

Abstract
The advent of magnetic resonance imaging (MRI) has contributed to increase the interest and awareness in childhood white matter disorders. Pediatric inflammatory demyelinating diseases of the central nervous system (CNS) are clinically heterogeneous with respect to their mode of presentation, clinical severity, rate of progression, and prognosis. Acute disseminated encephalomyelitis (ADEM) is an immune-mediated inflammatory disorder of the CNS, typically transitory and self-limiting. The highest incidence of ADEM is observed during childhood. It is characterized by an acute encephalopathy with polyfocal neurological deficits. In the absence of specific biological markers the diagnosis of ADEM is still based on clinical features and MRI evidence of widespread demyelination, after ruling out other possible explanations for an acute encephalopathy. Over the past decade, many retrospective patient studies have focused on clinical and neuroimaging features, in an attempt to define specific diagnostic criteria.
The occurrence of relapses in children with ADEM poses diagnostic difficulties in its differentiation from multiple sclerosis (MS) and neuromyelitis optica (NMO). With the widespread use of high-dose steroids, the long-term prognosis of ADEM with regard to functional and cognitive recovery is favorable. This chapter summarizes the available literature on ADEM in children, including the proposed consensus definitions for its monophasic and relapsing variants.
Introduction
Acute disseminated encephalomyelitis (ADEM) is an immune-mediated inflammatory demyelinating disease of the central nervous system (CNS), which is typically transitory and self-limiting. It is characterized by an acute or subacute encephalopathy with polyfocal neurological deficits, and magnetic resonance imaging (MRI) evidence of widespread demyelination that predominantly involves the white matter of the brain and spinal cord (Wingerchuk, 2003, Menge et al., 2007, Tenembaum et al., 2007). In the absence of specific biological markers, the diagnosis of ADEM is still based upon a combination of clinical and neuroimaging features and exclusion of diseases that mimic ADEM.
The present chapter will review the clinical, laboratory, neuroimaging, and biological characteristics of pediatric ADEM, including a comprehensive analysis of the differential diagnosis, and the current understanding of its pathophysiology.
Epidemiology
ADEM can occur at any age, but usually affects children and young adults. The mean age of clinical presentation in pediatric cohorts ranges from 5 to 8 years (Dale et al., 2000, Hynson et al., 2001, Tenembaum et al., 2002, Anlar et al., 2003). Rare cases in older adults have been reported. The diagnosis is often made in the setting of a defined viral illness or vaccination.
A seasonal distribution in the winter and spring months has been reported in at least three studies (Dale et al., 2000, Murthy et al., 2002, Leake et al., 2004). The highest incidence rates have been reported from San Diego County, USA (0.4/100 000 persons/year) and Fukuoka, Japan (0.64/100 000 persons/year) (Leake et al., 2004, Torisu et al., 2010). Similar studies conducted in Canada and Germany reported lower incidence rates of 0.2/100 000 and 0.07/100 000 children, respectively (Pohl et al., 2007, Banwell et al., 2009).
The disorder may be classified as either postvaccinal or postinfectious. However, absence of a clear precedent event has been reported in up to 26% of patients (Tenembaum et al., 2002). Vaccinations associated with ADEM include hepatitis B, pertussis, diphtheria, measles, mumps, rubella, pneumococcus, varicella, influenza, Japanese B encephalitis, smallpox, poliomyelitis, and human papillomavirus (Noorbakhsh et al., 2008). Some regional cases of ADEM have been linked to specific vaccines produced in neural tissue culture, in particular the Semple form of the rabies vaccine and Japanese B encephalitis vaccine. However, there has been a marked drop in postvaccination ADEM since the introduction of recombinant protein vaccines, which replaced vaccines cultured from CNS tissue.
Postinfectious forms of ADEM typically begin within 2–21 days after an infectious event; however, longer intervals have also been described. Viral infections commonly associated with ADEM include influenza virus, enterovirus, measles, mumps, rubella, varicella-zoster, Epstein–Barr virus, cytomegalovirus (CMV), herpes simplex virus, hepatitis A, and coxsackievirus. Bacterial triggers include Mycoplasma pneumoniae, Borrelia burgdorferi, Leptospira, and beta-hemolytic Streptococcus. Acute hemorrhagic leukoencephalomyelitis (AHLE) typically follows influenza or upper respiratory infection.
Although no clear gender predominance has been identified, a slight male preponderance has been described in some pediatric ADEM cohorts (Murthy et al., 2002, Tenembaum et al., 2002, Anlar et al., 2003, Banwell et al., 2009). These results are in contrast with the female predominance frequently described in patients with postpubertal onset of multiple sclerosis (MS).
Clinical features
Clinical presentation
Initial symptoms and signs of ADEM usually begin within 2 days to 4 weeks after a viral infection or vaccination, and include a rapid onset encephalopathy (behavioral change or altered consciousness) associated with a combination of multifocal neurological deficits, leading to hospitalization within a week.
Typically ADEM presents with systemic symptoms such as fever, malaise, headache, nausea, and vomiting, which may occur shortly before the appearance of neurological signs and symptoms. The clinical course is rapidly progressive, developing maximum deficits within a few days (mean 4.5 days) (Tenembaum et al., 2002). A wide variety of neurological deficits have been described in pediatric patients with ADEM, including: obtundation and depressed consciousness (invariable); unilateral or bilateral long tract signs (60–95%); acute hemiplegia (76%); ataxia (18–59%); meningismus (26–31%); seizures (13–35%); spinal cord involvement (24%); visual involvement (7–23%); and speech impairment or aphasia (5–21%). Cerebellar mutism and prolonged focal motor seizures in the context of ADEM have been mainly reported in children younger than 5 years of age (Dale et al., 2000, Hynson et al., 2001, Tenembaum et al., 2002, Anlar et al., 2003, Leake et al., 2004; Mikaeloff et al., 2007).
Acute combined peripheral nervous system demyelination is not rare in children with ADEM whereas it is more frequently described in adult patients, with a reported frequency of 44% in one study.
A particular ADEM phenotype affecting young children has been reported in association with group A beta-hemolytic streptococcal infection. Prominent behavioral disturbances, dystonic movements, and basal ganglia abnormalities on MRI (in addition to typical white matter lesions) characterize this syndrome (Dale et al., 2001).
There is a wide variation in the severity of the neurological involvement in ADEM. Children may present a subtle disease, with nonspecific irritability, headache, or somnolence lasting more than 1 day, or may show a rapid progression of symptoms and signs to coma and decerebrate rigidity (Wingerchuk, 2003). Respiratory failure secondary to brainstem involvement or severe impaired consciousness occurs in 11–16% of reported cases (Tenembaum et al., 2002, Wingerchuk, 2003). Clinically relevant features of pediatric ADEM are summarized in Table 132.1 .
Table 132.1.
Prominent clinical features of acute disseminated encephalomyelitis
| Age at clinical onset | Prepubertal children (median 5–8 years of age) |
|---|---|
| Clinical presentation | Frequent precedent viral infection or immunization Acute onset (over days) Headache, fever Acute encephalopathy Polyfocal neurological deficits |
| CSF | Lymphocytic pleocytosis (variable) Mild to moderate increased protein Oligoclonal banding 0–29% (usually transient) |
| Brain MRI features | Should be abnormal in all cases Large, multifocal, ill-defined white matter lesions Deep gray matter frequently involved Absence of previous lesions |
| Spinal MRI features | Large, confluent lesions |
| Clinical course | Improvement; there may be residual deficits Monophasic course Occasionally recurrent and multiphasic variants may occur |
| MRI follow-up | Complete or partial lesion resolution No new lesions appear on follow-up |
Neuroimaging features
MRI has become the most sensitive paraclinical marker of acute demyelination, showing lesions that are best detected on T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences as patchy, heterogeneous, and poorly marginated areas of increased signal intensity, as depicted in Fig. 132.1 . White matter lesions are typically multiple, bilateral, asymmetrical, and random in distribution, involving cerebral hemispheres, cerebellum, brainstem, and spinal cord (Kesselring et al., 1990, Hynson et al., 2001, Wingerchuk, 2003). Deep gray matter lesions tend to be symmetrical and often involve the thalamus and basal ganglia (Tenembaum et al., 2002). Lesions confined to the corpus callosum are less common. However, large demyelinating lesions of the adjacent white matter may extend into the corpus callosum. Spinal cord involvement in ADEM has been described in 11–28% of children as swollen and confluent intramedullary lesions with variable enhancement.
Fig. 132.1.
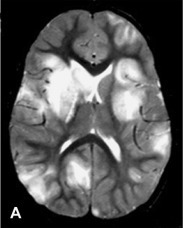
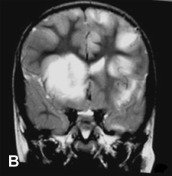
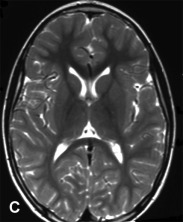
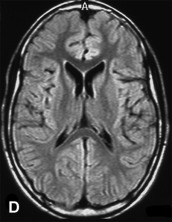
A previously healthy, developmentally normal, 3-year-old boy presented at a local emergency department after 48 hours of emesis, intermittent fever, and lethargy, 1 week after an upper respiratory viral infection. The subsequent neurological involvement included general weakness, drooling, and inability to walk, and he required admission to hospital. One day after admission, the patient presented left-sided hemiparesis followed by status epilepticus. A brain MRI was performed revealing widespread, hyperintense lesions in central and juxta-cortical white matter (A, axial T2-weighted image). Tumefactive lesions involving deep gray and white matter on the right with mass effect were also identified (B, coronal T2-weighted image). ADEM was diagnosed based on clinical and MRI picture and CSF studies including negative viral cultures and viral PCR assays. The child showed rapid improvement following intravenous corticosteroid treatment. Follow-up MRI performed at 6 months, 1 year, and 5 years (C, axial T2-weighted image; and D, axial FLAIR image) after the acute event revealed complete resolution of demyelinating lesions.
The reported frequency of gadolinium enhancing lesions on T1-weighted sequences throughout all groups is quite variable (8–100%), depending on the stage of inflammation. However, brain lesions in ADEM tend to be at the same stage of lesion formation. Different patterns of enhancement have been described: complete or incomplete ring-shaped, nodular, gyral, or diffuse-patchy.
The identification of radiological signs of mass effect on brain MRI of children with ADEM is extremely important before performing a lumbar puncture. Delayed MRI changes for several days and even weeks after the clinical onset have been exceptionally described in children and adult patients with ADEM.
Five patterns of cerebral involvement have been proposed to classify the MRI findings in children with ADEM (Tenembaum et al., 2002, Tenembaum et al., 2007):
-
1.
ADEM with small lesions (less than 5 mm);
-
2.
ADEM with large, confluent, or tumefactive lesions, with frequent extensive perilesional edema and mass effect;
-
3.
ADEM with additional symmetrical bithalamic involvement;
-
4.
acute hemorrhagic encephalomyelitis (AHEM), when the presence of blood degradation products can be identified within the large T2 hyperintense lesions; and
-
5.
ADEM with pseudo-leukodystrophic pattern, with a diffuse, bilateral, symmetrical, and usually nonenhanced white matter involvement (Kesselring et al., 1990).
The imaging pattern on conventional MRI does not appear to correlate with any particular outcome or disability as observed in a large pediatric cohort, since most lesions tend to resolve on follow-up studies (Tenembaum et al., 2002). However, these brain MRI patterns may be helpful when considering the differential diagnosis of ADEM (see Differential diagnosis, below).
Conventional MRI studies do not capture all the pathological features of the demyelinating process. Advanced neuroimaging techniques may provide a better assessment of the underlying histopathology process. A small study using diffusion tensor imaging (DTI) found restricted diffusion in studies performed during the acute stage of ADEM, and free diffusion in those performed in the subacute stage (Balasubramanya et al., 2007). Magnetization transfer and diffusion tensor MRI may also be helpful in identifying involvement of the so-called “normal-appearing white matter,” as these tend not to show any abnormalities in ADEM, in contrast to MS. Studies using proton magnetic resonance spectroscopy in children with ADEM have shown major elevation of lipids and reduction in myoinositol/creatine ratio during the acute phase, followed by a reduction in lipid peak and elevation in myoinositol/creatine ratio during the chronic phase.
The demonstration of lesion resolution on MRI serial studies plays a key role in supporting the diagnosis of ADEM. Complete resolution on sequential follow-up MRI has been described in 37–75%, and partial resolution in 25–53% of patients with ADEM. Conversely, the appearance of new lesions on serial MRI studies is unlikely in ADEM, but it is consistent with the dissemination in time criteria, a powerful predictor of MS diagnosis (McDonald et al., 2001).
Laboratory tests
The cerebrospinal fluid (CSF) findings of patients with ADEM are nonspecific and include mild lympho-monocytic pleocytosis, slightly increased protein level, and normal glucose levels with sterile cultures.
Intrathecal oligoclonal IgG bands (OCBs) by isoelectric focusing may be found as an acute inflammatory manifestation with published frequencies of 0–29% in patients with ADEM (Tenembaum et al., 2002, Dale and Pillai, 2007, Pohl et al., 2007, Dale and Brilot, 2010). However, OCBs in ADEM are usually transient, as opposed to those observed in MS. A mirrored pattern of OCB (presence of the same IgG clones in both CSF and serum) has also been described in ADEM (Dale et al., 2000).
Elevation of CSF neopterin has also been described in ADEM patients as well. However, neopterin is a nonspecific biomarker as it is only an indicator of Th1-cellular immune activation (Dale and Brilot, 2010).
Serology for suspected organisms, CSF viral, fungal, and bacterial cultures, as well as CSF viral polymerase chain reaction assay are further required to rule out a direct CNS infectious condition.
Proposed definitions
The lack of a uniform definition and clear diagnostic criteria previously led to the incorrect classification of any event of CNS demyelination as ADEM (Tenembaum et al., 2007). An international panel has proposed operational definitions for acquired CNS demyelinating disorders of childhood, including monophasic and relapsing variants of ADEM, which are as follows (Krupp et al., 2007):
-
1.
Acute disseminated encephalomyelitis (ADEM): A first clinical and self-limiting event including an acute or subacute encephalopathy with polyfocal deficits and widespread hyperintense lesions predominantly affecting the CNS white matter; no evidence of previous destructive white matter changes should be present on MRI; and no history of a previous clinical episode with features of a demyelinating event. If a relapse takes place within 4 weeks of tapering steroid treatment or within the first 3 months from the initial event, it is considered related to the same acute condition.
-
2.
Recurrent disseminated encephalomyelitis (RDEM): Initial ADEM event followed by a new one fulfilling diagnostic criteria for ADEM, occurring at least 3 months after the initial event and at least 4 weeks after completing steroid therapy. The subsequent attack has to reproduce the original clinical syndrome, completely or in part.
-
3.
Multiphasic disseminated encephalomyelitis (MDEM): Refers to one or more ADEM relapses, including encephalopathy and multifocal deficits, but involving new areas of the CNS on MRI and neurological exam. Relapses take place at least 3 months after initial ADEM attack and at least 4 weeks after completing steroid therapy.
The proposed criteria were designed to improve the clinical characterization of acquired demyelinating disorders, including ADEM, as it has been shown in some pediatric cohorts where the consensus definitions were applied (Dale and Pillai, 2007, Alper et al., 2009).
Relapsing variants of acute disseminated encephalomyelitis
ADEM has traditionally been described as a monophasic disorder, with symptoms and signs that usually resolve over time. However, patients classified as having ADEM but showing recurrences have been known since 1932, when van Bogaert published “ADEM with relapses.” Several studies have consistently described a proportion of patients who have further demyelinating events at different rates, ranging from 5.5% (Murthy et al., 2002) to 33% (Anlar et al., 2003). However, in more recently published series using the 2007 definitions, multiphasic ADEM is reported infrequently.
The reported clinical outcome in pediatric cohorts with MDEM and long-term follow-up is also favorable (Dale et al., 2000, Tenembaum et al., 2002, Mar et al., 2010).
Distinguishing acute disseminated encephalomyelitis from ms and neuromyelitis optica
The international pediatric panel defines MS in children and adolescents as an initial event of discrete CNS demyelination without encephalopathy followed by dissemination in time demonstrated by a new clinical event or new MRI lesions (Krupp et al., 2007), as specified for adults (McDonald et al., 2001) but including patients under 10 years of age. Initial demyelinating events (IDE) in pediatric patients may take on different clinical phenotypes, such as ADEM or discrete clinical syndromes, usually named clinically isolated syndromes (CIS). Although 45% of all children and adolescents with an IDE will have a second episode (Mikaeloff et al., 2004b), only 20% of patients who are initially diagnosed as having ADEM will later receive the diagnosis of MS (Mikaeloff et al., 2007).
In the special circumstance of a child whose ICE was classified as ADEM, a second attack not meeting ADEM criteria is considered not enough for a definite MS diagnosis and additional evidence of further dissemination in time is required, either on MRI with emergence of new lesions after at least 3 months, or a new clinical attack (Krupp et al., 2007).
There are no clear prognostic factors that determine whether a child with a first event of either ADEM or discrete demyelination will eventually develop MS. There is a wide range of reported risk of developing MS after ADEM in the pediatric population (Mikaeloff et al., 2004a, Mikaeloff et al., 2004b; Mar et al., 2010).
Different sets of MRI criteria have been proposed to assess the risk of relapses following an IDE. Within the KIDMUS (French National Kids with Multiple Sclerosis network) cohort, brain MRI characteristics associated with an increased risk of MS were the presence of periventricular lesions, lesions perpendicular to the long axis of the corpus callosum, and the presence of well-defined lesions (Mikaeloff et al., 2004b). In a retrospective study of brain MRI scans obtained at first attack from children subsequently diagnosed with MS and ADEM (Callen et al., 2009), the presence of any two of the following MRI features indicated an increased risk of developing MS: (1) absence of a diffuse bilateral lesion pattern; (2) presence of black holes; and (3) presence of two or more periventricular lesions. More recently, the presence of at least one T1-weighted hypointense lesion (hazard ratio 20.6, 95% confidence interval (CI) 5.46–78.0) or at least one periventricular lesion (3.34, 1.27–8.83) on the initial brain scan was associated with an increased risk of MS diagnosis, with a sensitivity of 84%, specificity of 93%, positive predictive value 76%, and negative predictive value 96% (Verhey et al., 2011). And the risk for MS diagnosis was highest when both parameters were present (hazard ratio 34.27, 95% CI 16.69–70.38).The predictive value of the proposed brain MRI criteria needs to be confirmed and validated in different pediatric MS cohorts.
Positive OCBs in the CSF have been reported in 64–95% of pediatric patients with MS, and in 0–29% of pediatric patients with ADEM followed for a variable number of years (Dale et al., 2000, Hynson et al., 2001, Tenembaum et al., 2002, Pohl et al., 2007). Within the KIDMUS cohort, 94% of children with positive OCBs went on to develop MS, indicating high specificity. However, only 40% of patients with established MS in this study had positive OCBs, indicating a low sensitivity for this test. Furthermore, OCBs may also be produced intrathecally during infections, in which case the IgG targets the etiological agent.
A definite distinction between an initial discrete event of CNS demyelination and ADEM, and between relapsing forms of ADEM (recurrent or multiphasic), relapsing-remitting MS, and recurrent neuromyelitis optica (NMO) and NMO spectrum disorders, has important prognostic and therapeutic implications. The long-term management of MS with immunomodulatory or immunosuppressive drugs is inappropriate for ADEM (Krupp et al., 2007, Poser and Brinar, 2007). This is the rationale for the conservative approach of the 2007 consensus definitions (Krupp et al., 2007), definitions that may eventually require a revision as more information is gathered regarding the predictability of developing MS after an IDE (see Chapter 133).
Neuromyelitis optica is an inflammatory demyelinating disease of the CNS characterized by recurrent episodes of transverse myelitis and optic neuritis. Although early diagnostic criteria for NMO suggested that the brain MRI should be relatively free of white matter lesions, recent publications provide evidence of a broader spectrum, including brain lesions consistent with ADEM and MS. The most widely accepted diagnostic criteria for NMO (Wingerchuk et al., 2006) are based on the clinical manifestations and additional specificity criteria that discriminate patients with NMO from those with MS, the most specific of which is an IgG autoantibody biomarker. The discovery of antibodies directed against the water channel aquaporin 4 (AQP4), NMO-IgG, has allowed a broadening of the NMO spectrum, which recognizes the involvement of CNS structures beyond the optic nerves and spinal cord. Indeed, a pattern of brain involvement has been delineated in which the hypothalamic and periventricular areas are mainly affected, particularly in children presenting with an ADEM-like event (Banwell et al., 2008, Lotze et al., 2008), with large brain MRI lesions with no enhancement, or showing a “cloud-like enhancement,” a finding described as specific to NMO.
Differential diagnosis
The association of an acute encephalopathy and demyelination of the CNS in a child represents a diagnostic challenge. A large number of inflammatory and noninflammatory disorders may have a similar clinical and radiological presentation and should be considered in the diagnostic evaluation (Table 132.2 ).
Table 132.2.
Differential diagnosis of pediatric ADEM by disease subtype
| A. CNS infectious conditions |
|
| B. CNS inflammatory-demyelinating disorders |
|
| C. CNS vascular disorders |
|
| D. Intracranial mass lesion |
|
| E. Toxic, nutritional, and metabolic disorders |
|
| F. Miscellaneous |
|
The exclusion of acute CNS infections should be the first and most important diagnostic step to be considered, by lumbar puncture and further microbiological laboratory tests of CSF and serum. Nevertheless, in a patient presenting with features of acute encephalitis, associated clinical signs such as optic neuritis, myelitis, or polyradiculoneuropathy should raise the suspicion of a possible acute demyelinating process.
Neuroimaging may play a particularly helpful role in the differential diagnosis. A standard MRI scan of the brain and spinal cord, with and without gadolinium enhancement, will be important to define the lesion appearance and the regional distribution of demyelination.
A lesion pattern with predominant posterior white matter involvement may indicate a posterior reversible encephalopathy syndrome. Brain malignancies, inflammatory vasculitis, and brain abscesses should be considered when MRI shows large focal tumor-like lesions. However, large and tumefactive plaques disseminated throughout the hemispheric white matter are typical features of Marburg disease, an acute variant of MS.
A lesion pattern with symmetrical bithalamic involvement may be seen in children with ADEM after Japanese B encephalitis vaccination, as well as in children with deep cerebral venous thrombosis, hypernatremia, and extrapontine myelinolysis. Symmetrical bithalamic involvement is also the hallmark of acute necrotizing encephalopathy (ANE), a distinct form of acute encephalopathy triggered by respiratory infections, particularly influenza A and B.
Bilateral basal ganglia involvement may be observed in poststreptococcal ADEM, but also in organic acidurias, infantile bilateral striatal necrosis, Mycoplasma pneumoniae infection, and potassium channel antibody-associated encephalopathy. The presence of complete ring-enhanced lesions in the cerebral white matter is unusual in ADEM, and should prompt consideration of brain abscess, tuberculomas, neurocysticercosis, toxoplasmosis, and histoplasmosis.
Lesions restricted to one hemisphere, or showing a predominantly cortical–subcortical distribution, are unlikely in ADEM, and obliterating vascular conditions should be investigated. The identification of microinfarcts in the central portion of the corpus callosum but sparing the periphery (also called “snowballs”) in an encephalopathic patient can be useful in assigning the diagnosis of Susac syndrome. This autoimmune endotheliopathy consists of the clinical triad of encephalopathy, branch retinal artery occlusion (BRAO), and hearing loss. An aggressive and sustained immunosuppressive treatment is recommended for the encephalopathic form of Susac syndrome (Rennebohm et al., 2008).
Children with atypical bilateral symmetrical white matter changes on MRI (pseudo-leukodystrophic pattern) should undergo an extensive workup for metabolic causes of leukodystrophy.
A clinical syndrome with psychosis-like symptoms, followed by seizures, language disintegration, and abnormal movements should evoke the diagnosis of anti-N-methyl-D-aspartate-receptor autoimmune encephalitis. This syndrome has been recently expanded to children and adolescents, usually with a preceding viral infection. Clinical recovery has been reported in 75% of patients treated with immunotherapy, including intravenous corticosteroid, immunoglobulin (IVIg), and plasma exchange. However, recovery is usually slow and patients may need second-line therapy (Vincent et al., 2011).
In children with a progressive leukoencephalopathy and neurological decline despite treatment, diagnoses such as inherited leukodystrophies, progressive multifocal leukoencephalopathy, subacute sclerosing panencephalitis, mitochondrial disorders, CNS malignancies, or nutritional deficiency (vitamin B12) should be considered. Neurosarcoidosis should also be considered in children with progressive neurological deficits, diencephalic dysfunction, and signs of leptomeningeal inflammation (see Chapter 131). The diagnosis of macrophage activation syndrome may also apply to young children with an unresolved persistent or progressive encephalopathy, multiorgan involvement, or recurrent fever, particularly during the first 2 years of life (Janka, 2007).
Recurrent events of CNS demyelination should raise the potential diagnosis of MS or NMO when recurrences predominantly involve optic nerves and spinal cord. Systemic vasculitides and collagen vascular diseases, like systemic lupus erythematosus, neuro-Behçet, and Sjögren disease, should also be considered in pediatric patients with recurrent events (see Chapter 131).
Immunopathogenesis
Histologically, ADEM is characterized by perivenular inflammatory infiltrates consisting of T cells and macrophages, associated with perivenular demyelination that is usually confined to a narrow rim around these infiltrated areas. Although ADEM is typically described as demyelination with relative preservation of axons, axonal damage confined to the perivenular area has been described. Lesions typically involve the cerebral white matter bilaterally, but can also involve the cortex and deep gray matter structures, in addition to brainstem, cerebellum, and spinal cord. Lesions appear of similar histological age. No evidence of direct viral, bacterial, fungal, or parasitic infection is found in pathological samples.
In the hemorrhagic variants of ADEM (acute hemorrhagic and acute necrotizing hemorrhagic leukoencephalitis) demyelination is often more widespread throughout the CNS, with a pronounced neutrophilic infiltrate. Areas of demyelination and acutely hemorrhagic or necrotic tissue containing inflammatory cells surround the small blood vessels.
The pathogenesis of ADEM is still unclear; however, given its histological features and typically monophasic disease course, it has been likened to the animal model of experimental autoimmune encephalomyelitis (EAE). EAE is an autoimmune demyelinating disease that can be induced in a variety of animal species by immunization with myelin proteins or peptides derived from myelin proteins. This results in a monophasic, autoimmune demyelinating syndrome of weakness with diffuse CNS demyelination. The postvaccinal form of ADEM associated with the Semple rabies vaccine reinforces this analogy.
The molecular mimicry hypothesis in ADEM suggests that, due to partial structural or amino-acid sequence homologies, antigenic epitopes are shared between certain pathogens or vaccines and host CNS myelin antigens. The pathogens have the capacity to activate myelin-reactive T cell clones and can thereby elicit a CNS-specific autoimmune response. Thus, it has been suggested that microbial infections or vaccinations may elicit a cross-reactive antimyelin response through molecular mimicry, resulting in ADEM. Various sequences of myelin peptides have been shown to resemble several viral sequences, and in some cases cross-reactive T cell responses have been demonstrated. Examples of cross-reactive T cell responses with myelin basic protein (MBP) antigens include human herpesvirus (HHV) 6, coronavirus, influenza virus, and Epstein–Barr virus. Proteolipid protein (PLP) shares common sequences with Hemophilus influenzae, and Semliki Forest virus peptides mimic myelin oligodendrocyte glycoprotein (MOG).
There is another hypothesis in ADEM, in which a direct CNS infection with a secondary inflammatory cascade is involved. The resulting tissue damage is thought to disrupt the blood–brain barrier, exposing CNS-confined autoantigens into the circulation where they are processed by the peripheral immune system. This process leads to a breakdown of tolerance, resulting in a self-directed autoimmune attack against the CNS driven by encephalitogenic T cells. Theiler's murine encephalomyelitis virus-induced demyelinating disease (TMEV-IDD) may be a good model for postinfectious etiology. TMEV-IDD is induced by direct CNS infection with the neurotropic TMEV picornavirus in genetically susceptible mice, inducing diffuse CNS inflammation and demyelination. Initially the primary immune-mediated reaction involves TMEV-specific CD4 and CD8 T cells. During the chronic stages of the disease, T cell reactivity to host CNS myelin peptides has been observed. This suggests that epitope spreading may have occurred with the appearance of secondary T cell responses to myelin breakdown products. The TMEV model highlights the phenomenon of epitope spreading which is initiated by a destructive CNS viral infection resulting in a secondary autoimmune response to myelin components.
The role of autoantibodies directed against the various CNS myelin antigens has been studied in ADEM. Several studies have demonstrated elevated anti-MBP antibody titers in patients with postvaccinal ADEM associated with the Semple rabies vaccine. Antibasal ganglia antibodies were detected in the sera of patients with poststreptococcal ADEM (Dale et al., 2001). Elevated titers of serum antibodies to MOG-peptide using a tetramer-based approach have been demonstrated to be highly elevated in 20–47% of children with acute CNS demyelination, with the highest reactivities observed in patients with ADEM. Although the presence of anti-MOG antibodies at disease onset do not differentiate ADEM from MS (O'Connor et al., 2007, Dale and Brilot, 2010), the time course seems to be different. Anti-MOG antibodies continuously declined in all analyzed children with ADEM, but they tended to persist in children with MS (Pröbstel et al., 2011).
Studies of cytokines and chemokines in ADEM have shown various upregulated patterns related to activation of macrophages and microglia. CSF analysis has demonstrated a bias towards Th2-type chemokines (CCL17, CCL22) in adult patients with ADEM compared to those with MS. Chemokines relevant in the migration of eosinophils and neutrophils were also found to be elevated in the CSF of patients with ADEM. Elevated CSF levels of the proinflammatory cytokines interleukin (IL)-6 and tumor necrosis factor (TNF)-α, as well as the Th2-cytokine IL-10, have been described in another study (Menge et al., 2007, Wingerchuk and Lucchinetti, 2007).
A possible genetic background has also been considered in ADEM. ADEM was associated with the major histocompatibility complex (MHC) class II alleles HLA-DRB1*1501, as well as HLA-DRB5*0101 in a Korean study. The same study showed an association of HLA-DRB3*0202 and HLA-DQB1*0502 with acute necrotizing forms of encephalopathy. In a Russian study, ADEM was associated with the class II alleles HLA-DRB1*01 and HLA-DRB*03(017). HLA-DRB1*1501 has been shown to bind effectively the immunodominant epitope of myelin basic protein, thus suggesting a link to the myelin hypothesis of MS pathogenesis. Recent studies established a link between genetics and viral diseases. Most cases of ANE, a distinct form of acute encephalopathy triggered by influenza A infection, are sporadic. The identification of recurrent and familial cases of ANE due to a mutation in the RANBP2 gene provided new evidence of the importance of genetic risk factors for an environmentally triggered neurological disease. Large-scale studies are required to assess definitively the association of various class II alleles in patients with ADEM compared to those with MS.
Treatment
There is no standard therapy for ADEM and current treatment has developed from expert opinion and small case series. Supportive care in the acute stage is critical and early antiviral treatment with acyclovir (30 mg/kg/day) is highly recommended on admission, considering that viral encephalitis and particularly herpes simplex encephalitis is the usual primary diagnosis in a child with fever, encephalopathy, seizures, and focal neurological signs. The treatment for ADEM includes primarily the management of acute attacks.
Corticosteroid treatment at high doses is the most widely reported therapy. Most authors recommend a brief (3–5 days) high-dose intravenous steroid course, usually methylprednisolone given at 20–30 mg/kg/day to a maximum dose of 1 g/day, or dexamethasone given at 1 mg/kg/day, followed by oral prednisone taper for 4–6 weeks (Dale et al., 2000, Hynson et al., 2001, Tenembaum et al., 2002, Tenembaum et al., 2007; Pohl and Tenembaum, 2012). Reported treatment approaches show a wide variety in the specific steroid formulation employed, as well as in the dosing, routes of administration, and tapering regimens. Treatment with corticosteroids requires careful monitoring of blood pressure, urine glucose, and serum potassium, and administration of gastric protection.
The use of IVIg has been reported in several case studies either alone or in combination with corticosteroids. The recommended total dose of IVIg is 2 g/kg, administered over 2–5 days. The usefulness of IVIg has been reported both as a second-line treatment in steroid-unresponsive ADEM cases and in patients showing recurrent or steroid-dependent demyelination.
The use of plasmapheresis (therapeutic plasma exchange, TPE) has been recently established as possibly effective and it may be considered as escalation therapy for steroid-unresponsive acute fulminant demyelinating CNS diseases including ADEM, MS, NMO, and transverse myelitis, with class II evidence (Cortese et al., 2011). Plasmapheresis should be started as soon as the treatment failure is recognized. The use of this procedure in ADEM has been reported in only a small number of severe cases. A median number of seven exchanges (range 2 to 20) was reported in one study (Keegan et al., 2002). Moderate to severe anemia, symptomatic hypotension, hypocalcemia, potential transfusion reactions or transmission of transfusion-related diseases, and heparin-associated thrombocytopenia have been described in relation to TPE. There is also a risk of catheter-related complications, including thrombosis, septic infections or pneumothorax.
Acute hemorrhagic leukoencephalitis is often considered the most acute and severe form of ADEM, with a universally fatal course within hours to days after the onset of neurological symptoms without treatment. Survival in pediatric patients has been reported with combined therapy including high-dose intravenous corticosteroid, IVIg, TPE, and decompressive craniotomies.
Aggressive treatment strategies such as surgical decompression have to be considered and performed in patients with fulminant variants of ADEM, with evidence of continued clinical deterioration due to increased intracranial pressure, unresponsive to conventional medical treatment and critical care measures (Payne et al., 2007; Pohl and Tenembaum, 2012).
Prognosis and outcome
With currently available treatment regimes, the long-term prognosis of ADEM is usually favorable. Full recovery occurs in about 57–89% of patients, and between 20% and 30% may show minor residual deficits. Following initiation of treatment a rapid improvement is usually observed, sometimes within hours, although recovery typically evolves over days. The reported average time period to full recovery ranges between 1 and 6 months.
The most frequently reported residual disabilities are: (a) focal motor deficits, ranging from mild clumsiness and ataxia to severe hemiparesis; (b) visual problems, from mild diminished visual acuity to blindness; and (c) the development of seizures following ADEM resolution (Tenembaum et al., 2002).
Neurocognitive outcome
Neurocognitive deficits following CNS demyelination in childhood is an important area of clinical and research investigation. Even children thought to have full recovery from ADEM can demonstrate subtle neurocognitive deficits in attention, executive function, and behavior when reevaluated more than 3 years after ADEM. Lower IQ and educational achievement as well as behavioral problems have been particularly observed in children younger than 5 years at ADEM diagnosis (Jacobs et al., 2004). Additional studies are required further to characterize neurocognitive deficits following ADEM in order to facilitate appropriate educational interventions.
References
- Alper G., Heyman R., Wang L. Multiple sclerosis and acute disseminated encephalomyelitis diagnosed in children after long-term follow-up: comparison of presenting features. Dev Med Child Neurol. 2009;51:480–486. doi: 10.1111/j.1469-8749.2008.03136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anlar B., Basaran C., Kose G. Acute disseminated encephalomyelitis in children: outcome and prognosis. Neuropediatrics. 2003;34:194–199. doi: 10.1055/s-2003-42208. [DOI] [PubMed] [Google Scholar]
- Balasubramanya K.S., Kovoor J.M., Jayakumar P.N. Diffusion-weighted imaging and proton MR spectroscopy in the characterization of acute disseminated encephalomyelitis. Neuroradiology. 2007;49:177–183. doi: 10.1007/s00234-006-0164-2. [DOI] [PubMed] [Google Scholar]
- Banwell B., Tenembaum S., Lennon V.A. Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology. 2008;70:344–352. doi: 10.1212/01.wnl.0000284600.80782.d5. [DOI] [PubMed] [Google Scholar]
- Banwell B., Kennedy J., Sadovnick D. Incidence of acquired demyelination of the CNS in Canadian children. Neurology. 2009;72:232–239. doi: 10.1212/01.wnl.0000339482.84392.bd. [DOI] [PubMed] [Google Scholar]
- Callen D.J.A., Shroff M.M., Branson H.M. Role of MRI in the differentiation of ADEM from MS in children. Neurology. 2009;72:968–973. doi: 10.1212/01.wnl.0000338630.20412.45. [DOI] [PubMed] [Google Scholar]
- Cortese I., Chaudhry V., So Y.T. Evidence-based guideline update: plasmapheresis in neurologic disorders: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2011;76:294–300. doi: 10.1212/WNL.0b013e318207b1f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale R.C., Brilot F. Biomarkers of inflammatory and auto-immune central nervous system disorders. Curr Opin Pediatr. 2010;22:718–725. doi: 10.1097/MOP.0b013e3283402b71. [DOI] [PubMed] [Google Scholar]
- Dale R.C., Pillai S.C. Early relapse risk after a first CNS inflammatory demyelination episode: examining international consensus definitions. Dev Med Child Neurol. 2007;49:887–893. doi: 10.1111/j.1469-8749.2007.00887.x. [DOI] [PubMed] [Google Scholar]
- Dale R.C., de Sousa C., Chong W.K. Acute disseminated encephalomyelitis, multiphasic disseminated encephalomyelitis and multiple sclerosis in children. Brain. 2000;123:2407–2422. doi: 10.1093/brain/123.12.2407. [DOI] [PubMed] [Google Scholar]
- Dale R.C., Church A.J., Cardoso F. Post streptococcal acute disseminated encephalomyelitis with basal ganglia involvement and auto-reactive antibasal ganglia antibodies. Ann Neurol. 2001;50:588–595. doi: 10.1002/ana.1250. [DOI] [PubMed] [Google Scholar]
- Hynson J.L., Kornberg A.J., Coleman L.T. Clinical and neuroradiologic features of acute disseminated encephalomyelitis in children. Neurology. 2001;56:1308–1312. doi: 10.1212/wnl.56.10.1308. [DOI] [PubMed] [Google Scholar]
- Jacobs R.K., Anderson V.A., Neale J.L. Neuropsychological outcome after acute disseminated encephalomyelitis: impact of age at illness onset. Pediatr Neurol. 2004;31:191–197. doi: 10.1016/j.pediatrneurol.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Janka G.E. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2007;166:95–109. doi: 10.1007/s00431-006-0258-1. [DOI] [PubMed] [Google Scholar]
- Keegan M., Pineda A.A., McClelland R.L. Plasma exchange for severe attacks of CNS demyelination: predictors of response. Neurology. 2002;58:143–146. doi: 10.1212/wnl.58.1.143. [DOI] [PubMed] [Google Scholar]
- Kesselring J., Miller D.H., Robb S.A. Acute disseminated encephalomyelitis: MRI findings and the distinction from multiple sclerosis. Brain. 1990;113:291–302. doi: 10.1093/brain/113.2.291. [DOI] [PubMed] [Google Scholar]
- Krupp L.B., Banwell B., Tenembaum S., for the International Paediatric MS Study Group Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology. 2007;68:S7–S12. doi: 10.1212/01.wnl.0000259422.44235.a8. [DOI] [PubMed] [Google Scholar]
- Leake J.A.D., Albani S., Kao A.S. Acute disseminated encephalomyelitis in childhood: epidemiologic, clinical and laboratory features. Pediatr Infect Dis J. 2004;23:756–764. doi: 10.1097/01.inf.0000133048.75452.dd. [DOI] [PubMed] [Google Scholar]
- Lotze T.E., Northrop J.L., Hutton G.J. Spectrum of pediatric neuromyelitis optica. Pediatrics. 2008;122:e1039–e1047. doi: 10.1542/peds.2007-2758. [DOI] [PubMed] [Google Scholar]
- Mar S., Lenox J., Benzinger T. Long-term prognosis of pediatric patients with relapsing acute disseminated encephalomyelitis. J Child Neurol. 2010;25:681–688. doi: 10.1177/0883073809343320. [DOI] [PubMed] [Google Scholar]
- McDonald W.I., Compston A., Edan G. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50:121–127. doi: 10.1002/ana.1032. [DOI] [PubMed] [Google Scholar]
- Menge T., Kieseier B.C., Nessler S. Acute disseminated encephalomyelitis: an acute hit against the brain. Curr Opin Neurol. 2007;20:247–254. doi: 10.1097/WCO.0b013e3280f31b45. [DOI] [PubMed] [Google Scholar]
- Mikaeloff Y., Suissa S., Vallée L., KIDMUS study group First episode of acute CNS inflammatory demyelination in childhood: prognostic factors for multiple sclerosis and disability. J Pediatr. 2004;144:246–252. doi: 10.1016/j.jpeds.2003.10.056. [DOI] [PubMed] [Google Scholar]
- Mikaeloff Y., Adamsbaum C., Husson B. MRI prognostic factors for relapse after acute CNS inflammatory demyelination in childhood. Brain. 2004;127:1942–1947. doi: 10.1093/brain/awh218. [DOI] [PubMed] [Google Scholar]
- Mikaeloff Y., Caridade G., Husson B., on behalf of the Neuropediatric KIDSEP study group of the French Neuropediatric Society Acute disseminated encephalomyelitis cohort study: prognostic factors for relapse. Eur J Paediatr Neurol. 2007;11:90–95. doi: 10.1016/j.ejpn.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Murthy S.N., Faden H.S., Cohen M.E. Acute disseminated encephalomyelitis in children. Pediatrics. 2002;110:21–28. doi: 10.1542/peds.110.2.e21. [DOI] [PubMed] [Google Scholar]
- Noorbakhsh F., Johnson R.T., Emery D. Acute disseminated encephalomyelitis: clinical and pathogenesis features. Neurol Clin. 2008;26:759–780. doi: 10.1016/j.ncl.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor K.C., McLaughlin K.A., De Jager P. Self-antigen tetramers discriminate between myelin autoantibodies to native or denatured protein. Nat Med. 2007;13:211–217. doi: 10.1038/nm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne E.T., Rutka J.T., Ho T.K. Treatment leading to dramatic recovery in acute hemorrhagic leukoencephalitis. J Child Neurol. 2007;22:109–113. doi: 10.1177/0883073807299971. [DOI] [PubMed] [Google Scholar]
- Pohl D., Hennemuth I., von Kries R. Paediatric multiple sclerosis and acute disseminated encephalomyelitis in Germany: results of a nationwide survey. Eur J Pediatr. 2007;166:405–412. doi: 10.1007/s00431-006-0249-2. [DOI] [PubMed] [Google Scholar]
- Pohl D., Tenembaum S. Treatment of acute disseminated encephalomyelitis. Curr Treat Options Neurol. 2012;14:264–275. doi: 10.1007/s11940-012-0170-0. [DOI] [PubMed] [Google Scholar]
- Poser C.M., Brinar V.V. Disseminated encephalomyelitis and multiple sclerosis: two different diseases – a critical review. Acta Neurol Scand. 2007;116:201–206. doi: 10.1111/j.1600-0404.2007.00902.x. [DOI] [PubMed] [Google Scholar]
- Pröbstel A.K., Dornmair K., Bittner R. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology. 2011;77:580–588. doi: 10.1212/WNL.0b013e318228c0b1. [DOI] [PubMed] [Google Scholar]
- Rennebohm R.M., Egan R.A., Susac J.O. Treatment of Susac's syndrome. Curr Treat Options Neurol. 2008;10:67–74. doi: 10.1007/s11940-008-0008-y. [DOI] [PubMed] [Google Scholar]
- Tenembaum S., Chamoles N., Fejerman N. Acute disseminated encephalomyelitis: a long-term follow-up study of 84 pediatric patients. Neurology. 2002;59:1224–1231. doi: 10.1212/wnl.59.8.1224. [DOI] [PubMed] [Google Scholar]
- Tenembaum S., Chitnis T., Ness J., for the International Pediatric MS Study Group Acute disseminated encephalomyelitis. Neurology. 2007;68:S23–S36. doi: 10.1212/01.wnl.0000259404.51352.7f. [DOI] [PubMed] [Google Scholar]
- Torisu H., Kira R., Ishizaki Y. Clinical study of childhood acute disseminated encephalomyelitis, multiple sclerosis, and acute transverse myelitis in Fukuoka Prefecture, Japan. Brain and Development. 2010;32:454–462. doi: 10.1016/j.braindev.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Verhey L.H., Branson H.M., Shroff M.M. MRI parameters for prediction of multiple sclerosis diagnosis in children with acute CNS demyelination: a prospective national cohort study. Lancet Neurol. 2011;10:1065–1073. doi: 10.1016/S1474-4422(11)70250-2. [DOI] [PubMed] [Google Scholar]
- Vincent A., Bien C.G., Irani S.R. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol. 2011;10:759–772. doi: 10.1016/S1474-4422(11)70096-5. [DOI] [PubMed] [Google Scholar]
- Wingerchuk D.M. Postinfectious encephalomyelitis. Curr Neurol Neurosci Rep. 2003;3:256–264. doi: 10.1007/s11910-003-0086-x. [DOI] [PubMed] [Google Scholar]
- Wingerchuk D.M., Lucchinetti C.F. Comparative immunopathogenesis of acute disseminated encephalomyelitis, neuromyelitis optica, and multiple sclerosis. Curr Opin Neurol. 2007;20:343–350. doi: 10.1097/WCO.0b013e3280be58d8. [DOI] [PubMed] [Google Scholar]
- Wingerchuk D.M., Lennon V.A., Pittock S.J. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–1489. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]