

Abstract
IQ motif-containing GTPase-activating protein 1 (IQGAP1) is a scaffold protein that interacts with numerous binding partners and thereby regulates fundamental biological processes. The functions of IQGAP1 are modulated by several mechanisms, including protein binding, self-association, subcellular localization, and phosphorylation. Proteome-wide screens have indicated that IQGAP1 is ubiquitinated, but the possible effects of this post-translational modification on its function are unknown. Here we characterized and evaluated the function of IQGAP1 ubiquitination. Using MS-based analysis in HEK293 cells, we identified six lysine residues (Lys-556, -1155, -1230, -1465, -1475, and -1528) as ubiquitination sites in IQGAP1. To elucidate the biological consequences of IQGAP1 ubiquitination, we converted each of these lysines to arginine and found that replacing two of these residues, Lys-1155 and Lys-1230, in the GAP-related domain of IQGAP1 (termed IQGAP1 GRD-2K) reduces its ubiquitination. Moreover, IQGAP1 GRD-2K bound a significantly greater proportion of the two Rho GTPases cell division cycle 42 (CDC42) and Rac family small GTPase 1 (RAC1) than did WT IQGAP1. Consistent with this observation, reconstitution of IQGAP1-null cells with IQGAP1 GRD-2K significantly increased the amount of active CDC42 and enhanced cell migration significantly more than WT IQGAP1. Our results reveal that ubiquitination of the CDC42 regulator IQGAP1 alters its ability to bind to and activate this GTPase, leading to physiological effects. Collectively, these findings expand our view of the role of ubiquitination in cell signaling and provide additional insight into CDC42 regulation.
Keywords: scaffold protein, ubiquitylation (ubiquitination), CDC42, cell signaling, Ras-related C3 botulinum toxin substrate 1 (Rac1), GAP-related domain, IQGAP1, small GTPase
Introduction
The IQ motif-containing GTPase-activating protein (IQGAP)2 family of scaffold proteins has three members, termed IQGAP1, IQGAP2, and IQGAP3 (1). IQGAPs contain several domains that mediate protein-protein interactions. These domains include a calponin homology domain (CHD), a region containing two tryptophans (WW), four IQ motifs (IQ), a Ras GTPase-activating protein-related domain (GRD), and a RasGAP C terminus (RGCT) (2). The multidomain composition of IQGAPs mediates the formation of protein complexes required for cellular processes (1). By scaffolding multiple proteins, IQGAP1, the most well-characterized member of the IQGAP family, integrates signaling pathways and coordinates cellular activities including cell migration (3), cell proliferation (4), intracellular signaling (5), vesicle trafficking (6), and cytoskeletal dynamics (2, 7).
The Rho GTPases cell division cycle 42 (CDC42) and Rac family small GTPase 1 (RAC1) are among the best characterized IQGAP1-binding partners (8). These GTPases cycle between a GTP-bound active state and a GDP-bound inactive state (9). Guanine nucleotide exchange factors (GEFs) promote the exchange of GDP for GTP, whereas GAPs enhance the intrinsic GTPase activity of the small G proteins, promoting hydrolysis of the bound GTP. The activities of these GEFs and GAPs are regulated by signaling pathways, including pathways initiated by ligand binding to cell-surface receptors (10). For example, platelet-derived growth factor and epidermal growth factor (EGF) promote GEF activity and therefore GTPase activation (11–13). Active CDC42 and RAC1 modulate actin filament reorganization, which contributes to numerous biological processes such as cell polarity, cell adhesion, the formation of lamellipodia and filopodia, and cell migration (14, 15).
Although similar in sequence to the catalytic domain of RasGAPs, the GRD of IQGAP1 does not have GAP activity (16). Instead, the GRD of IQGAP1 binds to GTP-bound CDC42 and RAC1, and inhibits their intrinsic GTPase activity (17, 18). As a consequence, IQGAP1 stabilizes CDC42 and RAC1 in their active forms and induces reorganization of the actin cytoskeleton (8, 19). Consistent with these observations, overexpression of IQGAP1 increases the amounts of active CDC42 and RAC1 in cells, promoting cell motility (3). Moreover, CDC42 and RAC1 participate in IQGAP1-stimulated proliferation and augmentation of tumorigenesis of human breast epithelial cells (20). In addition, through its interaction with CDC42, IQGAP1 regulates invasion of Salmonella (21). Thus, the association of IQGAP1 with CDC42 and RAC1 has biological relevance.
Post-translational modifications increase the functional diversity of the proteome by the covalent addition of functional groups to proteins. Reported modifications on IQGAP1 include phosphorylation (22), acetylation (23), ISG (interferon-stimulated gene 15)-ylation (24, 25), SUMOylation (26), and ubiquitination (27–30). Many of the studies that have identified these post-translational modifications, used high throughput MS methods to identify proteome-wide modifications, with little subsequent functional analysis.
Ubiquitination results from enzymatic linkage of the polypeptide ubiquitin to a lysine residue on target proteins. Following monoubiquitination, ubiquitin can itself be modified on any of its seven lysine residues or at its N terminus, leading to formation of polymeric ubiquitin chains (31). The role of ubiquitination has been extensively studied in the ubiquitin proteasome system where substrate-linked ubiquitin provides a signal for proteasomal degradation of target proteins (32). In addition, ubiquitination regulates nonproteolytic processes, including protein activity, localization, and interaction with other proteins (33, 34). Ubiquitinated IQGAP1 peptides have been identified in large-scale MS analyses of several cellular proteomes (27–30). However, the sites found globally in these studies often reflect ubiquitination of newly synthetized proteins that are misfolded and therefore targeted for immediate degradation (27). We are not aware of any studies where a detailed characterization of IQGAP1 ubiquitination was carried out or where the functional consequences of IQGAP1 ubiquitination was determined.
In this study, we investigated ubiquitination of IQGAP1 and its functional consequences. Using biochemical and MS analysis we demonstrate that IQGAP1 is ubiquitinated in HEK293 cells and identify the ubiquitination sites. Replacement of ubiquitinated lysine residues with arginine reduces ubiquitination of IQGAP1 in cells. In addition, mutation of the ubiquitinated lysines in the GRD increases the interaction of IQGAP1 with CDC42 and RAC1, and increases the amount of active CDC42 in cells.
Results
IQGAP1 is ubiquitinated in cells
Ubiquitination of IQGAP1 was assessed in HEK293 cells. Cells were transfected with Myc-IQGAP1 and/or His-ubiquitin. Proteins were resolved by SDS-PAGE and the amount of ubiquitination was determined by Western blotting with anti-ubiquitin antibodies. Incubation with the proteasome inhibitor MG132 increases ubiquitination of numerous cellular proteins (Fig. 1A, upper left panel). Probing lysates with anti-IQGAP1 antibodies revealed a single IQGAP1 band in lysates from cells transfected with Myc-IQGAP1 alone. In contrast, multiple higher molecular weight bands were observed in lysates of cells that express both Myc-IQGAP1 and His-ubiquitin (Fig. 1A, lower left panel). The multiple bands are likely due to multiple ubiquitination sites and/or polyubiquitination of IQGAP1, which is enhanced by the expression of His-ubiquitin. Immunoprecipitation of Myc-IQGAP1 and Western blotting with anti-ubiquitin antibodies confirmed that IQGAP1 is ubiquitinated in cells transfected with ubiquitin (Fig. 1A, upper right panel, compare second and fourth lanes). MG132 increased ubiquitination of samples where IQGAP1 was immunoprecipitated from cells expressing His-ubiquitin (Fig. 1A, upper right panel).
Figure 1.

Ubiquitination of IQGAP1. A, HEK293 cells were transfected with (+) or without (−) Myc-IQGAP1 and/or His-ubiquitin (Ub). Where indicated, cells were incubated with MG132 (+) or DMSO (−) (vehicle) for 4 h. Equal amounts of protein lysate were loaded directly onto gels or immunoprecipitated (IP) with anti-Myc beads. Proteins were analyzed by SDS-PAGE and immunoblotting (IB) using anti-ubiquitin and anti-IQGAP1 antibodies. B, HEK293 cells were transfected with (+) or without (−) His-ubiquitin and incubated with MG132 (+) or DMSO (−). His-ubiquitin was pulled-down (PD) by incubating equal amounts of protein lysate with TALON beads. Lysates and complexes were resolved by Western blotting. The PVDF membranes were probed with anti-ubiquitin and anti-IQGAP1 antibodies. All data are representative of at least three independent experiments.
In a second approach, IQGAP1 ubiquitination was studied using a pulldown assay. His-ubiquitin, expressed in HEK293 cells, was isolated with TALON beads. Enhanced ubiquitination of multiple proteins was observed in both the lysates and pulldowns when cells were incubated with MG132 (Fig. 1B). Western blots revealed that endogenous IQGAP1 was pulled down with His-ubiquitin. Moreover, additional bands that migrated above the main IQGAP1 band were observed in the pulldowns probed with anti-IQGAP1 antibody (Fig. 1B, upper panel, fifth and sixth lanes). Taken together, the data strongly suggest that IQGAP1 is ubiquitinated.
Identification of IQGAP1 ubiquitination sites
We used MS to confirm that IQGAP1 is ubiquitinated and to identify the specific sites of ubiquitin conjugation. Samples were prepared from HEK293 cells, transfected with or without His-ubiquitin, and incubated with or without MG132. Endogenous IQGAP1 was immunopurified and a portion of the samples resolved by Western blotting using anti-IQGAP1 and anti-ubiquitin antibodies (Fig. 2A, middle and bottom panels). The remainder of each sample was resolved by SDS-PAGE and proteins were stained with Colloidal Coomassie. Three gel sections were excised, digested with trypsin, and analyzed by LC-MS/MS sequencing. One region (section 3) corresponds to IQGAP1 itself, whereas the other two (sections 1 and 2) are higher molecular weight regions (Fig. 2A) that would be expected to contain ubiquitinated IQGAP1. Peptides containing six specific lysine residues were identified as sites of ubiquitination, namely Lys-556, Lys-1155, Lys-1230, Lys-1465, Lys-1475 and Lys-1528. A representative MS/MS spectrum of the ubiquitinated IQGAP1 peptide 1517–1532, containing ubiquitinated Lys-1528 is shown in Fig. 2B. Spectra of peptides containing all six identified ubiquitinated lysines are shown in Fig. S1. All six ubiquitinated peptides were identified in both gel sections 1 and 2 (Fig. 2A), but they were 2–9–fold less abundant in the highest molecular weight region (section 1). In sections 1 and 2 of the gel, we also identified tryptic peptides corresponding to polyubiquitin chains on Lys-29 and Lys-48 of ubiquitin. Here again, these peptides were less abundant in section 1 than in section 2. We were unable to determine the type of ubiquitination (mono- or polyubiquitination) at each reported IQGAP1 site as this information was lost following tryptic digestion. Taken together, these data suggest that, under our assay conditions, ubiquitin does not form lengthy polyubiquitin chains on IQGAP1.
Figure 2.

Identification of ubiquitination sites on IQGAP1. A, HEK293 cells were transfected with (+) or without (−) His-ubiquitin (Ub) and incubated with MG132 (+) or DMSO (−). Lysates were IP with anti-IQGAP1 polyclonal antibodies and resolved by SDS-PAGE. Three gel sections corresponding to IQGAP1 and the regions above IQGAP1 (1–3; red rectangles) were excised, digested with trypsin, and analyzed by LC-MS/MS as described under “Experimental procedures.” A portion of the immunoprecipitated lysate was analyzed by Western blotting and probed with anti-IQGAP1 and anti-ubiquitin antibodies. Aliquots of lysate not subjected to immunoprecipitation were processed in parallel (lysate). B, LC-MS/MS analysis of the in-gel tryptic digested samples allowed the identification of ubiquitinated lysine residues. A representative MS/MS spectrum of a ubiquitinated peptide is shown for peptide IQGAP1 1517–1532 containing ubiquitinated Lys-1528. C, schematic representation of IQGAP1 showing the six ubiquitination sites identified by LC-MS/MS and the tryptic peptides in which each is located. CC, coiled-coil; WW, tryptophan-containing domain; IQ, IQ domain.
Lys-556 is located in the coiled-coil region, Lys-1155 and Lys-1230 are in the GRD, and Lys-1465, Lys-1475, and Lys-1528 are in the RGCT (Fig. 2C). Each of these sites has been previously identified in more than five high-throughput proteomic experiments, where ubiquitinated peptides have been enriched from whole cell lysates (35). Moreover, three of the sites, namely Lys-1230, Lys-1465, and Lys-1528, have also been reported as sites of lysine acetylation (35). Because these modifications (ubiquitination and acetylation) are mutually exclusive and can have functional consequences, we searched for acetylation on all six lysines residues, but found no evidence for it.
The ubiquitinated peptides detected with or without MG132 were identical. This observation raises the possibility that ubiquitination of the sites we identified may not be regulated by the proteasome pathway. In accordance with this hypothesis, the amount of endogenous IQGAP1 proteins in cells incubated with MG132 is essentially identical to that in cells incubated with vehicle (Fig. S2).
Mutation of selected lysine residues reduces ubiquitination of IQGAP1
To elucidate the effect of IQGAP1 ubiquitination on its function, we developed constructs in which the ubiquitinated lysine residues identified in our MS analysis were replaced with arginine. Four Myc-tagged point mutant IQGAP1 constructs were generated, namely IQGAP1 CC-1K (K556R), IQGAP1 GRD-2K (K1155R and K1230R), IQGAP1 RGCT-3K (K1465R, K1475R, and K1528R), and IQGAP1–6K (K556R, K1155R, K1230R, K1465R, K1475R, and K1528R) (Fig. 3A). To avoid interference from endogenous IQGAP1, we expressed the IQGAP1 constructs in HEK293 cells that have stable IQGAP1-knockdown (Fig. 3B). The amount of Myc-IQGAP1 expressed in knockdown HEK293 cells was similar to that of endogenous IQGAP1 in control cells (Fig. 3C). In addition, similar amounts of each IQGAP1 constructs are expressed in the reconstituted cells (Fig. 3D). The extent of IQGAP1 ubiquitination was determined by quantifying ubiquitin bands and correcting for the total amount of immunopurified IQGAP1 in the same sample. Both WT IQGAP1 and all the lysine mutant proteins exhibited some ubiquitination (Fig. 3, D and E). The extent of ubiquitination of IQGAP1 GRD-2K, IQGAP1 RGCT-3K, and IQGAP1–6K was significantly (∼40–50%) lower than that of WT IQGAP1. Mutation of Lys-556 (IQGAP1 CC-1K) did not significantly reduce the extent of ubiquitination (Fig. 3, D and E). These data reveal that mutation of the identified ubiquitination sites within the GRD or the RGCT domain leads to a decrease in the overall ubiquitination of IQGAP1.
Figure 3.

Mutation of selected lysine residues of IQGAP1 reduces its ubiquitination. A, schematic representation of IQGAP1 mutant constructs. Individual lysine residues are replaced with arginine for each construct: CC-1K (Lys-556 is replaced with Arg), GRD-2K (K1155R; K1230R), RGCT-3K (K1465R; K1475R; K1528R), and 6K (K556R; K1155R; K1230R; K1465R; K1475R; K1528R). All plasmids contain a Myc-tag. B, equal amounts of protein lysate from HEK293 control (+/+) and IQGAP1-knockdown (−/−) cells generated with the CRISPR/Cas9 system were resolved by Western blotting. PVDF membranes were probed with anti-IQGAP1 and anti-actin (loading control) antibodies. A representative blot is shown. C, WT Myc-IQGAP1 was expressed in IQGAP1-knockdown HEK293 cells (−/−). Equal amounts of protein lysate from control (+/+) and knockdown cells were resolved by Western blotting using anti-IQGAP1, anti-Myc, and anti-actin antibodies. A representative blot is shown. D, IQGAP1-knockdown HEK293 cells were transfected with vector (V), WT IQGAP1 or the indicated IQGAP1 mutant constructs. Samples were IP using anti-Myc beads. Lysates and immunopurified proteins were resolved by Western blotting. Membranes were probed with anti-IQGAP1 and anti-ubiquitin antibodies. Data are representative of at least four independent experiments. E, the ubiquitin bands were quantified with Image Studio 2.0 and corrected for the amount of immunoprecipitated IQGAP1 in the corresponding sample. Data are expressed as mean ± S.D. (n = 5 to 6), with cells transfected with WT IQGAP1 set as 1. Each mutant protein was compared with WT IQGAP1 using Welch's t test. *, p < 0.05.
Reducing IQGAP1 ubiquitination enhances its interactions with CDC42 and RAC1
The GRD of IQGAP1 binds to the Rho GTPases CDC42 and RAC1 (17, 36, 37). Because two ubiquitination sites are located in the GRD (Fig. 2), we evaluated the impact of IQGAP1 ubiquitination on its interaction with CDC42 and RAC1. HEK293 cells lacking IQGAP1 were transfected with WT or mutant IQGAP1 constructs. Lysates were incubated with purified GST-CDC42(Q61L) (constitutively active form), and complexes were detected by Western blotting. As expected, transfected WT IQGAP1 binds CDC42 (Fig. 4A). Binding of IQGAP1 to GST alone is minimal, verifying the specificity of the interaction with CDC42. The three lysine mutations in the RGCT did not significantly impact the binding of IQGAP1 to CDC42 (Fig. 4, A and B). Binding of IQGAP1 GRD-2K to CDC42 was significantly enhanced; quantification revealed a 1.6-fold increase over WT IQGAP1. IQGAP1–6K, which contains the same mutations in the GRD as IQGAP1 GRD-2K, also had 1.6-fold more binding than WT IQGAP1 to CDC42 (Fig. 4, A and B). By contrast, mutation of Lys-556 in the coil-coiled region (CC-1K) decreased the interaction between IQGAP1 and CDC42 by 35%.
Figure 4.

Mutation of ubiquitination sites in the GRD of IQGAP1 enhances its binding to CDC42 and RAC1. A, IQGAP1-knockdown HEK293 cells were transfected with WT IQGAP1 or the indicated mutant IQGAP1 proteins and incubated with MG132. GST-CDC42(Q61L) or GST alone was incubated with equal amounts of protein from cell lysates. Complexes were isolated with GSH-Sepharose. Lysates and pulled down (PD) proteins were analyzed by Western blotting using anti-Myc and/or anti-GST antibodies. B, Myc-IQGAP1 in GST pulldowns was quantified with Image Studio 2.0 (LI-COR) and corrected for the amount of GST-CDC42 in the same sample. Data are expressed as mean ± S.D. (n = 5), with cells transfected with WT IQGAP1 set as 1. Each mutant protein was compared with WT IQGAP1 using Welch's t test. *, p < 0.05. C, RAC1 pulldown was performed as described above for CDC42, except GST-RAC1(Q61L) was used. D, Myc-IQGAP1 was quantified as described for panel B, except data were corrected for the amount of GST-RAC1 in the same sample. Data were analyzed as described for panel B (n = 3, except for IQGAP1–6K with n = 2). *, p < 0.05.
Similar experiments were performed using GST-tagged RAC1(Q61L) (constitutively active form). Analogous to the observations with CDC42, binding of IQGAP1 GRD-2K and IQGAP1–6K to RAC1 was 3.3- and 2.7-fold, respectively, greater than that of WT IQGAP1 (Fig. 4, C and D), although these differences did not reach statistical significance. IQGAP1 CC-1K exhibited reduced RAC1 binding. Unexpectedly, lysine mutation within the RGCT region decreased by 29% the binding of IQGAP1 to RAC1. Collectively, these data strongly suggest that ubiquitination of the GRD of IQGAP1 modulates its interaction with CDC42 and RAC1.
Mutation of ubiquitination sites within the IQGAP1 GRD enhances activation of CDC42
Previous publications from both our laboratory (18, 38) and other investigators (39) documented that IQGAP1 maintains CDC42 in its active (GTP-bound) state by inhibiting its intrinsic GTPase activity. Moreover, expression of an IQGAP1 construct with deletion of the GRD reduces the amount of active CDC42 in cells (18). To evaluate the impact of ubiquitination in IQGAP1 GRD on CDC42 activation, active CDC42 was quantified in cell lysates. Analysis was performed in IQGAP1-knockdown HEK293 cells transfected with WT IQGAP1, IQGAP1 GRD-2K, or vector alone. Western blotting revealed that the amount of IQGAP1 protein expressed was approximately the same for all the constructs (Fig. 5A). Rescue of IQGAP1-knockdown cells with WT IQGAP1 increased the amount of active CDC42 in serum-starved cells by 2.4-fold (Fig. 5, A and B). Transfecting cells with IQGAP1 GRD-2K further augmented active CDC42; these cells had 3.7-fold more active CDC42 than vector-transfected cells (Fig. 5B). Moreover, under serum-free conditions, cells reconstituted with IQGAP1 GRD-2K had significantly (1.6-fold) more active CDC42 than cells transfected with WT IQGAP1. These findings strongly suggest that ubiquitination of the GRD of IQGAP1 regulates activation of CDC42 in cells.
Figure 5.
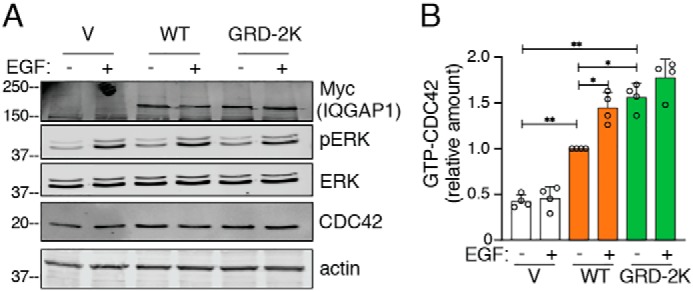
Mutation of ubiquitination sites in the IQGAP1 GRD enhances activation of CDC42. A, IQGAP1-knockdown HEK293 cells were transfected with vector (V), WT IQGAP1, or IQGAP1 GRD-2K. Cells were starved of serum overnight and stimulated with (+) or without (−) 100 ng/ml of EGF for 10 min. Equal amounts of protein lysate were analyzed by Western blotting using anti-Myc, anti-pERK, anti-ERK, and anti-actin (loading control) antibodies. B, equal amounts of protein lysate were used to quantify active CDC42 using GTPase-specific ELISA (G-LISA) as described under “Experimental procedures.” The amounts of GTP-CDC42 are expressed as mean ± S.D. (n = 4, each performed in triplicate), with unstimulated cells expressing WT IQGAP1 set as 1. *, p < 0.05; **, p < 0.001; Welch's t test.
Stimulation of cells with EGF has been demonstrated to increase the amount of active CDC42 (12). Therefore, we examined the effect of EGF on the amount of active CDC42 in serum-starved HEK293 cells. We validated that EGF activates EGF receptor signaling in these cells by observing increased ERK phosphorylation (pERK) (Fig. 5A). Interestingly, EGF was unable to augment the amount of active CDC42 in IQGAP1-knockdown HEK293 cells (Fig. 5). Reconstitution of IQGAP1-knockdown cells with WT IQGAP1 enabled EGF to significantly increase the amount of active CDC42. By contrast, addition of EGF to serum-starved cells rescued with IQGAP1 GRD-2K did not produce a significant increase in active CDC42 (Fig. 5).
Mutation of ubiquitination sites within the IQGAP1 GRD enhances cell migration
To investigate the physiological effects of IQGAP1 ubiquitination, we developed IQGAP1−/− MEFs that stably express WT IQGAP1 or IQGAP1 GRD-2K under control of a tetracycline promoter. The amount of IQGAP1 protein expressed in the reconstituted IQGAP1−/− MEFs is similar to that of endogenous IQGAP1 in control (IQGAP1+/+) MEFs (Fig. 6A). Moreover, similar amounts of WT IQGAP1 and IQGAP1 GRD-2K are expressed in these cells. We routinely see two bands of GFP-tagged IQGAP1 (Fig. 6A), presumably due to cleavage of one GFP molecule from the dual GFP-tagged IQGAP1 constructs (40).
Figure 6.

Mutation of ubiquitination sites in the IQGAP1 GRD enhances cell migration. A, expression of GFP-tagged WT IQGAP1 (WT) or IQGAP1 GRD-2K (GRD-2K) was induced in IQGAP1-null MEF cells (−/−) by adding doxycycline (Dox). Equal amounts of protein lysate from IQGAP1-null (−/−), control (+/+), and reconstituted MEFs were resolved by Western blotting. Membranes were probed with anti-IQGAP1 and anti-tubulin antibodies. A representative blot is shown. B, wound healing assays were performed using IQGAP1−/− MEFs reconstituted with WT IQGAP1 or IQGAP1 GRD-2K. After serum starvation, cells were incubated with medium containing 0.1% FBS, 100 ng/ml of doxycycline, and 25 ng/ml of EGF, and a wound was generated. Images were taken every 30 min for 24 h. Representative wounds are shown at 0, 4, 8, 16, and 24 h (scale bar, 100 μm). A video of the migration is provided in the supporting information (Video S1). C, open wound areas were analyzed with Fiji/ImageJ as described under “Experimental procedures.” Data are expressed as mean ± S.D. (1 or 2 fields from at least 6 independent wells were analyzed; WT IQGAP1 n = 9; IQGAP1 GRD-2K n = 14). Means between the two groups are significantly different at 3 h (p < 0.05) and reached p < 0.001 at 4.5 h (Welch's t test).
Cell motility was evaluated by a wound healing assay using a confluent monolayer of cells. The cells along the wound edge were imaged by time-lapse microscopy over a 24-h time period. Cells reconstituted with WT IQGAP1 or IQGAP1 GRD-2K migrated into the wound by cell spreading, not cell division (Fig. 6B and Video S1). Importantly, cells expressing IQGAP1 GRD-2K reduced the width of the wound more quickly than cells expressing WT IQGAP1. Quantification of the cell-free area of the wound revealed that cells expressing IQGAP1 GRD-2K reduced the wound area by 19 ± 6.3% (mean ± S.D., n = 14) 3 h after the wound was generated (Fig. 6C). By contrast, cells reconstituted with WT IQGAP1 achieved 14 ± 2.6% (mean ± S.D., n = 9) closure at 3 h. At 16 h, cells expressing IQGAP1 GRD-2K had 99 ± 0.7% closure, whereas WT IQGAP1 had closed the wound by only 59 ± 10.3%. These data strongly suggest that ubiquitination of IQGAP1 elicits effects on cell function.
Ubiquitination of the GRD likely blocks CDC42 binding
We used a computational approach to depict the spatial organization of the interaction between the ubiquitinated GRD and CDC42. The crystal structure of the IQGAP2 GRD (GRD2) bound to CDC42 has been solved (17). The structure revealed that four molecules of CDC42-GTP bind to two GRD2 molecules, which in turn bind to each other, giving an overall CDC42-to-GRD2 stoichiometry of 2:1. Two CDC42 molecules bind to the GRD2 “extra” subdomains from each GRD2 (Ex mode binding), whereas two CDC42 molecules bind the middle region of each GRD2 (RasGAP mode binding). Although the crystal structure of the isolated GRD of IQGAP1 (GRD1) has been solved (36), the complex of GRD1 with CDC42 has not been crystallized. The 384 residues that comprise the GRDs of IQGAP1 and IQGAP2 are over 75% identical in sequence with the CDC42-contacting residues having >80% identity. This suggests that the interactions of GRD1 and GRD2 with CDC42 are similar (17). This argument is supported by isothermal titration calorimetry, mutagenesis, and modeling considerations, which indicate that GRD1 binds two molecules of CDC42, in a manner very similar to GRD2 (17). Therefore, we used the GRD2/CDC42 structure as a guide to evaluate the impact of ubiquitination of GRD1 on CDC42 binding (Fig. 7).
Figure 7.

Ubiquitination of Lys-1155 and Lys-1230 in the GRD of IQGAP1 would impair CDC42 binding. A, two CDC42 molecules (green with red switch regions) bind to the GRD (yellow). One CDC42 molecule binds to the extra subdomain (Ex mode binding), whereas the other CDC42 molecule binds the middle region of GRD (RasGAP mode binding). B, ubiquitin (orange) conjugation at Lys-1155 tethers a large molecule very close to the CDC42 Ex mode-binding site; shown here occupied by CDC42. Inset, a magnified view of the interaction, with Gly-75 and Gly-76 of ubiquitin shown in stick representation. C, conjugation of ubiquitin at Lys-1230 within the GRD of IQGAP1. D, the same orientation of the GRD with CDC42 occupying the RasGAP-binding site. E, significant steric overlap would prevent simultaneous ubiquitin conjugation and CDC42 binding to the GRD.
Like the GRD2/CDC42 complex, the GRD1/CDC42 complex is formed by the binding of two CDC42 molecules (Ex mode binding and RasGAP mode binding) to one GRD1 molecule (Fig. 7A). The probable effect of ubiquitin conjugation at Lys-1155 on the binding of CDC42 to GRD1 via the Ex mode is depicted in Fig. 7B. In GRD2, the residue equivalent to Lys-1155 is surface-exposed Leu-1068, which in the GRD2/CDC42 structure makes favorable van der Waals contacts with Lys-131 and Lys-133 of CDC42 helix α3 (17). In GRD1, Lys-1155 would likely adopt a rotamer that would allow for a water-mediated interaction with the backbone oxygen of Ala-130, also within CDC42 helix α3. Considering the close proximity of Lys-1155 to the Ex mode-binding site, it is likely that covalent attachment of an 8-kDa ubiquitin would significantly reduce CDC42 binding at that site through a combination of steric clash and a dynamic screening effect by the bound yet mobile ubiquitin moiety (Fig. 7B). Nevertheless, we cannot completely exclude the possibility that a specific rotamer conformation for Lys-1155 along with minimal interference by the attached ubiquitin might allow for both Lys-1155 ubiquitination and CDC42 Ex mode binding.
Lys-1230 (Lys-1143 in GRD2) is located almost in the center of the RasGAP mode-binding site. In the GRD2/CDC42 complex, this lysine is hydrogen bonding to the carbonyl oxygen of Phe-37 of CDC42 and within hydrogen bonding distance of two bound waters trapped at the GRD2/CDC42 interface (17). When ubiquitin attaches to Lys-1230, different Lys-1230 rotamers (if these are in fact sampled through time) would have the effect of re-positioning and re-orienting the attached ubiquitin with respect to the GRD. However, because of the central location of Lys-1230 within the surface required by CDC42 for RasGAP mode binding, it appears that ubiquitin conjugation would preclude binding of CDC42 independent of any Lys-1230 rotamer sampling.
Mutation of Lys-1230 to arginine would very likely not be disruptive and would still allow for favorable van der Waals or polar interactions with CDC42. To verify this, we compared the abilities of pure WT IQGAP1 and IQGAP1 GRD-2K to bind to pure CDC42 in vitro (Fig. S3). GST-pulldown analysis revealed no difference in the amount of CDC42 bound (Fig. S3A). Essentially identical data were obtained with binding of the IQGAP1 constructs to RAC1 (Fig. S3B), verifying that replacement of Lys-1155 and Lys-1230 with arginine does not influence the interactions of IQGAP1 with CDC42 or RAC1.
In addition, in an effort to determine whether our conservative mutations of lysine-to-arginine would have a deleterious effect on the folding of the GRD, we used SWISS-MODEL to make an energy-minimized model containing the two arginine residues at positions 1068 and 1143. We found that the overall root-mean-square deviation/displacement for 360 Cα carbon positions is 0.136 Å with an average positional deviation of 0.121 Å and a maximum positional deviation of 0.363 Å. The Cα displacements at positions 1068 and 1143 are 0.068 and 0.062 Å, respectively. These small differences between the structures suggest that the introduction of arginine residues would not have a deleterious effect on the proper folding of the domain. Collectively, it appears that reduced binding of CDC42 is accomplished by mono- or poly-ubiquitin simply blocking access to the GRD. However, we cannot rule out the possibility that ubiquitination also induces conformational changes in the GRD that reduce CDC42 binding.
Discussion
Post-translational modifications are important mechanisms for regulation of protein function (32–34). Several post-translational modifications of IQGAP1 have been identified by MS (35), but only a few studies have demonstrated functional effects resulting from these changes. Phosphorylation of Ser-1441 and Ser-1443 of IQGAP1 promotes neurite outgrowth (22), whereas SUMOylation of Lys-1445 enhances proliferation and migration of colorectal carcinoma cells (26). Ubiquitination of IQGAP1 has been detected in proteome-wide ubiquitin screens (27–30), yet biological role(s) for ubiquitinated IQGAP1 have not been previously identified.
Although initially thought to modulate only proteolysis, subsequent findings revealed that ubiquitination has important functions in cell signaling (33, 34, 41). Using MS, we identified six lysine residues in IQGAP1 that are ubiquitinated. To explore the biological relevance of IQGAP1 ubiquitination, we considered the locations of the sites at which ubiquitin is attached. These are in three regions of IQGAP1, namely the coiled-coil region, the GRD, and the RGCT (see Fig. 2C). Two ubiquitinated residues, Lys-1155 and Lys-1230, are in the GRD. The GRD of IQGAP1 is the region that binds to the GTPases CDC42 and RAC1 (17, 37). These interactions have been the focus of considerable attention, with numerous papers characterizing these associations (16, 18, 19, 37, 42–44). We hypothesized that ubiquitination in the GRD might influence the binding of IQGAP1 to CDC42 and RAC1.
To test this, we replaced Lys-1155 and Lys-1230 of IQGAP1 with arginine. Removing these lysine residues decreases IQGAP1 ubiquitination in cells. Moreover, IQGAP1 GRD-2K binds significantly more CDC42 than the WT protein. These observations suggest that ubiquitination of Lys-1155 and Lys-1230 attenuates the interaction of IQGAP1 with CDC42. Illustration of the GRD/CDC42 complex supports this interpretation. Ubiquitination of Lys-1230 creates steric hindrance that precludes CDC42 RasGAP mode binding (see Fig. 7). Although ubiquitin attached to Lys-1155 of IQGAP1 does not directly overlap with CDC42 binding at the Ex mode site, the ubiquitin polypeptide is close to the helical insert of the CDC42 molecule. Dynamically, ubiquitin attached to Lys-1155 may shift into positions where it might exclude CDC42 binding by continually disturbing its interaction with IQGAP1. Moreover, only monoubiquitination was assessed. Polyubiquitination of the GRD would likely create a complete hindrance, abrogating CDC42 binding. In summary, the illustration strongly suggests that ubiquitination of Lys-1155 and Lys-1230 prevents the binding of two molecules of CDC42. In vitro analysis with pure ubiquitinated IQGAP1 and pure CDC42 is required to unequivocally establish this.
Alignment of the GRDs of IQGAP1, IQGAP2, and IQGAP3 shows that neither Lys-1155 nor Lys-1230 is conserved in IQGAP3 (Fig. S4). Only one of the two lysine residues is conserved in IQGAP2; Lys-1143 aligns with Lys-1230. No ubiquitination of Lys-1143 in IQGAP2 has been reported, suggesting that Lys-1155 and Lys-1230 of IQGAP1 may be unique sites of IQGAP ubiquitination.
By interacting with IQGAP1, CDC42 is stabilized in its active GTP-bound form (3, 39). Stable knockdown of IQGAP1 by 80% reduced the amounts of GTP-bound CDC42 in MCF-7 human breast epithelial cells (20). In accordance with these findings, we observed that HEK293 cells lacking IQGAP1 contain significantly less active CDC42 than cells reconstituted with WT IQGAP1. Furthermore, consistent with the CDC42 binding data, cells expressing IQGAP1 GRD-2K had significantly more active CDC42 than cells expressing WT IQGAP1.
CDC42 is known to have a central role in cell migration (15). Importantly, we previously documented that IQGAP1 promotes cell motility via CDC42 (3). Therefore, we compared the effects of WT IQGAP1 and IQGAP1 GRD-2K on cell migration using a wound-healing assay. Cells expressing IQGAP1 GRD-2K closed the wound significantly more quickly than cells expressing WT IQGAP1. Collectively, our data strongly suggest that ubiquitination of Lys-1155 and Lys-1230 impairs the binding of IQGAP1 to CDC42, thereby modulating the amount of active CDC42 in the cells. Importantly, this post-translational modification influences at least one of the fundamental biological processes in which IQGAP1 participates.
Analogous to CDC42, RAC1 binding to IQGAP1 is modulated by ubiquitination. Mutation of Lys-1155 and Lys-1230 of IQGAP1 increased its interaction with RAC1. In contrast to CDC42, mutation of the three ubiquitinated lysine residues (Lys-1465, Lys-1475, and Lys-1528) in the RGCT decreased its interaction with RAC1. The molecular mechanism underlying the difference is not known. Although the structure of the IQGAP1/RAC1 complex has not been solved, indirect evidence by calorimetry (17) and thermodynamic analysis (42) imply that the CDC42 and RAC1-binding sites on IQGAP1 are not identical. Consistent with these published data, initial binding studies with different fragments of IQGAP1 suggest that, in addition to the GRD, RAC1 interacts with a site located at the C-terminal region of IQGAP1, which includes the RGCT.3 Other investigators also suggest that IQGAP1 residues distal to the GRD are important for binding to RAC1 (45). Together, these data suggest that post-translational modifications in the RGCT may serve as a regulatory mechanism for RAC1 binding to IQGAP1. In addition, to activating CDC42, IQGAP1 increases the amount of active RAC1 in cells (20). It is therefore likely that ubiquitination of IQGAP1 will modify RAC1 signaling. Additional work is required to elucidate the potential consequences of IQGAP1 ubiquitination on RAC1 function. Similarly, it seems reasonable to postulate that ubiquitination of IQGAP1 alters its interaction with and modulation of several other binding partners. We look forward to future studies that explore this area of investigation that we hope will be stimulated by our provocative findings presented here.
Mutation of Lys-556 in the coiled-coil region of IQGAP1 decreased CDC42 and RAC1 binding. Neither CDC42 nor RAC1 binds directly to this region (16, 17, 37). The only protein known to associate with the coiled-coil region is the transcriptional regulator FOXO-1 (46). Binding of FOXO-1 disrupts IQGAP1-MAPK pathway interactions (46). Because MAPK components bind to IQGAP1 outside of its coiled-coil region (47–49), the effect of FOXO-1 on MAPK signaling is thought to be due to altered IQGAP1 conformation (46). In a similar manner, ubiquitination of Lys-556 might alter the ability of another protein to bind to the coiled-coil region of IQGAP1, influencing CDC42 and RAC1 interactions. Allosteric regulation of CDC42 binding has been previously documented. When Ca2+/calmodulin binds to the IQ region of IQGAP1, CDC42 is unable to bind to the GRD (38). Finally, ubiquitination of Lys-556 might impair dimerization of IQGAP1, which would prevent CDC42 activation (40).
Several extracellular stimuli, including EGF (12, 50, 51) activate CDC42. Consistent with the published literature, we observed that EGF increases active CDC42 in HEK293 cells. However, this effect was abrogated when IQGAP1 was knocked-down in the cells. The molecular mechanism by which IQGAP1 facilitates this effect of EGF is unknown. We previously documented that IQGAP1 binds to the EGFR and is required for maximal activation of the receptor by EGF (52). Moreover, we showed that IQGAP1 scaffolds components of the phosphoinositide 3-kinase (PI3K) signaling cascade and is required for maximal activation of this pathway by EGF (53). Both EGFR and PI3K have been reported to participate in activation of CDC42 (54, 55). Whether these scaffolding functions of IQGAP1 have a role in the EGF-mediated increase in GTP-CDC42 is not known. Regardless of the mechanism, our data reveal that IQGAP1 is essential for EGF to increase the amount of active CDC42 in HEK293 cells.
Unexpectedly, we observed that EGF did not significantly increase the amount of active CDC42 in cells in which endogenous IQGAP1 is replaced by IQGAP1 GRD-2K. This observation implies that ubiquitination of Lys-1155 and Lys-1230 of IQGAP1 may be important for this effect. Additional studies are required to elucidate the mechanism.
Both Rho GTPases and their canonical regulators, GEFs and GAPs, are regulated by post-translational modifications. Phosphorylation and sumoylation regulate GTPase activity, whereas ubiquitination modulates protein levels of Rho GTPases, GEFs, and GAPs via the ubiquitin proteasome system (56). In addition, ubiquitination of some GTPases alters their interactions with regulators or effectors. For example, ubiquitination of Rab5 disrupts its association with downstream effector proteins, such as EEA1 and Rabaptin5 (57). Similarly, ubiquitination of Ras impedes its interaction with GAP (58) and leads to a preferential association with PI3K and Raf (59). Our data extend the findings of these prior publications to identify an additional role of ubiquitination, namely that ubiquitination of a GTPase-binding protein influences GTPase signaling. We demonstrated that ubiquitination of IQGAP1 modulates activation of CDC42 by altering its binding to this GTPase. To the best of our knowledge, this is the first demonstration that ubiquitination of a GTPase regulator alters CDC42 activation independently of the proteasome pathway. A recent study suggests ISGylation of IQGAP1 as a post-translational modification that regulates IQGAP1 function (24). Analogous to our observations with ubiquitination, reduction of ISGylation of IQGAP1 increased GTP-bound CDC42 in cells.
Although regulation of protein degradation by ubiquitination has been well-characterized (32), the roles of protein ubiquitination in cell signaling is less understood. In this study, we identified ubiquitination sites on IQGAP1 and describe a previously uncharacterized role for ubiquitinated IQGAP1. Our findings strongly suggest that ubiquitination of IQGAP1 regulates its association with CDC42, thereby modulating activation of the GTPase, resulting in physiological effects. These observations expand our comprehension of ubiquitination signaling and provide additional insight into CDC42 regulation.
Experimental procedures
Antibodies and reagents
The pMPT107-His-ubiquitin plasmid was described previously (60). The EZVIEW Red anti-c-Myc affinity gel was from Sigma-Aldrich (catalogue number E6654). GSH-Sepharose and protein A-Sepharose beads were purchased from GE Healthcare. MG132 (catalogue number 17-485), LipofectamineTM 2000 Reagent (catalogue number 11668019), Lipofectamine LTX Reagent (catalogue number 15338500), hygromycin (catalogue number 10687010), doxycycline (catalogue number BP26531), and Colloidal Blue Staining Kit (catalogue number LC6025) were obtained from Thermo Fisher Scientific. InstantBlue Protein stain was purchased from Expedeon (catalogue number ISB1L). Human EGF (catalogue number SRP3027) was obtained from Sigma-Aldrich. Antibodies and dilutions used are listed in Table 1. Blocking buffer and IR dye-conjugated (IRDye) secondary antibodies were purchased from LI-COR Biosciences.
Table 1.
Antibodies used in this study
| Protein detected | Reference | Dilution for immunoblots |
|---|---|---|
| Actin | Santa Cruz, sc-8432 | 1:2000 |
| CDC42 | Cytoskeleton, ACD03 | 1:500 |
| GST | Santa Cruz, sc-138 | 1:1000 |
| pERK1/2 | Cell Signaling, 4377S | 1:1000 |
| ERK1/2 | Cell Signaling, 9107S | 1:1000 |
| IQGAP1 | Rabbit antiserum (38) | 1:1000 |
| Myc | Millipore, 06–549 | 1:1000 |
| RAC1 | Cytoskeleton, ARC03 | 1:500 |
| Tubulin | Sigma, T5201 | 1:2000 |
| Ubiquitin | Life Sensors, VU101 | 1:1000 |
Cell culture and transfection
HEK293 cells were purchased from American Type Culture Collection. Knockdown of IQGAP1 in HEK293 cells using the CRISPR/Cas9 strategy has been previously described (61). Immortalized MEFs isolated from embryos of IQGAP1−/− and control mice were described previously (47). HEK293 and MEF cells were grown at 37 °C in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin. Cells were transfected with plasmids using Lipofectamine 2000 according to the manufacturer's instructions. Briefly, plasmids expressing Myc-IQGAP1 and/or His-ubiquitin were incubated with Lipofectamine 2000 in minimal essential medium (Opti-MEM, Gibco) for 20 min at 22 °C, then added to the cells. Then 6 h later the medium was replaced with 10% FBS in DMEM. After 48 h, cells were washed twice with phosphate-buffered saline (PBS) and lysed with 500 μl of lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 1% Triton X-100, containing protease and phosphatase inhibitors (Thermo Scientific)). Lysates were subjected to two rounds of sonication for 10 s each, and insoluble material was precipitated by centrifugation at 20,000 × g for 10 min at 4 °C. Supernatants were frozen at −80 °C before experiments.
Purification of CDC42 and RAC1
pGEX-CDC42-Q61L (residues 1–184) and pGEX-RAC1-Q61L (residues 1–192) were generously donated by Darerca Owen, University of Cambridge, Cambridge, UK (42). The Q61L mutation stabilizes the active GTP-bound form of CDC42 or RAC1, which constitutively activates these GTPases. GST fusion proteins were expressed in Escherichia coli BL-21 and isolated using GSH-Sepharose chromatography, essentially as previously described (38). His-CDC42(Q61L) and His-RAC1(Q61L) were generated from pGEX-CDC42-Q61L and pGEX-RAC1-Q61L by excision and insertion into pRSET as previously described (62).
Myc-IQGAP1 constructions
Site-directed mutagenesis was performed on IQGAP1 to replace selected lysine residues with arginine. Mutagenesis was performed on pBlueScript vectors containing fragments of IQGAP1; these are the IQGAP1 PacI/ClaI (amino acids 544–1193) or IQGAP1 ClaI/XbaI (amino acids 1193–1657) regions, essentially as previously described (63). Mutations K556R, K1155R, and K556R/K1155R were made in the PacI/ClaI region. Mutations K1230R, K1465R/K1475R/K1528R, and K1230R/K1465R/K1475R/K1528R were made in the ClaI/XbaI region. These pBlueScript vectors were digested by the enzyme pairs PacI/ClaI or ClaI/XbaI. The resultant fragments were inserted into the pcDNA3-Myc-IQGAP1 plasmid at PacI/ClaI or ClaI/XbaI sites to generate six individual Myc-IQGAP1 constructs. The constructs were given the following abbreviations: IQGAP1 CC-1K (K556R), replacement of Lys-556 in the coiled-coil region; IQGAP1 GRD-2K (K1155R; K1230R), replacement of Lys-1155 and Lys-1230 in the GRD; IQGAP1 RGCT-3K (K1465R; K1475R; K1528R), replacement of Lys-1465, Lys-1475, and Lys-1528 in the RGCT; IQGAP1–6K (K556R; K1155R; K1230R; K1465R; K1475R; K1528R), replacement of Lys-556, Lys-1155, Lys-1230, Lys-1465, Lys-1475, and Lys-1528. The sequence of all constructs was confirmed by DNA sequencing.
GFP-IQGAP1 constructions
To make GFP-IQGAP1 constructs, the plasmid pEGFPx2-IQGAP1, previously described (40), was cut with NheI. The fragment generated, containing the EGFPx2-IQGAP1-N sequence, was inserted into vector pEN-TmiRc3 at SpeI site. Then, plasmid pCDNA3-myc-IQGAP1 (WT IQGAP1 or IQGAP1 GRD-2K) was cut with PacI/XbaI. The resultant fragment, containing the EGFPx2-IQGAP1-C sequence, was inserted into pEN-EGFPx2-IQGAP1-N at PacI/XbaI sites to generate full-length pEN-EGFPx2-IQGAP1 (WT IQGAP1 or IQGAP1 GRD-2K). Recombination of pEN-EGFPx2-IQGAP1 (WT or GRD-2K) and pSLIK-hygro (gift from I. D. Fraser (64)) was accomplished using a Gateway IR Clonase II Enzyme Mix kit (Invitrogen). The pSLIK-hygro-EGFPx2-IQGAP1 (WT IQGAP1 or IQGAP1 GRD-2K) sequence was confirmed by DNA sequencing. These proteins migrated to the expected positions on SDS-PAGE.
GST-IQGAP1 constructions
The construction of GST-WT IQGAP1, generated from pGEX2T-IQGAP1, was described previously (38). To generate GST-IQGAP1-GRD-2K, pEGFPx2-IQGAP1–2K was cut with PacI/KpnI. The resultant fragment was inserted into pGEX2T-IQGAP1 at PacI/KpnI sites. The sequence pGEX2T-IQGAP1–2K was confirmed by DNA sequencing and the expressed protein migrated to the expected position on SDS-PAGE.
GST pulldown assays
For pulldown from cell lysates, IQGAP1-knockdown HEK293 cells were transfected with Myc-tagged IQGAP1 (WT or mutant constructs) or Myc vector as described above. After 24 h, cells were incubated with 10 μm MG132 for 4 h at 37 °C. Cells were washed twice with PBS and lysed with 500 μl of lysis buffer as described above. Equal amounts of protein lysate were precleared with GSH-Sepharose beads for 1 h at 4 °C, then incubated for 3 h at 4 °C with 5 μg of GST-CDC42(Q61L), GST-RAC1(Q61L), or GST alone. All GST proteins were bound to GSH-Sepharose beads. After washing the beads five times with lysis buffer, samples were resolved by SDS-PAGE and transferred to a PVDF membrane. The membrane was incubated with blocking buffer for 1 h at 22 °C, and then probed with anti-GST and anti-Myc antibodies. After washing, the membrane was incubated with anti-rabbit and anti-mouse IRDye-conjugated secondary antibodies for 1 h at room temperature. Antigen-antibody complexes were detected using the Odyssey imaging system (LI-COR Biosciences). The amounts of Myc-tagged proteins were quantified and corrected for the amounts of GST-CDC42 or GST-RAC1 in the same sample.
For pulldown of pure proteins, 10 μg of GST-WT IQGAP1, GST-IQGAP1 GRD-2K, or GST alone, bound to GSH-Sepharose beads, was incubated for 2 h at 4 °C with 3 μg of His-CDC42(Q61L) or His-RAC1(Q61L). After washing the beads five times with lysis buffer, samples were resolved by SDS-PAGE. The gel was cut at ∼50 kDa. The upper section of the gel was stained with InstantBlue Protein Stain, whereas the lower section was evaluated by Western blotting probed with anti-CDC42 and anti-RAC1 antibodies.
Immunoprecipitation of Myc-tagged proteins
HEK293 cells were transfected with Myc-tagged IQGAP1 (WT or mutant constructs) or Myc vector with or without His-tagged ubiquitin. After 48 h, cells were incubated with 10 μm MG132 for 4 h at 37 °C and lysed as described above. Samples were precleared with GSH-Sepharose beads for 1 h. Protein concentrations were determined with the DC Protein Assay (Bio-Rad). Equal amounts of protein lysate were incubated for 2 h at 4 °C with anti-Myc antibodies covalently bound to agarose beads. Samples were washed five times with lysis buffer, then resolved by SDS-PAGE and Western blotting. Membranes, probed with anti-ubiquitin and anti-IQGAP1 antibodies, were visualized as described above. The amounts of ubiquitinated proteins were quantified and corrected for the amounts of corresponding immunopurified Myc-tagged proteins in the same sample.
His-tag pulldown assays
HEK293 cells were transfected with His-tagged ubiquitin plasmids as described above and incubated with 10 μm MG132 for 4 h at 37 °C. Cells were washed twice with PBS and lysed with 500 μl of HisTALON xTractor buffer (catalogue number 635651, Clontech Laboratories) containing protease and phosphatase inhibitors. Lysates were subjected to two rounds of sonication for 10 s each, and insoluble material was precipitated by centrifugation at 20,000 × g for 10 min at 4 °C. Supernatants were precleared with GSH-Sepharose beads for 1 h. Equal amounts of protein lysate were incubated with TALON Resin (catalogue number 635509, Clontech Laboratories) for 2 h at 4 °C. After 2 washes with equilibration buffer (catalogue number 635651, Clontech Laboratories), samples were transferred to a 2-ml gravity-flow column (catalogue number 635606, Clontech Laboratories). After washing columns once with equilibration buffer, samples were eluted with 50 μl of Elution buffer (catalogue number 635651, Clontech Laboratories) and resolved by SDS-PAGE and Western blotting. Membranes, probed with anti-ubiquitin and anti-IQGAP1 antibodies, were visualized as described above.
Immunoprecipitation of endogenous IQGAP1 proteins for MS analysis
Untransfected or His-ubiquitin–transfected HEK293 cells were incubated with DMSO or 10 μm MG132 for 4 h at 37 °C. Cells were washed, lysed, and spun down as described above. Supernatants were precleared with GSH-Sepharose beads for 1 h at 4 °C. Equal amounts of protein lysate were incubated with anti-IQGAP1 polyclonal antibodies at 4 °C. After 3 h, samples were incubated with protein A-Sepharose beads for 2 h at 4 °C. Each sample was washed five times with lysis buffer and analyzed by Western blotting and probed with anti-ubiquitin and anti-IQGAP1 antibodies.
Mass spectrometry analysis
Endogenous IQGAP1, immunopurified from HEK293 cells, was separated by SDS-PAGE and the gel was stained with Colloidal Coomassie. For each condition (untransfected and His-ubiquitin–transfected HEK293 cells incubated with either DMSO or 10 μm MG132), three bands (corresponding to IQGAP1 (190 kDa) and the region above) were excised from the stained gel, reduced with 10 mm tris(2-carboxyethyl)phosphine, alkylated with 10 mm iodoacetamide and digested overnight with trypsin at 37 °C. The resultant peptide samples were injected on an Easy-nLC 1000 UHPLC system (Thermo Scientific). The nanoLC was interfaced to a Q-Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific). Tryptic peptides were separated on a 25 cm × 75-μm inner diameter, PepMap C18, 2-μm particle column (Thermo Scientific) using a 40-min gradient of 2–30% acetonitrile, 0.2% formic acid and a flow of 300 nl/min. MS-based peptide sequencing data were acquired by MS using a top 10 data-dependent LC-MS/MS method. Full MS data were collected in profile mode from 400 to 2000 m/z at a resolution of 70,000 using an AGC target value of 1e6 with a maximum IT of 200 ms. The top 10 peptide ions were then isolated using a 1.5 m/z window and fragmented with an normalized collision energy of 30. MS/MS data were collected using an AGC target value of 5e4 with a maximum IT of 200 ms and a fixed first mass at 145 Da. A dynamic exclusion of 15 s was applied. These uninterpreted tandem MS spectra were processed using Mascot Distiller (Matrix Science version 2.7.1.0) and searched for peptide matches against the UniProt_Human (version 2014_03) protein sequence database using Mascot (Matrix Science version 2.6.0). 65,630 total protein sequences were searched using trypsin enzyme specificity with a possibility of 2 missed cleavages and a precursor mass tolerance of ±5 ppm and a fragment mass tolerance of 0.02 Da. Carbamidomethylation was selected as fixed modification on Cys residues. Oxidation on Met and Gly-Gly on Lys (indicative of ϵ-amine ubiquitination following trypsin digestion) were selected as variable modifications. Identified IQGAP1 ubiquitination sites were further validated using targeted PRM LC-MS/MS. For PRM data, full MS scans were collected in profile mode from 350 to 1600 m/z at a resolution of 35,000 using an AGC target value of 3e6 with a maximum IT of 200 ms. 10 specific m/z targets were then sequentially isolated using a 1.2 m/z window and fragmented with an normalized collision energy of 30. MS/MS data were collected using an AGC target value of 2e5 with a maximum IT of 250 ms at a fixed first mass of 100 Da. The resulting MS/MS spectra were manually reviewed to confirm ubiquitination site identification (Fig. S1).
CDC42 activation assays
The CDC42 activation assay was performed with GTPase-specific ELISA (G-LISA) activation kits (catalogue number BK127, Cytoskeleton) according to the manufacturer's instructions. IQGAP1-knockdown HEK293 cells were transfected with Myc-tagged WT IQGAP1, IQGAP1 GRD-2K, or vector alone. After overnight serum starvation, cells were incubated with or without 100 ng/ml of EGF for 10 min at 37 °C. Cells were washed with ice-cold PBS, lysed, and the G-LISA assay was performed. Briefly, equal amounts of protein lysate were incubated in the wells of the G-LISA 96-well-plate that was coated with CDC42-GTP-binding proteins. Only GTP-CDC42 binds to the coated well. A positive control (constitutively active CDC42) included in the kit was processed in parallel. After washing, the amount of GTP-bound CDC42 was quantified via an enzyme-linked antibody and colorimetric substrate, which absorbs light at 490 nm. Absorbance readings were performed with a Biotek Instruments Synergy 4 plate reader. An aliquot of cell lysate processed in parallel was analyzed by SDS-PAGE and Western blotting. Membranes, probed with anti-Myc, anti-pERK, anti-ERK, and anti-actin antibodies were visualized as described above.
Generation of IQGAP1-null MEF cells with stable expression of IQGAP1
IQGAP1-null MEF cells were transfected with the pSLIK-hygro-EGFPx2-IQGAP1 plasmids (WT IQGAP1 or IQGAP1 GRD-2K) using Lipofectamine LTX following the manufacturer's instructions. After 48 h, cells were selected using 200 μg/ml of hygromycin. Expression of EGFPx2-IQGAP1 proteins was induced with 1 μg/ml of doxycycline and confirmed by Western blotting.
Migration assay
Expression of WT IQGAP1 or IQGAP1 GRD-2K in IQGAP1-null MEF cells was induced by adding 1 μg/ml of doxycycline to DMEM containing 10% FBS. Cells were suspended to a concentration of 3.0 × 105 cells/ml and 70 μl of cell suspension (21,000 cells) was added to each chamber of an Ibidi Culture Insert (Ibidi, catalogue number 81176). Cells were allowed to attach to the dish and reach confluence. After serum starvation for 12 h, the silicone inserts were removed with sterile forceps and the medium was replaced with DMEM containing 0.1% FBS, 100 ng/ml of doxycycline, and 25 ng/ml of EGF. Confocal images were acquired every 30 min for 24 h using a Zeiss LSM780 microscope equipped with a ×20 plan-apochromat (N.A. 0.8) objective lens, transmitted light detector (T-PMT), and OKO Labs Bold Line stage top incubator to control the temperature at 37 °C, CO2 at 5%, and humidity. Differential interference contrast images were collected simultaneously with confocal images of E fluorescence using 0.461 μm x-y pixel size, 5.0-μm optical section thickness, and 12-bit data depth. Image analysis was performed using FIJI/ImageJ (65). GFP-labeled cells were processed using a 10 pixel kernel Gaussian blur, followed by a 10 pixel kernel variance filter. The edges of the cell monolayer were then segmented by intensity and filtering for particles >1000 μm2. The empty area between the two edges was quantified at each time point.
Molecular illustration
We previously determined the structure of CDC42 bound to GRD of IQGAP2 (GRD2) (17). This structure features two binding sites for CDC42. The very high level of sequence conservation between the GRD of IQGAP1 (GRD1) and the GRD of IQGAP2 (GRD2) (Fig. S4), coupled with calorimetry-based binding experiments using CDC42, RAC1, and both mutant and WT GRDs, suggest that CDC42 engages GRD1 in a manner very similar to its binding to GRD2. Therefore, we have illustrated interactions between CDC42 and GRD1 using information from our previous study (17). We used the molecular graphics program O (66) for in silico mutation of GRD2 (Chain E; PDB ID 5CJP) residues at positions 1068 and 1143 (equivalent to GRD1 positions 1155 and 1230, respectively) to lysines or arginines for the purpose of distance measurements and identification of probable steric clashes using an expanded side chain rotamer database (67). The program O was also used to move the coordinates of human monoubiquitin (PDB ID 1UBQ) such that the C terminus of this ubiquitin was within covalent-bonding distance to the lysine side chain nitrogens on GRD2. Once the ubiquitin Gly-76 carbonyl carbon had been positioned ∼1.3 Å from the lysine side chain nitrogens, the coordinates were output from O and used in the program PyMOL (68) to generate the cartoons shown in Fig. 7. No molecular dynamics or energy-minimization was performed on the GRD/ubiquitin coordinates. The purpose of this figure is for illustrative purposes only.
The homology model of GRD2 containing arginine residues at positions 1068 and 1143 was made using the SWISS-MODEL server (SCR_018123). To do this, we used the GRD2 sequence with arginines at 1068 and 1143, and the coordinates of GRD2 chain E (Protein Data Bank ID 5CJP; Chain E) as the structural template. To address poor geometry, steric clashes, and chemically disfavorable interactions, SWISS-MODEL performs energy minimization (69). We then used the CCP4 program LSQKAB (70) to compare the “mutant” GRD2 model to the WT GRD2 model acquired via protein crystallography.
Statistical analysis
All statistical analysis was performed using Welch's t test by Prism7 (GraphPad). Western blotting images were quantified with Image Studio 2.0 (LI-COR Biosciences) according to the manufacturer's instructions. After quantifying the bands, we calculated the ratio of each IQGAP1 band (WT or mutant) to the relevant loading control band (actin, tubulin, or GST, according to the experiment) in the same sample. We normalized data by dividing each ratio by the ratio of WT IQGAP1 to its loading control. Thus, WT IQGAP1 equaled 1.
Author contributions
L. G., F. Z., A. C. H., and R. S. A. data curation; L. G., Z. L., F. Z., A. C. H., R. S. A., and D. B. S. formal analysis; L. G., Z. L., C. D. W., D. K. W., F. Z., and A. C. H. investigation; L. G., Z. L., C. D. W., F. Z., A. C. H., and D. B. S. methodology; L. G. and D. B. S. writing-original draft; L. G., C. D. W., D. K. W., F. Z., A. C. H., R. S. A., and D. B. S. writing-review and editing; Z. L. and F. Z. validation; R. S. A. and D. B. S. resources; R. S. A. and D. B. S. supervision; D. B. S. conceptualization; D. B. S. funding acquisition; D. B. S. project administration.
Supplementary Material
Acknowledgments
We thank Michael Kruhlak, Andy Tran, and Langston Lim (CCR Microscopy Core Facility, National Institutes of Health) for expert assistance with confocal microscopy and image analysis. We thank Miles Houslay for encouragement in the initial stages of the project.
This work was supported by the National Institutes of Health Intramural Research Program. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The mass spectrometry data reported in this paper have be submitted to the Massive database under project ID MSV000084656.
This article contains Figs. S1–S4 and Video S1.
D. Worthylake, unpublished observations.
- IQGAP1
- IQ motif–containing GTPase–activating protein
- CDC42
- cell division cycle 42
- CHD
- calponin homology domain
- DMEM
- Dulbecco's modified Eagle's medium
- EGF
- epidermal growth factor
- ERK
- extracellular signal-regulated kinase
- FBS
- fetal bovine serum
- GAP
- GTPase-activating protein
- GEF
- guanine nucleotide exchange factor
- EGFP
- enhanced green fluorescent protein
- GRD
- Ras GTPase-activating protein-related domain
- PI3K
- phosphoinositide 3-kinase
- RAC1
- Rac family small GTPase 1
- RGCT
- RasGAP_C-terminal domain
- IB
- immunoblot
- IP
- immunoprecipitate
- PDB
- Protein Data Bank
- GST
- glutathione S-transferase
- MAPK
- mitogen-activated protein kinase
- G-LISA
- GTPase-specific ELISA
- MEF
- mouse embryonic fibroblast
- PVDF
- polyvinylidene difluoride.
References
- 1. Hedman A. C., Smith J. M., and Sacks D. B. (2015) The biology of IQGAP proteins: beyond the cytoskeleton. EMBO Rep. 16, 427–446 10.15252/embr.201439834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Briggs M. W., and Sacks D. B. (2003) IQGAP proteins are integral components of cytoskeletal regulation. EMBO Rep. 4, 571–574 10.1038/sj.embor.embor867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mataraza J. M., Briggs M. W., Li Z., Entwistle A., Ridley A. J., and Sacks D. B. (2003) IQGAP1 promotes cell motility and invasion. J. Biol. Chem. 278, 41237–41245 10.1074/jbc.M304838200 [DOI] [PubMed] [Google Scholar]
- 4. Brown M. D., and Sacks D. B. (2006) IQGAP1 in cellular signaling: bridging the GAP. Trends Cell Biol. 16, 242–249 10.1016/j.tcb.2006.03.002 [DOI] [PubMed] [Google Scholar]
- 5. White C. D., Erdemir H. H., and Sacks D. B. (2012) IQGAP1 and its binding proteins control diverse biological functions. Cell Signal. 24, 826–834 10.1016/j.cellsig.2011.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Osman M. (2010) An emerging role for IQGAP1 in regulating protein traffic. ScientificWorldJournal 10, 944–953 10.1100/tsw.2010.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mateer S. C., Wang N., and Bloom G. S. (2003) IQGAPs: integrators of the cytoskeleton, cell adhesion machinery, and signaling networks. Cell Motil. Cytoskeleton 55, 147–155 10.1002/cm.10118 [DOI] [PubMed] [Google Scholar]
- 8. Smith J. M., Hedman A. C., and Sacks D. B. (2015) IQGAPs choreograph cellular signaling from the membrane to the nucleus. Trends Cell Biol. 25, 171–184 10.1016/j.tcb.2014.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bourne H. R., Sanders D. A., and McCormick F. (1990) The GTPase superfamily: a conserved switch for diverse cell functions. Nature 348, 125–132 10.1038/348125a0 [DOI] [PubMed] [Google Scholar]
- 10. Bishop A. L., and Hall A. (2000) Rho GTPases and their effector proteins. Biochem. J. 348, 241–255 10.1042/0264-6021:3480241, 10.1042/bj3480241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dise R. S., Frey M. R., Whitehead R. H., and Polk D. B. (2008) Epidermal growth factor stimulates Rac activation through Src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G276–G285 10.1152/ajpgi.00340.2007 [DOI] [PubMed] [Google Scholar]
- 12. Tu S., Wu W. J., Wang J., and Cerione R. A. (2003) Epidermal growth factor-dependent regulation of Cdc42 is mediated by the Src tyrosine kinase. J. Biol. Chem. 278, 49293–49300 10.1074/jbc.M307021200 [DOI] [PubMed] [Google Scholar]
- 13. Liu B. P., and Burridge K. (2000) Vav2 activates Rac1, Cdc42, and RhoA downstream from growth factor receptors but not β1 integrins. Mol. Cell Biol. 20, 7160–7169 10.1128/MCB.20.19.7160-7169.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nobes C. D., and Hall A. (1999) Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 144, 1235–1244 10.1083/jcb.144.6.1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ridley A. J. (2015) Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 36, 103–112 10.1016/j.ceb.2015.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hart M. J., Callow M. G., Souza B., and Polakis P. (1996) IQGAP1, a calmodulin-binding protein with a rasGAP-related domain, is a potential effector for cdc42Hs. EMBO J. 15, 2997–3005 10.1002/j.1460-2075.1996.tb00663.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. LeCour L. Jr, Boyapati V. K., Liu J., Li Z., Sacks D. B., and Worthylake D. K. (2016) The structural basis for Cdc42-induced dimerization of IQGAPs. Structure 24, 1499–1508 10.1016/j.str.2016.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Swart-Mataraza J. M., Li Z., and Sacks D. B. (2002) IQGAP1 is a component of Cdc42 signaling to the cytoskeleton. J. Biol. Chem. 277, 24753–24763 10.1074/jbc.M111165200 [DOI] [PubMed] [Google Scholar]
- 19. Noritake J., Watanabe T., Sato K., Wang S., and Kaibuchi K. (2005) IQGAP1: a key regulator of adhesion and migration. J. Cell Sci. 118, 2085–2092 10.1242/jcs.02379 [DOI] [PubMed] [Google Scholar]
- 20. Jadeski L., Mataraza J. M., Jeong H. W., Li Z., and Sacks D. B. (2008) IQGAP1 stimulates proliferation and enhances tumorigenesis of human breast epithelial cells. J. Biol. Chem. 283, 1008–1017 10.1074/jbc.M708466200 [DOI] [PubMed] [Google Scholar]
- 21. Brown M. D., Bry L., Li Z., and Sacks D. B. (2007) IQGAP1 regulates Salmonella invasion through interactions with actin, Rac1, and Cdc42. J. Biol. Chem. 282, 30265–30272 10.1074/jbc.M702537200 [DOI] [PubMed] [Google Scholar]
- 22. Li Z., McNulty D. E., Marler K. J., Lim L., Hall C., Annan R. S., and Sacks D. B. (2005) IQGAP1 promotes neurite outgrowth in a phosphorylation-dependent manner. J. Biol. Chem. 280, 13871–13878 10.1074/jbc.M413482200 [DOI] [PubMed] [Google Scholar]
- 23. Vetterkind S., Lin Q. Q., and Morgan K. G. (2017) A novel mechanism of ERK1/2 regulation in smooth muscle involving acetylation of the ERK1/2 scaffold IQGAP1. Sci. Rep. 7, 9302 10.1038/s41598-017-09434-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cerikan B., Shaheen R., Colo G. P., Gläßer C., Hata S., Knobeloch K. P., Alkuraya F. S., Fässler R., and Schiebel E. (2016) Cell-intrinsic adaptation arising from chronic ablation of a key Rho GTPase regulator. Dev. Cell 39, 28–43 10.1016/j.devcel.2016.08.020 [DOI] [PubMed] [Google Scholar]
- 25. Durfee L. A., Lyon N., Seo K., and Huibregtse J. M. (2010) The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol. Cell 38, 722–732 10.1016/j.molcel.2010.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liang Z., Yang Y., He Y., Yang P., Wang X., He G., Zhang P., Zhu H., Xu N., Zhao X., and Liang S. (2017) SUMOylation of IQGAP1 promotes the development of colorectal cancer. Cancer Lett. 411, 90–99 10.1016/j.canlet.2017.09.046 [DOI] [PubMed] [Google Scholar]
- 27. Kim W., Bennett E. J., Huttlin E. L., Guo A., Li J., Possemato A., Sowa M. E., Rad R., Rush J., Comb M. J., Harper J. W., and Gygi S. P. (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 10.1016/j.molcel.2011.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oshikawa K., Matsumoto M., Oyamada K., and Nakayama K. I. (2012) Proteome-wide identification of ubiquitylation sites by conjugation of engineered lysine-less ubiquitin. J. Proteome Res. 11, 796–807 10.1021/pr200668y [DOI] [PubMed] [Google Scholar]
- 29. Udeshi N. D., Mani D. R., Eisenhaure T., Mertins P., Jaffe J. D., Clauser K. R., Hacohen N., and Carr S. A. (2012) Methods for quantification of in vivo changes in protein ubiquitination following proteasome and deubiquitinase inhibition. Mol. Cell Proteomics 11, 148–159 10.1074/mcp.M111.016857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wagner S. A., Beli P., Weinert B. T., Nielsen M. L., Cox J., Mann M., and Choudhary C. (2011) A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell Proteomics 10, M111.013284 10.1074/mcp.M111.013284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Komander D., and Rape M. (2012) The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 10.1146/annurev-biochem-060310-170328 [DOI] [PubMed] [Google Scholar]
- 32. Hochstrasser M. (1995) Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr. Opin. Cell Biol. 7, 215–223 10.1016/0955-0674(95)80031-X [DOI] [PubMed] [Google Scholar]
- 33. Chen Z. J., and Sun L. J. (2009) Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell 33, 275–286 10.1016/j.molcel.2009.01.014 [DOI] [PubMed] [Google Scholar]
- 34. Mukhopadhyay D., and Riezman H. (2007) Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 315, 201–205 10.1126/science.1127085 [DOI] [PubMed] [Google Scholar]
- 35. Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 10.1093/nar/gku1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kurella V. B., Richard J. M., Parke C. L., Lecour L. F. Jr, Bellamy H. D., and Worthylake D. K. (2009) Crystal structure of the GTPase-activating protein-related domain from IQGAP1. J. Biol. Chem. 284, 14857–14865 10.1074/jbc.M808974200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mataraza J. M., Briggs M. W., Li Z., Frank R., and Sacks D. B. (2003) Identification and characterization of the Cdc42-binding site of IQGAP1. Biochem. Biophys. Res. Commun. 305, 315–321 10.1016/S0006-291X(03)00759-9 [DOI] [PubMed] [Google Scholar]
- 38. Ho Y. D., Joyal J. L., Li Z., and Sacks D. B. (1999) IQGAP1 integrates Ca2+/calmodulin and Cdc42 signaling. J. Biol. Chem. 274, 464–470 10.1074/jbc.274.1.464 [DOI] [PubMed] [Google Scholar]
- 39. Zhang B., Wang Z. X., and Zheng Y. (1997) Characterization of the interactions between the small GTPase Cdc42 and its GTPase-activating proteins and putative effectors. Comparison of kinetic properties of Cdc42 binding to the Cdc42-interactive domains. J. Biol. Chem. 272, 21999–22007 10.1074/jbc.272.35.21999 [DOI] [PubMed] [Google Scholar]
- 40. Ren J. G., Li Z., Crimmins D. L., and Sacks D. B. (2005) Self-association of IQGAP1: characterization and functional sequelae. J. Biol. Chem. 280, 34548–34557 10.1074/jbc.M507321200 [DOI] [PubMed] [Google Scholar]
- 41. Hochstrasser M. (2009) Origin and function of ubiquitin-like proteins. Nature 458, 422–429 10.1038/nature07958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Owen D., Campbell L. J., Littlefield K., Evetts K. A., Li Z., Sacks D. B., Lowe P. N., and Mott H. R. (2008) The IQGAP1-Rac1 and IQGAP1-Cdc42 interactions: interfaces differ between the complexes. J. Biol. Chem. 283, 1692–1704 10.1074/jbc.M707257200 [DOI] [PubMed] [Google Scholar]
- 43. Kuroda S., Fukata M., Kobayashi K., Nakafuku M., Nomura N., Iwamatsu A., and Kaibuchi K. (1996) Identification of IQGAP as a putative target for the small GTPases, Cdc42 and Rac1. J. Biol. Chem. 271, 23363–23367 10.1074/jbc.271.38.23363 [DOI] [PubMed] [Google Scholar]
- 44. Kuroda S., Fukata M., Nakagawa M., Fujii K., Nakamura T., Ookubo T., Izawa I., Nagase T., Nomura N., Tani H., Shoji I., Matsuura Y., Yonehara S., and Kaibuchi K. (1998) Role of IQGAP1, a target of the small GTPases Cdc42 and Rac1, in regulation of E-cadherin-mediated cell-cell adhesion. Science 281, 832–835 10.1126/science.281.5378.832 [DOI] [PubMed] [Google Scholar]
- 45. Nouri K., Fansa E. K., Amin E., Dvorsky R., Gremer L., Willbold D., Schmitt L., Timson D. J., and Ahmadian M. R. (2016) IQGAP1 Interaction with RHO family proteins revisited: kinetic and equilibrium evidence for multiple distinct binding sites. J. Biol. Chem. 291, 26364–26376 10.1074/jbc.M116.752121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pan C. W., Jin X., Zhao Y., Pan Y., Yang J., Karnes R. J., Zhang J., Wang L., and Huang H. (2017) AKT-phosphorylated FOXO1 suppresses ERK activation and chemoresistance by disrupting IQGAP1-MAPK interaction. EMBO J. 36, 995–1010 10.15252/embj.201695534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ren J. G., Li Z., and Sacks D. B. (2007) IQGAP1 modulates activation of B-Raf. Proc. Natl. Acad. Sci. U.S.A. 104, 10465–10469 10.1073/pnas.0611308104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Roy M., Li Z., and Sacks D. B. (2005) IQGAP1 is a scaffold for mitogen-activated protein kinase signaling. Mol. Cell Biol. 25, 7940–7952 10.1128/MCB.25.18.7940-7952.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bardwell A. J., Lagunes L., Zebarjedi R., and Bardwell L. (2017) The WW domain of the scaffolding protein IQGAP1 is neither necessary nor sufficient for binding to the MAPKs ERK1 and ERK2. J. Biol. Chem. 292, 8750–8761 10.1074/jbc.M116.767087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tu S., and Cerione R. A. (2001) Cdc42 is a substrate for caspases and influences Fas-induced apoptosis. J. Biol. Chem. 276, 19656–19663 10.1074/jbc.M009838200 [DOI] [PubMed] [Google Scholar]
- 51. Sinha S., and Yang W. (2008) Cellular signaling for activation of Rho GTPase Cdc42. Cell Signal. 20, 1927–1934 10.1016/j.cellsig.2008.05.002 [DOI] [PubMed] [Google Scholar]
- 52. McNulty D. E., Li Z., White C. D., Sacks D. B., and Annan R. S. (2011) MAPK scaffold IQGAP1 binds the EGF receptor and modulates its activation. J. Biol. Chem. 286, 15010–15021 10.1074/jbc.M111.227694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Choi S., Hedman A. C., Sayedyahossein S., Thapa N., Sacks D. B., and Anderson R. A. (2016) Agonist-stimulated phosphatidylinositol-3,4,5-trisphosphate generation by scaffolded phosphoinositide kinases. Nat. Cell Biol. 18, 1324–1335 10.1038/ncb3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Itoh R. E., Kiyokawa E., Aoki K., Nishioka T., Akiyama T., and Matsuda M. (2008) Phosphorylation and activation of the Rac1 and Cdc42 GEF Asef in A431 cells stimulated by EGF. J. Cell Sci. 121, 2635–2642 10.1242/jcs.028647 [DOI] [PubMed] [Google Scholar]
- 55. Liu L., Zhang L., Zhao S., Zhao X. Y., Min P. X., Ma Y. D., Wang Y. Y., Chen Y., Tang S. J., Zhang Y. J., Du J., and Gu L. (2019) Non-canonical Notch signaling regulates actin remodeling in cell migration by activating PI3K/AKT/Cdc42 pathway. Front. Pharmacol. 10, 370 10.3389/fphar.2019.00370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hodge R. G., and Ridley A. J. (2016) Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 17, 496–510 10.1038/nrm.2016.67 [DOI] [PubMed] [Google Scholar]
- 57. Shin D., Na W., Lee J. H., Kim G., Baek J., Park S. H., Choi C. Y., and Lee S. (2017) Site-specific monoubiquitination downregulates Rab5 by disrupting effector binding and guanine nucleotide conversion. Elife 6, e29154 10.7554/eLife.29154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Baker R., Lewis S. M., Sasaki A. T., Wilkerson E. M., Locasale J. W., Cantley L. C., Kuhlman B., Dohlman H. G., and Campbell S. L. (2013) Site-specific monoubiquitination activates Ras by impeding GTPase-activating protein function. Nat. Struct. Mol. Biol. 20, 46–52 10.1038/nsmb.2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sasaki A. T., Carracedo A., Locasale J. W., Anastasiou D., Takeuchi K., Kahoud E. R., Haviv S., Asara J. M., Pandolfi P. P., and Cantley L. C. (2011) Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci. Signal. 4, ra13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li L., Li Z., Howley P. M., and Sacks D. B. (2006) E6AP and calmodulin reciprocally regulate estrogen receptor stability. J. Biol. Chem. 281, 1978–1985 10.1074/jbc.M508545200 [DOI] [PubMed] [Google Scholar]
- 61. Sayedyahossein S., Li Z., Hedman A. C., Morgan C. J., and Sacks D. B. (2016) IQGAP1 binds to Yes-associated protein (YAP) and modulates its transcriptional activity. J. Biol. Chem. 291, 19261–19273 10.1074/jbc.M116.732529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ozdemir E. S., Jang H., Gursoy A., Keskin O., Li Z., Sacks D. B., and Nussinov R. (2018) Unraveling the molecular mechanism of interactions of the Rho GTPases Cdc42 and Rac1 with the scaffolding protein IQGAP2. J. Biol. Chem. 293, 3685–3699 10.1074/jbc.RA117.001596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li Z., and Sacks D. B. (2003) Elucidation of the interaction of calmodulin with the IQ motifs of IQGAP1. J. Biol. Chem. 278, 4347–4352 10.1074/jbc.M208579200 [DOI] [PubMed] [Google Scholar]
- 64. Shin K. J., Wall E. A., Zavzavadjian J. R., Santat L. A., Liu J., Hwang J. I., Rebres R., Roach T., Seaman W., Simon M. I., and Fraser I. D. (2006) A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc. Natl. Acad. Sci. U.S.A. 103, 13759–13764 10.1073/pnas.0606179103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jones T. A., Zou J. Y., Cowan S. W., and Kjeldgaard M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119 10.1107/S0108767390010224 [DOI] [PubMed] [Google Scholar]
- 67. Lovell S. C., Word J. M., Richardson J. S., and Richardson D. C. (2000) The penultimate rotamer library. Proteins 40, 389–408 [DOI] [PubMed] [Google Scholar]
- 68. DeLano W. L. (2002) Pymol: An open-source molecular graphics tool. CCP4 Newsletter On Protein Crystallography 40, 82–92 [Google Scholar]
- 69. Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F. T., de Beer T. A. P., Rempfer C., Bordoli L., Lepore R., and Schwede T. (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.