

Transmission electron microscopy, a popular diagnostic adjunct in the 1970s and early 1980s, has been largely supplanted by new immunohistochemical methods and, to a lesser extent, cytogenetic and molecular techniques. However, many surgical pathologists have come to the realization that these new methods cannot resolve all their diagnostic problems, leading to a resurgence of interest in transmission electron microscopy as an ancillary diagnostic modality.1. , 2. , 3. , 4. , 5. , 6. , 7. , 8. The current appropriate and cost-effective use of electron microscopy for the diagnostic evaluation of non-neoplastic and neoplastic diseases is the main subject of this chapter.
THE TRANSMISSION ELECTRON MICROSCOPE
To the pathologist, the transmission electron microscope is like the equivalent of a high-magnification, high-resolution light microscope capable of visualizing small intracellular and extracellular structures in great detail. Some examples include mitochondria (organelles); melanosomes and various types of secretory granules (inclusions); microtubules, microfilaments (e.g., actin), and intermediate filaments (cytoskeleton); cilia, microvilli, and intercellular junctions (cell surface specializations); and extracellular constituents such as basement membranes, collagen, and amyloid.1. , 4. , 9. , 10. The high-resolution capability of the electron microscope is due to the small wavelength of the electron, approximately 0.004 nm for a 100-keV electron, compared with approximately 500 nm for visual light. The resolution of the modern electron microscope is 0.2 nm; in contrast, that of a good light microscope is 200 nm (these figures are based on Abbe's and de Brogli's fundamental equations).1 It is important for pathologists to realize that the electron microscope is a morphologic instrument similar to the light microscope.
All kinds of cell preparations and tissues can be proffered for electron microscopic evaluation. The most commonly submitted specimens are obtained from surgical and biopsy procedures, including percutaneous fine-needle aspiration biopsies (primarily solid tissue).11 For best results, freshly extirpated specimens are preferred to formaldehyde-fixed or paraffin-embedded tissues. Specimens are initially placed in a buffered glutaraldehyde-based fixative, post-fixed in osmium tetroxide, dehydrated in graded alcohols, and embedded in an epoxy resin. Thick (1 µm) epoxy sections are stained with a buffered 1% toluidine blue solution to localize appropriate material for ultrastructural evaluation. Thin sections (90 to 120 nm) are cut with a diamond knife, sequentially stained with uranyl acetate and lead citrate, and then placed on copper grids and studied in the electron microscope.1
Although there are many different types of electron microscopes (e.g., high-voltage, scanning, analytic), the transmission electron microscope is most commonly used for diagnostic pathology (and is the type of electron microscope referred to throughout this chapter, unless otherwise noted). Modern electron microscopes are partially computerized and easier to operate than older models. A high vacuum can be achieved in approximately 5 to 10 minutes by use of a turbomolecular pump in place of the much slower and contaminating oil diffusion pump. Many of the latest instruments have zoom magnification, axis image rotation, and data imprinting and storage on hard drives or CD-ROMs. Future electron microscopes will be substantially smaller and will be capable of being operated in a lighted room. A high-refresh-rate charge-coupled device (CCD) that can transfer the image to a high-resolution flat-panel computer screen will replace the current fluorescent screen and the mechanically complex sheet film camera. The microscope should have one accelerating voltage (80 keV) and a realistic maximum magnification of 100,000. Captured digital images can be manipulated by using one of the many available photo-editing programs and printed on a high-resolution inkjet printer. With the availability of rapid ultrasonic and microwave tissue processing techniques and instant photography, there is no reason why a diagnostic transmission electron microscopic study cannot be completed within 24 hours or less. When properly maintained, a transmission electron microscope should last upward of 30 years. The only major expense of current scopes is, ironically, the service contract.
PROCEDURE FOR EVALUATING DIAGNOSTIC PROBLEMS
Pathologists can contribute to the reduction of health care costs by ordering expensive ancillary diagnostic tests (e.g., electron microscopy, immunohistochemistry, cytogenetic, and molecular genetic procedures) only when they are absolutely necessary for an accurate diagnosis and will have an impact on the patient's care.12 The large majority of human diseases, including common neoplasms such as breast and colon carcinoma, can be evaluated with hematoxylin-eosin (H&E)-stained slides and an occasional histochemical stain, such as mucicarmine. When an ancillary procedure is necessary to solve a diagnostic problem, most pathologists order a specific panel of immunohistochemical stains. However, the extensive use of immunostaining procedures has revealed a number of serious pitfalls and limitations, including a paucity of absolute organ- or tumor-specific antibodies; antigen diffusion problems (e.g., thyroglobulin, myoglobulin); anomalous or unexpected immunostaining results (e.g., keratin expression in astrocytomas13 and in malignant bone and soft tissue neoplasms14); and the selection of an inappropriate and often expensive immunophenotyping panel based on a false diagnostic presumption and an inability to detect small amounts of antigen (e.g., in poorly differentiated neuroendocrine carcinomas). Perhaps the most important problem with immunohistochemical procedures is a lack of standardized methodology and quality control. For example, different antigen retrieval methods, such as enzymatic and citrate treatments, microwave and ultrasonic procedures, and especially the newer heat-induced epitope retrieval (HIER) method, can alter antigen epitopes and staining patterns.15
Cytogenetic identification of specific reciprocal chromosome translocations and gene rearrangement studies, once used primarily to determine the lineage and clonality of leukemias and lymphomas, are now applicable to the diagnosis of a growing number of soft tissue tumors, notably adipose neoplasms,16 alveolar soft part sarcomas,17 and synovial sarcomas.18 Promising new methods for the detection of specific chimeric transcripts resulting from gene fusions are currently being developed and perfected in well-equipped and well-staffed molecular pathology laboratories. Some of these procedures are fluorescence in situ hybridization (FISH), reverse transcriptase polymerase chain reaction, DNA-based polymerase chain reaction, laser capture microdissection, confocal scanning laser microscopy, and atomic force microscopy (see Chapter 7). As with electron microscopy and immunodiagnostic methods, problems and shortcomings in cytogenetic and molecular procedures limit their diagnostic usefulness. These methodologies are constantly evolving, require sophisticated equipment and highly trained personnel, and are not available in many pathology laboratories.
From the preceding discussion, it should be obvious that the pathologist can choose from a variety of ancillary diagnostic procedures to solve a specific diagnostic problem. If possible, tissue should be available for ultrastructural evaluation in case the initial diagnostic approach (e.g., immunohistochemical panel of antibodies) does not provide a definitive answer. Many technical problems can be avoided if these procedures are performed and the specimens evaluated in central laboratories that are well equipped and staffed by highly qualified personnel.12 The remainder of this chapter discusses the current role of electron microscopy in the diagnostic evaluation of a select number of non-neoplastic and neoplastic diseases. It is mandatory that ultrastructural findings be correlated with a thorough clinicopathologic workup.
NON-NEOPLASTIC DISEASES
Ultrastructural studies contribute to the diagnosis of a wide variety of non-neoplastic diseases, including a number of glomerulopathies, microbial diseases (notably viral), cilia abnormalities, microvillous inclusion disease, lysosomal storage diseases, bullous skin diseases, CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy), peripheral neuropathies, and striated muscle (heart and skeletal muscle) diseases. The ultrastructural features of a large number of non-neoplastic diseases are summarized in an excellent book by Papadimitriou and colleagues.19
Glomerulopathies
The most common use of electron microscopy is for the evaluation of glomerulopathies. It is customary to divide percutaneous renal biopsies obtained from patients with nephrotic syndrome for light microscopic (H&E, periodic acid-Schiff [PAS], silver methenamine, and trichrome stains), immunofluorescent, and ultrastructural studies, because establishing a correct diagnosis has therapeutic and prognostic implications.20. , 21. The electron microscope is invaluable for resolving the constituents of the glomerulus, including the glomerular basement membrane (GBM), which ranges in thickness from 150 nm at birth to approximately 300 nm to 400 nm in adulthood; the blood capillary endothelial cell; the foot processes of the visceral epithelial cell (podocyte), with their “filtration slit pores” that line the urinary space; the mesangium (axial region of the glomerulus); and Bowman's capsule, which is lined on its inner surface by flattened parietal epithelial cells.
Primary and secondary (systemic) glomerular diseases in which ultrastructural studies are crucial to an accurate diagnosis include (1) minimal change nephritic syndrome (also called nil disease or lipoid nephrosis), which is characterized by extensive fusion of podocyte foot processes; (2) benign familial recurrent hematuria with marked thinning of the GBM (<180 nm in children; <250 nm in adults), also called thin basement membrane disease; (3) Alport's syndrome (a hereditary glomerulopathy), which results in marked thickening, attenuation, and lamellarization of the GBM (a result of injury and repair); and (4) Berger's disease (immunoglobulin A [IgA] nephropathy), exhibiting focal splitting and disruption of the GBM as well as finely granular mesangial deposits. Electron microscopy is also helpful for visualizing small peri-GBM, GBM, and mesangial deposits in, for example, early (stage 1) membranous glomerulonephritis, in which only scattered, small, subepithelial, dense immunoglobulin or light-chain deposits are evident (Fig. 6-1 ), and thrombotic microangiopathy (hemolytic uremic syndrome), with subendothelial deposits of an electron-lucent substance and microfibrils, formation of a thin endothelial basement membrane, mesangial interposition, and intraluminal (capillary) thrombi.
Figure 6-1 ▪.

Early (stage 1) membranous glomerulonephritis in the left kidney of a 59-year-old man. Two small subepithelial kappa light-chain deposits are evident (arrows). The capillary lumen is at the top. (Magnification ×46,200.)
More recently, a number of glomerulopathies with organized deposits have been recognized.22 These include the rare fibrillar, immunotactoid, and cryoglobulinemic glomerulopathies and the more common amyloidosis. The differential diagnosis of the three rare types depends on the ultrastructural identification of disease-specific glomerular deposits. The pertinent diagnostic features (primarily ultrastructural) of these diseases can be summarized as follows. Amyloidosis (proteinuria-nephrotic syndrome) often occurs in myeloma (plasma cell dyscrasia) patients; the usual type found is AL amyloidosis (abnormal light chains are present). Ultrastructurally, numerous 8- to 12-nm nonbranching fibrils are randomly distributed in the glomerulus. Fibrillary glomerulopathy is characterized by a ribbon-like pattern of IgA and C3 and a primarily subepithelial deposition of 20- to 30-nm fibrils with an amyloid P component. In immunotactoid glomerulopathy, IgG, C3, and characteristic clusters of spherical microtubular structures with a 30- to 40-nm diameter are also found in the mesangium. The cryoglobulinemic glomerulopathies are divided into types I, II, and III and are associated with B-cell lymphoplasmacytoid malignancies. Type I is identified by IgG and 80-nm-wide bundles of rigid fibrils or “fingerprint” arrays of tubular structures, all of which are distributed throughout the glomerulus. In types II and III, mixed cryoglobulins and 25-nm microtubules are found in thrombi and in the subepithelial and mesangial regions of the glomerulus. For more detailed information on all the glomerulopathies, see Chapter 29 and the cited references.20. , 21. , 22. , 23.
Microbial Diseases
The recent emergence of microbial pathogens, primarily viruses, that are highly infectious and contagious and capable of causing epidemics (or even pandemics) of potentially fatal diseases, as well as their possible use as bioterrorism agents,24 is of great concern. Some examples include a new variant of the cold-causing coronavirus that appeared in China in 2002 and is the cause of the deadly and highly infectious pneumonia known as severe acute respiratory syndrome (SARS; see later); Marburg and Ebola viruses (Filoviridae family), which replicate in the cytoplasm of white blood cells and cause hemorrhagic fever; another African virus, West Nile virus (Flaviviridae family), which can cause a fatal encephalitis25. , 26. , 27. ; and human immunodeficiency virus (HIV), a retrovirus that causes acquired immunodeficiency syndrome (AIDS).28
Because of the potential for deadly epidemics, it is imperative that the disease-causing agent (usually a virus) be quickly identified so that a vaccine can be produced. Many DNA- or RNA-containing virus families can be identified by mixing body fluids (e.g., sputum), blister contents, feces, or lesion scrapings with a 2% phosphotungstic acid, 0.5% uranyl acetate, or ammonium molybdate solution; placing the specimen on a coated nickel or copper grid; drying by touching the edge of the drop with filter paper; and then examining the grid in the electron microscope. These electron-dense stains permeate the interstices of the surface viral capsomeres, thus allowing for the quick recognition of many viral families.29. , 30. This procedure is called “negative staining” and should be performed only in a biohazard or biocontainment facility such as those at the Centers for Disease Control and Prevention (CDC) in Atlanta, Georgia. Recently, this rapid technique was used concurrently at the CDC and in Hong Kong to identify a new variant of coronavirus, recognized by the presence of club-shaped projections on the virus surface (Fig. 6-2 ), as the cause of SARS.31. , 32. A coronavirus was initially suspected based on thin-section electron microscopy and verified by negative staining.
Figure 6-2 ▪.
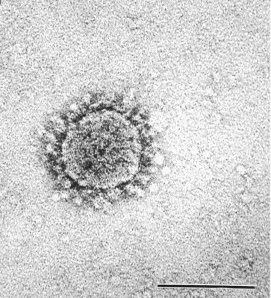
Negatively contrasted (2% methylamine tungstate) coronavirus (inoculated Vero E6 cell culture) in a 46-year-old man. The typical coiling of the coronavirus nucleocapsid results in circumferential club-shaped surface projections. (Bar = 100 nm.)
(Courtesy of Dr. Charles D. Humphrey; from Ksiazek TG, Erdman D, Goldsmith CS, et al: A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348:1953-1966, 2003. © 2003 Massachusetts Medical Society. All rights reserved.)
Other microbial agents such as intestinal spirochetes, rickettsia, protozoa, and bacteria can be identified using traditional thin-section electron microscopy (viruses can also be studied this way). Some examples include the protozoan microsporidia Encephalitozoon intestinalis 33 and Encephalitozoon hellem, which is the cause of ocular infections in immunocompromised individuals receiving chemotherapy and in HIV/AIDS patients.34
Cilia Abnormalities
Cross sections of normal cilia reveal an axoneme consisting of nine outer microtubule doublets with an inner and outer dynein-dynactin arm projecting from one of each of the doublets and two central tubules that are connected to the doublets by radial spokes (9 + 2 axoneme). Biopsy (either brush or forceps) is often performed to determine whether the cilia are structurally abnormal and presumably immotile in patients, particularly young children, with idiopathic chronic upper and lower respiratory tract infections.35. , 36. It is assumed that immotile cilia lack inner and outer dynein arms, as occurs in Kartagener's syndrome (sinusitis, bronchiectasis, and situs inversus).
Along with other investigators,19. , 35. I have found that most patients have secondary cilia defects that result from recurrent chronic respiratory tract infections and allergies. These defects include primary ciliary aplasia (loss of ciliated cells or cilia), compound cilia and megacilia, and abnormal microtubule patterns, such as an extra central tubule (9 + 3) and supernumerary outer tubules (9 + 2 + 1) (Fig. 6-3 ). Patients with primary (mainly hereditary) immotile cilia generally have a low or absent ciliary beat frequency and reduced or absent outer dynein arms.35 All the cilia should be affected. Most patients with chronic sinusitis variously show a loss of ciliated cells, respiratory tract basal cell metaplasia, or even a total loss of the surface epithelium, with a thickened basement membrane resting on a thick meshwork of collagen fibrils.36
Figure 6-3 ▪.

Chronic sinusitis in the nasal turbinate of a 57-year-old woman. Brush biopsy specimen illustrates a cilium with an extra outer microtubule (long arrow) and another cilium with three central microtubules (short arrow). (Magnification ×114,000.)
Microvillous Inclusion Disease
Microvillous inclusion disease (MID) is a rare but lethal congenital disorder characterized by intractable watery diarrhea beginning from birth to early infancy.37. , 38. MID is primarily a disease of the small intestines, but it has also been found in the large intestines and a number of other organs. By light microscopy, MID is characterized by diffuse hypoplastic villous atrophy, loss of the brush border, and absence of inflammatory changes. Fine cytoplasmic vacuolization is notable in the apical regions of the surface epithelial cells (enterocytes). Positive immunostaining for villin is evident on the surface of the epithelial cells and within the cytoplasmic vacuoles.
Because there are a number of causes of severe watery diarrhea in newborns and infants, a definitive diagnosis of MID requires ultrastructural examination of a biopsy specimen, which reveals shortened, poorly developed, and disorganized surface (brush border) epithelial cells that occasionally lack surface microvilli. Intestinal-type microvilli-lined intracytoplasmic inclusions, often surrounded by small vesicular bodies, are characteristically present in the apical cytoplasm of enterocytes (Fig. 6-4 ). A careful search may be required, because these diagnostic microvillous inclusions are not present in all surface enterocytes.
Figure 6-4 ▪.

Microvillous inclusion disease in the small intestine of a 5-month-old Navajo boy. A centrally located, microvilli-lined intracytoplasmic inclusion is evident. Note the intestinal-type microvilli rootlets extending into the cytoplasm. (Magnification ×15,000.)
(Courtesy of Gary Miereau, PhD, Children's Hospital, Denver.)
Lysosomal Storage Diseases
Characteristic inclusions in the cells of patients with various types of lysosomal storage diseases can be readily identified by electron microscopy and suggest a specific enzyme deficiency and stored metabolite.19 For example, intralysosomal collections of glucosylceramide-containing tubules are found in the splenic histiocytes of patients with Gaucher's disease, an acid β-glucosamide deficiency (Fig. 6-5 ). Other examples include the lipopigment fingerprint inclusions in neuronal ceroid-lipofuscinosis; needle and tubelike lactosylceramide inclusions in myelin sheaths from patients with globoid cell (Krabbe's) leukodystrophy; zebra bodies (large lamellar inclusions of dermatan and heparan sulfates) in Hurler's and Hurler-Scheie disease, a mucopolysaccharidosis; and accumulations of glycogen in striated muscle cells in Pompe's disease, a type II glycogenosis (acid maltase deficiency).39 The inclusions generally result from the absence, deficiency, or overproduction of specific lysosomal hydrolytic enzymes.
Figure 6-5 ▪.

Splenic histioctye from a 62-year-old man with Gaucher's disease. Intralysosomal long tubular inclusions have a diameter of approximately 50 nm. (Magnification ×40,000.)
Bullous Skin Disorders
Ultrastructural studies are useful for determining the exact cutaneous cleavage site in patients with blistering skin disorders, notably epidermolysis bullosa. The epidermal-dermal junction (basement membrane zone) consists of the hemidesmosomes of the basal keratinocyte with its associated cytokeratin intermediate filaments: the laminin-containing anchoring filaments of the lamina lucida, the collagen IV–containing lamina densa, and the collagen VII anchoring fibrils of the lamina fibroreticularis (the three laminae constitute the basement membrane).1. , 9. , 19. Defects in or the absence of any of these structures determines the site or level of cleavage of a blister and serves as a rough guide to diagnosis of the subtype of epidermolysis bullosa: (1) simplex (intraepidermal), in which cytolysis occurs in the basal cell cytoplasm; (2) atrophic (junctional), in which separation occurs in the lamina lucida owing to the loss of hemidesmosomes and laminin 5 deficiency; and (3) dystrophic (dermolytic), with cleavage below the lamina densa due to the lysis of collagen VII anchoring fibrils (Fig. 6-6 ).19 Antigen-mapping techniques using specific antibodies (e.g., antibodies to laminin and collagen type IV) can also be used to detect deficiencies in basement membrane constituents.
Figure 6-6 ▪.

Epidermolysis dystrophica, dermolytic type, in the skin biopsy of a 50-year-old woman. Cleavage occurs below the lamina densa of the basement membrane as a result of lysis of type VII collagen anchoring fibrils. Note the blister below the basement membrane (top right). The cytoplasm of the basal keratinocytes contains tonofilaments (arrow) and electron-dense melanosomes. (Magnification ×12,000.)
CADASIL
CADASIL, or cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, is a newly discovered cerebral vasculopathy.40 It is a hereditary multisymptomatic disease of early middle age. Manifestations include multiple episodes of aura-associated migraine headaches, recurrent subcortical cerebral infarcts, mood changes, pseudobulbar paralysis, demyelination, focal neurologic defects, strokes, and dementia.40 CADASIL is difficult to distinguish from other central nervous system diseases with similar symptoms. Magnetic resonance imaging, which reveals subcortical infarcts and demyelination, is required for diagnosis.
Histologic studies reveal characteristic arterial thickening due to a PAS-positive eosinophilic deposit of unknown origin.41 Ultrastructural studies have shown that a dome-shaped inclusion, commonly referred to as granular osmiophilic material (GOM; although the inclusions are not osmiophilic), is found in small arteries and arterioles throughout the vascular system (Fig. 6-7 ).41. , 42. Ultrastructural examination of a skin biopsy from a patient with suspected CADASIL can confirm the diagnosis by revealing the presence of pathognomonic arterial smooth muscle basement membrane–associated deposits or GOM in the small arteries and arterioles in the dermis.42 The significance or origin of the granular deposits is not known. Marked destruction of smooth muscle cells results in a decrease of vascular wall thickness and the loss of perivascular extracellular matrix.
Figure 6-7 ▪.

CADASIL granular osmiophilic material inclusion in the skin biopsy of a 40-year-old-man with chronic migraine headaches. Detail of a diagnostic electron-dense deposit in the basement membrane of an arterial smooth muscle cell is shown. (Magnification ×70,000.)
(Courtesy of Steven C. Bauserman, MD, Bruckenridge Hospital, Austin, Tex.)
Peripheral Neuropathies
Electron microscopy is a useful adjunct for the examination of peripheral nerve (primarily sural nerve) biopsy specimens from patients with a variety of neuropathies.43 Some examples include the identification of incipient Büngner's band formation (compact, layered Schwann cell processes enclosed in an external lamina) in axonal atrophy (Fig. 6-8 ); onion bulbs (concentric Schwann cells), a feature of demyelination and remyelination seen in Dejerine-Sottas, hypertrophic Charcot-Marie-Tooth, and Refsum's diseases; uncompacted myelin lamellae found in POEMS syndrome (polyneuropathy, oganomegaly, endocrinopathy, M protein, and skin lesions); and randomly organized arrays of 10-nm amyloid deposits in amyloidosis.
Figure 6-8 ▪.

Axonal atrophy in a sural nerve biopsy from a 50-year-old man with motor neuron disease of unknown cause. Early formation of layers of Schwann cell processes—Büngner's bands (arrow)—is due to the loss of axons (A). Note that the Schwann cells and unmyelinated nerve (N) are coated by a continuous external lamina. (Magnification ×20,000.)
Striated Muscle Diseases
Although the majority of ultrastructural changes seen in non-neoplastic muscle diseases are nonspecific, Kyriacou and associates44 found that electron microscopic examination of skeletal muscle biopsy specimens contributes to the elucidation and diagnosis of three main groups of muscle disorders: vacuolar, metabolic, and congenital myopathies. In the diverse group of vacuolar myopathies, electron microscopy is helpful in identifying the contents of the vacuoles common to these diseases. For example, tubules and filaments ranging in diameter from 15 to 18 nm are found in inclusion body myopathies. Abnormal lipid and glycogen metabolism, ion channel disorders, and so-called mitochondrial myopathies (pleomorphic mitochondria and paracrystalline cristae or matrical inclusions) constitute the metabolic myopathies. The congenital myopathies include nemaline “rod body” myopathy (elongate electron-dense aggregates of filaments that resemble streaming Z-discs); those exhibiting abnormal cytoplasmic structures (e.g., myofilaments, organelles, sarcoplasmic reticulum, T-tubules) and inclusions such as fingerprint and zebra bodies; and those with centrally located abnormal aggregates of sarcomere constituents (“target fibers”) such as central core disease, neurogenic atrophy, and inflammatory myopathies accompanying recent denervation (Fig. 6-9 ). Ultrastructural studies are indicated in approximately 20% of muscle biopsy specimens following standard light microscopy, special stains, and appropriate enzyme histochemical stains.44
Figure 6-9 ▪.

Inflammatory myopathy in a gastrocnemius muscle biopsy from a 70-year-old man. Portions of a so-called target fiber, consisting of aggregates of sarcomere constituents (top), are located in the center of the striated myocyte. (Magnification ×8000.)
Ultrastructural examination of endomyocardial biopsy specimens obtained from the right interventricular septum using a bioptome inserted into the right internal jugular vein and threaded to this structure is particularly useful for monitoring anthracycline-induced cardiotoxicity by the Billingham grading system (grades 0, 1, 1.5, 2, 2.5, and 3, with increasing myofibrillar loss, cytoplasmic vacuolization, and myocyte necrosis); detecting early amyloid deposition; and monitoring heart transplant patients for early signs of rejection, cardiomyopathy, and myocarditis.1. , 9. , 19.
NEOPLASTIC DISEASES
The main principle of tumor diagnosis using transmission electron microscopy is to identify a structure in neoplastic cells indicative of the line of differentiation, not histogenesis, of the neoplasm.1. , 2. , 3. , 4. , 6. , 7. , 8. , 10. , 11. , 12. For example, ultrastructural studies can be used for “quick diagnosis” by identifying myosin-ribosome complexes or rudimentary sarcomeres in an embryonal rhabdomyosarcoma (Fig. 6-10 ) or stage 2 elliptical melanosomes (premelanosomes) with a striated or zigzag filamentous core in variable numbers of cells composing an amelanotic malignant melanoma (Fig. 6-11 ). Because these structures are either absent or difficult to find in poorly differentiated rhabdomyosaromas (e.g., the solid alveolar variant) and in amelanotic malignant melanomas, an immunophenotyping panel of antibodies should be the first approach to accurate diagnosis (i.e., MyoD1, myogenin, skeletal muscle–specific actin, desmin, and S-100 protein and HMB-45, respectively). However, in a double-blind comparative study of the utility of phenotyping and electron microscopy in the diagnosis of childhood round cell tumors, Mierau and associates46 found that electron microscopy had an 89% efficiency rate versus 71% for immunohistochemistry. The rate rose to 95% when both procedures were used.
Figure 6-10 ▪.

Embryonal rhabdomyosarcoma in the deltoid muscle of a 21-year-old woman. Rigid myosin-ribosome complexes are randomly distributed throughout the cytoplasm. The Z-disc substance is indicated by an arrow. (Magnification ×24,000).
Figure 6-11 ▪.
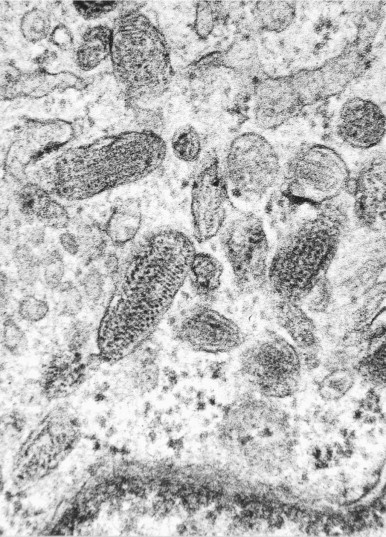
Lung metastasis from amelanotic malignant melanoma in a 75-year-old woman. Cytoplasmic elliptical premelanosomes have a striated or zigzag filamentous substructure. (Magnification ×74,400.)
The initial approach to diagnosis is often dictated by the laboratory facilities available at a specific pathology department. Nonetheless, if possible, a specimen from an undifferentiated tumor or other potentially difficult-to-diagnose problem should be placed in electron microscopy fixative in case another approach to diagnosis (usually immunophenotyping) fails.12 It is also important to maintain a collection of classic examples of specific nosologic entities for reference. For example, cells in all stages of myocyte differentiation are found in more well-differentiated rhabdomyosarcomas. The following sections discuss specific cases in which electron microscopy is important to tumor diagnosis. Ultrastructural studies are most commonly used to clarify a differential diagnosis.
Mesothelioma or Adenocarcinoma
The distinction between epithelial mesothelioma and adenocarcinoma is important for both therapeutic and medicolegal reasons. Although a variety of special stains and antibodies are being used for this important differential diagnosis (e.g., mucicarmine, carcinoembryonic antigen antibody, calretinin, Ber Ep4, B72.3, thrombomodulin, cytokeratin 5), not all of them are 100% specific for these two entities.47. , 48. , 49. , 50. Battifora and Gown51 recently reported that none of the antibody panels currently used for this differential diagnosis have sufficient specificity for either mesothelioma or carcinoma. For example, I recently studied a pleural tumor that contained a significant number of mucicarmine-positive cells. Ultrastructural examination, however, revealed the presence of numerous long, curving microvilli devoid of a surface glycocalyx (Fig. 6-12 ), which are diagnostic for epithelial mesothelioma.52 No mucigen granules were identified in the cytoplasm of the neoplastic mesothelial cells. A medium-density secretory substance, most likely hyaluronic acid, often coats the microvilli. Mucin-positive mesotheliomas have also been reported by other pathologists.49
Figure 6-12 ▪.

Epithelial mesothelioma in the pleura of a 69-year-old man. Profuse long, thin microvilli devoid of a surface glycocalyx cover the surface of a neoplastic mesothelial cell. (Magnification ×24,000.)
Current guidelines recommend that electron microscopy rather than expensive, large antibody panels be used for the differential diagnosis of epithelial mesothelioma and pulmonary adenocarcinoma.47 Electron microscopy should not be used to distinguish between reactive mesothelial cells and mesothelioma or for the diagnosis of sarcomatoid mesothelioma (immunophenotyping is more reliable). Immunohistochemistry is the more expensive procedure for this differential diagnosis.47
Soft Tissue Tumors
A specimen for possible electron microscopic examination should be taken from all spindle cell and epithelioid cell soft tissue neoplasms that cannot be diagnosed by the initial, routine gross and light microscopic (e.g., frozen sections) studies. Following are some examples of soft tissue tumors in which ultrastructural evaluation is important for the correct diagnosis. However, combined immunohistochemical and electron microscopic studies are often required to reach an accurate diagnosis.1. , 53. , 54.
Two variants of fibroblastic neoplasms—myofibroblastic fibrosarcoma (myofibroblastoma and myofibrosarcoma) and pleomorphic fibrosarcoma—require ultrastructural examination for diagnostic confirmation. The fibroblast is a remarkable cell with a large differentiation repertoire and no distinct immunoprofile or karyotype.9 The myofibroblast is the prototypical and best-known example of fibroblastic cell plasticity (see later). Myofibroblasts are primarily fibroblasts with the additional presence of peripheral arrays of actin microfilaments with dispersed fusiform densities, a fibronexus, and small desmosome-like and gap junctions. The immunohistochemical identification of a myofibroblast (especially neoplastic) is doubtful because five immunophenotyes have been identified: (1) vimentin and α-smooth muscle actin (α-SMA); (2) vimentin, α-SMA, and desmin; (3) vimentin and desmin; (4) vimentin; and (5) vimentin, α-SMA, and smooth muscle myosin heavy chain with or without desmin.55 It is thus obvious that myofibroblastic tumors—myofibroblastoma and myofibrosarcoma56—can be accurately diagnosed only by electron microscopy. Ultrastructural studies are often necessary to distinguish between myofibrosarcoma (peripheral actin filaments and a prominent rough endoplasmic reticulum) and poorly differentiated leiomyosarcoma (more randomly distributed actin filaments, numerous mitochondria, and a poorly developed rough endoplasmic reticulum).57 “Epithelioid myofibroblasts” are rare compared with smooth muscle neoplasms.
Ultrastructural studies in several laboratories, including two that examined a large number of cases,58. , 59. have clearly demonstrated that so-called malignant fibrous histiocytomas (a term commonly used as a diagnostic wastebasket)—notably, the more common storiform-pleomorphic and rare myxoid subtypes—are actually fibroblastic neoplasms and should be designated storiform-pleomorphic fibrosarcoma and storiform-pleomorphic myxoid fibrosarcoma, respectively. The predominant cells in the storiform areas are spindle-shaped neoplastic fibroblasts with a prominent branching and often dilated rough endoplasmic reticulum, variable arrays of vimentin filaments, and a large nucleolus. The giant rounded cells in the pleomorphic regions characteristically have a multisegmented nucleus with large nucleoli and occasional pseudoinclusions, arrays of vimentin intermediate filaments (prominent in the “rhabdoid” cells), and a well-developed rough endoplasmic reticulum (Fig. 6-13 ). Other cell types that are variably identified include myofibroblasts (see earlier), histiofibroblasts with the additional presence of variable numbers of lysosomes (these are more commonly seen in the pleomorphic tumor cells), cells with various combinations of all the preceding features, and undifferentiated mesenchymal cells that are variably present in all poorly differentiated sarcomas. On the basis of these findings, I propose a new pleomorphic fibrosarcoma classification (Table 6-1 ). Pleomorphic fibrosarcoma (originally designated by Arthur Purdy Stout) is a patternless neoplasm analogous to other pleomorphic sarcomas (e.g., pleomorphic liposarcoma).59 Pleomorphic myxoid fibrosarcoma replaces the inappropriate appellation myxofibrosarcoma. Current pathology nomenclature uses an adjective preceding the variant of a particular nosologic entity (e.g., sclerosing hemangioma, myxoid liposarcoma, plexiform schwannoma). Most myxoid fibrosarcomas are low-grade, poorly cellular neoplasms containing relatively few pleomorphic tumor cells. However, if these tumors are not completely excised, they may progress to high-grade neoplasms. I have not seen a storiform-pleomorphic myxoid fibrosarcoma in my practice. The term malignant giant cell tumor of soft parts is an appropriate general designation, because not all these neoplasms are fibroblastic.
Figure 6-13 ▪.

Storiform-pleomorphic fibrosarcoma (malignant fibrous histiocytoma) in the thigh of an 82-year-old man. A portion of a giant pleomorphic fibroblastic tumor cell with a multisegmented nucleus, large nucleolus (upper left), and well-developed rough endoplasmic reticulum is evident. (Magnification ×8800.)
TABLE 6-1.
Pleomorphic Fibrosarcoma Classification
| Pleomorphic fibrosarcoma |
| Storiform-pleomorphic fibrosarcoma* |
| Pleomorphic myxoid fibrosarcoma |
| Storiform-pleomorphic myxoid fibrosarcoma* |
| Giant cell fibrosarcoma with osteoclast-type giant cells* |
Neoplasms formerly designated variants of malignant fibrous histiocytoma.
Hemangiopericytoma is a commonly overdiagnosed neoplasm because hemangiopericytomatous patterns are seen in a variety of tumors (e.g., mesenchymal chondrosarcoma, solitary fibrous tumor). True cases of hemangiopericytoma also lack a distinctive immunoprofile.1 Although the generally plump spindle cells composing this controversial neoplasm lack unique ultrastructural features,60 a characteristic finding is the presence of basement membrane substance in the often narrow intercellular spaces that is continuous with the periendothelial basement membrane of the abundant, often dilated blood capillaries.
Ultrastructural studies are often helpful to confirm a suspected diagnosis of monophasic synovial sarcoma. A swirling pattern of ovoid and spindle cells variably immunoreactive for cytokeratins, epithelial membrane antigen, and S-100 protein is often found on light microscopic examination. Ultrastructural features of monophasic synovial sarcoma include arrays of closely packed fusiform tumor cells with tapering bipolar cytoplasmic processes that are joined by generally sparse rudimentary cell junctions, foci of intercellular basement membrane substance, inconspicuous organelles, occasional tonofilaments, and intercellular microvilli.1. , 61. Small lumens with microvilli and rudimentary junctional complexes are identified in more differentiated tumors. When possible, molecular genetic studies should be performed to identify the translocation t(X;18) resulting from the fusion of SYT at (18q11) with either SSX1 or SSX2, which are both at (Xp11).18 This translocation is found in essentially all cases of synovial sarcoma, biphasic and monophasic.
Rare primary benign soft tissue perineurioma, including its histologic variants,62 and perineurial cells in benign (notably neurofibroma) and malignant peripheral nerve sheath tumors63 can be easily identified by ultrastructural examination (see later). The principal peripheral nerve sheath cells are the S-100 protein-immunoreactive Schwann cell and the epithelial membrane antigen-reactive perineurial cell. Most nerve sheath tumors (e.g., benign schwannomas) show Schwann cell differentiation.63 By electron microscopy, Schwann cells possess elaborate cytoplasmic processes that are coated by a continuous external lamina. In contrast, perineurial cells have long, thin, generally straight or curving cytoplasmic processes that contain numerous pinocytotic vesicles (usually attached to the cell membrane) and are coated by a discontinuous external lamina (Fig. 6-14 ).1. , 63. The cytoplasmic processes are often joined by tight junctions.
Figure 6-14 ▪.

Soft tissue perineurioma in the right maxilla of a 59-year-old woman. Layers of thin perineurial cell processes are evident. Also note the pinocytotic vesicles (short arrows), external lamina (long arrow), and stromal collagen fibrils. (Magnification ×24,000.)
Soft tissue perineuriomas are difficult to recognize in histologic sections because they resemble a variety of low-grade fibroblastic and myofibroblastic neoplasms (see earlier), smooth muscle tumors, and neurofibromas.63 Although soft tissue perineuriomas have a characteristic immunophenotype (vimentin, epithelial membrane antigen with concentrated antibody, and collagen type IV and laminin, with no reactivity for S-100 protein) and a partial chromosome 22 deletion (M-ber locus at 22q;11), they can be quickly diagnosed by electron microscopy.1. , 62. , 63. , 64.
Gastrointestinal Stromal Tumors
The designation gastrointestinal stromal tumor (GIST) encompasses a variety of nonepithelial neoplasms most commonly originating in the wall of the stomach and small intestine, most of which were formerly considered to be smooth muscle tumors (e.g., leiomyoblastoma, leiomyosarcoma).65 Although the majority of GISTs show smooth muscle differentiation (see earlier), Herrera and associates66 in 1984 reported a “plexosarcoma,” a gastrointestinal tract mesenchymal tumor with ultrastructural features of autonomic nervous system (enteric plexus) differentiation (e.g., neuritic processes, sparse synapses, and 110-nm neurosecretory-type granules). A more commonly used term for plexosarcoma is gastrointestinal autonomic nerve tumor (GANT).
Recent studies have shown that all GANTs are immunoreactive for the kit tyrosine-kinase receptor CD117, which is also expressed by the intestinal pacemaker cells of Cajal in the gastrointestinal tract.67. , 68. , 69. Histologically, all GISTs—with the possible exception of smooth muscle neoplasms (this is controversial; see later) and other rare specific nosologic entities—are composed primarily of spindle-shaped cells with long bipolar, relatively thick cytoplasmic processes devoid of fine structural phenotypic markers. These cells characteristically contain few organelles or inclusions. Fine filaments are occasionally present in the cytoplasmic processes that are joined by rudimentary, tight, and gap (communicating) junctions. It is my opinion that normal interstitial cells of Cajal lack specific cytoplasmic markers and are difficult to identify.
Ultrastructural studies are often required to identify the GANT variant of GIST. In GANTs, long “neuritic” processes are variably present, in addition to Cajal-type cells. These processes contain variable numbers of microtubules, dense-core neurosecretory-type granules, and occasional synapses (Fig. 6-15 ). The synapses often contain both round and oblong neurosecretory granules and vesicles.65. , 66. The elongated cytoplasmic processes are primarily joined by tight and gap junctions, and so-called skeinoid fibers (tangled balls of collagen fibers) are occasionally present in the intercellular stroma. In addition to CD117, the cells composing GANTs express vimentin, CD34, and neuron-specific enolase, with occasional immunoreactivity for S-100 protein, neurofilaments, chromogranin, synaptophysin, and various peptides.65. , 66. The tumor cells thus resemble the neuritis of Auerbach and Meissner enteric plexosarcoma.66 On the basis of reports that virtually all smooth muscle tumors (GISTs) immunostain with kit (CD117) antibodies, a Cajal cell marker, Kindblom and colleagues67 proposed that GISTs be designated gastrointestinal pacemaker cell tumors (GIPacts). Lee and associates68 found that the clinicopathologic, histologic, immunohistologic, and molecular features of GANT are similar to GIST, indicating that GANT merely represents a phenotypic variant of GIST.
Figure 6-15 ▪.
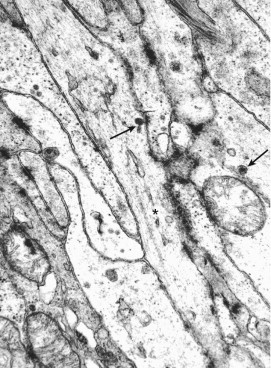
Gastrointestinal autonomic nerve tumor in the duodenum of a 57-year-old man. Arrays of neurite-like and Cajal cell processes are joined by rudimentary and tight cell junctions. Note the microtubules (asterisk) and the dense-core neurosecretory-type granules (arrows) with a diameter of 110 nm. (Magnification ×29,100.)
There is also a less common epithelioid cell variant of GIST (formerly designated leiomyoblastoma). Wardelmann and colleagues69 found that c-kit mutations were absent in seven cases they examined with an epithelioid component, whereas all GISTs with a spindle cell histology expressed this mutation. From all these studies, it is evident that further investigations are required to resolve the issue of the origin of these intriguing neoplasms.
Clear Cell Ependymoma
Electron microscopy is recommended to differentiate rare clear cell ependymoma from oligodendroglioma, central neurocytoma, and glioneurocytoma.70 These supratentorial glial fibrillary acidic protein–positive tumors often lack the classic light microscopic features of ependymoma. However, diagnostic hallmarks of ependymoma (e.g., microrosette formation, surface microvilli and cilia, long intercellular junctions) were identified in all eight cases reported by Min and Scheithauer.70 The diagnosis of clear cell ependymoma requires neuroimaging, histologic examination, and ultrastructural confirmation.
Dendritic Reticulum Cell Sarcoma
Langerhans cells, follicular dendritic cells, and interdigitating dendritic (reticular) cells are accessory cells of the lymphoid system that capture and present antigens to B cells (follicular dendritic cells) and T cells (interdigitating dendritic cells and Langerhans cells).71. , 72. These cells have a specific immunophenotype and ultrastructural features. For example, the Langerhans cell, the principal cells of Langerhans cell (eosinophilic) granulomatosis (these cells are not histiocytes), is characterized by a deeply cleaved or pseudomultisegmented nucleus and a unique organelle, the Birbeck granule, with its striated core and immunoreactivity for CD1c (Leu 6), CDw75, and S-100 protein.73
Follicular dendritic cell tumor is a rare, primarily intranodal neoplasm consisting of sheets and fascicles of oval to spindle-shaped cells with eosinophilic cytoplasm that are intimately admixed with small lymphocytes. These tumors resemble and behave like low-grade soft tissue sarcomas (they can originate in soft tissue). On ultrastructural examination, the spindle cells have long, occasionally interdigitating cytoplasmic processes that are joined by desmosomes and are immunoreactive for CD21 (IF8), CD23, CD35 (Ber-Mac-DCR), R4/23, and Ki-Myp.71 These tumors, which often have a focal storiform pattern and whorls, can be misdiagnosed as malignant melanoma, fibroblastic tumor, ectopic meningioma, orthotopic thymoma, or large cell lymphoma.
Rare cases of interdigitating dendritic cell sarcoma are found primarily in adult lymph nodes. Interdigitating dendritic cells are found in the T-cell portions of peripheral lymph node tissue, including the deep cortex and paracortex, and are responsible for stimulating resting T cells in the primary immune response. By light microscopy, the neoplasms are composed of a variable mixture of large and spindle-shaped cells with an abundant eosinophilic cytoplasm admixed with chronic inflammatory cells. The immunophenotype is S-100, CD68, CD45RO, and ATPase positive. Ultrastructurally, the tumor cells have long, interdigitating cytoplasmic extensions. No tonofilaments, desmosomes, Birbeck granules, dense-core granules, melanosomes, or basement membranes have been identified in any cases.72
More recently, my colleagues and I reported a cytokeratin-positive malignant tumor that may be a subtype of fibroblastic reticulum neoplasia arising from the interfollicuar fibroblastic reticular cells of mesenchymal origin.74 It is possible that they function to direct cell migration within the lymph node. These cells have a moderately well-developed rough endoplasmic reticulum in addition to interdigitating cytoplasmic processes. Following our report, Jones and colleagues75 published a case report of a clinically aggressive reticulum cell sarcoma with intermediate differentiation between follicular dendritic cells and fibroblastic reticular cells. Histologically, a multifocal proliferation of epithelioid and spindle cells, with prominent admixed lymphocytes and a high mitotic index, was found. Although ultrastructural examination revealed elongated cells with large nucleoli and interdigitating cytoplasmic processes joined by desmosomes, the immunohistochemical studies showed no expression of follicular dendritic cell antigens. It is obvious that these diverse neoplasms arise from lymph node stromal (reticular) cells, including mixed cell types. Combined ultrastructural and immunohistochemical studies are important for their characterization.71. , 72. , 73. , 74. , 75.
True Oncocytomas and “Granular” Renal Epithelial Tumors
True oncocytomas, unlike mitochondria-rich neoplasms, are characterized by numerous, often closely packed mitochondria with frequently stacked lamellar cristae that fill the cytoplasm.1 These epithelial tumors, which originate in many epithelial organs, can be readily identified by electron microscopy. Tumors consisting of cells that contain a moderate number of mitochondria (mitochondria-rich cells) are often incorrectly designated oncocytoma.
Mitochondrial morphology is useful for distinguishing a potentially malignant eosinophilic-granular “oncocytic” variant of renal chromophobe cell carcinoma (a tumor with distinctive cytoplasmic microvesicles discovered by electron microscopy) from benign renal oncocytoma.76 The cells composing renal chromophobe cell carcinomas, including mitochondria-rich cells, generally possess mitochondria with tubulovesicular cristae (twisting lamellar cristae seen in cross section) and lamellar cristae (Fig. 6-16 ), whereas oncocytomas have only lamellar cristae. The cells composing classic renal clear cell carcinoma (prominent cytoplasmic glycogen and lipid droplets) also have distinct long, pleomorphic mitochondria with lamellar cristae and an electron-dense matrix.
Figure 6-16 ▪.

Renal chromophobe cell carcinoma in the kidney of a 47-year-old man. A portion of the cytoplasm of a chromophobe cell illustrates mitochondria with tubulovesicular cristae and characteristic microvesicles (arrow). (Magnification ×20,800.)
The Unknown Primary
An article in the New York Times 77 reported that about 31,000 cancer patients (2% to 4% of all cancer patients) will be diagnosed with a neoplasm, usually a carcinoma, whose primary site cannot be identified, even after a complete workup that includes ancillary diagnostic procedures such as electron microscopy, immunohistochemistry, and molecular pathology. Many of these neoplasms are poorly differentiated (even undifferentiated) and are found in the lymph nodes, bones, skin, liver, lungs, and brain. Clinicians prefer an exact diagnosis, which enables them to choose the most effective therapy protocol and establish a prognosis.
Ultrastructural studies can occasionally help determine the primary site of a metastatic tumor, notably a carcinoma.1. , 78. , 79. , 80. For example, intestinal-type microvilli with anchoring microfilamentous rootlets, a surface branching glycocalyx, and glycocalyceal bodies are most often found in colorectal adenocarcinomas (Fig. 6-17 ). However, intestinal-type microvilli that are usually devoid of glycocalyceal bodies are also found in mucin-producing adenocarcinomas originating in other gastrointestinal sites, including associated organs and their ducts; pulmonary adenocarcinoma; rare enteric-type adenocarcinoma of the nasal cavity; and urachal-type mucinous carcinoma of the urinary bladder. Similarly, staghorn-like branching microvilli are see primarily in ovarian carcinomas, myelinosomes in bronchioalveolar carcinomas (notably the uncommon alveolar tumors), and abundant cytoplasmic glycogen particles and lipid droplets in renal clear cell carcinomas.
Figure 6-17 ▪.
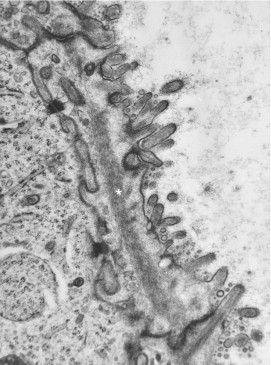
Skin metastasis of colonic adenocarcinoma in a 47-year-old man. Shown is the luminal surface of a neoplastic cell. Note the stubby intestinal-type microvilli with an external branching filamentous glycocalyx, gycocalyceal bodies (lower right), and a microfilamentous core that inserts into the terminal web (asterisk). (Magnification ×24,000.)
Allen M. Gown (personal communication) recommends a six-step immunohistochemical approach to determine the primary site of metastatic carcinoma: (1) antibodies to high- and low-molecular-weight cytokeratins, (2) cytokeratins 7 and 20 and CEA (CD66e, monoclonal antibody II-7), (3) vimentin coexpression with cytokeratin antibodies, (4) neuroendocrine markers (e.g., chromogranin, synaptophysin), (5) so-called specific markers (e.g., thyroglobulin, prostate-specific antigen), and (6) steroid hormone (estrogen and progesterone) receptors. For example, cytokeratin 20 positivity suggests colorectal or transitional epithelial origin, whereas cytokeratin 7 expression signifies a probable pulmonary (bronchial) primary. A marked elevation of CA125 levels in the blood of a patient with extensive abdominal adenocarcinoma suggests an ovarian primary.
Ultrastructural examination determined the primary site of a metastatic tumor at Memorial Sloan-Kettering Cancer Center in the following two examples. The first patient, an adult woman, presented with an enlarged cervical lymph node. A neoplasm that resembled a carcinoid tumor with an organoid growth pattern was identified by routine light microscopy, including special stains. The electron microscopic identification of round, oblong, and angulated endosecretory granules suggested a midgut primary tumor. At laparotomy, a small, asymptomatic carcinoid tumor was found in the midileum. The second patient, an adult man with extensive abdominal disease, had a biopsy of a thigh mass that revealed an undifferentiated round cell tumor devoid of significant immunoreactivity. Ultrastructural examination showed that the tumor cells had a moderately well-developed smooth endoplasmic reticulum, indicative of adrenocortical origin. A large tumor (most likely the primary) was found in the left adrenal gland (the testes contained no tumor).
In conclusion, it is obvious from these examples that ultrastructural pathology still contributes to the accurate diagnosis of a significant number of non-neoplastic and neoplastic diseases, although it is now used less often than in the 1970s and 1980s.
REFERENCES
- Erlandson RA. Raven Press; New York: 1994. Diagnostic Transmission Electron Microscopy, with Clinicopathological, Immunohistichemical, and Cytogenetic Correlations. [Google Scholar]
- Dickersin GR. Springer-Verlag; New York: 2000. Diagnostic Electron Microscopy: a Text/Atlas. [Google Scholar]
- Ghadially FN. 2nd ed. Butterworths-Heinemann; Boston: 1998. Diagnostic Ultrastructural Pathology: A Self-Evaluation and Self-Teaching Manual. [Google Scholar]
- Dardick I, Robb I. Society for Ultrastructural Pathology and Pathology Images; Ottawa, Canada: 2005. Primer on Electron Microscopy for Pathologists-in-Training [CD-ROM] [Google Scholar]
- Rosai J. The continuing role of morphology in the molecular age. Mod Pathol. 2001;14:258–260. doi: 10.1038/modpathol.3880295. [DOI] [PubMed] [Google Scholar]
- Ordonez NG, Mackay B. Electron microscopy in tumor diagnosis: Indications for its use in the immunohistochemical era. Hum Pathol. 1998;29:1403–1411. doi: 10.1016/s0046-8177(98)90008-9. [DOI] [PubMed] [Google Scholar]
- Hammar SP. Immunohistochemistry and electron microcopy in the diagnosis of neoplasms. Case Rev. 2002;7:201–208. [Google Scholar]
- Turbat-Herrera EA, D'Agostino H, Herrera GA. The use of electron microscopy to refine diagnosis in the daily practice of cytopathology. Ultrastruct Pathol. 2004;28:55–66. doi: 10.1080/01913120490430418. [DOI] [PubMed] [Google Scholar]
- Ghadially FN. 4th ed. Butterworths-Heinemann; Boston: 1997. Ultrastructural Pathology of the Cell and Matrix. [Google Scholar]
- Eyden B. Igaku-Shoin; New York: 1996. Organelles in Tumor Diagnosis: An Ultrastructural Atlas. [Google Scholar]
- Yazdi HM, Dardick I. Igaku-Shoin; New York: 1992. Guides to Clinical Aspiration Biopsy: Diagnostic Immunohistochemistry and Electron Microscopy. [Google Scholar]
- Erlandson RA, Rosai J. A realistic approach to the use of electron microscopy and other ancillary diagnostic techniques in surgical pathology [editorial] Am J Surg Pathol. 1995;19:247–250. doi: 10.1097/00000478-199503000-00001. [DOI] [PubMed] [Google Scholar]
- Kriho VK, Yang H-Y, Mookal JR. Keratin expression in astrocytoma: An immunofluorescent and biochemical reassessment. Virchows Arch. 1997;431:139–147. doi: 10.1007/s004280050080. [DOI] [PubMed] [Google Scholar]
- Swanson PE, Dehner LP, Sirgi KE. Cytokeratin immunoreactivity in malignant tumors of bone and soft tissue: A reappraisal of cytokeratins as a reliable marker in diagnostic immunohistochemistry. Appl Immunohistochem. 1994;2:103–112. [Google Scholar]
- Swanson PE. HIERanarchy: The state of the art in immunohistochemistry [editorial] Am J Clin Pathol. 1997;108:139–140. doi: 10.1093/ajcp/107.2.139. [DOI] [PubMed] [Google Scholar]
- Dei Tos AP, Del Cin P. The role of cytogenetics in the classification of soft tissue tumours. Virchows Arch. 1997;431:83–94. doi: 10.1007/s004280050073. [DOI] [PubMed] [Google Scholar]
- Ladanyi M, Lui MY, Antonescu CR. The der(17)t(X;17) (p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001;20:45–57. doi: 10.1038/sj.onc.1204074. [DOI] [PubMed] [Google Scholar]
- Ladanyi M. Fusion of the SYT and SSX genes in synovial sarcoma. Oncogene. 2001;20:5755–5762. doi: 10.1038/sj.onc.1204601. [DOI] [PubMed] [Google Scholar]
- Papadimitriou JM, Henderson DW, Spagnolo DV. Churchill Livingstone; Edinburgh: 1992. Diagnostic Ultrastructure of Non-Neoplastic Diseases. [Google Scholar]
- Churg J, Bernstein J, Glassock RJ. 2nd ed. Igaku-Shoin; New York: 1995. Renal Disease: Classification and Atlas of Glomerular Disease. [Google Scholar]
- Fogo ABS, Kashgarian M. WB Saunders; Philadelphia: 2005. Diagnostic Atlas of Renal Pathology. [Google Scholar]
- Howell DN, Gu X, Herrera GA. Organized deposits and look-alikes. Ultrastruct Pathol. 2003;27:295–312. [PubMed] [Google Scholar]
- Hass M. A re-evaluation of routine electron microscopy in the examination of native renal biopsies. J Am Soc Nephrol. 1997;8:70–76. doi: 10.1681/ASN.V8170. [DOI] [PubMed] [Google Scholar]
- Miller SE. Surveillance of bioterrorism agents: Considerations for electron microscopy laboratories. Microsc Today. 2004;56:57. [Google Scholar]
- Southam CM, Shipkey FH, Babcock VI. Virus biographies I: Growth of West Nile and Guaroa virus in tissue culture. J Bacteriol. 1964;88:187–199. doi: 10.1128/jb.88.1.187-199.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention Provisional surveillance summary of West Nile virus epidemic, United States, January-November. MMWR. 2002;51:1128–1133. [Google Scholar]
- Guarner J, Shieh W-S, Hunter S. Clinicopathologic study and laboratory diagnosis of 23 cases of West Nile encephalitis. Hum Pathol. 2004;35:983–990. doi: 10.1016/j.humpath.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Orenstein JM. Ultrastructure of HIV/AIDS. Ultrastruct Pathol. 2002;26:245–250. doi: 10.1080/01913120290104502. [DOI] [PubMed] [Google Scholar]
- Curry A. Electron microscopy as a tool for identifying new pathogens. J Infect. 2000;40:107–115. doi: 10.1053/jinf.2000.0634. [DOI] [PubMed] [Google Scholar]
- Hazelton PR, Gelderblom HR. Electron microscopy for the rapid diagnosis of infectious agents in emergent situations. Emerg Infec Dis. 2003;9:294–303. doi: 10.3201/eid0903.020327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith CS, Tattik M, Ksiazek TG. Ultrastructural characteristics of SARS coronavirus. Emerg Infect Dis. 2004;10:320–326. doi: 10.3201/eid1002.030913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek TG, Erdman D, Goldsmith CS. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- Chu P, West BA. Encephalitozoon (Septata) intestinalis: Cytologic, histologic, and electron microscopic features of a systemic intestinal pathogen. Am J Clin Pathol. 1996;106:606–614. doi: 10.1093/ajcp/106.5.606. [DOI] [PubMed] [Google Scholar]
- Orenstein JM. Diagnostic pathology of microsporidiosis. Ultrastruct Pathol. 2003;29:141–149. doi: 10.1080/01913120309938. [DOI] [PubMed] [Google Scholar]
- de Longh RU, Rutland J. Ciliary defects in healthy subjects, bronchiectasis, and primary ciliary dyskinesia. Am J Respir Crit Care Med. 1995;151:1559–1567. doi: 10.1164/ajrccm.151.5.7735615. [DOI] [PubMed] [Google Scholar]
- Al-Rawi MM, Edelstein DR, Erlandson RA. Changes in the nasal epithelium in patients with severe chronic sinusitis: A clinicopathologic and electron microscopic study. Laryngoscope. 1998;108:1816–1823. doi: 10.1097/00005537-199812000-00010. [DOI] [PubMed] [Google Scholar]
- Cutz E, Rhoads JM, Drumm B. Microvillous inclusion disease: An inherited defect of brush-border assembly and differentiation. N Engl J Med. 1989;320:646–651. doi: 10.1056/NEJM198903093201006. [DOI] [PubMed] [Google Scholar]
- Phillips AD, Schmitz J. Familial microvillous atrophy: A clinicopathological survey of 23 cases. J Pediatr Gastroenterol Nutr. 1992;14:380–396. [PubMed] [Google Scholar]
- Mierau GW, Weeks DA. Role of electron microscopy in the diagnosis of metabolic storage diseases affecting the nervous system of children. Ultrastruct Pathol. 1997;21:345–354. doi: 10.3109/01913129709021932. [DOI] [PubMed] [Google Scholar]
- Chabriat H, Vahedik K. Clinical spectrum of CADASIL: A study of 2 families. Lancet. 2005;346:934–939. doi: 10.1016/s0140-6736(95)91557-5. [DOI] [PubMed] [Google Scholar]
- Goubel HH, Meyermann R, Rosin R, Schlote W. Characteristic morphologic manifestations of CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, in skeletal muscle and skin. Muscle Nerve. 1992;20:625–627. doi: 10.1002/(sici)1097-4598(199705)20:5<625::aid-mus17>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Ruchoux MM, Maurage CA. CADASIL: Cerebral autosomal dominant arteriopathy with subacute infarcts and leukoencephalopathy. J Neuropathol Exp Neurol. 1997;56:947–964. [PubMed] [Google Scholar]
- Lampert PW, Schochet SS., Jr . Ultrastructural changes in peripheral nerve. In: Trump BF, Jones RT, editors. vol 2. John Wiley & Sons; New York: 1979. pp. 309–350. (Diagnostic Electron Mircroscopy). [Google Scholar]
- Kyriacou K, Kassianidies B, Hadjisavas A. The role of electron microscopy in the diagnosis of nonneoplastic muscle diseases. Ultrastruct Pathol. 1997;21:243–252. doi: 10.3109/01913129709021920. [DOI] [PubMed] [Google Scholar]
- Mierau GW, Weeks DA, Hicks MJ. Role of electron microscopy and other special techniques in the diagnosis of childhood round cell tumors. Hum Pathol. 1998;29:1347–1355. doi: 10.1016/s0046-8177(98)90002-8. [DOI] [PubMed] [Google Scholar]
- Mierau GP, Berry PJ, Malot RL. Appraisal of the comparative utility of immunohistochemistry and electron microscopy in the diagnosis of childhood round cell tumors. Ultrastruct Pathol. 1996;20:507–517. doi: 10.3109/01913129609016355. [DOI] [PubMed] [Google Scholar]
- Comin CE, de Klerk NH, Henderson DW. Malignant mesothelioma: Current conundrums over risk estimate and whither electron microscopy for diagnosis [editorial] Ultrastruct Pathol. 1997;21:315–320. doi: 10.3109/01913129709021929. [DOI] [PubMed] [Google Scholar]
- Riera JR, Astengo-Osana C, Longmate JA. The immunohistochemical diagnostic panel for mesothelioma: A reevaluation after heat-induced epitope retrieval. Am J Surg Pathol. 1997;21:1409–1419. doi: 10.1097/00000478-199712000-00003. [DOI] [PubMed] [Google Scholar]
- Hammar SP, Bockus DE, Remington FL. Mucin-positive epithelial mesothelioma: A histochemical, immunohistochemical and ultrastructural comparison with mucin-producing pulmonary adenocarcinomas. Ultrastruct Pathol. 1996;20:293–325. doi: 10.3109/01913129609016331. [DOI] [PubMed] [Google Scholar]
- Ordonez NG. The immunohistochemical diagnosis of mesothelioma: A comparative study of epithelial mesothelioma and lung adenocarcinoma. Am J Surg Pathol. 2003;27:1031–1051. doi: 10.1097/00000478-200308000-00001. [DOI] [PubMed] [Google Scholar]
- Battifora HA, Gown AM. Do we need two more mesothelial markers [editorial] Hum Pathol. 2005;36:451–452. doi: 10.1016/j.humpath.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Dury TD, Hammar SP, Roggli VL. Ultrastructural features of diffuse malignant mesothelioma. Hum Pathol. 1998;29:1382–1392. doi: 10.1016/s0046-8177(98)90006-5. [DOI] [PubMed] [Google Scholar]
- Erlandson RA, Woodruff JM. Role of electron microscopy in the evaluation of soft tissue neoplasms with emphasis on spindle cell and pleomorphic tumors. Hum Pathol. 1998;29:1372–1381. doi: 10.1016/s0046-8177(98)90005-3. [DOI] [PubMed] [Google Scholar]
- Fisher C. The value of electron microscopy and immunohistochemistry in the diagnosis of neoplasms. Case Rev. 2002;7:201–208. [Google Scholar]
- Schurch W, Seemayer TA, Gabbiani G. Myofibroblast. In: Sternberg SS, editor. Histology for Pathologists. 2nd ed. Lippincott-Raven; Philadelphia: 1997. pp. 129–166. [Google Scholar]
- Montgomery E, Goldblum JR, Fisher C. Myofibrosarcoma: A clinicopathologic study. Am J Surg Pathol. 2001;25:219–225. doi: 10.1097/00000478-200102000-00010. [DOI] [PubMed] [Google Scholar]
- Zukerberg LR, Cinti S, Dickersin GR. Mitochondria as a feature of smooth muscle differentiation: A study of 70 smooth muscle tumors. J Submicrosc Cytol Pathol. 1990;22:335–344. [PubMed] [Google Scholar]
- Suh CH, Ordonez NG, Mackay B. Malignant fibrous histiocytoma: An ultrastructural perspective. Ultrastruct Pathol. 2000;24:243–250. doi: 10.1080/01913120050176699. [DOI] [PubMed] [Google Scholar]
- Erlandson RA, Antonescu CR. The rise and fall of malignant fibrous histiocytoma. Ultrastruct Pathol. 2004;28:283–289. doi: 10.1080/019131290882150. [DOI] [PubMed] [Google Scholar]
- Dardick I, Hammar SP, Scheithauer BW. Ultrastructural spectrum of hemangiopericytoma: A comparative study of fetal, adult, and neoplastic pericytes. Ultrastruct Pathol. 1989;13:111–154. doi: 10.3109/01913128909057440. [DOI] [PubMed] [Google Scholar]
- Lopes JM, Bjerkehagen B, Sobrino-Simoes M. The ultrastructural spectrum of synovial sarcomas: A study of the epithelial differentiation of the primary tumors, recurrences, and metastases. Ultrastruct Pathol. 1993;17:137–151. doi: 10.3109/01913129309084034. [DOI] [PubMed] [Google Scholar]
- Hornick JL, Fletcher CDM. Soft tissue perineurioma: Clinicopathologic analysis of 81 cases including those with atypical histologic features. Am J Surg Pathol. 2005;29:845–855. doi: 10.1097/01.pas.0000155166.86409.d2. [DOI] [PubMed] [Google Scholar]
- Scheithauer BW, Woodruff JM, Erlandson RA. Atlas of Tumor Pathology. AFIP; Washington, DC: 1999. Tumors of the peripheral nervous system. 3rd ser, fasc 24. [Google Scholar]
- Giannini C, Scheithauer BW, Jenkins RB. Soft tissue perineurioma: Evidence for an abnormality of chromosome 22, criteria for diagnosis, and a review of the literature. Am J Surg Pathol. 1997;21:164–173. doi: 10.1097/00000478-199702000-00005. [DOI] [PubMed] [Google Scholar]
- Erlandson RA, Klimstra DS, Woodruff JM. Subclassification of gastrointestinal stromal tumors based on evaluation by electron microscopy and immunohistochemistry. Ultrastruct Pathol. 1996;20:373–393. doi: 10.3109/01913129609016340. [DOI] [PubMed] [Google Scholar]
- Herrera GA, Pinto de Moraes H, Grizzle WE. Malignant small bowel neoplasia of enteric plexus derivation (plexosarcoma): Light and electron microscopic study confirming the origin of the neoplasm. Dig Dis Sci. 1984;29:275–284. doi: 10.1007/BF01296263. [DOI] [PubMed] [Google Scholar]
- Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPact): Gastrointestinal stromal tumors show phenotypic characteristics of interstitial cells of Cajal. Am J Pathol. 1998;152:1259–1269. [PMC free article] [PubMed] [Google Scholar]
- Lee JR, Joshi V, Griffin JW., Jr Gastrointestinal autonomic nerve tumor: Immunohistochemical and molecular identity with gastrointestinal stromal tumor. Am J Surg Pathol. 2001;25:979–987. doi: 10.1097/00000478-200108000-00001. [DOI] [PubMed] [Google Scholar]
- Wardelmann E, Neidt I, Bierhoff E. c-kit Mutations in gastrointestinal stromal tumors occur preferentially in the spindle rather than in the epithelial cell variant. Mod Pathol. 2002;15:125–136. doi: 10.1038/modpathol.3880504. [DOI] [PubMed] [Google Scholar]
- Min K-W, Scheithauer BW. Clear cell ependymoma: A mimic of oligodendroglioma: Clinicopathologic and ultrastructural considerations. Am J Surg Pathol. 1997;21:820–826. doi: 10.1097/00000478-199707000-00010. [DOI] [PubMed] [Google Scholar]
- Perez-Ordonez B, Erlandson RA, Rosai J. Follicular dendritic cell tumor: Report of 13 cases of a distinct entity. Am J Surg Pathol. 1996;20:944–955. doi: 10.1097/00000478-199608000-00003. [DOI] [PubMed] [Google Scholar]
- Pilar K, Salomon R, Daurbenton JD, Sinclair-Smith CC. Interdigitating dendritic cell sarcoma: A report of four paediatric cases with a review of the literature. Histopathology. 2004;44:283–291. doi: 10.1111/j.0309-0167.2004.01816.x. [DOI] [PubMed] [Google Scholar]
- Lieberman PH, Jones CR, Steinman RM. Langerhans cell (eosinophilic) granulomatosis: A clinicopathologic study encompassing 50 years. Am J Surg Pathol. 1996;20:519–552. doi: 10.1097/00000478-199605000-00001. [DOI] [PubMed] [Google Scholar]
- Chan ACL, Serrano-Olmo J, Erlandson RA, Rosai J. Cytokeratin positive malignant tumors with reticulum cell morphology: A subtype of fibroblastic reticulum neoplasms? Am J Surg Pathol. 2000;24:107–116. doi: 10.1097/00000478-200001000-00013. [DOI] [PubMed] [Google Scholar]
- Jones D, Amin M, Ordonez NG. Reticulum cell sarcoma of lymph node with mixed dendritic and fibroblastic features. Mod Pathol. 2001;14:1059–1067. doi: 10.1038/modpathol.3880436. [DOI] [PubMed] [Google Scholar]
- Erlandson RA, Shek TWH, Reuter VE. Diagnostic significance of mitochondria in four types of renal epithelial neoplasms: An ultrastructural study. Ultrastruct Pathol. 1997;21:409–417. doi: 10.3109/01913129709021939. [DOI] [PubMed] [Google Scholar]
- Brody JE: A perplexing cancer, with no fixed address. New York Times, Science Times, Personal Health, Dec 7, 2004, p F7.
- Hammar SP. Metastatic adenocarcinoma of unknown primary origin. Mod Pathol. 1998;29:1393–1402. doi: 10.1016/s0046-8177(98)90007-7. [DOI] [PubMed] [Google Scholar]
- Hammar S, Bokus D, Remington F. Metastatic tumors of unknown origin: An ultrastructural analysis of 265 cases. Ultrastruct Pathol. 1987;11:209–250. doi: 10.3109/01913128709048325. [DOI] [PubMed] [Google Scholar]
- Bely M, Szabo TS, Kapp P. Ultrastructural identification of primary tumor site from bone metastases. Ultrastruct Pathol. 2003;27:163–186. doi: 10.1080/01913120309939. [DOI] [PubMed] [Google Scholar]