

Abstract
P-glycoprotein (P-gp; also known as MDR1 or ABCB1) is an ATP-driven multidrug transporter that extrudes various hydrophobic toxic compounds to the extracellular space. P-gp consists of two transmembrane domains (TMDs) that form the substrate translocation pathway and two nucleotide-binding domains (NBDs) that bind and hydrolyze ATP. At least two P-gp states are required for transport. In the inward-facing (pre-drug transport) conformation, the two NBDs are separated, and the two TMDs are open to the intracellular side; in the outward-facing (post-drug transport) conformation, the NBDs are dimerized, and the TMDs are slightly open to the extracellular side. ATP binding and hydrolysis cause conformational changes between the inward-facing and the outward-facing conformations, and these changes help translocate substrates across the membrane. However, how ATP hydrolysis is coupled to these conformational changes remains unclear. In this study, we used a new FRET sensor that detects conformational changes in P-gp to investigate the role of ATP binding and hydrolysis during the conformational changes of human P-gp in living HEK293 cells. We show that ATP binding causes the conformational change to the outward-facing state and that ATP hydrolysis and subsequent release of γ-phosphate from both NBDs allow the outward-facing state to return to the original inward-facing state. The findings of our study underscore the utility of using FRET analysis in living cells to elucidate the function of membrane proteins such as multidrug transporters.
Keywords: ABC transporter, ATPase, multidrug transporter, membrane protein, fluorescence resonance energy transfer (FRET), conformational change, ABCB1, MDR1, P-glycoprotein
Introduction
P-glycoprotein (P-gp),3 a member of the ATP-binding cassette (ABC) transporter family, is an ATP-dependent efflux pump that transports various hydrophobic compounds (1, 2). Its substrates include therapeutic drugs, alkaloids, flavonoids, and other hydrophobic natural toxic compounds. Accordingly, P-gp is an essential component of the protective physiological barriers in important organs (3), such as the brain and testis. The protein consists of four core domains: two transmembrane domains (TMDs), which create the translocation pathway for the substrates, and two nucleotide-binding domains (NBDs), which bind and hydrolyze ATP to power the transport process. Numerous studies have been conducted (4–8) following the discovery of P-gp (9–11) to reveal how ATP binding and ATP hydrolysis are coupled to conformational changes of P-gp and drug transport.
Recently, we reported a pair of structures of the P-gp orthologue (CmABCB1) from the thermophilic unicellular eukaryote Cyanidioschyzon merolae at resolutions of 2.4 and 1.9 Å, of an inward-facing apo-state (12) and an outward-facing state with bound nucleotides, respectively (13). Conformational changes from the inward-facing to the outward-facing state during drug transport could be clearly visualized at these high resolutions. In the inward-facing state (pre-drug transport), a large inner cavity was formed in the center of the TMDs, extending from the middle of the lipid bilayer to the cytosol, and the two NBDs were separated by about 26 Å. In the outward-facing state (post-drug transport), the NBDs were in close proximity, resulting in the formation of nucleotide-binding sites composed of a Walker A motif (P-loop) in one NBD and a signature motif (LSGGQ) in the other NBD. Conformational changes in the NBDs caused by nucleotide binding mediated TMD movements. The outward-facing structure at 1.9 Å resolution (13) revealed that a relay of van der Waals interactions and hydrogen-bond networks formed by ATP binding propagate from the NBDs to the TMDs and make the whole molecule move as a rigid body. In contrast, the inward-facing structure (12) contains two layers of networks, a hydrogen bond network and an aromatic hydrophobic network, at the top of the molecule, which stabilize the inward-facing state, but the networks between the NBDs and TMDs were not observed, which is unlike the outward-facing conformation. These structural features suggest that ATP binding in both NBDs drives conformational movements from the flexible inward-facing conformation (where the networks between the NBDs and TMDs are not formed) to the rigid outward-facing conformation (where the networks between the NBDs and TMDs are formed) of P-gp, and that ATP hydrolysis releases the rigid outward-facing conformation to the inward-facing conformation. This mechanism, in which ATP hydrolysis triggers conformational change from the outward-facing to the inward-facing state, is supported by a study using mouse P-gp reconstituted in lipid nanodiscs composed of a scaffold protein and lipid bilayer (7) and the cryo-EM structure of human P-gp (8). However, recent double electron–electron resonance (DEER) studies of purified mouse P-gp suggested that ATP binding is insufficient for the formation of tight NBD dimer and that ATP hydrolysis powers the isomerization of P-gp from the inward-facing to the outward-facing conformation (14, 15). Consequently, the role of ATP hydrolysis in the conformational movements of P-gp remains controversial. One possible reason for different theories is that studies have utilized different artificial membrane environments such as detergent micelles (12–14), artificial lipid bilayers (5, 7, 15, 16), or membrane vesicles derived from P-gp–overexpressing cells (17, 18). Because the membrane environment is very likely to affect the function of membrane-embedded P-gp, the detailed transport mechanism should be investigated in the native membrane environment. Thus, in this study, we analyzed the roles of ATP binding and ATP hydrolysis in the conformational changes of human P-gp in living cells by using fluorescence resonance energy transfer (FRET).
Results
FRET construct to detect the conformational change of human P-gp in living cells
The distances between the C terminus of NBD1 and NBD2 of human P-gp are estimated to be about 30 and 11 Å in the inward-facing and outward-facing structures, respectively (Movie S1). Because this difference in distance is assumed to be one of the largest between the inward-facing and outward-facing structures, we considered it suitable for FRET analysis in order to detect the conformational change of human P-gp in living cells in real time. We generated a FRET construct, P-gp–FRET, in which a monomeric (m)Cerulean (donor) was inserted after NBD1 and a monomeric (m)Venus (19) (acceptor) was fused after NBD2 of human P-gp (Fig. 1A). Another FRET construct, P-gp–VsCn, in which mVenus and mCerulean were fused tandemly after NBD2, was predicted to show a high level of FRET despite the conformation. P-gp–mCerulean, in which mCerulean was inserted after NBD1, and P-gp–mVenus, in which mVenus was fused after NBD2, were constructed as negative controls. When these constructs were transiently expressed in HEK293 cells, fluorescence signals were observed on the plasma membrane (Fig. 1B). Basal and strong signals were observed in the FRET channel with P-gp–FRET and P-gp–VsCn, respectively, whereas weak and almost no signals were observed with P-gp–mCerulean and P-gp–mVenus, respectively (Fig. 1B, right). The weak signals were due to the broad emission spectrum of mCerulean.
Figure 1.

Localization and function of fluorescent protein-tagged P-gp constructs. A, schematic representations of the fluorescent protein-tagged P-gps. a.a., amino acid. B, localization of the fluorescent protein-tagged P-gp constructs. Each P-gp construct was transiently expressed in HEK293 cells. Fluorescent images were obtained at 37 °C using an LSM700 confocal microscope (Zeiss). Scale bars, 10 μm. C, substrate transport activity. HEK293 cells transiently expressing each P-gp construct were incubated with 1 μm R6G with or without 25 μm inhibitor (PSC-833) for 30 min at 37 °C. R6G accumulation in the cells was measured by flow cytometry.
The substrate transport activity of P-gp constructs was evaluated by measuring the accumulation of the fluorescent substrate rhodamine 6G (R6G). Because hydrophobic R6G freely penetrates membranes and accumulates in cells, mock cells transfected by the empty vector showed high R6G accumulation (Fig. 1C, gray). However, when P-gp was expressed on the plasma membrane, R6G was extruded from the cells, and R6G accumulation was reduced (Fig. 1C, blue). However, when cells were co-incubated with PSC-833, a specific inhibitor of P-gp, R6G accumulated as much as in mock cells (Fig. 1C, red). These results suggest that the low R6G accumulation is due to the efflux activity of P-gp. All P-gp constructs caused a leftward shift of R6G fluorescence in the absence of PSC-833, whereas the degree of the shift was smaller in cells expressing P-gp constructs containing mVenus due to the leakage of mVenus fluorescence in the detection channel. These results indicated that neither the insertion nor fusion of fluorescent proteins impaired the plasma membrane localization or function of P-gp.
Transport substrate causes conformational changes in P-gp
Next, we investigated whether the addition of a transport substrate could affect the level of FRET. We defined the proximity ratio as the ratio of the corrected intensity of the FRET signal divided by the mCerulean intensity (20), which is shown in pseudocolor in Fig. 2A. The proximity ratio of P-gp–FRET on the plasma membrane (Fig. 2A, green) was low, and the signal of P-gp–VsCn (shown in yellow or red) was high in the absence of a transport substrate. When verapamil, a transport substrate of P-gp (21) that stimulates its ATPase activity (Fig. 5B) (22), was added to the cells, the proximity ratio of P-gp–FRET increased, whereas that of P-gp–VsCn was unchanged (Fig. 2A). Next, the plasma membrane was extracted by automatic membrane segmentation of the mVenus image of P-gp–FRET and -VsCn or the mCerulean image of P-gp–mCerulean (Fig. 2B), and the proximity ratio on the plasma membrane was calculated. The proximity ratio of P-gp–FRET increased about 1.5-fold upon the addition of 100 μm verapamil, which approximates the level of P-gp–VsCn (Fig. 2C). When cells were co-transfected with P-gp–mCerulean and P-gp–mVenus, the proximity ratio was negligible and unaffected by the addition of verapamil (Fig. 3A). The expression level of P-gp–FRET did not affect the proximity ratio either (Fig. 3B). These results suggested that intermolecular FRET did not affect the proximity ratio of P-gp–FRET with or without verapamil treatment. Indeed, verapamil increased the proximity ratio in a concentration-dependent manner, and the EC50 was 3.7 ± 0.9 μm (Fig. 3C). Moreover, the proximity ratio of P-gp–VsCn was unaffected by verapamil and that of P-gp–mCerulean was less than one-tenth that of P-gp–VsCn (Fig. 2C). These results suggest that verapamil causes conformational changes in P-gp.
Figure 2.

Effects of verapamil on the conformational changes of P-gp. A, proximity ratio pseudocolor images of HEK293 cells expressing fluorescent protein-tagged P-gp constructs. Cells were treated with 100 μm verapamil or DMSO (vehicle control) for 5 min at 37 °C. The proximity ratio is shown in an 8-color scale, and each hue has a range of 32 intensities based on the corrected intensity of the FRET image. Scale bars, 10 μm. B, region of the plasma membrane determined from automatically segmented mVenus images of P-gp–FRET and mCerulean images of P-gp–mCerulean. Scale bars, 5 μm. C, effect of verapamil on the proximity ratio of P-gp–FRET. Cells were treated with 100 μm verapamil (blue) or DMSO (empty) for 5 min at 37 °C. The proximity ratio is shown relative to that of P-gp–FRET without verapamil. 10–15 cells per sample; ***, p < 0.001; N.S., p > 0.05.
Figure 5.
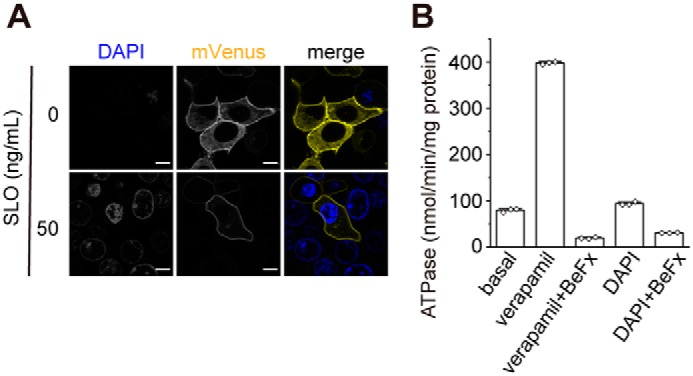
membrane permeabilized semi-intact cells. A, DAPI staining of cells expressing P-gp–mVenus permeabilized with 50 ng/ml SLO. Fluorescent images were obtained at 37 °C on a Nikon C2 confocal system equipped with a Plan Apochromat ×40/dry objective lens. Scale bars, 10 μm. B, effect of DAPI on the ATPase activity of human P-gp. Proteoliposomes were incubated for 30 min at 37 °C with 100 μm verapamil chloride or 2 μg/ml DAPI in the absence or presence of 1 mm beryllium fluoride (BeFx, an ATPase inhibitor). The values represent the means ± S.D. from three technical replicates.
Figure 3.

Concentration-dependent increase of the intramolecular FRET by verapamil. A, effect of verapamil on the proximity ratio of co-transfected P-gp–mCerulean and P-gp–mVenus. B, effect of the protein expression level on the proximity ratio of P-gp–FRET. C, dose-dependent increase of the proximity ratio by verapamil. Cells were treated with 100 μm verapamil (blue) or DMSO (empty) for 5 min at 37 °C. The proximity ratio is shown relative to that of P-gp–FRET without verapamil. 10–15 cells per sample; ***, p < 0.001; N.S., p > 0.05.
ATP binding- and hydrolysis-deficient mutants are trapped in the low- and high-FRET states, respectively
To investigate the involvement of ATP binding and ATP hydrolysis in the conformational changes induced by verapamil, we constructed P-gp(MM)–FRET (Fig. 8E), in which conserved lysine residues in the Walker A motif crucial for ATP binding (8, 13, 23) were replaced by methionines, and P-gp(QQ)–FRET (Fig. 8F), in which glutamate residues in the Walker B motif crucial for ATP hydrolysis (13, 24) were replaced by glutamines. Both P-gp(MM)–FRET and P-gp(QQ)–FRET properly localized to the plasma membrane (Fig. 4A) but had no R6G transport activity (Fig. 4B). The proximity ratio of P-gp(MM)–FRET was comparable with that of P-gp–FRET in the absence of a transport substrate but was unaffected by verapamil treatment (Fig. 4C). This result suggests that ATP binding is required for the increase of the proximity ratio induced by verapamil. However, the proximity ratio of P-gp(QQ)–FRET, which allows ATP binding but reduces the rate of ATP hydrolysis (24), was significantly higher than that of P-gp–FRET even in the absence of a transport substrate and was also unaffected by verapamil (Fig. 4D). This result suggests that ATP hydrolysis causes a transition from the high-FRET to the low-FRET state. Like P-gp–VsCn, control constructs P-gp(MM)–VsCn and P-gp(QQ)–VsCn exhibited a high-proximity ratio independent of verapamil.
Figure 8.

Schematic illustration of the ATP-dependent conformational changes of P-gp. A–K, transition equilibria between the inward-facing conformation (left), which generates basal FRET, and the outward-facing conformation (right), which generates strong FRET, of human P-gp are indicated by arrows. Rectangles and circles represent TMDs and NBDs, respectively. Blue and yellow cylinders represent mCerulean and mVenus, respectively. The replacement of Walker A lysine by methionine and replacement of catalytic glutamate by glutamine are shown as M and Q, respectively. ●, verapamil. The inward-facing conformation is stable in the absence of ATP or verapamil (A–C). The presence of ATP and verapamil causes the transition from the inward-facing to the outward-facing conformation and generates strong FRET (D). Replacement of the Walker A lysine by methionine prevents ATP binding and generates basal FRET (E). Replacement of the Walker B glutamate by glutamine in either NBD stabilizes the outward-facing state and generates strong FRET (F–H). Binding of MgAMP-PNP stabilizes the outward-facing state and generates strong FRET (I). Orthovanadate traps P-gp in the stable post-ATP hydrolysis transition state (probably in either NBD) to generate strong FRET (J). K, model of the transport cycle of P-gp caused by ATP. The inward-facing conformation is stable in the absence of ATP or verapamil (left and middle). The presence of ATP and verapamil causes the transition from the inward-facing to the outward-facing conformation (right). ATP hydrolysis and subsequent release of γ-phosphate from both NBDs allow P-gp to return to the inward-facing state. Mutations of the Walker A lysine prevents ATP binding. Mutations of the Walker B glutamate or binding of AMP-PNP inhibits ATP hydrolysis. Orthovanadate traps P-gp in the stable post-ATP hydrolysis transition state. The inward-facing conformation of mouse P-gp is shown in PDB 4m1m; the outward-facing conformation of human P-gp is shown in PDB 6c0v.
Figure 4.

Proximity ratio of ATP-binding–deficient and ATP hydrolysis-deficient mutants. A, fluorescent images of the mutants. Scale bars, 10 μm. B, substrate transport activity of the mutants. The proximity ratio in intact cells transiently transfected with the ATP-binding–deficient mutant MM (C) and ATP hydrolysis-deficient mutant QQ (D). Cells were treated with 100 μm verapamil (blue) or DMSO (empty) for 5 min at 37 °C. The proximity ratio is shown to that of P-gp–FRET without verapamil. n = 12–15 cells per sample; **, p < 0.01; ***, p < 0.001; N.S., p > 0.05.
ATP binding is required for the conformational change of P-gp
To further demonstrate the role of ATP binding on the conformational change of P-gp, intracellular ATP was depleted from cells by treating them with streptococcal toxin streptolysin O (SLO). SLO forms 25–30-nm aqueous pores within the plasma membrane, which allows the free passage of ions and small molecules (25). 4′,6-Diamidino-2-phenylindole (DAPI) hardly goes through the plasma membrane of intact cells. Therefore, cells with a DAPI-positive nucleus and permeabilized plasma membranes were used for the FRET analysis (Fig. 5A). DAPI is not a transport substrate of P-gp and does not stimulate its ATPase activity (Fig. 5B). In the presence of exogenously-added ATP, the proximity ratio of P-gp–FRET was increased by the addition of verapamil (Fig. 6A), consistent with the observations for intact cells (Fig. 2C). However, in the absence of ATP, the proximity ratio of P-gp–FRET was not increased even after the addition of verapamil. These results suggest that both ATP and substrate binding are required for enhancing the conformational change from the inward-facing state (Fig. 8, A–D).
Figure 6.

Effects of ATP binding and ATP hydrolysis on the conformational changes of P-gp. A and B, proximity ratio in permeabilized semi-intact cells treated with 100 μm verapamil (blue) or DMSO (empty) in the presence 4.5 mm MgATP. The proximity ratios in the absence of ATP are shown in the hatched plots. C, proximity ratio in permeabilized semi-intact cells in the presence of 4.5 mm MgATP (empty), 4.5 mm MgAMP-PNP (green), or 4.5 mm MgATP and 1 mm orthovanadate (magenta). The proximity ratio is shown relative to that of P-gp–FRET in the presence of MgATP. n = 12–15 cells per sample; **, p < 0.01; ***, p < 0.001; N.S., p > 0.05.
ATP hydrolysis at both NBDs resets the conformation of P-gp to the low FRET state
Next, to investigate the role of ATP hydrolysis in the conformational change of P-gp, we analyzed P-gp(QQ)–FRET, which binds ATP but has impaired ATP hydrolysis. The proximity ratio of P-gp(QQ)–FRET was high in the presence of exogenously-added ATP even without verapamil, but was low in the absence of ATP (Fig. 6B). In addition, when ATP-depleted cells were incubated with the nonhydrolyzable ATP analogue, AMP-PNP, the proximity ratio of P-gp–FRET was increased 1.5-fold even without verapamil treatment (Figs. 6C and 8I). It has been reported that P-gp is trapped in a state that mimics the post-hydrolysis transition state (26–28) when incubated with ATP and sodium orthovanadate (27, 28) and that the “vanadate trap” occurs at only one NBD randomly (27, 28). P-gp–FRET exhibited a high-proximity ratio in the presence of ATP and sodium orthovanadate (Figs. 6C and 8J). Finally, we generated two single Walker B glutamate mutants, P-gp(QE)–FRET and P-gp(EQ)–FRET (Fig. 8, G and H), in which the catalytic glutamate residue in only one NBD was substituted for glutamine. Both single Walker B mutants properly localized to the plasma membrane (Fig. 7A) but completely lost R6G transport activity (Fig. 7B). When these mutants were expressed in living cells, they exhibited a high-proximity ratio even in the absence of a transport substrate (Fig. 7C). These results suggest that ATP hydrolysis and subsequent release of γ-phosphate from both NBDs allow the outward-facing state to return to the original inward-facing state.
Figure 7.
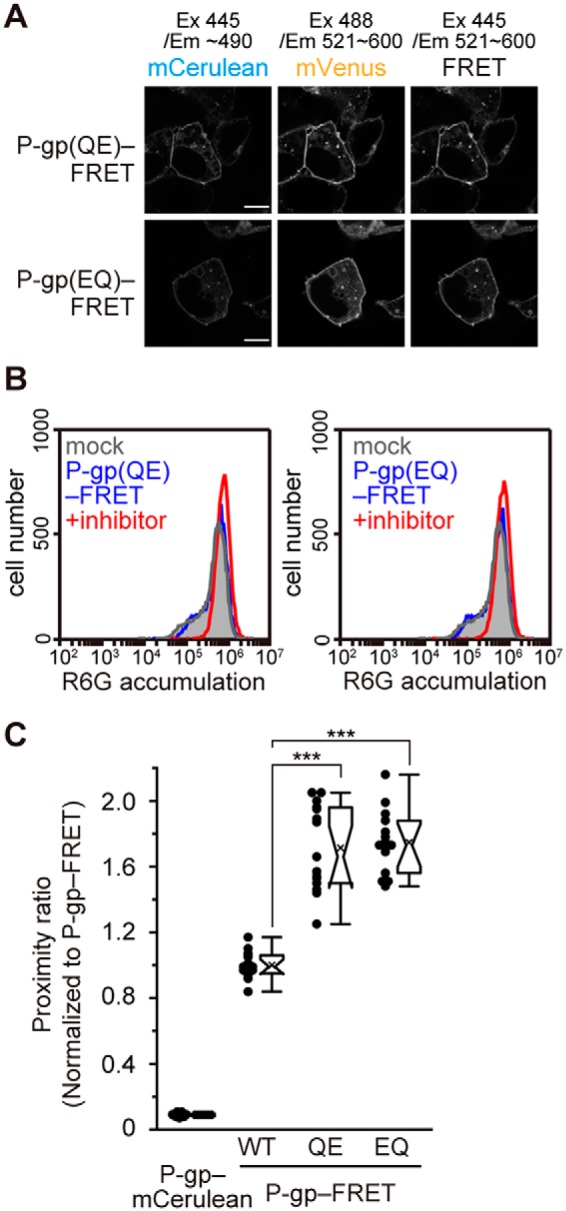
Proximity ratio of single Walker B mutants. A, fluorescent images of P-gp(QE)–FRET and P-gp(EQ)–FRET obtained using an LSM700 confocal microscope. Scale bars, 10 μm. B, substrate transport activity of the single Walker B mutants. C, proximity ratio in intact cells of the single Walker B mutants, shown relative to that of P-gp–FRET. n = 15 cells per sample; ***, p < 0.001.
Discussion
Since the discovery of P-gp (9–11), numerous studies have been conducted to clarify the transport mechanism of P-gp. Most of the studies have utilized artificial membrane environments such as lipid-detergent mixed micelles (14), artificial lipid bilayers (5, 7, 15, 16), or membrane vesicles derived from P-gp–overexpressing cells (17, 18). These conditions do not reflect physiological membrane environments in terms of lateral pressure, curvature, constituent lipid species, etc. Because these environmental factors can affect the function of P-gp, the detailed transport mechanism should be investigated in living cells. Although a conformation-sensitive mAb, UIC2, which recognizes the extracellular loops of P-gp and specifically binds to the inward-facing structure (29), has been used to evaluate the conformational changes of the extracellular region of P-gp in living cells (6, 30, 31), an analysis of the conformational changes in living cells is technically difficult. In this study, we detected the change in distance between two NBDs of human P-gp in living cells by using FRET-based noninvasive imaging. Because the NBDs move dramatically in P-gp and the insertion of fluorescent proteins after the NBDs did not compromise the proper localization or substrate transport activity of P-gp, we expected the constructs to be suitable for monitoring the intrinsic behavior of P-gp in living cells.
P-gp–FRET on the plasma membrane in living cells showed a basal FRET signal (Fig. 1B). Considering that the ATP binding–deficient mutant P-gp(MM)–FRET showed a similar level of FRET as P-gp–FRET (Fig. 4C), ATP binding did not contribute to basal FRET. Alternatively, because the distance between the C termini of NBD1 and of NBD2 of human P-gp is about 30 Å in the inward-facing conformation (Movie S1), it is possible that FRET occurs to some extent even in this conformation. The proximity ratio was increased by the addition of a transport substrate, verapamil, in a concentration-dependent manner, whereas the FRET signal of P-gp–VsCn was unchanged (Figs. 2, A and C, and 3C). The EC50 value for verapamil of the proximity ratio in intact cells (3.7 ± 0.9 μm) (Fig. 3C) was comparable with the Km value for verapamil (2.2 ± 0.2 to 4.1 ± 0.4 μm) in the ATPase activity of purified human P-gp (22). These results suggest that verapamil triggers the conformational change of P-gp and that mCerulean inserted after NBD1 and mVenus fused after NBD2 approach each other in close proximity via dimerization of the two NBDs to increase intramolecular FRET (Fig. 8D). This conclusion is consistent with a model proposed by structural studies (12, 13), in which a transport substrate interacts with the hydrophobic ceiling of the inner cavity and destabilizes it by a wedge-like action to induce the opening of the extracellular gate and accelerate the ATPase activity of P-gp. Although the distance between chromophores can be predicted from the proximity ratio, the flexible change of the orientation of fused fluorescent proteins makes it difficult to get an accurate estimation of the distance between two NBDs, which would explain the large variation of the proximity ratio especially in P-gp–VsCn (Fig. 2C). Therefore, we discuss proximity ratio only qualitatively in this work.
To examine the role of ATP binding in the conformational change of P-gp, intracellular ATP was depleted from living cells by using the streptococcal toxin SLO, a member of the cholesterol-dependent pore-forming toxins. After binding to the plasma membrane, SLO oligomerizes and creates large β-barrel pores (25). In the presence of exogenously added ATP, the proximity ratio of P-gp–FRET was increased by the addition of verapamil, but not in the absence of ATP (Fig. 6A). These results suggest that both ATP and substrate binding are required for enhancing the conformational change from the inward-facing state (Fig. 8, A–D). Because the addition of ATP did not affect the proximity ratio of P-gp–FRET in the absence of a transport substrate in experiments using semi-intact cells (Fig. 6A), the inward-facing structure of P-gp is considered to be very stable in the absence of a transport substrate. This feature is different from MRP1 (ABCC1) tagged with a FRET pair, as its proximity ratio was increased by exogenously-added ATP in a concentration-dependent manner even in the absence of a transport substrate (31). The requirement of ATP and substrate binding is consistent with the result of an ATP binding–deficient mutant P-gp(MM)–FRET in intact cells, not responding to verapamil (Fig. 4C). As with P-gp(QQ)–FRET, which we explain below, the results of WT and mutant P-gp are consistent with intact cells and SLO-treated and ATP-depleted cells, suggesting that the ATP-dependent conformational change of P-gp is not affected by SLO treatment.
When ATP-depleted cells were incubated with the nonhydrolyzable ATP analogue AMP-PNP, the proximity ratio of P-gp–FRET was increased even without verapamil treatment (Fig. 6C). The same effect was seen with the ATPase-deficient mutant P-gp(QQ)–FRET in intact cells and ATP-depleted cells. This mutant's proximity ratio was high in the presence of endogenously-added or exogenously-added ATP independent of verapamil but was low in the absence of ATP (Figs. 4D and 6B). These results suggest that human P-gp moves spontaneously between the inward-facing and outward-facing structures at low frequency and that ATP before hydrolysis stabilizes the dimerized NBDs and fixes P-gp to the outward-facing structure (Fig. 8, F and I).
Furthermore, single Walker B glutamate mutants, P-gp(QE)–FRET and P-gp(EQ)–FRET, exhibited high-proximity ratios even in the absence of a transport substrate (Fig. 7C), suggesting that inhibition of ATP hydrolysis at one NBD can stabilize dimerized NBDs and fixes P-gp to the outward-facing structure (Fig. 8, G and H). This is consistent with a variety of biochemical studies, in which mutations in a single ATP-binding site dramatically decreased the ATPase activity (6, 32, 33) and drug transport (34). This finding is also consistent with biochemical analyses that showed orthovanadate traps P-gp in a state that mimics the post-hydrolysis transition state before the release of γ-phosphate (26–28) and that the “vanadate trap” occurs at only one NBD randomly (23, 27, 28, 35). Accordingly, P-gp–FRET exhibited a high-proximity ratio in the presence of ATP and sodium orthovanadate (Fig. 6C).
To conclude, our results suggest that ATP binding provides the energy for the conformational change of P-gp to the rigid outward-facing state and that ATP hydrolysis and the subsequent release of γ-phosphate from both NBDs reset the conformation of P-gp from the outward-facing to the inward-facing state (Fig. 8K), which are in agreement with cryo-EM and luminescence studies (7, 8) but inconsistent with recent DEER studies (14, 15) performed under nonphysiological membrane environments. Our study underlines the necessity of experiments using living cells to understand the detailed mechanism of membrane proteins in addition to in vitro experiments.
Experimental procedures
Plasmid construction
All primer sequences are listed in Table 1. P-gp constructs were designed as MRP1 FRET constructs (36). pcDNA3.1P-gp was constructed by the insertion of cDNA encoding human P-gp in pcDNA3.1 (Thermo Fisher Scientific, Austin, TX). To engineer P-gp–mVenus, the pcDNA3.1(−)P-gp backbone and mVenus cDNA were PCR-amplified using the primer pairs 1–4, respectively. To engineer P-gp–mCerulean, the pcDNA3.1(+)P-gp backbone, mCerulean cDNA, and the following 10-amino acid linkers (GS repeats) were PCR-amplified using the primer pairs 5/6 and 7/8, respectively. The P-gp–FRET construct was generated by inserting mCerulean cDNA and the following 10-amino acid linkers into pcDNA3.1(−)P-gp–mVenus using the primer pairs 5/6 and 7/8, respectively. To engineer P-gp–VsCn, the pcDNA3.1(−)P-gp–mVenus backbone and mCerulean cDNA were PCR-amplified using the primer pairs 1/4 and 9/10, respectively. The amplified fragments were ligated using an In-Fusion HD cloning kit (Clontech). pcDNA3.1(−)P-gp(MM; K433M/K1076M)-FRET, pcDNA3.1(−)P-gp(QQ; E556Q/E1201Q), and other mutants were generated using the corresponding pcDNA3.1(−)P-gp mutant backbones. The cDNA of mCerulean was a kind gift from Dr. Shin-ich Aota (Osaka University, Japan).
Table 1.
Primers
| Primer no. | 5′–3′ |
|---|---|
| 1 | TGAGGATCCGAGCTCGGTAC |
| 2 | CTGGCGCTTTGTTCCAGCCTGGACACTGACCATTG |
| 3 | GGAACAAAGCGCCAGGTGAGCAAGGGCGAGGAG |
| 4 | GAGCTCGGATCCTCACTTGTACAGCTCGTCCATG |
| 5 | TGCTGTCTGCATTGTGACAAGTTTGAAGTAAATGCC |
| 6 | CAGGTTCTGGCTCAGGCTCTGGATCAGGAAATGAAG |
| 7 | ACAATGCAGACAGCAGTGAGCAAGGGCGAGGAG |
| 8 | CTGAGCCAGAACCTGATCCCTTGTACAGCTCGTCC |
| 9 | GACGAGCTGTACAAGGTGAGCAAGGGCGAGGAGC |
| 10 | GAGCTCGGATCCTCACTTGTACAGCTCGTCCATGC |
Flow cytometry
HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Nacalai Tesque, Kyoto, Japan) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific) at 37 °C under 5% CO2. HEK293 cells were seeded in 6-well plates (3.0 × 105 cells/well), coated with poly-l-lysine (Sigma-Aldrich), and incubated for 24 h. P-gp constructs were transfected using Lipofectamine LTX reagent (Thermo Fisher Scientific), and the transfected cells were incubated for an additional 24 h. Medium was replaced with serum-free DMEM containing 1 μm R6G with or without 25 μm PSC-833, a P-gp inhibitor (Abcam, Cambridge, MA), and incubated for 30 min at 37 °C under 5% CO2. Cells were washed twice with phosphate-buffered saline (PBS). After harvesting, the cells were washed with Hanks' balanced salt solution (HBSS; Thermo Fisher Scientific) and resuspended in HBSS. R6G accumulation was analyzed on an Accuri C6 flow cytometer (BD Biosciences).
FRET microscopy
HEK293 cells were seeded at 3.0 × 105 cells/well on glass-bottom dishes (AGC TECHNO GLASS, Shizuoka, Japan) coated with poly-l-lysine and incubated for 24 h. P-gp constructs were transfected using Lipofectamine LTX reagent and incubated for an additional 23 h. Medium was then replaced by FluoroBrite DMEM (Thermo Fisher Scientific) supplemented with 10% FBS, sodium pyruvate (Thermo Fisher Scientific), and GlutaMAX (Thermo Fisher Scientific) and incubated for another 1 h. Cells were further incubated with 100 μm verapamil chloride (Fujifilm Wako Pure Chemical Corp., Osaka, Japan) for 5 min at 37 °C under 5% CO2 as necessary. Fluorescence images were acquired at 37 °C under 5% CO2 by an LSM700 confocal microscope equipped with a Plan-Apochromat ×63/1.4 NA oil immersion objective lens (Carl Zeiss, Oberkochen, Germany). mCerulean was excited at 445 nm, and the fluorescence emission was collected using a short-pass filter (∼490 nm). mVenus was excited at 488 nm, and the fluorescence emission was collected using a bandpass filter (521–600 nm). For the FRET signal, the laser was set to 445 nm, and the sensitized emission was collected using a bandpass filter (521–600 nm).
Cell permeabilization
HEK293 cells were plated at 3.0 × 105 cells/well on glass-bottom–based dishes coated with poly-l-lysine and incubated for 24 h. P-gp constructs were transfected using polyethyleneimine “Max” (Mr 40,000) (PEI-MAX; Polysciences, Warrington, PA) and incubated for an additional 24 h. Cell permeabilization was performed as described previously (37) with slight modifications. Briefly, cells were washed twice with ice-cold PBS and incubated in serum-free DMEM containing 50 ng/μl SLO (BioAcademia, Osaka, Japan) for 5 min on ice, followed by three washes with ice-cold PBS. The cells were further incubated for 10 min at 37 °C under 5% CO2 in transport buffer (25 mm HEPES-KOH (pH 7.4), 115 mm KOAc, 4.5 mm MgCl2) pre-incubated at 37 °C and containing 2 μg/ml DAPI (Sigma-Aldrich) in the presence or absence of 5 mm ATP disodium salt (Oriental Yeast, Tokyo, Japan). The cells were then washed twice with transport buffer and soaked in transport buffer pre-incubated at 37 °C and supplemented with 5 mm ATP disodium salt, 5 mm AMP-PNP lithium salt (Sigma-Aldrich), or 5 mm ATP disodium salt and 1 mm orthovanadate (Fujifilm Wako), as indicated. The cells were then incubated with 100 μm verapamil chloride for 5 min at 37 °C under 5% CO2 as indicated. Only DAPI-positive cells were used for the FRET microscopy. FRET microscopy was performed on an LSM700 microscope as described above.
FRET calculation
FRET efficiency (proximity ratio) was calculated as shown in Equation 1 (20).
| (Eq. 1) |
Where IDA is the FRET signal or the sensitized emission of the acceptor during donor excitation (excitation 445 nm/emission 521–600 nm); IDD is the donor fluorescence during donor excitation (excitation 445 nm/emission ∼490 nm); and IAA is the acceptor fluorescence during acceptor excitation (excitation 488 nm/emission 521–600 nm). a and d are the cross-talk coefficients of the acceptor and donor, respectively. These constants were calculated from P-gp–mVenus and P-gp–mCerulean samples using the ImageJ plugin “FRET and Co-localization Analyzer.” The proximity ratio pseudocolor images were generated using MetaMorph software (Molecular Devices, San Jose, CA). For quantification of the proximity ratio, the plasma membrane was determined from automatically segmented mVenus images of P-gp–mVenus, P-gp–FRET, and P-gp–VsCn or mCerulean images of P-gp–mCerulean using ImageJ-based Fiji software. The proximity ratio was quantified on a cell-by-cell basis.
Protein expression and purification
Human P-gp cDNA was inserted into the expression vector pcDNA3.1(−). The C terminus of the DNA was modified by elimination of the natural termination codon and insertion of a NotI restriction cleavage site, tobacco etch virus protease-cleavage site (ENLYFQG), 10 histidine codons, a six-amino acid linker (GAAGTS), and the FLAG tag (DYKDDDDK), followed by a termination codon. FreeStyle 293-F cells (Thermo Fisher Scientific) were transfected with the resultant P-gp construct using PEI-MAX. After 48 h of transfection, the cells were harvested and resuspended in buffer A (50 mm HEPES-Na (pH 7.2), 150 mm NaCl, 50 mm KCl, 20% glycerol) containing 0.5 μm 2-mercaptethanol (Fujifilm Wako) and protease inhibitor (Roche Applied Science, Basel, Switzerland) and solubilized with 0.6% (w/v) n-dodecyl-β-d-maltopyranoside (DDM; Anatrace, Maumee, OH). Solubilized proteins were applied to FLAG-M2–agarose (Sigma-Aldrich) pre-equilibrated with buffer A. The mixture was rotated for 18 h. The resin was washed with a 4× bed volume of buffer A containing 0.6% DDM three times. The resin was further washed with a 4× bed volume of buffer A containing 0.1% DDM three times. The protein was eluted from the resin with 1× bed volume of buffer A containing 0.1% DDM, 300 μg/ml FLAG peptide (Sigma-Aldrich), and 3× FLAG peptide (Sigma-Aldrich). The eluate was concentrated by centrifugation using an Amicon Ultra (Merck, Kenilworth, NJ). Purified protein was stored at −80 °C. All purification steps were performed at 0–4 °C.
Reconstitution into proteoliposomes and measurement of ATPase activity
Purified protein was reconstituted in lipid, as described previously (38) with some modifications. Briefly, l-α-lecithin from egg yolk (Fujifilm Wako) was resuspended in reaction buffer (50 mm Tris-HCl (pH 7.4), 0.1 mm EGTA) at a final concentration of 20 mg/ml. The suspension was sonicated in a bath sonicator. The purified protein (2.1 μg) was mixed with 40 μg of lipid, and the mixture was incubated on ice for 20 min. Proteoliposomes were incubated at 37 °C for 30 min with 3 mm ATP disodium salt and 5 mm MgCl2 in 20 μl of reaction buffer. Verapamil chloride, DAPI, and BeFx (10 mm BeSO4, 50 mm NaF) were added to the reaction buffer at 100 μm, 2 μg/ml, and 1 mm, respectively. ATPase reaction was stopped by the addition of 20 μl of 20 mm EDTA. The ATPase activity was estimated by measuring the amount of released ADP by HPLC using a titanium column (39).
Statistical analysis
Multiple comparisons were evaluated using Tukey's post hoc test following one-way analysis of variance. All statistical analyses were performed using Origin2018 software (LightStone, Tokyo, Japan). Box and whisker plots show the following: boxes represent the 25th to 75th percentile range; the lines in the boxes represent the median; whiskers represent the maximum and the minimum points within the 1.5× interquartile range; notches represent the median with confidence intervals; and crosses represent mean values. Individual data points are shown on the left of the plots. All experiments were performed at least twice, and similar results were obtained.
Author contributions
R. F. investigation; R. F. writing-original draft; F. O. and N. K. data curation; T. I. and Y. K. methodology; A. K. visualization; Y. K. and N. K. validation; N. K. and K. U. supervision; K. U. conceptualization; K. U. funding acquisition; K. U. project administration; K. U. writing-review and editing.
Supplementary Material
This work was supported by JSPS KAKENHI Grants 18H05269 (to K. U.) and 18K19176 (to Y. K.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Movie S1.
- P-gp
- P-glycoprotein
- BeFx
- beryllium fluoride
- DAPI
- 4′,6-diamidino-2-phenylindole
- MgAMP-PNP
- Mg-5′-adenylyl imidodiphosphate
- PDB
- Protein Data Bank
- SLO
- streptolysin O
- TMD
- transmembrane domain
- NBD
- nucleotide-binding domain
- ABC
- ATP-binding cassette
- DEER
- double electron–electron resonance
- R6G
- rhodamine 6G
- AMP-PNP
- adenylyl-imidodiphosphate
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- DDM
- n-dodecyl-β-d-maltopyranoside
- HBSS
- Hanks' balanced saline solution
- PEI-MAX
- polyethyleneimine Max.
References
- 1. Ueda K., Cardarelli C., Gottesman M. M., and Pastan I. (1987) Expression of a full-length cDNA for the human “MDR1” gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc. Natl. Acad. Sci. U.S.A. 84, 3004–3008 10.1073/pnas.84.9.3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gottesman M. M., and Ling V. (2006) The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett. 580, 998–1009 10.1016/j.febslet.2005.12.060 [DOI] [PubMed] [Google Scholar]
- 3. Schinkel A. H., Smit J. J., van Tellingen O., Beijnen J. H., Wagenaar E., van Deemter L., Mol C. A., van der Valk M. A., Robanus-Maandag E. C., and te Riele H. P. (1994) Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood–brain barrier and to increased sensitivity to drugs. Cell 77, 491–502 10.1016/0092-8674(94)90212-7 [DOI] [PubMed] [Google Scholar]
- 4. Li J., Jaimes K. F., and Aller S. G. (2014) Refined structures of mouse P-glycoprotein. Protein Sci. 23, 34–46 10.1002/pro.2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moeller A., Lee S. C., Tao H., Speir J. A., Chang G., Urbatsch I. L., Potter C. S., Carragher B., and Zhang Q. (2015) Distinct conformational spectrum of homologous multidrug ABC transporters. Structure 23, 450–460 10.1016/j.str.2014.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bársony O., Szalóki G., Türk D., Tarapcsák S., Gutay-Tóth Z., Bacsó Z., Holb I. J., Székvölgyi L., Szabó G., Csanády L., Szakács G., and Goda K. (2016) A single active catalytic site is sufficient to promote transport in P-glycoprotein. Sci. Rep. 6, 24810 10.1038/srep24810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zoghbi M. E., Mok L., Swartz D. J., Singh A., Fendley G. A., Urbatsch I. L., and Altenberg G. A. (2017) Substrate-induced conformational changes in the nucleotide-binding domains of lipid bilayer-associated P-glycoprotein during ATP hydrolysis. J. Biol. Chem. 292, 20412–20424 10.1074/jbc.M117.814186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim Y., and Chen J. (2018) Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 359, 915–919 10.1126/science.aar7389 [DOI] [PubMed] [Google Scholar]
- 9. Juliano R. L., and Ling V. (1976) A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 455, 152–162 10.1016/0005-2736(76)90160-7 [DOI] [PubMed] [Google Scholar]
- 10. Chen C. J., Chin J. E., Ueda K., Clark D. P., Pastan I., Gottesman M. M., and Roninson I. B. (1986) Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 47, 381–389 10.1016/0092-8674(86)90595-7 [DOI] [PubMed] [Google Scholar]
- 11. Ueda K., Cornwell M. M., Gottesman M. M., Pastan I., Roninson I. B., Ling V., and Riordan J. R. (1986) The mdr1 gene, responsible for multidrug-resistance, codes for P-glycoprotein. Biochem. Biophys. Res. Commun. 141, 956–962 10.1016/S0006-291X(86)80136-X [DOI] [PubMed] [Google Scholar]
- 12. Kodan A., Yamaguchi T., Nakatsu T., Sakiyama K., Hipolito C. J., Fujioka A., Hirokane R., Ikeguchi K., Watanabe B., Hiratake J., Kimura Y., Suga H., Ueda K., and Kato H. (2014) Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc. Natl. Acad. Sci. U.S.A. 111, 4049–4054 10.1073/pnas.1321562111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kodan A., Yamaguchi T., Nakatsu T., Matsuoka K., Kimura Y., Ueda K., and Kato H. (2019) Inward- and outward-facing X-ray crystal structures of homodimeric P-glycoprotein CmABCB1. Nat. Commun. 10, 88 10.1038/s41467-018-08007-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Verhalen B., Dastvan R., Thangapandian S., Peskova Y., Koteiche H. A., Nakamoto R. K., Tajkhorshid E., and Mchaourab H. S. (2017) Energy transduction and alternating access of the mammalian ABC transporter P-glycoprotein. Nature 543, 738–741 10.1038/nature21414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dastvan R., Mishra S., Peskova Y. B., Nakamoto R. K., and Mchaourab H. S. (2019) Mechanism of allosteric modulation of P-glycoprotein by transport substrates and inhibitors. Science 364, 689–692 10.1126/science.aav9406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Verhalen B., Ernst S., Börsch M., and Wilkens S. (2012) Dynamic ligand-induced conformational rearrangements in P-glycoprotein as probed by fluorescence resonance energy transfer spectroscopy. J. Biol. Chem. 287, 1112–1127 10.1074/jbc.M111.301192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu R., and Sharom F. J. (1996) Site-directed fluorescence labeling of P-glycoprotein on cysteine residues in the nucleotide binding domains. Biochemistry 35, 11865–11873 10.1021/bi960823u [DOI] [PubMed] [Google Scholar]
- 18. Qu Q., and Sharom F. J. (2001) FRET analysis indicates that the two ATPase active sites of the P-glycoprotein multidrug transporter are closely associated. Biochemistry 40, 1413–1422 10.1021/bi002035h [DOI] [PubMed] [Google Scholar]
- 19. Kremers G. J., Goedhart J., van Munster E. B., and Gadella T. W. (2006) Cyan and yellow super-fluorescent proteins with improved brightness, protein folding, and FRET Förster radius. Biochemistry 45, 6570–6580 10.1021/bi0516273 [DOI] [PubMed] [Google Scholar]
- 20. Vanderklish P. W., Krushel L. A., Holst B. H., Gally J. A., Crossin K. L., and Edelman G. M. (2000) Marking synaptic activity in dendritic spines with a calpain substrate exhibiting fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. U.S.A. 97, 2253–2258 10.1073/pnas.040565597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kimura Y., Matsuo M., Takahashi K., Saeki T., Kioka N., Amachi T., and Ueda K. (2004) ATP hydrolysis-dependent multidrug efflux transporter: MDR1/P-glycoprotein. Curr. Drug Metab. 5, 1–10 10.2174/1389200043489090 [DOI] [PubMed] [Google Scholar]
- 22. Kimura Y., Kioka N., Kato H., Matsuo M., and Ueda K. (2007) Modulation of drug-stimulated ATPase activity of human MDR1/P-glycoprotein by cholesterol. Biochem. J. 401, 597–605 10.1042/BJ20060632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Szabó K., Welker E., Bakos, Müller M., Roninson I., Váradi A., and Sarkadi B. (1998) Drug-stimulated nucleotide trapping in the human multidrug transporter MDR1. Cooperation of the nucleotide binding domains. J. Biol. Chem. 273, 10132–10138 10.1074/jbc.273.17.10132 [DOI] [PubMed] [Google Scholar]
- 24. Tombline G., Bartholomew L. A., Urbatsch I. L., and Senior A. E. (2004) Combined mutation of catalytic glutamate residues in the two nucleotide binding domains of P-glycoprotein generates a conformation that binds ATP and ADP tightly. J. Biol. Chem. 279, 31212–31220 10.1074/jbc.M404689200 [DOI] [PubMed] [Google Scholar]
- 25. Stewart S. E., D'Angelo M. E., Paintavigna S., Tabor R. F., Martin L. L., and Bird P. I. (2015) Assembly of streptolysin O pores assessed by quartz crystal microbalance and atomic force microscopy provides evidence for the formation of anchored but incomplete oligomers. Biochim. Biophys. Acta 1848, 115–126 10.1016/j.bbamem.2014.10.012 [DOI] [PubMed] [Google Scholar]
- 26. Hofmann S., Januliene D., Mehdipour A. R., Thomas C., Stefan E., Brüchert S., Kuhn B. T., Geertsma E. R., Hummer G., Tampé R., and Moeller A. (2019) Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature 571, 580–583 10.1038/s41586-019-1391-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Urbatsch I. L., Sankaran B., Weber J., and Senior A. E. (1995) P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem. 270, 19383–19390 10.1074/jbc.270.33.19383 [DOI] [PubMed] [Google Scholar]
- 28. Urbatsch I. L., Tyndall G. A., Tombline G., and Senior A. E. (2003) P-glycoprotein catalytic mechanism: studies of the ADP-vanadate inhibited state. J. Biol. Chem. 278, 23171–23179 10.1074/jbc.M301957200 [DOI] [PubMed] [Google Scholar]
- 29. Alam A., Küng R., Kowal J., McLeod R. A., Tremp N., Broude E. V., Roninson I. B., Stahlberg H., and Locher K. P. (2018) Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc. Natl. Acad. Sci. U.S.A. 115, E1973–E1982 10.1073/pnas.1717044115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mechetner E. B., Schott B., Morse B. S., Stein W. D., Druley T., Davis K. A., Tsuruo T., and Roninson I. B. (1997) P-glycoprotein function involves conformational transitions detectable by differential immunoreactivity. Proc. Natl. Acad. Sci. U.S.A. 94, 12908–12913 10.1073/pnas.94.24.12908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Druley T. E., Stein W. D., and Roninson I. B. (2001) Analysis of MDR1 P-glycoprotein conformational changes in permeabilized cells using differential immunoreactivity. Biochemistry 40, 4312–4322 10.1021/bi001371v [DOI] [PubMed] [Google Scholar]
- 32. Müller M., Bakos E., Welker E., Váradi A., Germann U. A., Gottesman M. M., Morse B. S., Roninson I. B., and Sarkadi B. (1996) Altered drug-stimulated ATPase activity in mutants of the human multidrug resistance protein. J. Biol. Chem. 271, 1877–1883 10.1074/jbc.271.4.1877 [DOI] [PubMed] [Google Scholar]
- 33. Loo T. W., and Clarke D. M. (1995) Covalent modification of human P-glycoprotein mutants containing a single cysteine in either nucleotide-binding fold abolishes drug-stimulated ATPase activity. J. Biol. Chem. 270, 22957–22961 10.1074/jbc.270.39.22957 [DOI] [PubMed] [Google Scholar]
- 34. Azzaria M., Schurr E., and Gros P. (1989) Discrete mutations introduced in the predicted nucleotide-binding sites of the mdr1 gene abolish its ability to confer multidrug resistance. Mol. Cell. Biol. 9, 5289–5297 10.1128/MCB.9.12.5289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Urbatsch I. L., Beaudet L., Carrier I., and Gros P. (1998) Mutations in either nucleotide-binding site of P-glycoprotein (Mdr3) prevent vanadate trapping of nucleotide at both sites. Biochemistry 37, 4592–4602 10.1021/bi9728001 [DOI] [PubMed] [Google Scholar]
- 36. Iram S. H., Gruber S. J., Raguimova O. N., Thomas D. D., and Robia S. L. (2015) ATP-binding cassette transporter structure changes detected by intramolecular fluorescence energy transfer for high-throughput screening. Mol. Pharmacol. 88, 84–94 10.1124/mol.114.096792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ogasawara F., Kano F., Murata M., Kimura Y., Kioka N., and Ueda K. (2019) Changes in the asymmetric distribution of cholesterol in the plasma membrane influence streptolysin O pore formation. Sci. Rep. 9, 4548 10.1038/s41598-019-39973-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ishigami M., Tominaga Y., Nagao K., Kimura Y., Matsuo M., Kioka N., and Ueda K. (2013) ATPase activity of nucleotide binding domains of human MDR3 in the context of MDR1. Biochim. Biophys. Acta 1831, 683–690 10.1016/j.bbalip.2012.12.016 [DOI] [PubMed] [Google Scholar]
- 39. Kimura Y., Shibasaki S., Morisato K., Ishizuka N., Minakuchi H., Nakanishi K., Matsuo M., Amachi T., Ueda M., and Ueda K. (2004) Microanalysis for MDR1 ATPase by high-performance liquid chromatography with a titanium dioxide column. Anal. Biochem. 326, 262–266 10.1016/j.ab.2003.12.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.