activation of potentially self-reactive T-cell clones, which persist in the healthy immune repertoire in a dormant state.
Antigen presentation
The immune response is remarkable for its adaptability and specificity. It is flexible. Depending on the nature of a particular immunogen, the response may be mild or violent, use either chiefly cellular or humoral mechanisms, localize to a particular part of the body, and be very short-lived or long-lasting. These properties result from complex but robust intercellular regulation that starts and ends an immune response. The process is initiated by contact of immune cells with the immunogen. In the case of T cells, antigen recognition is not a simple binding phenomenon. Rather, it involves a complex interaction between the recognizing T cells and another player that presents the putative antigen in a recognizable fashion. T cells have receptors that intrinsically are unable to recognize intact foreign or self protein antigen. In order to be recognizable by T cells, proteins must be taken up by antigen-presenting cells, and cleaved into small peptide fragments (usually composed of 10–30 amino acids). The peptides are then bound into specifically shaped folds of MHC class I or class II proteins. The T-cell receptor recognizes the surface formed by the MHC protein and bound antigenic peptide.
The community of mature T cells using the αβ T-cell receptor falls into two major classes, each distinguished by a particular accessory recognition molecule – the CD4 and CD8 coreceptors. CD4 molecules bind to MHC class II proteins. Consequently, CD4+ T cells recognize antigen in the context of MHC class II. These proteins are specialized to pick up exogenous proteins (often bacterial structures), which are processed in particular intracellular compartments. CD4+ T cells participate in responses against particulate microbes (bacteria or fungi) and parasites, either controlling B-cell mediated antibody production or cellular responses of delayed-type hypersensitivity. In contrast, CD8 has an affinity for MHC class I proteins, which preferentially bind intracellular antigens (typically viral products). CD8+ T cells differentiate to cytotoxic (‘killer’) cells that are pivotal in protection from virus infections and tumour cells.
The immune synapse
Binding of MHC–peptide complexes to specific T-cell receptors is necessary, but by no means sufficient to trigger an immune response. In addition to the T-cell receptor, an impressive number of accessory molecules must participate in productive antigen recognition. Located near the T-cell receptor, they have to bind specific counterparts on the antigen-presenting membrane (Figure 11.3 ).
Figure 11.3.

Cell adhesion molecule interactions at the interface between a CD4+ T cell (bottom) and an antigen-presenting cell (APC; top). Antigen specificity is conferred by the clonotypic T-cell receptor, which recognizes an antigen peptide (arrowhead) bound to an MHC class II molecule. Signal transduction is mediated by the invariant proteins of the CD3 complex associated with the T-cell receptor. Various additional costimulatory signals are transmitted by interactions between costimulatory molecules and their ligands (B7-1 and B7-2 with CD28, CD2, TNF-α gene families and their receptors, CD40 and CD40-L, and CTLA-4). Binding between cell adhesion molecules (LFA-1–ICAM-1/2, VLA-4–VCAM) stabilizes the contact between T cell and antigen-presenting cell. ICAM = intercellular adhesion molecule; VCAM = vascular cell adhesion molecule; TCR = T-cell receptor; CTLA = cytotoxic T-lymphocyte antigen; LFA = leucocyte functional antigen; VLA = very late antigen; TMC = trimolecular complex; SLAM = signalling lymphocytic activation molecule. Kindly provided by Professor Reinhard Hohlfeld.
The elaborate structure of their intercellular contact area reflects the complexity of interactions between the recognizing T lymphocyte and antigen-presenting cell. This has rightly been dubbed the ‘immune synapse’. Like its neurobiological counterpart, the immune synapse is a highly elaborate structure organized in an amazingly ordered fashion (Figure 11.4 ). Moreover, both types of synapse are ephemeral. They form when and where needed and resolve after completion of their function. An immune synapse is initiated by the contact between T-cell receptor and MHC–peptide. This process leads to the local concentration of most available T-cell receptors to form a patch on the T-cell membrane, with a symmetrical accumulation of MHC–peptide molecules on the antigen-presenting cell side. During development of the synapse, additional molecules form concentric rings around the contact zone formed by T-cell receptor, MHC molecule and digested peptide. These include costimulatory molecules (which send additional signals for activation) as well as cell adhesion molecules (which stabilize the synaptic adhesion). Other molecules are sorted out of the synapse, such as CD45, a phosphatase, and CD43, a highly sialylated glycoprotein (A.S. Shaw 2001). Synapses are highly ordered not only on the membrane but also beneath the surface. In the case of synapses formed by cytotoxic T cells and their target cells, cytotoxic granules polarize, accumulate close to the contacts made by T-cell receptor, MHC and peptide, and are discharged towards the target, with subsequent cytoskeletal rearrangement of both cells (Dustin and Colman 2002).
Figure 11.4.

Comparison of the immune and neural synapses. Effector immune synapse linking CD8+ T cell with its target. (A) The neural synapse. (B) Inductive immune synapse formed between the CD8+ Tc cell as presynaptic and the antigen-presenting cell as postsynaptic. (C) Inductive immune synapse formed during antigen presentation between the antigen-presenting cell as presynaptic and the CD4+ Th cell as postsynaptic. (D) Diagram of the immune synapse formation. Green represents interactions between the T-cell receptor and peptide–MHC antigen. Red represents LFA-1–ICAM interaction (see Figure 11.3). Blue represents exocytic (secretory) vesicles. Yellow represents CD43 at the boundaries. Orange represents microtubules. Pale turquoise represents material in the synaptic cleft.
Adapted from Dustin and Colman (2002). © 2002, with permission from Science.
© 2006
A neurobiologist might object to the term ‘immune synapse’, suspecting a trick metaphor merely reflecting shallow similarity between neuronal and immunological contact interfaces, and representing the attempts of one discipline to stand (we might say ‘trample’) on the gigantic shoulders of another. As it now emerges, however, the analogies between both types of contact go deeper. Immune and neuronal synapses share remarkable features in common. They both concentrate specific cell adhesion molecules, primary adaptors, and use identical structures such as agrin, a proteoglycan involved in the aggregation and organization of synaptic receptors (A.A. Khan et al 2001). Both synapses exchange specific transmitter molecules through a synaptic cleft of around 30 nm (Donnadieu et al 2001), and there are also functional similarities. Finally, as in neuronal synapses, immune synapses are notable for their structural plasticity (Dustin and Colman 2002).
Immune synapses are central to the overall functioning of the immune system. Synaptic contacts are essential in the formation of the T-cell repertoire in the thymus, in homeostatic survival of T cells, in helper interactions between CD4+ T cells and B lymphocytes, and in the rejection of infected target cells by CD8+ T cells.
Antigen-presenting cells: professional and facultative
Although the first obvious prerequisite, MHC expression, is met by many cell types, not all are equally competent in presenting antigen to T cells. This is because, whilst MHC class I expression is constitutive in most tissues, this is not the case for expression of MHC class II. However, class II expression is readily induced on many cells by suitable proinflammatory stimuli – γ-interferon (IFN-γ) and tumour necrosis factor α (TNF-α) – even in the ‘immune privileged’ central nervous system (Wekerle 1994).
Given the relatively modest requirements of T cells, MHC expression may be sufficient for antigen presentation to experienced memory or effector T cells. Naive CD4+ T cells are more demanding. They require – apart from MHC–peptide presentation – an elaborate set of costimulatory elements, some found only on dendritic cells, the professional antigen-presenting cell. Another quite special type of antigen-presenting cell is the B lymphocyte. B cells are much more selfish. They present antigen in order to activate helper T cells, and then receive reciprocal instructions directing the further fate of their own antibody response. Prominent examples are helper T-cell-derived cytokines – IFN-γ, interleukin 4 (IL-4) and IL-10 – which control immunoglobulin isotype switching (see below).
Dendritic cells
Dendritic cells count among the most intriguing elements of the immune system. They are truly multifunctional and pivotal in their performances. They play central roles in shaping the immune repertoire, initiating the response, deciding its character, and bringing everything to a close. Dendritic cells maintain immunological memory and connect innate with adaptive immune reactivity.
The discovery of dendritic cells strictly dates from 1973 (Steinman and Cohn 1973). However, it should be noted that they have made other appearances, depending on location, wearing different disguises, such as interdigitating cells in the thymus (Kaiserling et al 1974) and veiled cells in the blood circulation (Hoefsmit et al 1982). Dendritic cells are the professional antigen-presenting cell par excellence. They carry out their manifold functions by presenting foreign or self antigen to specific T cells under very different conditions. As professional antigen-presenting cells, dendritic cells have the entire set of MHC antigens and costimulatory molecules required productively to engage any T lymphocyte – naive or memory, CD4 or CD8. Owing to their lineage diversity and functional plasticity, antigen presentation by dendritic cells may result in activation of an immune response, with formation of immune memory, or conversely the establishment of immune nonresponsiveness, or tolerance. Dendritic cells may also decide whether responding CD4+ T cells take the Th1 or the Th2 pathway of differentiation.
Dendritic cells are positioned strategically in practically all healthy tissues with the notable exception of the central nervous system parenchyma. Thus, they are the pioneer immune cells that make contact with a newly arrived pathogen. The antimicrobial response of a peripheral dendritic cell is multifaceted. First, microbial structures are sensed by the dendritic cell's innate pattern receptors (Toll-like or mannoside receptor). The dendritic cell responds to the stimulus by releasing chemotactic and proinflammatory soluble factors. These attract recirculating elements of the immune system, most prominently antigen-experienced memory T cells. At the same time, dendritic cells undergo further differentiation. They engulf and process the foreign structure, rendering it recognizable by T cells. Attraction of memory T cells and recognition of antigen on the dendritic cell are crucial aspects of immune surveillance.
The function of a dendritic cell is not exhausted by peripheral antigen presentation to memory cells. Antigen contact signals the dendritic cell to leave the tissue and to reach a local lymph node via its lymphatics. En route the dendritic cell responds to signals produced by ‘homeostatic’ chemokines. Once arrived at the lymphoid destination, it gets embedded in T-cell-rich compartments and starts to present the freshly acquired antigen mainly to naive T cells. It is important to note that only dendritic cells are able to activate naive T cells, whereas memory T cells can also respond to nonprofessional antigen-presenting cells. Thus, dendritic cells are sentinels distributed throughout the body. They sense and present foreign structures, but also take up and display dying cells or their debris from surrounding tissue. While, in most cases, such self presentations communicate tolerogenic signals to self-reactive T cells, there may be pathological conditions under which they activate autoreactive T cells and start the autoimmune disease process.
Dendritic cells are the progeny of bone marrow-derived progenitors. For lineage-specific differentiation, these precursors require stimulation by cytokines, including granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-4 and, for maturation, TNF-α. There are at least two sublineages of dendritic cells. Besides the classic variety, plasmacytoid cells were identified as an alternative lineage. These were already known to pathologists, and described in inflamed lymph nodes as ‘T cells with plasmacytoid features’ (Vollenweider and Lennert 1983). Plasmacytoid cells were discovered for immunology as a cell type with dendritic morphology, exhibiting some, but not all, T-cell markers (Grouard et al 1997). The cells gained particular attention as a major source of type I IFNs (M. Cella et al 1999; Siegal et al 1999).
It appears that classic dendritic cells and plasmacytoid cells have complementary functions. They differentiate from distinct progenitor cells – dendritic cells from the monocytic lineage and plasmacytoid cells from lymphoid cells. Each has distinct Toll-like receptor profiles for microbial determinants, and they possess somewhat different cytokine repertoires (Shortman and Liu 2002). Possibly, either cell is able to determine the Th1/Th2 lineage decision during the initial phase of a T-cell response (Rissoan et al 1999).
In short, dendritic cells are pivotal elements in the immune system. As professional antigen-presenting cells they contribute to the establishment of central self-tolerance in the thymic medulla. And they recognize foreign (microbial) antigens in the peripheral tissues, by linking innate to adaptive immune responses.
Antigen-presenting B cells
B cells express surface MHC class I and II antigens, along with a number of costimulatory determinants, and thus qualify for antigen presentation to T cells. B cells are special antigen-presenting cells. First, with their antigen-specific membrane immunoglobulin receptors, B cells are able to bind and focus specific protein antigens present even at low concentration. The immunoglobulin-bound antigen is engulfed by the lymphocyte and processed to become presentable in the MHC class I or II context (Abbas et al 1996). Hence B cells are especially efficient antigen-presenting cells specialized to present diluted soluble antigens. Moreover, B cells display sets of cytokines and surface costimulatory molecules, which radically differ from their counterparts in dendritic cells. B cells are not only the objects of instruction by T cells but, conversely, also influence their partners’ function and fate. The nature and molecular context of antigen presentation determines the character of a resulting T-cell response, including Th1/Th2 polarity. With such different cytokine and costimulatory molecule repertoires from dendritic cells, B lymphocytes drive antigen-recognizing CD4+ T cells preferentially towards the Th2 lineage (Abbas et al 1996; Finkelman 1995).
B lymphocytes produce humoral immunoglobulin antibodies, but they are not autonomous and they require help from CD4+ T cells in order to create an optimal immunoglobulin response. T cells contribute to the processes that result in formation of antibodies having the best antigen-binding fit and immunoglobulin isotype, and they control duration of the humoral response. Regulation of B-cell activity by T cells is the result of a T-cell– B-cell interaction that hinges around antigen presentation by B cells.
T-cell differentiation in the thymus: self recognition shaping the (auto)immune T-cell repertoire
Most T cells are formed and reach their maturity in the thymus, the central organ of the immune system. Immature progenitor cells, coming from the bone marrow, reach the thymus where they undergo rapid proliferation before leaving as immunocompetent T lymphocytes. The thymus is thus the site where T-cell receptor diversity and the differentiation of T-cell lineages are generated. Both result from sequential interactions between immature thymic T cells and the various cellular milieus formed by the thymic stroma.
Intrathymic T-cell differentiation is a complex process requiring profound developmental change involving induction of genes within single cells as well as radical selection processes on a population basis. The primitive bone marrow-derived progenitor cells reach the thymus via the bloodstream. They first settle in cortical areas located just beneath the thymic capsule. During differentiation, the maturing T-cell progenitors move through different compartments towards the thymic medulla, which they reach mostly as competent but naive T cells (Van Ewijk 1991).
The thymus is composed of material that develops from several different sources. Until recently, it was assumed that, at least in the mouse, cortical epithelia are progeny of the third ectodermal cleft, whereas the medullary epithelium derives from the third endodermal pouch (Cordier and Haumont 1980). Now, this dichotomy appears too simple. Instead of being derived from one epithelial sheet, thymic medullary epithelium seems to be a mosaic of multiple and diverse epithelial islets, each derived from distinct progenitors invading the thymus in early embryonic development (Rodewald et al 2001). As will be discussed later, such a diversified origin of medullary cells would have profound implications for the shaping of autoimmune T-cell receptor repertoires. In addition to epithelial cells, stem cells immigrating from bone marrow differentiate into macrophages, dendritic cells (which act in the periphery as professional antigen-presenting cells) and lymphocytes (Owen and Jenkinson 1984).
Thymic microenvironments are strictly determined by local stromal cells but, in turn, their composition and character are controlled by local T lymphocytes. For example, mice with severe combined immunodeficiency, and those treated with ciclosporin A, have no differentiated thymic medulla. However, reconstitution with intact T cells regenerates the intact medullary milieu (Shores et al 1991). The relevant signals seem to be communicated through lymphotoxin receptors (Boehm et al 2003). This is not unique for the thymus. Induction of a specialized lymphoid tissue microenvironment by immigrating lymphocytes is also seen in peripheral immune organs. Here, activated B lymphocytes form the germinal centres with their typical follicular interdigitating cells, presumably by secreting proinflammatory cytokines such as lymphotoxin-α (Le Hir et al 1996). In general, however, it is the composition of stromal cells that determines actual function of the local microenvironment in T-cell differentiation. There must be particular sets of cell signalling molecules that fit best and encourage suitably differentiated T-cell maturation over a critical period.
The influences from membrane signals, along with soluble mediators, that characterize the individual thymic microenvironments and induce the next step of differentiation are finally being deciphered. Genetic studies of the athymic nude mutations in rat and mouse identify a particular transcription factor, Whn, as central in controlling differentiation of embryonic thymus epithelium right after the invasion of lymphoid progenitor cells. It regulates development of cortical, subcortical and medullary epithelia. Inactivation by mutation causes thymus aplasia, and in addition deficient hair growth (Nehls et al 1996). Other genes involved in creating the early thymic stromal environment are provided by the homeobox gene family, which act in early stages of endoderm/mesenchyme interactions (Hoxa3) and on the interaction between lymphocyte precursors and epithelium (Pax9: Manley 2000).
Clearly, epithelia must be acting in concert with bone marrow-derived stromal cells, such as thymic dendritic cells and macrophages. At least in the case of epithelium, regional diversity has been demonstrated using sets of monoclonal antibodies (Van Ewijk et al 1994). In any case, it is clear that the intact architecture of the thymus is absolutely essential for correct development and function of the immune system. Disruption of thymic structure, following infection or graft versus host attacks, leads to profound deficits in normal T-cell production. This has two equally undesirable consequences – compromised reactivity against microbial pathogens and autoimmune disease.
Gene expression during T-cell maturation
The developmental steps of T-cell differentiation have been defined by examining membrane markers and receptor-gene expression in thymic T-cell subsets during fetal development, after regeneration, and in transgenic animals. Obviously, productive rearrangement of the genes encoding T-cell receptor α and β chains is the first key event. Moreover, the progression of T-cell receptor expression is closely related to induction and surface expression of the ancillary molecules CD4 and CD8 on differentiating T lymphocytes.
There is consensus that T-cell progenitors migrating from the bone marrow to the thymus have neither rearranged T-cell receptors nor CD4 or CD8 on their membranes. At this stage, the T-cell receptor genes are still located in germline formation at individual loci on the chromosome. The T-cell receptor β-chain genes are rearranged first. They appear on the cell membrane together with a primitive surrogate α chain. This signals several differentiation steps – induction of both CD4 and CD8, and subsequent rearrangement of the α-chain genes. The CD4+ and CD8+ T-cell receptor-expressing thymocytes are now ready to undergo selection events that result in the intact, functional T-cell repertoire composed of CD4+ and CD8+ single positive lymphocytes (Robey et al 1994).
Formation of the T-cell receptor repertoire: positive and negative selection events
The mature T-cell repertoire is generated in a sequence of intense interactions between the T-cell receptor of developing thymocytes and self-peptide-laden MHC products expressed on thymic stroma cells. It occurs in two separate global selection rounds. First, all thymocyte progenitors are pushed into proliferation by positive selection. At this stage they seem to generate a random spectrum of T-cell receptors. Some bind variously to self antigen with high or low affinity, whereas others do not bind anything present in the thymus. Most of these young T cells are doomed to die by programmed cell death. Only those that bind MHC products with low affinity are rescued and encouraged to expand. Non-binding T cells fail to receive a survival signal and die from neglect. Conversely, high-affinity binders receive a positive death signal. The nature of selecting MHC structures is not fully known. It appears, however, that, due to the low-affinity nature of positive selection, one particular complex of cortical MHC and peptide may select a broad range of (weakly) cross-reacting receptors.
The positively selected T-cell population now contains, inter alia, clones with receptors able to recognize self antigens in the periphery. Potentially, they could cause autoimmune disease (von Boehmer 1992). In a second, negative selection round, most truly self-reactive T-cell clones are eliminated. Negative selection takes place largely in the thymic medulla, as a result of interactions with medullary epithelium cells and bone marrow-derived dendritic cells. Interestingly, contact points between the differentiating T cells and selecting thymus epithelia are structured to form an immune synapse, reminiscent of that formed between mature T lymphocytes and antigen-presenting cells in the peripheral immune system (Bousso et al 2002; Hailman et al 2002; Richie et al 2002).
Very recent work supports an ancient hypothesis that the thymic medulla contains progenitor cells capable of producing and displaying a large diversity of antigenic structures specific for specialized tissues (J.R. Mackay and Goldstein 1967; Wekerle and Ketelsen 1977). Indeed, a large spectrum of putative tissue-specific antigens has been identified in the thymic medulla (Derbinski et al 2001). Transgenic mice expressing any one gene of interest under the control of a tissue-specific promoter express that gene not only in the target tissue but also in the thymus (Hanahan 1999). Most intriguingly, medullary expression of autoantigen seems to be controlled by epigenetic factors, in particular an autoimmune regulator, the AIRE (autoimmune regulator) gene, inactivation of which, either by spontaneous mutation or in transgenic mice, lowers the level of medullary autoantigen expression (Derbinski et al 2005). Such mutants suffer a multitude of autoimmune responses, through defective deletion of autoimmune T cells in the thymic medulla (Anderson et al 2002).
We cannot deny that T-cell differentiation in the thymus is an extremely complex event. It requires the sequential expression of genes within the differentiating cell. Positive and negative selection ultimately results in a T-cell population efficient in its reactions against foreign antigen and tolerant to self. The individual differentiation steps occur in specialized stromal microenvironments through which differentiating T cells pass until they enter the peripheral blood circulation. As we shall see later, mechanisms governing generation of the efficient immune repertoire, while safeguarding immunological self-tolerance, are not completely failsafe. Numerous organ-specific autoreactive T cells slip through negative selection in the intact thymus, and even fewer are held back in a pathologically altered thymus. These forbidden T cells are not only found in every healthy immune system but actually make up a substantial component of the entire population.
T-cell polarization: the Th1/Th2 dichotomy
We have mentioned that mature T lymphocytes can be considered in two major classes dependent on their expression of CD4 or CD8, two accessory molecules binding to MHC class II and I, respectively. Cytotoxic killer cells are characterized by the CD8 molecule, while helper T cells (required for full B-cell function and delayed-type hypersensitivity) express the CD4 molecule (Janeway 1992). However, the diversity of the T-cell population now goes further. Thus, the CD4+ T-helper lineage is composed of subsets producing different profiles of cytokines upon activation. In the classic study, a major part of the CD4+ cells was characterized by preferential secretion of IFN-γ and IL-2 (Mosmann and Coffman 1989). These T helper (Th) 1 cells were distinguished from another set of CD4+ T cells secreting IL-4 and IL-5, designated Th2 cells. Then, there are CD4+ T cells producing both IFN-γ and IL-4 (Th0 cells).
The Th1/Th2 dichotomy, firmly established for mouse lymphocytes, was also later outlined in principle for human T cells (Romagnani 1994). Although now considered rather too simple, the functional division of helper T lymphocytes into the Th1 and Th2 groups, and their corresponding cytokine release profiles, is still retained (Abbas et al 1996). Both cell populations also display distinct sets of functional marker structures on their surface (especially chemokine receptors) that can be used as differential markers (Figure 11.5 : Sallusto et al 1998). The Th1 and Th2 pathways are to some extent symmetrical, balancing the development and suppression of cell subpopulations controlling the immune response. Depending on effector function, the Th1-associated cytokines are IFN-γ, TNF-α and IL-2 with an autocrine effect of IL-2. Conversely, Th-2 cells deploy IL-3, IL-4, IL-5, IL-10 and IL-13 with an autocrine loop mediated by IL-4.
Figure 11.5.

Scheme showing the main chemokine receptors and their respective chemokine ligands. Adapted from Proudfoot (2002).
To reach its lineage fate, the naive T lymphocyte has to pass several checkpoints on its way to maturation as an effector cell. Decisions are taken at different sites and times during differentiation. While the CD4 versus CD8 lineage choice is made in the thymus, the Th1/Th2 decision is made within the peripheral immune system, following first contact with specific antigen (Figure 11.6 ). The circumstances that govern first contact of naive T cells with antigen – the mode of presentation – dictate the prospective cytokine pattern.
Figure 11.6.

(A) Scheme to show the dichotomous development of T-helper (Th) subpopulations from CD4+ T cells, and their respective cytokine profiles. T cells expressing the α/β T-cell receptor (α/β+ T cells) constitute >95% of the T cells in blood. The rare γ/δ+ T cells are slightly enriched in certain multiple sclerosis lesions. The α/β+ T cells can be subdivided into CD4+ and CD8+ cells, and the latter can be divided into Th1 and Th2 cells. (B) Differentiation of Th1 and Th2 cells. CD4+ T precursor cells mature into Th0 cells. Under the positive or negative influence of various cytokines, Th0 cells differentiate into Th1 or Th2 cells. Note that the scheme is an oversimplification. In reality, cells are generally less polarized in their cytokine spectrum than are murine Th cells. Bold arrows indicate differentiation pathways; thin arrows indicate positive or negative regulatory influences or cytokine secretion.
Adapted from Hohlfeld (1997).
© 2006
The quantity and quality of cytokines secreted by a particular antigen-presenting cell, and the spectrum of costimulatory molecules displayed on its membrane, together determine whether a Th1 or a Th2 response is generated upon antigen recognition. Thus, IL-12 secreted preferentially by dendritic cells or macrophages, induces production of IFN-γ in antigen-reactive CD4+ T cells, and at the same time suppresses IL-4 secretion. Under these conditions, Th1 differentiation would be expected. Conversely, antigen presentation by B cells tilts differentiation in the Th2 direction (Constant and Bottomly 1997). The cytokine pattern of an emerging CD4+ T-cell response is, however, not only imprinted by the instructing antigen-presenting cell but, in addition, depends strongly on the set of molecules recognized by the T-cell receptor and costimulatory molecules. The binding of B7-1 or B7-2 to T-cell CD28 structures may result in highly distinct cytokine responses, which can be reversed by soluble inhibitors (Reiser and Stadecker 1996), whereas binding to CTLA-4 has the opposite effect (Egen et al 2002). Furthermore, the antigenic peptide may itself determine distinct Th1/Th2 responses. Variation of peptide length or sequence of a given antigen affects the extent to which T cells are activated and, implicitly, their consequent cytokine secretion pattern (Kersh et al 1998).
Another, more indirect factor affecting this process is the microenvironment that surrounds antigen presentation. The composition of surrounding cells co-determines whether a naive CD4+ T cell assumes the Th1 or Th2 profile. The bystander cells – macrophages, mast cells, dendritic cells and a recently recognized natural killer NK1 T cell – shape ongoing T-cell responses both by surface structures and secreted cytokines (Coffman and von der Weid 1997).
At this point, innate immune reactivity comes into play. Recognition of microbial structures via Toll-like receptors activates cytokine responses in a number of cells, predominantly macrophages and dendritic cells, and thus influences activities of the maturing CD4+ T cell. Activation of these cells by bacterial DNA set free during infection, and acting on Toll-like receptor-9 receptors, creates a milieu favouring Th1 induction. This mechanism explains the immunogenic effect of microbial adjuvants, such as Freund's complete adjuvant, which contains bacterial components that drive the Th1 orientation (H. Wagner 2001).
T-cell memory and homeostasis
The adaptive immune system is created to identify specific antigen, respond with maximal efficiency and remember the encounter. Repeated exposure triggers much faster and stronger responses than the initial reaction. Here, our metaphor linking the immunological and neural synapse is again appropriate: immunological and cognitive memory are functions that depend on altered structure driven by experience (Huntley et al 2002). The anamnestic immune response is determined by generation and persistence of specially differentiated, long-lived and antigen-specific memory T cells. Immune memory is explained by Macfarlane Burnet's clonal selection theory in purely quantitative terms. The concept rests on the expansion of specific immune cell clones in response to antigen stimulation, and their persistence in the repertoire. More specific immune cells produce a vigorous memory response. It is now known that in addition to numerical clonal expansion, immune memory requires the differentiation of a particular set of antigen-specific memory T cells, which respond to antigen more vigorously and efficiently than their naive, antigen-inexperienced counterparts.
Primary contact of naive T lymphocytes triggers massive cell division, which usually proceeds to elimination of the target antigen. Thereafter, the overwhelming majority of all the new lymphocytes are quickly eliminated by programmed cell death. Only a small subset, the memory T cells, survive this attrition – sometimes for life (Jenkins et al 2001). Memory T cells and their naive progenitors are clearly distinct. Following antigen-induced differentiation, they assume a particular profile of surface markers, including elevated levels of cell adhesion molecules (such as CD44), sets of cytokine and chemokine receptors, and isoforms of the leucocyte common antigen CD45 (Dutton et al 1998). Although there is marked functional plasticity, CD4+ memory T cells seem to keep their Th1/Th2 lineage phenotype once this is acquired following initial antigen contact.
Most memory cells are migratory. In contrast to their naive counterparts, which travel from the thymus to peripheral immune organs and wait in expectation of specific antigen, a large set of memory T cells (termed effector memory cells by some investigators) indefatigably roam through the various organs in pursuit of their antigen. They go through blood and lymphatic vessels, and are attracted to putative antigen-presenting cells by chemotactic factors that are often induced by mechanisms of natural immune reactivity (Sprent and Surh 2002). A second type of memory cell, central memory cells, persist in lymphoid tissues where they wait for the eventual arrival of their antigen. The two types of memory cell are distinguishable by particular sets of surface markers, among these the chemokine receptor CCR-7 (Sallusto et al 1999) and by the intensity of their response to antigen. Both have a long life expectancy although survival is not part of their birthright and must be earned. It appears that memory T cells receive signals from neighbouring cells, especially in immunologically rich environments, that trigger survival responses and allow cells to escape programmed cell death. The signals that encourage T-cell survival are provided by cells forming the local microenvironment. Cytokines (IL-7 or IL-15), cell adhesion molecules and even MHC determinants presenting random (unspecific) peptides may each participate in homeostasis (Seddon and Zamoyska 2003). It appears that, as for antigen-dependent T-cell activation, antigen-independent homeostatic stimuli are derived from synapse-like intercellular contacts (Revy et al 2001). The signals instruct local T cells not to proliferate in an uncontrolled way, but to survive without mitosis. The composition of signals controlling long-term persistence of naive and memory T cells is not known in detail. Most probably, they include positive signals that ensure clonal persistence and self-renewal, or negative messages that prevent precocious cell activation. Examples of gene products involved in negative regulation of T cells are cytotoxic T lymphocyte antigen-4 (CTLA-4) members of the suppressor of cytokine signalling (SOCS) gene family and lung Krüppel-like factor (LKLF), each of which work at different levels of suppressive gene regulation.
CTLA-4 is expressed on the surfaces of activated T cells. It is a receptor for costimulatory molecules of the B7 family. In contrast to the alternative B7 receptor, CD28, CTLA-4 does not activate antigen-recognizing T cells. Instead, it suppresses cell activation. Blockade of CTLA-4 by recombinant inhibitor proteins or antagonist monoclonal antibodies leads to exuberant T-cell responses against foreign and self antigens (Karandikar et al 1996; Perrin et al 1996). Transgenic knockout mice lacking CTLA4 develop spontaneous organ infiltrations – presumably of an autoimmune nature – and die before adulthood (Tivol et al 1995). CTLA-4 limits T-cell activation at several levels: during primary or secondary antigen presentation and homeostatic regulation, acting both directly via interactions between T lymphocytes and antigen-presenting cells, or indirectly through CD4+CD25+ regulatory T cells (Sakaguchi 2000; Salomon and Bluestone 2001).
SOCS is a negative feedback inhibitor that acts on the signalling pathway triggered by cytokines. It interferes directly with activation of intracellular signalling molecules (jak/STAT families) through cytokine receptors. As a classic feedback inhibitor, it is induced by many proinflammatory cytokines, and reacts by suppressing the same cytokines (Alexander 2002). The pivotal regulatory role of SOCS in immune regulation is convincingly demonstrated by knockout mice, which, reminiscent of CTLA4 mutants, die early in life with exuberant inflammatory disease (Marine et al 1999).
Finally, LKLF exemplifies suppressive regulation acting at the transcriptional level. The molecule derives its bizarre name from a Drosophila gene, which shares structural similarities with LKLF in their zinc-finger segment. LKLF is expressed in memory cells, apparently keeping them in a homeostatically acceptable resting state. Following antigen presentation and activation, LKLF is transiently lost from the T cell only to reappear with acquisition of the new memory state (Di Santo 2001).
B LYMPHOCYTES
B lymphocytes are agents of the humoral immune response. They determine the production of soluble antibodies (immunoglobulins), which bind antigenic structures and prepare these for elimination, using either the lytic complement cascade or the activation of phagocytes. B lymphocytes are centrally involved in immune responses against bacteria. In fact, many traditional vaccination strategies focus on B-cell production of protective antimicrobial antibodies. However, there is also a dark side to B cells. They play pivotal roles in pathological processes such as allergies, and they mediate certain autoimmune diseases.
B lymphocytes recognize antigen via receptors formed by membrane-bound immunoglobulins. They are clonally diverse – each clone recognizing one particular antigenic structure via a single immunoglobulin type. Unlike T cells and the thymus, mammalian B lymphocytes do not develop within one specialized central immune organ. Instead, they differentiate in particular milieus within the bone marrow from primitive precursors. Having reached immunocompetence, these naive B cells leave the cradle and settle in special compartments within the peripheral immune organs. The B-cell lineage develops from pluripotential stem cells – the origin of all bone marrow haemopoietic cells. But like T cells, the differentiating B lymphocyte interacts with local stroma cells, which offer microenvironments permissive for distinct steps in differentiation leading to rearrangement of the immunoglobulin V regions and assembly of intact immunoglobulin on the surface membrane of naive cells (at least for IgM and IgD).
A completely new phase of B-cell differentiation is triggered by first contact with antigen. After this encounter, the courtship with T cells leads to dichotomous differentiation pathways – relatively long-lived memory B cells and short-lived immunoglobulin-producing plasma cells. Fundamental changes take place within germinal centres of peripheral immune organs. Here, naive B cells encounter specific antigen presented by follicular dendritic cells (not to be confused with the professional antigen-presenting cells that interact with T cells) – the enigmatic stroma cell of germinal centres. In addition, some T-helper cells are present in these areas to provide a microenvironment facilitating maturation of the B-cell response by inducing somatic hypermutation of immunoglobulin complementarity-determining regions and isotype switching.
Experimental work indicates that B lymphocytes have diverse and complex roles in the pathogenesis of autoimmune disease. In addition to antibody production, they have ancillary functions in presenting autoantigen to T cells. B cells express costimulatory molecules (B7-1 and B7-2) on their cell membrane and are thus able effectively to activate resting specific T lymphocytes. By presenting antigen, B cells direct the responding T cell towards the Th2 pattern of cytokine secretion (Lenschow et al 1996). Having recognized their antigen on B cells, T cells are prone preferentially to secrete IL-4, IL-5 and IL-10 – the cytokines that, incidentally, are required for T-cell help for antibody-producing B lymphocytes.
Immunoglobulins
Immunoglobulins play several pivotal roles in B-cell immune responses. First, inserted in the surface membrane, they act as receptors, binding specific antigen and transmitting activation signals that initiate the immune response. In secreted, soluble form, they act as antibodies, binding and earmarking antigen for destruction by macrophages or proteases (via complement), thus exerting effector functions of the immune response. In both situations, immunoglobulins use the variable (V) segments, which specifically bind antigens having complementary structures. The immunoglobulin V region and corresponding antigenic epitope fit together no less tightly than the proverbial key and its lock. The immunoglobulin V region is formed in the course of B-cell differentiation by a complex series of diversification events. The steps in naive B-cell development include recombination of germline genes, addition of non-germline (N) encoded elements, and somatic mutation to improve further the fit (affinity) of immunoglobulin after encounter with the actual antigen.
Typical monomeric immunoglobulin is composed of two light and two heavy chains interconnected by disulphide bonds. A monomeric immunoglobulin possesses two identical antigen-binding sites, each formed by the V regions of adjacent light and heavy chains (see also Chapter 3). The immunoglobulin V regions are thus the structural basis for antibody diversity. Immunoglobulin V regions are composed of framework segments with genetically conserved sequences and interspersed hypervariable regions (sequences characteristic for each individual specific immunoglobulin). They combine to form the molecular surface of an antigen-binding site. As for T-cell receptors, structural similarity between these hypervariable immunoglobulins is reflected by their denomination as complementarity-determining regions. Structural genes for the immunoglobulin V region fall into three sets: variable (V), diversity (D) and joining (J) segments clustered on the chromosome as linearly arranged gene segments. Each group contains a large number of individual genes and the human heavy chain has literally hundreds of V, dozens of D and several J genes (Rajewsky 1996).
Early mechanisms of B-cell immunoglobulin diversification
Mature, antigen-binding immunoglobulins are formed in a complex process. As a first step, single members of each set are selected during gene transcription from the diverse gene cluster joined at random (Figure 11.7 ). This recombination of individual germline genes is brought about by a sequence of events directed by special enzymes known as recombinases (RAG proteins) expressed both in B and T cells, but only in the narrow time windows of lymphoid differentiation. Random recombination of preformed gene elements alone potentially offers many thousands of possible V regions but, in reality, antibody diversity is much higher. In stark contrast to T-cell receptors, which remain unchanged throughout the life of a T cell, immunoglobulins undergo somatic modifications, which improve antigen affinity, and switch to the ideal isoform. For example, imprecise joining of genes further increases variety of the B-cell repertoire. Furthermore, additional nucleotides (P and N region, neither encoded in the germline) are added. Nucleotides are added by terminal deoxynucleotidyl transferase (TdT). Like recombinases, this is only expressed transiently during B- and T-cell development. Interestingly, N-addition, especially prominent in the mature immune system, is almost lacking in neonates (Rajewsky 1996).
Figure 11.7.

Rearrangement of germline genes in the formation of the immunoglobulin heavy chain. DNA encoding the variable region of the heavy chain contains variable (VH), diversity (DH) and joining (JH) functional gene segments. The constant region genes code for the different immunoglobulin isotypes (e.g. Cµ for IgM, Cδ for IgD, etc.). A complete heavy chain V region gene is assembled by somatic recombination events that first join the D and J segments, and then join the V gene segment to the combined DJ sequence. The heavy chain C region sequences are spliced to the variable domain sequences during processing of the heavy chain gene RNA transcript. The mRNA coding for the two types of immunoglobulin light chain (κ and λ) is constructed by similar mechanisms (not shown).
Adapted from Hohlfeld (1997).
© 2006
Nothing lasts forever and freshly formed B-cell immunoglobulin receptors are no exception. A second round of B-cell receptor formation often takes place in the bone marrow. In a penultimate stage of B-cell maturation, certain immunoglobulin may happen to bind antigen, presumably expressed on unidentified bone marrow stroma, vetoing further B-cell production of its preferred receptor and rebooting the immunoglobulin recombination machinery. A new and distinct immunoglobulin chain is then formed providing the B cell with an amended, and more acceptable, antigen specificity – a process termed receptor revision or editing (Nemazee 2000), which presumably acts to avoid the formation of pathogenic autoantibodies.
Affinity maturation and immunoglobulin class switches
At this point in ontogeny, generation of the immunoglobulin repertoire is independent of exogenous antigen. Once activated, further changes affect immunoglobulin-binding sites on the B cell. During a primary reaction, the originally formed hypervariable complementarity-determining region sequences are further modified and diversified by somatic mutation. These improve the binding fit of antibodies (immunoglobulin affinity) through the ongoing immune response. Affinity maturation of immunoglobulin takes place in the germinal centres of secondary immune organs and involves complex intercellular responses between the differentiating B cell, helper T cells and dendritic cells (MacLennan 1994). Apparently, the selecting antigen is presented by follicular dendritic cells, which signal B cells with the best-fitting immunoglobulin receptors to amplify and persist, whilst those less fortunate die from neglect (Kelsoe 1996).
Immunoglobulins come in five different classes – IgD, IgM, IgG, IgA and IgE. These are defined by constant (C) regions of the heavy chain. Each B-cell clone is identified by its antigen specificity (V region) whilst still able to undergo a change in immunoglobulin class thereby grafting the same V regions to different heavy chain isotypes. Switches are not random but follow exact rules – IgM to IgA or IgG, but never in the reverse direction. This sequence follows the location of isotype structural genes on chromosome 12. Switch events are controlled by the interaction of B cells and helper T cells. Cytokines secreted by the T lymphocytes seem to provide the signals required to select individual isotypes. Thus, IL-4 favours production and secretion of IgE and IgG1, whilst IgG2a is preferentially induced by IFN-γ and IgA by TGF-β. Although the molecular mechanisms of affinity maturation and class switching are not yet fully characterized, there is good evidence that the activation-induced cytidine deaminase (AID) functions in both processes (Muramatsu and Honjo 2001).
AUTOIMMUNITY AND SELF-TOLERANCE IN THE CENTRAL NERVOUS SYSTEM
As emphasized before, the immune system is programmed to search out and remove suspicious organisms. This function is life saving, but at the same time carries a deadly risk. Immune cells with receptors for healthy tissues have the potential to divert immune responses against self, and thereby cause organ-specific disease. Immune cell clones with such a self-destructive potential should clearly be forbidden. Self-reactive, forbidden clones should be eliminated from the immune system early in development, ideally during embryonic life, as proposed by Burnet (Burnet 1959): no self-recognition, no autoimmune disease.
Burnet was partly right. Physical elimination of T-cell clones with receptors recognizing self antigen in the thymus was shown in pioneering experiments (von Boehmer et al 1989). This work was based on the construction of transgenic mice with a highly simplified, autoimmune-prone immune repertoire. The mice expressed a rearranged T-cell receptor transgene encoding a receptor for the male-specific H-Y autoantigen. In its absence (e.g. in females), most emerging T cells used the transgenic receptor. However, in the presence of autoantigen (as in male mice), recognition of H-Y peptides in the context of MHC class I protein led to thymic elimination of autoreactive T cells.
Clonal elimination is not absolute and, thus, additional ways of securing global self-tolerance must exist. In some cases, potentially autoreactive T lymphocytes that have escaped censure acquire profound nonreactivity against self antigen – a state of anergy. This concept stems from studies of transgenic mouse models, in which an experimental transgenic autoantigen is expressed at high level in a particular peripheral tissue, but only weakly in the thymus. In some (but not all) cases, anergy of self-reactive T cells is matched by downregulation of the self-reactive T-cell receptor (Arnold et al 1993).
The most intriguing, enigmatic and, for clinical immunology, important feature of tolerance, however, is the persistence of fully reactive, self-recognizing T-cell clones in the healthy immune repertoire. Under normal conditions, these remain harmless, but self-reactive T cells may escape control and become autoaggressive. One explanation offered for innocuous persistence of self-reactive T cells in the immune repertoire is clonal ignorance. This describes the situation in which a particular tissue remains secluded from blood and lymph circulation by a dense barrier. Therefore, whilst the immune repertoire contains T cells with receptors for this tissue, they are barred from entry and exert no effect until an accidental breach of the shielding barrier provides the portal for a less than friendly autoimmune encounter. As it turns out, this was always a naive concept, and truly secluded autoantigens are rare, if they exist at all. Few, if any of these antigens are ‘tissue specific’, and most are found outside the organ in question, and represented in the more distributed immune system.
Spectacular examples of autoimmune lymphocyte clones in the intact immune repertoire are T cells recognizing the brain myelin component, myelin basic protein. First isolated from lymph nodes of healthy rodents, these T cells unfolded a lethal autoaggressive potential when transferred in an activated state into healthy animals of the same strain (Schlüsener and Wekerle 1985). Later, myelin basic protein specific T-cell lines were also isolated from the peripheral blood of patients with multiple sclerosis and healthy volunteers (Pette et al 1990a). At first, inaccessibility of central nervous system target autoantigen was held responsible for the persistence of autoimmune T cells in the absence of obvious pathological consequences. Myelin basic protein and other brain autoantigens are, however, quite commonly produced outside the central nervous system and within the immune system, where they are readily accessible (K. Kojima et al 1997; Pribyl et al 1993; Zelenika et al 1993). Clonal ignorance cannot explain self-tolerance against central nervous system autoantigens. There must also be regulatory mechanisms, positive or negative, that normally hold autoimmune disease in check.
Now, we introduce the nature of autoreactive T cells and regulatory pathways that keep them from igniting the autoimmune process. We portray experimental autoimmune encephalomyelitis as proving phenomenally instructive as a model for certain aspects of multiple sclerosis and one that also offers essential insights into the cellular organization of immunological self- tolerance and immune reactivity within the immune system. We proceed by examining individual features of autoimmune reactivity targeted against the central nervous system, revealed by experimental autoimmune encephalomyelitis, and verify their contribution to the pathogenesis of multiple sclerosis.
Experimental autoimmune encephalomyelitis: a model of multiple sclerosis and more
Organ-specific autoimmune diseases are caused by autoimmune T cells that attack the body's own tissues. Thus, T cells that recognize brain structures can assault brain tissues, while T cells with receptors for pancreatic islet cells may cause immune diabetes. Autoreactive T cells are the pathogens of autoimmune disease, much like microbes qualify as pathogens in infectious diseases. The identification of autoimmune T cells consequently follows the rules established by Robert Koch in his search for pathogenic bacteria. A pathogen, autoimmune or microbial, must satisfy Koch's postulates: it must be regularly present in a particular tissue lesion; it must be isolated from the lesion and be propagated in pure culture; and, when transferred back into a healthy tissue, the pathogen must create tissue changes similar to the original lesion. Koch's postulates were first successfully applied to myelin autoreactive T cells, the pathogens of experimental autoimmune encephalomyelitis – often dubbed ‘the’ model of multiple sclerosis. The disorder has been invaluable in revealing the basic rules of organ-specific autoimmune reactions, and beyond that, the organization of immune reactivity in the central nervous system.
The classic experimental autoimmune encephalomyelitis models
Neurological complications observed at the turn of the century in the context of rabies vaccination, first suggested that autoimmunity targeted against nervous tissue elements can induce brain inflammation (Remlinger 1928). This concept was formally proven in experimental animals by the intentional induction of inflammatory demyelinating lesions after active immunization with brain tissue (Rivers et al 1933). Experimental autoimmune encephalomyelitis later became one of the most intensively studied experimental systems in autoimmune research, profoundly shaping our understanding of basic T-cell immunology and the pathogenesis of autoimmune inflammation. Experimental autoimmune encephalomyelitis can be induced in virtually all mammalian species, including humans. In the human, acute inflammatory demyelinating disease was reported after subcutaneous injection of brain tissue in the course of vaccinations (Uchimura and Shiraki 1957) and following unconventional cell therapy (Seitelberger et al 1958). The disease induced by sensitization with whole central nervous system tissue extracts in adjuvant has a complex pathogenesis and involves both cell-mediated and humoral immune mechanisms. It can usefully be considered as having several basic components (Lassmann 1983).
T lymphocytes, reactive against central nervous system proteins, are responsible for the induction of disease. Autoimmune-mediated inflammation of the brain can be transferred to naive recipients by T lymphocytes from sensitized donors (Paterson 1960) and even by monospecific T-lymphocyte lines and clones (Ben-Nun et al 1981b). Experimental autoimmune encephalomyelitis induced by T-cell lines is, in most models, an acute monophasic disease characterized pathologically by intensive inflammation (Figure 11.8 ). The disease develops in a very characteristic pattern, typically starting in lower parts of the spinal cord and progressing finally to reach the base of the brain. This is true for the development of inflammatory lesions, and reflected by the (motor) neurological deficits, which start with paralysis of tail muscles routinely spreading to affect hind and front limbs and, in severe cases, manifesting as brainstem disorders.
Figure 11.8.

Inflammatory reaction in the central nervous system induced by T lymphocytes directed against different antigens. (A) Spinal cord inflammation in experimental autoimmune encephalomyelitis induced by transfer of an MBP-reactive T-lymphocyte line; ×30. (B) Perivascular inflammation with spread of leucocytes into the central nervous system parenchyma; ×400. (C) Inflammatory reaction in the cerebral cortex after transfer of an S-100-reactive T-cell line; ×100. (D) Prominent perivascular inflammatory reaction with little infiltration of leucocytes into the parenchyma; ×500. (E) Inflammation in the cerebellar white matter after transfer of a myelin oligodendrocyte glycoprotein-reactive T-cell line; ×100. (F) Inflammatory cells of cerebellar white matter are mostly located in the perivascular space; sections are stained with haematoxylin/eosin; ×500.
Demyelination and associated tissue damage are minimal or absent in most monophasic models. Nevertheless, in some animal strains, chronic progressive or relapsing disease variants may develop after a single transfer of encephalitogenic T cells (Mokhtarian et al 1984). In this situation, the inflammation may be accompanied by some demyelination and nonspecific tissue damage. Even in these models, and in contrast to the lesions of multiple sclerosis, inflammation is dominant and the sparse demyelination mainly restricted to the perivascular central nervous system parenchyma. These are the pathological features of acute disseminated leucoencephalomyelitis (ADEM; see Chapter 12).
In contrast to these purely T-cell mediated models of experimental autoimmune encephalomyelitis, sensitization with whole central nervous system tissue may result in prolonged chronic progressive or relapsing disease, in which the pathological appearances are inflammation, widespread primary demyelination and gliosis (Figures 11.9 and 11.10 ) (Raine et al 1974b; S.H. Stone and Lerner 1965). These models approximate more closely the pathology of multiple sclerosis, but are difficult to induce reproducibly. Their fickle nature depends upon many different factors, including the mode of sensitization and genetic background of the animal (Lassmann 1983). However, it is well established that chronic demyelinating experimental autoimmune encephalomyelitis models can nevertheless be induced in many different animal species and strains.
Figure 11.9.
Demyelination and remyelination in chronic experimental autoimmune encephalomyelitis. (A) Early demyelinated plaque with complete demyelination; ×300. (B) Some macrophages contain myelin degradation products; ×900. (C) Late, partly remyelinated lesion; the axons are surrounded by thin myelin sheaths; ×900. (D) Toluidine blue-stained plastic section; ×300.
Figure 11.10.

Astroglia reaction in demyelinated lesions in chronic experimental autoimmune encephalomyelitis. (A) Large hypertrophied astrocytes, some with multiple nuclei; ×300. (B) Occasional degradation products in the astrocyte cytoplasm; toluidine blue-stained plastic section; ×1000.
In addition to T-cell mediated inflammation, autoantibodies against myelin components seem to play a major role in the pathogenesis of chronic demyelinating variants of experimental autoimmune encephalomyelitis. Very similar disease and pathology can be induced when intravenous injection of anti- myelin antibodies is timed to coincide with the onset of the inflammatory reaction induced by transfer of encephalitogenic T lymphocytes (Linington et al 1988; Schlüsener et al 1987). Furthermore, in most models of chronic experimental autoimmune encephalomyelitis associated with widespread primary demyelination, significant serum titres of demyelinating antibodies can be detected (Linington and Lassmann 1987). Large confluent areas of demyelination develop, especially in DA and BN guinea pigs that had been immunized against recombinant myelin oligodendrocyte (MOG) protein, a minor protein component of myelin located on the surface of myelin-forming oligodendrocytes and myelin sheaths (Raine et al 1974b).
Experimental autoimmune encephalomyelitis in transgenic mice
Historically, most experimental autoimmune encephalomyelitis models used the Lewis rat or mouse strains PL/J (or B10PL for myelin basic protein-induced experimental autoimmune encephalomyelitis) and SJL/J (for proteolipid protein-induced experimental autoimmune encephalomyelitis), respectively. Many transgenic mice were generated and bred on the background of 129/J or C57BL strains. Unfortunately, neither was susceptible to the induction of experimental autoimmune encephalomyelitis. Thus, attempts to use available transgenic mice for experimental autoimmune encephalomyelitis research resorted to breeding the transgene onto a susceptible background – a time-consuming, expensive and bothersome procedure. Relief came when the group of Avi Ben-Nun identified the p35–55 peptide of myelin oligodendrocyte protein as a reliable and efficient encephalitogenic antigen in the C57BL mouse (Mendel et al 1995). Experimental autoimmune encephalomyelitis driven by the p35–55 epitope of myelin oligodendrocyte glycoprotein is now the most popular transgenic mouse system for modelling neuroimmunological disease (Owens et al 2001).
Spontaneous models of experimental autoimmune encephalomyelitis
These models are all induced by immunization of healthy animals with components of central nervous system protein. Spontaneously occurring experimental autoimmune encephalomyelitis has been observed only in transgenic mice that express genes encoding the myelin-specific T-cell receptor from an encephalitogenic T-cell clone. The first report was of a transgenic mouse in which most T cells used a myelin basic protein-specific T-cell receptor. Up to 40% of such transgenic mice developed spontaneous experimental autoimmune encephalomyelitis during the first 8 months of life. However, spontaneous experimental autoimmune encephalomyelitis emerged only in animals that were kept under conventional (‘dirty’) conditions. Animals of the same strain raised in a clean unit did not fall ill spontaneously (Goverman et al 1993).
High frequencies of spontaneous experimental autoimmune encephalomyelitis were observed in transgenic mice that had a comparable myelin basic protein-specific T-cell receptor, but lacked intact RAG genes. Since RAG genes are indispensable for generation of diverse T- and B-cell repertoires, these double transgenics possessed a monoclonal immune repertoire, comprising CD4+ T cells with the transgenic anti-myelin basic protein receptor. Practically all of these mice developed experimental autoimmune encephalomyelitis within 12 months of age (Lafaille et al 1994). As will be discussed later, a deficit of regulatory T cells appeared to be the pivotal event in determining the high frequency of autoimmune disease (Furtado et al 2001). It is worth mentioning that spontaneous experimental autoimmune encephalomyelitis is not limited to transgenic models of myelin basic protein-driven experimental autoimmune encephalomyelitis. Similar disease and in equivalent proportions was noted in transgenic mice with proteolipid protein-specific receptors (Waldner et al 2000). Optic neuritis developed in transgenics with myelin oligodendrocyte glycoprotein-specific T-cell receptors (Bettelli et al 2003).
Central nervous system-specific T cells as pathogenic agents of experimental autoimmune encephalomyelitis
Paterson (1960) provided formal proof that experimental autoimmune encephalomyelitis is an autoimmune disease mediated by cells and not humoral antibodies. At a time when immunologists were unaware of the distinction between T and B lymphocytes, he transferred experimental autoimmune encephalomyelitis from actively immunized rats to untreated recipients using large numbers of primed lymphocytes. The transferred cells were clearly organ specific and autoimmune, because they attacked the host brain tissue and ignored all others including the peripheral nervous system. Remarkably, these pathogenic lymphocytes came from donor rats that had been perfectly normal before immunization with myelin basic protein. Many questions immediately arose. Had the autoimmune encephalitogenic lymphocytes been formed de novo as a consequence of autoimmunization? Were they forbidden clones in the Burnetian sense? Or, alternatively, were encephalitogenic lymphocytes the progeny of precursors, pre-existing in the healthy rodent immune system but previously causing no harm because their target tissue, the brain, was inaccessible and shielded by the tight endothelial blood–brain barrier? We now know that none of these hypotheses provides a full explanation. Instead, healthy vertebrate immune systems contain ample numbers of potentially autoaggressive lymphocyte clones (T cells as well as B cells) that, once activated, readily access and attack their target tissues (see above).
General characteristics of encephalitogenic T cells
Most isolated encephalitogenic T-cell lines share essential characteristics. First, they are all members of the CD4+ subset of Th cells and, as such, recognize target autoantigen in the molecular context of MHC class II proteins (Ben-Nun and Cohen 1982). Only very recently have CD8+ killer T cells been described (Huseby et al 2001; D. Sun et al 2001). Secondly (and by definition), encephalitogenic T cells are principally able to transfer experimental autoimmune encephalomyelitis to naive syngeneic recipient animals. It is, however, crucial to note that activation is required to mediate disease by myelin-specific T cells. The same cells are harmless in the resting state. Interestingly, although encephalitogenic T cells qualify as CD4+ T helper cells, many have a remarkable cytotoxic potential. In the Lewis rat, for example, myelin basic protein-specific encephalitogenic T-cell lines readily lyse all target cells that present the target encephalitogenic peptide in a recognizable MHC class II context. In sharp contrast, T-cell lines with nonencephalitogenic specificity (targeting ovalbumin or mycobacterial antigens) are not cytotoxic (D. Sun and Wekerle 1986). This behaviour is seen also in experimental autoimmune encephalomyelitis-inducing, myelin basic protein-specific T cells from SJL/J mice (Fallis and McFarlin 1989), and in human T-cell lines with the same specificity (J. Burns et al 1991; Martin et al 1990). Cytotoxic behaviour of encephalitogenic T cells correlates closely with their cytokine profile. Myelin basic protein-specific T cells transferring experimental autoimmune encephalomyelitis secrete IFN-γ and IL-2, but not IL-4 and thus qualify as members of the Th1 subset (Ando et al 1989). More precisely, these cells seem to belong to a subset of Th1 lymphocytes which is shaped by the cytokine IL-23 (Langrish et al 2005). Most, if not all encephalitogenic T-cell lines show these properties. Interestingly, the encephalitogenic potential is lost when CD4+ T cells are manipulated to change from a Th1 to Th2-like cytokine pattern in vivo or in vitro (Racke et al 1994). Th1 bias is characteristic for adult myelin basic protein-specific T cells. In contrast, exposure of neonatal cells to the target autoantigen seems preferentially to induce Th2-like cells, and these are associated with lifelong resistance to experimental autoimmune encephalomyelitis (Forsthuber et al 1996).
However, it would be rash to conclude that Th2-like myelin-specific T cells are nonencephalitogenic, or even protective, under all circumstances. In fact, myelin basic protein-specific transgenic T cells can be re-educated in vitro to assume the Th2 phenotype. They mediate a vigorous central nervous system inflammatory disease when transferred into immunodeficient RAG knockout mice. Such lesional infiltrates are, however, dominated by polymorphonuclear leucocytes and thus thoroughly differ from classic Th1-dependent lesions (Lafaille et al 1997). Indeed, an ‘anaphylactic’ response was also observed in regular mice after challenge during recuperation from a preceding episode of experimental autoimmune encephalomyelitis. The allergic response was explained as pathological exaggeration of the physiological Th2-biased disposition prevailing during recovery from experimental autoimmune encephalomyelitis (Pedotti et al 2001).
Potentially pathogenic T-cell clones in the healthy immune repertoire
The fact that encephalitogenic T-cell lines can be isolated from autoimmunized rodents is in itself rather stunning. Ultimately, it indicates the presence of potentially pathogenic autoimmune T-cell clones in the regular immune repertoire. After all, it is safe to assume that, before immunization, the donor animal had enjoyed perfect health and possessed a normal immune system. Since, in striking contrast to B-cell immunoglobulin receptors, T-cell receptors are not somatically mutated at any stage of activation or proliferation (Ikuta et al 1985), encephalitogenic T lines must have been derived from clonal progenitors that pre-existed, without doing harm, in the donor's normal immune system. In fact, central nervous system-specific autoimmune T-cell clones have been demonstrated even more directly in healthy immune repertoires. Encephalitogenic T cells were first isolated from completely naive, nonimmunized Lewis rats. Their myelin basic protein-specific T cells shared all functional and structural properties with conventional T-cell lines extracted from preimmunized animals. They were CD4+, recognized the same peptide epitopes presented in an identical MHC class II antigen context, and used similar T-cell receptors. Most importantly, myelin basic protein-reactive T cells from naive rats roll out the same encephalitogenic potential as T cells derived from presensitized animals (Schlüsener and Wekerle 1985).
The existence of potentially autoaggressive T-cell clones in healthy immune systems is a general phenomenon, not restricted to myelin-specific T cells or Lewis rats. T-cell lines recognizing a virtually unlimited range of organ-specific autoantigens – including synovial, thyroid, retinal and pancreatic determinants (Cohen and Miller 1994) – have been raised in rodents by many laboratories. Encephalitogenic, myelin basic protein-specific T-cell lines were established from naive monkeys (Genain et al 1994; Meinl et al 1997) and similar T cells abound in the immune system of healthy human donors (see below). The relative number of myelin basic protein autoreactive T cells in immune compartments of naive rats is not known. There is, however, evidence that their concentration in the thymus is considerably higher than in peripheral immune organs (Lannes-Vieira et al 1995). One could argue that the frequency of myelin basic protein-specific T cells in the peripheral immune repertoire is insufficient to mount a spontaneous lifelong autoimmune attack. This, however, would not be the case in transgenic mice with myelin basic protein-specific TCR transgenes. The immune repertoires of these mice comprise >70% myelin basic protein-specific T cells and yet, kept under clean conditions, very few of these animals ever develop spontaneous experimental autoimmune encephalomyelitis (Goverman et al 1993). We argue later that regulatory control mechanisms seem to guarantee the nonreactivity of these potentially autoreactive T cells.
Thymic generation of the myelin autoreactive T-cell repertoire
It has been known for a long time that experimental autoimmune encephalomyelitis can be actively induced only in animals with an intact thymus. Thymectomized or congenitally athymic rodents fail to develop disease upon encephalitogenic immunization (Ortiz-Ortiz and Weigle 1976), although they are fully susceptible to experimental autoimmune encephalomyelitis transferred by activated myelin basic protein-specific CD4+ helper T lymphocytes (Hinrichs and Humphres 1983). We have noted that the thymus affects susceptibility to autoimmune diseases (including experimental autoimmune encephalomyelitis) by controlling the development of mature immunocompetent T cells and eliminating many autoreactive T cells from the repertoire. Traditional theories explain tolerance by physical deletion of self-reactive lymphocytes from the immune repertoire – best guaranteed by the absence of a potentially autoreactive T-cell clone. Thereafter, the induction of autoimmune disease would depend on complex mechanisms, including pathological mutation of forbidden clones (Burnet 1959), or de novo exposure of sequestered autoantigens.
There is little doubt that deletion of self-reactive T-cell clones is a developmental feature of the immune repertoire. In transgenic mice with a self-reactive T-cell receptor, most T cells are removed during intrathymic development upon contact with the relevant autoantigen (von Boehmer et al 1989). More recent work has qualified that position. Self, presented in the context of MHC, plays a role in the positive selection of T-cell clones responding with low avidity. Conversely, the same peptide–MHC antigen product seems to select against T-cell clones with high avidity antigen receptors (Jameson et al 1995). Why then would brain-specific T-cell clones escape negative selection? The obvious explanation is clonal ignorance. It might be assumed that encephalitogenic proteins are localized exclusively within the central nervous system parenchyma, an immune privileged location secluded from the periphery by the intact blood–brain barrier (Wekerle et al 1986). In that situation, brain autoantigen would fail to reach the thymus and negative selection would not occur. Reality is, however, more complicated. Surprisingly, many encephalitogenic proteins are identified in thymic compartments. The first examples were unusual isoforms of myelin basic protein (‘golli-protein’), a component of myelin appearing during postnatal development and in the thymus before birth as mRNA (Grima et al 1992; Pribyl et al 1993) and protein (Mathisen et al 1993). Myelin basic protein is by no means the only encephalitogen expressed in the thymus. Proteolipid protein has also been observed (Pribyl et al 1996), apparently within thymic macrophages. Furthermore, the calcium-binding S100b protein of astrocytes, the target autoantigen of an astrocyte directed experimental autoimmune encephalomyelitis model (Kojima et al 1994), is also expressed within the healthy adult thymus, where it directly coexists with S100b-specific encephalitogenic T-cell clones (Kojima et al 1997). Thus, the mere presence of autoantigen in the thymus by no means rules out the generation of autoimmune T-cell clones and their emigration to the periphery.
Encephalitogenic T cells must slip through a gap in the thymic negative-selection meshwork. While it is not clear how the cells manoeuvre their escape, there is evidence in favour of active central nervous system autoantigen-directed negative selection within the thymus. In the SJL mouse, the thymus mainly produces only the short isoform of proteolipid protein, DM-20, and not full-length protein. Most T cells recognizing DM-20 epitopes are retained in the thymus, whereas T cells specific for epitopes located on the extra sequences of full-length proteolipid protein, not covered by DM-20, reach the peripheral immune repertoire in substantial numbers (A.C. Anderson et al 2000; L. Klein et al 2000). Evidence from mice that lack particular myelin proteins due to natural mutation or transgenic inactivation complements these data. PLPI knockout mice lacking the entire proteolipid protein gene, and thus not expressing any protein variant in the central nervous system and thymus, display a much broader myelin protein-specific T-cell repertoire than wild-type animals (L. Klein et al 2000). Similar observations are available in myelin oligodendrocyte glycoprotein knockout (Delarasse et al 2003) and shiverer mutant mice, deficient in myelin basic protein (Targoni and Lehmann 1998). Both murine variants possess higher numbers and broader repertoires of myelin oligodendrocyte glycoprotein or myelin basic protein-specific T-cell clones.
The thymus should not, however, be viewed simply as a filter holding back (many) autoimmune T-cell clones. It plays an additional role in shaping the autoimmune T-cell receptor repertoire. Myelin basic protein-specific T cells in the Lewis rat use a very particular set of T-cell receptors. Instead of utilizing a broad set of available V genes, most cells are restricted to the Vβ8.2 gene along with a narrow spectrum of J chain genes. Unusually, most receptors have an exceptionally short MHC/peptide-binding segment (CDR3). The formation of this biased T-cell receptor repertoire depends on an intact, natural thymus. The chimeric thymus, composed experimentally of lymphocytes and dendritic cells from Lewis rats but epithelium from another animal, produces myelin basic protein-specific T cells that are fully functional but do not show the Vβ8.2 bias, and their complementarity-determining region 3 sequences are of conventional lengths (Wekerle et al 1996).
What could be the role of self-reactive T cells in the healthy immune system? Is their presence just the reflection of a porous regulatory mechanism? Or, could autoreactive T lymphocytes exert a positive homeostatic function, as postulated in the concept of the immunological homunculus (I.R. Cohen 1992)? The evidence supports a positive function for naturally self-reactive T cells – especially in tissue regeneration. Through their self-reactive receptors, lymphocytes are able to identify specific tissues, especially those undergoing degenerative or inflammatory changes, and provide molecules – such as neurotrophins – that mediate repair (Kerschensteiner et al 2003). Such beneficial autoimmunity has been proposed as a treatment (Schwartz and Kipnis 2001). These are evidently properties of autoreactive CD4+ T lymphocytes, but it is not yet known whether cytotoxic CD8+ T cells also participate in tissue regeneration.
Myelin-specific T-cell clones in the human immune repertoire
The existence of myelin-specific T-cell clones in the healthy immune repertoire is by no means a curiosity peculiar to rodent immune repertoires. The human immune system shows even higher proportions of myelin basic protein-specific T-cell lines (Burns et al 1983; Ota et al 1990; Pette et al 1990a). The data indicate that, with few exceptions, all are CD4+ helper T lymphocytes recognizing autoantigenic epitopes with molecular restriction by DR products of the class II MHC, often of the DR2 haplotype (DRB1*1501 and DRB5*0101: Martin et al 1990; Ota et al 1990; Pette et al 1990b). However, it should be noted that, in principle, every haplotype may act as an antigen-restricting element. In addition to DR, myelin basic protein presentation is described in the context of DQ and DP (Martin et al 1992a).
The cytokine patterns secreted by myelin basic protein-specific human T cells correspond to the Th1 subset of CD4+ T cells. Upon antigen-dependent activation, these cells release large amounts of IL-2, IFN-γ and TNF-α (Voskuhl et al 1993). The Th1-like nature of human myelin basic protein-specific T cells has been confirmed by immunospot assays determining the cytokine release pattern of individually plated T lymphocytes (T. Olsson et al 1990b). An analysis of myelin basic protein-reactive T cells in the context of multiple sclerosis shows differences between cases and controls. These T cells have higher IFN-γ and IL-4 production, but do not polarize towards a distinct Th1 or Th2 profile, and clones produce IL-2, IL-4, TNF-α, IFN-γ and IL-10 but not IL-6 (Hermans et al 1997). As in the Lewis rat, many if not all human myelin basic protein-specific CD4+ T-helper lymphocytes are in fact efficient cytotoxic killer cells. They destroy any antigen-presenting cell displaying myelin basic protein peptides in the appropriate MHC context (Martin et al 1990; J.R. Richert et al 1989; J. Zhang et al 1992).
The human T-cell repertoire contains not only clones reactive against myelin basic protein, but virtually all central nervous system autoantigens tested. Several groups have described healthy donor-derived T-cell lines specific for myelin oligodendrocyte glycoprotein (Lindert et al 1999), 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase: Rösener et al 1997) and proteolipid protein (Markovic-Plese et al 1995; Ohashi et al 1995).
Autoantigen recognition by central nervous system-specific T cells
The isolation of central nervous system autoantigen-specific T cells from human donors has raised enormous hopes for a better understanding of the autoimmune pathogenesis and, implicitly, for the design of new, specific and efficient immune therapies. The study of such T cells should help elucidate the precise mechanisms of autoantigen recognition, the first critical step leading to the development of the inflammatory lesion. Then, therapies interfering with this recognition could be developed and used to curb ongoing plaque formation in the initial stage.
The dynamics of T-cell recognition: epitope and determinant spreading in experimental autoimmune encephalomyelitis
Comparison of myelin basic protein-specific T-cell lines isolated from primed or naive Lewis rats reveals that the overwhelming majority recognize the sequence p68–86 in the context of the RT1.Al MHC class II product (Vandenbark et al 1985). Indeed, this is the fragment found to possess the strongest encephalitogenic potential in earlier active immunization experiments (Kibler et al 1977). A second, minor epitope at position p85–99 is presented in the Lewis rat in the context of an alternative MHC class II product, RT1.Dl. Immunization with intact myelin basic protein very rarely activates T cells specific for p85–99, but these can readily be selected after priming with this peptide. p85–99, is thus defined as a cryptic epitope (Mor and Cohen 1995).
To a greater or lesser extent, epitope dominance in encephalitogenic T-cell responses is seen in many experimental systems. There is considerable epitope dominance in experimental autoimmune encephalomyelitis induced by myelin basic protein in PL/J mice (and other strains exhibiting H-2u). Most myelin basic protein-specific T cells from PL/J mice recognize the acetylated form of p1–11, with a minority responding to an epitope on p35–47 (Zamvil and Steinman 1990). The important encephalitogenic response of C57BL mice against myelin oligodendrocyte glycoprotein seems to be dominated by sequence 35–55, positioned on the extracellular portion of the molecule (Mendel et al 1995). The myelin basic protein epitope spectrum is, however, considerably broader in the SJL/J mouse, with multiple determinants nested in the central and N-terminal portions of the protein. Furthermore, in most rodents tested, the T-cell response against the second classic encephalitogenic myelin protein – proteolipid lipoprotein – is dissipated amongst several epitopes (Greer et al 1996), as is the response to myelin oligodendrocyte glycoprotein (Amor et al 1994).
The epitope response pattern in encephalitogenic T-cell responses is remarkably dynamic. In a landmark paper, Lehmann et al (1993) concluded that Ac1-11 is dominant only during the early active phase of experimental autoimmune encephalomyelitis in PL/J mice immunized against myelin basic protein. After remission and with the onset of chronic disease, additional but normally cryptic epitopes seem to be newly recognized by freshly recruited T cells – a phenomenon now known as determinant spreading and reported in the Lewis rat by some (Mor and Cohen 1993) but not all (Y. Matsumoto and Abo 1994) investigators. Although similar observations have been made in diabetes mellitus (D. L. Kaufman et al 1993; Tisch et al 1993), the universality of this phenomenon in autoimmune reactions remains to be established. What can be said is that similar broadening of epitope recognition is not seen in response to foreign antigens (Gammon et al 1990).
There is no ultimate explanation for intramolecular determinant spreading, and its relevance for the dynamics of relapse and remission is still debated. As we discuss below, it is possible that the first wave of encephalitogenic T cells, specific for the initially dominant epitope, enter the central nervous system parenchyma and are there eliminated by apoptosis. As a replacement, T cells recognizing minor or cryptic determinants then expand and dominate the demonstrable repertoire (J. Bauer et al 1995). Other hypotheses should also be considered. In fact, several reports highlight the late appearance of T cells reacting against proteolipid protein-induced chronic relapsing models of experimental autoimmune encephalomyelitis (L. L. Perry et al 1991). This trans-molecular determinant spreading is best explained by de novo immunization of T-cell populations as a consequence of local inflammatory responses. In this situation, primary encephalitogenic T cells could invade the central nervous system and trigger an inflammatory autoimmune attack, creating a proinflammatory milieu in which local debris is taken up by antigen-presenting cells (microglia, immigrant macrophages or dendritic cells) and offered to T cells already responsive to additional autoantigens (Vanderlugt and Miller 2002). In models of chronic relapsing experimental autoimmune encephalomyelitis, relapses can be stopped by treatments that block costimulatory receptors. Blindfolding B7-1 structures by blocking antibodies abrogates relapses and stops epitope spreading (S.D. Miller et al 1995b). Activation of B7-1 has the opposite effect (Vanderlugt et al 1997). It will be important for understanding relapses in multiple sclerosis to learn whether a causal relationship exists between epitope spreading and new episodes.
Human T-cell reactivity against brain autoantigens
The brain proteins known to provoke experimental autoimmune encephalomyelitis responses in animals have been examined for their immunogenicity to immune cells harvested from patients with multiple sclerosis. Myelin basic protein was the first brain autoantigen to be investigated. This putative autoantigen owes its candidature (and hence its devoted popularity amongst neuroimmunologists) to several properties. It is a small protein (main isoform about 18.5 kDa) positioned on the inner surface of the myelin membrane (see Chapter 10) but quantitatively a major component of the myelin membrane (30–40% dry weight). Due to its basic charge and solubility in water, it is easy to isolate to purity – in contrast to most other myelin proteins, which are either highly lipophilic or only minor components of myelin. Early attempts to study myelin basic protein reactivity in samples from patients with multiple sclerosis relied on simple proliferation or cytokine release tests confronting peripheral blood lymphocytes from patients or healthy control donors with purified antigen in vitro. The results of primary response assays were, however, often ambiguous or hardly reproducible despite the best efforts of competent investigators. It is now clear that simple bulk proliferation assays are not sufficiently sensitive to detect subtle differences in the myelin-specific repertoire of patients with multiple sclerosis. Detailed study of the disease-specific immune reactivity to brain determinants requires a technology allowing a large number of autoantigen-specific T-cell lines to be isolated from each patient. This has become possible with the development of the split-well cloning system, in which a primary limiting dilution approach allows the selection of mono- or oligoclonal T-cell lines from freshly isolated peripheral blood lymphocytes (Figure 11.11 : Pette et al 1990a; 1990b). The principle is simple. Peripheral blood mononuclear cells are distributed into multiple microcultures containing decremental cell numbers. Addition of the putative autoantigen results in the activation and proliferation of individual autoreactive T cells contained within each well. At limiting (low) cell dilutions, some but not all parallel cultures respond by cell division. Initially, selectively presented antigen drives proliferation, but later this is sustained by cytokines such as IL-2. Microcultures containing a proliferating lymphocyte colony are selected and transferred in equal parts (split) to two culture wells. Autoantigen is added only to one split well. Colonies that require the presence of autoantigen for their further proliferation are, by definition, selected and propagated as autoantigen-specific T-cell lines. Often, but not always, this response takes its origin in the multiplication of one autoantigen-responsive T cell contained within the originally seeded lymphocyte population. In this case, the resulting cell line is truly monoclonal.
Figure 11.11.

Establishment of autoantigen-specific oligoclonal T-cell lines (clonoids) by primary limiting dilution (split-well method). Ag = antigen; APC = antigen-presenting cell.
In terms of their membrane phenotype and cytokine secretion pattern, human peripheral blood-derived myelin basic protein-specific T lymphocytes closely resemble their rodent counterparts. However, they differ strikingly in their interactions with target autoantigen. Epitope recognition pattern and T-cell receptor repertoire usage are much more complex in human than rodent T-cell responses. Whilst the myelin basic protein response in the Lewis rat focuses on a narrow segment of the molecule, the variety of epitopes recognized by human T cells is remarkably broad. The large body of data can be summarized: each individual human immune system contains T cells recognizing many epitopes distributed along the entire myelin basic protein molecule; any given MHC class II product (e.g. the DR2 molecule) can bind and present a variety of peptides (Wucherpfennig et al 1994a). Conversely, most peptides actually bind to different DR molecules and can therefore be presented in varied contexts (Martin et al 1991; Valli et al 1993); lastly, the central segment p87–106 and the C-terminal sequence p144–163 show relative immunodominance, recruiting more specific T-cell lines than most other segments (Martin et al 1992b; Ota et al 1990; Pette et al 1990b).
The epitope pattern characteristically found in the majority of human blood donors (with or without multiple sclerosis) showing reactivity to myelin basic protein is broad but not completely random. In a study of monozygotic twins, for example, disease concordant pairs showed remarkably uniform epitope recognition patterns, whilst there was more discrepancy between discordant twins (Utz et al 1993). Perhaps even more striking, and much in contrast to the majority of human anti-myelin T-cell repertoires, there is a small number of patients with multiple sclerosis who display an anti-myelin basic protein T-cell response focused against a narrow, dominant peptide segment reminiscent of the Lewis rat. Meinl et al (1993) describe a set of patients whose T lymphocytes (represented by panels of CD4+ T-cell lines) almost exclusively recognize epitope(s) nested within one narrow myelin basic protein sequence – in one case with stability over 7 years. Similar patterns have been described in other patients using T-cell cloning (Lovett-Racke et al 1997; Salvetti et al 1993; Wucherpfennig et al 1994a), and polymerase chain reaction-based spectratype analyses (Goebels et al 2000; Muraro et al 2003; Musette et al 1996). The nature of these unusual human T-cell responses is obscure. They certainly do not result from isolated proliferation in vivo of one single T-cell clone, as indicated by T-cell receptor sequence analyses. However, some unrecognized antigen-driven oligoclonal proliferation event is not entirely excluded.
In multiple sclerosis, reactivity to the other mass protein of myelin, proteolipid protein, was first demonstrated using the immunospot assay. Olsson et al (1990b) reported a relative increase of reactivity to this and myelin basic protein, showing the presence of IFN-γ-secreting T cells both in peripheral blood and cerebrospinal fluid from patients with multiple sclerosis. Greer et al (1997) reported that immunodominant reactivity of peripheral blood mononuclear cells to proteolipid protein in patients with relapsing or progressive multiple sclerosis is confined to two overlapping peptides (PLP184–199 and PLP190–209) – these responses increase with disease duration and disability. At present, the number of antigen-specific T cells remains far lower than the availability of myelin basic protein-reactive T-cell lines, thereby limiting the opportunity to evaluate epitope dominance and T-cell receptor utilization. However, thus far, there is no evidence for unusual characteristics in proteolipid protein-specific human T-cell repertoires. And, most importantly, none of the reported features is specific for multiple sclerosis. All this is reminiscent of investigations into myelin basic protein-specific T cells. Proteolipid protein-specific T cells mainly secrete the Th1 cytokine profile (Correale et al 1995a). Those harvested from patients with multiple sclerosis seem to express IL-2 receptors (J. Zhang et al 1994), have a broad spectrum of epitope recognition (Markovic-Plese et al 1995; Ohashi et al 1995), and offer no evidence for highly dominant T-cell receptor V gene usage (Kondo et al 1996).
A particularly intriguing candidate autoantigen in multiple sclerosis is myelin oligodendrocyte glycoprotein, first identified independently by Linington et al (1984) and Lebar et al (1986) as a minor myelin component exclusively located within the central nervous system. Two properties make it a particularly interesting molecule. First, the gene for myelin oligodendrocyte glycoprotein maps to the MHC (Pham-Dinh et al 1993). Secondly, it is an unusual member of the immunoglobulin supergene family with a long extracellular domain that can readily be accessed by humoral mediators of immunity (Kroepfl et al 1996). Myelin oligodendrocyte glycoprotein-specific humoral antibodies thus can readily bind myelin membranes, and in cooperation with complement components and/or Fc-receptor bearing phagocytes, propagate large areas of demyelination (Linington et al 1988; Schlüsener et al 1987). In addition, myelin oligodendrocyte glycoprotein possesses highly autoimmunogenic T- and B-cell epitopes. In rodents, immunization with myelin oligodendrocyte glycoprotein peptides or recombinant protein causes activation of encephalitogenic T and B cells, mediating a disease that, in its similarity to multiple sclerosis, is strikingly distinct from myelin basic protein-induced experimental autoimmune encephalomyelitis (Johns et al 1995; Kerlero de Rosbo et al 1995).
The few reports on human myelin oligodendrocyte glycoprotein-specific T cells have raised considerable interest. In contrast to other brain autoantigens, their reactivity in patients with multiple sclerosis has been shown by primary proliferation responses in vitro. In one study using native myelin oligodendrocyte glycoprotein, proliferation responses were consistently stronger in T cells from patients with multiple sclerosis than from matched healthy donors (Kerlero de Rosbo et al 1993). While the original testing was done using native, brain-derived myelin oligodendrocyte glycoprotein, these findings were repeated using recombinant material (Kerlero de Rosbo et al 1997). Similar findings come from immunospot studies detecting cytokine release from myelin oligodendrocyte glycoprotein-specific T lymphocytes and specific autoantibodies from B cells. In both cases, the frequencies of positive lymphocytes were enhanced in peripheral blood mononuclear lymphocytes and cerebrospinal fluid from patients with multiple sclerosis (J. Sun et al 1991b). Definitive investigation of myelin oligodendrocyte glycoprotein-reactive immune cells appears hopeful but awaits confirmation by larger collections of cell lines and experiments.
Is there any evidence for epitope spreading in human T cells, as in rodent models of myelin autoimmunity? In principle, owing to the generally unfocused human anti-myelin response patterns, it is technically difficult to identify true epitope spreading over time. In some patients, however, epitope recognition is subject to marked changes. The number of epitopes recognized may increase but, in other cases, the range contracts (Goebels et al 2000; Mazza et al 2002; Tuohy et al 1998) and epitope dynamics are often unconnected to the clinical course of multiple sclerosis (Ristori et al 2000). This may come as no surprise since, to date, the pathogenic target autoantigen in patients is itself unidentified. Thus, no clear linkage exists between epitope patterns and clinical courses in multiple sclerosis.
Myelin-specific T-cell receptors
Isolation of encephalitogenic T-lymphocyte lines from immune organs or central nervous system infiltrates of autoimmune rats using the protocol developed by Ben-Nun et al (1981a) paved the way for studies of the myelin autoimmune T-cell receptor repertoire. The initial descriptions were tantalizing. Myelin basic protein T cells both in Lewis rats and PL/J (or B10PL) mice used, almost uniformly, the Vβ8.2 gene along with a very narrow spectrum of Vα genes, and peculiar short complementarity-determining region 3 sequences. In particular, the preferential usage of Vβ8.2 by encephalitogenic T cells in diverse animals led to a ‘V region disease hypothesis’, which predicted selective use of particular T-cell receptor V genes in autoimmune disease (Heber-Katz and Acha-Orbea 1989). It later turned out that dominance of individual Vβ genes is the exception rather than the rule. In particular, the gene repertoire required to form functional T-cell receptors on human myelin basic protein-specific T lymphocytes is very broad and does not duplicate at all the unique arrangements of the Lewis rat. This may look unfortunate to creative therapists, because lack of T-cell receptor restriction undermines vaccination strategies directed at T-cell receptors or blocking of responses using peptides and small molecules (see Chapter 18).
The myelin-specific T-cell receptor repertoire in experimental autoimmune encephalomyelitis
The first studies of T-cell receptors used for recognition of central nervous system autoantigens were carried out using myelin basic protein-specific encephalitogenic T-cell clones isolated from Lewis rats. These indicated that anti-central nervous system autoimmune responses were characterized by their monotonous character and restriction to a minority of receptor subtypes. Thus, the Lewis rat T-cell response against myelin basic protein utilizes a strikingly simplified repertoire of structural genes. The earliest studies of myelin basic protein-specific T-cell receptors noted an almost complete utilization of Vβ8.2 genes, often combined with a limited set of Jβ elements (Burns et al 1989; Chluba et al 1989), and paired with an equally restricted repertoire of Vα genes. Most myelin basic protein T-cell receptors have complementarity-determining region 3 sequences – the receptor segments primarily dictating peptide specificity – encoded by unusually short base sequences (Kääb et al 1998; G. Kim et al 1998; X-M. Zhang and Heber-Katz 1992). Furthermore, myelin basic protein-specific complementarity-determining region 3 sequences are dominated by the Asp-Ser sequence, and they have a deficit of N-region inserts (Gold et al 1991; X-M. Zhang and Heber-Katz 1992). All these peculiarities are seen not only in T-cell lines isolated from myelin basic protein-primed rats but also in lines cloned out from naive Lewis rat thymus (Lannes-Vieira et al 1995). Short, N-insert-deficient complementarity-determining region 3 sequences are typical of immature T-cell responses (Bogue et al 1991). Strongly biased use of Vβ8.2 genes for myelin basic protein-specific T-cell receptors is also the rule in PL/J and B10.LP mice, both having the H-2u haplotype (Acha-Orbea et al 1988; Urban et al 1988).
The dominant use of Vβ8.2 and biased occurrence of one simple complementarity-determining region sequence motif in the encephalitogenic T-cell response raised high expectations for immunospecific therapy of brain autoimmunity. But, disappointingly, this seems to be an exception and not the rule. Even in the Lewis rat, Vβ8.2 dominance changes over time. It is strict in early phases of the encephalitogenic response but has a tendency to dissipate over time (Offner et al 1993). Furthermore, this bias seems restricted to T-cell responses against the dominant epitope p68–84 and not minor encephalitogenic determinants (Offner et al 1992; D. Sun et al 1992), or truncated versions of the dominant peptide where loss of a few key amino acids completely alters the Vβ8.2 focus (D. Sun et al 1995). Encephalitogenic T-cell responses have been analysed in many species and with many brain components as target autoantigens. Most data indicate diversity for T-cell receptor repertoires other than those directed at myelin basic protein. This applies especially to proteolipid protein (Kuchroo et al 1992) and myelin oligodendrocyte glycoprotein (Mendel et al 1996).
Autoantigen-reactive T-cell receptors in the human repertoire
Initial studies based on a limited number of myelin basic protein-specific T-cell lines suggested that preferential T-cell receptor gene usage is not uncommon, at least within the repertoire of an individual patient (Ben-Nun et al 1991; Kotzin et al 1991; Wucherpfennig et al 1990). Careful scrutiny of the accumulated data, however, does not show much conformity in T-cell receptor gene dominance or clonal expansion amongst patients with multiple sclerosis (Figure 11.12 : Hafler et al 1996). Overall, there is no generally applicable and multiple sclerosis-associated T-cell receptor-α/β gene usage (comparable to Vβ8.2 in the Lewis rat or PL/J mouse). Furthermore, this repertoire is not fixed, as indicated by studies of bone-marrow transplant recipients (Muraro et al 2005). Whilst some studies indicate enhanced usage of certain Vβ genes (Vβ5.2 and Vβ6.1 in one study of T-cell lines: Kotzin et al 1991) in polymerase chain reaction material amplified from the lesions of multiple sclerosis (Oksenberg et al 1993), similar genes are not strikingly dominant in other series (Hafler et al 1996). Broad usage of the V gene repertoire has also been noted in T-cell line aggregates sorted for recognition of individual peptide epitopes and/or class II restricting elements (such as DR2 related molecules; Hafler et al 1996; Meinl et al 1993). In a few individuals, several independently isolated T-cell lines use the same receptor – based on identical complementarity-determining region 3 sequences – indicating clonal expansion that appears stable over years (Meinl et al 1993; Wucherpfennig et al 1994b).
Figure 11.12.
T-cell receptor genes used by human myelin basic protein-specific T-cell lines. (A) Analysis of Vβ family usage in human T-cell clones reactive to various myelin basic protein epitopes. Human myelin basic protein-reactive clones were grouped according to antigen fine specificity into three general categories: ‘80–105’ includes clones reactive to 80–99, 80–105, 84–102, and 86–105; ‘139–168’ includes clones reactive to 139–153, 143–168 and 152–162; ‘other’ includes clones reactive to all other epitopes. The number of clones in each category is indicated. (B) Analysis of Vα family usage in human T-cell clones reactive to various myelin basic protein epitopes. (C) Analysis of Jβ family usage in human T-cell clones reactive to various myelin basic protein epitopes. All three results show that there are no significant differences between groups.
Adapted from Hafler et al (1996). © 1996, with permission from Elsevier.
Are the few putatively expanded T-cell clones related to the pathogenesis of multiple sclerosis? How might they change during clinical relapse? Would they expand further or, conversely, be eliminated? Would their elimination by therapeutic manipulation (T-cell receptor peptide therapies) demonstrably affect the clinical course of multiple sclerosis? These questions all remain to be answered, but the available results do not seem sufficiently coherent to advance strong guidelines for T cell-specific therapies. Antigen specificity of the T-cell receptor is mediated by the hypervariable complementarity-determining region 3 segment, which contacts antigenic peptide embedded within the central groove of the MHC product (Y. Li et al 2000; 2001; K. J. Smith et al 1998). The complementarity-determining region 3 sequence thus strongly determines peptide specificity of a T lymphocyte. Are there complementarity-determining region 3 sequence motifs related to recognition of myelin basic protein fragments? There is indirect evidence for structural sequence-specific constraints. One example is provided by two T-cell lines isolated from different individuals (one with multiple sclerosis and one healthy), each displaying the same unusual fine specificity. Both lines recognized the myelin basic protein peptide (p139–153) when presented either in the context of DR2 (DRB1*1501) or DR1 (DRB1*0101) using β chains identical at the protein, though not at the mRNA, level (Giegerich et al 1992). Obviously, it is tempting to relate the fine specificity of these cell lines to their T-cell receptor β-chain sequence and, more specifically, to the identical complementarity-determining region 3 sequence region.
Unfortunately, the search for sequence motifs predicting myelin basic protein epitope specificity is not yet conclusive. In one intriguing study, a T-cell receptor sequence (Vβ5.2-LRGA) amplified from post-mortem brain tissue had been previously found in a myelin basic protein-specific T-cell line from a different individual with multiple sclerosis (Oksenberg et al 1993). However, this identity may be an exceptional phenomenon. Most myelin-related motifs were identified by their similarity to complementarity-determining region 3 sequences from Lewis rat T cells, which recognize myelin basic protein peptides embedded in rat, not human, MHC class II proteins. Interestingly, myelin-related sequence motifs were noted in a panel of T cells selected from the blood of patients with multiple sclerosis for somatic mutation in the hprt (hypoxanthine guanine phosphoribosyltransferase) gene, a marker of extensive prior cell proliferation (see below; Allegretta et al 1990; Lodge et al 1995). This would indeed possibly relate myelin basic protein reactivity of CD4+ T cells to ongoing clonal expansion. On the other hand, at least two of the putative myelin basic protein-related complementarity-determining region 3 motifs (LRG, LGG) were also seen in a study of T-cell receptors from peripheral blood-isolated CD8+ T lymphocytes of patients with multiple sclerosis (Monteiro et al 1996). Several studies have been performed of T-cell receptor repertoires selected in response to antigens other than myelin basic protein. In the case of anti-proteolipid protein, one study observed a broad V gene utilization (Correale et al 1995b), whereas a more restricted, epitope-related pattern was noted in another ethnically distinct population (Kondo et al 1996). Also the myelin basic protein-specific T-cell repertoire seems to use a broad spectrum of T-cell receptor V genes (Lindert et al 1999; Van der Aa et al 2003).
Over the past few years, new molecular approaches have evolved that are poised to illuminate the immune pathogenesis of multiple sclerosis. ‘Humanized’ transgenic mice have been constructed, with transgene inserts encoding key elements of the human myelin basic protein-specific T-cell recognition machinery – the two paired T-cell receptor chains, the peptide-presenting MHC class II protein (DR2), and the human coreceptor molecule CD4 (Madsen et al 1999). The behavioural properties of these triple transgene mice mimic transgenic mice with a murine anti-myelin basic protein T-cell receptor. About 5% of otherwise normal mice expressing this T-cell receptor develop spontaneous experimental autoimmune encephalomyelitis. In contrast, virtually all T-cell receptor transgenic mice bred on an immunodeficient, RAG knockout background become spontaneously unwell. Presumably, these lack the regulatory cells that prevent most autoreactive T cells from mounting autoimmune injury in wild-type animals.
‘Spectratyping’ – a polymerase chain reaction-based technique – allows the identification of individual T-cell clone expansions within immune populations, and permits the fate of such clones to be followed in individual patients over time. The method makes use of the highly variable lengths of MHC/peptide-recognizing T-cell receptor-αβ complementarity-determining region 3 regions, selecting individual sequences, depicting the relevant transcripts as individual peaks within the overall spectrum, and mapping these sequentially (Pannetier et al 1995). Musette et al (1996) first applied spectratyping to the study of myelin basic protein-specific T lymphocytes in patients with multiple sclerosis. They found complementarity-determining region 3 sequences suggestive of myelin basic protein specificity, and T cells bearing such receptors were expanded in some patients but not in matched controls. Spectratyping also documented the unusual longevity of myelin basic protein-specific T cells in patients with multiple sclerosis for up to seven years (Goebels et al 2000). Expanded cells were present both in peripheral blood and cerebrospinal fluid (Matsumoto et al 2003).
Very recently, a new tool became available to analyse T-cell function in fixed tissue preparations. ‘Laser microdissection’ allows the excision of single T cells from the lesions of multiple sclerosis. DNA segments and mRNA encoding both T-cell receptor chains of such individual cells can be amplified by combining complex elaborations of the polymerase chain reaction. The receptor genes can then be introduced into cell lines or even transgenic mice, thus making them accessible to study of their specificity and function. This approach literally permits the restoring to life of dead cells. To date, this has not only been restricted to the T-cell receptor β chain (Babbe et al 2000), but to the isolation of complete paired T-cell receptors of CD8 T cells infiltrating human multiple sclerosis brain tissue (Dornmair et al 2003).
Cytokine patterns of encephalitogenic T cells
Soon after their first description, encephalitogenic T lymphocytes sprung something of a surprise. Although clearly members of the CD4+ helper T-cell subset, myelin basic protein-specific T-cell lines were shown to behave like CD8+ cytotoxic killer lymphocytes. Encephalitogenic T cells effectively destroyed any cell presenting relevant myelin basic protein epitope in a recognizable fashion. Presentation was antigen dose-dependent, and restricted by the MHC class II product, RT1.B1 (D. Sun and Wekerle 1986). This finding collided with the ruling dogma, but was seen with great regularity in most models of experimental autoimmune encephalomyelitis. Now, it is clear that classic encephalitogenic T cells are not only cytotoxic but also respond to activation by secreting IL-2 and IFN-γ (the Th1 response: Ando et al 1989).
It appears that the Th1-like cytokine pattern of encephalitogenic T cells is not merely associated with their pathogenic potential but may actually help generate the lesions (Table 11.2 ). Many current therapies for T-cell autoimmune disease are thus aimed at redirecting the Th1 cytokine production of autoreactive T cells to a Th2 profile. This can be achieved by changing the conditions of immune stimulation, presenting peptide variants rather than whole protein (Nicholson et al 1995), blocking costimulatory molecules (Khoury et al 1995), or changing modes of autoantigen administration through oral tolerance induction (Benson et al 2000; Y. Chen et al 1994). The nature of antigenic peptide presented by a suitable MHC protein determines polarity of the Th1 or Th2 immune response. This applies particularly to myelin protein fragments and potentially encephalitogenic T cells – a relationship that offers real therapeutic opportunities. Indeed, in experimental autoimmune encephalomyelitis models, sequence variants of encephalitogenic peptides have been successfully used to divert a pathogenic Th1 response to a protective Th2 orientation. Careful study of peptide interactions with MHC class II and the T-cell receptor surface, in experimental autoimmune encephalomyelitis induced by immunization against proteolipid protein peptide 139–151 in SJL mice, led to the discovery of an altered peptide ligand that is not only unable to induce experimental autoimmune encephalomyelitis but actively protects from experimental autoimmune encephalomyelitis induction. The altered peptide ligand acts by activating proteolipid protein-specific T cells to produce Th2, instead of Th1, cytokines (Nicholson et al 1995). A number of studies have confirmed the protective effects of altered peptides, including natural ligands (Ruiz et al 1999).
Table 11.2.
Mediators associated with experimental autoimmune encephalomyelitis
| Nature of associationa |
|||||
|---|---|---|---|---|---|
| Cytokines | Active disease/relapse (reference*) | Remission/suppression (reference*) | Disease modulated by antibody or inhibitors (reference)* | ||
| IL-1 | + | *1 | − | Yes | *22 |
| IL-2 | + | 2,3 | − | NRb | |
| IL-3 | + | 4 | − | NR | |
| IL-4 | − | + | − | ||
| IL-6 | + | 1 | − | NR | |
| IL-10 | − | + | *1,5 | ||
| IL-12 | + | 5 | − | Yes | 12 |
| IL-13 | − | + | 13 | ||
| IL-18 | + | 6 | − | Yes | 14 |
| IFN-γ | + | 1,3 | +c | Yesd | 3,15 |
| TNF-α | + | 1,5,7 | − | Yes | 16 |
| LT-α | + | 8 | − | Yes | 8 |
| TGF-β | − | + | 17,18 | ||
| IFN-α | ? | + | 19 (directly inhibited EAE) | ||
| Chemokinesd | |||||
| MCP-1 | + | 9 | − | Yes | 20,21 |
| MIP-1α | + | 9,10 | − | Yes | 10,20,21 |
| MIP-1β | + | 9 | − | NR | |
| RANTES | + | 9 | − | No | 20,21 |
| IP-10 | + | 9 | − | Yes | 20 |
| C10 | + | 11 | − | NR | |
| MIP-2 | − | − | 10,20 | ||
| TCA-3 | +e | 9 | |||
References: 1 = Okuda et al (1995); 2 = Litzenburger et al (1998); 3 = Owens et al (1994); 4 = Campbell et al (1998); 5 = Issazadeh et al (1995); 6 = Jander and Stoll (1998); 8 = Hjelstrom et al (1998); 9 = Stalder et al (1998); 10 = Karpus (1995); 11 = Bruce et al (1996); 12 = Leonard et al (1995); 13 = Cash et al (1994); 14 = Wildbaum et al (1998); 15 = Popko et al (1997); 16 = Selmaj and Raine (1995); 17 = Johns and Sriram (1993); 18 = Racke et al (1991); 19 = Billian et al (1988); 20 = Ransohoff (1999); 21 = Youssef et al (1998); 22 = Martin and Near (1995).
References cited are as far as possible those that identified the mediators in question in the CNS, at or near sites of pathology, or reviews that summarize those data.
NR: No reports.
The viewpoint that IFN-γ suppresses experimental allergic encephalomyelitis is not consistent with recent data, but was a logical interpretation of antibody and knockout experiments in their time.
The literature on this topic is broad, so only a few reviews have been cited.
Association in this case was on the basis of expression by encephalitogenic T cells.
Reproduced fromT. Owens et al (2001).
© 2006
Needless to say, altered peptide ligand approaches have been translated into therapies for multiple sclerosis (see Chapter 18). Thus, a phase II trial of peptide 83–93, an analogue of myelin basic protein, showed positive effects in some patients (L. Kappos et al 2000), whereas other patients experienced exacerbations or hypersensitivity responses (Bielekova et al 2000). Clearly the best-known therapy based on altered peptide ligand peptide logic is glatiramer acetate or Copaxone, which behaves like a myelin basic protein altered peptide ligand, activating Th2 cells that may neutralize the activity of pathogenic myelin-specific Th1 lymphocytes (Duda et al 2000; Gran et al 2000b; Neuhaus et al 2000).
Other therapies aiming to drive Th1 to Th2 immune diversion include oral tolerization strategies (Bitar and Whitacre 1988; Higgins and Weiner 1988; Weiner 2000; Weiner et al 1993b). Oral application of myelin basic protein protects rats from subsequent attempts to induce experimental autoimmune encephalomyelitis. The mechanisms underlying oral tolerance induction include (in theory) deletion, anergy, regulation and activation of autoimmune T cells that secrete anti-inflammatory cytokines (IL-4, IL-10 and TGF-β) and therefore suppress the autoaggressive potential of Th1 effector T cells.
Maintenance and breakage of T-cell self-tolerance
It is difficult to understand in detail how the ability of autoreactive T cells to cause autoimmune disease is held in check so reliably. A number of distinct factors must cooperate normally to guarantee immunological health, and, conversely, more than one trigger is required for the development of a clinical autoimmune reaction. One essential prerequisite is activation of autoimmune T cells. Studies of experimental autoimmune encephalomyelitis proved that myelin basic protein-specific T cells only mediate autoimmune central nervous system inflammation after fresh activation. Resting cells neither reach nor attack their target tissue. However, lack of activation is probably not the only mechanism maintaining tolerance. There is additional, indirect evidence for active control through positive regulatory intercellular signals. For example, in rodent experimental autoimmune encephalomyelitis removal of CD8+ T cells from mice by monoclonal antibody depletion (H. Jiang et al 1992) increases the incidence of spontaneous relapse, but does not interfere with recovery from the initial disease episode. Likewise, in transgenic mice lacking CD8+ T cells, relapses in experimental autoimmune encephalomyelitis are more frequent than in control mice (Koh et al 1992). Also, relapses can be prompted by discontinuing immunosuppression previously achieved with ciclosporin A, which may result in an imbalance of immune regulatory circuits (Polman et al 1988). Formal demonstration of central nervous system-specific suppressor T-cell circuits has not been reported. Finally, spontaneous experimental autoimmune encephalomyelitis is rare in transgenic mice with myelin-specific T-cell receptors but otherwise intact immune systems; however, it happens regularly in mice having an immunodeficient genetic background.
We have to consider several mechanisms that could lead to autoimmune reactions. First, pathological mutation might lead to the appearance of forbidden lymphocyte clones in the immune repertoire. This is an improbable mechanism for T cells not using somatic mutation to shape their antigen receptors. A second possibility is the sudden gain of access to previously sequestered antigen. This can also be relegated to unimportant since most putatively sequestered antigens, including those encased within the central nervous system, turn out to be quite accessible. Accidental exposure, for example following central nervous system trauma, does not often result in autoimmune responses. More probable mechanisms revolve around activation, or derepression of self-reactive T cells. For example, microbial ‘superantigens’ have been shown semispecifically to activate relapses of experimental autoimmune encephalomyelitis (Brocke et al 1993; Schiffenbauer et al 1993). Next is antigenic mimicry of microbial structures sharing structural motifs with encephalitogenic proteins, thus engaging and activating the T-cell receptor. Fujinami and Oldstone (1985) showed that hepatitis virus polymerase shares the aa sequence with myelin basic protein and immunization with virus peptide caused experimental autoimmune encephalomyelitis in some rabbits. Finally, local microenvironments can be intensely stimulatory, using their cytokine content rather than specific cognate interactions to activate autoimmune T cells (Segal et al 1997).
Molecular mimicry
George Snell, the pioneer of MHC research and winner of the 1980 Nobel Prize for Physiology or Medicine, coined the concept of molecular mimicry. He reasoned that certain (viral) agents persist because they produce antigenic structures indistinguishable from self determinants and are thus invisible to immune cells. But what would happen if microbial antigen that mimicked self, activated potentially autoreactive immune cells? This scenario has long preoccupied immunologists. Originally, the search concentrated on contiguous peptide sequences shared between microbial and brain proteins (Jahnke et al 1985). Indeed, in their classic and often quoted study, Fujinami and Oldstone (1985) identified a peptide of hepatitis B virus polymerase sharing a contiguous sequence of six amino acids with myelin basic protein p69–75. Immunization of seven (outbred) rabbits against the viral peptide resulted in measurable anti-myelin basic protein antibody titres. Some animals had lymphocyte responses both to virus and myelin, and perivascular central nervous system infiltrates were observed at autopsy.
With improved understanding of antigen processing and presentation, it became clear that molecular mimicry at the T-cell level does not depend strictly and exclusively on the segregated identity of these two individual proteins. First, much rests on intracellular processing of (auto)antigenic proteins by the antigen-presenting cell. Intracellular proteases determine which particular peptides are excised from the parent protein and, among these, only those preferentially binding MHC products are selected for actual presentation on the membrane (Früh and Yang 1999; Geuze 1998; Villadangos and Ploegh 2000).
Sharing of MHC-binding motifs, and of structural profiles recognized by the T-cell receptor, is the basis for cross-reactivity. Thus, one investigation of synthetic peptide variants found that among the encephalitogenic peptides for PL/J mice (Ac1–11) only four amino acid positions are crucial for activation of encephalitogenic T cells, and with only two in continuity (Gautam et al 1994). Obviously, a multitude of microbial peptides could in theory fulfil these loose criteria but, as shown by other work, few qualify for a cross-stimulating mimicry reaction (Wucherpfennig 2001). Thus, mere structural resemblance is not sufficient for a peptide to qualify as a molecular agonist in central nervous system autoimmunity. Ideally, for this to be established, evidence that a given peptide will initiate experimental autoimmune encephalomyelitis (Gautam et al 1998), cross-prime an animal but fail directly to induce (Carrizosa et al 1998), or even protect against (Ruiz et al 1999), experimental autoimmune encephalomyelitis induction is required from direct testing in experimental models.
Two intriguing potential examples of molecular mimicry are mentioned here but also discussed in Chapters 2 and 5. Lang et al (2002) examined a cloned T-cell receptor previously considered simply to cross-recognize myelin basic protein (p85–99) and Epstein–Barr virus polymerase (p627–641) presented by the same human class II protein, DR2. However, the cross-reactive T-cell receptor turned out to recognize the two distinct peptides presented in separate molecular contexts – although each formed part of the DR2 haplotype – but nevertheless forming an antigenic surface complex that this T-cell receptor could not distinguish. Preliminary evidence from serology and cerebrospinal fluid suggested more often exposure to Chlamydia pneumoniae in people with multiple sclerosis compared with controls (Sriram et al 1999; S. Yao et al 2001), but this anti-bacterial response has been attributed to nonspecific polyclonal B-cell activation (Derfuss et al 2001; Gieffers et al 2001). Yet, Chlamydia pneumoniae does seem to affect anti-myelin autoimmune responses in rodent experimental autoimmune encephalomyelitis models. In the Lewis rat, immunization against a chlamydial peptide, which shares seven amino acid positions with a dominant myelin basic protein epitope, resulted in severe experimental autoimmune encephalomyelitis, suggesting a molecular mimicry response. Further, injection of living Chlamydia pneumoniae (but not other chlamydial organisms) into an autoimmunized mouse worsened the developing experimental autoimmune encephalomyelitis (Du et al 2002). And a crystallographic study of myelin-associated glycoprotein established structural similarity with a chlamydial protein of unknown function (Breithaupt et al 2003).
Finally, it should be pointed out that molecular mimicry is not limited to microbial antigens and autoantigens. In an intriguing set of studies, mimicry between a myelin autoantigen and nutritional milk component was found in the DA rat. Myelin-associated glycoprotein-specific encephalitogenic T cells recognized a peptide derived from butyrophilin, a component of cow's milk. Further immunization of these rats with butyrophilin led to mild experimental autoimmune encephalomyelitis, establishing symmetrical cross-reactivity between myelin and milk proteins (Stefferl et al 2000). Bovine butyrophilin, a member of a family of proteins that interestingly includes B7 costimulatory molecules (Tazi-Ahnini et al 1997), shares 50% sequence homology with the mouse myelin-associated glycoprotein extracellular domain (Gardinier et al 1992). One gene cluster for butyrophilin is located very close to MHC class I and class II regions in human and mouse (Stammers et al 2000). Whilst the concept of mimicry between myelin-associated glycoprotein and butyrophilin remains unproven, an increased frequency of butyrophilin-binding antibodies is described in patients with multiple sclerosis (De March et al 2003).
Microbial superantigens
Microbial products have evolved a second, semispecific means of activating autoreactive T cells. Microbial superantigen can activate central nervous system reactive T lymphocytes and thus initiate experimental autoimmune encephalomyelitis without structurally mimicking encephalitogenic peptide epitopes. Like specific peptide antigens, microbial superantigens activate T cells via the T-cell receptor and associated signal transduction pathways, and they also only activate T cells when complexed with MHC class II products. However, in contrast to antigenic peptides, the superantigens do not react with the hypervariable complementarity-determining region 3 portion of the receptor (which mediates peptide specificity) but bind to relatively constant determinants typical for Vβ segments of the receptor (Herman et al 1991). Superantigens thus do not activate T cells in a clonally specific fashion but activate larger subsets of T-cell clones – those populations carrying the same relevant Vβ determinant.
Activation of encephalitogenic T cells by bacterial superantigen was first shown in culture through triggering of Lewis rat myelin basic protein-specific Vβ8.2 T cells by the staphylococcal enterotoxin E, which also binds Vβ8.2 (Rott et al 1992) or enterotoxin D (Matsumoto and Fujiwara 1993). In vivo, the superantigen paradigm was validated by treatment of myelin basic protein-primed mice with staphylococcal enterotoxin B, another classic bacterial superantigen activating murine T-cell receptor containing Vβ8.2 determinants. Mice, recovered from experimental autoimmune encephalomyelitis mediated by Vβ8.2+ T cells, were then exposed to staphylococcal enterotoxin B and underwent a new relapse. Similar treatment of unprimed mice did not trigger experimental autoimmune encephalomyelitis (Brocke et al 1993; Schiffenbauer et al 1993). Dormant, potentially encephalitogenic T cells must have been reactivated by the semispecific bacterial proteins. But, as discussed in Chapter 12, additional effects presumably contribute to the encephalitogenic activity of superantigens. By activating large populations of T cells, substantial doses of proinflammatory cytokines are released (Miethke et al 1992), which may activate cerebral endothelial cells and thus contribute to enhanced formation of brain infiltrates. Human endogenous retroviruses have been isolated from central nervous system tissues of people with multiple sclerosis (Perron et al 1997: see Chapter 2). Under certain conditions, these may produce superantigens, which in turn could activate autoimmune T lymphocytes (Sutkowski et al 2001). Their specific role in multiple sclerosis is disputed and awaits definitive validation (Brahic and Bureau 1997).
Bystander activation in inflammatory milieus: the link to innate immunity
Models of chronic virus infection exist that do not seem to involve the recruitment and activation of myelin-specific encephalitogenic T cells in response to molecular mimicry and superantigen responses. Coronavirus infection of Lewis rat brain, for example, may take a subacute course with round cell infiltration of central white matter (Watanabe et al 1983). Myelin basic protein (but not coronavirus virus)-specific CD4+ T-cell lines isolated from infected donor rats mediate comparable disease. Mechanisms underlying the apparent activation of encephalitogenic T cells in the course of a slow virus infection of the brain remain to be elucidated. In another demyelinating central nervous system disease model, Theiler virus-induced encephalomyelitis, a chronic autoimmune response follows an acute phase of inflammation. While virus-specific T cells control the initial phase, the inflammatory response is subsequently driven by myelin-specific T cells (S.D. Miller et al 1995a). Release of brain autoantigen in concert with locally produced proinflammatory cytokines may be the key factor in virus-associated autoimmune encephalitis (Vanderlugt and Miller 2002).
The discovery of links between innate and adaptive immunity add a new component to our understanding of the role played by local tissue milieus in development and course of autoimmune disease. By activating mechanisms of innate immunity, microbial infections may create a local environment that is particularly supportive of T-cell activation (Bachmann and Kopf 2001). We have already described activation of innate immune mechanisms by the binding of microbial structures to Toll-like and other pattern receptors. Activation of dendritic cells by bacterial lipopolysaccharide, double-stranded RNA, CpG DNA or heat-shock proteins binding to Toll-like receptor induces costimulatory antigens in dendritic cells, and triggers secretion of proinflammatory cytokines including IL-12 (Reis e Sousa 2001). Alternatively, recent investigations suggest that the triggering of innate immune responses by engagement of Toll-like receptor may result in the neutralization of CD4+CD25+ regulatory T cells (Treg cells). The process apparently does not act via costimulatory structures and antigen presentation, but involves signalling through soluble IL-6. Functional inactivation of the regulatory T cells allows suppressed autoimmune T cells to become activated (Pasare and Medzhitov 2003). Radically different is the result of signalling via Toll-like receptor expressed on the Treg cells themselves where, rather than being blocked, the suppressive activity is enhanced (Caramalho et al 2003).
Taken together, the present evidence suggests that innate antibacterial immune responses promote the development of autoimmunity by creating a proinflammatory milieu, which strongly enhances the presentation of foreign or self antigen to T cells, or even facilitates ‘bystander’ activation of local T cells independent of antigen.
T cells carrying two sets of receptors
Both T and B lymphocytes normally only use genes from one of the two available alleles to code their antigen receptor, a phenomenon termed allelic exclusion (Nemazee 2000). This rule is firm but not unfailing. Violation of allelic exclusion, the expression of two types of receptor, was first described for T-cell receptor α chains in human (Padovan et al 1993) and transgenic mouse T cells (Heath and Miller 1993). Later, double expression of β chains was also reported (Balomenos et al 1995; Davodeau et al 1995; Padovan et al 1995). The theoretical implications of double T-cell receptor expression for the initiation of autoimmune disease are considerable. This might give rise to T-cell clones coexpressing self- and non-self-reactive T-cell receptors. Immunogenic ligation of non-self T-cell receptor by the appropriate foreign (microbial) antigen would activate the T cell and, at the same time, animate its autoimmune potential – defined and executed by the second autoreactive receptor. This mode of autoimmune activation is no doubt intellectually attractive, but so far unproven in clinical practice. The available experimental data are also not strongly supportive. Studies of double transgenic mice with two distinct T-cell receptor transgenes did identify a minor population of T cells with two T-cell receptors, one recognizing self and the other foreign antigen. In these dual receptor-expressing T cells, binding of a foreign ligand resulted in silencing of the cell, instead of activating its pathogenic potential (Dittel et al 1999; Fossati et al 1999). Activation of the autoimmune potential by recognition of microbial antigen remains to be demonstrated.
B-cell tolerance and autoimmunity
Thanks to B-cell tolerance, the production of pathogenic autoantibodies is prevented or suppressed, although our body fluids contain plenty of immunoglobulin binding harmlessly to self structures. Much knowledge of B-cell self-tolerance comes from transgenic mouse models. These animals have B cells, most of which use the transgenic autoantibodies as surface receptors. Autoreactive immunoglobulin production is restricted to the B lymphocyte subclass B-1. In normal animals, this population commonly makes polyreactive autoantibodies that are not autoaggressive. Release of pathogenic autoantibodies is triggered by microbial activation (Murakami and Honjo 1995). In classic B cells, however, self-tolerance is obligatory. It is effected by several mechanisms, and these are not necessarily mutually exclusive. Depending on the concentration of soluble self antigen, autoreactive B lymphocytes may either be physically eliminated from the immune repertoire by apoptosis or silenced to a state of profound nonreactivity or anergy (Goodnow 1992). Other B cells respond to the encounter of self antigen with replacement of their original, self-reactive immunoglobulin receptor by a newly arranged, non-self-reactive immunoglobulin chain – a mechanism termed immunoglobulin ‘editing’ (Radic and Zouali 1996). Receptor editing occurs in a late stage of B-cell maturation (Chen et al 1995b; Melamed and Nemazee 1997; Pelanda et al 1997), and may involve either the immunoglobulin H (C. Chen et al 1995a) or L chain (Brard et al 1999; Casellas et al 2001; H. Li et al 2001). Autoimmune prone strains, such as MRL-lpr/lpr mice, seem to have an editing machinery insufficient to delete autoimmune B-cell clones, and even allow their functional maturation by somatic mutation (Brard et al 1999; Pelanda et al 1997).
Autoimmune B lymphocytes in experimental autoimmune encephalomyelitis
So far, it has become clear that autoimmune T cells, especially CD4 T cells, are the pathogenic effectors in experimental autoimmune encephalomyelitis. They determine the location, activity and functional character of the central nervous system lesion. But this by no means excludes B cells from central nervous system autoimmune diseases. On the contrary, we shall see that B cells have essential functions in several stages of the unfolding experimental autoimmune encephalomyelitis lesion. Central nervous system autoantigen-specific B cells may help activate T lymphocytes as professional antigen-presenting cells, able to concentrate autoantigen due to their immunoglobulin surface receptors. Further, with their particular set of secreted cytokines, they are able to push antigen-recognizing CD4 T cells into particular functional pathways, usually along the Th2 pathway. Finally, binding of B-cell secreted anti-myelin autoantibodies to exposed surface structures of the myelin sheath or the myelin-forming oligodendrocyte is definitely one paramount mechanism of autoimmune demyelination.
Anti-myelin autoantibodies
Early studies on experimental autoimmune encephalomyelitis revealed that sera from affected animals may demyelinate organotypic central nervous system tissue cultures (Bornstein and Appel 1961). A similar demyelinating effect was later observed in vivo after injection of such sera into the central nervous system parenchyma or cerebrospinal fluid (Lassmann et al 1981b). The demyelinating factor turned out to be immunoglobulin – both IgG and IgM – and demyelination was mediated either via complement activation alone or in combination with activated macrophages. These observations suggest that antibodies against myelin can, at least in part, be responsible for demyelination in models of acute and chronic experimental autoimmune encephalomyelitis. This is further substantiated by studies of chronic experimental autoimmune encephalomyelitis, which show good correlation between the antibody response and amount of demyelination (Linington and Lassmann 1987).
Characterization of the fine specificity of demyelinating sera from experimental autoimmune encephalomyelitis animals first suggested that antibodies against certain glycolipids, such as galactocerebroside, are responsible for the demyelinating effect (Dubois-Dalcq et al 1970). However, the correlation between anti-glycolipid antibody titres and demyelinating activity of respective sera is generally poor. Much better correlation is found with antibody titres against myelin oligodendrocyte glycoprotein (Linington and Lassmann 1987). To date, no other myelin protein has been identified that serves as a target of demyelinating antibodies. The pathogenic role of demyelinating antibodies in vivo has formally been proven by the intravenous transfer of anti-myelin oligodendrocyte glycoprotein antibodies to animals in whom T-cell-mediated experimental autoimmune encephalomyelitis had already been induced (Figure 11.13 : Linington et al 1988; Schlüsener et al 1987). In this disease paradigm, the encephalitogenic T cells are required to prime the central nervous system parenchyma and open the blood–brain barrier to allow myelin oligodendrocyte glycoprotein-reactive monoclonal antibodies to enter the central nervous system. The antibodies cause demyelination, probably with the help of accessory macrophages and/or complement (Linington et al 1989; Piddlesden et al 1993). Rat experimental autoimmune encephalomyelitis mediated by double transfer of encephalitogenic T cells plus myelin-specific autoantibodies is certainly quite an artificial model of autoimmunity in the central nervous system. However, very similar inflammatory and demyelinating lesions have been created by very simple immunization with recombinant or myelin-derived myelin oligodendrocyte glycoprotein in adjuvant in rats (Johns et al 1995; Storch et al 1998b; Figures 11.14 and 11.15 ), mice (Amor et al 1994) and primates (Genain et al 1996). However, not all anti-myelin oligodendrocyte glycoprotein autoantibodies are demyelinating: only those recognizing a conformational epitope on myelin oligodendrocyte glycoprotein, but not linear peptide epitopes, are able to cause demyelination in vivo (Brehm et al 1999; von Büdingen et al 2002). At least in the mouse, production of anti-conformational autoantibodies seems to be controlled genetically by loci close to the MHC (Bourquin et al 2003).
Figure 11.13.

Demyelination in T-cell-mediated experimental autoimmune encephalomyelitis is augmented by the presence of demyelinating anti-myelin basic protein antibodies. (A) Injection of anti-myelin oligodendrocyte glycoprotein antibodies 3 days after transfer of myelin basic protein-reactive T cells; perivenous inflammation is associated with extensive perivascular loss of myelin; Klüver/PAS myelin stain; ×50. (B) Repeated cotransfer of myelin basic protein-reactive T cells and anti-myelin oligodendrocyte glycoprotein antibodies leads to confluent demyelinated plaques with loss of oligodendrocytes; myelin is stained by immunocytochemistry for proteolipid protein (blue) and oligodendrocytes by in situ hybridization for proteolipid protein mRNA (black cells); ×50. (C) Transfer of T lymphocytes in the absence of anti-myelin oligodendrocyte glycoprotein antibodies leads to perivenous inflammation without demyelination; Klüver/PAS myelin stain; ×300. (D) Extensive perivenous demyelination after cotransfer of myelin basic protein-reactive T cells and anti-myelin oligodendrocyte glycoprotein antibodies; Klüver/PAS myelin stain; ×500. (E) Higher magnification (×250) of the plaque in (B) with demyelination and oligodendrocyte loss; double staining for proteolipid protein immunocytochemistry and in situ hybridization.
Figure 11.14.

Patterns of demyelination in chronic experimental autoimmune encephalomyelitis in a DA rat induced by active sensitization with myelin oligodendrocyte glycoprotein. (A) Large inflammatory demyelinating lesion affecting most of the cerebellar white matter (hypercellular lesion); haematoxylin/eosin; ×10. (B) Extensive loss of myelin; Klüver/PAS myelin stain; ×10. (C) Topography of demyelinated lesions in the central nervous system of this DA rat; only two lesions are found – one in the cerebellar white matter, the other in the optic nerve; ×500. (D) Patterns of oligodendrocyte loss in demyelinating cerebellar lesion; periplaque white matter with normal density of oligodendrocytes; ×500. (E) Actively demyelinating area with granular myelin degradation products in macrophages (red granules) and nearly complete loss of proteolipid protein mRNA-reactive oligodendrocytes; ×500. (F) Inactive centre of the lesion with demyelination and a large number of proteolipid protein mRNA-positive oligodendrocytes, possibly recruited from the pool of progenitor cells; immunocytochemistry for proteolipid protein (red) and in situ hybridization for proteolipid protein mRNA (black cells); ×500. (G) Deposition of complement C9 in the demyelinating lesion; fibrillar staining of myelinated fibres at the actively demyelinating edge of the lesion; ×500. (H) Granular C9-reactive degradation products in macrophages; immunocytochemistry for C9; ×500.
Figure 11.15.

Remyelinating ‘shadow plaque’ in the optic chiasm of the same animal shown in Figure 11.13. (A) Increased cell density in the lesion occupying the left side of the optic chiasm; haematoxylin & eosin; ×45. (B) Reduced density of myelin but increased numbers of oligodendrocytes in the lesion; immunocytochemistry for proteolipid protein (red) and in situ hybridization for proteolipid protein mRNA (black cells); ×45. (C) Reduced axonal density in the affected portion of the optic chiasm; Bielschowsky silver impregnation for axons; ×45. (D) Reduced density of myelin in the affected portion of the optic chiasm (left half of the micrograph); Klüver/PAS myelin stain; ×300. (E) Higher magnification of (C) showing the reduced axonal density; Bielschowsky silver impregnation for axons; ×300.
Transgenic models of central nervous system-specific B cells
Transgenic mice with T-cell receptors for encephalitogenic autoantigen have provided models for understanding myelin self-recognition and autoimmunity. The same can be expected from transgenic mice with myelin autoimmune B lymphocytes (Litzenburger et al 1998). In contrast to T-cell receptor transgenics, these were gene replacement (‘knock-in’) mutant mice. Instead of randomly inserting the transgene into germline DNA, the germline J(H) locus was excised and replaced by the rearranged immunoglobulin H chain V gene of a pathogenic myelin oligodendrocyte glycoprotein-specific monoclonal antibody. It should be noted that expression of the transgenic H chain, in the absence of the paired transgenic L chain, was sufficient to produce a large proportion of myelin oligodendrocyte glycoprotein-binding B cells (>30% of all B cells in the repertoire) and high titres of myelin oligodendrocyte glycoprotein-binding serum immunoglobulins. Further, due to correct insertion of the H chain transgene, all essential features of immunoglobulin affinity maturation and isotype switching were recapitulated in transgenic B cells.
Study of the anti-myelin oligodendrocyte glycoprotein H chain knock-in mouse showed that the mere presence of myelin autoreactive B cells does not cause spontaneous experimental autoimmune encephalomyelitis or demyelination. However, the same transgenic B cells and their autoantibody products became highly pathogenic in cooperation with transferred encephalitogenic T cells (Litzenburger et al 1998). The autoreactive B cells persisted throughout life in the transgenic mouse, and in high numbers. In contrast, B-cell differentiation was blocked in the bone marrow at a late pre-B-cell stage in double transgenic mice expressing both the myelin oligodendrocyte glycoprotein-reactive H and L chain. B cells underwent receptor editing, with replacement of the transgenic L chain by an endogenous L chain freshly created by renewed rearrangement (Litzenburger et al 2000). Clearly, it will be instructive to investigate transgenic mice with a myelin oligodendrocyte glycoprotein-biased immune repertoire, both at the T- and B-cell levels.
Autoimmune B cells in multiple sclerosis
In human multiple sclerosis an active participation of B cells in the immune pathogenesis has been postulated for many years. In fact, as will be detailed later, abnormal oligoclonal immunoglobulins in the cerebrospinal fluid (CSF), a fluid that normally contains no demonstrable immunoglobulins at all, is a diagnostic hallmark of multiple sclerosis. The antigen specificity of these immunoglobulins is completely obscure to date; curiously they do not bind any of the myelin candidate autoantigens. Remarkably, however, there is increasing evidence of specific antibody binding within particular types of multiple sclerosis plaques, and it appears plausible but remains yet unproven that such locally bound antibodies are indeed autoimmune and contribute to local myelin destruction.
Anti-myelin autoantibodies
Despite good evidence implicating B lymphocytes in the pathogenesis of multiple sclerosis, their precise role remains to be clarified. It has been known for many years that most, if not all, patients have abnormal oligoclonal immunoglobulin bands, reflecting intrathecal synthesis, in the cerebrospinal fluid. Although not specific, their detection is used as one laboratory criterion supporting the clinical diagnosis of multiple sclerosis. The specificity of the oligoclonal antibodies in cerebrospinal fluid remained enigmatic for many years, and attempts to identify myelin or other central nervous system structures as target antigens met with limited success (Cortese et al 1996). This uncomfortable situation may be changing. Several groups have described autoantibodies against myelin oligodendrocyte glycoprotein in the serum and cerebrospinal fluid of patients with multiple sclerosis (Lindert et al 1999; Reindl et al 1999; Xiao et al 1991). Others dispute this clarification (Karni et al 1999). When tested for the structural characteristics of binding epitopes, most myelin oligodendrocyte glycoprotein-reactive immunoglobulins were found to bind linear, rather than conformational epitopes (Haase et al 2001) – making the point that only autoantibodies directed against conformational determinants have a pathogenic demyelinating potential (see above: Brehm et al 1999; von Büdingen et al 2001). Nevertheless, a recent study correlated serum and cerebrospinal fluid myelin oligodendrocyte glycoprotein antibodies with disease severity (T. Berger et al 2003). An active role for anti-myelin associated glycoprotein autoantibodies in the pathogenesis of multiple sclerosis is further supported by morphological analysis. Certain lesion types are characterized by myelin debris decorated with activated complement (Storch et al 1998a), possibly involving a humoral anti-myelin associated glycoprotein reaction (Genain et al 1999).
B cells in the central nervous system of patients with multiple sclerosis
The inflammatory infiltrates of active lesions commonly contain a relatively minor B lymphocyte or plasma cell infiltrate within a majority of T cells and macrophages (see Chapter 12). The B cells are often within plaque areas, rather than in the peripheral zones (Esiri 1980). It is tempting to assume that plaque-infiltrating B cells participate in inflammatory demyelination, perhaps by secreting anti-myelin oligodendrocyte glycoprotein autoantibodies. However, such a pathogenic function has not been demonstrated to date, perhaps for technical reasons. Early attempts to characterize B cells in tissue from individuals with multiple sclerosis used Elispot methods and B cells harvested from cerebrospinal fluid (Storch et al 1998a). This approach led to the identification of B cells specific for a range of myelin autoantigens, and semiquantification in various compartments at different stages of the disease (Archelos et al 2000). But cellular mechanisms underlying the antibody responses remained obscure.
Molecular approaches, especially those using polymerase chain reaction techniques, now seem poised to help out. The screening of cDNA libraries established from B cells infiltrating tissue favours an antigen-driven immune response. Comparison of antibody H chains documented extensive somatic mutagenesis. Individual sequence could be sorted as pedigrees probably derived from a few progenitor B cells (Owens et al 1998). A very similar observation was made using B cells or short lived plasmablasts (Cepok et al 2005a) from the cerebrospinal fluid of patients with multiple sclerosis. Expansion of a few individual B-cell clones, with strong somatic mutations, was again observed (Y. Qin et al 1998). These original reports were confirmed by more recent studies using immunoglobulin spectratyping (Baranzini et al 1999), and methods of single-cell gene cloning (A. M. Ritchie et al 2004).
REGULATION OF CENTRAL NERVOUS SYSTEM AUTOIMMUNE RESPONSES
We have stressed that the immune system achieves its well-dosaged reactivity against foreign structures by tight intercellular regulation. No individual immune cell, be it a T or a B lymphocyte, acts autonomously, but is subject to a chain of control exerted by neighbouring cells, which can be lymphocytes, macrophages, dendritic cells or components of the surrounding stroma. The principle of cell-to-cell regulation governs especially immunological self-tolerance. A failure of regulation may result in autoimmunity. Investigations into experimental autoimmune encephalomyelitis have revealed how failed immune regulation may cause or support autoimmune reactivity against the central nervous system, and studies of multiple sclerosis patients now confirm such failure in the pathogenesis of human disease. It now appears that an important part of the regulation is effected by check-and-balance interactions between two competing T-cell subsets, the Th1 and Th2 cells. This has been discussed above. In addition, however, there are specialized regulatory T cells that keep effector cells in check. These limit an ongoing immune response to its necessary duration, and help prevent activation of self-reactive T cells to warrant self-tolerance.
Chronic relapsing experimental autoimmune encephalomyelitis: a failure of regulation?
Although the mechanisms that control inflammation in acute monophasic experimental autoimmune encephalomyelitis are tolerably well understood, much less is known about pathogenic factors that operate in the induction of chronic disease or individual relapses. Most animal species and strains are resistant to re-induction of experimental autoimmune encephalomyelitis after a disease episode that follows active sensitization. However, certain mouse strains (SJL/J or Biozzi mice) are more exuberant and react with new inflammatory episodes after each fresh antigenic challenge (Kozlowski et al 1987). In these mice, a spontaneously developing chronic relapsing disease can be induced by single transfer of myelin-reactive T lymphocytes (Mokhtarian et al 1984; Zamvil et al 1985). Furthermore, in contrast to most animals with monophasic disease, in this situation the encephalitogenic reaction shifts from the originally transferred T cells towards an immune response directed against other cryptic determinants of myelin basic protein or even proteolipid protein (A. H. Cross et al 1993; Lehmann et al 1992). This indicates that liberation of antigen at the site of brain inflammation and/or its transport to local lymphatic tissue may in some animal species result in antigenic drift and uncontrolled autoimmunity. The reason for this lack of immune regulation in certain animal species is unknown. Suggestions include increased persistence of autoimmune effector cells in the central nervous system and immune system (Fritz et al 1998), participation of γδ-T-cell receptor cells (Rajan et al 1996) and increased expression of variant golli myelin basic protein in immune organs (MacKenzie-Graham et al 1997).
By active sensitization, chronic experimental autoimmune encephalomyelitis can be induced in a large variety of different animal species. In these models, chronic persistence of local antigen, made available from initial sensitization or repeated challenge, is critical for maintaining chronicity (Rivers et al 1933; Tabira et al 1984). By comparison with the transfer models described above, even less is known about the immunoregulatory events that lead to chronicity and relapse after active sensitization. Although no overt differences between active and inactive phases of the disease are noted in delayed-type skin responses against myelin basic protein (Tabira et al 1983), in one study increased antigen-specific suppression of T-cell activation was found during disease remission (Lyman et al 1981). In terms of the central nervous system lesions, there is little difference in the composition of inflammatory infiltrates between acute and chronic experimental autoimmune encephalomyelitis, or even between relapses and remissions (Figures 11.8, 11.16 and 11.17 ). Whether changes in local cytokine patterns per se can explain disease activity or remission in chronic experimental autoimmune encephalomyelitis models, such as described for acute experimental autoimmune encephalomyelitis (Kennedy et al 1992), remains to be determined.
Figure 11.16.

Composition of inflammatory infiltrates in T-cell-mediated experimental autoimmune encephalomyelitis. (A) Perivascular inflammation and tissue infiltration with T cells; immunocytochemistry with W3/13; ×200. (B) Large numbers of inflammatory cells express a lysosomal macrophage marker; immunostaining with ED1; ×200. (C) Many inflammatory cells (mainly T lymphocytes) show the typical alterations of apoptosis (nuclear condensation, margination of chromatin and nuclear fragmentation); haematoxylin & eosin stain; ×1000.
Figure 11.17.

Inflammatory reaction in chronic demyelinating experimental autoimmune encephalomyelitis. (A) Leucocytes bind to the luminal surface of venules and migrate through the vessel wall; toluidine blue; ×1000. (B) Macrophages with myelin debris are located in the central nervous system parenchyma and perivascular space in actively demyelinating lesions; toluidine blue; ×1000. (C) Inactive lesion macrophages are mainly located in the perivascular Virchow–Robin space; toluidine blue; ×1000. (D) Inactive, remyelinating lesions show pronounced perivascular fibrosis of parenchymal vessels; toluidine blue; ×1000.
Also relevant to the induction of chronic experimental autoimmune encephalomyelitis is the ability of animals to mount a humoral autoimmune response. In mice with identical MHC backgrounds, differences in the incidence of chronic experimental autoimmune encephalomyelitis can be traced to their production of high-affinity antibodies (Devey et al 1990). As discussed below, autoantibodies against antigens exposed on the surface of myelin sheaths play an important role in the induction of demyelination in chronic experimental autoimmune encephalomyelitis models (Linington and Lassmann 1987; Linington et al 1988). Furthermore, subclinical encephalitis induced by suboptimal numbers of encephalitogenic T lymphocytes can become clinically manifest in the presence of such autoantibodies (Lassmann et al 1988).
Regulatory CD8 T lymphocytes
Classic MHC class I restricted CD8+ T cells are commonly equated with cytotoxic killer lymphocytes, which act as effectors in the context of tumour rejection, autoimmune inflammation and viral infection. Indeed, such cells may play an important role in mediating demyelination, axonal injury and global tissue destruction in the lesions of multiple sclerosis (see Chapter 12). Less clear is whether there is another type of CD8+ T cell, with regulatory functions, bringing to a close otherwise ongoing cellular (auto)immune responses. The regulatory function of CD8+ T cells has been revealed in two models of mouse experimental autoimmune encephalomyelitis where, in the absence of functional CD8+ T cells, immunization against myelin basic protein enhances the encephalitogenic response. This has been shown, for example, after ablation of CD8+ T cells by in vivo infusion of monoclonal antibodies directed against CD8. Mice depleted of CD8+ T cells show more severe relapses, but their recovery from the individual attack is unimpaired (H. Jiang et al 1992). Furthermore, transgenic mice lacking CD8+ T cells due to disruption of the CD8 gene also suffer increased severity of relapses, although the disease course in general seems milder (D-R. Koh et al 1992). Recent work indicates that suppressive CD8+ T cells are specific for peptide epitopes from T-cell receptors of CD4+ effector T cells that dominate the encephalitogenic immune response (H. Jiang et al 2001), and recognize these epitopes in the unusual context of Qa antigens – atypical class I-related MHC products (class Ib) (Rodgers and Cook 2005). They thus can be defined as idiotype-specific regulatory cells.
The first reports of putative CD8+ regulatory T cells were based on histological evidence. In the Lewis rat model of experimental autoimmune encephalomyelitis, CD8+ T-cell infiltrates in the spinal cord persisted beyond clinical recovery, indicating a suppressive function for these lymphocytes (Hickey and Gonatas 1984). However, later work showed that depletion of CD8+ T cells from the immune system altered neither the severity nor course of experimental autoimmune encephalomyelitis in the same strain (Sedgwick 1988). Regulatory CD8+ T cells were first isolated from the spleens of Lewis rats that had previously received transfers of syngeneic encephalitic CD4+ T-cell lines. Their transfer efficiently protected naive rats against the induction of experimental autoimmune encephalomyelitis by the relevant CD4+ line (Lider et al 1988; D. Sun et al 1988). The cells are exquisitely specific for the inducing T-cell line and do not cross-react with or affect actively induced experimental autoimmune encephalomyelitis. They neutralize encephalitogenic target T cells in vivo, and specifically lyse these in vitro (D. Sun et al 1988).
Another CD8+ T cell with potential regulatory function is described in studies of oral tolerization against myelin basic protein. Low-dose oral administration of antigen leads to the activation of CD8+ T cells that suppress activation of encephalitogenic CD4+ T lymphocytes in the host immune system (Lider et al 1989) by secreting anti-inflammatory TGF-β (A. Miller et al 1992). Thus far, activation of these T cells depends on pharmacological manipulations, and a physiological role remains to be defined. In a much quoted paper, Reinherz et al (1980) reported a dramatic loss of CD8+ T lymphocytes from peripheral blood in patients with multiple sclerosis. In active disease, CD8+ T cells (determined by cytofluorometry) were reduced to 5% of peripheral T cells compared with normal values of around 20%. Intriguingly, patients in remission showed much less pronounced changes. The interpretation seemed obvious. According to the prevailing view, active disease in multiple sclerosis was associated with, and possibly due to, the loss of CD8+ suppressor T lymphocytes. Indeed, loss of negative immune regulation was corroborated in studies using functional suppressor assays. For example, Antel et al (1979), using a mitogen-driven T-cell suppressor assay, showed loss of suppressor activity during phases of active disease with restoration to normal levels during remission. Considered in their day to be pivotal results, they triggered a plethora of CD8/suppressor T-cell analyses using ever more elaborate (and expensive) panels of monoclonal antibodies as these became available. These accumulated data now seem to indicate much weaker disease-related effects than were initially claimed (Antel et al 1985), and this also holds true for the evidence of an inverse correlation between CD4+/CD8+ ratios and functional suppressor assays (P. J. Hughes et al 1988). Whilst it would be easy to conclude that this was a blind alley in multiple sclerosis research, active participation of regulatory CD8+ T cells in the pathogenesis is by no means ruled out, and the functional significance of the markers used to define lymphocyte phenotypes has since systematically been refined. As suggested by the lessons from experimental autoimmune encephalomyelitis (see above), CD8+ T cells have been observed after vaccination with attenuated myelin basic protein-specific CD4+ T cells (J. Zhang et al 1995). They may participate both in immune regulation and cellular autoimmune attack. This topic should move forward with the ability to isolate CD8+ T lymphocytes specific for myelin basic protein and proteolipid protein peptide determinants in the context of MHC class I molecules from the blood of patients with multiple sclerosis (Biddison et al 1997; Tsuchida et al 1995). New methods, such as recombinant MHC class I proteins complexed with (auto)antigenic peptides, now allow identification of specific CD8+ T cells in blood and tissues. These approaches, together with single cell polymerase chain reaction techniques (Dornmair et al 2003), ought to improve our understanding of multiple sclerosis-related CD8+ T cells, be they regulatory or effector cells (Appay and Rowland-Jones 2002; Skinner and Haase 2002).
Gamma/delta (γδ) T cells
In addition to the majority of T lymphocytes, which use classic antigen receptors of the αβ class, the immune system also harbours T cells with γδ receptors. Both populations are clonally diverse with antigen receptors composed of various gene segments linked by mechanisms of somatic recombination, sharing essential structural features of antigen-binding protein. However, the function(s) of γδ T cells are still largely obscure. They are by no means a homogeneous cell population and contain numerous subpopulations with diverse antigen recognition properties, cytokine spectra and tissue localizations. Curiously, while only few γδ T cells reside in peripheral lymph organs, they preferentially settle within epithelia, especially in the gut. Most γδ T cells lack CD4 and CD8, but some express one or the other of these markers (Hayday 2000).
The antigens recognized by γδ T cells are unusual and also diverse. Different subsets using distinct patterns of γδ receptors recognize small non-peptidic molecules (most prominently phosphorylated antigens from mycobacteria) without the need for a particular antigen-presenting molecule, heat-shock proteins, or more conventional peptides bound to variant MHC class Ib proteins (Allison and Garboczi 2002). This and additional evidence suggests that γδ T cells participate in antimicrobial immune responses, but other functions, such as regulation of immune responses, have also been proposed. Evidence for γδ T cells participating in autoimmune responses, as in the generation of experimental autoimmune encephalomyelitis lesions, remains indirect. The course of experimental autoimmune encephalomyelitis has been related to increased frequencies of γδ T cells in central nervous system infiltrates. Disease activity correlates with their state of activation, especially in chronic relapsing experimental autoimmune encephalomyelitis models. Relatively high frequencies (>10% of total T cells) were noted in active stages, whereas the concentration of γδ T cells was very low during remission. Depletion of γδ T cells by monoclonal antibody treatment increased experimental autoimmune encephalomyelitis susceptibility and severity, establishing the functional relevance of these cells (Rajan et al 1996).
γδ T cells may have an accessory role in experimental autoimmune encephalomyelitis but, unlike their αβ T-cell receptor-bearing counterparts, do not seem to be active mediators of disease. Transgenic mice lacking functional αβ T cells have unimpaired γδ T-cell receptor repertoires (Elliott et al 1996) but are completely resistant to active induction of experimental autoimmune encephalomyelitis. Conversely, transgenic mice lacking T-cell receptor γδ cells are no less susceptible to T-cell-transferred experimental autoimmune encephalomyelitis than their wild-type counterparts (Clark and Lingenheld 1998). Possibly γδ T cells regulate cytokine/chemokine production required for recruiting effector T cells into the central nervous system (Rajan et al 1999). It should also be mentioned that γδ T cells have been implicated only in selective models of experimental autoimmune encephalomyelitis, but seem to have no role in others. For example, their depletion in Lewis rats has no demonstrable effect on the induction of experimental autoimmune encephalomyelitis (Matsumoto et al 1998).
As in experimental autoimmune encephalomyelitis, most neuroimmunological studies of autoreactive T lymphocytes in multiple sclerosis have focused on T cells with αβ receptors, the agents of conventional cellular immune responses. There are, however, good reasons not to neglect γδ receptor T cells in the human disease. Selmaj et al (1991a) first observed notable proportions of γδ cells in the lesions of multiple sclerosis. These infiltrates commonly co-localize with enhanced expression of heat-shock proteins – a point of interest since these protein determinants are specifically recognized by some, but by no means all, γδ T cells (Chien et al 1996). What is the role of γδ T cells in multiple sclerosis? Are they, as we assume to be the case for their myelin-specific T-cell receptor αβ colleagues, primarily involved in the pathogenesis of individual lesions? Do they attack local brain cells? Or could they have a beneficial role, participating in the attempt to limit or restore the autoimmune injury?
To resolve these questions, and confirm or refute the pathogenic significance of γδ T-cell accumulation in multiple sclerosis lesions, and the reactivity of peripheral blood to heat-shock proteins, antigen-specific γδ T-cell lines are needed to facilitate the direct study of their T-cell receptor gene usage, antigen recognition, cytokine repertoire and cytotoxic potential (Pon and Freedman 2003). These reagents have proved more difficult to generate than T-cell receptor αβ-expressing cell lines. To obtain insights into the gene repertoire used by γδ T-cell receptor cells in selecting and responding to antigen, several groups have isolated this subpopulation from blood or cerebrospinal fluid of patients with multiple sclerosis (Nick et al 1995; Shimonkevitz et al 1993; Stinissen et al 1995). It should be noted that these γδ T-cell receptor cell lines were established using nonspecific polyclonal proliferation stimuli (such as anti-CD3 monoclonals or plant mitogens) rather than specific antigen. A set of short-term lines isolated from the cerebrospinal fluid of patients with early active multiple sclerosis contained an increased proportion of Vδ1- and Vδ2-bearing T cells. In this collection, some T-cell lines used identical receptor sequences (including complementarity-determining region 3 segments) and there was evidence for limited clonal expansion (Shimonkevitz et al 1993). Biased utilization of Vδ1 was also noted in short-term lines isolated from cerebrospinal fluid in patients with multiple sclerosis compared with peripheral blood (Nick et al 1995). An analysis combining immunocytochemistry and polymerase chain reaction also identified Vδ1- and Vδ2-bearing T cells in acute plaques with possible expansion of some individual clones (Wucherpfennig et al 1992b), while dominant usage of Vδ2 was described in another report (Battistini et al 1995).
Thus, there seems to be some bias in the Vδ gene repertoire used by brain-associated T cells in patients with multiple sclerosis. The significance of this is unclear, especially considering the limited gene usage typically seen in several physiological γδ T-cell receptor cell compartments, and the age-dependent shifts observed in responsiveness (Bluestone et al 1995). Unfortunately, the target epitopes of T-cell lines derived from patients with multiple sclerosis remain largely unknown – with the possible exception of some γδ cells that recognize brain membrane determinants produced, amongst others, by glioma cells (Nick et al 1995), and Vδ2 T cells isolated from peripheral blood and cerebrospinal fluid, which react with heat-shock protein 70 (Stinissen et al 1995). The key issue of whether the infiltrates of γδ receptor-expressing T cells attack myelin-forming cells in patients with multiple sclerosis, as suggested by the in vitro experiments (M.S. Freedman et al 1991), or actually limit the autoimmune process, remains unanswered.
T cells reactive against glycolipids (NK1 T cells)
With identification of the natural killer (NK1) T-cell subset, a newly identified and promising regulatory cell type arrived on the immunological stage. NK1 T cells share essential properties with natural killer cells and with classic T lymphocytes. Like T cells, some NK1 T cells express CD4, others CD8, while a third group expresses neither (Godfrey et al 2000). Besides NK receptors, they use regular T-cell receptors, mostly αβ but also the γδ type. The T-cell receptor collection used by NK1 T cells is highly simplified and differs from conventional repertoires. In humans, practically all NK1 T cells use the Vα24 chain, often together with a paired Vβ11 chain. The murine correlates are receptors composed of Vα14 mostly with Vβ8.2 chains (Godfrey et al 2000). However, unlike T lymphocytes, NK1 T cells display markers of the natural killer cell lineage on their membrane.
These monomorphic receptors recognize unusual target antigens. Observations using transgenic mice indicate that NK1 T cell development and function require β2-microglobulin (a component of MHC class I and class Ib molecules, but not the TAP peptide transporters; see Chapter 3: Vicari and Zlotnik 1996). Rather, it turns out that instead of responding to peptide bound to conventional MHC class I or II proteins, NK1 T-cell receptors bind Ib variants of the CD1 series. These present glycolipids but not peptides. Even more remarkable, the most powerful antigen recognized by NK1 T cells is an ‘exotic’ small glycolipid, α-galactosylceramide, the product of a marine sponge. No corresponding antigen has yet been found in vertebrates (Hong et al 1999).
Whereas there are several CD1 subtypes in humans (CD1a–e), the mouse has only CD1d. Like classic MHC class I proteins, they are composed of one heavy chain that bears the antigen-binding site, and the monomorphic β1-microglobulin. Crystallography shows an antigen-binding groove conformed so as to bind glycolipid antigen rather than short peptides. Interestingly, like MHC class II antigens, CD1 seems to use the invariant (ii) chain for full structural maturation, membrane expression and antigen presentation to NK1 T cells (Moody and Porcelli 2001). Some NK1 T cells mature within the thymus (Schulz et al 1996), others in the periphery. Thymic NK1 T cells seem to differentiate following local interaction with CD1 antigens.
A possible connection of NK1 T cells with autoimmunity was first observed in nonobese diabetic (NOD) mice, which spontaneously develop autoimmune diabetes mellitus. They show a deficit of NK1 T cells, which is most notable in the thymus (Gombert et al 1996). The involvement of NK1 T cells was shown more formally in transgenic mice. Animals that express the Vα14 transgene of NK1 T cells, and thus possess increased numbers of these cells, are protected from disease (LeHuen et al 1998). Consequently, CD1 knockout mice completely lack NK1 T cells and are more susceptible to autoimmune diabetes (B. Wang et al 2001). In support of these findings, activation of endogenous NK1 T cells by their ligand α-galactosylceramide suppresses diabetes development in non-NOD mice (Hong et al 2001). The mechanism of NK1 T-cell-mediated protection is not yet clear. There is evidence that these cells act on autoimmune effector T cells, suppressing production of proinflammatory cytokines but without deletion of cells (Beaudoin et al 2002).
NK1 T cells play an indisputable role in different models of experimental autoimmune encephalomyelitis. NOD mice that overexpress NK1 T cells and are free from diabetes, are also resistant to the induction of experimental autoimmune encephalomyelitis (Mars et al 2002). Transfer of NK1 T cell populations decreases experimental autoimmune encephalomyelitis induction in transgenic mice (Fritz and Zhao 2001). Further, several groups have shown that activation of NK1 T cells by α-galactosylceramide (A. K. Singh et al 2001) or agonistic synthetic derivatives (Miyamoto et al 2001) protects mice from subsequent attempts to induce experimental autoimmune encephalomyelitis. It should, however, be noted that protection depends much on the treatment protocol. Altered regimens may even result in paradoxical exacerbation of disease (Jahng et al 2001). A comparable role for NK1 T cells in multiple sclerosis remains to be demonstrated but may be expected considering one study that showed a deficit of these cells in central nervous system plaque infiltrates, which could indicate an insufficient immune regulation (Illés et al 2000).
CD4+CD25+ regulatory T cells (Treg)
One hallmark of the adaptive immune response is regulation of its intensity and temporal course. The start of an immune response is marked by immune cell activation and expansion of the immune clones involved but, at the conclusion of this mission, activated cells either die by programmed cell death or are downregulated to the resting state. Either way, the population contracts. One mechanism contributing substantially to the termination of immune responses relies on suppressor T cells. They respond to antigen recognition by silencing, not activating, immune effector mechanisms. This concept enjoyed extreme popularity two decades back, but was then all but abandoned by most immunologists. Remarkably, specific programmed counter-regulatory T cells were later resurrected, now wearing the new colours of T regulatory (Treg) cells (Bach 2003).
Tregs were discovered in mice that had been thymectomized shortly after birth. While growing to adulthood, these partly immunodeficient animals commonly develop spontaneous immune cell infiltrates in particular tissues, such as stomach, thyroid, pancreatic islets, adrenals or reproductive organs (Sakaguchi and Sakaguchi 2000). Interestingly, central or peripheral nervous tissues are missing from the list. The selection of affected organs within one particular strain of rodents is surprisingly stable. It is controlled by genetic factors that remain to be identified. Similar changes are seen in animals recovering from transient immunosuppression (ciclosporin), or after transfer of T cells into T cell-deficient animals. The key observation was that thyroiditis could be prevented by the transfer of spleen or thymus cells from normal syngeneic mice (A. Kojima et al 1976). Later, Sakaguchi and Sakaguchi (2000) showed that the protective anti-autoimmune cells are members of the CD4+ subset of T lymphocytes. Tregs represent a particular set of T cells characterized by the expression of CD25 (the IL-2 receptor α chain) along with CD4 (Sakaguchi et al 1995).
The mechanisms used by Tregs for suppression of the ongoing immune response are not yet fully characterized. One is downregulation of effector T cells via the CTLA-4 molecule. CTLA-4 expression on Tregs is constitutive and, with CD4 and CD25, is a characteristic marker of this cell type (Sakaguchi 2000). The functional phenotype of Tregs is under the control of a transcriptional regulator of the Forkhead gene family, FoxP3. Mutant or transgenic mice lacking functional FoxP3 develop an autoimmune syndrome, characterized by general immune hyper-reactivity and immune cell infiltration of diverse tissues. The syndrome, which also occurs in a few humans with mutant FoxP3, is typically associated with a deficit of CD25+CD4+ Treg cells (Wildin et al 2003). Conversely, FoxP3 is specifically expressed in Treg cells. Introduction of the gene into regular CD4+ T cells transforms them into specialized Tregs. These are not rare cells. They constitute around 10% of all immune competent CD4+ T cells in the thymus and peripheral immune organs (Sakaguchi 2000). The way in which Tregs silence autoimmune effector T cells and participate in general immune responses is not well understood. First, the T-cell receptor specificity of Tregs remains to be established. The indirect evidence suggests that Tregs, possessing the relevant antigen specificity, are efficient regulators of organ-specific autoimmune responses. Thus, old results from neonatally thymectomized mice suggested that regulatory cells from male donors suppress autoimmunity in male (Taguchi and Nishizuka 1981; 1987) better than female gonads with corresponding inverse differential regulatory activities of female T cells (Taguchi and Nishizuka 1980). More recent work has established that tolerogenic treatment of T-cell receptor transgenic mice (using oral administration of autoantigen) involves expansion of antigen-specific Tregs, which downregulate effector or helper cells of identical antigen specificity (X. Zhang et al 2001).
Tregs depend on IL-2 for survival. Mice that lack either IL-2 or IL-2 receptor (including its α chain, CD25) develop spontaneous autoimmune disease, affecting especially the gastrointestinal tract (Sadlack et al 1993; Strober and Ehrhardt 1993). It appears that classic immunoregulatory cytokines such as IL-4, IL-10 or TGF-β do not play a pivotal role in Treg-mediated suppression (Bach 2003). Instead, direct cell-to-cell contacts, possibly involving the downregulatory molecule CTLA-4, are involved (Salomon and Bluestone 2001). Recent work has shown that the regulatory function of Tregs is linked directly and indirectly to elements of the innate immune system. Activation of dendritic cells by ligation of Toll-like receptor seems to neutralize the suppressive potential of Tregs, presumably acting via soluble mediators that include IL-6 (Pasare and Medzhitov 2003). Tregs also express and use Toll-like receptors on their own membrane, and their activation has opposite results. Lipopolysaccharide activation of Tregs is reported further to enhance their suppressive action on other immune cells (Caramalho et al 2003).
Treg cells occur in several spontaneous and induced animal models of autoimmune disease. In addition to their effect in partly immune-deprived animals (neonatal thymectomy), Tregs protect against spontaneous and antigen-induced models of autoimmunity. Spontaneous autoimmune diabetes mellitus, which commonly develops in (female) NOD mice within the first 4 months of life, seems to coincide with a decline of CD4+CD25+ Treg cells. Their transfer from prediabetic NOD mice has a beneficial effect on the onset of diabetes in recipients of the same strain. The self-protective effect of Treg-like lymphocytes in anti-myelin autoimmunity was first demonstrated in monoclonal transgenic mice expressing a myelin basic protein-specific T-cell receptor on all T cells. The receptor came from an antigen-specific encephalitogenic T-cell clone isolated from a myelin basic protein-immunized wild-type mouse. (If a transgenic receptor is expressed in otherwise regular mice, the majority, but not all, T cells express the transgene, while the rest use receptors from the internal receptor gene repertoire: see Figure 11.17.) Most such transgenic mice with myelin basic protein-specific receptors rarely develop spontaneous experimental autoimmune encephalomyelitis (Lafaille et al 1994). This is in contrast to strictly monoclonal mice, which have the same receptor transgene but with a defective recombination machinery that prevents production of endogenous T- and B-cell receptors. Practically all of these monoclonal myelin basic protein-reactive mice eventually develop experimental autoimmune encephalomyelitis. This spontaneous autoimmune disease is prevented by transfer of Treg cells, most prominently those expressing both CD4 and CD25 (Furtado et al 2001). Unfortunately, as with other models of autoimmunity, the receptor specificity of the self-protective Tregs remains undefined.
Regulatory defects in multiple sclerosis
There are excellent reasons for immunologists to search for defective immune regulation in the pathogenesis of multiple sclerosis. One example is the undisputed presence of brain-specific autoreactive T cells in the healthy immune repertoire. Even though these must be activated in order to trigger autoimmune disease in the central nervous system, mere lack of activation seems too paltry a basis for lifelong self-tolerance. One would expect safeguards from active regulatory mechanisms to keep the potential autoimmune time bombs unexploded. Their identification in experimental autoimmune encephalomyelitis encourages the belief that similar regulatory mechanisms would directly lead to new and specific therapeutic approaches in human disease. We have discussed above early attempts to identify CD8+ T lymphocytes as suppressor cells responsible for maintaining immunological self-tolerance. We noted that simple T-cell subset enumeration or functional suppressor assays did not illuminate the matter, although an ever increasing number of publications seems to indicate a defect of regulatory cells (for example, Viglietta et al 2004). We omitted discussion of T-cell regulation via recognition of T-cell receptors (or their components). The concept of T–T-cell interactions as regulatory events in brain autoimmunity takes its origin from an observation made by Cohen and colleagues, namely that myelin basic protein-specific T lymphocytes not only act as pathogenic agents mediating the transfer of experimental autoimmune encephalomyelitis, but are themselves the targets for counter-regulatory cellular responses in the host immune system. Rats receiving myelin basic protein-specific T cells inactivated by irradiation not only fail to develop experimental autoimmune encephalomyelitis but also are protected against subsequent attempts to induce disease – the first example of vaccination against a T-cell-mediated organ-specific autoimmune disease (Ben-Nun et al 1981b). We referred to later experiments indicating that the vaccinating autoimmune T cells activate counter-regulatory T lymphocytes – mainly of the CD8+ population – which then anticipate and destroy the activated encephalitogenic clones. Some indirect evidence suggests that the vaccination effect is directed against determinants of the brain-specific T-cell receptor (Lider et al 1988). A second, and unambiguously T-cell receptor-directed vaccination strategy was successfully used later by immunizing rats against peptide analogues to receptors typically used by encephalitogenic T cells (Howell et al 1989; Vandenbark et al 1989). Obviously, it is now known that profound differences exist between rodent and primate immune systems. But is there evidence for T-cell receptor-directed idiotypic T–T-cell interactions in the human immune system? T cells recognize receptor fragments of other T cells, but the relevance of these interactions for the pathogenesis and treatment of multiple sclerosis remains to be established.
T-cell receptor-specific human T-cell lines were first described by Saruhan-Direskeneli et al (1993), who used synthetic peptides representing sequences of the complementarity-determining region 3 and V regions of a known myelin basic protein-specific T-cell receptor β chain to select anti-idiotypic T-cell receptor-specific cells. Myelin basic protein-specific T cells were obtained from peripheral blood of the same (healthy) donor. A small panel of antigen- and receptor-specific T cells, all CD4+ and restricted by DR2 products, proved strongly cytotoxic against antigen-presenting target cells. Importantly, all lines recognized specifically the relevant T-cell receptor-derived peptides when offered by antigen-presenting cells, but none of the anti-T-cell receptor lines showed any demonstrable interaction with the myelin basic protein-specific T cells. Following contemporary logic, the anti-T-cell receptor-specific T cells were recognizing cryptic T-cell receptor epitopes (Saruhan-Direskeneli et al 1993). More recently, complementarity-determining region 3 peptide-specific anti-idiotype reactive T cells were confronted in vitro with full-length recombinant T-cell receptor V chains. Some, but not all, of the peptide-specific T cells co-recognized their epitope when derived from the intact protein (Zipp et al 1998). While the anti-T-cell receptor-specific T cells were of the CD4 class, other investigators have isolated comparable anti-T-cell receptor-reactive T cells using shorter peptides. CD8+ T cells were obtained that killed T cells with the suitable complementarity-determining region 3 and T-cell receptor sequence (Zang et al 2003b).
Two clinical studies support T-cell receptor-directed immune regulation controlling the T-cell response to myelin basic protein. We describe the preliminary therapeutic effects in Chapter 18, but here rehearse implications for understanding the immunology of multiple sclerosis. J. Zhang et al (1993) first directly applied Irun Cohen's T-cell vaccination scheme to multiple sclerosis. They isolated myelin basic protein-specific T cells from the peripheral blood of patients with multiple sclerosis, expanded these as lines, and transferred the T cells back to the original donors after irradiation. The autovaccinated patients underwent remarkable changes in their immune repertoire. For periods of several years, all myelin basic protein-reactive T cells were lost from the peripheral blood. During the same periods, CD8+ T cells capable of specifically destroying the immunizing T cells developed in a clonotypic fashion (J. Zhang et al 1995). It is tempting to speculate, but premature to conclude, that elimination of myelin autoreactive T cells indeed affects the autoimmune pathogenesis of multiple sclerosis. An alternative strategy relied on vaccination of patients with synthetic peptides representing V region sequences from human myelin basic protein-specific T-cell receptors. This approach was based on the assumption of dominant usage of one particular Vβ gene (Vβ8.2) by human T cells recognizing myelin basic protein in the context of DR2 (DRB1*1501: Vandenbark et al 1996a). Such a biased repertoire was noted in some groups of patients (Chou et al 1994; Kotzin et al 1991; Oksenberg et al 1993), but not others (Afshar et al 1998; Hafler et al 1996), somewhat limiting general applicability of the therapy.
Vaccination of patients with multiple sclerosis using T-cell receptor peptide resulted in a partial loss of myelin basic protein-reactive T cells in patients who appeared to respond by entering disease remission. The effect of T-cell receptor peptide vaccination was attributed to the activation of Th2-like T cells suppressing pathogenic brain-reactive T cells in a bystander fashion (Vandenbark et al 2001). Unfortunately, like many other immunotherapeutic strategies, T-cell receptor-targeted vaccination has not yet yielded a satisfactory therapeutic success (Hohlfeld and Wiendl 2001). Taken together, the present data are compatible with idiotypic regulation of myelin-specific T cells, but the active role of Tregs in the pathogenesis of multiple sclerosis – plausible as a hypothesis – remains unproven.
IMMUNE REACTIVITY IN THE CENTRAL NERVOUS SYSTEM
Traditionally, the tissues of the nervous system have been considered exempt from immunological surveillance and devoid of immune reactivity. Barker and Billingham (1977) coined the term ‘immune privilege’ to describe this status. The concept carries with it the implication that immune surveillance, which protects most other tissues, does not extend to the nervous system. It follows that immunosurveillant lymphocytes, patrolling elsewhere in the organism, are excluded from brain and nerves. In its strict original version, the notion of immune privilege in the nervous system stood on somewhat shaky foundations. The brain parenchyma was considered to be totally secluded from the systemic circulation by a tight endothelial blood–brain barrier, excluding all recirculating immune cells. The central nervous system was thought to lack any lymphatic drainage, since fully differentiated lymphatic vessels, which permit transport of antigenic material from a primary site of infection into the local lymph node, were not found. In contrast to most other tissues, the healthy brain parenchyma was considered not to express MHC products, the essential prerequisite for antigen presentation to T lymphocytes. And, finally, professional antigen-presenting cells were thought not to be present in the normal brain. It is now clear that most of these assumptions, although not actually wrong, were somewhat exaggerated. It turns out that the central nervous system is far from absolutely immune privileged, although its behaviour differs from the classic pattern of immune reactivity seen in most other organs. Immune surveillance is restricted to a small set of activated lymphocytes, and antigen presentation can be induced in a number of brain cells by proinflammatory stimuli, such as IFN-γ (Wekerle et al 1986).
Lymphocyte traffic into the central nervous system
We have mentioned that the central nervous system tissues are secluded by a specialized endothelial blood–brain barrier that tightly seals the parenchyma from the bloodstream. The central nervous system endothelium differs indeed from endothelia lining the blood vessels in other organs, because it is lined by tight junctions that bar plasma molecules and circulating cells from entry. A few molecules that are required to feed the tissue, such as glucose or lipids, are taken up by transport systems on the luminal side and transferred in a controlled fashion into the tissue. For blood cells, such transportation is unknown. Yet, there must be some cellular traffic between blood and central nervous system. It is known that bone marrow-dependent progenitor cells slowly replace at least those microglial cells positioned close to the microvessels. Furthermore, there must be conditions that allow certain immune and inflammatory cells to enter the central nervous system. Without such mechanisms, the inflammatory lesions of multiple sclerosis would not occur (Sospedra and Martin 2005).
Migration of T cells into the central nervous system
Passage through the blood–brain barrier has been shown by metastasizing tumour cells (Subramaniam et al 2003), bone marrow-derived stem cells (Priller et al 2001), and – of significance in the present context – lymphocytes, provided that they are in a state of high activation (Wekerle et al 1986). Within the first few hours after transfer, a few highly activated T cells cross the resting blood–brain barrier and migrate into the central nervous system parenchyma. Assuming that brain antigen is not recognized, this T-cell infiltration is transient and lasts only a few hours. However, when the T-cell population is directed against a central nervous system antigen, the cells persist within the brain tissue (Hickey et al 1991; Wekerle et al 1986). After approximately 2–3 days, a sudden new influx of inflammatory cells occurs, which results in clinical disease associated with histological evidence for meningoencephalitis (Figures 11.8 and 11.16).
It is probable that the entry of activated lymphocytes through the initially resting blood–brain barrier is governed by the combination of chemotactic cytokines and special patterns of cell adhesion molecules displayed on the inner surface of brain microvascular endothelial cells. Prevailing wisdom distinguishes three steps in the attachment of lymphocytes to endothelial surfaces (Springer 1994). First, mutual contacts are made through selectins on one cell and glycoproteins on the other (L-selectins bind to GlyCAM, and P-selectin to the sLex glycoprotein). Although this selectin-mediated adhesion is relatively weak, selectin-bound lymphocytes roll along the endothelial surface directed by the streaming blood. In the second phase, the lymphocyte becomes activated either by selectin-glycoprotein binding or stimuli provided by chemokines. Additional cell adhesion molecules, including members of the integrin and immunoglobulin superfamilies, are quickly induced to form stronger bonds, tethering the T cell firmly to the endothelial membrane (Figure 11.17: von Andrian and Mackay 2000). Using proteases and glycosidases, in a third phase, the migrant T cells then puncture the blood–brain barrier and pass directly into the central nervous system parenchyma, presumably attracted by gradients of locally produced chemotactic factors (Butcher and Picker 1996; Kunkel and Butcher 2002).
There are observations suggesting that the general rules of leucocyte recirculation are not competely represented in the passage of activated lymphocytes across resting cerebral endothelial cells. One study combined fluorochrome labelling of activated T lymphocytes with in vivo microcinematography. It showed that, in contrast to regular lymphocyte penetration through blood vessel walls, activated T cells did not roll along the lining of cerebral vessels but directly adhered to the endothelium (Vajkoczy et al 2001). Initial adhesion involved binding of the VLA-4 integrin to vascular cell adhesion molecule 1 (VCAM-1), which seems to be constitutively expressed on resting cerebral endothelia, in contrast to most other endothelial cells. Importantly, interaction between VLA-4 and VCAM-1 leads to the enhanced production and T-lymphocyte secretion of gelatinases – enzymes that help the lymphocyte to break up the perivascular basal lamina and allow entry to the surrounding tissue (Hartung and Kieseier 2000).
VLA-4 was previously identified as a target of antibody-mediated immunotherapy (J. L. Baron et al 1993; Yednock et al 1992). Now, there is evidence that anti-VLA-4 antibodies act in a dual fashion. Initially they reduce the number of effector T cells entering the central nervous system in the early phase. Later, they block the influx of additional, nonspecific inflammatory cells (Brocke et al 1999). However, therapy using anti-VLA-4 blockade should be used with caution (see Chapter 18; Hohlfels and Wekerle 2005; Steinman 2005). Although it blocks entry of effector and accessory cells into the central nervous system when given early in a response, exposure at later time points may paradoxically cause exacerbation of chronic disease (Theien et al 2001). This has to be remembered in planning therapeutic trials, which, to date, have produced favourable clinical results (see Chapter 18: D. H. Miller et al 2003a; Tubridy et al 1999).
Cell migration through the primed blood–brain barrier: the second wave of immigration
Activation of the endothelial blood–brain barrier is central to the initiation of clinical experimental autoimmune encephalomyelitis. In T cell-transferred experimental autoimmune encephalomyelitis in the Lewis rat, mass invasion of the central nervous system by mononuclear cells coincides with acute onset of neurological defects. At this time, brain endothelia show notable changes. They become permeable, allowing influx of plasma macromolecules, and thus promote the formation of oedema (Butter et al 1991; Juhler et al 1984; Koh et al 1993). At the same time, the expression of MHC class I determinants and several cell adhesion molecules increases (Cannella et al 1991). The activated blood–brain barrier allows passage of mononuclear cells, including naive T cells, which are otherwise excluded by the resting unactivated blood–brain barrier (Krakowski and Owens 2000). Thus, activation of the blood–brain barrier endothelium is an essential prerequisite for formation of the pathogenic infiltrate in experimental autoimmune encephalomyelitis.
It remains unresolved where exactly in the central nervous system (and peripheral immune system) effector T cells are located, as the inflammatory infiltrate is assembled in the brain parenchyma, and how they influence the central nervous system tissue. The application of novel biological fluorochrome markers should help. Our position is that early effector T cells enter the central nervous system parenchyma, release proinflammatory cytokines and chemokines, and prime the local tissue. Accordingly, they are supposed to activate cerebral endothelia to express an inflammatory phenotype, and induce immune molecules in glia cells required for successful presentation of local autoantigens (Cross et al 1991; Wekerle et al 1986). The proportion and distribution of antigen-specific T-cell blasts in the lesions remain controversial. It was generally believed that antigen-specific cells in the lesion were rather sparse, representing only a few percent of the total T-cell population (A. H. Cross et al 1990; Fallis et al 1987; Stohl and Gonatas 1978), although others report a much higher proportion (Körner et al 1997; Matsumoto and Fujiwara 1988; Skundric et al 1994; Zeine and Owens 1992). We have used transgenic markers (J. Bauer et al 1998) and retroviral engineering (Flügel et al 1999) to trace transferred encephalitogenic T lymphocytes in the inflammatory infiltrates. As previously described, the small number of T cells arriving in the initial wave after transfer are all derived from the transferred antigen-specific pool. At the onset of clinical disease, 3 days after transfer, a massive influx of the transferred antigen-specific T cells is seen in the nervous system, and these constitute >90% of all CD4 T cells in the fresh infiltrate. As shown by recent two-photon imaging, the T cells rapidly cruise through the central nervous system parenchyma, apparently in search of the specific autoantigen (Kawakami et al 2005). This initial phase, however, is transient, and the antigen-specific T cells are gradually replaced in the lesions by secondarily recruited, host-derived T lymphocytes (Flügel et al 2001a).
One may assume that the activated blood–brain barrier surface per se is insufficient to attract inflammatory cells to the developing lesion. Soluble signals emanating from chemokines, perhaps produced by perivascular dendritic cells (Greeter et al 2005), should also play a vital role. In fact, an ever growing list of chemokines attracting T cells and monocytic cells has been demonstrated in the early phase of experimental autoimmune encephalomyelitis (Ransohoff et al 2003). Among the many chemokines making an appearance in the developing experimental autoimmune encephalomyelitis lesion, macrophage inflammatory protein MIP-1α seems especially important since its neutralization by antibody stops disease (Karpus et al 1995). The chemokine attracts T cells and macrophages, but also participates in orchestrating the entire chemokine symphony. In general, chemokines such as MIP-1α, macrophage chemotactic protein MCP-1, RANTES and interferon induced protein IP-10 determine an inflammatory milieu, which controls the number and composition of inflammatory infiltrates in experimental autoimmune encephalomyelitis (Ransohoff 1999). It should be noted that chemokines contributing to the central nervous system infiltrate include not only those induced in response to inflammatory stimuli, but also molecules produced under homeostatic conditions. CCL21 (secondary lymphoid organ chemokine, SLC) and CCL19 (EBI-1 ligand chemokine, ELC), for example, chemokines that govern ordered migration of immune cells into and through lymphoid tissues, appear in chronic relapsing experimental autoimmune encephalomyelitis, although with different distributions from those characterizing the acute phases (Columba-Cabezas et al 2003).
Clearance of central nervous system inflammation
Experimental autoimmune encephalomyelitis in the Lewis rat (and other strains) is self-limiting and follows a monophasic course. Clinically, neurological signs develop, reach a peak, and thereafter resolve, often fully. The histological infiltrates usually, but not invariably, run a parallel course (Källen and Nilsson 1986; Levine and Sowinski 1980). Typically, the lesions of experimental autoimmune encephalomyelitis in the Lewis rat vanish within 2–3 weeks of disease onset. Clearance of inflammation in the central nervous system and recovery from an episode of experimental autoimmune encephalomyelitis are controlled by systemic immune regulatory mechanisms, as well as by events taking place within the nervous tissue. The severity of clinical disease and the time course of brain inflammation reflect the genetically determined ability of animals to mount a vigorous corticosteroid response (D. Mason 1991). In addition, adrenalectomy, or the blockade of glucocorticoid receptors, augments experimental autoimmune encephalomyelitis. Recovery from experimental autoimmune encephalomyelitis is also associated with changes in cytokine pattern within the circulation and local brain environment. In particular, cytokines associated with the suppression of T-cell reactivity (TGF-β and IL-10) are increased within the central nervous system lesions themselves (M. K. Kennedy et al 1992; Khoury et al 1992; Racke et al 1992). These cytokines have also been shown to suppress experimental autoimmune encephalomyelitis when systemically administered during the course of the disease (Racke et al 1991; 1994). Other data, however, indicate that local dysregulated expression of TGF-β may also augment inflammation (Wyss-Coray et al 1997).
How do inflammatory cells leave the central nervous system? Is this via blood vessels or other pathways? Alternatively, do they die and undergo local degradation? So far there is no convincing evidence for emigration of intraparenchymal T cells via a vascular route or cerebrospinal fluid (Wekerle 1993). Instead, several groups have found that antigen-activated T cells arriving in brain tissue undergo apoptosis at very high rates. Recovery from experimental autoimmune encephalomyelitis is associated with extensive apoptotic destruction of T and B lymphocytes in inflammatory brain lesions (Figure 11.16: Pender et al 1991; A. T. White et al 2000). In the late stages of T cell-transferred EAE, up to 50% of the local T-cell population may synchronously be destroyed by this mechanism (Schmied et al 1993). To illustrate the significance of this high percentage it has to be understood that execution of the full apoptosis programme in vitro lasts for approximately 3 hours. A 50% apoptosis rate, observed within a given tissue section, suggests that about three times the number of cells present in the lesion are destroyed by programmed cell death within 24 hours. This clearance process is therefore so effective that an acute inflammatory lesion in the central nervous system can only be maintained by a continuous supply of new inflammatory T cells from the circulation.
T-cell apoptosis mainly affects the cell population that infiltrates the brain parenchyma, leaving inflammatory cells in the perivascular space and meninges unaffected (J. Bauer et al 1998). In experimental autoimmune encephalomyelitis induced by sensitization with myelin basic protein, most apoptotic T cells in the parenchyma carry the Vβ8.2 T-cell receptor, at least during the early phases of brain inflammation (Tabi et al 1994). Since this is also the receptor subtype of the transferred encephalitogenic T-cell fraction, it was originally suggested that the pool of autoreactive T cells dies selectively in the central nervous system by activation-induced cell death (Pender 1999). However, by transferring prelabelled T cells in the induction of experimental autoimmune encephalomyelitis, autoantigen-specific as well as irrelevant, it is clear that secondarily recruited T cells are also destroyed in the central nervous system lesions by apoptosis (J. Bauer et al 1998). Several different immunological mechanisms may be responsible for the induction of this T-cell apoptosis in inflammatory brain lesions. Programmed cell death of lymphocytes can be induced by the glucocorticoid response associated with recovery from experimental autoimmune encephalomyelitis. Indeed, myelin basic protein-specific T cells can synchronously be driven to apoptotic death when glucocorticoids are added in vitro at a late stage after T-cell activation (D. P. Gold et al 1991). Furthermore, high-dose corticosteroids significantly increase the rate of T-lymphocyte apoptosis in the lesions of experimental autoimmune encephalomyelitis (McCombe et al 1996; J. Schmidt et al 2000) and neuritis (Zettl et al 1995). Corticosteroids induce apoptosis not only in the Vβ8.2 positive, putative autoreactive T cells, but also in other T lymphocytes and microglia (McCombe et al 1996; Nguyen et al 1997).
Another possible mechanism of apoptosis induction in T cells lies in the action of TGF-β. The expression of this cytokine is massively upregulated in the recovery stage of experimental autoimmune encephalomyelitis, and TGF-β has been noted to have a potent suppressive effect on experimental autoimmune encephalomyelitis. In vitro, T cells can be driven into apoptosis by exposure to TGF-β (Weller et al 1994). On the other hand, it is known that apoptotic T cells themselves release substantial amounts of TGF-β (W. Chen et al 2001), making it difficult to determine whether enhanced cytokine levels are a cause or consequence of local apoptosis. In fact, the evidence suggests that neither the steroid response nor cytokines alone are responsible. Adrenalectomy does not prevent T-cell death in the brain lesions of experimental autoimmune encephalomyelitis, although it massively potentiates disease and mortality (T. Smith et al 1996). In addition, T-cell apoptosis in experimental autoimmune encephalomyelitis lesions is mainly found in the brain parenchyma and – at least in the early stages of the inflammatory process – predominantly involves the antigen-specific T-cell population. Thus, antigen-specific mechanisms may also play a central role in the induction of T-cell apoptosis in experimental autoimmune encephalomyelitis.
Antigen-specific induction of T-cell apoptosis could result from unbalanced signal transduction mediated by local nonprofessional antigen-presenting cells such as microglia or astrocytes. In line with this interpretation, apoptosis of antigen-specific T cells in vitro is readily induced by astrocytes (R. Gold et al 1996) and freshly isolated microglia (Ford et al 1996). Furthermore, the rate of T-cell apoptosis in the lesions in vivo is massively increased following treatment of animals with the respective soluble antigenic peptide (Ishigami et al 1998; Weishaupt et al 1997). On the other hand, it is unlikely that antigen presentation by local nonprofessional presenting cells alone is responsible for the induction of T-cell apoptosis, since its rate in brain lesions is identical when experimental autoimmune encephalomyelitis is induced in bone marrow chimeras or fully competent Lewis rats. In the former, antigen presentation on local tissue elements such as astrocytes or microglia cannot take place because of MHC antigen mismatch (J. Bauer et al 1998).
Since T-cell destruction by apoptosis is not restricted to the transferred antigen-specific cell population – being prominent in inflammatory lesions of the central (Schmied et al 1993) and peripheral nervous systems (Zettl et al 1994) but virtually absent in inflammatory lesions elsewhere (Schmied et al 1993; Schneider et al 1996) – should we conclude that the nervous systems have special mechanisms that control inflammation through effective destruction and elimination of T cells? One possible mechanism is by the Fas/Fas-L pathway. Activation of Fas through its specific ligand triggers an intracellular cell death pathway that can drive cells into apoptosis (Nagata and Goldstein 1995). In the central nervous system, Fas is widely expressed during inflammation, whereas Fas-ligand complex is mainly present on invading leucocytes, microglia (Dowling et al 1996; D'souza et al 1996b) and possibly on astrocytes (Bechmann et al 2002; Kohji and Matsumoto 2000). When the Fas/Fas-L interaction is blocked, either pharmacologically or through genetic deletion of one binding partner, the rate of T-cell apoptosis is reduced in various in vitro and in vivo models (Bechmann et al 2002; Ciusani et al 2001; Flügel et al 2000). But in one other study, deletion of Fas or Fas-L had no effect on T-cell apoptosis in experimental autoimmune encephalomyelitis lesions, whereas, in the absence of tumour necrosis factor-receptor 1 (TNF-R1) signalling, T-cell apoptosis was reduced by 50% (Bachmann et al 1999). Taken together, there is good evidence that the network of microglia and astrocyte processes expressing Fas-L or TNF-α represents a hostile environment for activated Fas- or TNF-receptor-positive lymphocytes that enter the central nervous system from the circulation or become activated locally. Apoptosis of T cells in inflammatory brain lesions is further augmented by IFN-γ. In the absence of IFN-γ production in the central nervous system, T-cell apoptosis is massively reduced in experimental autoimmune encephalomyelitis lesions, and the clearance of CD4+ T lymphocytes from the central nervous system is disturbed (Chu et al 2000). Conversely, overproduction of IFN-γ in the central nervous system results in increased rates of lymphocyte apoptosis in animals with experimental autoimmune encephalomyelitis, reflected by reduced disease severity and earlier recovery (Furlan et al 2001). This may in part explain the unexpected effect of manipulating IFN-γ in the context of experimental autoimmune encephalomyelitis.
Whatever mechanisms are responsible for the induction of T-cell apoptosis in inflammatory brain lesions, dead cells are taken up locally by macrophages, microglia and (less effectively) astrocytes (Magnus et al 2002; Nguyen and Pender 1998). Uptake and digestion of apoptotic T cells by microglia leads to downregulation of their activation state (Magnus et al 2001).
The mechanisms described above may account for the removal of T cells from autoimmune lesions of the nervous system and consequently for downregulation of the inflammatory process. They do not, however, elucidate how secondarily recruited effector cells leave the lesion. Although in rare instances apoptotic macrophages and microglia are found in experimental autoimmune encephalomyelitis lesions (Nguyen et al 1994; White et al 1998), their low number does not offer a satisfactory explanation for effector cell clearance (T. Smith et al 1996). Macrophages containing tissue debris tend to accumulate in the perivascular space (Figure 11.17), from where they are able to reach the cerebrospinal fluid, suggesting that there is lymphatic drainage of the central nervous system (Cserr et al 1992; Weller et al 1996). Particulate substances injected into the central nervous system can be traced along the perivascular space into the leptomeninges (Weller et al 1992). From there, the material can further be transported along cranial and spinal nerve roots to reach the epidural space through channel systems in the arachnoid membranes of root pockets (Zenker et al 1994) and large brain vessels, as well as through the lamina cribrosa (Weller et al 1992). Consistent with the possible existence of lymphatic drainage of the central nervous system is the observation that in chronic experimental autoimmune encephalomyelitis in primates, myelin-reactive material ingested in macrophages or dendritic cells can be found in the deep cervical lymph nodes (De Vos et al 2002).
These data suggest that, in the normal brain and conditions of acute inflammation, T and B lymphocytes are effectively and rapidly cleared from the central nervous system by programmed cell death, while macrophages and microglia cells persist in the lesions for much longer. These cells may also be slowly removed by apoptosis. Alternatively, they have the capacity to migrate out of the central nervous system and reach regional lymph nodes. Apoptosis of T lymphocytes has been observed in similar quantity in the lesions of acute disseminated leucoencephalitis (Bauer et al 1999), suggesting that the basic mechanisms of T-cell clearance from inflammatory central nervous system lesions are similar between rodents and humans. However, in the lesions of multiple sclerosis, the rate of T-cell apoptosis is very low (Ozawa et al 1994). This may reflect the chronic course of the disease, in which a synchronous and simultaneous apoptosis of lymphocytes cannot be expected. In line with this interpretation is the low apoptosis rate of lymphocytes in chronic models of Toxoplasma encephalitis (Schlüter et al 2002). Alternatively it has been suggested that a genetically determined failure of activation-induced apoptosis of autoreactive T cells may underlie the chronic disease in multiple sclerosis (Pender 1998).
Regulation of major histocompatibility antigen expression in the brain
While it is clear that the healthy central nervous system displays very little MHC class I or class II antigen, it is equally apparent that many, if not all, brain cells can be induced under suitable conditions to express these molecules. And from in vitro studies, we know that there is a clear hierarchy of MHC inducibility – microglia, astrocytes, endothelia, oligodendrocytes and then neurons (Wekerle 1994). MHC determinants are also induced in vivo under a variety of pathological conditions. Ectopic MHC expression is commonly noted in inflammatory responses, virus infections, tumour, development, and degenerative disorders. Whilst proinflammatory cytokines, such as IFN-γ, have a predominant role in MHC induction during inflammation of the central nervous system, mechanisms that result in the gene expression associated with noninflammatory neurodegenerative processes are less clear. Recent observations indicate that neurons have a crucial role in regulating MHC expression in the central nervous system. Electrically active neurons strongly suppress MHC expression on surrounding glia cells (H. Neumann et al 1996). However, neuronal degeneration following axotomy results in efficient induction of MHC class I components in vivo (Finsen et al 1993; Lindå et al 1998). Thus, paralysis of neuronal activity leads to the prompt inducibility of MHC determinants on all central nervous system cell classes. It is open to debate whether this negative signalling is mediated by diffusible neurotransmitters (glutamate, vasoactive intestinal peptide or catecholamines), or is the effect of electrical activity (H. Neumann et al 1996). Class I expression in the central nervous system is not an experimental curiosity, but is the hallmark of (human) neurodegenerative disorders. Alzheimer's disease (Tooyama et al 1990), Parkinson's disease (McGeer et al 1988), amyotrophic lateral sclerosis (Kawamata et al 1992) and brain trauma (Kaur et al 1995) are all associated with upregulation of MHC proteins, especially on microglia cells.
A particularly intriguing finding is the inducibility of MHC class I antigens on neurons. Traditionally, these were considered as the only cell types absolutely devoid of MHC antigens. Indeed, even high doses of IFN-γ, one of the strongest inducers of MHC, fail to induce class I molecules in electrically active neurons. However, after paralysis with sodium channel blockers, the same neurons become fully inducible (H. Neumann et al 1995; 1997). Some MHC class I expression appears during neuronal development (Huh et al 2000).
Neuronal MHC class I proteins are fully functional. They can present viral peptides to virus-specific class I restricted cytotoxic T cells. Recognition triggers cytolytic mechanisms, which start by disrupting axonal processes (Medana et al 2001b) and, with some delay, end up killing the entire neuron (Medana et al 2001a).
The central nervous system is therefore a conditionally privileged organ; it is accessible to immune surveillance and accessory inflammatory machinery; but this is well controlled so as not to threaten neurons, which are especially vulnerable and cannot be regenerated.
T-cell interaction with local glia
Once through the blood–brain barrier, activated T cells find themselves in contact with local glia (and neuronal) cells. By secretion of proinflammatory cytokines (IFN-γ, TNF-α and others), glia are induced to synthesize and express MHC products (Male et al 1987; Traugott and Lebon 1988c; Vass and Lassmann 1990), cell-adhesion molecules (Cannella et al 1991; Male et al1994) and costimulatory molecules (Issazadeh et al 1998; Soos et al 1999; Williams et al 1994) needed to process and present self and foreign protein antigens to patrolling T cells. However, not all glia cells are equally responsive to the activating signals communicated by T lymphoblasts, and not all central nervous system cells are equally efficient antigen presenters. Perivascular cells and microglia, which scan the central nervous system tissue with fast moving processes (Davalos et al 2005; Nimmerjahn et al 2005), are undoubtedly the fastest and most proficient cytokine responders, whereas astrocytes and ependymal cells require more intense signals to become immunologically activated (Vass and Lassmann 1990).
The onset of overt brain inflammation is associated with a variety of immunopathological changes in the local tissue. First, different cytokines are produced locally in a time-dependent manner (Issazadeh et al 1995a; 1995b). The first cytokine to be expressed in the inflammatory lesion is IL-12 followed after a few hours by IFN-γ and TNF-α. Parallel with the expression of these cytokines, extensive upregulation of adhesion molecules and histocompatibility antigens takes place. In particular, endothelial cells of cerebral vessels express a variety of adhesion molecules – selectins, intercellular adhesion molecules (ICAMs), VCAMs and others – and endothelial cells acquire an activated phenotype (Cannella et al 1991; Lossinsky et al 1989; Wilcox et al 1990). These endothelial changes apparently are instrumental for the secondary recruitment of inflammatory cells (T cells and monocytes/macrophages) into the established lesions (Figure 11.17). Histocompatibility antigens are primarily expressed on meningeal and perivascular monocytes (Hickey and Kimura 1988; Matsumoto and Fujiwara 1987). In addition, a less pronounced expression of class I antigen is found on endothelial cells and, with more severe proinflammatory stimuli, on all other neuroectodermal elements in the lesions (Vass and Lassmann 1990). Class II MHC antigen expression in established lesions is more restricted and rarely found on elements other than leucocytes and resident microglia (Vass et al 1986). However, in rare instances, some expression is seen on cerebral endothelial cells, astrocytes and ependymal cells (Deckert-Schlüter et al 1994; Hickey et al 1985; Steiniger and Van der Meide 1988; Vass and Lassmann 1990). Although much emphasis has been placed on the possible role of class II MHC antigen expression and antigen presentation by neuroectodermal cells, studies in bone marrow chimeras clearly show that the expression of MHC antigens on local resident cells of the central nervous system, including astrocytes, ependymal cells, endothelial cells and resident microglia, is not required for the development of experimental autoimmune encephalomyelitis (Hickey and Kimura 1988; Lassmann et al 1993; Y. Matsumoto and Fujiwara 1988). In these animals, a model system is created in which experimental autoimmune encephalomyelitis is induced in a host environment where only meningeal and perivascular monocytes and haematogenous macrophages carry the specific MHC haplotype recognized by the transferred encephalitogenic T lymphocytes (Hickey et al 1992). Results obtained in this model indicate that induction, maintenance and downregulation of brain inflammation do not require antigen presentation on resident tissue elements. It has, however, yet to be determined whether local antigen presentation modifies experimental autoimmune encephalomyelitis, for example in the chronic models.
Is autoimmunity beneficial?
Traditionally, and throughout this text so far, autoimmunity has been treated as an unwanted upheaval of the immune system working against the organism – an illicit and pathological aberration. Consequently, early theories of immunity invoked emotive terms such as ‘horror autotoxicus’ (Ehrlich and Morgenroth 1901) to describe the catastrophic effects of immune reactions directed against self. Immune cells driving such responses were assigned the status of ‘forbidden clones’, having no place in health and therefore ripe for removal from the immune repertoire (Burnet 1959).
A dramatic revision of this thinking began with the discovery of organ-specific T-cell clones as normal components of the healthy immune system (Schlüsener and Wekerle 1985). Clearly, such T cells have receptors for antigenic structures on the body's own tissues and, yet, in most individuals they appear harmless throughout life. How then should we view these self-recognizing T-cell populations; can they be badged as flawed products of a sloppily evolved immune system? Irun Cohen formulated a theory that assigned a positive and physiological function to immunological self-reactivity. Briefly, his concept of the ‘immunological homunculus’ postulates that throughout the body, all major antigenic structures are reflected by complementary self-reactive T-cell clones in the immune system (Cohen 1992). These self-reactive T cells would be beneficial in more than one way. First, as proposed by Cohen, by activating a regulatory network, they would hold down exuberant, potentially pathogenic immune responses. More recently, tissue-specific T-cell activation occurring in response to trauma in the central nervous system was suggested to represent a mechanism of attempted tissue regeneration (Schwartz et al 1999). Indeed, a detailed series of experimental studies from Michal Schwartz and her group provides impressive evidence in support of this concept. Transfer of central nervous system autoreactive T-cell lines improves regeneration of severed central nervous system tissues in the optic nerve, spinal cord and retina (Schwartz and Kipnis 2001). In a key experiment, activated T cells were infused into Lewis rats after a unilateral optic nerve crush trauma. Myelin basic protein autoreactive, but not ovalbumin specific, T cells protected local axons from degeneration, as reflected by a threefold higher survival rate of relevant retinal ganglion cells (Moalem et al 1999). Active immunization against myelin basic protein or transfer of myelin basic protein-specific activated T cells, protected rats from degeneration of spinal cord motor neurons after contusion (Hauben et al 2000a) or ventral root avulsion (Hammarberg et al 2000). Intriguingly, neuroprotection derives not only from classically autoreactive T cells, specific to ‘true’ autoantigens such as myelin basic protein, but also from T cells responding to altered peptide ligands. Vaccination with a partially agonistic myelin basic protein ligand protected spinal cord motor neurons from crush-induced degeneration (Hauben et al 2001). Similar therapeutic effects were obtained by vaccination with copolymer-1 (Kipnis et al 2000), which probably acts as a super-altered peptide ligand variant of myelin basic protein (see Chapter 18).
Therapeutic vaccination in neurodegeneration is an intriguing concept that is not yet fully understood or free from debate (Popovich and Jones 2003). Several properties of the central nervous system-directed autoimmune response act in favour of the approach. First, autoimmune T cells accumulate preferentially in central nervous system areas with ongoing degeneration, a behaviour that helps to target the therapy. This has been known ever since S. Levine and Hoenig (1968) showed in rat experimental autoimmune encephalomyelitis that immune cells strongly invade brain areas exposed to thermal injury but not the unlesioned tissue. Selective T-cell infiltration is also noted in central nervous system areas affected by peripheral nerve axotomy. Clipping of a facial nerve, for example, leads to retrograde degeneration of the facial nucleus embedded in the brainstem. In unilaterally axotomized rats, myelin-specific T cells infiltrate the lesioned facial nucleus, but avoid the contralateral intact side (Flügel et al 2000; Maehlen et al 1989). T-cell migration targeted to degenerative tissues has also been noted in the optic nerve (Hickey 1991; Konno et al 1990).
The signals that lure T cells into degenerative lesions are poorly understood. Endothelial cells of the local blood–brain barrier may be activated following neuronal injury, expressing profiles of cell adhesion molecules that favour inflammatory cell entry (Andjelkovic and Pachter 1998). No less important for degenerative lesions are the membrane profiles that support immune responses assumed by glial cells. Facial nerve axotomy leads to production and expression of MHC antigens, costimulatory structures, cell adhesion molecules and soluble mediators that all promote presentation of local (auto)antigen to T cells (Raivich et al 1999). The central nervous system milieu becomes immune friendly, allowing efficient processing of central nervous system autoantigen from cellular debris and its presentation to invading autoimmune T cells – as in antigen spreading following central nervous system infection (Vanderlugt and Miller 2002).
How might autoimmune T cells protect neurons from death and promote cell regeneration? It is notable that activated T cells produce and secrete a plethora of soluble mediators, cytotoxic as well as proregenerative. For example, T cells release neurotrophic factors including nerve growth factor (Ehrhard et al 1993) and brain-derived nerve growth factor (Kerschensteiner et al 1999), neurotrophin-3 (Besser and Wank 1999), and glia cell-derived neutrotrophic factor (GDNF) (Kerschensteiner et al 2003). Local deposition of neurotrophins could reduce cyto- toxic inflammation, as has been found using nerve growth factor-engineered T-cell lines (Kramer et al 1995), and at the same time encourage survival and regeneration of central nervous system cells (Hammarberg et al 2000; Moalem et al 2000). In addition, proinflammatory mediators, like TNF-α, are known to play a dual role in the central nervous system. At high concentrations, TNF-α clearly damages central nervous system cells in vivo (Probert et al 1995), while in vitro (Nicholas et al 2002) it plays a role in tissue protection (see Chapter 10). In explant cultures, TNF-α protects hippocampus cells against the toxic effect of β-APP (Barger et al 1995) and glutamate (Cheng et al 1994). TNF-α also protects neurons in vivo from traumatic damage (Sullivan et al 1999).
To conclude, the concept of beneficial autoimmunity in the central nervous system and its application to therapy of central nervous system degeneration are fascinating. At present, the most formidable hurdle preventing practical application is the encephalitogenic potential of autoreactive T cells. Research must find ways to tune down the autoaggressive power of central nervous system reactive cells, while preserving or even amplifying the beneficial capacity of these cells. Newly emerging techniques of genetic engineering may realize that goal (T.N.M. Schumacher 2003).
PATHOGENESIS OF DEMYELINATION AND TISSUE DAMAGE
A variety of different mechanisms have been defined in vitro that lead, directly or indirectly through an effect on oligodendrocytes, to the destruction of myelin sheaths. These can loosely be considered as having immunological or neurobiological bases. We refer readers to Chapter 10 for additional discussion, especially of the latter category, but reiterate the point that the division is to some extent artificial, the evidence indicating complexity and interplay with, for example, an altered growth factor environment in the nervous system influencing the outcome of superimposed inflammatory injury. That said, the immunological mechanisms described here include both antigen-specific and nonspecific immune processes targeted against several components of the axon-glial unit. T-cell cytotoxicity is mediated either by αβ (D'souza et al 1995) or γδ T lymphocytes (M.S. Freedman et al 1991), as well as antibody-mediated immune reactions targeting antigens expressed at the surface of myelin or oligodendrocytes (Linington et al 1988). Although it is easy to define such mechanisms in vitro, their role in the complex in vivo situation is more difficult to characterize.
Inflammation and tissue injury induced by Th1 and Th2 cells
MHC class II-restricted T lymphocytes, polarized to the production of classic proinflammatory cytokines such as IFN-γ and TNF-α (Th1), enter the central nervous system compartment in the process of immune surveillance and, encountering specific antigen, become reactivated and start an inflammatory process. These Th1 cells can themselves be cytotoxic and kill targets in the process of antigen presentation (D. Sun and Wekerle 1986). This direct cytotoxicity, however, appears to be limited in vivo, due to the low or absent MHC class II expression on glia cells within the lesions (Vass and Lassmann 1990). Th1 cells produce high amounts of proinflammatory cytokines, which attract haematogenous macrophages and activate the local microglia population.
Experimental models, induced by the transfer of autoreactive Th1 cells, are characterized in general by intense inflammation with rather limited tissue injury (Ben-Nun et al 1981a). An exception to this general rule is Th1-mediated brain inflammation in mice. In this species, a chronic disease with quite extensive demyelination and tissue destruction is induced even after a single transfer of myelin basic protein-reactive encephalitogenic Th1 cells (Mokhtarian et al 1984). Clinical disease and tissue damage in Th1-mediated encephalitis correlates with the extent of macrophage, but not T-cell infiltration (T. Berger et al 1997).
Class II-restricted T cells, polarized to the production of IL-4, IL-5 and IL-10, were initially regarded as having a role in downregulation of brain inflammation (during recovery from experimental autoimmune encephalomyelitis: Issazadeh et al 1995a; 1995b). Under favourable conditions, Th2 cells can themselves induce brain inflammation (Lafaille et al 1997). However, brain lesions in this model are fundamentally different compared with those induced by Th1 cells. They are characterized by massive nonselective tissue damage, associated with dense tissue infiltration by granulocytes, including basophils (Lafaille et al 1997). Although this model is rather artificial, requiring profound immunosuppression of the recipients, it shows that central nervous system-specific Th2 cells have, in principle, a pathogenic potential. Indirect evidence suggests that this may be the case in experimental autoimmune encephalomyelitis induced with myelin oligodendrocyte glycoprotein, where sensitization procedures that preferentially stimulate a Th2 response influence severity of disease and the extent of tissue damage (Genain et al 1996; Stefferl et al 1999). In these conditions, autoantibodies play a major role in the pathogenesis of lesions (Stefferl et al 1999). Infiltration of eosinophilic granulocytes (Storch et al 1998b) and the particular profile of antibody isotypes, indicate a Th2-driven immune response (Tsunoda et al 2000). Although such a spectrum of changes is rare in classic multiple sclerosis, it is found in the pathology of Devic's type of neuromyelitis optica (Lucchinetti et al 2002). As with Th1 cells, the damage induced by Th2 cells is most probably mediated by activated effector cells rather than the T cells themselves.
Tissue destruction through class I MHC-restricted cytotoxic T cells
There is recent evidence pointing to CD8+ cytotoxic T cells as effectors in the lesions of multiple sclerosis (Friese and Fugger 2005). Cell destruction through cytotoxic T cells can be mediated through either the release of cytotoxic granules (mainly perforin and granzyme B) or the activation of cell death receptors on the target cells, such as Fas or other receptors of the TNF receptor family. All central nervous system neuroectodermal cells can be induced to express MHC class I molecules and are lysed by cytotoxic class I MHC-restricted T cells in vitro (H. Neumann et al 2002). Although cell death generally ensues through the induction of apoptosis, specific attack by CD8+ T cells may exclusively affect the axon, leaving the rest of the neuron intact (Medana et al 2001b). The pathway of cell destruction mediated by cytotoxic granules or death ligands depends on the activation state of T cells as well as properties of the target cells. Thus under comparable in vitro conditions, in contrast to astrocytes, neurons may be protected against granule-mediated cytotoxicity, whilst still sensitive to lysis mediated through the activation of death receptors (Medana et al 2000). Transgenic models, in which virus antigens are selectively expressed in different central nervous system cells, show that class I MHC-restricted T cells can reach and destroy their targets in vivo (Cabarrocas et al 2003; C. F. Evans et al 1996; Oldstone and Southern 1993). In addition, new models of brain inflammation have been developed, induced by passive transfer of autoreactive class I MHC-restricted T cells (Huseby et al 2001; D. Sun et al 2001). Polarization to Tc1 cells (production of IFN-γ) and activation prior to transfer was required to induce brain inflammation and disease (Cabarrocas et al 2003; Huseby et al 2001; D. Sun et al 2001).
Brain inflammation induced by cytotoxic CD8+ T cells differs in several essential aspects from that mediated by class II MHC-restricted cells. In the situation of mild inflammation associated with exquisitely selective destruction of antigen-containing target cells (and in the absence of any bystander damage of other tissue elements), inflammation is reflected by T-cell infiltration and microglial activation, with limited recruitment of haematogenous macrophages (Cabarrocas et al 2003). When the inflammatory reaction is more vigorous, tissue injury is induced, which in many respects resembles that seen in hypoxic brain damage (Huseby et al 2001). This may be due to liberation of antigen within the lesions and its diffusion to brain vessels. There, it may possibly be directly recognized by cytotoxic T cells at the luminal surface of endothelial cells, leading to vasculitis and thrombotic vessel occlusion. Alternatively, cytotoxic T cells may damage the nervous tissue by a nonspecific bystander mechanism. It has recently been shown that, after brain infection with JHM virus (mouse hepatitis virus strain JHM) in immunodeficient animals, the transfer of activated CD8+ but not CD4+ T cells directed against an irrelevant epitope of a completely unrelated virus may induce focal demyelination. Perhaps, these activated nonspecific cytotoxic T cells are recruited to sites of brain infection and induce tissue damage through activation of the local microglia population (Haring et al 2002; Haring and Perlman 2003).
The importance of class I MHC-restricted cytotoxic T cells in mediating clinical disease and tissue damage in the central nervous system is highlighted in the model of Theiler's virus-induced demyelinating encephalomyelitis. Studies on the genetic susceptibility to this disease (Altintas et al 1993; Rodriguez et al 1986) and disease induction in β2-microglobulin-deficient mice (Rivera-Quinones et al 1998) show that the clinical and pathological phenotypes depend on a class I MHC-restricted T-cell response. Disease can also be blocked by treatment with anti-CD8 antibodies (Rodriguez and Sriram 1988). Specific deletion of a virus peptide-specific class I-restricted T-cell response preserves motor function in infected animals (K.P. Johnson et al 2001). Cytotoxic T cells appear also to play a major role in the pathogenesis of demyelination and tissue damage in multiple sclerosis (H. Neumann et al 2002; see Chapter 12). CD8+ cells dominate the T-cell infiltrates in all lesions, and clonal expansion within the central nervous system and cerebrospinal fluid mainly reflects this subset. Furthermore, a large proportion of CD8+ T cells in actively demyelinating lesions express granzyme B. They are seen in close contact with injured oligodendrocytes and axons.
Antibody-mediated demyelination and tissue injury
We have illustrated the ways in which antibody plays a major role in the induction of demyelination and tissue injury in the central nervous system. This is particularly evident in models of chronic experimental autoimmune encephalomyelitis in rats, guinea pigs and primates, where a good correlation exists between the antibody response and amount of demyelination (Linington and Lassmann 1987; Stefferl et al 1999). The requirement for antibody to induce demyelination is epitope expression on the surface of myelin or oligodendrocytes. So far, only very few myelin antigens fulfil this criterion. More prominent are glycolipids (Dubois-Dalcq et al 1970) and myelin oligodendrocyte glycoprotein (Linington and Lassmann 1987). When demyelinating antibodies are injected intravenously into animals with experimental autoimmune encephalomyelitis, induced by encephalitogenic T cells at the point when T-cell-mediated inflammation becomes apparent in the central nervous system, they massively augment clinical disease and induce widespread demyelinating lesions restricted to the central nervous system (Linington et al 1988; Schlüsener et al 1987). Furthermore, size and shape of the demyelinating lesions depend upon the balance between encephalitogenic T lymphocytes and demyelinating antibodies (Lassmann et al 1988). A high T-cell response together with low antibody titres leads to inflammatory infiltrates ubiquitously distributed throughout the central nervous system, and associated with a small rim of perivascular demyelination. This pattern is similar to that found in acute disseminated encephalomyelitis. In contrast, a mild T-cell-mediated encephalomyelitis together with a pronounced demyelinating antibody response results in the occurrence of few focal plaques with extensive, confluent demyelination. This pattern more closely resembles that found in acute or chronic multiple sclerosis. Furthermore, when such cotransfers of encephalitogenic T cells and demyelinating antibodies are repeated several times, persistent demyelinating lesions with extensive gliosis and impaired remyelination are found, in most respects resembling those found in patients with multiple sclerosis (Figure 11.13: Linington et al 1992). In this situation, encephalitogenic T cells are required to prime the central nervous system parenchyma and open the blood–brain barrier to allow the entry of myelin oligodendrocyte glycoprotein monoclonal antibodies. Demyelination is due to antibody assisted by accessory macrophages and/or complement (Linington et al 1989; Piddlesden et al 1993). Depletion of macrophages strongly reduces the demyelinating effect of cotransferred anti-myelin-associated glycoprotein antibody (Huitinga et al 1995), while intrathecal injection of macrophage-activating IFN-γ amplifies antibody-dependent demyelination (Vass et al 1992). Depletion of complement ameliorates the demyelination (Piddlesden et al 1991; 1994). A similar disease is induced by direct sensitization of susceptible animals with myelin oligodendrocyte glycoprotein (Figures 11.13 and 11.15). Antibody converts monophasic and nondemyelinating experimental autoimmune encephalomyelitis in the Lewis rats immunized with myelin basic protein to a chronic relapsing variant characterized by large-scale demyelination (Johns et al 1995). Similar disease is inducible by myelin oligodendrocyte glycoprotein in some mouse strains (Amor et al 1994; Mendel et al 1995) and, as it now turns out, primates (Genain et al 1996). Thus, to date, myelin oligodendrocyte glycoprotein is the only myelin component that elicits B- and T-cell autoimmune responses that combine to establish histological changes reproducing many features typical of the lesions seen in multiple sclerosis (Storch et al 1998a).
An issue of major clinical and diagnostic relevance is therefore which myelin oligodendrocyte glycoprotein antibodies can induce demyelination. Myelin oligodendrocyte glycoprotein is a small folded glycoprotein that lies deeply embedded in the oligodendrocyte cell membrane (Kroepfl et al 1996). Thus, only a very small domain of the myelin oligodendrocyte glycoprotein molecule is exposed on the cell surface. Most antibodies binding peptide sequences of myelin oligodendrocyte glycoprotein or denatured protein in an ELISA or Western blot do not recognize intact antigen on the cell surface (Brehm et al 1999).
Direct evidence for a role of anti-myelin oligodendrocyte glycoprotein antibodies in the pathogenesis of demyelination has recently been described in primates with experimental autoimmune encephalomyelitis and in a subset of multiple sclerosis patients (Genain et al 1999). Here, the authors used labelled myelin-associated glycoprotein in glutaraldehyde-fixed and plastic-embedded tissue to detect antibody within actively demyelinating lesions, and found binding to damaged myelin. However, in support of this interpretation, O'Connor et al (2005) recently eluted anti-myelin oligodendrocyte glycoprotein immunoglobulin from the lesions of multiple sclerosis. This was interpreted as direct evidence for opsonization of degenerate myelin by anti-myelin-associated glycoprotein antibodies. That leaves unanswered the question of why labelled myelin-associated glycoprotein sticks selectively to damaged myelin sheaths, but not the more abundant supply in B lymphocytes and plasma cells. Furthermore, the studies were performed with labelled peptides – epitopes not recognized by demyelinating antibodies in intact oligodendrocytes or myelin. Amplification of tissue damage by antibodies within inflammatory brain lesions may not be restricted to demyelination and oligodendrocyte injury. In the search for other antibodies capable of damaging the axon-glial unit, circulating antibodies against AN-2 (an antigen expressed on oligodendrocyte progenitor cells) have been detected in patients with multiple sclerosis (Niehaus et al 2000). Clearly, such antibodies have the potential to eliminate progenitor cells from the lesions of multiple sclerosis in those lesions where shortage of progenitors contributes to the failure of remyelination.
Antigen-independent tissue injury by toxic macrophage products
Macrophages and activated microglia play an important role in the induction of tissue damage in the central nervous system. They are highly activated within acute and chronic inflammatory lesions. In most experimental autoimmune encephalomyelitis models, the extent of tissue damage correlates with macrophage infiltration (T. Berger et al 1997). In general, macrophage depletion ameliorates clinical disease and tissue damage (Huitinga et al 1990). Macrophages destroy tissue through the action of secreted toxic products in an antigen-independent and nonselective manner. Nevertheless, damage to different cellular components of the nervous system by macrophage toxins follows a hierarchical pattern. Myelin and oligodendrocytes are most susceptible followed by neurons and axons. In contrast, astrocytes and microglia are relatively resistant. Thus, a classic delayed-type hypersensitivity reaction, in which tissue injury is mainly mediated by activated macrophages and microglia, is reflected by a pseudoselective pattern of tissue damage showing primary demyelination with a variable extent of axonal and neuronal injury (Matyszak et al 1997; T.A. Newman et al 2001). It follows that selective primary demyelination associated with an inflammatory central nervous system lesion does not necessarily reflect a specific immune reaction against myelin antigens.
Proteases
Macrophages and microglia cells can induce tissue damage through a variety of different toxic molecules, including proteases, complement components, cytotoxic cytokines, reactive oxygen and nitrogen intermediates, and even excitotoxins. Proteases play a major role in the pathogenesis of inflammatory lesions in the central nervous system (Cuzner and Opdenakker 1999). This is best exemplified by the beneficial effects of protease inhibitors in the treatment of experimental autoimmune encephalomyelitis (Brosnan et al 1980; Clements et al 1997; Gijbels et al 1994) or other conditions of T-cell-mediated brain inflammation (Matyszak and Perry 1996c; T.A. Newman et al 2001). Proteases are produced by macrophages and microglia (Clements et al 1997; Teesalu et al 2001) and by astrocytes (Teesalu et al 2001). In general, they are secreted as inactive precursors requiring cleavage in order to become biologically active. Activity is strictly controlled, for example by tissue inhibitors of metalloproteases (TIMPs).
Proteases serve different functions in inflammation of the central nervous system. They are instrumental in the migration of inflammatory cells through vascular barriers and extracellular space by dissolving intercellular junctions or the extracellular matrix. It follows that inhibition of proteases has a direct anti-inflammatory effect. The decrease of enhancing MRI lesions in patients with multiple sclerosis treated with IFN-β may in part be due to inhibition of matrix metalloproteases (Leppert et al 1996). By cleaving proteins already released into the extracellular space, proteases increase the amount of peptides available for antigen presentation and therefore augment the immune reaction (Fabry et al 1994; Opdenakker and Van Damme 1994). In addition, proteases are directly involved in the induction of tissue damage, such as demyelination and axonal injury (Anthony et al 1998; Matyszak and Perry 1996c; T.A. Newman et al 2001).
Complement
There is good agreement that components of the complement system enter the brain under conditions of blood–brain barrier damage. Complement is also produced locally in pathological conditions by resident central nervous system cells (Morgan et al 1997). Deposition of complement components, including the lytic C9 neoantigen, characterizes a subset of multiple sclerosis patients (D.A.S. Compston et al 1989; Lucchinetti et al 2000; Prineas et al 2001; Storch et al 1998a). In general, experimental autoimmune encephalomyelitis induced in complement-deficient animals shows attenuated disease severity by comparison with wild-type controls (Mead et al 2002; Nataf et al 2000; G. T. Tran et al 2002). Complement may influence tissue injury in several ways. The induction of antibody-mediated demyelination is largely mediated through complement activation. Thus, demyelination and the subsequent axonal injury are effectively blocked in myelin oligodendrocyte glycoprotein-associated experimental autoimmune encephalomyelitis, induced in complement-deficient mice (Mead et al 2002). However, there are also effects of complement other than those attributable to the effect of antibody. The proinflammatory effects of leucotactic fragments, released in the course of complement activation, may explain why complement inhibition or deficiency not only blocks demyelination, but can also ameliorate inflammation in models of autoimmune encephalomyelitis – even in the absence of pathogenic antibodies (Nataf et al 2000; Piddlesden et al 1991; G. T. Tran et al 2002). In addition, complement may be activated in the central nervous system by components of myelin in the absence of antibody following the interaction of myelin oligodendrocyte glycoprotein and C1q (T.G. Johns and Bernard 1997). Activated complement can lyse rat oligodendrocytes in vitro without anti-myelin antibodies (Piddlesden and Morgan 1993). Oligodendrocytes are more sensitive to this effect than other central nervous system cells (Benn et al 2001). Human oligodendrocytes, however, are less susceptible to complement-mediated damage since they express at least some cell surface complement inhibitory proteins (Scolding et al 1998a; Zajicek and Compston 1995). Complement also plays a role in the phagocytosis of myelin fragments through an interaction with the complement receptor 3 on microglia (Reichert and Rotshenker 2003).
Tumour necrosis factor α
TNF-α has pleotropic functions. It is expressed within the active lesions both of experimental autoimmune encephalomyelitis and multiple sclerosis (Issazadeh et al 1995a; Selmaj et al 1991b), produced by activated microglia and astrocytes (Frei et al 1987; Lieberman et al 1989). TNF-α exerts systemic immunoregulatory functions, but may act locally within lesions to act as a proinflammatory cytokine directly destroying oligodendrocytes (Probert et al 2000; Selmaj and Raine 1988). In transgenic animals overexpressing TNF-α under central nervous system-specific promoters, the pathology of lesions depends on the cellular source and amount of TNF-α produced. With low expression levels, no spontaneous disease occurs, but inflammation and demyelination are augmented in pre-existing experimental autoimmune encephalomyelitis (Taupin et al 1997). High levels in immature oligodendrocytes lead to spontaneous primary demyelination, while the expression in astrocytes is associated with very severe inflammation, demyelinating lesions and profound axonal injury (Akassoglou et al 1998). These effects are all mediated by signalling through the TNF-R1 pathway. Large vessel vasculitis with thrombosis and brain infarction induced in TNF-α transgenic animals, results in signalling exclusively through the TNF-R2 pathway (Akassoglou et al 2003).
These results in transgenic animals clearly highlight the pathogenic potential of TNF-α in the central nervous system and suggest that therapeutic blockade of this cytokine may have a beneficial effect in multiple sclerosis. Many studies have dealt with the effect of TNF-α blockade in experimental autoimmune encephalomyelitis either through pharmacological intervention or gene deletion. Although a beneficial effect has often been found, the results remain diverse and not easy to formulate (Willenborg et al 1995). Our synthesis is that, without TNF-α or TNF-R1, the extent of tissue injury is ameliorated, but with a variable effect on inflammation. That said, in some studies inflammation is augmented (Eugster et al 1999) and clearance of T-cell infiltration by apoptosis decreased (R. Bachmann et al 1999). Furthermore, major variations are found in the effects of TNF-R1 deletion on experimental autoimmune encephalomyelitis depending on mouse strain (Kassiotis et al 1999; Körner et al 1997; J. Liu et al 1998). Thus, TNF-α appears to be an important cytotoxic cytokine within inflammatory brain lesions, and blockade of its action may be beneficial. Conversely, the same molecule appears instrumental in immune regulation, involving the elimination of pathogenic T cells by apoptosis. Blockade of this function may augment inflammation and promote clinical disease (Probert et al 2000). Finally, TNF-α may also promote the survival and proliferation of oligodendrocyte progenitor cells, potentially contributing to remyelination (Arnett et al 2001).
Reactive oxygen and nitrogen intermediates
Reactive oxygen and nitrogen species, both produced by activated macrophages and microglia in their defence against pathogenic microorganisms, are highly cytotoxic molecules that can induce cell membrane damage and apoptosis. They can act synergistically through the formation of more toxic molecules such as peroxynitrite. Within the central nervous system they mediate a variety of toxic effects, and these can be highly relevant for the pathogenesis of demyelination and axonal injury (K.J. Smith and Lassmann 2002; K. J. Smith et al 1999; Willenborg et al 1999). Both sets of molecules are cytotoxic for oligodendrocytes and, as with most other macrophage toxins, selectively expose the myelin/oligodendrocyte complex (Y.S. Kim and Kim 1991; Merrill et al 1993; Noble et al 1994). However, as discussed in Chapter 13, nitric oxide may induce conduction block and degeneration when axons are electrically active (Kapoor et al 1999; K.J. Smith et al 2001a) due to modification of ion channels (Bielefeldt et al 1999) and impaired mitochondrial function (Bolanos et al 1997).
These observations identify reactive oxygen and nitrogen species as attractive candidates for therapeutic intervention in inflammatory demyelinating disease. Perhaps we should have expected that pharmacological blockade of nitric oxide production, or genetic deletion of inducible nitric oxide synthase, would yield the variety of conflicting results in experimental autoimmune encephalomyelitis – in many instances, aggravating, not settling the disease process (K.J. Smith and Lassmann 2002; Willenborg et al 1999). The explanation for this paradox lies in the additional roles of nitric oxide in regulating the inflammatory process. Nitric oxide inhibits T-cell activation (Albina and Henry 1991), downregulates endothelial adhesion mole-cule expression (Kubes et al 1991) and induces T-lymphocyte apoptosis (Okuda et al 1997; Zettl et al 1997). It is not surprising that inhibition of these activities augments the inflammatory process and exaggerates clinical manifestations of the disease.
Excitotoxins
Several studies suggest that excitotoxins contribute to tissue damage in inflammatory demyelinating lesions. First, the NMDA (N-methyl-d-aspartate) receptor inhibitor memantine was shown to inhibit experimental autoimmune encephalomyelitis (Wallstrom et al 1996). Later, blockade of AMPA/kainate receptors was shown to modify clinical disease and reduce demyelination as well as axonal and neuronal injury in animals with experimental autoimmune encephalomyelitis (Pitt et al 2000; T. Smith et al 2000). Excitotoxins are released from neurons as a consequence of brain damage. Microglia also represent a potent source of glutamate and the neurotoxin quinolinic acid (Espey et al 1997; Heyes et al 1996; Lehrmann et al 2001). Glutamate homeostasis is also implicated in the lesions of multiple sclerosis, as reflected by increased glutaminase expression in macrophages or microglia and reduced expression of glutamate dehydrogenase in oligodendrocytes (Werner et al 2001). Finally, downregulation of glutamate transporters, as seen in the lesions of experimental autoimmune encephalomyelitis, may also increase the extracellular concentration of excitotoxins (Ohgoh et al 2002). Oligodendrocytes are highly susceptible to damage mediated by excitotoxins in vitro and in vivo (Cammer 2002; Matute et al 2001). Excitotoxic mechanisms are implicated in inflammatory demyelinating lesions targeting neurons in grey matter (T. Smith et al 2000).
Taken together, the immune system evidently packages a large repertoire of mechanisms for eliminating pathogens. However, the same molecules are also able to attack the body's own tissue in the context of autoimmunity. Since many of these mechanisms act in parallel, it follows that therapeutic blockade of one component may inadvertently affect another, yielding a mixed package of responses. In addition, several mechanisms instrumental in mediating tissue injury – for instance TNF-α or nitric oxide – are simultaneously used by the immune system in immune regulation, seeing off inflammatory cells once their task is fulfilled. In this context, the net effect of therapeutic blockade is often to exaggerate inflammation and the expression of disease – an inherently difficult problem to overcome in the treatment of immune-mediated diseases. But (as we discuss in Chapter 12) it remains possible that, in a disease like multiple sclerosis, one or other mechanism is dominant – generally or in particular groups – and so can be selectively targeted for disease-modifying effects.
PERIPHERAL BLOOD BIOMARKERS FOR MULTIPLE SCLEROSIS AND DISEASE ACTIVITY
Clinical and experimental investigations into multiple sclerosis and its experimental models provide an avalanche of data, in particular with regard to immune aspects of the disease. Clinicians and patients can be forgiven for expecting that much of this information can be applied to the diagnosis of human disease and in the assessment of treatment efficacy. But, sadly, this is not the case. In fact, many neurologists have been disappointed by the halting progress in this field. Up to now, there are only very few immunological biomarkers that are accepted as reliable and practicable parameters for determining the nature and state of activity of a given patient's affliction, or that follow the influence of any one drug on the disease process and its activity. At present, we use certain biomarkers to complement and confirm clinical, imaging or other findings, but there is no immunological test that convincingly diagnoses multiple sclerosis.
Biomarkers are needed to illuminate different aspects of the disease and its mechanisms. Apart from the initial diagnosis, they are required to monitor the course, potentially to help classify particular subsets of the disease and, ideally, to select optimal treatment and determine its efficacy over time. The current status of biomarkers for diagnosis and management of multiple sclerosis patients has been admirably summarized in a critical and comprehensive review by Bielekova and Martin (2004). The main results of this survey are shown in Table 11.3 .
Table 11.3.
Biomarkers for diagnosis and management of multiple sclerosis
| Evaluated biomarkers according to categories | Biomarkers with potential for further development | Biological rationale | Correlation with disease activity | Correlation with disability progression | Correlation with treatment effect | Notesa |
|---|---|---|---|---|---|---|
| Biomarkers reflecting alteration of the immune system | Unlikely candidates for surrogate end points; may prove useful in studying disease heterogeneity and in developing of new therapies | |||||
|
The most extensively studied biomarkers in multiple sclerosis | |||||
| IL-1, IL-2, IL-6, IL-10, IL-12, IL-18, TNF-α, LT-α/β, TGF-β, CD25 | IL-6 (+ soluble interleukin sIL-6R and soluble glycoprotein sgp130) | +++b | ++/– | + | +/+ | Candidate cytokine system linking innate immune system with both arms of adaptive immune responses (T and B cells) |
| IL-10 | ++ | ++/– | + | +/+ | Candidate immunoregulatory cytokine | |
| IL-12 (p70)/IL-23 | +++ | ++ | ++ | +/+/+ | Suggested as biomarker that can differentiate between relapsing-remitting and secondary progressive stages of multiple sclerosis | |
|
Biomarkers that may aid in studying disease heterogeneity and on proof of principle in therapy trials | |||||
| CCR5, CXCR3, CXCL10, CCR2/CCL2 | CCR5 | ++ | ndc | +/– | Suggested as a candidate biomarker of Th1 T cells | |
| CXCR3/CXCL10 | ++ | ++ | nd | − | Marker of activated T cells | |
|
The least systematically studied category with some interesting novel markers; e.g. diagnostic relevance of antibody in neuromyelitis optica. These biomarkers need systematic development and standardization of techniques | |||||
| CSF IgG index, κ light chains, oligoclonal bands, anti-MBP Ab, anti-MOG Ab | Anti-MBP and anti-MOG Ab | +++ | nd | nd | nd | Suggested as a possible diagnostic tool for predicting the development of definite multiple sclerosis after first clinical symptom (clinically isolated demyelinating syndrome) |
|
Biomarkers needed for assessment of disease heterogeneity (based on pathological classification of multiple sclerosis lesions) and for development of novel therapies | |||||
|
Activated neo-C9 | ++ | + | + | nd | Biomarker reflecting formation of membrane-attack complex (MAC) that is expected to contribute to demyelination at least in a subgroup of multiple sclerosis patients |
|
It is unlikely that these biomarkers would become more useful than MRI-based markers of blood–brain barrier dysfunction | |||||
| E-selectin, L-selectin, ICAM-1, VCAM-1, CD31, surface expression of LFA-1 and VLA-4 | ||||||
|
Very important category, little explored; needs further development for multiple sclerosis | |||||
| CD40/CD40L, CD80, CD86, heat-shock proteins (hsp) | CD40/CD40L | ++ | + | nd | + | Suggested as candidate biomarker that can differentiate between relapsing–remitting and secondary progressive stages of multiple sclerosis |
| hsp | + | nd | nd | nd | Dysregulation in the heat-shock protein system is the most prominent and consistent result of gene expression studies in multiple sclerosis and other autoimmune diseases | |
|
Markers reflecting activation of the innate immune system would contribute to studies of disease heterogeneity and aid in selection and screening of prospective novel immunomodulatory agents | |||||
| CD26, CD30, CD71, perforin, OX-40 (CD134), osteopontin, macrophage-related proteins MRP-8 and MRP-16, neopterin, amyloid A protein, somatostatin | Neopterin | ++ | ++ | nd | +/– | |
|
Very important category of biomarkers because they may reflect both defects in regulation of immune cells as well as proapoptotic properties of central nervous system components | |||||
| Fas (CD95) and Fas-L, FLIP, Bcl-2, TRAIL | FLIP | ++ | + | + | + | Anti-apoptotic protein overexpressed in multiple sclerosis |
| TRAIL | +/? | nd | nd | + | Suggested as biomarker reflecting clinical response to IFN-β therapy in multiple sclerosis | |
|
Potentially very interesting biomarkers that need to be developed further; would contribute to disease heterogeneity studies and to development of process-specific therapies | |||||
| BDNF expression | ||||||
|
Markers studied predominantly in the past; many should be reassessed by new, more precise techniques | |||||
| NK cells, Vα24+ NKT cells, CD4+/CD25bright and IL-10-producing immunoregulatory T cells, CSF cells, CD45RA−/RO+/CD4+ (memory) T cells | CD4+/CD25bright T cells and IL-10-producing regulatory T cells, regulatory NK cells and NKT cells | +++ | + | nd | +/nd | These cellular subpopulations were shown to have important immunoregulatory roles in animal models and other human autoimmune disorders and they merit careful evaluation in multiple sclerosis |
|
Although potentially very interesting, these assays are very tedious and therefore are likely to remain restricted to early phases of drug development and to proof-of-principle clinical trials | |||||
| Proliferation assays (Ag-specific and polyclonal), cytokine-secretion assays, cytotoxic assays | ||||||
| Biomarkers of blood–brain barrier disruption | It is unlikely that these biomarkers would become more useful than MRI-based markers of blood–brain barrier dysfunction | |||||
| MMPs and their inhibitors (TIMP), platelet-activating factor, thrombomodulin | ||||||
| Biomarkers of demyelination | Would greatly enhance the understanding of MRI/pathological correlations and have a potential for partial surrogacy | |||||
| MBP and MBP-like material, proteolytic enzymes, endogenous pentapeptide QYNAD, gliotoxin | QYNAD – endogenous peptide with Na-channel blocking properties | ++ | nd | nd | nd | QYNAD is an endogenous substance in cerebrospinal fluid that probably originates from proteolytic cleavage during inflammation; deserves further evaluation |
| Biomarkers of oxidative stress and excitotoxicity | Very important biomarkers from the standpoint of disease heterogeneity and potentially for development of novel therapies; need to be developed further | |||||
| NO and its stable metabolites (nitrite NO2− and nitrate NO3−), uric acid, isoprostane, marker for hypoxia-like tissue damage in multiple sclerosis | NO (+ NO2− and NO3−) | ++ | +/– | –/nd | nd | May help in disease heterogeneity studies |
| Uric acid | ++ | ++ | + | +/– | Strong natural peroxynitrate scavenger | |
| Isoprostane | ++ | nd | + | + | Interesting candidate marker that merits further studies | |
| Marker for hypoxia-like tissue damage in multiple sclerosis | + | nd | nd | nd | Described in pivotal study as an endogenous epitope that is cross-recognized by monoclonal antibody against canine distemper virus and may become a diagnostic tool to identify specific multiple sclerosis subtype | |
| Biomarkers of axonal/neuronal damage | Most likely category of biomarkers with surrogate potential in multiple sclerosis | |||||
| Cytoskeletal proteins (actin, tubulin and neurofilaments), tau protein | Neurofilaments: light subunit (NF-L) | +++ | + | + | nd | Might be the most likely candidate for surrogacy; its development warrants further efforts |
| Tau protein | ++ | ++ | nd | nd | Also potentially very useful biomarker that needs further development | |
| Biomarkers of gliosis | May be useful for disease heterogeneity studies but with unpredictable surrogacy potential | |||||
| GFAP, S-100 proteins | ||||||
| Biomarkers of remyelination and repair | Much-needed biomarkers that would guide development of repair-promoting strategies in multiple sclerosis and aid in disease heterogeneity studies | |||||
| NCAM, CNTF, microtubule-associated protein-2 exon 13 (MAP-2 + -13), protein 14-3-3, CPK-BB, peptidylglycine α-amidating monooxygenase (PAM), neural-specific enolase (NSE) | NCAM – neural cell adhesion molecule |
+ |
+ |
nd |
nd |
Very sparse data on both biomarkers. However, because these are so far the only potential candidates, their evaluation warrants further effort |
| CNTF – ciliary neurotrophic factor | ++ | nd | nd | nd | ||
Ab = antibody; Ag = antigen; CNTF = ciliary neurotrophic factor; CSF = cerebrospinal fluid; FLIP = Fas-associated death domain-like interleukin-1β-converting enzyme inhibitory protein; GFAP = glial fibrillary acidic protein; hsp = heat-shock protein; ICAM-1 = intracellular adhesion molecule-1; IFN = interferon; Ig = immunoglobulin; IL = interleukin; LP = lumbar puncture; LT-α/β = lymphotoxin α/β; MAC = membrane-attack complex; MBP = myelin basic protein; MOG = myelin oligodendrocyte glycoprotein; MMP = matrix metalloproteinase; NCAM = neural cell adhesion molecule; NF-L = neurofilament light subunit; NK cells = natural killer cells; NKT cells = NK-like T cells; NSE = neuron-specific enolase; OCB = oligoclonal bands; PAM = peptidylglycine α-amidating monooxygenase; TGF-β = transforming growth factor β; TIMP = tissue inhibitor of matrix metalloproteinases; TNF-α = tumour necrosis factor α; VCAM-1 = vascular cell adhesion molecule 1.
BDNF = brain derived neurotrophic factor
CPK-BB = creatine phosphokinase-BB
LFA = lymphocyte function antigen
TRAIL = tumour necrosis factor related apoptosis-inducing ligand
VLA = very late appearing antigen
Simplified summary of reviewed biomarkers in multiple sclerosis taken from Bielekova and Martin (2004).
Brief opinion of the authors about the potential use of the biomarkers and the need for further developments.
We attempted to grade the strength of supportive evidence for each characteristic of the biomarker as low (+), medium (++) and high (+++). In treatment effects +/– implies positive correlation with one type of therapy and negative with another.
No reliable data.
Markers for the diagnosis of multiple sclerosis
multiple sclerosis presents a stunning variety of clinical symptoms, and histological lesion patterns, a complexity that often mimics other conditions of the central nervous system. Therefore a doubtfree diagnosis in an early stage of the disease is particularly difficult. Definite multiple sclerosis is often diagnosed only after repeated bouts, that is, only after some time following disease onset. Then, diagnosis is mainly based on a combination of clinical features with imaging, and cerebrospinal fluid abnormalities (oligoclonal immunoglobulin bands). The latter changes can be counted among immunological biomarkers, but they are not specific for multiple sclerosis. Additional supportive immune biomarkers are badly needed for the initial diagnosis but, to date, there is no such test that could be used on a routine basis.
Markers for disease diversity (pathogenesis)
As has been emphasized repeatedly, multiple sclerosis can evolve in distinct patterns and these variations in the clinical course may, in turn, reflect distinct pathogenic processes. Making these distinctions would assume additional importance if it were to be shown that the various disease subtypes require different therapeutic approaches. However, classification of lesion subtypes in multiple sclerosis has mainly emerged from morphological studies of post-mortem tissue samples with rather few available biopsies during the lifetime of the patient who, we hypothesize, might benefit from knowledge of their specific pathological features. For obvious ethical reasons, such biopsy material can be accessed only in very rare cases. Pieces of the central nervous system cannot be taken merely to confirm suspected multiple sclerosis, but the procedure may be justified to exclude other disorders, such as tumours.
Hence there is an urgent need for easily accessible biomarkers. As an obvious candidate, autoantibodies could serve as markers for the putative subtype mediated by humoral immunity. As described in Chapter 16, a subset of patients responds to plasma exchange, perhaps through the removal of circulating autoantibodies.
Lennon et al (2004) recently reported a possible immune biomarker for neuromyelitis optica (Devic's disease). A tight correlation was demonstrated between an IgG antibody binding to mouse brain sections, identified as acquaporin-4 (Lennon et al 2005), and the diagnosis of neuromyelitis optica. Time will tell whether this finding becomes of generalized use for diagnosis of this particular subset, and whether similar strategies will help to identify other categories of patients with multiple sclerosis.
Markers for the disease course
Especially in the relapsing–remitting phase, it would be of practical value to gain insight into the degree, distribution and consequences of the pathogenic inflammatory mechanisms. Is there a smouldering process that may evolve into overt clinical disease or is the force driving inflammation all but extinguished? Specific information would be invaluable for tailoring optimal treatment schemes according to the patient's current needs. Furthermore, relapse might be predicted and intercepted before the clinical manifestations erupted. Numerous activity-related blood and cerebrospinal fluid markers have been proposed and investigated, in particular those that relate to an ongoing inflammatory process (see box below).
Methods for detecting potential biomarkers.
Soluble molecules (released into body fluids)
-
•
ELISA (enzyme-linked immunosorbent assay)
-
•
Western blotting (electrophoresis-separated proteins identified by specific antibody binding)
Cellular markers
-
•
FACS cytofluorometry (membrane markers, soluble factors)
-
•
Elispot (cytokine released by individual cells, bound to surfaces and identified by specific antibodies)
Molecular markers
-
•
TPCR-related gene amplification
More hopeful are studies that use anti-myelin autoantibodies as a potential biomarker. Recently, Villar et al (2005) identified lipid-binding IgM antibodies in the oligoclonal cerebrospinal fluid bands, which (in their trial) predicted a particularly severe clinical course. T. Berger et al (2003) measured anti-myelin oligodendrocyte glycoprotein autoantibodies in a series of freshly diagnosed cases with possible/probable multiple sclerosis. High antibody titres were associated with a more severe disease course compared to cases with low titres or no antibody responses. Another study correlated the level of autoantibody titres against native glycosylated myelin oligodendrocyte glycoprotein with disease activity (Gaertner et al 2004). Both studies are intriguing, but it should be noted that the findings have not been repeated in other patient populations studied with similar technology (Lampasona et al 2004; Lim et al 2005). Without more information, we are unable to reconcile whether technical or other factors are responsible for the discrepancies.
Markers of therapeutic response
As a rule, drug treatments have diverse effects – some good, others not; they all require monitoring. Most obvious is the intended therapeutic effect, which ideally would be matched by clinical improvement. For this purpose, biomarkers are selected to resemble those used to follow the natural history of the disease. In addition, for most treatments, especially in the case of immunosuppressive or disease-modifying therapies, adverse effects must be recognized and, where necessary, quantified. Finally, biomarkers inform directly on the successful introduction and function of biological agents. The classic example is the MxA protein (‘myxovirus resistance protein A’), the product of a gene induced by type I interferons. MxA-detecting immune assays are now commonly used as a sensitive parameter for bioavailability of therapeutic interferons (von Wussow et al 1990).
Biomarkers: assays and accessibility
To be useful, biomarkers should be clinically informative, correlated with a clinical outcome, easily measurable, and routinely accessible. On a more theoretical level, the biomarker might either relate to a mechanism that directly contributes to development and course of the disease, or serve as a marker of mechanisms that protect from or reduce the disease process.
The choice of suitable methods to monitor biological structures is not the limiting factor: a large repertoire of modern assays exists that is capable of measuring molecular and cellular components of body structures (Table 11.3). These allow the fast and reliable identification and quantification of soluble proteins circulating in body fluids, or of structures located on the surface of tissues or single cells.
A real problem in the context of multiple sclerosis is accessibility of biologically meaningful samples. Unfortunately, the most informative sample, namely the pathological lesion itself, is inaccessible, although the cerebrospinal fluid (see below) provides indirect access to markers of the disease process. But, even cerebrospinal fluid sampling at lumbar puncture requires a clear indication and cannot be repeated at the whim of the investigator. Cerebrospinal fluid originates from the interstitial fluid that drains tissues in the central nervous system. Therefore it contains some, but by no means all, soluble molecules interchanged between the brain parenchyma and cerebrospinal fluid-filled spaces (Teunissen et al 2005). Many details of its composition remain enigmatic. Thus, the character of cerebrospinal fluid antibodies, which form the characteristic oligoclonal bands, remains a matter of debate. Some investigators relate these proteins to processes that are considered relevant to the development of lesions in the central nervous system; other commentators, by contrast, maintain that the pathogenic autoantibodies are retained in the affected tissue, and only irrelevant antibodies are shed into the accessible cerebrospinal fluid. The same is true for the immune cells that are found in this compartment, and for their soluble products such as cytokines.
Other body fluids – such as blood, urine and saliva – are freely accessible to monitoring, but remote from the local pathogenic process. These fluids may display changes reflecting local processes powerful enough to spill over into the systemic circulation where, however, they may be overshadowed by unrelated events happening outside the central nervous system. Further, at least in theory, there may be systemic changes that have a direct impact on activity of the disease process in multiple sclerosis. Examples include concomitant infections with their multiple possibilities to act on pathogenic autoaggressive T cells (reviewed in this chapter). Such procedures have not, however, become part of routine clinical practice.
Markers that have been proposed and studied
Table 11.3 summarizes a large number of biomarkers, compiled by Bielekova and Martin (2004). The list contains:
-
•
markers of immune activity – cytokines, chemokines, soluble membrane structures, proteases, peripheral immune cells, autoantigen reactivity, T-cell receptor profile, myelin autoantibodies, transcriptomics, Th1/Th2 profiles
-
•
markers of neuronal damage (S100, neuronal filaments, etc.) and myelin damage (soluble myelin proteins)
-
•
indicators of increased permeability through the blood– brain barrier in the form of blood-derived proteins.
Monitoring of these markers has been proposed for diagnosis, disease course (state of activity), therapeutic efficiency and, to a modest degree, subtype identification.
In addition, markers for drug bioavailability and drug-induced adverse effects are of importance. In the case of IFN-β, the interferon-induced MxA protein, which indicates the presence of biologically active interferon in the patient's system, and (conversely) the detection of interferon neutralizing antibodies (a response that can interfere with therapeutic efficiency) have entered clinical practice. In the case of treatment with glatiramer acetate, several groups recommend ELISPOT assays, which document cytokine conversion of T cells from Th1 to Th2 profiles.
Whilst there is perfect agreement that the identification of immunological biomarkers would be of enormous help for diagnosis, classification, and monitoring of natural course and therapeutic efficiency, only very few such markers are used widely in clinical practice. One example is the detection of oligoclonal immunoglobulin in the cerebrospinal fluid, a time-honoured assay. Other tests, based on the detection of specific antibodies in the central nervous system, have raised much interest but have yet to stand the test of time.
MARKERS OF MULTIPLE SCLEROSIS AND DISEASE ACTIVITY IN CEREBROSPINAL FLUID
The central nervous system is encased in strong layers of bone and connective tissue. These barriers protect the brain from traumatic injury but also make it inaccessible to the investigator. This inconvenience has impeded research into multiple sclerosis and other neurological diseases, but two windows permit some insight into the process of brain inflammation. One is the eye, which is commonly affected and where ophthalmoscopy can directly illuminate pathological processes (see Chapter 13: W.I. McDonald 1986). The other is cerebrospinal fluid, routinely sampled by lumbar puncture. It provides information that supplements clinical evidence for the diagnosis (see Chapter 7), whilst also offering clues to the pathogenesis. Typically, in patients with multiple sclerosis, cerebrospinal fluid contains abnormal levels of immunoglobulins distributed as oligoclonal bands, and inflammatory cells, which are mainly CD4+ T lymphocytes and macrophages/monocytes. Immunoglobulins and T cells are both implicated in the autoimmune pathogenesis of multiple sclerosis. Although their direct roles and relative contributions remain to be determined, at the very least each contains signatures of the disease process and its activity.
However, the cerebrospinal fluid only partially reflects events taking place within central nervous system lesions. In immunological terms, the meningeal and subarachnoid compartment is different from the brain parenchyma (Matyszak and Perry 1996b; Schnell et al 1999). Furthermore, due to the narrow extracellular space, diffusion of immunological mediators is restricted, and thus cerebrospinal fluid does not necessarily mirror the composition of extracellular fluid in the parenchyma, especially when lesions are buried deep in the parenchyma. Pathogenic leucocytes, antibodies and inflammatory mediators are preferentially trapped within lesions, while those not directly involved in lesion pathogenesis may remain in the perivascular and meningeal compartments (Flügel et al 2000). For these reasons, effective spillover of inflammatory components into the cerebrospinal fluid will only take place in the presence of multiple lesions or when they touch the meningeal or ventricular surface, and when massive oedema and widening of the extracellular space facilitate diffusion to the extracellular space. This is obviously not the case in many patients with multiple sclerosis, and direct correlation between cerebrospinal fluid alterations and lesional pathology is frequently disappointing. Thus, interpretation of cerebrospinal fluid changes in multiple sclerosis requires consideration of the basic physiology of its production and turnover.
Physiology of cerebrospinal fluid
The brain and spinal cord are bathed in cerebrospinal fluid. This reservoir fills the inner ventricles and outer subarachnoid space, including its cisterns. This compartment is sealed from the dural and epidural space by a tight layer of arachnoid membrane and separated, in turn, from the systemic circulation by the blood–brain barrier. There is communication with the extracellular space of the brain and spinal cord, although diffusion is limited by the narrow extracellular space in central nervous system tissue and by cellular layers of the ependyma and glia limitans (J.C. Lee 1972; see also Chapter 10).
Production, turnover and resorption
The molecular composition of cerebrospinal fluid and its sites of production, physiological turnover and resorption are reviewed in detail by Bradbury (1979) and Fishman (1992). Whereas its ionic composition is consistent with a plasma ultrafiltrate, the low protein content is explained by a stringent blood–brain barrier for nonpolar high molecular weight solutes (Felgenhauer 1974). The choroid plexus is the major source of cerebrospinal fluid. About 10% is of local extracellular origin, draining primarily into the perivascular (Virchow–Robin) spaces (Cserr 1984). From there, diffusion occurs into the subarachnoid compartment. In periventricular white matter, the brain extracellular fluid accumulates along fibre tracts and then passes through the ependymal lining of the ventricular walls (Weller et al 1992). It has to be remembered, however, that cerebrospinal fluid proteins are not exclusively derived from the choroid plexus and the brain extracellular space. A variety of proteins (enzymes, transport proteins, growth factors and cytokines) can be directly synthesized by leptomeningeal cells (Ohe et al 1996) and by leucocytes present in all central nervous system compartments (Renno et al 1994; Waage et al 1989).
Under pathological conditions, exchange between the brain extracellular space and cerebrospinal fluid appears much more pronounced, largely for two reasons. First, in conditions of acute pathology, the extracellular space is enlarged by vasogenic oedema, which facilitates the bulk flow of fluid into this compartment (J.C. Lee 1972). Secondly, as a result of tissue damage and scar formation, the perivascular spaces are enlarged, due to perivascular fibrosis (see Chapter 12). This may lead to the formation of connective tissue channels similar to lymphatic capillaries in other tissues (Prineas 1979). Since the central nervous system extracellular space is in continuity with the cerebrospinal fluid, alterations of the extracellular milieu in the brain and spinal cord are, in principle, reflected in the composition of the fluid. There are, however, important limitations. Due to the narrow extracellular space in the nervous tissue, diffusion of molecules and migration of cells are somewhat limited (Cserr et al 1992) – the rate at which molecules move depending on radius (in a hydrated form) and charge. In general, diffusion is most rapid for small anionic or uncharged molecules. Large or cationic material moves more slowly. For example, after intrathecal injection, neutral protein tracers freely disperse in the extracellular space, whereas cationic molecules, such as myelin basic protein, largely remain trapped in the meningeal compartment (Vass et al 1984). In addition, molecules liberated in the central nervous system extracellular space may be removed by local uptake in macrophages (Broadwell and Salcman 1981) and also in neurons and glia. The uptake of different serum proteins into local macrophages in the lesions of multiple sclerosis lesions is well documented (see Chapter 12). Even more specific and effective uptake occurs with autoantibodies that are involved in the pathogenesis. These may be absorbed from cerebrospinal fluid through complexing with respective target brain antigens. For example, demyelinating activity is generally lower using cerebrospinal fluid than corresponding serum samples from patients with multiple sclerosis (S.U. Kim et al 1970). It is to be expected that antibody directed against any surface component of normal or pathological central nervous system tissue will largely be removed from the cerebrospinal fluid. Conversely, antibody directed against intracellular antigens, or targets not present in the central nervous system, will persist and may be enriched in the cerebrospinal fluid. In other words, the antigen specificity of cerebrospinal fluid antibodies shows a spectrum reflecting bystander reactions, not necessarily of relevance to the pathogenesis. One method for detecting antibodies that may directly be involved in tissue damage is to define antigen specificity by analysing antibodies secreted in vitro by B lymphocytes harvested from cerebrospinal fluid (J. Sun et al 1991a; 1991b) rather than by direct determination of antibody titres (Xiao et al 1991).
Taken together, the evidence suggests that – with respect to proteins and small molecular weight solutes (and acknowledging major inherent limitations) – cerebrospinal fluid offers a useful diagnostic window allowing the indirect study of pathological events taking place in the central nervous system.
Source and turnover of cells
As in the central nervous system parenchyma, a small number of mononuclear cells (0.3–6.2/μL) are normally present in the cerebrospinal fluid. These mainly consist of lymphocytes (86%) and monocytes/macrophages (12%) (Sörnäs and Östlund 1972). In a concise review, Oehmichen et al (1982) concluded that the normal turnover of cells in cerebrospinal fluid is below the limits of detection by transfer of labelled leucocytes, with a low rate of cell division, in vivo and in vitro, and a small number of cells undergoing necrosis – together indicating extremely low turnover under normal conditions and mainly resulting from proliferation and death of cells already present. In addition, the influx of haematogenous cells into the cerebrospinal fluid compartment in the context of inflammation is also supplemented by up to 25% local cell proliferation.
This traditional view now requires modification in the light of more recent experimental data. Activated T lymphocytes can pass through the normal blood–brain and cerebrospinal fluid barriers suggesting that, following peripheral immune activation, T cells selectively enter the cerebrospinal fluid compartment. In terms of numbers, five- to tenfold more lymphocytes express activation markers, such as IL-2 receptors and MHC class II antigens, in cerebrospinal fluid compared with peripheral blood. But the percentage of activated T lymphocytes in cerebrospinal fluid does not differ between patients with inflammatory and noninflammatory neurological diseases (Hafler et al 1985b; Noronha et al 1980; Tournier-Lasserve et al 1987). Since it is unlikely that lymphocytes are locally activated in the central nervous system compartment in noninflammatory diseases of the nervous system, these data suggest that lymphocytes enter the cerebrospinal fluid already activated, but are rapidly removed whilst continuing to express late activation markers. Kivisäkk et al (2003) used a large panel of antibodies against chemokine receptors and adhesion molecules, postulating that the normal cerebrospinal fluid mainly contains activated central memory T cells, which enter through the choroid plexus and the meninges in a P-selectin dependent manner. Conversely, the entry and turnover of monocytes may be much slower. It takes several months for donor-derived cells to repopulate the meninges in radiation bone marrow chimeras (Hickey and Kimura 1988; Hickey et al 1992). Slow turnover of cerebrospinal fluid monocytes is further supported by the observation that phenotypically they show a lower degree of activation than those in the peripheral circulation, and no differences in activation of monocytes are found in patients with inflammatory compared with noninflammatory neurological diseases (Salmaggi and Sandberg-Wollheim 1993). Little is known about the migratory properties and turnover in the cerebrospinal fluid of other inflammatory cells, such as B lymphocytes or natural killer cells. Granulocytes are extremely rare in normal cerebrospinal fluid, despite the local availability of lipopolysaccharide, cytokines and chemokines that trigger a granulocyte-dominated inflammatory reaction in the meningeal space and cerebrospinal fluid compartments (Andersson et al 1992; Bell et al 1996; Quagliarello and Scheld 1992). By comparison with the brain parenchyma, meningeal vessels appear more permeable both to proteins (Westergaard and Brightman 1973) and leucocytes (Perry et al 1995). This principle is clearly exemplified by T-cell-mediated brain inflammation in experimental autoimmune encephalomyelitis, in the many variants of which there is a dominance of meningeal over brain inflammation when the disease is induced by low numbers of encephalitogenic T cells (T. Berger et al 1997; Perry et al 1995). Furthermore, the dynamics and extent of brain inflammation are much more vigorous when foreign antigen or lipopolysaccharide is injected into cerebrospinal fluid compared with inoculation of the brain parenchyma (Matyszak and Perry 1996b; Perry et al 1995). This is also reflected by the presence of cells within the meninges and choroid plexus, which express dendritic cell antigens (Matyszak and Perry 1996c; McMenamin 1999; Pashenkov et al 2003b) and may be the origin of dendritic cells identified in cerebrospinal fluid (Pashenkov et al 2001).
In pathological conditions of the nervous system, leucocytes found in cerebrospinal fluid are derived not only from inflamed meningeal vessels but also as a result of cell migration from central nervous system lesions into the subarachnoid space or ventricles. It was noted in the earliest pathological studies on multiple sclerosis that debris containing macrophages in the spinal plaques of multiple sclerosis can pass directly into the superficial glia limitans to enter the spinal meninges and subarachnoid space (Marburg 1906). A similar exchange of inflammatory cells through the ependymal lining of the ventricles has been found in periventricular lesions of chronic experimental autoimmune encephalomyelitis (Lassmann et al 1981c). In addition, cellular drainage of deep white matter lesions can occur through the distended perivascular Virchow–Robin spaces (Prineas 1979). An additional barrier of leptomeningeal cells seals the perivascular from the subarachnoid space. Thus, although this barrier is permeable to fluid, solutes and the active migration of activated leucocytes, erythrocytes do not readily disperse into the Virchow–Robin space following subarachnoid haemorrhage (Hutchins and Weller 1986).
Lymphatic drainage
Labelled cells, erythrocytes or particulate material, injected into the cerebrospinal fluid, can reach regional lymph nodes (Oehmichen et al 1979; Weller et al 1992). Furthermore, haemosiderin-containing macrophages are found in peritracheal cervical lymph nodes in patients with subarachnoid haemorrhage (Oehmichen et al 1982). Antigen injected into the brain or cerebrospinal fluid may elicit specific immune reactions, predominantly in regional lymph nodes (Cserr et al 1992). de Vos et al (2002) described the appearance of macrophages with myelin degradation products in the cervical lymph nodes of animals with experimental autoimmune encephalomyelitis. These findings indicate active drainage of cells and antigens from the central nervous system to the lymphatic system. Several anatomical pathways have been identified. They include arachnoid channels in the cribriform plate (Weller et al 1992), tracking along cranial nerve roots (Bradbury and Cserr 1985), and additional pathways exiting through spinal root pockets (Zenker et al 1994). There are, however, several aspects that suggest much more complex drainage mechanisms. First, tracers injected on one side of the brain usually drain to ipsilateral lymph nodes (Yamada et al 1991). This argues against a diffuse drainage mechanism through the cerebrospinal fluid, and rather suggests the existence of more specialized lymphatic-like channels within the leptomeninges. These have recently been defined in periarteriolar meningeal sheaths (Preston et al 2003). The continuous leptomeningeal cell layer also inhibits diffusion of particulate material and nonactivated cells, even without sealing by continuous tight junction ridges. No detailed anatomical mapping of adhesion molecule expression, needed to enable the passage of activated cells, in meninges of the normal and diseased central nervous system is available. Meningeal cells from some mouse strains constitutively express P-selectin, and these show enhanced T-cell immune surveillance by comparison with animals lacking P-selectin expression in the meningeal compartment (Carrithers et al 2002). Finally, in contrast to the brain parenchyma proper, leucocyte apoptosis, as a mechanism for clearing inflammatory cells, rarely occurs in the meningeal space (J. Bauer et al 1998).
Multiple sclerosis-related cellular changes in cerebrospinal fluid
An increase in inflammatory cells in the cerebrospinal fluid is characteristic of multiple sclerosis. The total count may be 5–50 cells/mm3 in acute disease, of which the majority are T lymphocytes. Many express late activation markers, such as CD26 (formerly referred to as Ta1; Hafler et al 1985b). Cell adhesion molecules that are potentially involved in interaction with cerebrovascular endothelium (e.g. VLAs and other integrins) are also expressed on lymphocytes present in cerebrospinal fluid (Svenningsson et al 1993). On the basis of their chemokine receptor profile, these represent activated memory T cells of the CD4+ and CD8+ subset (Giunti et al 2003; Kivisäkk et al 2003). We interpret this lymphocyte marker profile on the basis that activated T cells are selectively recruited from the periphery to the central nervous system, and thus to the cerebrospinal fluid. It is worth considering that these lymphocyte populations, accumulating in the cerebrospinal fluid, include activated T cells effecting physiological immune surveillance of the central nervous system (Wekerle et al 1986). This interpretation is consistent with the finding that (notwithstanding differences in absolute count) a predominance of activated T cells is found in samples from individuals who are healthy as well as those with neurological disease (Hedlund et al 1989; Vrethem et al 1998). It has, however, to be emphasized that the preferential recruitment of activated memory T cells into the cerebrospinal fluid is more pronounced in normal controls than in patients with inflammatory brain disease, in whom there is apparently also a secondary recruitment of leucocytes more closely reflecting cell populations in the circulation (Kleine et al 1999). Thus the appearance of nonactivated naive T cells in the cerebrospinal fluid is associated with active inflammation or disease activity (Kraus et al 2000b). As in the brain parenchyma of patients with multiple sclerosis, it is the CD8+ T cell population in cerebrospinal fluid that undergoes preferential oligoclonal expansion (Jacobsen et al 2002).
B cells represent a small fraction of lymphocytes present in cerebrospinal fluid (Sandberg-Wollheim 1983). Whilst little information is available on their state of activation, a considerable number of B cells expressing cell surface CD5 is described in multiple sclerosis, although the functional status of these CD5+ cells is undefined (Mix et al 1990). They may be members of the primordial B-1 population (containing a large proportion of B cells producing polyreactive natural autoantibodies and giving rise to B-cell lymphomas: Kantor and Herzenberg 1993) or regular activated B lymphocytes (R.A. Miller and Gralow 1984; Werner et al 1989).
In a systematic study of cerebrospinal fluid cytology during the evolution of multiple sclerosis, Cepok et al (2001) described inter-individual heterogeneity of cerebrospinal fluid cytological profiles, independent of stage and activity of the disease. While all patients showed a dominance of T lymphocytes in the cerebrospinal fluid, others showed a predominance of B cells or monocytes. Patients with high B-cell counts and intrathecal IgG production followed a more rapidly progressive course than those in whom monocytes were mainly found (Cepok et al 2001). These findings raise the possibility that high B-cell responses carry a worse prognosis. It is claimed that early conversion to definite multiple sclerosis is associated with the presence of anti-myelin antibodies at presentation with a first demyelinating episode (T. Berger et al 2003). These differences in clinical behaviour are also consistent with the evidence from pathological analyses for disease heterogeneity (see Chapter 12).
Immune function of cerebrospinal fluid lymphocytes
If immunosurveillant T cells do accumulate in the healthy central nervous system, one would expect a similar (or even increased) accumulation in cerebrospinal fluid in patients with multiple sclerosis at times of disease activity. The immunological properties of lymphocytes recovered from cerebrospinal fluid have therefore been intensively scrutinized. This work relies on several methodological approaches, including establishment of lymphocyte lines and characterization of products secreted by single lymphocytes using immunospot and in situ hybridization assays. Primary limiting dilution experiments using polyclonal mitogen (PHA) as the proliferating stimulus yield clonal T-cell lines, but none of these responds to known myelin autoantigens (Hafler et al 1985a). This result, superficially disappointing, presumably reflects a relatively low frequency of brain-specific T cells within the general T-cell repertoire contained in cerebrospinal fluid. In fact, application of more refined techniques, primarily selecting for autoantigen specificity, has led to the isolation of myelin-specific T-cell lines. In particular, the split well technique (Figure 11.11: Pette et al 1990b) helped to establish panels of myelin basic protein-reactive T-cell lines from the cerebrospinal fluid of patients with multiple sclerosis. The enrichment of autoreactive T cells within the overall population is estimated to be about tenfold higher than in peripheral blood. Many of the cells recognize a myelin basic protein epitope dominant response, both from patients with multiple sclerosis and healthy controls, involving the centrally located p84–102 and the C-terminal p143–168 fragments (Zhang et al 1994). γδ T cells, the lymphocytes implicated in lesions of multiple sclerosis that are seen to regenerate, have been isolated by several groups. Vδ1, often in conjunction with Vγ1 usage, was noted in one study of cerebrospinal fluid-derived T-cell lines from patients with multiple sclerosis. Some showed reactivity against human glioma determinants (Nick et al 1995). Another study demonstrated reactivity of γδ T cell receptor cell lines from patients with multiple sclerosis to heat-shock protein 70 (but not heat-shock protein 65; Stinissen et al 1995).
A second approach, pioneered by the groups of Tomas Olsson and Hans Link, for studying the functional repertoire contained within the cerebrospinal lymphocyte pool, relies on single-cell assays for secreted protein products (immunoglobulins or cytokines) and immunologically relevant mRNA. T lymphocytes isolated from peripheral blood or cerebrospinal fluid of patients with multiple sclerosis are exposed in vitro to putative autoantigens to allow specific activation of autoreactive lymphocytes. Cultured cells are then tested for release of activation-dependent cytokines by direct immunostaining. This extremely sensitive immunospot approach has demonstrated remarkably large proportions of myelin basic protein-specific T cells in peripheral blood, and even higher frequencies in the cerebrospinal fluid of patients with multiple sclerosis (Olsson et al 1990a). Enrichment of autoreactive T cells in cerebrospinal fluid was also found for other putative myelin autoantigens, such as proteolipid protein (J. Sun et al 1991a), myelin associated glycoprotein (Söderström et al 1994b) and myelin oligodendrocyte glycoprotein (Sun et al 1991b). The particular T-cell response profile is seen early in the course of the disease in the individual patient, and seems stable over time (Söderström et al 1994b). Although it is not surprising that myelin basic protein-reactive T cells are also enriched in optic neuritis (because of its close relationship to multiple sclerosis; Söderström et al 1994b), an unexpected elevation of such cells was documented in cerebrospinal fluid from patients with purely cerebrovascular diseases (W-Z. Wang et al 1992). Whilst these observations rely on the determination of secreted IFN-γ, the prototypic marker for Th1 cells, comparable analyses have more recently been extended to other cytokines such as IL-4, IL-10 and TGF-β (Söderström et al 1995) through the use of in situ hybridization. Preliminary data indicate a relative prevalence of IL-4 or TGF-β transcribing T cells in mild relapsing–remitting multiple sclerosis, with a reduction in clinically more severe cases (Link et al 1994).
Are cerebrospinal fluid lymphocytes involved in the pathogenesis of multiple sclerosis?
Close proximity of cerebrospinal fluid to the brain parenchyma, and the relative increase of myelin-specific autoreactive T lymphocytes within the mononuclear cell population in both compartments, might reasonably suggest that these T cells are autoaggressive immune cells. However, this is by no means proven. In fact, research using experimental models of myelin-specific autoimmune diseases (see above) shows that most, if not all, pathogenic T lymphocytes infiltrating the central nervous system die locally by apoptosis. Recirculation from the central nervous system to the periphery via the cerebrospinal fluid is assumed but not proven. Indeed, the origin of these cerebrospinal fluid cells is ambiguous. In experimental autoimmune encephalomyelitis, studies using cellular markers indicate that pathogenic T cells accumulate at high frequency in the parenchymal infiltrates, with many fewer cells in the perivascular and meningeal infiltrates. As in all inflammatory areas, post-activated T lymphocytes are additionally attracted to these locations in an antigen-independent manner. Thus, the enrichment of myelin-specific T cells in cerebrospinal fluid, both in multiple sclerosis and experimental autoimmune encephalomyelitis, may reflect their post-activated state rather than their recent participation in the demyelinating process.
Intrathecal immunoglobulin synthesis and oligoclonal bands
The central nervous system parenchyma is clearly not a physiological site for B-cell responses and antibody production, but individual B cells evidently can cross the blood–brain barrier, survive in the central nervous system and produce substantial amounts of immunoglobulin (Hickey 2001). Indeed B lymphocytes are readily demonstrable in a number of neuropathological contexts – such as viral encephalitides and lymphomas. In multiple sclerosis, small but consistent numbers of B lymphocytes at different stages of differentiation are present in inflammatory round cell infiltrates (see Chapter 12), as well as in the cerebrospinal fluid. B-lymphocyte infiltrations are also seen, although in variable degrees, in other pathological conditions,
Cellular basis of intrathecal immunoglobulin synthesis
Although the direct evidence is patchy, it seems likely that recirculating B lymphocytes share essential features with their T-cell counterparts (Hickey 2001). In the resting nonactivated stage, B lymphocytes are excluded by the specialized blood–brain barrier endothelium. However, after antigen-driven activation, B lymphoblasts cross into the central nervous system. In immunocompromised individuals, most primary intracerebral lymphomas are Epstein–Barr virus (EBV)-transformed B-cell lymphomas (Morgello 1995). When transferred to immunodeficient mice, these lymphomas grow well after injection via the intracerebral but not the intravenous route (Bashir et al 1991; Nakamine et al 1991). This suggests that the brain parenchyma is not completely hostile to (xenogeneic) B cells and that specific contact-dependent interaction between recirculating B cells and the cerebral endothelium is required for correct lymphocyte migration through the blood–brain barrier. In xenogeneic (mouse/human) combination, this necessary fit of cell adhesion molecules is not reproduced. Rather, EBV-transformed B cells express a number of lymphocyte activation markers – most prominently MHC class II, costimulatory factors (B7-1 and -2) and cell-adhesion molecules, including LFA-1, LFA-3 and the ICAM family (Clark and Lane 1991). Although not clearly defined, a particular profile of surface adhesion molecules along with activation-dependent membrane proteases and chemokines must work together, selectively to allow activated B lymphocytes access to the brain parenchyma across the blood–brain barrier (Ambrosini et al 2003; Columba-Cabezas et al 2003).
The central nervous system lacks all stroma cells – such as follicular dendritic cells (which shape the germinal centres of secondary lymphoid follicles), and the fibroblast-like sinus cells that support B-lymphocyte formation in the bone marrow that normally make up the microenvironment of B-cell tissues (Weissman 1993). But even in the absence of these specialized lymphoid cells, there is no shortage of available cytokines in the central nervous system for survival and differentiation of antibody-forming B lymphocytes. While helper T cell-dependent cytokine signals are delivered by T-lymphocyte inflammatory infiltrates (e.g. IL-3, IL-4 and IFN-γ), local brain cells produce an impressive range of cytokines involved in B-cell biology. These include the proinflammatory mediators IL-1, TNF-α and IL-6; the chemokines MIP-1 and MIP-2; the B cell survival factor BAFF (Krumbholz et al 2005) and other cytokines of the epidermal, fibroblast and TGF families (Hopkins and Rothwell 1995). Each affects different aspects of B-cell function. Thus, for example, IL-6 was originally described as a B-cell growth factor (BGF-2) before its pleiotropic functions became evident. IL-6 is readily induced in brain microglia but also as part of the neuronal response to endotoxin, trauma, ischaemia and other insults. Besides acting on local brain cells, it has the capacity to maintain differentiation and activation of brain-infiltrating B cells up to the terminal plasma cell stage. IL-4, IFN-γ and TGF-β, and the chemokines MCP-1 and -2, influence immunoglobulin isotype switching and B-cell entry into the central nervous system.
Significance of intrathecal immunoglobulin for disease pathogenesis
In health, human cerebrospinal fluid contains little immunoglobulin or other plasma proteins. The appearance of immunoglobulin signifies pathological changes, reflecting either increased permeability of the blood–brain barrier or B cell-related immune processes active within the brain parenchyma (E.J. Thompson 1995). This is especially characteristic of samples taken from individuals with multiple sclerosis, which typically show immunglobulin distributed in oligoclonal patterns (see Chapter 7). Although not specific for multiple sclerosis, oligoclonal immunoglobulin bands have become a standard in providing laboratory support for the diagnosis of multiple sclerosis. Immunoglobulin permeating into the cerebrospinal fluid through a leaky endothelial barrier can be distinguished from that released by active plasma cells positioned within the brain or meningeal infiltrates by comparing the ratios of immunoglobulin and albumin concentrations in cerebrospinal fluid and plasma (the IgG index; see Chapter 7). Since albumin must reach the central nervous system from sources in the liver through the blood circulation, the albumin ratio can be considered as a pure permeability marker. Various formulas have been proposed for denoting the origin of immunoglobulins present in cerebrospinal fluid (Blenow et al 1994).
The elevated IgG index and presence of oligoclonal bands are general features of chronic brain inflammation and have achieved paraclinical diagnostic usefulness in multiple sclerosis (Fishman 1992; see also Chapter 7). It is the rare patient with multiple sclerosis who does not demonstrate oligoclonal bands and, in longitudinal studies, their absence may only be transient (Zeman et al 1996). However, the proportion lacking oligoclonal bands in the cerebrospinal fluid may be greater in cases with late-onset disease or chronic progressive disease variants (Pirttila and Nurmikko 1995) – perhaps reflecting contamination of these series with patients having other diagnoses. But, as discussed in Chapters 5, 7 and 8, the presence of oligoclonal bands is clearly low in other demyelinating disorders of the central nervous system, especially the optico-spinal form in Oriental peoples and in neuromyelitis optica (Fukazawa et al 1993; Wingerchuk et al 1999). Decoding the antigen specificity of oligoclonal IgG in multiple sclerosis has proved frustrating, and attempts to date have not illuminated our understanding of lesion pathogenesis. A large number of studies have focused on the occurrence of specific autoantibodies and found reactivity against myelin or oligodendrocyte proteins, glycolipids, axonal antigens, endothelial components and stress proteins (Archelos et al 2000; Ilyas et al 2003). Whether these antibodies are pathogenic is still unresolved.
Although brain specificity of oligoclonally distributed immunoglobulins has not regularly been shown, immunospot assays measuring myelin-specific immunoglobulin secretion by B lymphocytes harvested from cerebrospinal fluid in patients with multiple sclerosis show enhanced production of antibodies against a number of myelin proteins (M. Lu et al 1996). The groups of Hans Link and Tomas Olsson have used the immunospot assay to identify single B cells secreting brain-specific autoantibodies. Their results indicate that samples from patients with multiple sclerosis contain elevated numbers of myelin basic protein-specific B lymphocytes, but these do not change substantially with the clinical course (Link et al 1990; Olsson et al 1990a). As with T cells, the autoreactive B lymphocyte patterns are remarkably stable over time and do not parallel the clinical course (Link et al 1990). The B-cell response to myelin basic protein is directed against sequence p70–89. Reactions against proteolipid protein and myelin-associated glycoprotein have also been demonstrated (Baig et al 1991; Sellebjerg et al 1994). The antibody response against myelin-associated glycoprotein in the cerebrospinal fluid of patients with multiple sclerosis deserves special comment. These antibodies initiate myelin destruction in the presence of complement or macrophage-activating cytokines when injected into the cerebrospinal fluid of experimental animals (Vass et al 1992). Intrathecal production of anti-myelin-associated glycoprotein antibodies in patients with multiple sclerosis could therefore be directly involved in the pathogenesis of demyelination. By simple ELISA screening, anti-myelin-associated glycoprotein antibodies were detected only in a small number of patients with multiple sclerosis (Xiao et al 1991), perhaps because such antibodies are absorbed from cerebrospinal fluid by the excess antigen in brain tissue. With more sensitive techniques, such antibodies were found in the majority of individuals with multiple sclerosis but also in those with other inflammatory neurological diseases (Markovich et al 2003; Padberg et al 2001; Reindl et al 1999). For example, B lymphocytes secreting anti-myelin basic protein were present at high frequency in 8/10 patients with multiple sclerosis (J. Sun et al 1991b), and intrathecal synthesis of anti-myelin oligodendrocyte glycoprotein antibodies in multiple sclerosis is also reflected by an increased anti-myelin oligodendrocyte glycoprotein Ig index in cerebrospinal fluid (Reindl et al 1999). These data indicate that antibodies with demyelinating potential are produced intrathecally in patients with multiple sclerosis. However, anti-myelin oligodendrocyte glycoprotein antibodies, recognizing an epitope expressed on the surface of myelin and oligodendrocytes and therefore able to induce demyelination, may represent a small fraction of only the anti-myelin basic protein reactivity detected by ELISA or Western blot. Thus, the majority of such antibodies, present in cerebrospinal fluid, may not be directly involved in the demyelinating process (Haase et al 2001).
Using a broad-based new system of phage library screening for antigen specificity, no stereotyped pattern of intrathecal antibody production has been disclosed (Cortese et al 1996; 2001). For example, oligoclonal IgG in patients with multiple sclerosis contains reactivities against a variety of different virus antigens. But the patterns of antigen specificity are inconsistent and the virus-specific antibodies do not have the high affinity expected in the presence of specific antigen (Luxton et al 1995; Sindic et al 1994). More recent attempts to characterize oligoclonal bands include screening pooled cerebrospinal fluid from patients with multiple sclerosis against a murine oligodendrocyte precursor cell line-derived phage protein expression library. Five of seven positive clones, from amongst a pool of 1 × 106, contained an identical seven amino acid Alu repeat sequence spanning the B-cell epitope that reacted with 24/54 (44%) samples from patients with clinically definite multiple sclerosis, compared with <18% in patients with a variety of other neurological diseases. These differences were not seen when screening for reactivity to heat-shock protein (>50% in both groups), although antibody titres to both antigens were higher in the patients with multiple sclerosis (Archelos et al 1998). Finally, a recent study using expression arrays, identified Epstein–Barr virus proteins as common targets (Cepok et al 2005b). Taken together, it is likely that antibody responses in the cerebrospinal fluid of patients with multiple sclerosis, and contained in the oligoclonal response, represent a bystander reaction rather than direct markers of immune response driving the disease.
Soluble immune mediators in cerebrospinal fluid
Since multiple sclerosis is a chronic inflammatory disease of the nervous system, one would expect the cerebrospinal fluid to contain a variety of soluble immune mediators, such as cytokines, adhesion molecules and toxic effector molecules. By summarizing the data from a large number of studies on this topic, several general conclusions can be reached. First, a similar range and concentration of soluble immune mediators occur in the cerebrospinal fluid from patients with multiple sclerosis as in other chronic T-cell-mediated inflammatory diseases of the central nervous system and, to date, no disease-specific patterns have been revealed. Secondly, in general, the concentration of immune mediator proteins in cerebrospinal fluid correlates with the cellular reaction, indicating that inflammatory cells are their major source. Thirdly, cytokine patterns, as well as the appearance of antigens associated with inflammation in cerebrospinal fluid, may be useful in determining immunological activity of the disease process. Last, although not backed by much evidence, the patterns of toxic immune effector molecules in cerebrospinal fluid may help to identify mechanisms of tissue injury in these lesions.
Cytokines
Although early studies on cytokine patterns in cerebrospinal fluid from patients with multiple sclerosis were hampered by technical problems, improved assay sensitivity now makes it possible to detect practically all cytokines and chemokines (Navikas and Link 1996). There is a growing consensus that proinflammatory cytokines characterize cerebrospinal fluid during active stages of the disease, whereas anti-inflammatory mediators dominate the profile in remission (Rieckmann et al 1995). TNF-α appears to be particularly important during disease activity (Chofflon et al 1992; Hauser et al 1990; Maimone et al 1991b; Sharief and Thompson 1992; Spuler et al 1996; Tsukada et al 1991); levels correlate not only with clinical disease activity but also with measures of blood–brain barrier damage acting as a surrogate for acute inflammation (Sharief and Thompson 1992). This cytokine may be important not only for propagation of brain inflammation but also for playing a direct role in the destruction of myelin and oligodendrocytes. In addition, other cytokines that are important in regulation of T-cell-mediated inflammation, such as IL-2 (Gallo et al 1991; Sharief and Thompson 1993), IL-1 (Rovaris et al 1996), IL-10 (Navikas et al 1995), IL-15 (Kivisäkk et al 1998b), IL-18 (Losy and Niezgoda 2001) and lymphotoxin (Navikas et al 1996a), have been found. To this list can be added IL-4, IL-6 and Il-10 as indicators of intrathecal B-cell stimulation and antibody production (Nakashima et al 2000; Navikas et al 1996b; Perez et al 1995). But, again we should emphasize that the presence of proinflammatory cytokines in the cerebrospinal fluid is by no means specific for multiple sclerosis (Navikas and Link 1996).
Soluble adhesion molecules
Migration of inflammatory cells through the blood–brain barrier depends on the interaction between adhesion molecules on leucocytes and endothelial cells. Adhesion molecules in general are expressed on the surface of cells upon activation, but they may subsequently be shed and secreted. These soluble forms are not only reliable markers for cell activation in immunological processes, but they also function competitively to inhibit cell contact and migration. It is thus not surprising that practically all adhesion molecules, implicated in cell traffic through the blood–brain barrier, have been identified in soluble form in the cerebrospinal fluid (Dore Duffy et al 1995; S.J. Lee and Benveniste 1999; Mossner et al 1996; Sharief et al 1993; Tsukada et al 1993; 1995) and their concentrations differ with relapsing or progressive forms of the disease (Elovaara et al 2000). Serum levels of soluble ICAM-1 but not VCAM-1 correlate to some degree with MRI findings in relapsing–remitting and secondary progressive multiple sclerosis, and thus may offer an additional surrogate marker for disease progression (Giovannoni et al 1997). Soluble adhesion molecules are shed by leucocytes and resident meningeal cells (Jander et al 1993; Trojano et al 1996). Their concentration therefore correlates with pleocytosis (Sharief et al 1993) and is reduced after steroid treatment (Elovaara et al 2000). But, as with cytokines, the presence of soluble adhesion molecules in cerebrospinal fluid is not specific for multiple sclerosis (Rieckmann et al 1993; Mossner et al 1996).
Chemokines and chemokine receptors
Chemokines and their receptors are instrumental in the migration of inflammatory cells from the circulation to the central nervous system. Their patterns of expression within multiple sclerosis lesions reflect the composition of inflammatory infiltrates as well as activity and stage of the lesions (see Chapter 12). Although few members of the large family of chemokines and their receptors have so far been analysed in the cerebrospinal fluid of patients with multiple sclerosis, several general conclusions can already be reached. Overall, the patterns reflect the spectrum of inflammatory cells preferentially recruited into the central nervous system under normal and inflammatory conditions. Some apparently serve as useful markers for ongoing inflammatory activity. Furthermore, the patterns of chemokine and chemokine receptor expression in cerebrospinal fluid reflect those present within parenchymal lesions.
The cerebrospinal fluid from patients with multiple sclerosis contains elevated levels of CXCL 10 (previously called IFN-γ inducible protein 10; IP-10), and one of its receptors (CXCR 3) is expressed on the majority of lymphocytes present in cerebrospinal fluid (Narikawa et al 2004; T.L. Sorensen et al 1999). CXCR 3 is present on both T and B cells (T.L. Sorensen et al 1999; 2002b), and more detailed mapping of chemokine receptor expression in relation to other leucocyte markers shows that the population of activated memory cells is preferentially recruited (Giunti et al 2003; Kivisäkk et al 2003). This pattern of expression further suggests a bias in the recruitment of T cells polarized as Th1 and Tc1 cells (Giunti et al 2003; Misu et al 2001). Both are highly enriched in the active lesions of multiple sclerosis, but the same cell types also dominate the cerebrospinal fluid leucocyte population in controls (Kivisäkk et al 2002; 2003). These studies suggest that local production of IFN-γ in the central nervous system compartment induces the production of CXCL 10, which is then responsible for the dominant recruitment of Th1/Tc1 polarized T cells.
CCL2 (formerly monocyte chemoattractant protein 1; MCP-1) is present at increased concentration in cerebrospinal fluid from patients with multiple sclerosis and neuromyelitis optica compared with controls, and correlates inversely with disease activity (Franciotta et al 2001; Mahad et al 2002; Narikawa et al 2004; Scarpini et al 2002; T.L. Sorensen et al 1999). However, RANTES (regulated on activation, normal T cell expressed and secreted) is elevated in active lesions and may serve as a ligand for CCR 5 on T cells and monocytes (T.L. Sorensen et al 1999). Additional signals for cell recruitment are provided by the interaction of fractalkine (CX3CL1) and its receptor (Kastenbauer et al 2003) and from CCL 17, CCL 19 and CCL 12 (Narikawa et al 2004; Pashenkov et al 2003a). The latter may play a role in retention and recruitment of dendritic cells in the cerebrospinal fluid (Pashenkov et al 2003b).
Although these studies define the major cell populations recruited into the cerebrospinal fluid of patients with multiple sclerosis, it remains to be determined whether the cerebrospinal fluid reflects the subtleties of chemokine and chemokine receptor expression within lesions (see Chapter 12), and whether their subtypes are mirrored by characteristic chemokine patterns in the cerebrospinal fluid.
Markers of inflammation and effector molecules
Macrophages are the dominant cells in actively demyelinating multiple sclerosis lesions. Thus, despite relatively low numbers in cerebrospinal fluid, their products can be liberated in lesions and are then readily detected. As a rather nonspecific marker of macrophage activation, cerebrospinal fluid neopterin levels are elevated in patients with multiple sclerosis – in particular during active stages of the disease (Fredrikson et al 1987; Ott et al 1993; Shaw et al 1995). Neopterin is released by cytokine-stimulated cells of the macrophage-monocyte lineage and may appear not only in cerebrospinal fluid but also in urine. Serial measurement shows urinary neopterin/creatinine ratios of 187, 187 and 218 in patients with relapsing–remitting and primary or secondary progressive multiple sclerosis, respectively, compared with 134 in controls. More days showed elevated ratios amongst patients than controls, and there were sustained elevations following relapses and identified infections (Giovannoni et al 1997). Elevated levels of β2-microglobulin (Bjerrum et al 1988; Us et al 1989) and the soluble alpha-chain of class I are also found in cerebrospinal fluid (Fainardi et al 2002).
Activated macrophages produce a variety of other toxic effector molecules that may be directly involved in the formation of multiple sclerosis lesions. Protease activity, which can be attributed to different specific proteases (Akenami et al 1996; Gijbels et al 1992; Liuzzi et al 2002) and their inhibitors, is increased in cerebrospinal fluid from patients with multiple sclerosis (Banik 1992; Price and Cuzner 1979; Richards and Cuzner 1978). As with cytokines and adhesion molecules, the concentration of proteases correlates with pleocytosis (Gijbels et al 1992) and is also elevated in other inflammatory neurological diseases (Akenami et al 1996). Treatment with corticosteroids may reduce matrix metalloproteinase and increase proteinase inhibitor activity in the cerebrospinal fluid (Rosenberg et al 1996). Prostaglandins and leucotrienes have been identified in cerebrospinal fluid from patients with multiple sclerosis (Dore-Duffy et al 1991; Neu et al 1992).
Studies of cerebrospinal fluid provide evidence for complement activation in multiple sclerosis. Complement is implicated both as an amplifier of the inflammatory process and mediator of demyelination. The concentrations of C2 (Delasnerie-Laupretre et al 1981), C4 (Jans et al 1984) and C9 (D.A.S. Compston et al 1986; Halawa et al 1989; Morgan et al 1984) are decreased, whereas soluble C5-9 complexes can be detected (Mollnes et al 1987; Sanders et al 1986). These observations indicate intrathecal complement activation. Others have not reproduced the alterations in late complement component concentrations comparing samples from multiple sclerosis patients with controls (M. Rodriguez et al 1990) and, at best, there is a wide concentration range for cerebrospinal fluid C5-9 between patients (Sanders et al 1986). One interpretation is that such measurements identify a subgroup of patients with an antibody-dependent complement-mediated pathway of myelin destruction (see Chapter 12). The situation, however, appears more complex, and complement activation in the cerebrospinal fluid may merely reflect local interaction with soluble immune complexes (Jans et al 1984; Rudick et al 1985). In addition, lack of complement inhibitory proteins in the cerebrospinal fluid may by itself lead to complement activation when the blood–brain barrier is damaged. In line with this observation, a similar activation of terminal complement complexes has been found in patients with subarachnoid haemorrhage as that reported in multiple sclerosis (Lindsberg et al 1996). Nevertheless, activation of terminal complement complexes correlates significantly with neurological disability, suggesting that patients with severe and destructive lesions have higher complement activation in the cerebrospinal fluid (Sellebjerg et al 1998a).
The potential role of reactive oxygen intermediates in the pathogenesis of demyelination and axonal pathology in multiple sclerosis is reflected by the appearance of lipid peroxidation products (Calabrese et al 1998; Hunter et al 1985) and increased levels of isoprostane in the cerebrospinal fluid (Greco et al 1999). Even more intriguing is the observation of increased nitric oxide-producing cells (Xiao et al 1996). A component of the cerebrospinal fluid of patients with multiple sclerosis, apparently unrelated to proinflammatory cytokines (Köller et al 1996), has been reported to block sodium channel activity (Chapter 13: Brinkmeier et al 1993; 1996). Later, a large number of studies looked for nitrite and nitrate as footprints of nitric oxide production in the cerebrospinal fluid of patients with multiple sclerosis. Both are elevated, especially in patients with active disease (Brundin et al 1999; Danilov et al 2003; Giovannoni 1998; Sveningsson et al 1999; Yamashita et al 1997), their concentrations correlating with the extent of blood–brain barrier damage (Acar et al 2003; Giovannoni et al 1998). Clinically, high nitrite and nitrate levels are associated with prolonged relapse duration and a more pronounced response to treatment with corticosteroids (Sellebjerg et al 2002). Other nitrosylation-induced metabolites, as well as nitric oxide synthase activity, are enhanced in the cerebrospinal fluid of patients with multiple sclerosis (Calabrese et al 2002). In cooperation with reactive oxgen species, nitric oxide induces mitochondrial dysfunction leading to hypoxia-like tissue injury (see Chapter 12; K.J. Smith and Lassmann 2002). Recently a new marker (D-110) has been identified, which selectively labels hypoxic tissue. The D-110 protein is liberated into the cerebrospinal fluid and detected by ELISA or Western blot. It is increased in a subset of patients with multiple sclerosis, and preliminary evidence correlating tissue biopsy with cerebrospinal fluid measurement indicates that it may indeed identify the subset of patients with hypoxia-like tissue damage (Lassmann et al 2003).
Finally, the cerebrospinal fluid of patients with multiple sclerosis possesses neurotoxic or gliotoxic activity. A factor, toxic for glia cells both in vitro and in vivo, has been identified. This is a small protein encoded by endogenous retrovirus sequence (Benjelloun et al 2002; Menard et al 1997) and found mainly in patients with active disease (Pierig et al 2002). It is present in urine and reported to show 91% sensitivity and 97% specificity for multiple sclerosis (Malcus-Vocanson et al 1998). Although as yet unidentified, a further factor is described in the cerebrospinal fluid of patients with multiple sclerosis that is able to induce neuronal apoptosis in vitro in a caspase-dependent manner (Cid et al 2002; 2003). This factor is mainly enriched in samples from patients with active disease and its activity seems to correlate with the destructiveness of lesions, as visualized by MRI (Cid et al 2002).
Markers of central nervous system damage
Damage to central nervous system tissue liberates intracellular proteins, which may diffuse through extracellular space into the cerebrospinal fluid compartment. These proteins offer an opportunity for monitoring acute tissue destruction in the course of multiple sclerosis. The first myelin protein analysed for this purpose was CNPase, since this is fairly specific for myelin and its enzyme activity provides a simple detection system. Activity was increased in active stages of multiple sclerosis (Banik et al 1979; Eickhoff and Heipertz 1979) but not during remission (Banik et al 1979). However, a more reliable marker for demyelination is myelin basic protein (Cohen et al 1978). This parallels the increase in CNPase (Suda et al 1984). The validity of myelin basic protein as a marker of myelin destruction is supported by several investigations, although, not unexpectedly, it has become clear that its presence in cerebrospinal and other body fluids is not disease specific but also occurs in other conditions resulting in white matter destruction (Mukherjee et al 1985; Whitaker et al 1980). The detection of myelin basic protein in urine has not been adopted as a routine assay for assessing disease activity in multiple sclerosis.
Increased levels of myelin basic protein are present in cerebrospinal fluid during rather a short window of lesional activity (Gupta 1987; Lamers et al 1998; Sellebjerg et al 1998a; Thompson et al 1985). Detailed epitope mapping of myelin basic protein molecules in cerebrospinal fluid reveals that not all portions of the molecule are detected, and neoepitopes of degraded myelin basic protein, not recognized in the intact molecule, may appear (Whitaker 1998; Whitaker et al 1986). Whereas the liberation of myelin basic protein appears restricted to the stage of active demyelination, S-100 protein also remains elevated in the cerebrospinal fluid during remission (Massaro et al 1985). In contrast, glial fibrillary acidic protein, which is also released into the cerebrospinal fluid in patients with multiple sclerosis, correlates more closely with the extent of disability than with lesion activity (Rosengren et al 1995), and neural cell adhesion molecule (NCAM) peaks at the time of recovery, possibly indicating synaptic remodelling (Massaro et al 1987). Furthermore, release of neurofilament protein into the cerebrospinal fluid in multiple sclerosis (Lycke et al 1998; Semra et al 2002) and optic neuritis (E.T. Lim et al 2004) may reflect axonal damage in lesions. In addition, antibodies against neurofilament protein were found to correlate with disease progression and brain atrophy (Eikelenboom et al 2003; Silber et al 2002). For diagnostic purposes a combined analysis of different structural central nervous system components (myelin basic protein, S-100 and neuron-specific enolase) in the cerebrospinal fluid may be useful (Lamers et al 1995). Levels of the cytoskeleton protein tau and 14.3.3 in cerebrospinal fluid may reflect neuronal and axonal damage in the active lesions of multiple sclerosis (Bartosik-Psujek and Archelos 2004; Martinez-Yelamos et al 2001; Sussmuth et al 2001), although this has not been reproduced in another study (Jiminez-Jiminez et al 2002). A recent study describes the presence of Nogo-A antibodies in the cerebrospinal fluid (Reindl et al 2003). Antibody titres against this antigen were particularly high during the early stage of the disease, in patients with a relapsing disease course. They may be induced in the course of myelin and oligodendrocyte destruction within active lesions. It is not clear whether such Nogo-A antibodies are functionally active and may in part counteract the regeneration blockade mediated by Nogo.
Conclusion
With the introduction of MRI, the impact of cerebrospinal fluid analysis for the diagnosis of multiple sclerosis has declined, although, as we argue in Chapter 7, these laboratory investigations provide qualitatively different yet complementary information. In many institutions the view now prevails that lumbar puncture is no longer necessary in patients with multiple sclerosis. We hope that this attitude will change since, as we discuss here and in Chapter 12, the pathways involved in inflammation, demyelination and axonal destruction are diverse and require definition in each patient if treatment is to be individualized to suit people with specifically different pathological substrates. These classifications require an integrated approach combining clinical, neuroradiological and immunological analyses. In this respect, the analysis of cerebrospinal fluid offers a unique opportunity to define autoimmune responses in the central nervous system and to identify immunological pathways of myelin and axonal destruction.