

Abstract
Microtubule-severing enzymes katanin, spastin and fidgetin are AAA ATPases important for the biogenesis and maintenance of complex microtubule arrays in axons, spindles and cilia. Because of a lack of 3D structures, their mechanism has remained poorly understood. Here we report the first X-ray structure of the monomeric AAA katanin module and cryo-EM reconstructions of the hexamer in two conformations. These reveal an unexpected asymmetric arrangement of the AAA domains mediated by structural elements unique to severing enzymes and critical for their function. Our reconstructions show that katanin cycles between open spiral and closed ring conformations, depending on the ATP occupancy of a gating protomer that tenses or relaxes inter-protomer interfaces. Cycling of the hexamer between these conformations would provide the power stroke for microtubule severing.
INTRODUCTION
Cells constantly assemble and disassemble their microtubule cytoskeleton through the concerted action of microtubule polymerases, depolymerases, crosslinkers and severing enzymes. Microtubule severing enzymes spastin, katanin and fidgetin generate internal breaks in microtubules thus modulating their dynamics and organization1. The mechanism used by these enzymes to destabilize the microtubule, a polymer with a flexural rigidity comparable to Plexiglas®2 is still poorly understood. Microtubule severing enzymes are critical in a wide range of cell biological processes including biogenesis of neuronal and non-centrosomal microtubule arrays3–7, phototropism5,7, spindle scaling7–11, chromosome segregation10,12, and control of centriole and cilia numbers13–15. They are found throughout the animal kingdom, plants and protozoa, and mutations in microtubule severing enzymes cause severe neurodegenerative and neurodevelopmental disorders1,14–16. All known microtubule-severing enzymes belong to the family of ATPases associated with various cellular activities (AAA ATPases).
Katanin was the first microtubule-severing enzyme discovered17,18. Fittingly, it was named after the Japanese for sword. Katanin is composed of a catalytic (p60) and a regulatory (p80) subunit. The catalytic subunit contains the AAA ATPase motor and is sufficient for microtubule severing10,19. The p80 subunit regulates association with the centrosome and enhances microtubule binding18–21. ATP hydrolysis is required for severing and katanin p60 ATPase is stimulated by the microtubule19. The p60 AAA ATPase domain is connected to an N-terminal microtubule interacting and trafficking (MIT) domain through a poorly conserved linker, a sequence arrangement shared with the microtubule-severing enzymes spastin and fidgetin1. In large part due to a lack of 3D structures, the mechanism of microtubule severing by this class of enzymes has remained poorly understood 25 years after the discovery of the first microtubule enzyme17,18. Here we report the first X-ray structure of the monomeric AAA katanin module and cryo-EM reconstructions of the hexamer in two conformations. These reveal an unexpected asymmetric arrangement of the AAA domains mediated by structural elements unique to microtubule severing enzymes and that we show are critical for their function. Cryo-EM reconstructions at 4.4Å and 6Å resolution of the katanin hexamer reveal an open spiral and a closed ring conformations of the AAA core, depending on the nucleotide occupancy of a gating protomer that closes a ~ 40Å wide gate in the katanin hexamer. Together with solution small-angle X-ray scattering (SAXS) reconstructions, our integrated structural study supports a model whereby katanin makes multivalent interactions with the microtubule through its AAA core, flexible MIT domains and a newly defined linker element that crowns the AAA ring, and engages the C-terminal tails of tubulin through conserved pore loops that gradually pull tubulin dimers out of the microtubule lattice by cycling between open spiral and closed AAA ring conformations. Due to the high sequence homology, a similar mechanism is likely shared by all microtubule-severing enzymes. Lastly, our integrated study also provides insight into the many katanin mutants identified from classic genetic studies on meiosis where the katanin gene (also known as mei-1) was first identified as well as disease mutations found in microtubule severing enzymes.
RESULTS
Solution structure of the katanin hexamer
Both microtubule severing enzymes katanin and spastin assemble into a hexamer in their active, ATP-bound state1,22. This hexamer is labile and falls apart at lower protein concentrations22. Consequently, the high-resolution architecture of an assembled microtubule-severing enzyme has remained elusive. Analytical ultracentrifugation (AUC) shows that the catalytic subunit of Caenorhabditis elegans katanin (MEI-1) populates a mixture of oligomerization states as a function of concentration and nucleotide. At the highest concentration (24 μM) and in the presence of ATP, the dominant species is a hexamer that dissociates at lower concentrations (Supplementary Figs. 1a, b). Introduction of a commonly used mutation in the Walker B element that retains ATP binding, but prevents hydrolysis, stabilizes the hexamer22 to concentrations as low as 3 μM (Supplementary Fig. 1c). Solution SAXS measurements of this species (hereafter referred to as katanin) reveal an elongated molecule with a maximal diameter (Dmax) of ~ 250Å (Fig. 1a–c). SAXS ab initio reconstructions generated by imposing six-fold symmetry reveal a disc structure ~130Å in diameter with slender arms that fan out at variable angles (Fig. 1a and Supplementary Fig. 1d, e, f, h and Methods) indicating a high-degree of flexibility. This architecture is reminiscent of the one reported earlier for spastin23. The central ring dimensions are compatible with a hexameric assembly of AAA ATPase domains23,24. The MIT domains reside on average at the tips of the extended arms, as a deletion construct that misses this domain (hereafter ΔMIT katanin, Fig. 1d) has a Dmax of ~180Å and shorter arms (Fig. 1b, c and Supplementary Fig. 1g, h).
Figure 1. Architecture of monomeric and assembled katanin from X-ray diffraction, solution SAXS and cryo-EM structures.
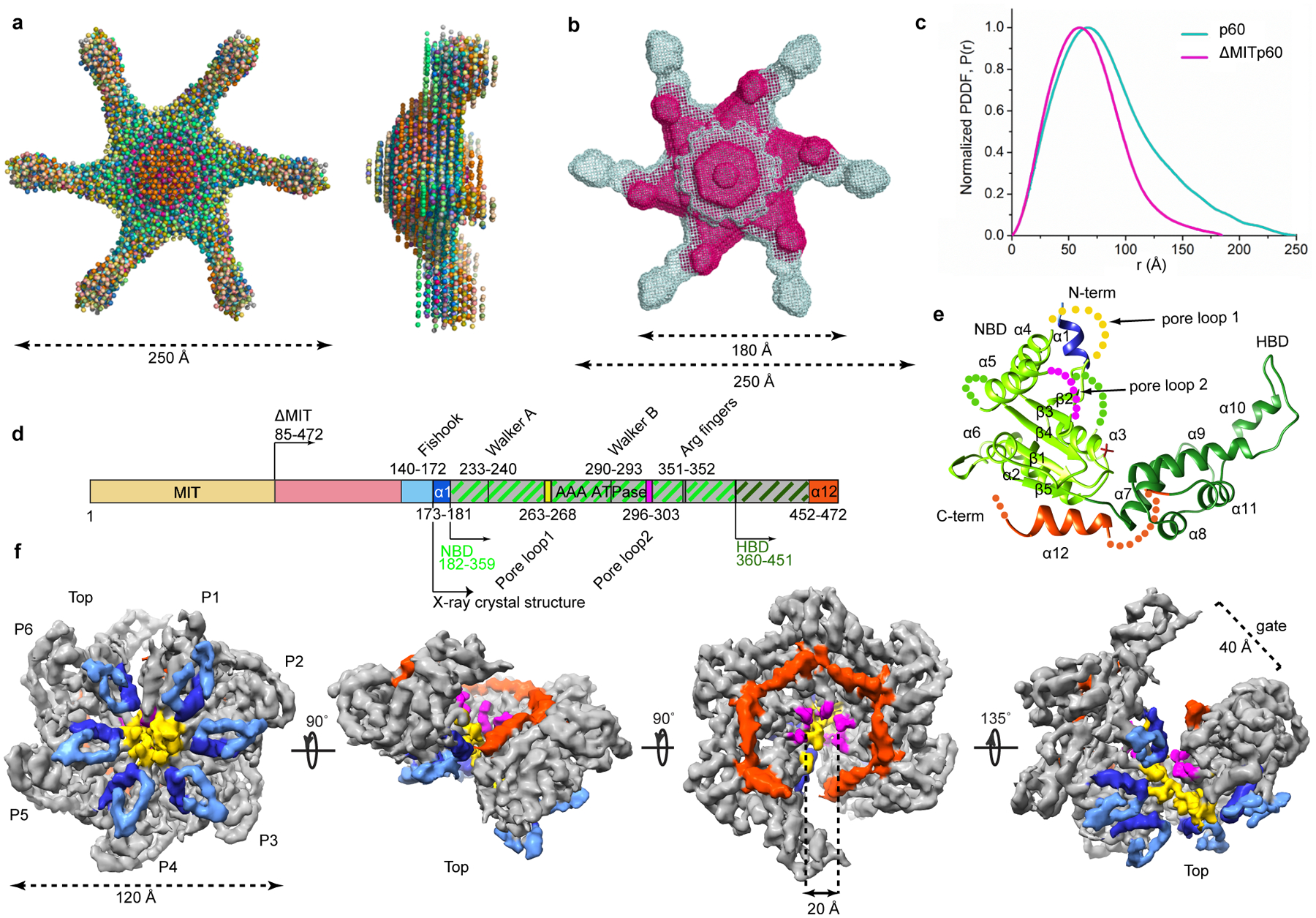
(a) Top and side views of twenty superimposed ab initio bead models of full-length katanin p60 calculated from solution SAXS data (Supplementary Fig. 1 and Methods). (b) Top view of an overlay of representative models for full-length (grey) and ΔMIT katanin (magenta). Approximate dimensions shown. (c) Pair distance distribution functions for full-length and ΔMIT katanin. (d) Domain diagram of katanin p60; MIT, microtubule interacting and trafficking domain, yellow; flexible linker, pink; AAA domain, grey; fishhook-shaped linker element, light-blue; α1, dark-blue; α12 and 468–472 C-terminal residues, dark-orange. NBD and HBD are highlighted by light green and dark green hatches, respectively as in panel (e). Residue numbers for C. elegans katanin. (e) Cartoon representation of the katanin AAA core crystal structure; N-terminal helix α1, blue; NBD, light green; HBD, dark green; C-terminal helix α12, dark-orange; sulfate ion as stick representation; unresolved residues shown as spheres. (f) Views of the final sharpened 3D density map (13 σ) of the katanin hexamer filtered to 4.4 Å resolution showing a spiral architecture (rotation angles between the different views indicated with arrows). Top, microtubule-binding face23. Approximate dimensions shown. Structural elements color-coded as in (d). Bracket indicates open gate between P1 and P6.
X-ray and cryo-EM structures reveal asymmetric architecture of the katanin hexamer
To obtain a higher-resolution view of katanin, we employed a hybrid structural approach combining X-ray crystallography of the monomer and cryo-EM of the hexamer. We solved the X-ray structure of the nucleotide-free monomer by X-ray crystallography at 3.3Å resolution (Fig. 1e and Table 1). This showed well-resolved density only for the AAA domain (residues 173–467). It is comprised of an enzymatic, α/β nucleotide binding domain (NBD) that contains the Walker A and B motifs involved in coordinating ATP, and a helix bundle domain (HBD). The katanin AAA domain is distinguished from that of other AAA ATPases like ClpX, N-ethylmaleimide sensitive factor (NSF) and Heat-shock protein (Hsp) 104 by two helices (N-terminal α1 and C-terminal α12) that augment the NBD and are characteristic of severing enzymes23 (Fig. 1d and Supplementary Fig. 2).
Table 1.
Data collection and refinement statistics
| Katanin AAA apo (5WC1) | |
|---|---|
| Data collection | |
| Space group | P65 |
| Cell dimensions | |
| a, b, c (Å) | 99.4,99.4,76.6 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å) | 50.0–3.2 (3.26–3.20) |
| Rsym (%) | 9.8 (56.7) |
| I/σ(I) | 36.7(3.4) |
| Completeness (%) | 98.7(100.00) |
| Redundancy | 9.8 (10.2) |
| Refinement | |
| Resolution (Å) | 46.0 – 3.3 |
| No. reflections | 7090 |
| Rwork / Rfree | 30.7/25.8 |
| No. atoms | |
| Protein | 1793 |
| Ion (SO4) | 10 |
| Water | 0 |
| B factors | |
| Protein | 107.6 |
| Ligand/ion | 177.2 |
| Water | |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.001 |
| Bond angles (°) | 0.359 |
Data collected from one crystal
Values in parentheses are for highest-resolution shell.
We also obtained structures of the assembled full-length katanin hexamer by single particle cryo-EM (Table 2, Fig. 1f, Supplementary Fig. 3 and Methods). The data were refined and classified without imposing symmetry, yielding two reconstructions of distinct conformational states at 4.4Å and 6.0Å resolution (FSC=0.143 criterion), respectively (Supplementary Figs. 3, 4 and Methods). Well-resolved secondary structure elements in both reconstructions allowed derivation of atomic models by flexibly fitting the X-ray structure of the AAA monomer followed by real-space refinement (Table 2, Figs. 2, 3, and Methods). The 4.4Å reconstruction represents the dominant class (49% of particles sorted by 3D classification; Supplementary Fig. 4). It reveals a hexamer with a right-handed spiral arrangement of subunits with a ~ 5Å rise and ~60° twist per protomer (Fig. 2) that results in a ~30Å offset between the first and last protomers (hereafter P1 and P6), and a ~ 40Å wide gate between them (Fig. 1f, Fig. 2). Notably, the microtubule severing enzyme spastin and other AAA ATPases were proposed to form functional helical assemblies during their mechanochemical cycles based on their propensity to form helical arrangements in crystals25. Both katanin and spastin form helices in crystals (Supplementary Fig. 5) that are left-handed and much more open (~13–16 Å rise and ~60° twist per crystallographic copy) than the right-handed spiral arrangement we see in our cryo-EM reconstruction thus resulting in a larger opening between the boundary subunits (~75Å), consistent with the more limited crystal packing interactions between the AAA domains. The second conformation identified by our cryo-EM analysis (24% of particles sorted by 3D classification) reveals a planar ring arrangement of the protomers that results in the closure of the P1–P6 gate (Fig. 3 and Supplementary Fig. 4).
Table 2.
Cryo-EM data collection, refinement and validation statistics
| #1 Spiral conformation (EMD-8794, PDB 5WC0) | #2 Ring Conformation (EMD-8796, 5WCB) | |
|---|---|---|
| Data collection | ||
| Microscope | FEI Titan Krios | FEI Titan Krios |
| Camera | Gatan K2 Summit | Gatan K2 Summit |
| Magnification | 22,500 | 22,500 |
| Voltage (kV) | 300 | 300 |
| Electron dose (e−/Ǻ2) | 51 | 51 |
| Defocus range (μm) | −1.0 to −3.5 | −1.0 to −3.5 |
| Pixel size (Ǻ) | 1.31 | 1.31 |
| Reconstruction and Model Refinement | ||
| Particles | 38,072 | 16,185 |
| Map sharpening B factor (Ǻ2) | −98.36 | −95.45 |
| Resolution (Å) | 4.4 | 6.0 |
| FSCaverage | 0.884 | 0.90 |
| Model composition | ||
| Non-hydrogen atoms | 11477 | 11422 |
| Protein residues | 1660 | 1662 |
| Ligands (ATP) | 6 | 5 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.003 | 0.004 |
| Bond angles (°) | 0.74 | 0.83 |
| Validation | ||
| MolProbity score | 1.77 | 2.18 |
| Clashscore | 3.91 | 6.76 |
| Poor rotamers (%) | 0.00 | 0.00 |
| Ramachandran plot | ||
| Favored (%) | 92.57 | 87.86 |
| Allowed (%) | 7.37 | 12.14 |
| Disallowed (%) | 0.06 | 0.00 |
Figure 2. Cryo-EM maps and three-dimensional models for the katanin hexamer in the spiral conformation.
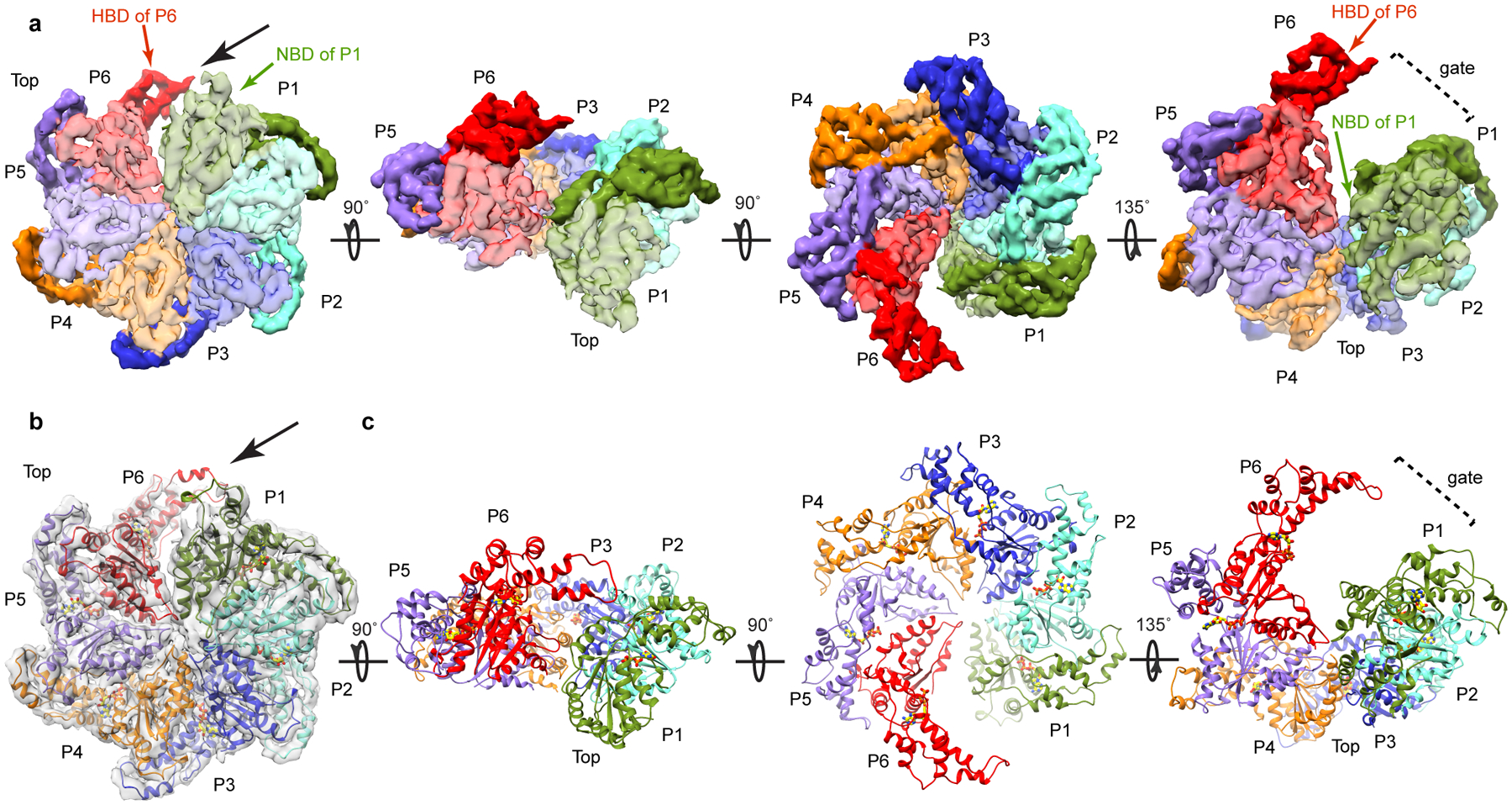
(a) Views of the katanin spiral conformation (rotation angles between the different views indicated with arrows). Protomer P1, green; P2, cyan; P3, blue; P4, orange; P5, purple; P6, red. NBD, HBD, light and dark hue, respectively. Black arrow indicates the open gate between protomers P1 and P6. (b) Top view of the spiral conformation with fitted atomic model. Cryo-EM map, shown as a transparent gray isosurface. The resolution of the map precluded de novo building of the fishhook linker element. Protomers colored as in (a). (c) Views of the atomic models for the katanin spiral conformation in the orientations shown above in panel (a).
Figure 3. Cryo-EM maps and three-dimensional models for the katanin hexamer in the ring conformation.
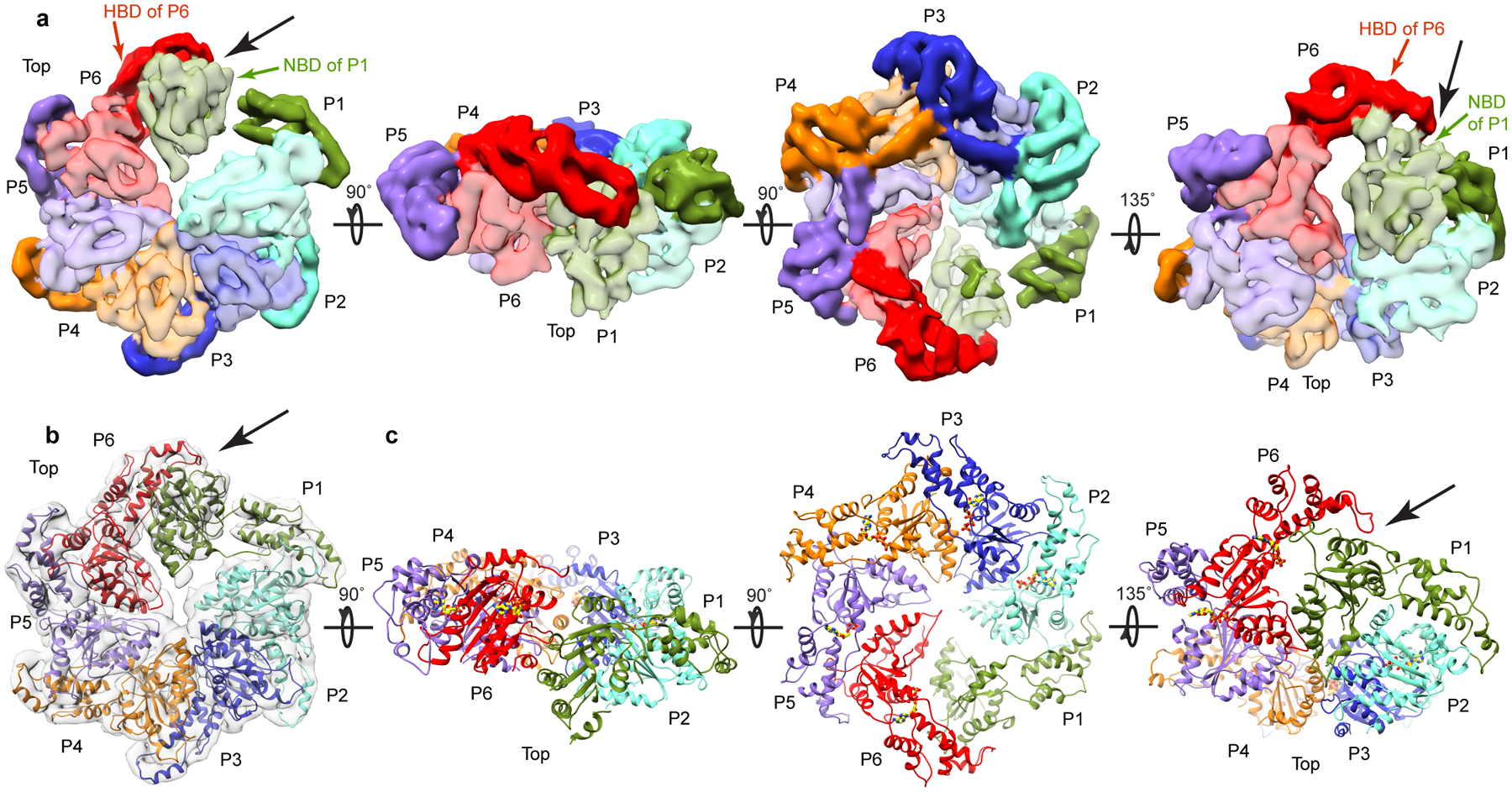
(a) Views of the katanin ring conformation (rotation angles between the different views indicated with arrows). Protomer P1, green; P2, cyan; P3, blue; P4, orange; P5, purple; P6, red. NBD, HBD, light and dark hue, respectively. Black arrow indicates the newly formed P1–P6 interface. (b) Top view of the ring conformation with fitted atomic model. Cryo-EM map, shown as a transparent gray isosurface. Protomers colored as in (a). (c) Views of the atomic models for the katanin ring conformation in the orientations shown above in panel (a).
The cryo-EM reconstructions lack density for the MIT domains and the N-terminal half of the linker (Fig. 1d), suggesting that these parts of katanin are flexible and thus averaged during image processing, consistent with our SAXS analyses that show a large distribution of angles for the arms protruding from the AAA ring (Fig. 1 and Supplementary Fig. 1). The dimensions of the AAA ring in the cryo-EM reconstructions are consistent with those obtained from our low-resolution SAXS envelopes (Fig. 1). The C-terminal ends of the linkers that immediately precede the α1 helices (shown in dark-blue in Figs. 1d, f) form fishhook-shaped structures (shown in light-blue in Figs. 1d, f). The fishhooks and the α1 helices crown the N-terminal entry to a 20 Å-wide axial pore that opens up into the 40Å wide gate between protomers P1 and P6 formed due to their spiral offset (Fig. 1f). Well-defined density reveals a spiral arrangement of the pore loops 1 and 2 which are essential for katanin and spastin severing activity26,23. These loops are poorly ordered in the monomer katanin X-ray structure (Fig. 1e) indicating that they are stabilized upon hexamerization. Pore loop 1 contains a conserved aromatic residue characteristic to all AAA ATPase polypeptide unfoldases that is critical for direct interaction and translocation of the substrate, while pore loop 2 consists of positively charged residues that are highly conserved among severing enzymes (Supplementary Fig. 2 and ref.23).
The NBD and HBD of the AAA core of each protomer form two lobes of a crescent. Protomers P2 through P5 make canonical convex to concave AAA interactions between successive protomers27–29 (Fig. 2a). For a given protomer, the convex face of its NBD interacts with both the NBD and HBD of one neighboring protomer, while its concave face interacts with the NBD of the other neighbor. In the spiral arrangement, the gate is opened by the loss of convex and concave interactions for P1 and P6, respectively (Fig. 2). In the ring conformation, protomers P2 through P5 retain their helical arrangement (~ 5Å rise and ~60° twist per protomer as for P1 through P6 in the spiral conformation) with the canonical AAA ring interactions (Fig. 3). However, because of a 44° rotation, the P1 NBD makes near-canonical interactions with the HBD in P6 and non-canonical interactions on both its convex and concave interfaces with the NBD of P6 and P2, respectively, thus closing the P1–P6 gate (Fig. 3).
Katanin hexamer assembly is mediated by structural elements unique to microtubule severing enzymes
Katanin augments this mode of inter-protomer association with interactions mediated by structural elements characteristic to microtubule severing enzymes: the fishhook-shaped linker element, helix α1 and helix α12 (Fig. 1d, f, Supplementary Fig. 2). Helix α1 (shown in dark-blue in Fig. 1f) of the concave face of the NBD of one protomer makes contacts with the linker that crowns the NBD of the neighboring protomer (Fig. 1f). Additional contacts are mediated by the α1-α2 and α5-β4 loops that are positioned to sense conformational changes in the nucleotide-binding pocket and become ordered upon ATP-dependent hexamer formation (Supplementary Fig. 6a). Notably, mutation of conserved Ile 195 to lysine in the α1-α2 loop inactivates katanin in C. elegans30,31. α12 helices form part of a belt around the C-terminal side of the axial channel (shown in orange in Fig. 1f). This arrangement is mediated by a tight interaction between the conserved α11-α12 linker of one protomer and the four C-terminal residues of the adjacent protomer that immediately follow helix α12 (Supplementary Fig. 6b). These four terminal residues are highly conserved among microtubule severing enzymes and are disordered in the monomeric crystal structures of both katanin (Fig. 1e) and spastin23,25, consistent with their role in hexamer assembly. They include invariant Phe469 and Gly470 (Supplementary Figs. 2 and 6b). Mutation of Gly470 to Asp impairs katanin activity in C. elegans30,31, while mutation of Phe469 impairs both ATPase and microtubule severing activity of katanin in vitro (Supplementary Table 1 and Methods). Mutation of the equivalent aromatic residue in spastin also has a drastic effect on microtubule severing activity23.
Nucleotide state of a gating subunit mediates transition between katanin spiral and ring conformations
In the spiral conformation, all nucleotide-binding sites are occupied by ATP (Fig. 4), with clear density visible for all three phosphates as indicated by difference map analysis between the cryo-EM map and the nucleotide-free atomic model (Figs. 4c–e, Supplementary Fig. 7a and c). ATP binds at the NBD-HBD junction (Fig, 4a). Accordingly, the angle between the NBD and HBD is similar in all six protomers (Cα rmsd ~ 0.3Å; Fig. 4a). AAA ATPases contain arginine residues at the inter-protomer interface, termed “Arg fingers” that stimulate hydrolysis of ATP in trans around their rings32. In katanin, invariant Arg351 and Arg352 from protomers P2 through P6 are ~ 4Å away from the ATP β- and γ-phosphates of the neighboring protomer, yielding a catalytically competent arrangement of the nucleotide binding pockets in protomers P1 through P5 (Figs. 5a, b, Supplementary Fig. 6c). The exception is protomer P6 where the ATP is ~ 36Å from the arginine fingers in P1 (Fig. 5c). Mutation of both Arg351 and Arg352 drastically reduces katanin ATPase and inactivates microtubule severing (Supplementary Table 1).
Figure 4. Different nucleotide occupancies in the spiral and ring conformations of the katanin hexamer.
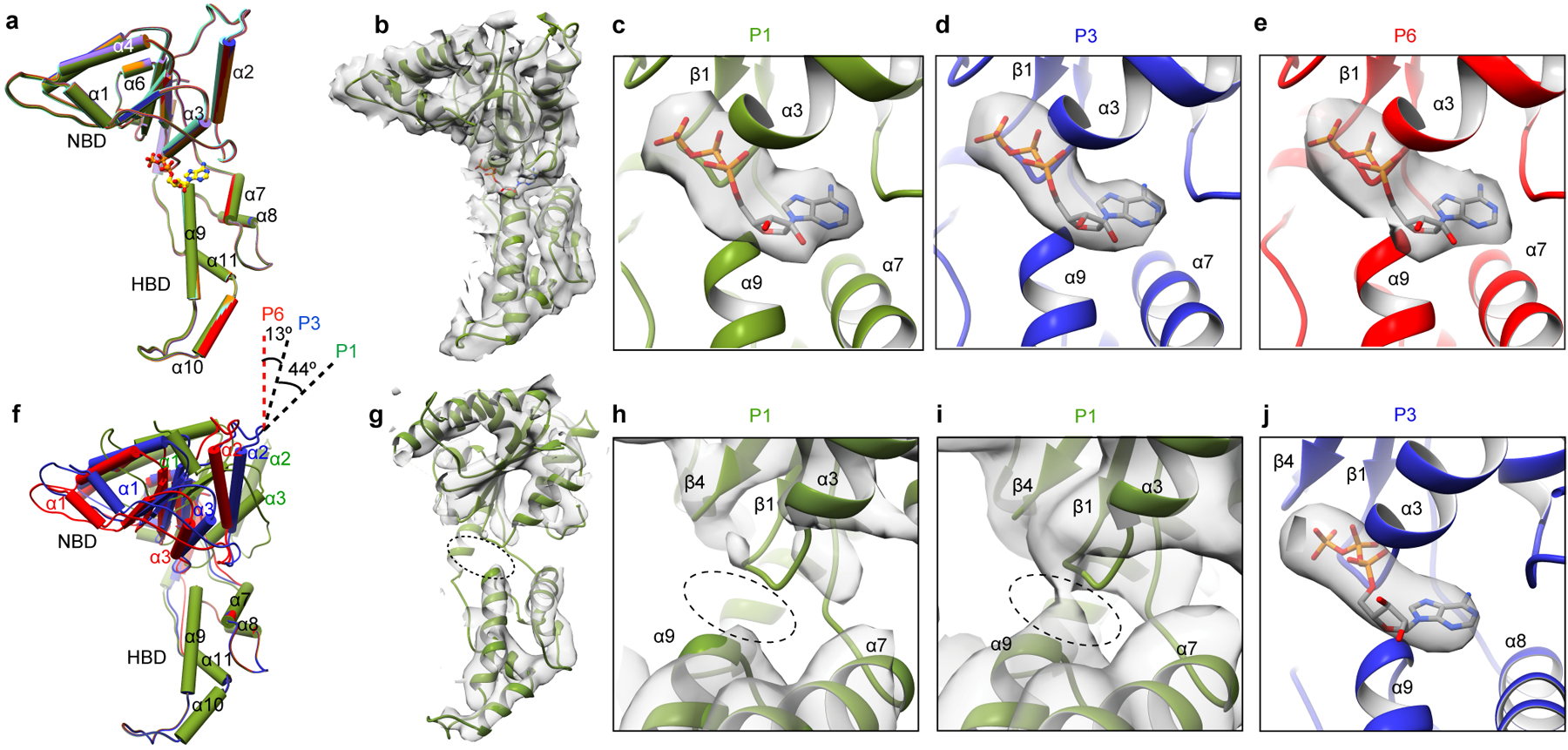
(a) Superposition of protomers P1 through P6 in the spiral conformation. (Cα overall r.m.s.d. ~ 0.3Å). Protomers colored as in Fig. 2a. (b) P1 structure in the spiral conformation. Bound ATP shown as stick representation. (c-e) Enlarged views of the nucleotide-binding pocket showing bound ATP for protomers P1, P3 and P6 in the spiral conformation with the difference map as transparent gray isosurface (Supplementary Fig. 7a, c and Methods). (f) Superposition of P1, P3 and P6 in the ring conformation showing the 44° rotation of the NBD in the P1 protomer. (g) P1 structure in the ring conformation. Oval highlights ATP-binding region. (h, i) Enlarged views of the nucleotide-binding pocket at two contour levels, 15 σ (h) and 10 σ (i) showing absence of nucleotide in P1. (j) Enlarged view of the nucleotide-binding pocket showing bound ATP for protomer P3 in the ring conformation with the difference map as transparent gray isosurface (Supplementary Fig. 7b, d).
Figure 5. Protomer-protomer interface rearrangements between the spiral and ring conformations: transition between tense and relaxed states of the P1 gating protomer.
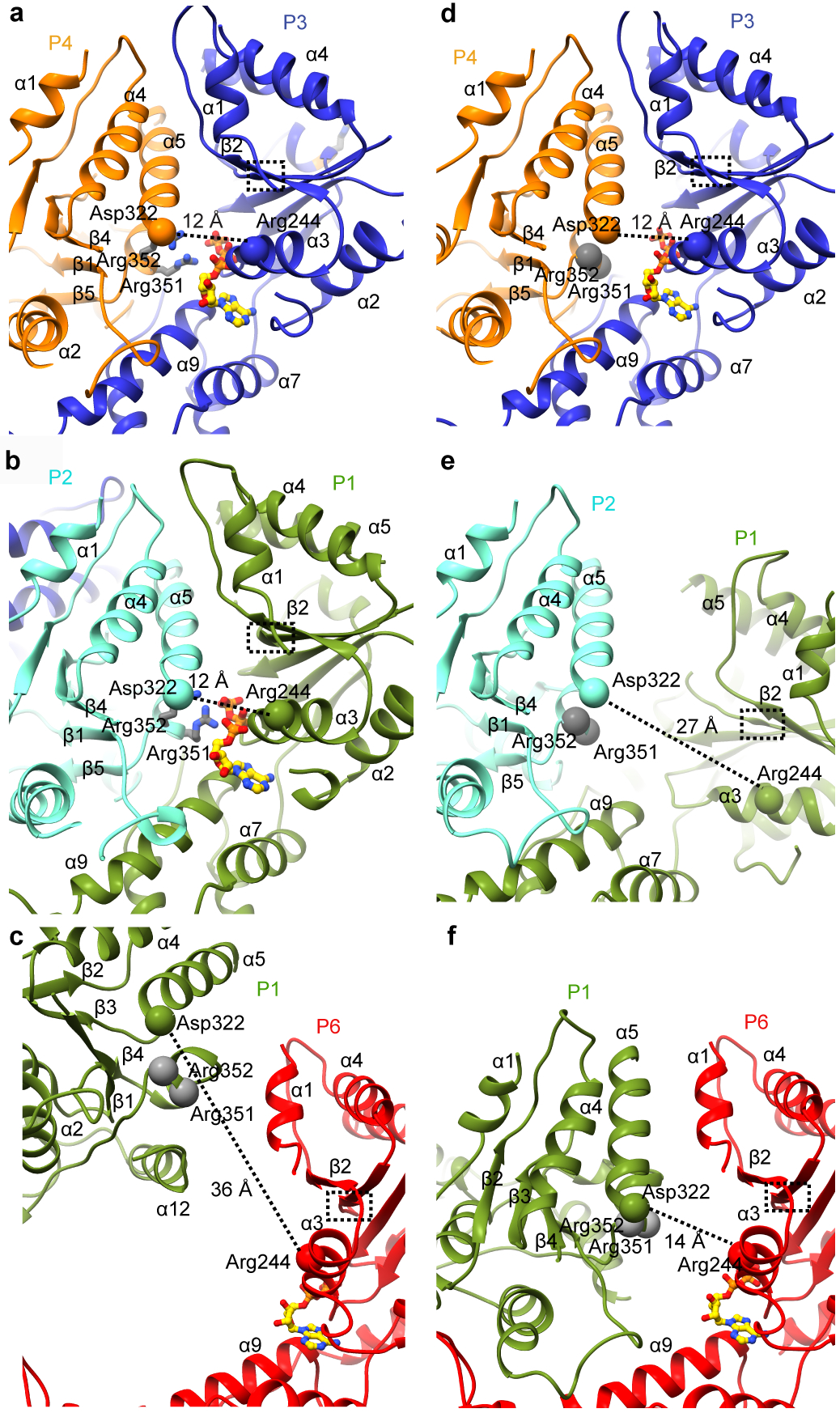
(a, b) Canonical P3–P4 (a) and P1–P2 (b) interfaces in the spiral conformation highlighting the potential salt bridge between invariant Arg244 and Asp322 (Cα shown as spheres). (c) Lack of contacts between the P6 and P1 protomers in the spiral conformation. (d) Canonical P3–P4 interface in the ring conformation highlighting the potential salt bridge between invariant Arg244 and Asp322 (Cα shown as spheres) (e) The relaxed non-canonical P1–P2 interface in the ring conformation. (f) The P6–P1 interface in the ring conformation. Walker B region highlighted by a dashed-line box in all panels and arginine fingers are represented as gray ball and stick (a, b) or Cα spheres (c-f).
In the ring conformation, no nucleotide density is visible in the P1 protomer (Figs. 4h, i and Supplementary Figs. 7b). Protomers P2 through P6 have bound ATP (Fig. 4j and Supplementary Fig. 7b and d). Consistent with the absence of ATP at the P1–P2 interface, the Arg fingers that would be supplied in trans by P2 are ~ 12Å from the nucleotide-binding pocket (Figs. 5b, and e). The loss of ATP in P1 widens the angle between the NBD and HBD by ~18 ° (Fig. 4f). As a result, the P1–P2 interface relaxes. It loses NBD-NBD contacts and the interface is formed only by interactions between the P1 HBD and P2 NBD (Fig. 3a, b). As a result, the salt bridge between invariant Arg244 in P1 and invariant Asp322 in P2, present in the spiral conformation (Fig. 5b) is lost (Fig. 5e). Consistent with its importance, mutation of Asp322 reduces ATPase by 96% and inactivates katanin in in vitro microtubule severing assays (Supplementary Table 1). The rotation of the P1 NBD brings it into contact with P6, thereby closing the gate (Figs. 5f, 3 and Supplementary Video 1). In the canonical interface, α5 is involved in extensive inter-protomer interactions, notably with the Walker B motif that senses nucleotide identity while α1 makes contact with the fishhook-shaped linker element of the neighboring protomer (Figs. 5a, b, d and Fig. 1f). The loss of nucleotide in P1 displaces α5 so that it packs against α1 in P6 and makes more limited inter-protomer contacts (Fig. 5f). Notably, mutations of residues in helix α5 of spastin are found in hereditary spastic paraplegia cases and impair microtubule-severing activity (Supplementary Fig. 2 and refs.23,33).
DISCUSSION
AAA proteins actively thread their substrate biopolymers through an axial channel with the help of conserved pore loops, leading to remodeling or disassembly of their substrates34. Katanin and spastin are thought to sever the microtubule by engaging the C-terminal tails of tubulin with their essential pore loops and pulling on them to dislodge tubulin subunits out of the microtubule lattice23,35. The lack of structural information on microtubule severing enzymes has however impeded the advancement of a mechanistic framework for the mechanochemical cycle for this class of enzymes. The spiral and ring conformations of the katanin AAA motor that we uncovered in our study suggest a power stroke that links ATP hydrolysis and nucleotide release to microtubule severing. The katanin hexamer binds the tubulin tails essential for severing18,35–38. In the spiral ATP- bound conformation, the pore loops stack sequentially to form a spiral ladder optimal for substrate translocation, with pore loop 1 of protomer P1 positioned at the bottom of the spiral close to the microtubule-binding surface, and the open gate between P1 and P6 facilitating tubulin C-terminal tail access to the axial pore. Moreover, the NBD of P1 is exposed in this conformation and accessible to the tubulin tail, which may thus directly stimulate its ATPase upon binding. Our structure reveals that the novel linker element, pore loops 1 and 2, and α12 form a contiguous interface through the axial channel that is optimal for substrate binding and translocation (Fig. 1f and Supplementary Video 1). The location of the fishhook linker element indicates that it is likely one of the first structural elements to interact with the microtubule surface. This structural element is absent in the vps4 hexamer of the meiotic clade of AAA ATPases39,40 and thus is a defining feature of katanin. Interestingly, linker residue Ser135, immediately proximal to the fishhook element, is phosphorylated by Aurora A kinase. This phosphorylation impairs katanin microtubule stimulated ATPase and severing, thus positively regulating spindle size in Xenopus laevis9,41. The additional negative charges proximal to the channel entrance likely interfere with the productive engagement of the negatively charged microtubule substrate. In spastin, mutations of residues in this region or deletion of this linker element impair microtubule severing23,35.
We propose that ATP hydrolysis and nucleotide release in P1 drives the transition to the ring state, thus displacing the linker and pore loop in P1 by ~ 20Å together with the bound tubulin substrate (Figs. 6a, b and Supplementary Video 1) from the bottom of the spiral and up and into the axial channel of the hexamer. The tubulin chain can be then further pulled into the axial channel by the engagement of loop 1 in protomer P2 for another cycle through a sequential mechanism of ATP hydrolysis in the AAA ring. This ~ 20Å displacement would correspond to a ~ 5 amino acid length translocation of the C-terminal tubulin tail from the microtubule surface towards the center of the axial pore.
Figure 6. Pore loop displacement of the gating protomer suggests power stroke for microtubule severing.
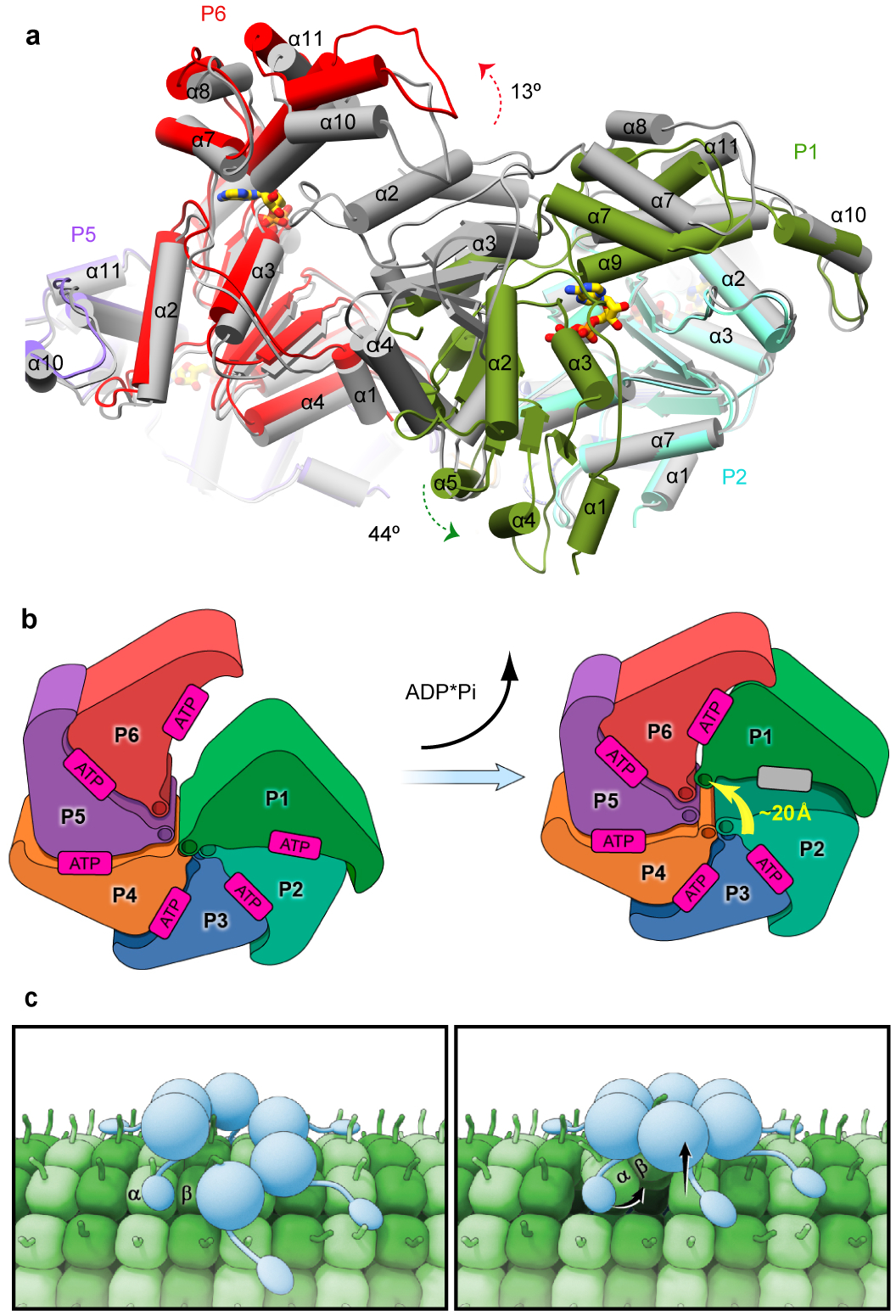
(a) Superposition of the katanin spiral and ring conformations shows the large movement of the gating protomer P1 (green arrow) and the smaller-scale movement of P6 (red arrow). Protomers are colored as in Fig. 2 in the spiral conformation and are gray in the ring conformation. (b) Cartoon depicting the movement of protomer P1 between the spiral and ring conformations. Protomers colored as in Fig. 2. Arrow highlights the translocation of loop 1 of the P1 gating protomer. (c) Cartoon illustrating proposed power stroke that extracts a tubulin dimer and initiates microtubule lattice breakdown and severing. Left panel, Katanin (blue) assembles as a hexamer with a spiral configuration of the AAA domains and the MIT domains emanating from the AAA motor core and making multivalent interactions with the microtubule (green). The flexible tubulin tail is engaged in the axial pore of the katanin hexamer. Right panel, ATP hydrolysis and release in the gating protomer P1 leads to closure of the AAA ring and a ~20Å displacement in the P1 loop that translocates with it the bound C-terminal tail of a tubulin subunit. The cycle is repeated until lattice contacts unravel and the microtubule severs.
The MIT domains bind the microtubule autonomously with low affinity42. Our SAXS analyses demonstrate they are located at the tip of flexible tethers that connect them to the AAA core. This flexibility is likely important to the ability of the MIT domains and the AAA ring to simultaneously engage the microtubule substrate through multivalent interactions and thus help the enzyme stay anchored to the microtubule as it remodels its AAA ring during the mechanochemical cycle and pulls on the tubulin tails with its pore loops (Fig. 6 c). The avidity-based interaction with the microtubule through the MIT domains could also help retain the assembled enzyme on the polymer for subsequent engagement of neighboring tubulin tails if the extraction of one tubulin subunit is not enough to initiate catastrophic breakdown of the microtubule lattice. Complete unfolding and axial translocation of the entire polypeptide chain as in the case of ClpX or Vps443,44 is not absolutely necessary as the microtubule could be destabilized by repeated “tugging” on the tubulin to apply a distorting force to the protofilament structure by cycling through the spiral and closed ring states in a mechanism more analogous to the one recently proposed for NSF45.
In addition to our results here on microtubule severing, structural studies of AAA ATPases that remodel protein complexes or aggregates such as Hsp10446, ClpX29 the archaeal homolog of p97 of unknown function, VAT47, the proteasome48,49 and Vps439,40 have identified asymmetric spiral architectures of AAA rings with pore loops stacked in a spiral suggestive of a relay of the substrate through their central pores. Recent cryo-EM studies identified both open and closed spiral configurations for VAT and Vps4 in the presence of substrate and as a function of nucleotide state40,47 suggesting a mechanism analogous to katanin, although there are differences in nucleotide occupancies between these structures, consistent with their different domain organization and function. The substrate-bound structures of Vps440 and VAT47 are asymmetric with five of the pore loops bound to the unfolded substrate polypeptide and arranged in a tight spiral and the sixth subunit in a different conformation and disengaged from the substrate. Thus, a common theme is emerging for some AAA ATPases whereby the power stroke for substrate translocation is generated by breaking the helical symmetry of a spiral arrangement of subunits by a boundary subunit in response to nucleotide hydrolysis. Such a mechanism for substrate translocation was originally advanced for nucleic acid AAA ATPases translocases such as the DNA helicase E150 and the RNA translocase Rho51,52. In contrast to these, the recent cryo-EM reconstructions of p97 show six-fold symmetric arrangements indicative of a concerted mechanism of substrate engagement and remodeling53, and highlight the mechanistic richness used by AAA ATPases, consistent with their large representation in the genome and diverse cellular substrates and functions. Thus, details of nucleotide occupancies and nucleotide hydrolysis mechanism are likely different between AAA ATPases and might even differ between substrates for the same enzyme as studies on ClpB and Hsp104 have shown different ATP hydrolysis mechanisms depending on the type of substrate they engage54,55. This is an interesting aspect to consider when comparing microtubule severing enzymes and other closely related AAA ATPases, as the former are able to disassemble one of the stiffest non-covalent polymer structures found in eukaryotic cells2.
The mechanistic complexity of AAA ATPases highlights the importance of obtaining high-resolution structural and kinetic information for each enzyme along its entire mechanochemical cycle and with its physiological substrates. Future structures of katanin and spastin in different nucleotide-bound states and with the microtubule substrate will be critical for elucidating how these enzymes generate the force needed to disrupt lattice interfaces in the microtubule polymer that ultimately lead to its catastrophic disassembly. Our structural study is an important first step towards establishing a mechanistic framework for these fascinating molecular machines.
ONLINE METHODS
Protein expression and purification
Caenorhabditis elegans katanin p60 was expressed in Escherichia coli as a maltose binding-fusion protein. Cultures were grown at 37°C to an OD600 of ~ 1.0 and katanin expression was induced with 0.5 mM IPTG at 16°C and for fourteen hours. Harvested cells were resuspended in 50 mM HEPES pH 7.5, 500 mM KCl, 10 mM MgCl2, 1 mM DTT, 1 mM PMSF, and lysed with a microfluidizer in the presence of a protease inhibitor cocktail (Roche). The supernatant was collected by centrifugation at 31,000 x g for 40 min and loaded onto amylose resin (New England Biolabs), equilibrated in 50 mM HEPES pH 7.5, 500 mM KCl, 10 mM MgCl2 and 1 mM DTT. Katanin was released from the resin by cleavage with Tobacco Etch Virus protease. Katanin was further purified by anion exchange chromatography. The sample was exchanged into buffer containing 20 mM HEPES pH 7.5, 300 mM KCl, 10 mM MgCl2, 1 mM TCEP and 15 % glycerol and flash-frozen in liquid nitrogen for storage at −80°C. Prior to conducting all analytical ultracentrifugation (AUC), solution small-angle x-ray scattering (SAXS) and electron microscopy (EM) analyses katanin was further purified by size exclusion chromatography on a Superose 6 Increase 10/300 GL column (GE Healthcare). In order to determine the stoichiometry of ATP to the katanin hexamer we chromatographed full-length and ΔMIT katanin p60 (100 μL of 100 μM) on a Superose 6 10/300 GL Increase column in the buffer containing excess ATP (100 μM). As the protein migrates through the gel filtration matrix, it depletes ATP from the buffer resulting in a negative peak. Bound ATP was determined from the area of the negative ATP depletion peak. The determined molar ratio of ATP per katanin hexamer was 5.6±0.3 (n=3). Mutants were generated using Quickchange (Stratagene).
Analytical ultracentrifugation
AUC experiments were conducted at 24, 12, 6 and 3 μM of katanin p60 concentrations in 20 mM HEPES pH 7.5, 300 mM KCl, 10 mM MgCl2, 1 mM TCEP with or without 100 μM ATP. Measurements were collected in a ProteomeLab XL-I analytical ultracentrifuge (Beckman Coulter, Indianapolis, IN) using absorption optics. To obtain a final sample optical density in the 0.25 to 1.00 range, cells of 3-mm or 12-mm optical path length were used. Loaded cells, placed in the four-hole AN-Ti rotor, were thermally equilibrated in the ultracentrifuge. After thermal equilibrium was reached at rest at 10°C or 20°C, the rotor was accelerated to 45,000 rpm to start the collection of intensity scans at 280 nm. The data were collected until no further sedimentation boundary movement was observed. Sedimentation velocity data analyses were performed with the program SEDFIT using a continuous c(s) model56. All accepted fits have root-mean-square deviations (RMSD) less than 0.008. Partial specific volume and buffer parameters were calculated with SEDNTERP (http://sednterp.unh.edu/). Sedimentation coefficient distribution values were corrected to standard conditions of water at 20°C, s20,w.
Crystallization and X-ray structure determination
For crystallization, katanin p60 (E293Q) eluted from the size execution column (see above) was supplemented with 1 mM ATP and concentrated to 10 mg/ml. Crystals grew by hanging drop vapor diffusion at room temperature from protein mixed 2:1 (v/v) with the crystallization solution containing 1.0 M ammonium sulfate and 0.1 M Tris pH 7.8. Crystals were cryoprotected with 25% glycerol before flash freezing in liquid nitrogen. X-ray diffraction data were collected at the Advanced Light Source (ALS), beamline 5.0.2. Crystals grew with the symmetry of space group P65 with one katanin protomer per asymmetric unit (Unit Cell: a = 99.4 Å, b = 99.4 Å, c = 76.6 Å). Most crystals diffracted poorly and were not suitable for structural determination. Diffraction data were reduced with HKL200057. Details of the experimental data collection and processing statistics are in Table 1. The structure was solved by molecular replacement using PHASER58 with the previously published structure of spastin as the search model (PDB entry 3B9P23). Multiple rounds of model building and refinement were performed with COOT59 and PHENIX60. The current refinement model for katanin p60 consists of 262 residues and one sulfate ion. Despite the presence of ATP in the crystallization conditions, no well-ordered nucleotide is visible in the maps. Six regions of the polypeptide chain (residues 1–172; 186–190, 263–269, 297–306, 324–332, 467–472) are not well-resolved and presumed disordered. Residues 173–183, 270–284 and 310–322 were built as poly-Ala as the sequence register is based on the sequence alignment of the spastin X-ray structure. The fishhook element visible in the cryo-EM map is also visible in the X-ray model, although less well defined in the latter because of the lack of stabilization by protomer-protomer contacts. The quality of the map in this region was not sufficient to allow confident de novo building in either the X-ray or cryo-EM map. This structural feature comprises ~ 25 residues N-terminal to helix α1. The current crystallographic model of the katanin protomer at 3.3 Å resolution has Rwork and Rfree of 30.7 % and 25.8 %, respectively (Table 1) with 95.1% of residues in favored regions and 0.8% in disallowed regions. Coordinate error is 0.53Å. Figures were prepared with UCSF Chimera61.
Solution small-angle X-ray scattering
Solution SAXS experiments of full-length and ΔMIT katanin p60 (E293Q) were performed at beamline 12-ID-B of the Advanced Photon Source (APS) at Argonne National Laboratory using an in-line FPLC AKTA micro setup with a Superose 6 Increase 10/300 GL size exclusion column. The wavelength, λ, of X-ray radiation was set to 0.886 Å. Scattered X-ray intensities were measured using a Pilatus 2M detector. The sample-to-detector distance was set such that the detecting range of momentum transfer q [=4π sinθ/λ, where 2θ is the scattering angle] was 0.004–0.55 Å−1. The sample passed through the FPLC column and was fed to a flow cell for SAXS measurements. The flow cell is a cylindrical quartz capillary 1.5 mm in diameter and 10 μm wall thickness, and is equipped with a Peltier cooling device that keeps the flow cell at 10°C. SAXS data were collected at the elution peak while background data were collected before and after the peak. The exposure time was set to <1 second to reduce radiation damage. More than forty images were acquired for each sample (around the elution peak) and background in order to obtain good signal-to-noise ratio values. The 2-D scattering images were converted to 1-D SAXS (I(q) vs q) curves through azimuthally averaging after solid angle correction and then normalizing with the intensity of the transmitted X-ray beam flux, using custom scripts. The sample concentration was ~2.0 mg/ml – 4.0 mg/ml at the elution peak. The SAXS profile of the protein was obtained by subtracting background from the sample data. The radius of gyration (Rg) was calculated using the Guinier equation62, i.e., , where Io is the forward scattering, an indicator of molecular weight63. The pair distance distribution function P(r) that is the inverse Fourier transform of X-ray scattering data and roughly a weighted histogram of atomic-pair distances in the molecule, was calculated using GNOM v4.664. Rg were calculated from the second momentum of the P(r) function and compared favorably to the initial value derived from the Guinier plots (fig. S1E), 67.7±0.4 Å versus 63 ± 5 Å for full-length katanin p60 and 51.4 ± 0.3 Å versus 51 ± 1 Å for ΔMIT katanin p60. Molecular weights were estimated directly from the background-subtracted SAXS data using the program SCATTER v2.3h65 and were 360 kDa and 260 kDa for katanin p60 and ΔMIT katanin p60, respectively. The expected molecular weights of the katanin p60 and ΔMIT katanin hexamers are 319 kDa and 252 kDa, respectively. Three-dimensional molecular envelopes were calculated from SAXS data up to q of 0.30 Å−1, using DAMMIF v1.1.2, a fast version of DAMMIN66. Twenty and twenty-four runs were performed for katanin p60 full-length and the ΔMIT construct, respectively. The reconstructions were aligned, averaged and filtered on the basis of occupancy using DAMAVER v5.067. P6 symmetry was applied in the DAMMIF and DAMAVER calculations. The overall size of the DAMAVER averaged bead model was significantly reduced comparing to the individuals, indicating that the regions beyond the central ring have a high degree of flexibility (Fig. 1a and Supplementary Figs. 1f, d, h). Therefore, both the individual and averaged models are reported, with the former being a better representation of molecular size.
Cryo-EM specimen preparation, data acquisition and analysis
Katanin p60 (E293Q) was diluted to 0.6 mg/ml in 20 mM HEPES-KOH pH 7.5, 300 mM KCl, 10 mM MgCl2, 1mM TCEP and 1mM ATP and spun at 69,800 x g for 15 min at 4°C. 5 μl was applied to a glow-discharged C-flat holey carbon grid with 1.2 μm hole, 1.3 μm space (C-flat™, Electron Microscopy Sciences). The grids were blotted for 5 seconds at 90% humidity and plunge-frozen in liquid ethane cooled by liquid nitrogen using Leica EM GP (Leica Microsystems, Germany). To assess sample stability and monodispersity, cryo-EM data were first collected on a Tecnai TF20 electron microscope (FEI) operated at 200 kV, equipped with a K2 Summit direct electron detector camera (Gatan) operated in electron counting mode. To obtain a preliminary reconstruction, ~800 images were recorded at a nominal magnification of 29,000x, corresponding to a physical pixel size of 1.27 Å/pixel. Images were recorded with a defocus range from −1.5 μm to −3.5 μm. With no specimen present, the rate of exposure of the detector was 6 e−/Å2/s. Exposure-fractionated movie stacks of 40 frames were recorded for 10 s with a total electron dose of 60 e−/Å2.
To obtain a preliminary reconstruction of katanin p60 (E293Q), images were aligned with each other using Unblur68. Defocus parameters were estimated from aligned sums of 5 frames using CTFFINF469. 90,000 particles were manually picked from movie sums with the total dose of 60 e/Å2 and extracted in Relion using a 300 pixel box size70. Images were downsampled to a pixel size of 2.54 Å and classified in 2D using Relion. An initial 3D reconstruction with C6 symmetry was generated using a subset of particles (40,000) in FREALIGN71, following a protocol described previously72. To obtain the initial reconstruction, images were downsalmpled to 5.08 Å/pixel and frequencies only up to 1/40 Å−1 in the initial rounds and up to 1/15 Å−1 in the final rounds of refinement were used. The most populated class was low-pass filtered to 60 Å and used as an initial reference for 3D classification of the full data set. For 2D and 3D classification in Relion, particles were re-extracted from the movie sums with the total dose of 35 e−/Å2. 3D classification was carried out without application of symmetry operators. Only spatial frequencies up to 1/10 Å−1 were used to avoid overfitting. After 50 rounds of 3D classification with 4 classes, particles in 2 classes that produced similar 3D reconstructions were combined for 3D auto-refinement. The 3D reconstruction from ~27,000 particles was resolved to 14.1 Å (FSC=0.143 criterion).
To obtain the final high-resolution 3D reconstructions, a dataset of ~2,100 images was collected on a Titan Krios microscope (FEI) operated at 300 kV, equipped with a K2 Summit direct electron detector camera, operated in electron counting mode. Micrographs indicated monodisperse distribution of particles in ice (Supplementary Fig. 3a). Movie stacks were recorded at a nominal magnification of 22,500x, corresponding to a physical pixel size of 1.31 Å/pixel at a defocus range from −1.0 μm to −3.5 μm. The rate of exposure was 2.91 e−/Å2/s. 50-frame movie stacks were recorded for 17.5 s with an exposure of 1.02 e−/Å2 per frame and a total electron dose of 51 e−/Å2. Data collection was automated with SerialEM73. Data collection statistics are in Table 2.
All image frames (51 e−/Å2 total dose) were aligned and summed with Unblur through the Relion pipeline68,70. Defocus parameters were estimated from aligned sums of 5 frames using CTFFINF469. Images were downsampled to a pixel size of 2.62 Å/pix and ~403,023 manually picked particles were classified in 2D using Relion. Reference-free 2D class averages showed fine molecular features and presented the complex in different orientations, suggestive of its structural order and the lack of preferential orientation in ice (Supplementary Fig. 3b). After 3 rounds of the 2D classification ~197,779 particles were 3D classified into 4 classes without application of symmetry operators (Supplementary Fig. 4). The reconstruction from the TF20 dataset was low-pass filtered to 60 Å and used as the initial reference for the 3D classification. 100 rounds of 3D classification with no applied symmetry produced two similar classes with a spiral conformation (~95,468 particles total, Classes 2 and 4 in Supplementary Fig. 4), one class with a ring conformation (~48,565 particles, Class 3 in Supplementary Fig. 4) with the rest of the particles classifying into a low-resolution class (~53,746 particles, Class 1 in Supplementary Fig. 4). The low-resolution class consists of broken or misaligned particles and looked structurally different from the three major classes. Further classification of particles in this class did not identify any dominant conformation and did not improve the overall quality of the map. The particles from the two classes with the spiral conformation were combined and reclassified into 3 classes. All three classes looked alike and contained a similar number of particles, indicating the structural homogeneity of this particle set. Consequently, the particle set was refined in 3D as a single class. Similarly, the particle subset that produced the reconstruction with the ring conformation was further classified into 2 classes. Both new classes looked similar and contained a similar number of particles indicating the structural homogeneity of this particle set. Consequently, the original particle subset was refined in 3D as a single class.
For the 3D refinement particles were re-extracted from unbinned image sums that showed tone rings to 5 Å or higher spatial frequencies. The 3D refinement was carried out using a created mask with a soft edge of 6 pixels to mask out flexible protein parts around the AAA ATPase hexameric core. Prior to application of the mask, no other well-defined features were visible outside the hexameric core. The 3D reconstruction of the spiral conformation was refined to 5.2 Å and the ring conformation to 7.3 Å (FSC=0.143 criterion). To improve the quality of the 3D maps further, particles were sorted based on their differences with their aligned, CTF-corrected references as described in refs.46,74 A subset of particles with z-scores ≤ 0.7 and ≤ 0.8 for the spiral and the ring conformations, respectively, were re-refined in 3D. This procedure improved the quality of the final 3D maps with the resolution of the spiral reconstruction calculated to 4.4 Å and that of the planar reconstruction to 6.0 Å after implementation of the PostProcessing procedure with automatic masking and negative Bfactor application. Both reconstructions were sharpened with the automatically determined negative B-factors of ~100 Å2 for both structures. 38,072 and 16,185 particles contributed to the final maps in the spiral and ring conformations, respectively. Data collection statistics and image-processing summary are in Table 2.
To generate the difference maps in Fig. 4 and Supplementary Fig. 7, the atomic models for the protein were converted to mrc format using the bgex routine in Bsoft75 and low-pass filtered using the proc3d routine in EMAN76. The difference maps for the spiral conformation between the cryo-EM reconstruction and the atomic model, low-pass filtered to either 5.5 Å for P1 and P6 or to 4.4 Å for P2 through P5, were generated using diffmap.exe (http://grigoriefflab.janelia.org/software). The resolution cut-off values for each protomer were determined by the blocres program in Bsoft75. Atomic models for the ring conformation were low-pass filtered to 8 Å for P1 and P6 and to 6 Å for P2 through P5. The difference maps were generated following the procedure described for the spiral conformation.
Model building and refinement.
Consistent with the estimated 4.4 Å resolution, we observe grooves in α-helixes, separated β-strands within β-sheets and densities for some bulky side chains. The X-ray structure of the katanin protomer was docked into the sharpened map using UCSF Chimera61 and its fit was improved with the collage option in Situs77. The atomic model was fit flexibly into the cryo-EM reconstructions using NAMD with Molecular Dynamics Flexible Fitting (MDFF)78. Secondary structure, chirality and cis-peptide restraints were applied to the model, and MDFF simulations were performed in implicit solvent at temperature T=300 K to enhance the protein conformational sampling. Energy minimization was performed for 200 steps. MDFF simulations were carried out for 150 ps with a coupling constant (gscale) = 0.3. MDFF simulation convergence was monitored by plotting the root mean square deviation over time. Additional adjustments to the backbone and side chains for this model were performed manually in COOT59, residue by residue. Densities for ATP were clearly visible in all six protomers of the spiral conformation (Fig. 4c–e and Supplementary Fig. 7a and c). The α11 – α12 linker, the C-terminus and pore loop1 were built de novo as they are disordered in the monomer X-ray structure. Weak residual density features are present near pore loop1 that were not interpreted at the current resolution limit. None of the side chains of the pore loop residues were built. The resulting model was subjected to real space refinement in PHENIX60. The final atomic model has an overall correlation to the map of 0.884. For the ring conformation, the X-ray structure was first rigidly docked using Situs and then flexibly fitted using MDFF and COOT into the cryo-EM map as described for the spiral conformation. Additional adjustments to the backbone for this model were performed manually in COOT. Densities for ATP were clearly visible in five of the six AAA protomers (Fig. 4j and Supplementary Fig. 7b and d). The model was subjected to real space refinement in PHENIX. The final atomic model has an overall correlation to the map of 0.90. MolProbity was used to evaluate model geometry79. Model statistics for both conformations are listed in Table 2.
ATPase and microtubule severing assays
ATPase assays were performed at room temperature in BRB80, 50 mM KCl, 2.5 mM MgCl2, 1 mM ATP, 10 μM taxol at 50 nM katanin concentration. ATPase activities were measured using an EnzChek Phosphate Assay (Life Technologies). Initial rates were calculated from the linear portion of the reaction profiles after addition of 1 mM ATP. ATPase rates were adjusted by subtraction of the measured release of phosphate in the absence of ATP. Rates reported are means from four separate reactions.
Chambers for TIRF microscopy were assembled as previously described80. GMPCPP brain microtubules containing 1% biotinylated tubulin and 20% HiLyte647-labeled tubulin were immobilized in the chamber with 2 mg/ml Neutravidin (Sigma) and imaged by TIRF or DIC microscopy in severing buffer (BRB80 buffer with 2 mg/ml casein, 14.3 mM 2-mercaptoethanol, 2.5% glycerol, 50 mM KCl, 2.5 mM MgCl2, 1 mM ATP, 1% Pluronic F127 (Life Technologies) and oxygen scavengers prepared as described in80). The chamber was then perfused with 20 nM katanin in severing buffer. Severing rates were calculated by manual counting of severing events as a function of time as described previously37.
A Life Sciences Reporting Summary accompanies this paper. All data and constructs used in this study are available upon request from the corresponding author. Protein Data Bank accession number for atomic coordinates and structure factor amplitudes for the X-ray structure is 5WC1. The EMDB accession codes for the spiral and ring maps are EMD-8794 and EMD-8796, respectively. The atomic coordinates for the cryo-EM models were deposited with PDBIDs 5WC0 and 5WCB for the spiral and ring conformation, respectively.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to N. Grigorieff for initial advice and access to the Krios for collection of one high-resolution dataset, R. Diaz-Avalos for data collection advice, J. Hinshaw and H. Wang for TF20 access and F. McNally (University of California, Davis) for a katanin expression plasmid. X-ray diffraction data were collected at beamline 502 of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. SAXS was performed at Beamline 12ID-B of the Advanced Photon Source, which is a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. A.R-M. is supported by the intramural programs of NINDS and NHLBI.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Roll-Mecak A & McNally FJ Microtubule-severing enzymes. Current opinion in cell biology 22, 96–103 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gittes F, Mickey B, Nettleton J & Howard J Flexural rigidity of microtubules and actin filaments measured from thermal fluctuations in shape. The Journal of cell biology 120, 923–934 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmad FJ, Yu W, McNally FJ & Baas PW An essential role for katanin in severing microtubules in the neuron. The Journal of cell biology 145, 305–315 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu W et al. The microtubule-severing proteins spastin and katanin participate differently in the formation of axonal branches. Molecular biology of the cell 19, 1485–1498 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Q, Fishel E, Bertroche T & Dixit R Microtubule severing at crossover sites by katanin generates ordered cortical microtubule arrays in Arabidopsis. Current Biology 23, 2191–2195 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Lindeboom JJ et al. A mechanism for reorientation of cortical microtubule arrays driven by microtubule severing. Science 342, 1245533 (2013). [DOI] [PubMed] [Google Scholar]
- 7.McNally K, Audhya A, Oegema K & McNally FJ Katanin controls mitotic and meiotic spindle length. The Journal of cell biology 175, 881–891 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cummings CM, Bentley CA, Perdue SA, Baas PW & Singer JD The Cul3/Klhdc5 E3 ligase regulates p60/katanin and is required for normal mitosis in mammalian cells. J Biol Chem 284, 11663–75 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loughlin R, Wilbur JD, McNally FJ, Nedelec FJ & Heald R Katanin contributes to interspecies spindle length scaling in Xenopus. Cell 147, 1397–1407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McNally K et al. Katanin maintains meiotic metaphase chromosome alignment and spindle structure in vivo and has multiple effects on microtubules in vitro. Mol Biol Cell 25, 1037–49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srayko M, O’Toole ET, Hyman AA & Muller-Reichert T Katanin disrupts the microtubule lattice and increases polymer number in C. elegans meiosis. Current biology : CB 16, 1944–1949 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Zhang D, Rogers GC, Buster DW & Sharp DJ Three microtubule severing enzymes contribute to the “Pacman-flux” machinery that moves chromosomes. J Cell Biol 177, 231–42 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma N et al. Katanin regulates dynamics of microtubules and biogenesis of motile cilia. The Journal of cell biology 178, 1065–1079 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu WF et al. Katanin p80 regulates human cortical development by limiting centriole and cilia number. Neuron 84, 1240–1257 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mishra-Gorur K et al. Mutations in KATNB1 cause complex cerebral malformations by disrupting asymmetrically dividing neural progenitors. Neuron 84, 1226–1239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yigit G et al. A syndrome of microcephaly, short stature, polysyndactyly, and dental anomalies caused by a homozygous KATNB1 mutation. American journal of medical genetics.Part A 170, 728–733 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Vale RD Severing of stable microtubules by a mitotically activated protein in Xenopus egg extracts. Cell 64, 827–839 (1991). [DOI] [PubMed] [Google Scholar]
- 18.McNally FJ & Vale RD Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell 75, 419–429 (1993). [DOI] [PubMed] [Google Scholar]
- 19.Hartman JJ et al. Katanin, a microtubule-severing protein, is a novel AAA ATPase that targets to the centrosome using a WD40-containing subunit. Cell 93, 277–287 (1998). [DOI] [PubMed] [Google Scholar]
- 20.McNally FJ, Okawa K, Iwamatsu A & Vale RD Katanin, the microtubule-severing ATPase, is concentrated at centrosomes. Journal of cell science 109 ( Pt 3), 561–567 (1996). [DOI] [PubMed] [Google Scholar]
- 21.Joly N, Martino L, Gigant E, Dumont J & Pintard L Microtubule-severing activity of AAA-ATPase Katanin is essential for female meiotic spindle assembly. Development (Cambridge, England) (2016). [DOI] [PubMed]
- 22.Hartman JJ & Vale RD Microtubule disassembly by ATP-dependent oligomerization of the AAA enzyme katanin. Science (New York, N.Y.) 286, 782–785 (1999). [DOI] [PubMed] [Google Scholar]
- 23.Roll-Mecak A & Vale RD Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature 451, 363–367 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scott A et al. Structural and mechanistic studies of VPS4 proteins. The EMBO journal 24, 3658–3669 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor JL, White SR, Lauring B & Kull FJ Crystal structure of the human spastin AAA domain. J Struct Biol 179, 133–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johjima A et al. Microtubule severing by katanin p60 AAA+ ATPase requires the C-terminal acidic tails of both alpha- and beta-tubulins and basic amino acid residues in the AAA+ ring pore. The Journal of biological chemistry 290, 11762–11770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caillat C et al. Asymmetric ring structure of Vps4 required for ESCRT-III disassembly. Nature communications 6, 8781 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lenzen CU, Steinmann D, Whiteheart SW & Weis WI Crystal structure of the hexamerization domain of N-ethylmaleimide-sensitive fusion protein. Cell 94, 525–36 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Glynn SE, Martin A, Nager AR, Baker TA & Sauer RT Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell 139, 744–756 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mains P, Kemphues K, Sprunger S, Sulston I & Wood W Mutations affecting the meiotic and mitotic divisions of the early Caenorhabditis elegans embryo. Genetics 126, 593–605 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark-Maguire S & Mains PE mei-1, a gene required for meiotic spindle formation in Caenorhabditis elegans, is a member of a family of ATPases. Genetics 136, 533–546 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wendler P, Ciniawsky S, Kock M & Kube S Structure and function of the AAA+ nucleotide binding pocket. Biochimica et biophysica acta 1823, 2–14 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Fonknechten N et al. Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia. Hum Mol Genet 9, 637–44 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Hanson PI & Whiteheart SW AAA+ proteins: have engine, will work. Nature reviews.Molecular cell biology 6, 519–529 (2005). [DOI] [PubMed] [Google Scholar]
- 35.White SR, Evans KJ, Lary J, Cole JL & Lauring B Recognition of C-terminal amino acids in tubulin by pore loops in Spastin is important for microtubule severing. The Journal of cell biology 176, 995–1005 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roll-Mecak A & Vale RD The Drosophila homologue of the hereditary spastic paraplegia protein, spastin, severs and disassembles microtubules. Curr Biol 15, 650–5 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Valenstein ML & Roll-Mecak A Graded Control of Microtubule Severing by Tubulin Glutamylation. Cell 164, 911–921 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailey ME, Sackett DL & Ross JL Katanin Severing and Binding Microtubules Are Inhibited by Tubulin Carboxy Tails. Biophysical journal 109, 2546–2561 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su M et al. Mechanism of Vps4 hexamer function revealed by cryo-EM. Sci Adv 3, e1700325 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Monroe N, Han H, Shen PS, Sundquist WI & Hill CP Structural basis of protein translocation by the Vps4-Vta1 AAA ATPase. Elife 6(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whitehead E, Heald R & Wilbur JD N-terminal phosphorylation of p60 katanin directly regulates microtubule severing. J Mol Biol 425, 214–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iwaya N et al. A common substrate recognition mode conserved between katanin p60 and VPS4 governs microtubule severing and membrane skeleton reorganization. The Journal of biological chemistry 285, 16822–16829 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olivares AO, Nager AR, Iosefson O, Sauer RT & Baker TA Mechanochemical basis of protein degradation by a double-ring AAA+ machine. Nat Struct Mol Biol 21, 871–5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang B, Stjepanovic G, Shen Q, Martin A & Hurley JH Vps4 disassembles an ESCRT-III filament by global unfolding and processive translocation. Nat Struct Mol Biol 22, 492–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao M et al. Mechanistic insights into the recycling machine of the SNARE complex. Nature 518, 61–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yokom AL et al. Spiral architecture of the Hsp104 disaggregase reveals the basis for polypeptide translocation. Nature structural & molecular biology 23, 830–837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ripstein ZA, Huang R, Augustyniak R, Kay LE & Rubinstein JL Structure of a AAA+ unfoldase in the process of unfolding substrate. Elife 6(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lander GC et al. Complete subunit architecture of the proteasome regulatory particle. Nature 482, 186–91 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wehmer M et al. Structural insights into the functional cycle of the ATPase module of the 26S proteasome. Proc Natl Acad Sci U S A 114, 1305–1310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Enemark EJ & Joshua-Tor L Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442, 270–5 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Thomsen ND & Berger JM Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell 139, 523–34 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomsen ND, Lawson MR, Witkowsky LB, Qu S & Berger JM Molecular mechanisms of substrate-controlled ring dynamics and substepping in a nucleic acid-dependent hexameric motor. Proc Natl Acad Sci U S A 113, E7691–E7700 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Banerjee S et al. 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science (New York, N.Y.) 351, 871–875 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Doyle SM, Hoskins JR & Wickner S DnaK chaperone-dependent disaggregation by caseinolytic peptidase B (ClpB) mutants reveals functional overlap in the N-terminal domain and nucleotide-binding domain-1 pore tyrosine. J Biol Chem 287, 28470–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DeSantis ME et al. Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 151, 778–93 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schuck P Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78, 1606–19 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Otwinowski Z & Minor W [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 58.McCoy AJ et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–32 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–12 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Glatter VO & Kratsky O Small angle x-ray scattering, (Academic Press, London, 1982). [Google Scholar]
- 63.Mylonas E & Svergun DI Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. Journal of Applied Crystallography 40, s245–s249 (2007). [Google Scholar]
- 64.Svergun D Determination of the regularization parameter in indirect-transform methods using perceptual criteria. Journal of Applied Crystallography 25, 495–503 (1992). [Google Scholar]
- 65.Rambo RP & Tainer JA Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496, 477–81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Svergun DI Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J 76, 2879–86 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Volkov VV & Svergun DI Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography 36, 860–864 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grant T & Grigorieff N Measuring the optimal exposure for single particle cryo-EM using a 2.6 A reconstruction of rotavirus VP6. Elife 4, e06980 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rohou A & Grigorieff N CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scheres SH RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–30 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grigorieff N FREALIGN: high-resolution refinement of single particle structures. J Struct Biol 157, 117–25 (2007). [DOI] [PubMed] [Google Scholar]
- 72.Grigorieff N Frealign: An Exploratory Tool for Single-Particle Cryo-EM. Methods Enzymol 579, 191–226 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Chua EY et al. 3.9 A structure of the nucleosome core particle determined by phase-plate cryo-EM. Nucleic Acids Res 44, 8013–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Heymann JB Bsoft: image and molecular processing in electron microscopy. J Struct Biol 133, 156–69 (2001). [DOI] [PubMed] [Google Scholar]
- 76.Ludtke SJ, Baldwin PR & Chiu W EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128, 82–97 (1999). [DOI] [PubMed] [Google Scholar]
- 77.Wriggers W, Milligan RA & McCammon JA Situs: A package for docking crystal structures into low-resolution maps from electron microscopy. J Struct Biol 125, 185–95 (1999). [DOI] [PubMed] [Google Scholar]
- 78.Trabuco LG, Villa E, Mitra K, Frank J & Schulten K Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 16, 673–83 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Davis IW et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35, W375–83 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ziolkowska NE & Roll-Mecak A In vitro microtubule severing assays. Methods in molecular biology (Clifton, N.J.) 1046, 323–334 (2013). [DOI] [PubMed] [Google Scholar]
- 81.Pintard L et al. The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature 425, 311–6 (2003). [DOI] [PubMed] [Google Scholar]
- 82.Clandinin TR & Mains PE Genetic studies of mei-1 gene activity during the transition from meiosis to mitosis in Caenorhabditis elegans. Genetics 134, 199–210 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McNally KP & McNally FJ The spindle assembly function of Caenorhabditis elegans katanin does not require microtubule-severing activity. Molecular biology of the cell 22, 1550–1560 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McNally KP, Bazirgan OA & McNally FJ Two domains of p80 katanin regulate microtubule severing and spindle pole targeting by p60 katanin. Journal of cell science 113 ( Pt 9), 1623–1633 (2000). [DOI] [PubMed] [Google Scholar]
- 85.Waterhouse AM, Procter JB, Martin DM, Clamp M & Barton GJ Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–91 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.