

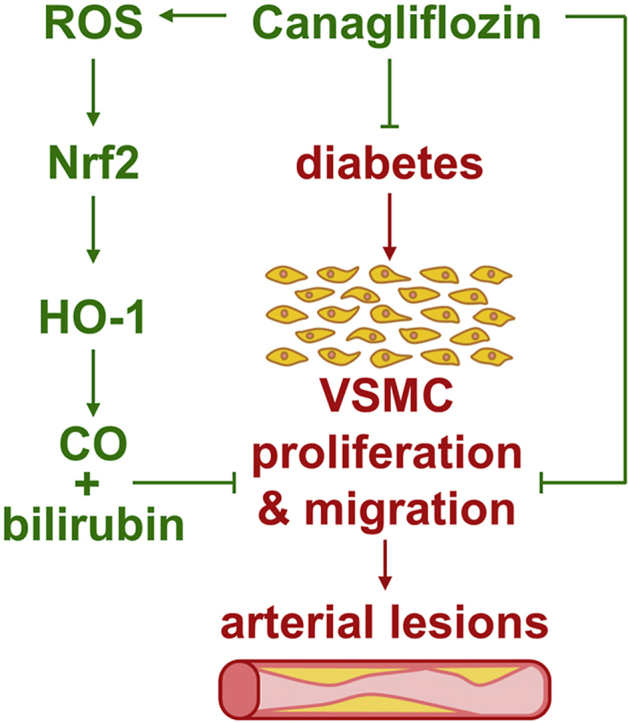
Abstract
Recent cardiovascular outcome trials found that sodium-glucose cotransporter-2 (SGLT2) inhibitors reduce cardiovascular disease and mortality in type 2 diabetic patients; however, the underlying mechanisms are not fully known. Since the proliferation and migration of vascular smooth muscle cells (SMCs) contributes to the development of arterial lesions, we hypothesized that SGLT2 inhibitors may exert their beneficial cardiovascular effects by inhibiting the growth and movement of vascular SMCs. Treatment of rat or human aortic SMCs with clinically relevant concentrations of canagliflozin, but not empagliflozin or dapagliflozin, inhibited cell proliferation and migration. The inhibition of SMC growth by canagliflozin occurred in the absence of cell death, and was associated with the arrest of SMCs in the G0/G1 phase of the cell cycle and diminished DNA synthesis. Canagliflozin also resulted in the induction of heme oxygenase-1 (HO-1) expression, and a rise in HO activity in vascular SMCs, whereas, empagliflozin or dapagliflozin had no effect on HO activity. Canagliflozin also activated the HO-1 promoter and this was abrogated by mutating the antioxidant responsive element or by overexpressing dominant-negative NF-E2-related factor-2 (Nrf2). The induction of HO-1 by canagliflozin relied on reactive oxygen species (ROS) formation and was negated by antioxidants. Finally, silencing HO-1 expression partially rescued the proliferative and migratory response of canagliflozin-treated SMCs, and this was reversed by carbon monoxide and bilirubin. In conclusion, the present study identifies canagliflozin as a novel inhibitor of vascular SMC proliferation and migration. Moreover, it demonstrates that canagliflozin stimulates the expression of HO-1 in vascular SMCs via the ROS-Nrf2 pathway, and that the induction of HO-1 contributes to the cellular actions of canagliflozin. The ability of canagliflozin to exert these pleiotropic effects may contribute to the favorable clinical actions of the drug and suggest an extra potential benefit of canagliflozin relative to other SGLT2 inhibitors.
Keywords: Canagliflozin, Smooth muscle, Proliferation, Migration, Heme oxygenase-1
Abbreviations: Heme oxygenase-1, HO-1; AdHO-1, Adenovirus expressing HO-1; AdGFP, Adenovirus expressing green fluorescent protein; CO, Carbon monoxide; SMCs, Smooth muscle cells; EDTA, Ethylenediaminetetraacetic acid; PBS, Phosphate buffered saline; Nrf2, NF-E2-related factor-2; SDS, Sodium dodecyl sulfate; SGLT2, Sodium-glucose cotransporter-2; CM-H2DCFDA, 5-(and 6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate acetyl ester; CORM2, Carbon monoxide-releasing molecule-2; Keap1, Kelch-like erythroid cell-derived protein-1
Graphical abstract
1. Introduction
Cardiovascular disease is the leading cause of morbidity and mortality in diabetes, and is a major contributor to health care costs in patients suffering from this metabolic disorder. Adults with diabetes have a two- to four-fold increased risk of cardiovascular disease relative to those without the disease, accounting for up to 80% of the premature excess mortality in diabetic patients [[1], [2], [3]]. Atherosclerotic cardiovascular disease, and its attendant clinical manifestations of coronary heart disease, ischemic heart disease, stroke, peripheral artery disease, and heart failure, is the principal cause of death in diabetes [4,5]. Compared to patients without diabetes, atherosclerosis occurs earlier in life, with greater severity, and with a more diffuse distribution in individuals with diabetes. In addition, diabetic patients display a greater incidence of restenosis following percutaneous coronary revascularization and stenting [[6], [7], [8]]. Large prospective clinical studies demonstrate a strong relationship between glycemia and diabetic microvascular disease; however, the role of hyperglycemia in the pathogenesis of macrovascular disease is less clear [[9], [10], [11]]. While numerous processes contribute to aberrant vascular remodeling in diabetes, excessive proliferation and migration of vascular smooth muscle cells (SMCs) plays a key role in the pathogenesis of arterial lesions [4,12].
Sodium-glucose cotransporter-2 (SGLT2) inhibitors are the newest approved class of oral hypoglycemic agents for patients with type 2 diabetes mellitus (T2DM) [13]. These drugs act by reducing glucose reabsorption in the proximal tubules of the kidney leading to glycosuria and decreases in both fasting and postprandial glycemia. Interestingly, large cardiovascular outcome trials such as the EMPA-REG-OUTCOME, the CANVAS Program, and the DECLARE-TIMI 58 have demonstrated that SGLT2 inhibitors (empagliflozin, canagliflozin, and dapagliflozin) reduce cardiovascular disease and/or mortality compared to other anti-hyperglycemic agents in patients with T2DM [[14], [15], [16]]. Furthermore, a meta-analysis of these three cardiovascular outcome trials found that SGLT2 inhibitors have a beneficial effect on adverse atherosclerotic cardiovascular events in patients with established atherosclerosis [17]. In addition, observational studies have confirmed that the administration of SGLT2 inhibitors is associated with a significantly decreased risk of cardiovascular mortality and major cardiovascular complications [18,19]. Moreover, experimental studies revealed that SGLT2 inhibitors reduce atherosclerotic lesions in atherosclerosis-prone mouse models and inhibit cuff-induced vascular remodeling in mice [[20], [21], [22], [23]].
Heme oxygenase-1 (HO-1) is a highly inducible antioxidant enzyme that catalyzes the degradation of heme into equimolar amounts of carbon monoxide (CO), biliverdin, and iron (see 24). Biliverdin is rapidly metabolized to bilirubin by biliverdin reductase, while iron is promptly sequestered by ferritin. HO-1 protects against the development of cardiovascular disease by dampening oxidative stress, apoptosis, platelet activation, and inflammation in arteries [[24], [25], [26]]. In addition, HO-1 is a potent inhibitor of vascular SMC proliferation and migration, and limits intimal hyperplasia following arterial injury by generating CO and bilirubin [[27], [28], [29]]. Interestingly, several vanguard drugs used to treat cardiovascular disease stimulate the expression of HO-1, and this may contribute to the clinical efficacy of these drugs [[30], [31], [32], [33]]. However, it remains to be established whether SGLT2 inhibitors regulate the expression of HO-1 in vascular SMCs.
The mechanism by which SGLT2 inhibitors confer their salutary effects on the cardiovascular system is not fully known; however, it does not involve differential improvements in glycemic control [[14], [15], [16]]. While reductions in body weight, adiposity, inflammation, blood pressure, and arterial stiffness have been implicated in bestowing the beneficial action of these drugs, direct effects of SGLT2 inhibitors on vascular SMC function have not been examined. Given the crucial role that SMCs play in promoting diabetes-associated vascular disease, the present study investigated the effect of three different SGLT2 inhibitors (canagliflozin, empagliflozin, and dapagliflozin) on vascular SMC proliferation and migration. In addition, since HO-1 directly influences SMC function, the ability of these SGLT2 inhibitors to stimulate the activity of this enzyme was explored. Moreover, the role of HO-1 in mediating the cellular actions of canagliflozin was also determined.
2. Materials and methods
2.1. Materials
Minimum essential medium, streptomycin, penicillin, gelatin, heparin, trypsin, EDTA, bovine serum, sodium dodecyl sulfate (SDS), NaOH, elastase, trichloroacetic acid, collagenase, MgCl2, phosphate-buffered saline (PBS), chloroform, CO-releasing molecule-2 (CORM2), KCl, HEPES, heme, MgCl2, glucose-6-phosphate, RNase, propidium iodide, mito-TEMPO, glycerol, Nonidet P-40, glucose-6-phosphate dehydrogenase, N-acetyl-l-cysteine (NAC), bromophenol blue, and mercaptoethanol were from Sigma-Aldrich (St. Louis, MO). Inactivated CORM2 (iCORM2) was generated by leaving CORM2 solutions at room temperature for two days and then purging solutions of any residual CO with nitrogen. Phenylmethylsulfonyl fluoride (PMSF), aprotinin, leupeptin, and pepstatin A were from Roche Applied Sciences (Indianapolis, IN). Lipofectamine and 5-(and 6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA) were from Life Technologies (Carlsbad, CA). Bilirubin was from Frontier Scientific (Logan, UT). The antibodies against NF-E2-related factor-2 (Nrf2) and β-actin were from Santa Cruz Biotechnology (Santa Cruz, CA) and the antibody against HO-1 was from Assay Designs (Ann Arbor, MI). [3H]Thymidine (20 Ci/mmol) was from PerkinElmer (Boston, MA). Canagliflozin, empagliflozin, and dapagliflozin were purchased from Selleck Chemicals (Houston, TX).
2.2. Cell culture
Rat aortic SMCs were isolated and characterized by morphological and immunological criteria, and serially cultured in minimum essential medium containing 10% bovine calf serum, Earle's salts, 2 mM l-glutamine, 100 U/ml of penicillin, and 100U/ml streptomycin as we previously described [34]. Human aortic SMCs were purchased from Cell Biologics (Chicago, IL) and grown in SMC basal medium with supplements containing insulin, epidermal growth factor, fibroblast growth factor, hydrocortisone, penicillin, streptomycin, and amphotericin B with 10% fetal bovine serum (Cell Biologics, Chicago, IL). Cells were propagated at 37 °C in a humidified atmosphere of 95% air and 5% CO2. For the proliferation and cell cycle experiments, SMC quiescence was induced by placing rat aortic SMCs on serum-free medium containing bovine serum albumin (1%) and human aortic SMCs on basal medium without growth supplements for 48 h before treatment.
2.3. Cell proliferation and DNA synthesis
Vascular SMCs were seeded (2-4 x 104 cells/well) onto 12-well plates in serum-containing media. Following quiescence, cells were treated with serum replete media in the absence or presence of SGLT2 inhibitors. During the treatment period, the media were replaced every 48 h with fresh serum-replete media containing the indicated concentrations of SGLT2 inhibitors. Cell number determinations were made at various times by dissociating cells with trypsin (0.05%):EDTA (0.53 mM) and counting cells using an automated cell counter (Moxi Z ORFLO Technologies, Ketchum, ID). As another index of cell proliferation, vascular SMC DNA synthesis was monitored. In brief, cells were pulsed with [3H]thymidine (1 μCi/mL [0.037 MBq]) for 4 h. Cells were then washed with ice-cold PBS, fixed with 10% trichloroacetic acid for 30 min at 4 °C, and DNA extracted with 0.2% SDS/0.2 N NaOH. Radioactivity was determined by scintillation counting (Tricarb liquid scintillation analyzer, model 2100, Packard, Meriden, CT).
2.4. Cell cycle analysis
Cell cycle progression was determined in vascular SMCs grown to 80% confluence by flow-activated cell sorting, as previously reported [35]. Following quiescence, cells were treated with serum-replete media in the absence and presence of SGLT2 inhibitors for 24 h. Cells were then collected, washed with PBS, and incubated overnight in 70% ethanol at 4 °C. Fixed cells were then treated with propidium iodide (50 μg/ml) and RNase A (100 μg/ml) for 60 min at room temperature, and DNA fluorescence monitored in a Beckman Coulter CyAN ADP Cytometer (Brea, CA). Flow cytometry data were evaluated using FlowJo™ software (FlowJo LLC, Ashland, OR).
2.5. Cell migration
Vascular SMC migration was assessed using a scratch wound assay [36]. Confluent monolayers of SMCs were scraped with a sterile pipette tip to generate a wound, cell debris cleared by several washes with PBS, and injured monolayers incubated in the absence or presence of SGLT2 inhibitors. Cell monolayers were photographed immediately and 20 h after scratch injury with a digital camera (Q-Imaging, QICAM; Hitschfel Instruments, Incorporated, St. Louis, MO), and the extent of wound closure determined by planimetry.
2.6. Cell viability
Vascular SMC viability was assessed by measuring lactate dehydrogenase activity in the culture media using the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega Life Sciences, Madison, WI). In brief, cells were seeded onto 12-well plates and incubated in the absence or presence of SGLT2 inhibitors. Media were then collected, centrifuged, and an aliquot of the supernatant transferred to a 96-well plate containing an equal amount of CytoTox 96 reagent. After 30 min of incubation at room temperature, the reaction was terminated by the addition of acetic acid (1 M) and absorbance at 490 nm measured by spectroscopy (μQuant spectrophotometer, Bio-Tek Instruments, Winooski, VT). Lactate dehydrogenase activity was reported as a percentage of control cells.
2.7. Western blotting
Vascular SMCs were harvested in electrophoresis buffer (125 mM Tris, pH 6.8, 12.5% glycerol, 2% SDS, and trace bromophenol blue), and proteins resolved by SDS-polyacrylamide gel electrophoresis. Following electrophoretic transfer to nitrocellulose, membranes were blocked with PBS and nonfat milk (5%) and then incubated overnight at 4 °C with primary antibodies against HO-1 (1:1500), Nrf2 (1:200) or β-actin (1:2000). Membranes were then washed extensively in PBS, incubated with horseradish peroxidase-labeled secondary antibodies for 60 min at room temperature, washed, and incubated with enhanced chemoluminescence substrate for horseradish peroxidase (GE Healthcare, Chicago, IL). Protein expression was quantified by densitometry and normalized with respect to β-actin [37].
2.8. Quantitative real-time PCR
Total RNA was extracted from SMCs with Trizol reagent (Thermo Fisher Scientific, Waltham, MA) and reverse transcribed to cDNA using a transcription kit and random hexamer primers (Thermo Fisher Scientific, Waltham, MA). Real-time PCR was performed with SYBR Green Supermix in a SYBER Green Cycler iQ 5RT-PCR detection system (Bio-Rad Laboratories, Hercules, CA). The primer sequences were: HO-1, forward CGTGCAGAGAGAATTCTGAGTTC and reverse AGACGCTTTACGTAGTGCTG and β-actin, forward CCTGTATGCCTCTGGTCGTA and reverse CCATCTCTTGCTCGAAGTCT. Transcript levels were quantified using the 2-ΔΔCT method and normalized to β-actin, as we previously described [38].
2.9. ROS measurement
The levels of intracellular ROS were measured by fluorimetry using the cell-permeable probe CM-H2DCFDA, as we previously described [39,40]. SMCs were washed with PBS three times and subsequently incubated with the dye (10 μM) for 30 min at 37 °C. Following washing of cells with PBS, fluorescence was quantified by microplate fluorimetry with excitation at 490 nm and emission at 530 nm. Mean values from each treatment were expressed relative to control cells.
2.10. HO activity
HO activity was measured by absorption spectroscopy. In brief, SMCs were sonicated in MgCl2 (2 mM) and phosphate (100 mM) buffer (pH 7.4) and centrifuged at 18,000 g at 4 °C for 15 min. Supernatants were collected and included in a reaction mixture consisting of NADPH (0.8 mM), glucose-6-phosphate (2 mM), glucose-6-phosphate dehydrogenase (0.2U), rat liver cytosol (2 mg), and the substrate heme (20 μM). Following a 1 h incubation, the reaction was terminated by the addition of chloroform. The extracted bilirubin was calculated by the difference in absorption between 464 and 530 nm with an extinction coefficient of 40mM−1cm−1 [38].
2.11. Luciferase reporter activity
HO-1 promoter activity was determined using luciferase constructs that included a wild-type enhancer (E1) consisting of three antioxidant responsive elements (ARE) linked to a minimum HO-1 promoter and a mutant enhancer (M739) that has mutations in its three ARE sequences [41]. To determine luciferase activity, cells were transfected with the wild-type or mutant enhancer using lipofectamine for 48 h and then exposed to canagliflozin for 8 h. Firefly luciferase activity was quantified using a Glomax luminometer (Promega, Madison, WI) and normalized for transfection efficiency by co-transfecting cells with a plasmid encoding Renilla luciferase.
2.12. Nrf2 activity
Vascular SMCs were harvested in lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.5 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 10 μg/ml aprotonin, 0.5 mM dithiothreitol, and 0.4% Nonidet P-40) and then centrifuged at 14,000×g for 3 min. Nuclear pellets were suspended in extraction buffer (20 mM HEPES, pH 7.9, 0.4 mM NaCl, 1.0 mM EDTA, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 10 μg/ml aprotonin, and 10% glycerol) and then spun at 14,000×g for 5 min. The supernatant was collected and Nrf2 activity determined by quantifying the binding of Nrf2 to the ARE using an ELISA-based assay (Active Motif, Carlsbad, CA). Bound protein was detected using a primary antibody specific to DNA-bound Nrf2 and a secondary horseradish peroxidase-conjugated antibody that generated a colorimetric reaction that was monitored by absorbance spectroscopy at 405 nm [38].
2.13. Gene silencing
Gene expression was silenced using siRNA targeting HO-1. The experimental and control, non-targeting siRNAs were obtained from Dharmacon (Lafayette, CO) and delivered to vascular SMCs (100 nM) using lipofectamine, as previously described [38].
2.14. Statistical analysis
Results are expressed as mean ± SEM. Statistical analyses were performed with the use of a Student's two tailed t-test and one way analysis of variance with the Holm-Sidak post-hoc test when more than two treatment regimens were compared. A value of P less than 0.05 was considered statistically significant.
3. Results
Treatment of rat aortic SMCs with canagliflozin resulted in a potent, concentration-dependent decrease in cell proliferation (Fig. 1A). The ability of canagliflozin to inhibit SMC proliferation was observed at a concentration (10 μM) that is attained in the plasma of diabetic patients taking canagliflozin [42]. A significant inhibition of SMC growth by canagliflozin was detected following one day of treatment and the anti-proliferative action of the drug was further amplified after four days of exposure (Fig. 1B). In contrast, clinically relevant concentrations of empagliflozin and dapagliflozin (1–2 μM) [43,44] failed to inhibit SMC proliferation (Fig. 1C and D). However, extremely high concentrations (30–50 μM) of empagliflozin and dapagliflozin moderately attenuated SMC growth. Inhibition of cell growth by these three SGLT2 inhibitors was independent of any change in cell viability, as monitored by lactate dehydrogenase activity measurements (Fig. 2). Furthermore, pharmacologically relevant concentrations of canagliflozin also inhibited the proliferation of human aortic SMCs, without affecting cell viability (Fig. 3A and B).
Fig. 1.

Effect of SGLT2 inhibitors on the proliferation of rat aortic SMCs. (A) Canagliflozin inhibits the proliferation of SMCs in a concentration-dependent manner. Cells were treated with canagliflozin (0–50 μM) for three days. (B) Canagliflozin inhibits the proliferation of SMCs in a time-dependent manner. Cells were treated with canagliflozin (50 μM) for one to four days. (C) High concentrations of empagliflozin inhibits the proliferation of SMCs. Cells were treated with empagliflozin (0–50 μM) for three days. D) High concentrations of dapagliflozin inhibits the proliferation of SMCs. Cells were treated with dapagliflozin (0–50 μM) for three days. Results are mean ± SEM (n = 6). *Statistically significant effect of SGLT2 inhibitors.
Fig. 2.

Effect of SGLT2 inhibitors on the viability of rat aortic SMCs. (A–C) Canagliflozin, empagliflozin, or dapagliflozin do not alter cell viability. Cells were incubated in the absence or presence of SGLT2 inhibitors (0–50 μM) for three days and cell viability determined by lactate dehydrogenase activity measurement. Results are mean ± SEM (n = 6).
Fig. 3.

Effect of canagliflozin on the proliferation and viability of human aortic SMCs. (A) Canagliflozin inhibits cell proliferation in a concentration-dependent manner. (B) Canagliflozin does not alter cell viability as determined by lactate dehydrogenase measurements. Cells were incubated in the absence or presence of canagliflozin (0–50 μM) for three days. Results are mean ± SEM (n = 6). *Statistically significant effect of canagliflozin.
Subsequently, the effect of canagliflozin on cell cycle progression was assessed. Administration of canagliflozin arrested rat aortic SMCs in the G0/G1 phase of the cell cycle, as illustrated by an increase in the percentage of cells in G0/G1 with a resultant decrease in the fraction of cells in S phase (Fig. 4A and B). Consistent with a decrease in the entry of cells into S phase, canagliflozin markedly repressed SMC DNA synthesis (Fig. 4C). We also investigated the effect of canagliflozin on SMC migration following scrape injury. While canagliflozin blocked the migration of rat aortic SMCs in a concentration-dependent fashion (Fig. 5A), neither empagliflozin nor dapagliflozin had any effect on vascular SMC motility (Fig. 5B and C). A similar anti-migratory action of canagliflozin was observed in human aortic SMCs (Fig. 5D).
Fig. 4.

Canagliflozin inhibits cell cycle progression and DNA synthesis in rat aortic SMCs. (A) Representative histograms of SMCs treated for 24 h in the absence or presence of canagliflozin (50 μM). (B) Effect of canagliflozin (50 μM) treatment for 24 h on the distribution of SMCs in the cell cycle. (C) Effect of canagliflozin treatment for 24 h on SMC DNA synthesis. Results are means ± SEM (n = 6). *Statistically significant effect of canagliflozin.
Fig. 5.

Effect of SGLT2 inhibitors on the migration of vascular SMCs. (A) Canagliflozin inhibits the migration of rat aortic SMCs in a concentration-dependent manner. (B) Empagliflozin has no effect on the migration of rat aortic SMCs. (C) Dapagliflozin has no effect on the migration of rat aortic SMCs. D). Canagliflozin inhibits the migration of human aortic SMCs. Cells were incubated in the absence or presence of SGLT2 inhibitors (0–50 μM) for 20 h. Results are mean ± SEM (n = 6). *Statistically significant effect of canagliflozin.
Since HO-1 is an established regulator of vascular SMC function [[27], [28], [29]], the effect of canagliflozin on the expression of this enzyme was examined. Treatment of rat aortic SMCs with canagliflozin for 24 h evoked a concentration-dependent increase in HO-1 protein (Fig. 6A). Canagliflozin also stimulated a time-dependent increase in HO-1 mRNA that peaked after 8 h of drug exposure (Fig. 6B). A significant rise in HO activity was also noted following canagliflozin treatment (Fig. 6C). In contrast, empagliflozin and dapagliflozin failed to stimulate HO activity (Fig. 6C). Canagliflozin also induced the expression of HO-1 in human aortic SMCs (Fig. 6D).
Fig. 6.

Canagliflozin (Cana) stimulates HO-1 expression in vascular SMCs. (A) Cana (0–50 μM) exposure for 24 h stimulates a concentration-dependent increase in HO-1 protein in rat aortic SMCs. (B) Cana (50 μM) stimulates a time-dependent increase in HO-1 mRNA expression in rat aortic SMCs. (C) Cana (50 μM), but not dapagliflozin (Dapa; 50 μM) or empagliflozin (Empa; 50 μM), exposure for 24 h increases HO activity in rat aortic SMCs. (D) Cana (50 μM) exposure for 24 h stimulates HO-1 protein expression in human aortic SMCs. Results are mean ± SEM (n = 3–6). *Statistically significant effect of Cana.
The ability of canagliflozin to induce HO-1 expression was mitigated by the transcriptional inhibitor actinomycin D, suggesting that de novo DNA synthesis is required for this response (Fig. 7A). Canagliflozin also activated the HO-1 promoter and this was negated by mutating the ARE sequences in the promoter (Fig. 7B). Modification of the ARE sequences also markedly reduced baseline promoter activity. Since previous work from our laboratory determined that Nrf2 plays a critical role in HO-1 gene transcription [38,40,41], the role of this transcription factor was analyzed. Indeed, transfection of vascular SMCs with a dominant-negative mutant of Nrf2 that has its activation domain deleted diminished basal and canagliflozin-mediated increases in HO-1 promoter activity (Fig. 7B). Furthermore, canagliflozin stimulated a time-dependent increase in Nrf2 protein expression as well as a concentration-dependent rise in Nrf2 activity (Fig. 7C and D).
Fig. 7.

Canagliflozin (Cana) stimulates HO-1 promoter activity via the Nrf2/ARE complex in vascular SMCs. (A) Cana-mediated HO-1 gene expression is dependent on de novo RNA synthesis. Rat aortic SMCs were treated with actinomycin D (ActD; 1 μg/ml) for 8 h in the absence or presence of Cana (50 μM). (B) Cana stimulates HO-1 promoter activity. Cells were transfected with a HO-1 promoter construct (E1) or a mutated HO-1 promoter construct (M739) and a Renilla luciferase construct, treated with Cana (50 μM) for 8 h, and then analyzed for luciferase activity. In some cases, a dominant-negative Nrf2 (dnNrf2) construct was co-transfected into cells. (C) Cana (50 μM) stimulates Nrf2 protein expression in a time-dependent manner. (D) Cana stimulates Nrf2 activity in a concentration-dependent manner. O. D.; optical density. Results are mean ± SEM (n = 3–5). *Statistically significant effect of Cana.
In follow-up experiments, the upstream signaling pathway responsible for the induction of HO-1 by canagliflozin was determined. Since oxidative stress is a potent inducer of HO-1 [[24], [25], [26]], the involvement of ROS was examined. Incubation of vascular SMCs with canagliflozin evoked a marked increase in intracellular ROS production that was not observed with either empagliflozin or dapagliflozin (Fig. 8A). However, treatment of SMCs with NAC or rotenone abolished the increase in ROS by canagliflozin (Fig. 8A). They also prevented the canagliflozin-mediated elevation in HO-1 protein and Nrf2 activity (Fig. 8C, D, and E). Similarly, incubation of SMCs with mito-TEMPO blocked the induction of HO-1 by canagliflozin (Fig. 8F)
Fig. 8.

Canagliflozin (Cana)-induced HO-1 protein expression is dependent on oxidative stress. (A) Cana (50 μM), but not empagliflozin (Empa; 50 μM) or dapagliflozin (Dapa; 50 μM), exposure for 24 h stimulates ROS production. (B) N-acetyl-l-cysteine (NAC; 10 mM) or rotenone (Rot; 2 μM) inhibits Cana (50 μM for 24 h)-mediated ROS production. (C) NAC (10 mM) inhibits Cana (50 μM for 8 h)-mediated Nrf2 activation. O.D.; optical density. (D) NAC (10 mM) inhibits Cana (50 μM for 24 h)-mediated HO-1 protein expression. (E) Rot (2 μM) inhibits Cana (50 μM for 24 h)-mediated HO-1 protein expression. (F) Mito-TEMPO (MT; 100 μM) inhibits Cana (50 μM for 24 h)-mediated HO-1 protein expression Results are mean ± SEM (n = 3–6). *Statistically significant effect of Cana.
Finally, the functional significance of the induction of HO-1 by canagliflozin was investigated by silencing HO-1 expression. Transfection of rat aortic SMCs with HO-1 siRNA blocked the expression of HO-1 by canagliflozin and partially rescued the proliferation and migration of canagliflozin-treated SMCs, whereas NT siRNA failed to modify the response of vascular SMCs to canagliflozin (Fig. 9). In the absence of canagliflozin, HO-1 or NT siRNA minimally affected SMC function. In addition, the exogenous administration of the CO donor (CORM2) or bilirubin could substitute for the loss of HO-1 and fully restore the negative effects of canagliflozin on SMC proliferation and migration. In contrast, the inactive CO donor (iCORM2) failed to restore the actions of canagliflozin.
Fig. 9.

HO-1 contributes to the anti-proliferative and anti-migratory actions of canagliflozin (Cana). (A) HO-1 expression in vascular SMCs transfected with HO-1 (100 nM) or NT (100 nM) siRNA and then exposed to Cana (50 μM) for 24 h. (B–C) HO-1 silencing reduces Cana-mediated inhibition of SMC DNA synthesis and migration. Cells were transfected with HO-1 (100 nM) or NT (100 nM) siRNA and then exposed to Cana (50 μM) in the absence or presence of bilirubin (BR; 20 μM), CORM2 (CM2; 20 μM), or iCORM2 (iCM2; 20 μM). Results are mean ± SEM (n = 3–6). *Statistically significant effect of Cana. +Statistically significant effect of HO-1 siRNA.
4. Discussion
The present study identifies canagliflozin as a novel inhibitor of vascular SMC proliferation and migration. These actions appear to be unique to canagliflozin as pharmacologically relevant concentrations of other SGLT2 inhibitors, including empagliflozin or dapagliflozin, minimally affect SMC function. The anti-proliferative and anti-migratory actions of canagliflozin are observed in vascular SMCs derived from both rat and human arteries. The study also found that canagliflozin induces HO-1 expression and HO activity in vascular SMCs, whereas empagliflozin or dapagliflozin had no effect on HO activity. The induction of HO-1 by canagliflozin requires ROS and is mediated by Nrf2. Significantly, the study shows that the induction of HO-1 contributes to the anti-proliferative and anti-migratory effects of canagliflozin by generating CO and/or bilirubin. The ability of canagliflozin to elicit these pleiotropic actions on SMCs may contribute to the reduced risk of cardiovascular complications and mortality noted in diabetic patients taking this drug, and suggest a potential additional benefit of canagliflozin compared to other SGLT2 inhibitors.
To our knowledge, this is the first study to demonstrate that canagliflozin blocks vascular SMC proliferation. The anti-proliferative action of canagliflozin is concentration-dependent and, importantly, is seen at concentrations (10 μM) that are attained in the plasma of patients treated with this SGLT2 inhibitor [42]. The inhibition of vascular SMC proliferation by canagliflozin is not due to cell death as disclosed by lactate dehydrogenase activity analysis and the absence of a sub G0/G1 fraction in the cell cycle histograms. Thus, canagliflozin functions in a cytostatic rather than cytotoxic fashion. The anti-proliferative action of canagliflozin was detected in both rodent and human vascular SMCs, indicating that the drug retards SMC growth across animal species. The inhibition of SMC growth occurs rapidly and is first detected after one day of canagliflozin treatment and the decrease in cell number is further magnified after four days of canagliflozin exposure. Finally, cell cycle analysis revealed that canagliflozin arrests SMCs in the G0/G1 phase of the cell cycle and blocks entry of SMCs into S phase, where DNA synthesis occurs. In agreement with these cell cycle findings, canagliflozin markedly attenuates SMC DNA synthesis.
The finding that canagliflozin inhibits SMC replication is in accord with previous work showing that canagliflozin blocks the proliferation of renal proximal tubule epithelial cells, endothelial cells, and various tumor cells [[45], [46], [47], [48], [49]]. The anti-proliferative action of canagliflozin has also been verified in vivo in murine cancer models [47,49,50]. The blockade of SMC growth by canagliflozin most likely signifies a compound-specific effect rather than a class effect since pharmacologically relevant concentrations of other SGLT2 inhibitors including empagliflozin and dapagliflozin (1–2 μM) [43,44] fails to inhibit SMC proliferation.
We also found that canagliflozin blocks the migration of vascular SMCs. The inhibition of SMC motility by canagliflizon is observed in both human and rodent cells, occurs in a concentration-dependent fashion, and is detected at a clinically relevant concentration (10 μM). However, the ability of canagliflozin to block SMC movement (⁓20% inhibition) is not as severe as its anti-proliferative effect (⁓80% inhibition), indicating that the drug preferentially targets SMC growth. Notably, the anti-migratory effect of canagliflozin is not detected with either empagliflozin or dapagliflozin, suggesting another pleiotropic action of canagliflozin on SMCs. Aside from directly retarding vascular SMC migration, canagliflozin may also indirectly influence SMC movement by inhibiting the secretion of chemotactic factors from perivascular tissue [23]. Interestingly, clinically achievable concentrations of canagliflozin, but not empagliflozin or dapagliflozin, block inflammatory responses in endothelial cells, indicating that off-target effects of canagliflozin extend beyond vascular SMCs [51]. The ability of canagliflozin to directly suppress SMC proliferation and migration, as well as endothelial inflammation may contribute to the beneficial cardiovascular actions of the drug by hindering the development and progression of atherosclerosis in patients with T2DM [4,5]. In addition, by blocking SMC growth and motility, canagliflozin may reduce the elevated risk of postangioplasty restenosis in the diabetic population [[6], [7], [8]]. Moreover, by maintaining vascular SMCs in a non-proliferative and non-motile state, canagliflozin may also favorably impact the development of vascular disease in non-diabetic patients.
The study also identified the enzyme HO-1 as a novel target of canagliflozin. Treatment of rat or human vascular SMCs with canagliflozin stimulates a concentration- and time-dependent increase in HO-1 expression, and a marked increase in HO activity that is not seen with either empagliflozin or dapagliflozin. The induction of HO-1 by canagliflozin is dependent on de novo RNA synthesis and likely involves the transcriptional activation of the gene as the drug directly stimulates HO-1 promoter activity. The stimulation of HO-1 gene transcription requires the presence of AREs since mutation of this element eliminates the activation of the reporter by canagliflozin. Interestingly, we found that canagliflozin increases Nrf2 protein levels and nuclear Nrf2 binding to the ARE. Furthermore, transfection of SMCs with a dominant-negative mutant of Nrf2 obviates the activation of the HO-1 promoter by canagliflozin. Thus, the activation of Nrf2 plays a necessary role in mediating HO-1 gene transcription in vascular SMCs.
The capacity of canagliflozin to stimulate HO-1 expression is dependent on oxidative stress. Incubation of SMCs with canagliflozin leads to a rise in ROS production that is not detected with empagliflozin or dapagliflozin. In addition, the antioxidant NAC inhibits the canagliflozin-mediated formation of ROS, the induction of HO-1, and the activation of Nrf2. Furthermore, it appears that ROS generation by the mitochondria is responsible for the induction of HO-1 by canagliflozin as the mitochondrial electron transport chain complex I inhibitor, rotenone, blocks both ROS formation and HO-1 expression. Moreover, the induction of HO-1 is suppressed by the mitochondrial-targeted antioxidant, mito-TEMPO. These findings are consistent with a recent study documenting the stimulation of ROS production by canagliflozin in renal proximal tubule epithelial cells secondary to the inhibition of mitochondrial complex I activity [46]. While the molecular mechanism that underlies the activation of Nrf2 by canagliflozin is not precisely known, it likely involves the oxidation of specific cysteine residues in Kelch-like erythroid cell-derived protein-1 (Keap1) that results in the release of Nrf2 from Keap1 and/or the inhibition of Keap1-dependent degradation of Nrf2 [52].
Our study adds to an expanding number of off-targets effects elicited by canagliflozin. Besides stimulating HO activity, canagliflozin has been reported to activate 5′-adenosine monophosphate-activated protein (AMP) kinase and block mitochondrial glutamate dehydrogenase and mitochondrial complex I activity [45,46,53]. In addition, clinically relevant concentrations of canagliflozin have been shown to inhibit the activity of uridine 5′-diphospho-glucuronosyltransferase and the sodium-hydrogen exchanger [54,55]. Collectively, these studies highlight the many non-specific actions of canagliflozin and the need to consider these pleiotropic effects when evaluating the clinical profile of the drug.
Significantly, the induction of HO-1 contributes to the anti-proliferative and anti-migratory actions of canagliflozin. Silencing HO-1 expression increases the proliferation and migration of canagliflozin-treated vascular SMCs. These results are in-line with previous work showing that HO-1 blocks the growth and motility of SMCs [[27], [28], [29]]. We also found that the exogenous administration of CO and bilirubin can substitute for HO-1 and fully restore the anti-proliferative and anti-migratory response of canagliflozin-challenged SMCs. These latter findings are in agreement with earlier studies demonstrating that both of these HO-1 products exert negative effects on SMC function [29,35,56,57]. Notably, silencing HO-1 expression does not fully restore SMC function, suggesting that other factors may also participate in driving the anti-proliferative and anti-migratory actions of canagliflozin. It is unlikely that inhibition of SGLT2 activity mediates the effects of canagliflozin on vascular SMCs since the transporter is not expressed in these cells [58]. Moreover, if canagliflozin inhibits SMC function via the inhibition of this transporter, similar effects would have been obtained with pharmacologically relevant concentrations of empagliflozin and dapagliflozin. However, it is possible that the activation of Nrf2 by canagliflozin leads to the induction of other antioxidant enzymes that block SMC function [59,60]. Intriguingly, a recent report found that clinically relevant concentrations of canagliflozin, but not empagliflozin or dapagliflozin, activates AMP kinase [53], which we previously identified as a potent inhibitor of vascular SMC proliferation and migration [36]. This provides for an alternative mechanism by which canagliflozin attenuates SMC function. In addition, canagliflozin may repress SMC growth and movement by blocking glutamine-dependent anaplerosis through the tricarboxylic acid cycle [46,61]. Thus, canagliflozin may curb SMC proliferation and migration via several distinct mechanisms.
The induction of HO-1 by canagliflozin may elicit many important effects in the cardiovascular system that contributes to the favorable pharmacological profile of the drug in T2DM. Aside from limiting arterial lesion formation by maintaining vascular SMCs in a quiescent state, HO-1 has been demonstrated to correct endothelial dysfunction in diabetes [62,63]. In addition, HO-1 improves oxidative stress and inflammation in blood vessels, and reduces hypertension in various animal models [26,64]. Moreover, HO-1 exerts numerous positive metabolic actions in the setting of T2DM: it increases insulin sensitivity, improves glucose tolerance, increases adiponectin levels, and reduces adiposity [65,66]. HO-1 and its products may also protect against several complications of diabetes, including diabetic nephropathy, cardiomyopathy, and retinopathy [[67], [68], [69]].
While our cell-based study provides important new cellular and mechanistic insight regarding the actions of canagliflozin in the vasculature, it does not fully replicate the intricate biochemical and biophysical interactions that occur in vivo. Going forward, it will be important to extend this work using animal models of arterial injury and repair. In addition, studies employing HO-1-deficient mice will be critical in establishing a role for this enzyme in mediating the vascular actions of canagliflozin. Moreover, the mechanism by which canagliflozin blocks vascular SMC proliferation and migration is not completely known. While HO-1 contributes to the anti-proliferative and anti-migratory effects of canagliflozin, other mediators are also involved and need to be identified. Finally, this work should be expanded to examine the effect of other SGLT2 inhibitors on vascular cell function and the action of canagliflozin on SMCs derived from the venous circulation and different tissues.
In conclusion, the present study identified canagliflozin as a novel inhibitor of vascular SMC proliferation and migration. The anti-proliferative and anti-migratory actions of canagliflozin are observed at clinically relevant concentrations in SMCs derived from both the rat and human arterial circulation. In addition, the study found that canagliflozin stimulates the expression of HO-1 in vascular SMCs via the activation of the ROS-Nrf2 pathway. Notably, all these actions are unique for canagliflozin and not seen with other SGLT2 inhibitors. This study also found that HO-1 contributes to the anti-proliferative and anti-migratory effects of the canagliflozin. The ability of canagliflozin to exert these pleiotropic effects on SMCs may contribute to the beneficial cardiovascular actions of the drug in patients with T2DM, and suggest an extra potential advantage of canagliflozin therapy relative to other SGLT2 inhibitors.
Declaration of competing interest
None.
Acknowledgement
This work was supported by the American Diabetes Association grant #1-17-IBS-290.
References
- 1.Gu K., Crowe C.C., Harris M.I. Mortality in adults with and without diabetes in a national cohort of the U.S. population, 1971-1993. Diabetes Care. 1998;21:1138–1145. doi: 10.2337/diacare.21.7.1138. [DOI] [PubMed] [Google Scholar]
- 2.Winer N., Sowers J.R. Epidemiology of diabetes. J. Clin. Pharmacol. 2004;44:397–405. doi: 10.1177/0091270004263017. [DOI] [PubMed] [Google Scholar]
- 3.Taylor K.S., Heneghan C.J., Farmer A.J., Fuller A.M., Adler Ai, Aronson J.K., Stevens R.J. All-cause and cardiovascular mortality in middle-aged people with type 2 diabetes compared with people without diabetes in a large U.K. primary care database. Diabetes Care. 2013;36:2366–2371. doi: 10.2337/dc12-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beckman J.A., Creager M.A., Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. J. Am. Med. Assoc. 2002;287:2570–2581. doi: 10.1001/jama.287.19.2570. [DOI] [PubMed] [Google Scholar]
- 5.Wang C.C.L., Hess C.N., Hiatt W.R., Goldfine A.B. Clinical update: cardiovascular disease in diabetes mellitus. Atherosclerotic cardiovascular disease and Heart Failure in Type 2 diabetes mellitus – mechanisms, management, and clinical considerations. Circulation. 2016;133:2459–2502. doi: 10.1161/CIRCULATIONAHA.116.022194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kip K.E., Faxon D.P., Detre K.M., Yeh W., Kelsey S.F., Currier J.W. Coronary angioplasty in diabetic patients. The national heart, lung, and blood institute percutaneous transluminal coronary angioplasty registry. Circulation. 1996;94:1818–1825. doi: 10.1161/01.cir.94.8.1818. [DOI] [PubMed] [Google Scholar]
- 7.West N.E.J., Ruygrok P.N., Disco C.M.C., Webster M.W.I., Lindeboom W.K., O'Neill W.W., Mercado N.F., Serruys P.W. Clinical and angiographic predictors of restenosis after stent deployment in diabetic patients. Circulation. 2004;109:867–873. doi: 10.1161/01.CIR.0000116750.63158.94. [DOI] [PubMed] [Google Scholar]
- 8.Lu C.H., Tsai M.L., Chen C.C., Hsieh M.J., Chang S.H., Wang C.Y., Lee C.H., Chen D.Y., Yang C.H., Hsieh I.C. Comparison of very long-term clinical and angiographic outcomes of bare metal stent implants between patients with and without type 2 diabetes. Prim Care Diabetes. 2017;11:445–452. doi: 10.1016/j.pcd.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 9.Diabetes Control and Complications Trial Research Group The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 10.UK Prospective Diabetes Study (UKPDS) Group Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- 11.Duckworth W., Abraira C., Moritz T., Reda D., Emanuele N., Reaven P.D., Zieve F.J., Marks J., Davis S.N., Hayward R., Warren S.R., Goldman S., McCarren M., Vitek M.E., Henderson W.G., Huang G.D., Investigators V.A.D.T. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 2009;360:129–139. doi: 10.1056/NEJMoa0808431. [DOI] [PubMed] [Google Scholar]
- 12.Creager M.A., Luscher T.F., Cosentino F., Beckman J.A. Diabetes and vascular Disease. Pathophysiology, clinical consequences, and medical therapy: Part I. Circulation. 2003;108:1527–1532. doi: 10.1161/01.CIR.0000091257.27563.32. [DOI] [PubMed] [Google Scholar]
- 13.Kalra S. Sodium glucose co-transporter 2 (SGLT2) inhibitors: a review of their basic and clinical pharmacology. Diabetes Ther. 2014;5:355–366. doi: 10.1007/s13300-014-0089-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zinman B., Wanner C., Lachin J.M., Fitchett D., Bluhmki E., Hantel S., Mattheus M., Devins T., Johansen O.E., Woerle H.J., Broedl U.C., Inzucchi S.E., EMPA-REG Outcome Investigators Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015;373:2117–2128. doi: 10.1056/NEJMoa1504720. [DOI] [PubMed] [Google Scholar]
- 15.Neal B., Perkovic V., Mahaffey K.W., de Zeeuw D., Fulcher G., Erondu N., Shaw W., Law G., Desai M., Matthews D.R., Canvas Program Collaborative Group Canagliflozin and cardiovascular and renal events in type 2 diabetes. N. Engl. J. Med. 2017;377:644–657. doi: 10.1056/NEJMoa1611925. [DOI] [PubMed] [Google Scholar]
- 16.Wiviott S.D., Raz I., Bonaca M.P., Mosenzon O., Kato E.T., Cahn A., Silverman M.G., Zelnicker T.A., Kuder J.F., Murphy S.A., Bhatt D.L., Leiter L.A., McGuire D.K., Wilding J.P.H., Ruff C.T., Gause-Nilsson I.A.M., Fredriksson M., Johansson P.A., Langkilde A.M., Sabatine M.S., for the Declare-TIMI 58 Investigators Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2019;380:347–357. doi: 10.1056/NEJMoa1812389. [DOI] [PubMed] [Google Scholar]
- 17.Zelnicker T.A., Wiviott S.D., Raz I., Im K., Goodrich E.L., Bonaca M.P., Mosenzon O., Kato E.T., Cahn A., Furtado R.H.M., Bhatt D.L., Leiter L.A., McGuire D.K., Wilding J.P.H., Sabatine M.S. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systemic review and meta-analysis of cardiovascular outcome trials. Lancet. 2019;393:31–39. doi: 10.1016/S0140-6736(18)32590-X. [DOI] [PubMed] [Google Scholar]
- 18.Birkeland K.I., Jørgensen M.E., Carstensen B., Persson F., Gulseth H.L., Thuresson M., Fenici P., Nathanson D., Nyström T., Eriksson J.W., Bodegård J., Norhammar A. Cardiovascular mortality and morbidity in patients with type 2 diabetes following initiation of sodium-glucose co-transporter-2 inhibitors versus other glucose-lowering drugs (CV-REAL-NORDIC): a multinational observational study. Lancet Diabetes Endocrinol. 2017;5:709–717. doi: 10.1016/S2213-8587(17)30258-9. [DOI] [PubMed] [Google Scholar]
- 19.Kosiborod M., Cavender M.A., Fu A.Z., Wilding J.P., Khunti K., Holl R.W., Norhammar A., Birkeland K.I., Jørgensen M.E., Thuresson M., Arya N., Bodegård J., Hammar N., Fenici P., CVD-Real Investigators and Study Group Lower risk of heart failure and death in patients initiated on sodium-glucose cotransporter-2 inhibitors versus other glucose-lowering drugs: the CVD-REAL Study (comparative effectiveness of cardiovascular outcomes in new users of sodium-glucose cotransporter-2 inhibitors) Circulation. 2017;136:249–259. doi: 10.1161/CIRCULATIONAHA.117.029190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han J.H., Oh Tj, Lee G., Maeng H.J., Lee D.H., Kim K.M., Choi S.H., Jang H.C., Lee H.S., Park K.S., Yim Y.B., Lim S. The beneficial effects of empagliflozin, an SGLT2 inhibitor, on atherosclerosis in ApoE-/- mice fed a western diet. Diabetologia. 2017;60:363–376. doi: 10.1007/s00125-016-4158-2. [DOI] [PubMed] [Google Scholar]
- 21.Nakatsu Y., Kokubo H., Bumdelger B., Yoshizumi M., Yamamotoya T., Matsunaga Y., Ueda K., Inoue Y., Inoue M.K., Fujishiro M., Kushiyama A., Ono H., Sakoda H., Asano T. Int. J. Mol. Sci. 2017;18:1704. doi: 10.3390/ijms18081704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nasiri-Ansari N., Dimitriadis G.K., Agrogiannis G., Perrea D., Kostakis I.D., Kaltsas G., Papavassiliou A.G., Randeva H.S., Kassi E. Canagliflozin attenutates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc. Diabetol. 2018;17:106. doi: 10.1186/s12933-018-0749-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mori K., Tsuchiya K., Nakamura S., Miyachi Y., Shiba K., Ogawa Y., Kitamura K. Ipragliflozin-induced adipose expansion inhibits cuff-induced vascular remodeling in mice. Cardiovasc. Diabetol. 2019;18:83. doi: 10.1186/s12933-019-0886-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ayer A., Zarjou A., Agarwal A., Stocker R. Heme oxygenases in cardiovascular health and disease. Physiol. Rev. 2016;96:1449–1508. doi: 10.1152/physrev.00003.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durante W. Targeting heme oxygenase-1 in vascular disease. Curr. Drug Targets. 2010;11:1504–1516. doi: 10.2174/1389450111009011504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durante W. Protective role of heme oxygenase-1 against inflammation in atherosclerosis. Front. Biosci. 2011;16:2372–2388. doi: 10.2741/3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duckers H.J., Boehm M., True A.L., Yet S.F., San H., Park J.L., Clinton Webb R., Lee M.E., Nagel G.J., Nagel E.G. Heme oxygenase-1 protects against vascular constriction and proliferation. Nat. Med. 2001;7:693–698. doi: 10.1038/89068. [DOI] [PubMed] [Google Scholar]
- 28.Tulis D.A., Durante W., Peyton K.J., Evans A.J., Schafer A.I. Heme oxygenase-1 attenuates vascular remodeling following balloon injury in rat carotid arteries. Atherosclerosis. 2001;155:113–122. doi: 10.1016/s0021-9150(00)00552-9. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez A.I., Gangopadhyay A., Kelly E.E., Pagano P.J., Zuckerbraun B.S., Bauer P.M. HO-1 and CO decrease platelet-derived growth factor-induced vascular smooth muscle cell migration via inhibition of Nox1. Arterioscler. Thromb. Vasc. Biol. 2010;30:98–104. doi: 10.1161/ATVBAHA.109.197822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Visner G.A., Lu F., Zhou H., Liu J., Kazemfar K., Agarwal A. Rapamycin induces heme oxygenase-1 in human pulmonary vascular cells: implications in the antiproliferative response to rapamycin. Circulation. 2003;107:911–916. doi: 10.1161/01.cir.0000048191.75585.60. [DOI] [PubMed] [Google Scholar]
- 31.Grosser N., Abate A., Oberle S., Vreman H.J., dennery P.A., Becker J.C., Pohle T., Seidman D.S., Schroder H. Heme oxygenase-1 induction may explain the antioxidant profile of aspirin. Biochem. Biophys. Res. Commun. 2003;308:956–960. doi: 10.1016/s0006-291x(03)01504-3. [DOI] [PubMed] [Google Scholar]
- 32.Lee T.S., Chang C.C., Zhu Y., Shyy J.Y. Simvastatin induces heme oxygenase-1: a novel mechanism of vascular protection. Circulation. 2004;110:1296–1302. doi: 10.1161/01.CIR.0000140694.67251.9C. [DOI] [PubMed] [Google Scholar]
- 33.Liu X.M., Peyton K.J., Wang X., Durante W. Sildenafil stimulates the expression of gaseous monoxide-generating enzymes in vascular smooth muscle cells via distinct signaling pathways. Biochem. Pharmacol. 2012;84:1045–1054. doi: 10.1016/j.bcp.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Durante W., Schini V.B., Catovsky S., Kroll M.H., Vanhoutte P.M., Schafer A.I. Plasmin potentiates induction of nitric oxide synthesis by interleukin-1 beta in vascular smooth muscle cells. Am. J. Physiol. 1993;264:H617–H624. doi: 10.1152/ajpheart.1993.264.2.H617. [DOI] [PubMed] [Google Scholar]
- 35.Peyton K.J., Ensenat D., Azam M.A., Keswani A.N., Kannan S., Liu X.M., Wang H., Tulis D.A., Durante W. Arginase promotes neointima formation in rat injured carotid arteries. Arterioscler. Thromb. Vasc. Biol. 2009;29:488–494. doi: 10.1161/ATVBAHA.108.183392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peyton K.J., Yu Y., Yates B., Shebib A.R., Liu X.M., Wang H., Durante W. Compound C inhibits vascular smooth muscle cell proliferation and migration in an AMPK-activated protein kinase-independent fashion. J. Pharmacol. Exp. Therapeut. 2011;338:476–484. doi: 10.1124/jpet.111.181784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peyton K.J., Shebib A.R., Azam M.A., Liu X.M., Tulis D.A., Durante W. Bilirubin inhibits neointima formation and vascular smooth muscle cell proliferation and migration. Front. Pharmacol. 2012;3:48. doi: 10.3389/fphar.2012.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu X.M., Peyton K.J., Durante W. Physiological cyclic strain promotes endothelial cell survival via the induction of heme oxygenase-1. Am. J. Physiol. Heart Circ. Physiol. 2013;304:H1634–H1643. doi: 10.1152/ajpheart.00872.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Durante W., Peyton K.J., Schafer A.I. Platelet-derived growth factor stimulates heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999;19:2666–2672. doi: 10.1161/01.atv.19.11.2666. [DOI] [PubMed] [Google Scholar]
- 40.Liu X.M., Durante Z., Peyton K.J., Durante W. Heme oxygenase-1 derived bilirubin counteracts HIV protease inhibitor-mediated endothelial cell dysfunction. Free Radic. Biol. Med. 2016;94:218–229. doi: 10.1016/j.freeradbiomed.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu XM, Peyton KJ, Ensenat DE, Wang H, Schafer AI, Alam J, and Durante W. Endoplasmic reticulum stress stimulates heme oxygenase-1 gene expression in vascular smooth muscle. Role in cell survival. J. Biol. Chem. 280: 872-877. [DOI] [PubMed]
- 42.Devineni D., Polidori D., Curtin C.R., Murphy J., Wang S.-S., Stieltjes H., Wajs E. Pharmacokinetics and pharmacodynamics of once-and twice-daily multiple-doses of canagliflozin, a selective inhibitor of sodium glucose co-transporter 2, in healthy participants. Int. J. Clin. Pharm. Ther. 2015;53:438–446. doi: 10.5414/CP202324. [DOI] [PubMed] [Google Scholar]
- 43.Kasichayanula S., Chang M., Hasegawa M., Liu X., Yamahira N., LaCreta F.P., Imai Y., Boulton D.W. Pharmacokinetics and pharmacodynamics of dapagliflozin, a novel selective inhibitor of sodium–glucose co‐transporter type 2, in Japanese subjects without and with type 2 diabetes mellitus. Diabetes Obes. Metabol. 2011;13:357–365. doi: 10.1111/j.1463-1326.2011.01359.x. [DOI] [PubMed] [Google Scholar]
- 44.Brand T., Macha S., Mattheus M., Pinnetti S., Woerle H.J. Pharmacokinetics of empagliflozin, a sodium glucose cotransporter-2 (SGLT-2) inhibitor, coadministered with sitagliptin in healthy volunteers. Adv. Ther. 2012;29:889–899. doi: 10.1007/s12325-012-0055-3. [DOI] [PubMed] [Google Scholar]
- 45.Villani L.A., Smith B.K., Marcinko K., Ford R.J., Broadfield L.A., Green A.E., Houde V.P., Muti P., Tsakiridis T., Steinberg G.R. The diabetes medication canagliflozin reduces cancer cell proliferation by inhibiting mitochondrial complex-I supported respiration. Mol Metab. 2016;5:1048–1056. doi: 10.1016/j.molmet.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Secker P.F., Beneke S., Schlichenmaier N., Delp J., Gutbier S., Leist M., Dietrich D.R. Canagliflozin mediated dual inhibition of mitochondrial glutamate dehydrogenase and complex I: an off-target adverse effect. Cell Death Dis. 2018;9:226. doi: 10.1038/s41419-018-0273-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaji K., Nishimura N., Seki K., Sato S., Saikawa S., Nakanishi K., Furukawa M., Kawaratani H., Kitade M., Moriya K., Namisaki T., Yoshiji H. Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int. J. Canc. 2018;142:1712–1722. doi: 10.1002/ijc.31193. [DOI] [PubMed] [Google Scholar]
- 48.Behnammanesh G., Durante Z.E., Peyton K.J., Martinez-Lemus L.A., Brown S.M., Bender S.B., Durante W. Canagliflozin inhibits human endothelial cell proliferation and tube formation. Front. Pharmacol. 2019;10:362. doi: 10.3389/fphar.2019.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hung M.H., Chen Y.L., Chen L.J., Chu P.Y., Hsieh F.S., Tsai M.H., Shih C.T., Chao T.I., Huang C.Y., Chen K.F. Canagliflozin inhibits the growth of hepatocellular carcinoma via blocking glucose-influx-induced β-catenin activation. Cell Death Dis. 2019;10:420. doi: 10.1038/s41419-019-1646-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shiba K., Tsuchiya K., Komiya C., Miyachi Y., Mori K., Shimazu N., Yamaguchi S., Ogasawara N., Katoh M., Itoh M., Suganami T., Ogawa Y. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci. Rep. 2018;8:2362. doi: 10.1038/s41598-018-19658-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mancini S.J., Boyd D., Katwan O.J., Stembitska A., Almabrouk T.A., Kennedy S., Palmer T.M., Salt I.P. Canagliflozin inhibits interleukin-1β-stimulated cytokine and chemokine secretion in vascular endothelial cells by AMP-activated protein kinase-dependent and –independent mechanisms. Sci. Rep. 2018;8:5276. doi: 10.1038/s41598-018-23420-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang D.D., Hannink M. Distinct cysteine residues in keap1 are required for keap-1-dependent ubiquitination of Nrf2 and for stabilizationof Nrf2 by chemopreventative agents and oxidative stress. Mol. Cell Biol. 2003;23:137–151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hawley S.A., Ford R.J., Smith B.K., Gowans G.J., Mancini S.J., Pitt R.D., Pitt R.D., Day E.A., Salt I.P., Steinberg G.R., Hardie D.G. The Na+/glucose cotransporter inhibitor canagliflozin activates AMPK by inhibiting mitochondrial function and increasing cellular AMP levels. Diabetes. 2016;65:2784–2794. doi: 10.2337/db16-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pattanawongsa A., Chau N., Rowland A., Miners J.O. Inhibition of human UDP-glucuronosyltransferase enzymes by canagliflozin and dapagliflozin: implications for drug-drug interactions. Drug Metab. Dispos. 2015;43:1468–1476. doi: 10.1124/dmd.115.065870. [DOI] [PubMed] [Google Scholar]
- 55.Uthman L., Baartscheer A., Bleijlevens B., Schumacher C.A., Fiolet J.W.T., Koeman A. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na+/H+ exchanger, lowering cytosolic Na+ and vasodilation. Diabetologia. 2018;61:722–726. doi: 10.1007/s00125-017-4509-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Otterbein L.E., Zuckerbraun B.S., Haga M., Liu F., Song R., Usheva A., Stachulak C., Bodyak N., Smith R.N., Csizmadia E., Tyagi S., Akamatsu Y., Flavell R.J., Billiar T.R., Tzeng E., Bach F.H., Choi A.M., Soares M.P. Carbon monoxide suppresses artheriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat. Med. 2003;9:183–190. doi: 10.1038/nm817. [DOI] [PubMed] [Google Scholar]
- 57.Ollinger R., Bilban M., Erat A., Froio A., McDaid J., Tyagi S., Csizmadia E., Graca-Souza A.V., Liloia A., Soares M.P., Otterbein L.E., Usheva A., Yamashita K., Bach F.H. Bilirubin: a natural inhibitor of vascular smooth muscle proliferation. Circulation. 2005;112:1030–1039. doi: 10.1161/CIRCULATIONAHA.104.528802. [DOI] [PubMed] [Google Scholar]
- 58.Wakisaka M., Nagao T., Yoshinari M. Sodium glucose cotransporter 2 (SGLT2) plays a physiological glucose sensor and regulates cellular contractility in rat mesangial cells. PloS One. 2016;11 doi: 10.1371/journal.pone.0151585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levonen A.L., Inkala M., Heikura T., Jauhiainen S., Jyrkkanen H.K., Kansanen E., Maatta K., Romppanen E., Turunen P., Rutanen J., Yla-Herttuala S. Nrf2 gene transfer induces antioxidant enzymes and suppresses smooth muscle cell growth in vitro and reduces oxidative stress in rabbit aorta in vivo. Arterioscler. Thromb. Vasc. Biol. 2007;27:741–747. doi: 10.1161/01.ATV.0000258868.80079.4d. [DOI] [PubMed] [Google Scholar]
- 60.Ashino T, Yamamoto M, Yoshida T, and Numazawa S. Redox-sensitive transcription factor Nrf2 regulates vascular smooth muscle cell migration and neointimal hyperplasia. Arterioscler. Thromb. Vasc. Biol. 33: 760-768. [DOI] [PubMed]
- 61.Peyton K.J., Liu X.M., Yu Y., Yates B., Behnammanesh G., Durante W. Glutaminase-1 stimulates the proliferation, migration, and survival of human endothelial cells. Biochem. Pharmacol. 2018;156:204–214. doi: 10.1016/j.bcp.2018.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang Y., Ying L., Chen Y., Shen Y., Guo R., Jin K., Wang L. Induction of heme oxygenase-1 ameliorates vascular dysfunction in streptozotocin-induced type 2 diabetic rats. Vasc. Pharmacol. 2014;61:16–24. doi: 10.1016/j.vph.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 63.Liu J., Wang L., Tian X.Y., Liu L., Wong W.T., Zhang Y., Han Q.B., Ho H.M., Wang N., Wong S.L., Cehn Z.Y., Yu Y., Ng C.F., Yao X., Huang Y. Unconjugated bilirubin mediates heme oxygenase-1-induced vascular benefits in diabetic mice. Diabetes. 2015;64:1564–1575. doi: 10.2337/db14-1391. [DOI] [PubMed] [Google Scholar]
- 64.Tiwari S., Ndisang J.F. Heme oxygenase system and hypertension: a comprehensive insight. Curr. Pharmaceut. Des. 2014;20:1354–1369. doi: 10.2174/13816128113199990558. [DOI] [PubMed] [Google Scholar]
- 65.Li M., Kim D.H., Tsenovoy P.L., Peterseon S.J., Rezzani R., rodella L.F., Aronow W.S., Ikehara S., Abraham N.G. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes. 2008;57:1526–1535. doi: 10.2337/db07-1764. [DOI] [PubMed] [Google Scholar]
- 66.Ndisang J.F., Jadhav A. Up-regulating the hemeoxygenase system enhances insulin sensitivity and improves glucose metabolism insulin-resistant diabetes in Goto-Kakizaki rats. Endocrinology. 2009;150:2627–2636. doi: 10.1210/en.2008-1370. [DOI] [PubMed] [Google Scholar]
- 67.Lever J.M., Boddu R., George J.F., Agarwal A. Heme oxygenase-1 in kidney health and disease. Antioxidants Redox Signal. 2016;25:165–183. doi: 10.1089/ars.2016.6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao Y., Zhang L., Qiao Y., Zhou X., Wu G., Wang L., Peng Y., Dong X., Huang H., Si L., Zhang X., Zhang L., Li J., Wang W., Zhou L., Gao X. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis, and enhancing autophagy. PloS One. 2013;8 doi: 10.1371/journal.pone.0075927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fan J., Xu G., Jiang T., Qin Y. Pharmacologic induction of heme oxygenase-1 plays a protective role in diabetic retinopathy in rats. Invest. Ophthalmol. Vis. Sci. 2012;53:6541–6556. doi: 10.1167/iovs.11-9241. [DOI] [PubMed] [Google Scholar]