

The renin-angiotensin-aldosterone system (RAAS) plays a critical role in the maintenance of salt and water homeostasis by the kidney, particularly in hypovolemic or salt-depleted states. The net effect of the unopposed activation of this system results in sodium retention, potassium loss, and an increase in blood pressure (1).
COMPONENTS OF THE RAAS
Angiotensin Generation
Renin is synthesized in the juxtaglomerular (JG) cells (smooth muscle cells in the walls of the afferent arteriole as it enters the glomerulus), and is stored as prorenin (2, 3, 4, 5). It is released as renin, and regulated by renal afferent arteriolar baroreceptors and the macula densa of the distal nephron of the kidney. Renin enzymatically causes the formation of angiotensin I (Ang I) from angiotensinogen. Ang I is acted upon by angiotensin converting enzyme (ACE) to form angiotensin II (Ang II). The rate limiting step in this sequence of events in humans is the release of renin, rendering it the most well regulated variable of all constituents of the Renin-Angiotensin System (RAS). Renin secretion/release is increased by three primary pathways: (i) stimulation of renal baroreceptors by a decrease in afferent arterial stretch (pressure) (6,7); (ii) stimulation of renal β1-adrenergic receptors, in part, through renal sympathetic nerve activity (8, 9, 10); and (iii) a decrease in sodium and chloride delivery to, and transport by, the macula densa (11,12). Renin secretion can also be regulated by several endocrine and paracrine hormones.
ACE acts on Ang I (Ang 1-10) to cleave off the active octapeptide, Ang II (Ang 1-8), which is a more potent vasoconstrictor than Ang I (13). Ang II can also be formed by non-ACE enzymes and non-renin enzymes, such as chymase, cathepsin G, cathepsin A and tissue plasminogen activator (t-PA) (14). This assumes greater significance in the organs where not all components of the RAS are expressed, providing alternate means of generation of Ang II, the physiological significance of which is still not well understood, and may be organ specific. For example, mast cells produce renin, and have chymases, which help form Ang II and may play a role in heart failure and generation of arrhythmias (15).
ACE2 is a recently identified human homolog of ACE, sharing about 42% sequence homology. ACE2 converts Ang II to Ang 1-7. Ang 1-7 has vasodilator, antigrowth and antiproliferative properties, and exerts counterregulatory effects on Ang II. ACE 2 also decreases the level of Ang II by converting Ang I to Ang 1-9, an inactive nonapeptide (16,17). Thus, it appears that ACE and ACE2 exert opposing roles on the effects of the RAS. Unlike ACE, ACE2 is unable to inactivate bradykinin, and is insensitive to currently available ACE inhibitors. ACE2 has been identified as the receptor for the SARS-corona virus (SARS-CoV), allowing entry of the SARS-CoV into the cell (18,19). ACE2 may contribute to the localized overproduction of Ang 1-7 in the kidney, observed during pregnancy, which might protect against a rise in blood pressure (20).
Synthesis of the components of the RAAS occurs to a greater extent in certain organs relative to others, such as angiotensinogen in the liver, renin in the kidney, ACE in the lung, and aldosterone in the adrenal glands, which function together as an endocrine system. Some or all of its components are expressed in the brain, heart, vasculature, adipose tissue, pancreas, placenta, and kidney, among others, exerting autocrine, intracrine, and paracrine effects. This adds complexity to our understanding of the modulatory effects of the RAAS in maintaining homeostasis (5). Renin can be produced in the heart, brain, adrenal gland, testes, submaxillary gland, and mast cells, in addition to JG cells. Angiotensinogen is produced in extrahepatic sites such as the kidney, brain, spinal cord, mesentery, adrenal gland, atria, lung, stomach, spleen, large intestine, and ovary, and is also expressed in the ventricle and conduction tissue of the heart (5,21). ACE is ubiquitously expressed; however, the conversion of circulating Ang I to Ang II by ACE occurs mainly in the lung. ACE2 has been identified in the human heart, kidney and testis, and may be present in other tissues as well (21,22).
Ang II stimulates aldosterone synthesis and secretion by zona glomerulosa cells of the adrenal gland (23). Extra-adrenal sites of aldosterone synthesis include brain neurons, where it stimulates thirst, and cardiac myocytes, where it plays a role in the ventricular remodeling associated with salt retention (24). In these areas, the effects of aldosterone oppose those of glucocorticoids (25).
Ang II and its Metabolites
Aminopeptidases cleave Ang II at different sites. Aminopeptidase A acts on Ang II to form the heptapeptide Ang III, which participates, along with Ang II, in the classical effects on body fluid and electrolyte homeostasis, such as drinking behavior, vasopressin release and sodium appetite, in brain centers (5,21,26). Ang IV is a hexapeptide (Ang 3-8), formed by the action of aminopeptidase N on Ang II (27,28). Ang IV negatively regulates aminopeptidase A and thus influences the generation of Ang III.
Gene Targeting of Angiotensin Synthesis
The net impact of the autocrine, endocrine, intracrine, and paracrine effects of the RAAS is understood best in experimental models of controlled hyper-expression or deletion of target genes of the RAAS, such that their effects are enhanced or abolished. Most of these studies have been done in mice. Some have been performed in rats. Mice are different from other species in that they can have one or two renin genes (Ren-1 and Ren-2), depending on the strain (29). Ren-2 may have arisen from gene duplication during evolution. Thus, C57/BL6 mice express one renin gene while 129 sv mice express two renin genes. Interestingly, the adenosine receptor gene knockout mice, regardless of the strain (30), always express both renin genes. The physiological significance of one versus two genes is unknown thus far: there is no significant difference in plasma renin levels or renal renin mRNA expression between the two.
Deficiency of individual components of the RAAS is associated with low blood pressure while over-expression of individual components of the RAAS increases blood pressure, confirming the essential role of the RAAS in maintaining blood pressure. Angiotensinogen-deficient mice exhibit profound hypotension (31,32). These mice also exhibit delayed glomerular maturation and small papillae and develop nephrosclerosis (33). Transgenic mice, which over-express the human angiotensinogen gene, develop hypertension and renal fibrosis; the latter is ameliorated by the administration of an ACE inhibitor, independent of its blood pressure lowering effects (34).
Tissue-specific targeted ablation helps to elucidate the paracrine and autocrine effects of tissue RAS. In hypertensive mice expressing human renin (hREN) and human angiotensinogen transgenes under the control of their own endogenous promoters, glial-specific deletion of angiotensinogen results in lowered blood pressure (35), indicating that the central nervous system also contributes to the regulation of blood pressure. Introduction of the mouse Ren-2 gene into normotensive rats creates a transgenic strain that expresses Ren-2 mRNA in the adrenal gland, and the kidney (36). This transgene provides a monogenic model for a form of sodium dependent malignant hypertension. The Ren-2 transgenic rat has hyperproreninemia, low plasma and renal renin, high adrenal renin, and increased adrenal corticosteroid production. The hypertension that occurs in this model is responsive to ACE inhibitors. ACE deficient mice have decreased blood pressure and renal disease characterized by perivascular infiltrates and impaired concentration ability, indicating a role of ACE in the development of nephron function (37). ACE2 deficient mice develop dilated cardiomyopathy, and have a hypertensive response to Ang II (38,39).
Angiotensin Receptors
The effects of the angiotensin ligands Ang II, Ang III, and Ang IV are mediated by their interaction with specific angiotensin receptors. Ang II is known to interact mainly with two receptors, AT1 and AT2. Ang II and Ang III are full agonists at the AT1 receptor, while Ang IV binds to the receptor with low affinity (27,28,33,40). Almost all the usual physiological effects of Ang II are mediated by the AT1 receptor. The human AT1 receptor gene is located in chromosome 3q21–q25. The human AT2 receptor is located in chromosome Xq22–q23. AT1 and AT2 receptors belong to the seven-transmembrane class of G protein-coupled receptors (27,28,33,41). Adult human renal vasculature, glomeruli, and tubules (proximal, ascending limb of Henle, and collecting duct) express AT1 receptors; AT2 receptors are expressed in the vasculature and glomeruli but not in the tubules (42).
Rodents, but not humans, have two AT1 receptors, AT1A and AT1B. They are 94% homologous, and are located in chromosome 17q12 and chromosome 2q24, respectively (43). These receptor subtypes are differentially expressed: the AT1A receptor is expressed in the hypothalamus while the AT1B receptor mRNA is expressed in the pituitary gland. Both AT1A and AT1B receptors are expressed in the adrenal gland. While aldosterone secretion is mediated by both AT1A and AT1B receptors in the zona glomerulosa, corticosterone secretion is mediated by AT1A in the zona fasciculata (44). Adult rat kidneys express AT1A receptors in blood vessels and glomeruli. More AT1A receptors are expressed in collecting ducts than in other nephron segments (45,46). The AT1B receptor is not expressed in the adult or fetal rat kidney (47). In adult rat kidneys, AT2 receptors are expressed to a greater extent in glomeruli and to a lesser extent in proximal tubules and blood vessels. Female rats express more AT2 receptors than male rats (48).
Occupation of the AT1 receptor by Ang II triggers the generation of various second messengers via heteromeric G-proteins, chiefly Gq/11. Phospholipase C (PLC) β1 is activated leading to the formation of 1,4,5-inositol trisphosphate (IP3) and diacylglycerol (DAG) from the hydrolysis of phosphatidylinositol-4,5-bisphosphonate (PIP2). IP3 activates IP3 receptors in the endoplasmic reticulum releasing Ca2+. Ca2+ released from the endoplasmic reticulum causes the Ca2+-sensing STIM1 protein to interact with Orai1 in the plasma membrane. This interaction and the activation of IP3 receptors at the plasma membrane allow the influx of extracellular calcium (49,50). The increase in intracellular calcium and the stimulation of protein kinase C (PKC) by DAG lead to vasoconstriction (51). Activation of the AT1 receptor stimulates growth factor pathways such as tyrosine phosphorylation and PLCγ activation, leading to activation of downstream proteins, including mitogen-activated protein (MAP) kinases, signal transducers and activators of transcription (STAT protein). These cellular proliferative pathways, mediated by the AT1 receptor, have been implicated in the proliferative changes seen in cardiovascular and renal diseases (52).
The AT1 and AT2 receptors are differentiated based on their affinity for various non-peptide antagonists (51,53). The AT2 receptor shares 32–34% amino acid homology with the AT1 receptor, but activates second messenger systems with opposite effects via various signal transduction systems, mainly Gi and Go proteins (27,28,33,41,49,54). Stimulation of the AT2 receptor leads to activation of various phosphatases, resulting in inactivation of extracellular signal-regulated kinase (ERK), opening of K+ channels and inhibition of T-type Ca2+ channels. The AT2 receptor has a higher affinity for Ang III than Ang II. AT2 receptors may mediate antiproliferative effects, apoptosis, differentiation, and possibly vasodilatation.
Gene Targeting of Angiotensin Receptors
Gene targeting experiments in mice have elucidated the effects of angiotensin receptors. AT1A knockout mice are hypotensive and have a mild reduction in survival, compared to angiotensinogen knockout mice (41,55,56). AT1A receptor knockout mice have minor anomalies in the renal inner medulla and papillary structure (41,55, 56, 57, 58, 59). AT1B receptor knockout mice show no specific abnormalities and have normal blood pressure. This indicates that the AT1A receptor can compensate for the absence of the AT1B receptor (60). However, mice lacking both AT1A and AT1B receptors show deficient urinary peristalsis during the perinatal period (61). AT2 receptor knockout mice have modest elevation of blood pressure, and an enhanced pressor response to Ang II (62, 63, 64).
Several proteins interact with the AT1 and AT2 receptors. Over-expression of the AT1 receptor-associated protein (ATRAP) decreases AT1 receptor number (65). In contrast, the AT2 receptor-interacting protein (ATIP1) cooperates with the AT2 receptor in the inactivation of receptor tyrosine kinases, thus playing a role in the initiation of a signaling cascade that inhibits cell growth (66).
Other Ang II Receptors
There are two other receptors for Ang II. The AT3 receptor represents an angiotensin binding site and is identified in a mouse neuroblastoma cell line (67). The AT3 receptor has a high affinity for Ang II but a low affinity for Ang III. The AT4 receptor is an angiotensin binding site, with a high affinity for Ang IV (27,40,68). Unlike the AT1 and AT2 receptors, the AT4 receptor is not coupled to heterotrimeric G proteins, and has been identified as an insulin-regulated transmembrane aminopeptidase (IRAP). A unique binding site for the heptapeptide Ang 1-7 (formed by the action of ACE2 on Ang I) has also been identified (Fig. 7-1 ).
Figure 7-1.
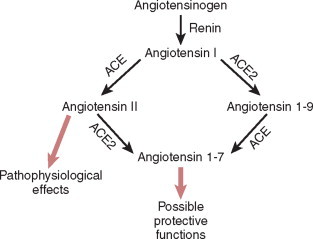
Pathways of ANG generation. ACE2-regulated pathways: both ACE and ACE2 are involved in the production of the biologically active peptides Ang II and Ang1-7 from Ang I. Elevated levels of Ang II are known to be detrimental to the function of the heart and kidney. Ang1-7 may function as a key peptide involved in cardioprotection and renoprotection. Genetic experiments suggest that ACE and ACE2 have complementary functions by negatively regulating different RAS products. The fine details of their regulatory function may differ depending on the local RAS environment.
(After Danilczyk U, Penninger M: Angiotensin-converting enzyme II in the heart and kidney. Circ Res, 98(4): 463–471, 2006.)
© 2008
Physiological Effects of Ang II
Ang II exerts most of its physiological effects via the AT1 receptor. Ang II has pleiotropic actions (69), including direct and indirect vasopressor effects. In response to sodium depletion, hypotension and/or hypovolemia, Ang II is formed, which causes immediate vasoconstriction of arteries and veins, increasing peripheral vascular resistance and venous return respectively, and raising blood pressure. The effect of Ang II on blood pressure secondary to increased ion transport by the renal tubule is more gradual. Ang II increases sodium and chloride reabsorption directly in several segments of the nephron. In the proximal tubule, low concentrations of Ang II play a central role in ion transport by activating luminal Na+/H+ exchanger (NHE), Na-glucose cotransporter (70), sodium phosphate cotransporter (NaPi) (71), basolateral Na+K+ATPase (72,73) and Na+-HCO3− exchanger (74). AT1 receptors stimulate NHE3 (75, 76, 77) but not NHE2 (78). High concentrations of Ang II can inhibit proximal tubule sodium transport via the stimulation of PLA2- and cytochrome P-450-dependent metabolites of arachidonic acid (74,79, 80, 81, 82). Ang II also affects ion transport in the medullary thick ascending limb of the loop of Henle in a biphasic manner (83). In this nephron segment, low concentrations of Ang II increase sodium, potassium, and chloride transport by stimulating NHE3 and NaK2Cl cotransporter activities (84). Ang II stimulates NHE1 activity in the macula densa (85). Ang II also stimulates NHE2 in the distal convoluted tubule and amiloride-sensitive Na+ transport (epithelial sodium channel, ENaC) in the collecting duct (86). All these effects are mediated by the AT1 receptors.
The role of AT2 receptors in influencing sodium transport is not well established. AT2 receptors may inhibit sodium transport. AT2 receptors are coupled to NHE6 (87), but NHE6 is not involved in sodium reabsorption (88). Ang 1-7 has been reported to inhibit Na+K+ATPase in pig outer cortical nephrons (89), and Na+-HCO3 − exchanger in mouse renal proximal tubules (90). However, an increase in sodium transport via AT2 receptors in rat proximal tubules has also been reported (91). These discrepancies may be related to the condition of the animal. For example, AT2 receptors inhibit Na+K+ATPase activity in renal proximal tubules of obese but not lean rats (92), and during AT1 receptor blockade (93).
Ang II indirectly increases sodium transport by stimulating the synthesis of aldosterone in the zona glomerulosa of the adrenal cortex. Aldosterone increases the density of ENaC in the cortical collecting duct by inducing the transcription of α-ENaC and redistribution of α-ENaC in the early cortical collecting duct from a cytoplasmic to an apical location. However, Ang II can stimulate the expression of α, β, and γ ENaC, independent of aldosterone (94). Aldosterone also activates Na+K+ATPase in the basolateral membrane of the principal cells of the cortical collecting duct. Aldosterone, like Ang II, can act in an autocrine and paracrine manner. Aldosterone has been reported to be produced by aldosterone producing cells other than the adrenal glomerulosa, such as neuronal glial cells and cardiac myocytes (95).
There is evidence that Ang I and Ang II may act, in ligand-independent ways, to affect cell signaling, cell–cell communication and growth, via their metabolism by the enzyme ACE2, mentioned above. They may also oppose the traditionally accepted effects of the RAAS (96). While AT1 receptors can be stimulated by stretch, independent of Ang II (97), Ang II can have intracellular effects independent of AT1 receptors (98).
Concepts and Controversies in our Current Understanding of the Renal Effects of the RAAS in Maintaining Fluid and Electrolyte Homeostasis and Blood Pressure
Feedback mechanisms in the kidney contribute to the maintenance of renal blood flow and glomerular filtration rate (GFR) in the face of fluctuations in blood pressure. This phenomenon of renal autoregulation was first recognized as early as 1932 (99). In some animals, autoregulation of renal blood flow is negligible at birth (100). In primates, in the immediate neonatal period, autoregulation of renal blood flow is present, but autoregulation of blood flow to other organs, such as brain, myocardium, and intestines, is not observed (101). Autoregulation of renal blood flow is observed in canine puppies as young as 14 days of age (102). However the set point is lower and the efficiency of autoregulation is less in younger than in older puppies.
Autoregulation is achieved by the interplay of two mechanisms: (i) tubuloglomerular feedback (TGF), which involves a flow-dependent signal from the macula densa and alteration of afferent arteriolar tone, which is mediated by adenosine and/or ATP (103, 104, 105); and (ii) myogenic response, which involves direct vasoconstriction of the afferent arteriole in response to increased transmural pressure. These two mechanisms act in concert to prevent acute fluctuations in renal blood flow and GFR in response to changes in blood pressure (Fig. 7-2 ) (106).
Figure 7-2.

The juxtaglomerular apparatus and tubuloglomerular feedback (TGF). TGF is achieved because of the anatomy of the nephron and juxtaglomerular (JG) apparatus. TGF is a phenomenon that occurs when changes in tubular fluid concentrations of Na+, Cl-, and K+ are sensed by the macula densa, via the luminal Na+-K+-2Cl cotransporter. Increases or decreases in luminal uptake of these ions cause reciprocal changes in GFR by alterations in vascular tone, mainly in the afferent arteriole.
(From Guyton AC, Hall JE: The kidneys and body fluids. In Guyton AC, Hall JE (eds): Textbook of Medical Physiology (Fig 26.14, p. 292). Philadelphia, PA: WB Saunders, 2000.)
Recent studies challenge pre-existing concepts and merit discussion. Glomerulotubular balance (GTB) is a negative feedback loop that occurs when proximal tubular reabsorption of sodium and water increases in response to increased GFR (107), and vice versa. Thus, GTB affects distal sodium delivery and, consequently, TGF. TGF and GTB may be more critical in the regulation of renal function in the neonate than in the adult as sodium intake is low in the newborn; sodium balance must be tightly regulated to achieve the net positive balance required for growth. Recent advances in our understanding of GTB, specifically in the neonate, will be discussed.
ONTOGENY
Development of the RAAS: Structure of the Kidney and Urinary Tract
Prenatal Changes in RAAS Structure and Function in Rodents and Humans
STUDIES IN HUMANS
In humans, angiotensin-related genes are activated at Stage 11 of the developing embryo (108), which corresponds to 23–24 days of gestation. AT1 and AT2 receptors are expressed very early (24 days of gestation), indicating that Ang II may play a role in organogenesis. The AT1 receptor is expressed in the glomeruli while the AT2 receptor is found in the undifferentiated mesonephros that surrounds the primitive tubules and glomeruli. AT2 receptor is maximally expressed at about 8 weeks of gestation, followed by decreasing, but persistent expression until about 20 weeks of gestation (109). At Stage 12–13, which corresponds to 25–29 days of gestation, angiotensinogen is expressed in the proximal part of the primitive tubule, while renin is expressed in glomeruli and JG arterioles. ACE is detected in the mesonephric tubules at stage 14. By 30–35 days, all components of the RAAS are expressed in the human embryonic mesonephros. Expression of these proteins in the future collecting system occurs later, at about 8 weeks of gestation.
ACE has a role in fetal growth and development. ACE inhibitor-induced fetopathy, consisting of oligohydramnios, intrauterine growth retardation, hypocalvaria, renal dysplasia, anuria, and death, has been described in mothers exposed to ACE inhibitors during the second and third trimesters of pregnancy (110). These effects were initially believed to arise from a decrease in organ perfusion (111). More recent evidence indicates that ACE inhibition has teratogenic effects during development. Fetuses exposed to ACE inhibitors during the first trimester have an increased risk of congenital malformations, with an incidence of 7.1%, compared to those with no maternal exposure to antihypertensive medications during the same time period. The congenital anomalies are major and include cardiovascular defects such as atrial septal defect, patent ductus arteriosus, ventricular septal defects and pulmonic stenosis; skeletal malformations, including polydactyly, upper limb defects and craniofacial anomalies; gastrointestinal malformations such as pyloric stenosis and intestinal atresia; central nervous system malformations such as hydrocephalus, spina bifida and microcephaly; and genitourinary malformations including renal malformations (112). Angiotensin receptor blockers (ARB) have also been reported to be fetotoxic (113). Inhibition of the activity of the RAAS in pregnancy may have effects on the fetus that manifest later on in life, such as hypertension, which are addressed later.
STUDIES IN RODENTS
The expression patterns of the RAS components in the embryonic kidney are similar in rodents and humans (108,114). Angiotensinogen is expressed in the proximal part of the primitive tubule in the mouse at embryonic day 12.5 (30 days’ gestation in humans) (115). Renin is widely expressed initially in the renal vasculature in the embryonic rat, but its expression becomes localized to the afferent arteriole with development. Renin secretion is greater during the early embryonic period and decreases gradually. Circulating renin levels are higher in the neonatal rodent than in the adult, and appear to be due to increased production (115). Rodent ACE follows a similar expression pattern as in humans. Ang II and AT1 receptors are important in nephron development and afferent arterial growth. AT1 receptors may guide Ang II to its target sites to induce cell growth and differentiation (116, 117, 118). AT1 receptor mRNA is strongly expressed during the early embryonic period (E17 to E20 in the rat), with increased expression in mature nephron segments by E17, persisting until adulthood (119). AT2 expression is seen in both the undifferentiated mesenchyme in the maturing kidney and the mature nephron, including the proximal tubule. Collectrin, a novel homolog of ACE2 (120) shares about 47% homology with ACE2 in humans, but lacks the carboxypeptidase function. Collectrin mRNA is expressed in the mouse embryo at day 13, is localized to the ureteric bud, the progenitor of the collecting duct, increases throughout development, and decreases after birth.
Mutations of genes encoding renin, angiotensinogen, ACE, or AT1 receptor are associated with skull ossification defects (121), as well as autosomal recessive renal tubular dysgenesis (122, 123, 124). AT1 receptor deficient mice do not develop a renal pelvis (122). AT2 receptor deficient mice have congenital abnormalities of the kidney and urinary tract (CAKUT), such as multicystic dysplastic, and aplastic kidneys (124). The more dramatic phenotypes seen in mice with deficient AT2 receptor, as compared to humans with similar defects, is attributed to the fact that human nephrogenesis is completed in utero, while maturation of rodent nephrons occurs for up to 10 days after birth. However, AT1 receptor gene deficient phenotypes are similar in rodents and humans. As indicated earlier, mutations in angiotensin-related genes in humans are associated with renal tubular dysgenesis (123). These studies demonstrate the importance of the RAAS in development and maturation of the kidney and collecting systems (115).
Postnatal Changes in RAAS Structure and Function in Rodents and Humans
STUDIES IN RODENTS
In rats, angiotensionogen mRNA expression is negligible immediately after birth, but increases dramatically by day 5, only to decrease again over the next few days, followed by a gradual increase to adult levels by 2 months of life. The newborn rat kidney can generate Ang II directly from angiotensinogen via a serine-protease that is induced in the newborn (125). Renin levels are higher in the neonatal than the adult rodent, and appear to be due to increased production (107). Ang II levels are high in the early neonatal period, and decrease with age.
STUDIES IN HUMANS
The RAAS is more highly activated in the neonatal period and infancy compared to later in childhood (126). Plasma renin activity and plasma aldosterone levels are markedly increased in preterm human infants in the first 3 weeks of life (127). While fetal ACE levels remain stable during gestation in rats (128), serum ACE activity has been reported by some to be higher in late fetal than in neonatal life in humans (128, 129, 130), lambs and guinea pigs (131,132). In contrast, renal ACE activity may increase with age, at least in pigs and horses (133,134). A similar pattern may be found in humans based on measurement of urine ACE isoforms (135). Recent studies measuring plasma Ang II levels in 46 normal and low birth weight human infants have shown increased Ang II levels at 7 days of life in very low birth weight infants (136). Maximum serum aldosterone levels are seen at 2 h of age, and gradually decrease over the first year of life (137).
Sodium Homeostasis in the Neonatal Period
Sodium intake is low in infants compared to adults, as milk is a poor source of sodium. However, a positive balance of sodium in the neonate is needed to sustain growth, in contrast to normal adults who are in sodium balance. The kidney of a full term infant filters 4–5% of the volume of plasma filtered by an adult (120 mL/min/1.73M2). Despite the relative paucity of sodium transporters, the full-term neonate does retain sodium, in part because of the low GFR (3). Although renal sodium transporters are reduced, neonates cannot excrete a sodium load compared to adults. This has been attributed to the increased activity of agents that increase sodium reabsorption in the neonatal period, including the RAAS and α-adrenergic receptors (117,138). Endogenous Ang II inhibits the natriuresis of acute expansion in the neonatal rat (139). The decreased effects of natriuretic factors (e.g., atrial natriuretic peptide, dopamine, nitric oxide) in the term neonate compared to the adult may also impair its ability to excrete a sodium load (140, 141, 142). Water transport across the proximal tubule also differs in adults and neonates. The adult tubule transports water by the water channels called aquaporins, which belong to the category of non-solute carrier-related genes. These are present in very low concentrations in the neonatal nephron. However, water is transported effectively in the neonatal nephron by paracellular and transmembrane mechanisms and by passage through non-aquaporin channels to maintain GTB (see below) (143). Aquaporin expression increased with age, an effect that is mediated by glucocorticoids (144).
Development of TGF
Growth and maturation affect the influence of distal ion delivery on the sensors of the macula densa in producing a vascular response. TGF responses are operative in neonatal rats, as early as 3.5 weeks of age (145,146). The inflection point for TGF may also be different between younger and older rats. Extracellular fluid volume and dietary protein affect the TGF response profoundly. The effect of dietary protein on the TGF response varies in different studies, with high protein diets stimulating or blunting the TGF response.
Despite a lower GFR, urinary sodium losses are highest in the most premature babies but fractional sodium excretion (FENa) is exponentially related to gestational age. The renal sodium loss in prematurity appears, in part, to result from the immaturity of the TGF mechanism. Postnatal age has been shown to have an independent effect on FENa but not on GFR. These findings indicate that in infants of greater than 33 weeks’ gestation, sodium conservation is possible because of adequate TGF. The rapid increase in sodium reabsorption in the first few postnatal days seems to be due to maturation of distal tubular function. Although this was initially attributed to aldosterone (147), the decreased amount of transporters that are targets of aldosterone makes this mechanism unlikely. An increase in maturity of the Na+K+ATPase in the distal tubule and a decrease in extracellular fluid volume may be contributory factors to postnatal maturation of sodium transport in the distal tubule.
Development of GTB
The essential regulatory mechanisms of tubule transport include GTB and neural and hormonal factors such as sympathetic innervation, Ang II, endothelin, parathormone, and other mediators. As defined earlier, GTB is the capacity of each segment of the nephron to reabsorb a constant fraction of the GFR, fluid and solute delivered to it, and is influenced by peritubular and intratubular capillary pressures and GFR (148). The capacity of the proximal tubule to reabsorb sodium, chloride and water, bicarbonate, glucose and organic substances is adjusted to GFR: the higher the GFR, the greater the reabsorption. The underlying physical mechanism of GTB is a flow-dependent reabsorption of ions and water across the renal proximal tubular luminal membrane in response to changes in GFR, which is independent of neural and hormonal systems. Maintenance of GTB is influenced by flow rate, substrate delivery and other unknown systems. It is signaled by the hydrodynamic torque (bending movement) on epithelial microvilli (149). Increases in luminal diameter have the effect of blunting the impact of flow velocity on microvillous shear stress and, thus, on microvillous torque. Variations in microvillous torque produce nearly identical fractional changes in sodium reabsorption. Furthermore, the flow-dependent sodium transport is enhanced by increasing luminal fluid viscosity, diminished in NHE3 knockout mice, and abolished by nontoxic disruption of the actin cytoskeleton. These data suggest that the ‘brush-border’ microvilli serve a mechanosensory function wherein fluid dynamic torque is transmitted to the actin cytoskeleton to modulate sodium absorption in renal proximal tubules.
Clinical studies have been conducted to study GTB in term and preterm human infants, which suggest that GTB is operational from about 33 weeks of gestational age (147). In a study of 70 infants of gestational ages 27–40 weeks and postnatal ages 3–68 days, 24-h sodium balance studies and creatinine clearance measurements showed that intrauterine age and extrauterine existence independently increased the maturation of this function. The incidence of hyponatremia was associated with a negative sodium balance, which varied directly with the degree of prematurity. Indeed, no babies born after 36 weeks had a negative sodium balance. Studies in rats also show that GTB is present in 4.5 month old rats. Thus, it appears that GTB is maintained during development by proportional maturation of GFR and tubular reabsorptive mechanisms (150).
CURRENT CONCEPTS AND CONTROVERSIES
The changes in preglomerular resistance that regulate renal blood flow and GFR in the face of changing blood pressure is attributed to TGF and the renal myogenic response, acting in concert, as mentioned earlier. This is the classic paradigm. However, over the last 20 years, the importance of TGF and renal autoregulation has been challenged, and the role of the myogenic response as a renoprotective mechanism to prevent renal damage due to increased blood pressure has been demonstrated in several studies.
There is evidence that renal protection is lost when autoregulation fails (151,152). The changes in glomerular capillary pressure to maintain GFR at a relatively constant level in the face of increasing or decreasing blood pressure is different from the vascular response needed to reduce GFR to limit pressure-induced increases, which cause renal injury. Thus, TGF and the myogenic response occur at different levels and may have different goals in the balance between renal autoregulation and renal protection. Classic studies in the two kidney, one clip model of hypertension (153,154) were followed by studies in the uninephrectomized deoxycorticosterone acetate (DOCA)/salt model of malignant nephrosclerosis (155), confirming the damage engendered by a dilated renal vascular bed in the face of hypertension. In a 5/6 ablation model of chronic kidney disease, the loss of autoregulation increases susceptibility to hypertensive renal injury.
There are different requirements for protection versus regulation. The myogenic response, which constricts the afferent arteriole, occurs after every 3–4 s in response to a blood pressure change. A TGF response takes up to 20 s, as it involves the sequence of increased distal delivery, sensing by the macula densa, release of mediator (now presumed to be adenosine/ATP) and generation of arteriolar afferent response (156). Thus, very brief perturbations of blood pressure have insignificant effects on renal blood flow and GFR. Such brief episodes should alter the myogenic response, but there are no convincing in vivo data available that support this. Similarly, there are no studies on the effects of spikes in systolic versus spikes in mean arterial blood pressure in generating a renal myogenic response. A delay in the onset of pressure-induced vasoconstriction has been reported in the intact kidney (157), with a longer delay in vasodilatation induced by decreasing blood pressure in the intact kidney and an even longer delay in a hydronephrotic kidney.
The advocates for TGF as the dominant mechanism argue that as GFR is influenced by several factors, including plasma colloid pressure, proximal tubular pressure and the filtration coefficient (158), a vascular response to changes in pressure alone may not be adequate for regulation. TGF, with its response to alteration in distal sodium delivery, may thus play the stronger autoregulatory role, while its immediate renoprotective role may be less important.
Differentiating these two closely aligned mechanisms is difficult. In the Fawn-Hooded rat, the Brown-Norway rat, and the Dahl salt-sensitive rat, the genetic defect in autoregulation seems to involve the myogenic response, with an intact or even enhanced TGF (159, 160, 161). Studies of renal injury in these models may shed more light on this issue. We do know that humans with uncomplicated, essential hypertension and intact renal autoregulation do not exhibit renal injury (151,152).
If autoregulation is essential for volume homeostasis, one would expect an unequivocal relationship between the two. However, there is little evidence that impaired autoregulation leads to impaired volume homeostasis. Hypertension is not clearly linked to loss of autoregulation. In the Brown-Norway rat, blood pressure is reduced and administration of DOCA or NaCl has little effect (162). In genetic knockout mice without the TGF mechanism, no overt volume disturbances are noted (163). Similarly, if distal delivery is manipulated, such as by the chronic use of loop diuretics, the effects should be catastrophic, since these also suppress TGF. However after an initial loss of volume, steady state adaptations occur within 3–4 days. GTB helps in this adjustment.
Mediators and Modulators of TGF
There are several recent studies aimed at identifying the mediator of the TGF mechanism, which is critical to autoregulation. As defined earlier, TGF is a phenomenon that occurs when changes in tubular ion transport by the macula densa cause reciprocal changes in GFR by alterations in vascular tone, mainly in the afferent arteriole (106). The initial step in the TGF mechanism is the sensing of the luminal signal by the Na+K+2Cl cotransporter at the macula densa. The activation of the Na+K+2Cl cotransporter generates adenosine, which stimulates A1 adenosine (A1) receptors, resulting in increased cytosolic calcium. Some investigators have suggested that ATP, activating P2X1 receptors, triggers the increase in cytosolic calcium. The calcium signal is propagated to extraglomerular mesangial cells, constricting vascular smooth muscle cells of the afferent arteriole and decreasing GFR. Renin secretion is also inhibited, which allows recovery of arteriolar flow and GFR. TGF is absent in A1 receptor knockout mice. In contrast, TGF response persists in mice in which the ACE, AT1 receptor, NOS1, or thromboxane receptor genes are disrupted (164). Vasoconstrictors, such as Ang II, increase the sensitivity of the TGF response, while vasodilators, such as nitric oxide, blunt the response (106). These studies indicate that TGF is modulated by Ang II, arachidonic acid metabolites, and nitric oxide, while the primary mediators are adenosine and/or ATP.
Adenosine
A recent study of cd73-/- mice, which cannot generate ATP/adenosine, suggests that a humoral factor, adenosine, may mediate TGF response. Thus, a lack of adenosine abrogates the change in GFR engendered by TGF, but does not affect distal reabsorption of sodium and water along the tubule, likely mediated by aldosterone (165). The cells of the macula densa display a role in regulation, by sensing increased NaCl delivery to the distal tubule, and activation of Na+K+2Cl cotransporter activity to reduce GFR, with adenosine most likely being the mediator of this response. Indeed, TGF is absent in A1 receptor knockout, mice (165). In anesthetized wild type, and A1 receptor knockout mice, GFR and renal blood flow were measured before and after reducing renal perfusion pressure by a suprarenal aortic clamp. A reduction in blood pressure produced a significantly greater fall in A1 receptor knockout mice compared to the wild type, indicating reduced regulatory responses in the knockout mice. This suggests that deficient autoregulation in the absence of the effector adenosine or A1 receptor is mediated by abrogation of the TGF response.
The administration of highly selective adenosine 1 receptor antagonist, CVT-124, results in marked diuresis and natriuresis, indicating failure of autoregulation (148). The diuresis and natriuresis are associated with a reduction in absolute proximal tubular reabsorption and its uncoupling from glomerular filtration. In addition, the response of the macula densa to increased distal delivery of sodium chloride and water is blunted, as there is no corresponding decrease in GFR. This indicates inhibition of the TGF mechanisms, and provides additional support for adenosine being the mediator of the TGF response. The lack of proximal tubular regulation in GTB could be indicative of adenosine being a player in GTB, in addition to the known and accepted peritubular, luminal and oncotic controls that regulate it. However, the mechanism underlying the increased formation of adenosine in response to an increase in Na+K+2Cl cotransporter activity remains to be determined.
ATP
Navar et al (166) make the case for ATP being the mediator of the TGF response based on their finding that ATP selectively affects renal afferent arteriolar tone, the demonstration of the presence of ATP-specific receptors P2X1 in the renal afferent arteriole, the absence of the TGF response in mice lacking the P2X1 receptor, and evidence that ATP directly stimulates the L-type voltage gated calcium channel, leading to calcium influx and vascular smooth muscle cell contraction (167,168).
Nitric Oxide (NO)
NO, derived from arginine, modulates TGF. Type I NO synthase (NOS) is expressed in the macula densa (169). Other NOS isoforms may be expressed in the mesangium and glomerular microvessels. These enzymes are strategically positioned to influence each step of the TGF process (170). However, micropuncture experiments using NOS antagonists have shown that NO does not mediate TGF. Instead, local NOS blockade causes the curve that represents TGF to shift leftward and become steeper. Changes in NO production in the macula densa may underlie the resetting of TGF, which is needed to maintain the TGF curve so that it adapts to different conditions of ambient tubular flow to accommodate physiologic circumstances and maintain homeostasis (171). Also, macula densa NO production may be substrate limited and dissociated from NOS protein content. The importance of NO to TGF resetting and the substrate dependence of NO production have both been found during changes in dietary salt. Changes in neuronal NOS (nNOS or NOS1) function have been shown to occur in the JGA of the spontaneously hypertensive rat (SHR) (172). NOS inhibition has been demonstrated to have a reduced effect on TGF in the SHR (173).
Reactive Oxygen Species
Reactive oxygen species and ongoing oxidative stress are increasingly implicated in the pathophysiology of vascular changes in many diseases, including hypertension. AT1 receptors increase the sensitivity of the TGF response, which could also be related to reactive oxygen species. While the superoxide anion may lead to vasoconstriction and an increase in myogenic tone, directly, and via many signaling pathways, it also influences vasoconstriction and vasodilatation as a consequence of TGF. These effects occur due to changes in macula densa cell function in response to changes in sodium chloride delivery to the distal tubule. The macula densa expresses nNOS (171), which is activated during sodium chloride reabsorption and has a vasodilatory effect; this blunts the vasoconstriction caused by the TGF in response to increased sodium chloride transport. Oxygen radicals enhance the TGF response and limit NO signaling from the macula densa (174). Reactive oxygen species react with NO, producing peroxynitrate, which impairs vasorelaxation and promotes hypertrophy (175, 176, 177): thus reactive oxygen species effectively counteract the effects of NO.
The interaction between NO and superoxide anion has been studied in microperfusion experiments. The infusion of a nitric oxide precursor into the macula densa caused a graded reduction in the TGF-mediated afferent arteriolar vasoconstriction; this response was more pronounced in the Wistar Kyoto (WKY) rat compared to the (SHR) (171). The thick ascending limb of the loop of Henle also produces NO, as it expresses endothelial NOS (eNOS or NOS III) (170,173). Here, studies have shown that NO decreases net absorption of chloride and bicarbonate in the isolated thick ascending limb (178), thus indirectly decreasing the TGF response by increasing delivery of these to the distal tubule. Superoxide anion activates 5′-nucleotidase, thereby increasing adenosine generation in the kidney (which we know to be a mediator of TGF). These studies implicate reactive oxygen species in the enhancement of TGF (106).
Calcium Wave
Intracellular calcium modulates vascular smooth muscle tone and is the mediator for stimulation of renin release. There is evidence for a calcium wave that spreads through the mesangial cell field and constricts the afferent arteriolar smooth muscle cells. It appears that both gap junctional communication and extracellular ATP are integral components of the TGF calcium wave. The finding that the calcium wave is generated by ATP, but not by adenosine, offers a new model for a direct effect of ATP, not necessarily mediated by adenosine, as the final common pathway of changes in vascular tone in response to signals from the JGA and macula densa. A recent study demonstrates this using ratiometric calcium imaging of the in vitro microperfused isolated JGA-glomerular complex dissected rabbits (179).
New Directions
Fetal Programming for Hypertension: Failure of renoprotection?
The association of low birth weight with the development of hypertension later in life has been validated epidemiologically (180) and, more recently, has been demonstrated experimentally in animal models (181). A suboptimal fetal environment may lead to maladaptive responses, including failure of renal autoregulation and the development of hypertension. A reduction in nephron number during development may contribute to a reduction in GFR, but this is not always borne out in experimental studies. Multiple factors may contribute to the development of hypertension, but we will restrict ourselves to the role of renal hemodynamics. The RAAS may play a more important role as it is expressed early and associated with nephrogenesis. Blockade of the AT1 receptor during the nephrogenic period after birth in the rat led to a decrease in nephron number, a reduction in renal function and hypertension (182). Protein restricted diets have also shown a rise in ACE levels in pregnant ewes (183) and a general stimulation of all components of the systemic RAS in other studies in response to protein restriction, which is blocked by treatment with an ACE inhibitor or an AT1 receptor blocker. Thus, the adverse environment in utero, which programs the fetus to develop hypertension could be critically linked to abnormalities in the RAS (182).
Unconventional Behavior of RAS Components
While the canonical scheme of activation of the RAS components, from renin to Ang II and its effects on AT receptors is well recognized, recent evidence of other effects merit discussion. Both Ang I and Ang II may lead to effects that are independent of, or even antagonistic to, the accepted effects of the RAS (96). ACE may also function as a ‘receptor’ that initiates intracellular signaling and influences gene expression (184). AT1 and AT2 receptors have been shown to form heterodimers with other 7-transmembrane receptors, and influence signal transduction pathways (185, 186, 187, 188). Intracellular Ang II exerts effects on cell communication, cell growth and gene expression via the AT1 receptor, but also has independent effects (189,190).
Clinical Aspects
As discussed earlier, the development of the kidney occurs during the first 35 days postconception in humans. The integrity of the RAAS is essential for normal development, and Ang II is essential for normal structural development of the kidney and collecting system. The complex interplay of GTB and TGF increases with gestational age. The excretory function of the kidney begins soon after clamping of the umbilical cord at birth and continues through the first year of life. It follows that the neonate is exquisitely sensitive to stressors, which activate the RAAS, and lead to profound oligoanuric states.
Ischemia and Asphyxia Generate Renin by Sympathetic Stimulation
Drugs such as furosemide increase distal delivery of sodium and water, but reduce Na+K+2Cl activity, which should decrease renin release. However, decreased cotransporter activity impairs the TGF and, in the face of continued ion and water excretion, may result in hypovolemia, activation of the RAS and may lead to anuria. Indomethacin, used to treat patent ductus arteriosus in the neonate, could lead to altered renal vasoregulation (191). Congenital abnormalities in steroid synthesis could lead to deficiencies in aldosterone production, and cause profound salt-wasting states (192). In pseudohypoaldosteronism Type 1, mutations in the sodium channel may cause profound neonatal salt wasting (193). To counter the heightened awareness of the RAAS in neonates, adenosine receptor antagonists may offer new avenues of therapy.
SUMMARY
In summary, the RAAS is an important developmental and physiological system that contributes to renal blood flow and GFR autoregulation. Together with the myogenic response, the RAAS serves to maintain volume homeostasis in sodium and volume depleted states, and renoprotection in hypertensive states. The relative contributions of these two systems continue to be an area of controversy and debate. We also know that central and peripheral sympathetic input influences all aspects of renal autoregulation. More studies in transplanted or denervated kidneys in animal models and humans will elucidate these areas of controversy. A better understanding of these mechanisms would translate to better care for premature and full term newborns with renal dysregulation, hyponatremia and oligoanuric renal failure.
REFERENCES
- 1.Guyton AC, Hall JE. Textbook of Medical Physiology. 10th edn. W.B. Saunders; Philadelphia, PA: 2001. Dominant role of the kidney in long-term regulation of arterial pressure and in hypertension: The integrated system for pressure control; pp. 201–203. [Google Scholar]
- 2.Gomez RA, McReddie TA, Everett AD. Ontogeny of renin and AT1 receptor in the rat. Pediatr Nephrol. 1993;5:635–638. doi: 10.1007/BF00852571. [DOI] [PubMed] [Google Scholar]
- 3.Chevalier RL. The moth and the aspen tree: sodium in early postnatal development. Kidney Int. 2001;59(5):1617–1625. doi: 10.1046/j.1523-1755.2001.0590051617.x. [DOI] [PubMed] [Google Scholar]
- 4.Persson AE, Ollerstam A, Liu R. Mechanisms for macula densa cell release of renin. Acta Physiol Scand. 2004;181:471–474. doi: 10.1111/j.1365-201X.2004.01320.x. [DOI] [PubMed] [Google Scholar]
- 5.Lavoie JL, Sigmund CD. Minireview: overview of the renin-angiotensin system—an endocrine and paracrine system. Endocrinology. 2003;144(6):2179–2183. doi: 10.1210/en.2003-0150. [DOI] [PubMed] [Google Scholar]
- 6.Krieger MH, Moreira ED, Oliveira EM. Dissociation of blood pressure and sympathetic activation of renin release in sinoaortic-denervated rats. Clin Exp Pharmacol Physiol. 2006;33(5–6):471–476. doi: 10.1111/j.1440-1681.2006.04389.x. [DOI] [PubMed] [Google Scholar]
- 7.Schweda F, Segerer F, Castrop H. Blood pressure-dependent inhibition of renin secretion requires A1 adenosine receptors. Hypertension. 2005;46(4):780–786. doi: 10.1161/01.HYP.0000183963.07801.65. [DOI] [PubMed] [Google Scholar]
- 8.Milavec-Krizman M, Evenou JP, Wagner H. Characterization of beta-adrenoceptor subtypes in rat kidney with new highly selective beta 1 blockers and their role in renin release. Biochem Pharmacol. 1985;34(22):3951–3957. doi: 10.1016/0006-2952(85)90371-5. [DOI] [PubMed] [Google Scholar]
- 9.DiBona GF. Neural regulation of renal tubular sodium reabsorption and renin secretion. Fed Proc. 1985;44(13):2816–2822. [PubMed] [Google Scholar]
- 10.Goldsmith SR. Interactions between the sympathetic nervous system and the RAAS in heart failure. Curr Heart Fail Rep. 2004;1(2):45–50. doi: 10.1007/s11897-004-0024-5. [DOI] [PubMed] [Google Scholar]
- 11.Bell PD, Lapointe JY, Peti-Peterdi J. Macula densa signaling. Annu Rev Physiol. 2003;65:481–500. doi: 10.1146/annurev.physiol.65.050102.085730. [DOI] [PubMed] [Google Scholar]
- 12.Lorenz JN, Greenberg SG, Briggs JP. The macula densa mechanism for control of renin secretion (1993) Semin Nephrol. 1993;13(6):531–542. [PubMed] [Google Scholar]
- 13.Erdos EG. Conversion of angiotensin I to angiotensin II. Am J Med. 1976;60:749–759. doi: 10.1016/0002-9343(76)90889-5. [DOI] [PubMed] [Google Scholar]
- 14.Urata H, Nishimura H, Ganten D. Mechanisms of angiotensin II formation in humans. Eur Heart J. 1995;16(Suppl N):79–85. doi: 10.1093/eurheartj/16.suppl_n.79. [DOI] [PubMed] [Google Scholar]
- 15.Le TH, Coffman TM. A new cardiac MASTer switch for the renin-angiotensin system. J Clin Invest. 2006;116(4):866–869. doi: 10.1172/JCI28312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chappel MC, Ferrario CM. ACE and ACE2: their role to balance the expression of angiotensin II and angiotensin-(1-7) Kidney Int. 2006;70:8–10. doi: 10.1038/sj.ki.5000321. [DOI] [PubMed] [Google Scholar]
- 17.Donoghue M, Hsieh F, Baronas E. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87(5):E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 18.Li W, Moore MJ, Vasilieva N. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han DP, Penn-Nicholson A, Cho MW. Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology. 2006;350(1):15–25. doi: 10.1016/j.virol.2006.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brosnihan KB, Neves LA, Joyner J. Enhanced renal immunocytochemical expression of ANG-(1-7) and ACE2 during pregnancy. Hypertension. 2003;42(4):749–753. doi: 10.1161/01.HYP.0000085220.53285.11. [DOI] [PubMed] [Google Scholar]
- 21.Gavras I, Gavras H. Angiotensin II as a cardiovascular risk factor. J Hum Hypertens. 2002;16(Suppl 2):S2–S6. doi: 10.1038/sj.jhh.1001392. [DOI] [PubMed] [Google Scholar]
- 22.Danilczyk U, Penninger JM. Angiotensin-converting enzyme II in the heart and kidney. Circ Res. 2006;98(4):463–471. doi: 10.1161/01.RES.0000205761.22353.5f. [DOI] [PubMed] [Google Scholar]
- 23.Romero DG, Plonczynski M, Vergara GR. Angiotensin II early regulated genes in H295R human adrenocortical cells. Physiol Genomics. 2004;19:106–116. doi: 10.1152/physiolgenomics.00097.2004. [DOI] [PubMed] [Google Scholar]
- 24.Declayre C, Swynghedauw B. Molecular mechanisms of myocardial remodeling. The role of aldostorone. J Mol Cell Cardiol. 2002;34:1577–1584. doi: 10.1006/jmcc.2002.2088. [DOI] [PubMed] [Google Scholar]
- 25.Colombo L, Dala Valle L, Fiore C. Aldosterone and the conquest of land. J Endocrinol Invest. 2006;29(4):373–379. doi: 10.1007/BF03344112. [DOI] [PubMed] [Google Scholar]
- 26.Veerasingham SJ, Raizada MK. Brain renin-angiotensin system dysfunction in hypertension: recent advances and perspectives. Br J Pharmacol. 2003;139:191–202. doi: 10.1038/sj.bjp.0705262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chai SY, Fernando R, Peck G. The angiotension IV/AT4 receptor. Cell Mol Life Sci. 2004;61:2728–2737. doi: 10.1007/s00018-004-4246-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saavedra JM, Ando H, Armando I. Brain angiotensin II, an important stress hormone: regulatory sites and therapeutic opportunities. Ann N Y Acad Sci. 2004;1018:76–84. doi: 10.1196/annals.1296.009. [DOI] [PubMed] [Google Scholar]
- 29.Pan L, Gross KW. Transcriptional regulation of renin: an update. Hypertension. 2005;45:3–8. doi: 10.1161/01.HYP.0000149717.55920.45. [DOI] [PubMed] [Google Scholar]
- 30.Schweda F, Wagner C, Kremer BK. Preserved macula densa dependent renin secretion in A1 adenosine receptor knockout mice. Am J Physiol Renal Physiol. 2003;284(4):F770–F777. doi: 10.1152/ajprenal.00280.2002. [DOI] [PubMed] [Google Scholar]
- 31.Kim HS, Krege JH, Kluckman KD. Genetic control of blood pressure and the angiotensinogen locus. Proc Natl Acad Sci USA. 1995;92(7):2735–2739. doi: 10.1073/pnas.92.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tanimoto K, Sugiyama F, Goto Y. Angiotensinogen deficient mice with hypotension. J Biol Chem. 1994;269(50):31334–31337. [PubMed] [Google Scholar]
- 33.Niimura F, Labosky PA, Kakuchi J. Gene targeting in mice reveals a requirement for angiotensin in the development and maintenance of kidney morphology and growth factor regulation. J Clin Invest. 1995;96(6):2947–2954. doi: 10.1172/JCI118366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fern RJ, Yesko CM, Thornhill BA. Reduced angiotensinogen expression attenuates renal interstitial fibrosis in obstructive nephropathy in mice. J Clin Invest. 1999;103(1):39–46. doi: 10.1172/JCI4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sherrod M, David DR, Zhou X. Glial-specific ablation of angiotensinogen lowers arterial pressure in renin and angiotensin transgenic mice. Am J Physiol Regul Integr Comp Physiol. 2005;289(6):R1763–R1769. doi: 10.1152/ajpregu.00435.2005. [DOI] [PubMed] [Google Scholar]
- 36.Wagner J, Thiele F, Ganten D. The renin-angiotensin system in transgenic rats. Pediatr Nephrol. 1996;10(1):108–112. doi: 10.1007/BF00863461. [DOI] [PubMed] [Google Scholar]
- 37.Bernstein KE. Views of the renin-angiotensin system: brilling, mimsy and slithy tove. Hypertension. 2006;47(3):509–514. doi: 10.1161/01.HYP.0000196266.23639.c6. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto K, Ohishi M, Katsuya T. Deletion of angiotensin-converting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II. Hypertension. 2006;47(4):718–726. doi: 10.1161/01.HYP.0000205833.89478.5b. [DOI] [PubMed] [Google Scholar]
- 39.Crackower MA, Sarao R, Oudit GY. Angiotensin-converting enzyme 2 is an essential regulator of heart functionfunction. Nature. 2002;417(6891):822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 40.Davis CJ, Kramar EA, De A. AT4 receptor activation increases intracellular calcium influx and induces a non-N-methyl-D-aspartate dependent form of long-term potentiation. Neuroscience. 2006;37:1369–1379. doi: 10.1016/j.neuroscience.2005.10.051. [DOI] [PubMed] [Google Scholar]
- 41.Crowley SD, Tharaux PL, Audoly LP. Exploring type I angiotensin (AT1) receptor functions through gene targeting. Acta Physiol Scand. 2004;181:561–567. doi: 10.1111/j.1365-201X.2004.01331.x. [DOI] [PubMed] [Google Scholar]
- 42.Mifune M, Sasamura H, Nakazato Y. Examination of Ang II type 1 and type 2 receptor expression in human kidneys by immunohistochemistry. Clin Exp Hypertens. 2001;23(3):257–266. doi: 10.1081/ceh-100102664. [DOI] [PubMed] [Google Scholar]
- 43.Iwai N, Inagamii T. Identification of two subtypes in the rat type 1 angiotensin II receptor. FEBS Lett. 1992;8(2–3):257–260. doi: 10.1016/0014-5793(92)80071-n. [DOI] [PubMed] [Google Scholar]
- 44.Naruse M, Tanabe A, Sugaya T. Deferential roles of angiotensin receptor subtypes in adrenocortical function in mice. Life Sci. 1998;63(18):1593–1598. doi: 10.1016/s0024-3205(98)00428-7. [DOI] [PubMed] [Google Scholar]
- 45.Bonnet F, Candida R, Carey RM. Renal expression of angiotensin receptors in long-term diabetes and the effects of angiotensin typ. 1 receptor blockade. J Hypertens. 2002;20(8):1615–1624. doi: 10.1097/00004872-200208000-00025. [DOI] [PubMed] [Google Scholar]
- 46.Imanishi K, Nonoguchi H, Nakayama Y. Type IA Ang II receptor is regulated differently in proximal and distal nephron segments. Hypertens Res. 2003;267:E260–E267. doi: 10.1291/hypres.26.405. [DOI] [PubMed] [Google Scholar]
- 47.Burson JM, Aguilera G, Gross KW. Differential expression of angiotensin receptor 1A and 1B in mouse. Am J Physiol. 1994;267:E260–E267. doi: 10.1152/ajpendo.1994.267.2.E260. [DOI] [PubMed] [Google Scholar]
- 48.Baiardi G, Macova M, Armando I. Estrogen upregulates Ang II AT1 and AT2 receptors in the rat. Regul Pept. 2005;124(1–3):7–17. doi: 10.1016/j.regpep.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 49.Dellis O, Dedos SG, Tovey SC. Ca2+ entry through plasma membrane IP3 receptors. Science. 2006;313:229–233. doi: 10.1126/science.1125203. [DOI] [PubMed] [Google Scholar]
- 50.Gill DL, Spassova MA, Soboloff J. Signal transduction. Calcium entry signals—trickles and torrents. Science. 2006;313:183–184. doi: 10.1126/science.1130811. [DOI] [PubMed] [Google Scholar]
- 51.Sandberg K, Ji H. Comparative analysis of amphibian and mammalian angiotensin receptors. Comp Biochem Physiol A Mol Integr Physiol. 2001;128:53–57. doi: 10.1016/s1095-6433(00)00297-x. [DOI] [PubMed] [Google Scholar]
- 52.Kalantarinia K, Okusa MD: The renin-angiotensin system and its blockade in diabetic renal and cardiovascular disease. Curr Diab Rep (1):8–16, 2006 [DOI] [PubMed]
- 53.De Gasparo M, Catt KJ, Inagami T. International union of pharmacology XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–472. [PubMed] [Google Scholar]
- 54.Zhang J, Pratt RE. The AT2 receptor selectively associates with Gia2 and Gia3 in the rat fetus. J Biol Chem. 1996;271:15026–15033. [PubMed] [Google Scholar]
- 55.Ito M, Oliviero P, Mannon C. Regulation of blood pressure by the type 1A receptor for angiotensin II. Proc Natl Acad Sci USA. 1995;92:3521–3525. doi: 10.1073/pnas.92.8.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schnermann J, Travnor T, Yang Y. Absence of tubuloglomerular feedback responses in AT1A receptor-deficient mice. Am J Physiol. 1997;273:F315–F320. doi: 10.1152/ajprenal.1997.273.2.F315. [DOI] [PubMed] [Google Scholar]
- 57.Matsusaka T, Nishimura H, Utsonomiya H. Chimeric mice carrying ‘regional’ targeted deletion of the angiotensin type 1A receptor gene. Evidence against the role for local angiotensin in the in vivo feedback regulation of renin synthesis in juxtaglomerular cells. J Clin Invest. 1996;98(8):1867–1877. doi: 10.1172/JCI118988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oliverio MI, Madsen K, Best CF. Renal growth and development in mice lacking the AT1A receptors for Angiotensin II. Am J Physiol. 1998;274:F43–F50. doi: 10.1152/ajprenal.1998.274.1.F43. [DOI] [PubMed] [Google Scholar]
- 59.Sugaya T, Nishimatsu S, Tanimoto K. Angiotensin II type 1a receptor-deficient mice with hypotension and hyperreninemia. J Biol Chem. 1995;270(32):18719–18722. doi: 10.1074/jbc.270.32.18719. [DOI] [PubMed] [Google Scholar]
- 60.Chen XW, Li H, Yoshida S. Targeting deletion of angiotensin type 1B receptor gene in the mouse. Am J Physiol. 1997;272:F299–F304. doi: 10.1152/ajprenal.1997.272.3.F299. [DOI] [PubMed] [Google Scholar]
- 61.Miyazaki Y, Tsuchida S, Nishimura H. Angiotensin induces the urinary peristaltic machinery during the perinatal period. J Clin Invest. 1998;102(8):1489–1497. doi: 10.1172/JCI4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hein LG, Barsh R, Pratt V. Behavioral and cardiovascular effects of disrupting the angiotensin II type-2 receptor gene in mice. Nature. 1995;377:744–747. doi: 10.1038/377744a0. [DOI] [PubMed] [Google Scholar]
- 63.Siragy HM, Inagami T, Ichiki T. sustained hypersensitivity to Ang II and its mechanism in mice lacking the subtype 2 (AT2) angiotensin receptor. Proc Natl Acad Sci USA. 1996;96(11):6506–6510. doi: 10.1073/pnas.96.11.6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ichiki T, Labosky PA, Shiota C. Effects on blood pressure and exploratory behaviour of mice lacking Ang II type-2 receptor. Nature. 1995;377(6551):748–750. doi: 10.1038/377748a0. [DOI] [PubMed] [Google Scholar]
- 65.Tanaka Y, Tamura K, Kkoide Y. The novel angiotensin II type 1 receptor (AT1R)-associated protein ATRAP downregulates AT1R and ameliorates cardiomyocyte hypertrophy. FEBS Lett. 2005;579(7):1579–1586. doi: 10.1016/j.febslet.2005.01.068. [DOI] [PubMed] [Google Scholar]
- 66.Nouet S, Amzallag N, Li JM. Transactivation of receptor tyrosine kinases by novel angiotensin II receptor-interacting protein, ATIP. J Biol Chem. 2005;279(28):28989–28997. doi: 10.1074/jbc.M403880200. [DOI] [PubMed] [Google Scholar]
- 67.Chaki S, Inagami T. Identification and characterization of a new binding site for angiotensin II in mouse neuroblastoma neuro-2A cells. Biochem Biophys Res Commun. 1992;182(1):388–394. doi: 10.1016/s0006-291x(05)80157-3. [DOI] [PubMed] [Google Scholar]
- 68.Albiston AL, McDowall SG, Matsacos D. Evidence that the angiotensin IV (AT4) receptor is the enzyme insulin-regulated aminopeptidase. J Biol Chem. 2001;276(52):48623–48626. doi: 10.1074/jbc.C100512200. [DOI] [PubMed] [Google Scholar]
- 69.Hunyadi L, Catt KJ. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of Ang II. Mol Endocrinol. 2006;20(5):953–970. doi: 10.1210/me.2004-0536. [DOI] [PubMed] [Google Scholar]
- 70.Garvin JL. Angiotensin stimulates glucose and fluid absorption by rat proximal straight tubules. J Am Soc Nephrol. 1990;1(3):272–277. doi: 10.1681/ASN.V13272. [DOI] [PubMed] [Google Scholar]
- 71.Xu L, Dixit MP, Chen R. Effects of angiotensin II on NaPi-II a co-transporter expression and activity in rat renal cortex. Biochim Biophys Acta. 2004;1667(2):114–121. doi: 10.1016/j.bbamem.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 72.Yingst DR, Massey KJ, Rossi NF. Angiotensin II directly stimulates the activity and alters the phosphorylation of Na-K-ATPase in rat proximal tubule with a rapid time course. Am J Physiol Renal Physiol. 2004;287(4):F713–F721. doi: 10.1152/ajprenal.00065.2004. [DOI] [PubMed] [Google Scholar]
- 73.Shah S, Hussain T. Enhanced angiotensin II-induced activation of Na+- K+- ATPase in the proximal tubules of obese Zucker rats. Clin Exp Hypertens. 2006;28:29–40. doi: 10.1080/10641960500386650. [DOI] [PubMed] [Google Scholar]
- 74.Horita S, Zheng Y, Hara C. Biphasic regulation of Na+ HCO3− cotransporter by angiotensin II type 1A receptor. Hypertension. 2002;40(5):707–712. doi: 10.1161/01.hyp.0000036449.70110.de. [DOI] [PubMed] [Google Scholar]
- 75.Noonan WT, Woo AL, Nieman ML. Blood pressure maintenance in NHE3-deficient mice with transgenic expression of NHE3 in small intestine. Am J Physiol Regul Integr Comp Physio. 2005;288(3):R685–R691. doi: 10.1152/ajpregu.00209.2004. [DOI] [PubMed] [Google Scholar]
- 76.Kolb RJ, Woost PG, Hopfer U. Membrane trafficking of angiotensin receptor type-1 and mechanochemical signal transduction in proximal tubule cells. Hypertension. 2004;44(3):352–359. doi: 10.1161/01.HYP.0000136645.90116.1a. [DOI] [PubMed] [Google Scholar]
- 77.Quan A, Chakravarty S, Chen JK. Androgens augment proximal tubule transport. Am J Physiol Renal Physiol. 2004;287(3):F452–F459. doi: 10.1152/ajprenal.00188.2003. [DOI] [PubMed] [Google Scholar]
- 78.Dixit MP, Xu L, Xu H. Effect of angiotensin II on renal Na+/H+ exchanger-NHE3 and NHE2. Biochim Biophys Acta. 2004;1664(1):38–44. doi: 10.1016/j.bbamem.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 79.Han HJ, Park SH, Kob HJ. Mechanism of regulation of Na+ transport by angiotensin II in primary renal cells. Kidney International. 2000;57:2457–2467. doi: 10.1046/j.1523-1755.2000.00104.x. [DOI] [PubMed] [Google Scholar]
- 80.Romero MF, Hopfer U, Madhun ZT et al. Angiotensin II actions in the rabbit proximal tubule: angiotensin II mediated signaling mechanisms and electrolyte transport in the rabbit proximal tubule. Ren Physiol Biochem 14(4–5):199–207, 1991 [DOI] [PubMed]
- 81.Houillier P, Chambrey R, Achard JM. Signaling pathways in the biphasic effect of angiotensin II on apical Na/H antiport activity in proximal tubule. Kidney Int. 1996;50(5):1496–1505. doi: 10.1038/ki.1996.464. [DOI] [PubMed] [Google Scholar]
- 82.Romero MF, Madhun ZT, Hopfer U. An epoxygenase metabolite of arachidonic acid, 5, 6 epoxy-eicosatetranoic acid mediates angiotensin-induced natriuresis in proximal tubular epithelium. Adv Prostaglandin Thromboxane Leukot Res. 1991;21A:205–208. [PubMed] [Google Scholar]
- 83.Good DW, George T, Wang DH. Angiotensin II inhibits HCO3 absorption via a cytochrome P450 dependent pathway in MTAL. Am J Physiol. 1999;276:F726–F736. doi: 10.1152/ajprenal.1999.276.5.F726. [DOI] [PubMed] [Google Scholar]
- 84.Kwon TH, Nielsen J, Kim H. Regulation of sodium transporters in the thick ascending limb of rat kidney: response to angiotensin II. Am J Physiol Renal Physiol. 2003;285(1):F152–F165. doi: 10.1152/ajprenal.00307.2002. [DOI] [PubMed] [Google Scholar]
- 85.Bell PD, Peti-Peterdi J. Angiotensin II stimulates macula densa basolateral sodium/hydrogen exchange via type 1 angiotensin II receptors. J Am Soc Nephrol Suppl. 1999;11:S225–S229. [PubMed] [Google Scholar]
- 86.Wang T, Geibisch G. Eeffects of angiotensin II on electrolyte transport in the early and late distal tubule in rat kidney. Am J Physiol. 1996;271:F143–F149. doi: 10.1152/ajprenal.1996.271.1.F143. [DOI] [PubMed] [Google Scholar]
- 87.Pulakat L, Cooper S, Knowle D. Ligand-dependent complex formation between the Angiotensin II receptor subtype AT2 and Na+/H+ exchanger NHE6 in mammalian cells. Peptides. 2005;26(5):863–873. doi: 10.1016/j.peptides.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 88.Bobulescu IA, Di Sole F, Moe OW. Na+/H+ exchangers; physiology and link to hypertension and organ ischemia. Curr Opin Nephrol Hypertens. 2005;14(5):485–494. doi: 10.1097/01.mnh.0000174146.52915.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lara LS, Cavalcante F, Axelband F. Involvement of Gi/o/cGMP/PKG pathway in the AT2-mediated inhibition of outer cortex proximal tubule Na+K+ATPase by Ang (1-7) Bichem J. 2006;395(1):183–190. doi: 10.1042/BJ20051455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Haithcock D, Jiao H, Cui XL. Renal proximal tubular AT2 receptor: Signaling and transport. J Am Soc Nephrol Suppl. 1999;11:S69–S74. [PubMed] [Google Scholar]
- 91.Quan A, Baum M. Effect of luminal angiotensin II receptor antagonists on proximal tubule transport. Am J Hypertens. 1999;12(5):499–503. doi: 10.1016/s0895-7061(99)00018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hakam AC, Hussain T. Angiotensin II type 2 receptor agonist directly inhibits proximal tubule sodium pump activity in obese but not in lean Zucker rats. Hypertension. 2006;47(6):1117–1124. doi: 10.1161/01.HYP.0000220112.91724.fc. [DOI] [PubMed] [Google Scholar]
- 93.Padia SH, Howell NL, Siragy HM. Renal angiotensin type 2 receptors mediate natriuresis via angiotensin III in the angiotensin II type 1 receptor-blocked rat. Hypertension. 2006;47(3):537–544. doi: 10.1161/01.HYP.0000196950.48596.21. [DOI] [PubMed] [Google Scholar]
- 94.Beutler KT, Masilamani S, Turban S. Long-term regulation of ENaC expression in kidney by angiotensin II. Hypertension. 2003;41(5):1143–1150. doi: 10.1161/01.HYP.0000066129.12106.E2. [DOI] [PubMed] [Google Scholar]
- 95.Davies E, McKenzie SM. Extra adrenal production of corticosteroids. Clin Exp Pharmacol Physiol. 2003;30(7):437–445. doi: 10.1046/j.1440-1681.2003.03867.x. [DOI] [PubMed] [Google Scholar]
- 96.Kurdi M, De Mello WC Working outside the system: an update on the unconventional behavior of the renin-angiotensin system components. Int J Biochem Cell Biol. 2005;37:1357–1367. doi: 10.1016/j.biocel.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 97.Zou Y, Akazawa H, Qin Y. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6(6):499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 98.Baker KM, Kumar R: Intracellular Ang II induces cell proliferation independent of AT1 receptor. Am J Physiol Cell Physiol 291(5):C995–1001, 2006 [DOI] [PubMed]
- 99.Rein H. Vasomotorische regulationen. Ergebn de Physiol. 1932;32:28–72. [Google Scholar]
- 100.Buckley NM, Brazeau P, Frasier ID. Renal blood flow autoregulation in developing swine. Am J Physiol. 1983;245(1):H1–H6. doi: 10.1152/ajpheart.1983.245.1.H1. [DOI] [PubMed] [Google Scholar]
- 101.Paton JB, Fisher DE. Organ blood flows of fetal and infant baboons. Early Hum Dev. 1984;10(1–2):137–144. doi: 10.1016/0378-3782(84)90120-8. [DOI] [PubMed] [Google Scholar]
- 102.Jose PA, Slotkoff LM, Montgomery S. Autoregulation of renal blood flow in the puppy. Am J Physiol. 1975;229(4):983–988. doi: 10.1152/ajplegacy.1975.229.4.983. [DOI] [PubMed] [Google Scholar]
- 103.Castrop H, Schnermann J. Impairment of tubuloglomerular feedback regulation of GFR in ecto-5′-nucleotidase/CD73-deficient mice. J Clin Invest. 2004;114:634–642. doi: 10.1172/JCI21851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Inscho EW, Cook AK, Imig JD. Physiological role for P2X1 receptors in renal microvascular autoregulatory behavior. JCI. 2003;112(12):1895–1905. doi: 10.1172/JCI18499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sun D, Samuelson LC, Yang T. Mediation of tubuloglomerular feedback by adenosine: evidence from mice lacking adenosine 1 receptors. Proc Natl Acad Sci USA. 2001;98(17):9938–9989. doi: 10.1073/pnas.171317998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wilcox CS. Redox regulation of the afferent arteriole and tubuloglomerular feedback. Acta Physiol Scand. 2003;179(3):217–223. doi: 10.1046/j.0001-6772.2003.01205.x. [DOI] [PubMed] [Google Scholar]
- 107.Bank N. Physical factors in glomerular tubular balance. Renal Physiol. 1979-1980;2:289–294. [Google Scholar]
- 108.Schutz S, Le Moullec JM, Corvoll P. Early expression of all the components of the renin-angiotensin system in human development. Am J Pathol. 1996;149:2067–2079. [PMC free article] [PubMed] [Google Scholar]
- 109.Niimura F, Kon V, Ichikawa I. The renin-angiotensin system in the development of congenital anomalies of the kidney and urinary tract. Curr Opin Pediatr. 2006;18(2):161–166. doi: 10.1097/01.mop.0000193288.56528.40. [DOI] [PubMed] [Google Scholar]
- 110.Tabacova S, Little R, Tsong Y. Adverse pregnancy outcomes associated with maternal enalapril antihypertensive treatment. Pharmacoepidemiol Drug Saf. 2003;12:633–646. doi: 10.1002/pds.796. [DOI] [PubMed] [Google Scholar]
- 111.Buttar HS. An overview of the influence of ACE inhibitors on fetal-placental circulation and perinatal development. Mol Cell Biochem. 1997;176(1–2):61–67. [PubMed] [Google Scholar]
- 112.Cooper WO, Harnandez-Diaz S, Arbogast PG. Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med. 2006;354:2443–2451. doi: 10.1056/NEJMoa055202. [DOI] [PubMed] [Google Scholar]
- 113.Schaefer C. Angiotensin II receptor antagonists: further evidence of fetotoxicity but not terratogenecity. Birth Defect Res. 2003;67:591–594. doi: 10.1002/bdra.10081. [DOI] [PubMed] [Google Scholar]
- 114.Niimura F, Okubo S, Togo A. Temporal and spatial expression of the angiotensinogen gene in mice and rats. Am J Physiol. 1997;272:142–147. doi: 10.1152/ajpregu.1997.272.1.R142. [DOI] [PubMed] [Google Scholar]
- 115.Gomez RA, Lynch KR, Sturgill BC. Distribution of renin mRNA and its distribution in the developing kidney. Am J Physiol. 1989;257:F850–F858. doi: 10.1152/ajprenal.1989.257.5.F850. [DOI] [PubMed] [Google Scholar]
- 116.Schieffer B, Bernstein KE, Marrero MB. The role of tyrosine phosphorylation in angiotensin II mediated intracellular signalling and cell growth. J Mol Med. 1996;74(2):85–95. doi: 10.1007/BF00196783. [DOI] [PubMed] [Google Scholar]
- 117.Marrero MB, Schieffer B, Paxton WG. The role of tyrosine phosphorylation in angiotensin II mediated intracellular signaling. Cardiovasc Res. 1995;30(4):530–536. [PubMed] [Google Scholar]
- 118.Sadoshima J, Izumo S. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res. 1993;73(3):413–423. doi: 10.1161/01.res.73.3.413. [DOI] [PubMed] [Google Scholar]
- 119.Norwood VF, Craig MR, Harris JM. Differential expression of angiotensin II receptors during early renal morphogenesis. Am J Physiol. 1997;272:R662–R668. doi: 10.1152/ajpregu.1997.272.2.R662. [DOI] [PubMed] [Google Scholar]
- 120.Zhang H, Wada J, Hida K. Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J Biol Chem. 2001;276(20):17132–17139. doi: 10.1074/jbc.M006723200. [DOI] [PubMed] [Google Scholar]
- 121.Kumar D, Moss G, Primhak R. Congenital renal tubular dysplasia and skull ossification defects similar to teratogenic effects of angiotensin converting enzyme (ACE) inhibitors. J Med Genet. 1997;34:541–545. doi: 10.1136/jmg.34.7.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gribouval O, Gonzales M, Neuhaus T. Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat Genet. 2005;37:964–968. doi: 10.1038/ng1623. [DOI] [PubMed] [Google Scholar]
- 123.Lacoste M, Cai Y, Guicharnaud L. Renal tubular dysgenesis, a not uncommon autosomal recessive disorder leading to oligohydramnios: role of the renin-angiotensin system. J Am Soc Nephrol. 2006;17(8):2253–2263. doi: 10.1681/ASN.2005121303. [DOI] [PubMed] [Google Scholar]
- 124.Nishimura H, Yerkes E, Hohenfellner K. Role of the angiotensin type 2 receptor l gene in congenital anomalies of the kidney and urinary tract, CAKUT, of mice and men. Mol Cell. 1999;3:1–10. doi: 10.1016/s1097-2765(00)80169-0. [DOI] [PubMed] [Google Scholar]
- 125.Yosypiv IV, El-Dahr SS. Role of the renin-angiotensin system in the development of the ureteric bud and renal collecting system. Pediatr Nephrol. 2005;20:1219–1229. doi: 10.1007/s00467-005-1944-3. [DOI] [PubMed] [Google Scholar]
- 126.Fiselier T, Monnens L, van Munster P. The renin-angiotensin-aldosterone system in infancy and childhood in basal conditions and after stimulation. Eur J Pediatr. 1984;143(1):18–24. doi: 10.1007/BF00442742. [DOI] [PubMed] [Google Scholar]
- 127.Sulyok E, Nemeth M, Tenyi I. Relationship between the postnatal development of the renin-angiotensin-aldosterone system and electrolyte and acid-base status of the NaCl-supplemented premature infants. In: Spizer A, editor. (ed.): The Kidney during Development Morphogenesis and Function. Masson; New York: 1982. p. 273. [Google Scholar]
- 128.Peleg E, Peleg D, Yaron A. Perinatal development of angiotensin-converting enzyme in the rat's blood. Gynecol Obstet Invest. 1988;25(1):12–15. doi: 10.1159/000293738. [DOI] [PubMed] [Google Scholar]
- 129.Walther T, Faber R, Maul B. Fetal, neonatal cord and maternal plasma concentrations of angiotensin converting enzyme (ACE) Prenat Diagn. 2002;22(2):111–113. doi: 10.1002/pd.254. [DOI] [PubMed] [Google Scholar]
- 130.Bender JW, Davitt MK, Jose P. Angiotensin-1-converting enzyme activity in term and premature infants. Biol Neonate. 1978;34(1–2):19–23. doi: 10.1159/000241100. [DOI] [PubMed] [Google Scholar]
- 131.Forhead AJ, Melvin R, Balouzet V. Developmental changes in plasma angiotensin-converting enzyme concentration in fetal and neonatal lambs. Reprod Fertil Dev. 1998;10(5):393–398. doi: 10.1071/rd98101. [DOI] [PubMed] [Google Scholar]
- 132.Raimbach SJ, Thomas AL. Renin and angiotensin converting enzyme concentrations in the fetal and neonatal guinea-pig. J Physiol. 1990;423:441–451. doi: 10.1113/jphysiol.1990.sp018032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Forhead AJ, Gulati V, Poore KR. Ontogeny of pulmonary and renal angiotensin-converting enzyme in pigs. Mol Cell Endocrinol. 2001;185(1–2):127–133. doi: 10.1016/s0303-7207(01)00623-2. [DOI] [PubMed] [Google Scholar]
- 134.O'Connor SJ, Fowden AL, Holdstock N. Developmental changes in pulmonary and renal angiotensin-converting enzyme concentration in fetal and neonatal horses. Reprod Fertil Dev. 2002;14(7–8):413–417. doi: 10.1071/rd02044. [DOI] [PubMed] [Google Scholar]
- 135.Hattori MA, Del Ben GL, Carmona AK. Angiotensin I-converting enzyme isoforms (high and low molecular weight) in urine of premature and full-term infants. Hypertension. 2000;35(6):1284–1290. doi: 10.1161/01.hyp.35.6.1284. [DOI] [PubMed] [Google Scholar]
- 136.Miyawaki M, Okutani T, Higuchi R et al: Plasma angiotensin II levels in the early neonatal period. Arch Dis Child Fetal Neonatal 91(5):F359–F362 [DOI] [PMC free article] [PubMed]
- 137.Sippell WG, Dorr HG, Bidlingmaier F. Plasma levels of aldosterone, corticosterone, 11-deoxycorticosterone, progesterone, 17-hydroxyprogesterone, cortisol, and cortisone during infancy and childhood. Pediatr Res. 1980;14:39–46. doi: 10.1203/00006450-198001000-00010. [DOI] [PubMed] [Google Scholar]
- 138.Felder RA, Pelayo JC, Calcagno PL. Alpha adrenoreceptors in the developing kidney. Pediatr Res. 1983;17(2):177–180. doi: 10.1203/00006450-198302000-00019. [DOI] [PubMed] [Google Scholar]
- 139.Chevalier RL, Thornhill BA, Belmonte DC. Endogenous angiotensin II inhibits natriuresis after acute volume expansion in the neonatal rat. Am J Physiol. 1996;270:R393–R397. doi: 10.1152/ajpregu.1996.270.2.R393. [DOI] [PubMed] [Google Scholar]
- 140.Pelayo JC, Fildes RD, Jose PA. Age-dependent renal effects of intrarenal infusion dopamine infusion. Am J Physiol. 1984;247:R212–R216. doi: 10.1152/ajpregu.1984.247.1.R212. [DOI] [PubMed] [Google Scholar]
- 141.Muchant DG, Thornhill BA, Belmonte DC. Chronic sodium loading augments natriuretic response to acute volume expansion in the preweaned rat. Am J Physiol. 1995;269:R15–R22. doi: 10.1152/ajpregu.1995.269.1.R15. [DOI] [PubMed] [Google Scholar]
- 142.Solhaug MJ, Dong XO, Adelman RD. Ontogeny of neuronal nitric oxide synthase NOS1, in the developing porcine kidney. Am J Physiol Regul Integr Comp Physiol. 2000;278(6):R1453–R1459. doi: 10.1152/ajpregu.2000.278.6.R1453. [DOI] [PubMed] [Google Scholar]
- 143.Mulder J, Baum M, Quigley R. Diffusional water permeability (PDW) of adult and neonatal rabbit renal brush border membrane vesicles. J Membr Biol. 2002;187:167–174. doi: 10.1007/s00232-001-0161-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mulder J, Chakravarty S, Haddad MN. Glucocorticoids increase osmotic water permeability (Pf) of neonatal rabbit renal brush border membrane vesicles. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1417–R1421. doi: 10.1152/ajpregu.00448.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Boberg U, Persson AE. Increased tubuloglomerular feedback activity in Milan hypertensive rats. Am J Physiol. 1986;250:F967–F974. doi: 10.1152/ajprenal.1986.250.6.F967. [DOI] [PubMed] [Google Scholar]
- 146.Dilley JR, Arendshorst WJ. Enhanced tubuloglomerular feedback activity in rats developing spontaneous hypertension. Am J Physiol. 1984;274:F672–F679. doi: 10.1152/ajprenal.1984.247.4.F672. [DOI] [PubMed] [Google Scholar]
- 147.Al-Dahhan J, Haycock GB, Chantler C. Sodium Homeostasis in term and preterm neonates. I. Renal aspects. Arch Dis Child. 1983;58(5):335–342. doi: 10.1136/adc.58.5.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wilcox CS, Welch WJ, Schreiner GF. Natriuretic and diuretic actions of a highly-selective adenosine A1 receptor antagonist. J Am Soc Nephrol. 1990;10:714–720. doi: 10.1681/ASN.V104714. [DOI] [PubMed] [Google Scholar]
- 149.Du Z, Duan Y, Yan Q. Mechanosensory function of microvilli of the kidney proximal tubule. Proc Natl Acad Sci USA. 2004;101(35):13068–13073. doi: 10.1073/pnas.0405179101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kon V, Highes ML, Ichikawa I. Physiological basis for the maintenance of glomerulotubular balance in young growing rats. Kidney Int. 1984;25(2):391–396. doi: 10.1038/ki.1984.29. [DOI] [PubMed] [Google Scholar]
- 151.Bidani AK, Griffin KA. Long-term renal consequences of hypertension for normal and diseased kidneys. Curr Opin Nephrol Hypertens. 2002;11:73–80. doi: 10.1097/00041552-200201000-00011. [DOI] [PubMed] [Google Scholar]
- 152.Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: implications for therapy. Hypertension. 2004;44:1–7. doi: 10.1161/01.HYP.0000145180.38707.84. [DOI] [PubMed] [Google Scholar]
- 153.Wilson C, Byrom FB. Renal changes in malignant hypertension. Lancet. 1939;1:136–139. [Google Scholar]
- 154.Wilson C, Byrom FB. The vicious circle in chronic Bright's disease. Experimental evidence from the hypertensive rat. Q J Med. 1941;10:65–93. [Google Scholar]
- 155.Hill GS, Heptinstall RH. Steroid-induced hypertension in the rat. A microangiopathic and histologic study on the pathogenesis of hypertensive vascular and glomerular lesions. Am J Pathol. 1968;52:1–39. [PMC free article] [PubMed] [Google Scholar]
- 156.Loutzenhiser R, Griffin K, Williamson G. Renal Autoregulation: new perspectives: the protective and regulatory roles of the underlying mechanisms. Am J Physiol Regul Integr Comp Physiol. 2006;290:1153–1167. doi: 10.1152/ajpregu.00402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Just A, Arendshorst WJ. Dynamics and contribution of mechanisms mediating renal blood flow autoregulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R619–R631. doi: 10.1152/ajpregu.00766.2002. [DOI] [PubMed] [Google Scholar]
- 158.Navar LG. Renal autoregulation; perspectives from whole kidney and single nephron studies. Am J Physiol Renal Fluid Electrolyte Physiol. 1978;234:F357–F370. doi: 10.1152/ajprenal.1978.234.5.F357. [DOI] [PubMed] [Google Scholar]
- 159.Karlsen FM, Anderson PP, Holstein-Rathlou NH. Dynamic autoregulation and renal injury in Dahl rats. Hypertension. 1997;30:975–983. doi: 10.1161/01.hyp.30.4.975. [DOI] [PubMed] [Google Scholar]
- 160.Takemaka T, Forster H, De Micheli A. Impaired myogenic responsiveness of renal microvessels in Dahl salt-sensitive rats. Circ Res. 1992;71:471–480. doi: 10.1161/01.res.71.2.471. [DOI] [PubMed] [Google Scholar]
- 161.Karlsen FM, Leysacc PP, Holstein Rathlou NH. Tubuloglomerular feedback in Dahl rats. Am J Physiol Renal Physiol. 1998;274:F1561–F1569. doi: 10.1152/ajpregu.1998.274.6.R1561. [DOI] [PubMed] [Google Scholar]
- 162.Churchill PC, Churchill MC, Bidani AK. Genetic susceptibility to hypertension-induced renal damage in the rat: evidence based on kidney specific genome transfer. J Clin Invest. 1997;100:1373–1382. doi: 10.1172/JCI119657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Castrop H, Huang Y, Hashimoto S. Impairment of tubuloglomerular feedback regulation of GFR in ecto-5′ nucleotidase/CD73-deficient mice. J Clin Invest. 2004;114:634–642. doi: 10.1172/JCI21851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Vallon V. Tubuloglomerular feedback in the kidney: insights from gene-targeted mice. Pflugers Arch-Eur J Physiol. 2003;445:470–476. doi: 10.1007/s00424-002-0952-4. [DOI] [PubMed] [Google Scholar]
- 165.Hashimoto S, Huang Y, Briggs J. Reduced autoregulatory effectiveness in adenosine 1 receptor-deficient mice. Am J Physiol Renal Physiol. 2006;290:888–891. doi: 10.1152/ajprenal.00381.2005. [DOI] [PubMed] [Google Scholar]
- 166.Nishayama A, Navar LG. ATP mediates tubuloglomerular feedback. Am J Physiol Regulatory Integrative Comp Physiol. 2002;283:R273–R275. doi: 10.1152/ajpregu.00071.2002. [DOI] [PubMed] [Google Scholar]
- 167.Inscho EW. PX2 receptors in regulation of renal microvascular function. Am J Physiol Renal Physiol. 2001;280:F927–F944. doi: 10.1152/ajprenal.2001.280.6.F927. [DOI] [PubMed] [Google Scholar]
- 168.Inscho EW, Belott TP, Mason MJ. Extracellular ATP increases cytosolic calcium in cultured rat renal arterial smooth muscle cells. Clin Exp Pharmacol Physiol. 1996;23:503–507. doi: 10.1111/j.1440-1681.1996.tb02769.x. [DOI] [PubMed] [Google Scholar]
- 169.Tojo A, Madsen KM, Wilcox CS. Expression of immunoreactive nitric oxide synthase isoforms in rat kidney: effects of dietary salt and losartan. Jpn Heart J. 1995;36:389–398. doi: 10.1536/ihj.36.389. [DOI] [PubMed] [Google Scholar]
- 170.Wilcox CS, Welch WJ, Murad F. Nitric oxide synthase in macula densa regulates glomerular capillary pressure. Proc Natl Acad Sci USA. 1992;89:11993–11997. doi: 10.1073/pnas.89.24.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Welch WJ, Wilcox CS, Thomson SC. Nitric oxide and tubuloglomerular feedback. Semin Nephrol 19(3):251–62, 1999 [PubMed]
- 172.Welch WJ, Tojo A, Lee Ju. Nitric oxide synthase in the JGA of the SHR: expression and role in tubuloglomerular feedback. Am J Physiol. 1999;277:F1301–F1308. doi: 10.1152/ajprenal.1999.277.1.F130. [DOI] [PubMed] [Google Scholar]
- 173.Thorup C, Persson AE. Impaired effect of nitric oxide synthesis inhibition on tubuloglomerular feedback in hypertensive rats. Am J Physiol. 1996;271:F246–F252. doi: 10.1152/ajprenal.1996.271.2.F246. [DOI] [PubMed] [Google Scholar]
- 174.Welch JW, Tojo A, Wilcox CS. Roles of NO and oxygen radicals in tubuloglomerular feedback in SHR. Am J Physiol Renal Physiol. 2000;278(5):F769–F776. doi: 10.1152/ajprenal.2000.278.5.F769. [DOI] [PubMed] [Google Scholar]
- 175.Chen YF, Li PL, Zuo AP. Oxidative stress enhances the production and actions of adenosine in the kidney. Am J Physiol Regul Integr Comp Physiol. 2001;281(6):R1808–R1816. doi: 10.1152/ajpregu.2001.281.6.R1808. [DOI] [PubMed] [Google Scholar]
- 176.McIntyre M, Bohr DF, Dominiczak AF. Endothelial function in hypertension: the role of superoxide anion. Hypertension. 1999;34:539–545. doi: 10.1161/01.hyp.34.4.539. [DOI] [PubMed] [Google Scholar]
- 177.Yang ZZ, Zhang AY, Yi FX. Redox regulation of HIF-1alpha levels and HO-1 expression in renal medullary interstitial cells. Am J Physiol Renal Physiol. 2003;284(6):F1207–F1215. doi: 10.1152/ajprenal.00017.2002. [DOI] [PubMed] [Google Scholar]
- 178.Ortiz PA, Garvin JL. Role of nitric oxide in the regulation of nephron transport. Am J Physiol Renal Physiol. 2002;282(5):F777–F784. doi: 10.1152/ajprenal.00334.2001. [DOI] [PubMed] [Google Scholar]
- 179.Peti-Peterdi et al. Calcium wave of tubuloglomerular feedback. Am J Physiol Renal Physiol 291(2):F473–802, 2006 [DOI] [PubMed]
- 180.Barker DJ, Osmond C, Golding J. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ. 1989;298:564–567. doi: 10.1136/bmj.298.6673.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Alexander BT. Fetal programming of hypertension. Am J Physiol Regul Integr Comp Physiol. 2006;290:1–10. doi: 10.1152/ajpregu.00417.2005. [DOI] [PubMed] [Google Scholar]
- 182.Woods LL, Rasch R. Perinatal Ang II programs adult blood pressure, glomerular number and renal function in rats. Am J Physiol Regul Integr Comp Physiol. 1998;275:R1593–R1599. doi: 10.1152/ajpregu.1998.275.5.R1593. [DOI] [PubMed] [Google Scholar]
- 183.Gilbert JS, Lang AL, Grant AR. Maternal nutrient restriction in sheep: hypertension and decreased nephron number in offspring at 9 months of age. J Physiol. 2005;565:137–147. doi: 10.1113/jphysiol.2005.084202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kohlstedt K, Brandis RP, Muller-Esterl W. Angiotensin-converting enzyme is involved in outside-in signaling in endothelial cells. Circ Res. 2002;94:60–67. doi: 10.1161/01.RES.0000107195.13573.E4. [DOI] [PubMed] [Google Scholar]
- 185.Zeng C, Wang Z, Hopfer U. Rat strain effects of AT1 receptor activation on D1 dopamine receptors in immortalized renal proximal tubule cells. Hypertension. 2005;46:799–805. doi: 10.1161/01.HYP.0000184251.01159.72. [DOI] [PubMed] [Google Scholar]
- 186.Zeng C, Hopfer U, Asico LD. Altered AT1 receptor regulation of ETB receptors in renal proximal tubule cells of spontaneously hypertensive rats. Hypertension. 2005;46:926–931. doi: 10.1161/01.HYP.0000174595.41637.13. [DOI] [PubMed] [Google Scholar]
- 187.AbdAlla S, Lother H, Quitterer U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature. 2000;407:94–98. doi: 10.1038/35024095. [DOI] [PubMed] [Google Scholar]
- 188.Abadir PM, Periasamy A, Carey RM. Angiotensin II type 2 receptor-bradykinin B2 receptor functional heterodimerization. Hypertension. 2006;48:316–322. doi: 10.1161/01.HYP.0000228997.88162.a8. [DOI] [PubMed] [Google Scholar]
- 189.Zhuo JL. Intracrine renin and angiotensin II: a novel role in cardiovascular and renal cellular regulation. J Hypertens. 2006;24:1017–1020. doi: 10.1097/01.hjh.0000226188.90815.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zou Y, Akazawa H, Qin Y, Sano M. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6:499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 191.Drukker A, Guignard JP. Renal aspects of the term and preterm infant: a selective update. Current Opin Pediatr. 2002;14:175–182. doi: 10.1097/00008480-200204000-00006. [DOI] [PubMed] [Google Scholar]
- 192.Menke DR, Bornstein SR. Congenital adrenal hyperplasia. Lancet. 2005;365:2125–2136. doi: 10.1016/S0140-6736(05)66736-0. [DOI] [PubMed] [Google Scholar]
- 193.Chang SS, Grunder S, Hanukoglu A. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. 1996;53:248–533. doi: 10.1038/ng0396-248. [DOI] [PubMed] [Google Scholar]