

Abstract
The HIV-1 virulence factor Nef promotes high-titer viral replication, immune escape, and pathogenicity. Nef interacts with interleukin-2–inducible T-cell kinase (Itk) and Bruton's tyrosine kinase (Btk), two Tec-family kinases expressed in HIV-1 target cells (CD4 T cells and macrophages, respectively). Using a cell-based bimolecular fluorescence complementation assay, here we demonstrate that Nef recruits both Itk and Btk to the cell membrane and induces constitutive kinase activation in transfected 293T cells. Nef homodimerization-defective mutants retained their interaction with both kinases but failed to induce activation, supporting a role for Nef homodimer formation in the activation mechanism. HIV-1 infection up-regulates endogenous Itk activity in SupT1 T cells and donor-derived peripheral blood mononuclear cells. However, HIV-1 strains expressing Nef variants with mutations in the dimerization interface replicated poorly and were significantly attenuated in Itk activation. We conclude that direct activation of Itk and Btk by Nef at the membrane in HIV-infected cells may override normal immune receptor control of Tec-family kinase activity to enhance the viral life cycle.
Keywords: human immunodeficiency virus (HIV), protein kinase, dimerization, signal transduction, protein–protein interaction, T-cell receptor (TCR), infectious disease, bimolecular fluorescence complementation (BiFC), Bruton's tyrosine kinase (BTK), HIV-1 Nef, interleukin-2-inducible kinase (ITK), Tec-family kinase
Introduction
Nef, one of four accessory proteins encoded by HIV-1, promotes viral infectivity (1), high-titer replication, and immune escape (2). Although Nef is not required for HIV-1 replication in most T-cell lines in vitro, it is essential for viral growth and pathogenicity in primate hosts. Early work showed that nonhuman primates infected with nef-defective SIV3 developed low viral loads and failed to progress to simian AIDS (3). Similarly, patients harboring nef-defective HIV-1 have been reported to remain disease-free for more than 10 years without antiretroviral therapy, demonstrating that Nef function is essential for AIDS progression (4, 5).
Nef is a relatively small, membrane-associated protein (27–34 kDa, depending on the subtype) and is conserved among the primate lentiviruses (6). Nef has no known intrinsic biochemical or enzymatic activities, functioning instead through interactions with numerous host proteins involved in signal transduction, endocytic trafficking, and host cell survival. For example, Nef down-regulates cell-surface receptors essential for viral entry (CD4, CXCR4, and CCR5) (7–9) via the endosome/lysosome pathway to prevent superinfection and antibody-dependent cell–mediated cytotoxicity (10). Nef also selectively down-regulates cell surface HLA-A and HLA-B to evade immune surveillance by cytotoxic CD8+ T cells and natural killer cells (11, 12). Nef enhances viral infectivity by suppressing incorporation of the SERINC5 restriction factor into progeny virions via AP-2–dependent endocytosis and lysosomal degradation (13, 14).
Nef hijacks multiple host-cell kinase signaling pathways to mimic T-cell and macrophage activation, resulting in enhanced HIV-1 replication and viral spread. In particular, Nef interacts with and activates nonreceptor tyrosine kinases of the Src and Tec families that are normally involved in immune receptor signaling. Among the Src-family members, Nef selectively interacts with Hck and Lyn through their SH3 domains, resulting in constitutive kinase activation (15, 16). Nef-mediated Src-family kinase activation is highly conserved across all M-group HIV-1 subtypes, supporting an important function in HIV-1 replication and pathogenesis (17, 18). Indeed, selective inhibition of Nef-induced Src-family kinase activity blocks Nef-dependent enhancement of HIV-1 infectivity and replication, supporting this idea (17, 19).
X-ray crystal structures of HIV-1 Nef in complexes with the SH3 domain of Fyn, as well as the SH3-SH2 regulatory unit from Hck, have been reported (20–22). Each complex crystallizes as a 2:2 dimer, with Nef forming the dimer interface through conserved residues in its αB helix. Subsequent solution studies using hydrogen–deuterium exchange MS have confirmed that the Nef αB helix is responsible for dimer formation in both complexes, despite important differences in their overall quaternary structures (23). These structural studies suggest that Nef homodimerization may activate Hck and other host-cell kinases through a mechanism that involves clustering of kinase domains for activation via trans-autophosphorylation.
Previous studies have linked the Tec family of nonreceptor tyrosine kinases to the HIV-1 life cycle through Nef. Tec-family kinases have essential roles in immune receptor signaling (24), and the interleukin-2–inducible T-cell kinase (Itk) and Bruton's tyrosine kinase (Btk) are both expressed in HIV-1 target cells (CD4+ T cells and macrophages, respectively). Readinger et al. (25) provided the first evidence that Itk, an essential component of the T-cell receptor signaling cascade, is required for multiple steps in the HIV-1 life cycle including viral entry, transcription, assembly, and egress. Subsequent work demonstrated that Nef links HIV-1 infection to Itk activation. In this previous study, it was demonstrated that Nef interacts with both Itk and Btk at the cell membrane and that a selective inhibitor of Itk kinase activity blocked Nef-dependent enhancement of viral infectivity and replication (26). Like the Src-family kinases, interaction with Itk was shared by Nef proteins representative of all major subtypes of HIV-1, supporting the activation of this T-cell kinase as a conserved and essential function. However, the mechanisms regulating Nef-dependent activation of Tec-family kinases are unknown.
In the present study, we unraveled the molecular mechanism by which HIV-1 Nef activates Itk and Btk and demonstrate that this pathway is essential for both kinase activation and HIV-1 replication in T cells. Using a cell-based fluorescence complementation assay, we show that HIV-1 Nef interacts with Itk and Btk at the cell membrane, resulting in constitutive kinase activity. Kinase activation, but not interaction with Nef, depends upon Nef homodimerization, suggesting that Nef directly activates Itk through a mechanism that bypasses the T-cell receptor and Lck, a Src-family member essential for Itk activation in response to antigen stimulation (27). HIV-1 infection also enhanced endogenous Itk activity in T cells in a manner dependent on Nef homodimer formation. HIV-1 expressing Nef mutants defective for homodimerization replicated poorly in a T-cell line and in donor PBMCs, demonstrating that Nef-dependent activation of Itk is essential for efficient viral replication. Our data support the idea that selective inhibitors of the Nef–Itk signaling pathway may provide a new approach to antiretroviral therapy.
Results
Nef induces constitutive activation of Itk and Btk at the cell membrane
Previous work has shown that Nef interacts with both Itk and Btk at the plasma membrane of transfected cells and that an Itk kinase inhibitor blocks Nef-dependent viral replication (26). However, whether Nef induces activation of Itk or other Tec-family kinases in the membrane compartment was not reported. To address this question directly, we combined bimolecular fluorescence complementation (BiFC) with antiphosphotyrosine immunofluorescence (IF) microscopy to visualize Nef·kinase complex formation and activation simultaneously. Itk or Btk and Nef were fused to complementary, nonfluorescent fragments of the Venus variant of YFP. The kinases were then expressed either alone or together with Nef in 293T cells, followed by immunostaining for Nef and kinase protein expression as well as protein-tyrosine phosphorylation with anti-pTyr antibodies. When expressed alone, both Itk (Fig. 1) and Btk (Fig. 2) showed a diffuse subcellular staining pattern with both the kinase and anti-pTyr antibodies. Co-expression with Nef resulted in a strong, membrane-localized BiFC signal with both kinases, indicative of interaction, as well as a strong anti-pTyr immunoreactivity that also localized to the membrane. Treatment of the cells with the Itk inhibitor BMS-509744 (28) or the Itk/Btk inhibitor ibrutinib (29) suppressed the anti-pTyr signal but did not affect interaction (BiFC), supporting the conclusion that interaction with Nef at the cell membrane is sufficient to stimulate kinase activity.
Figure 1.
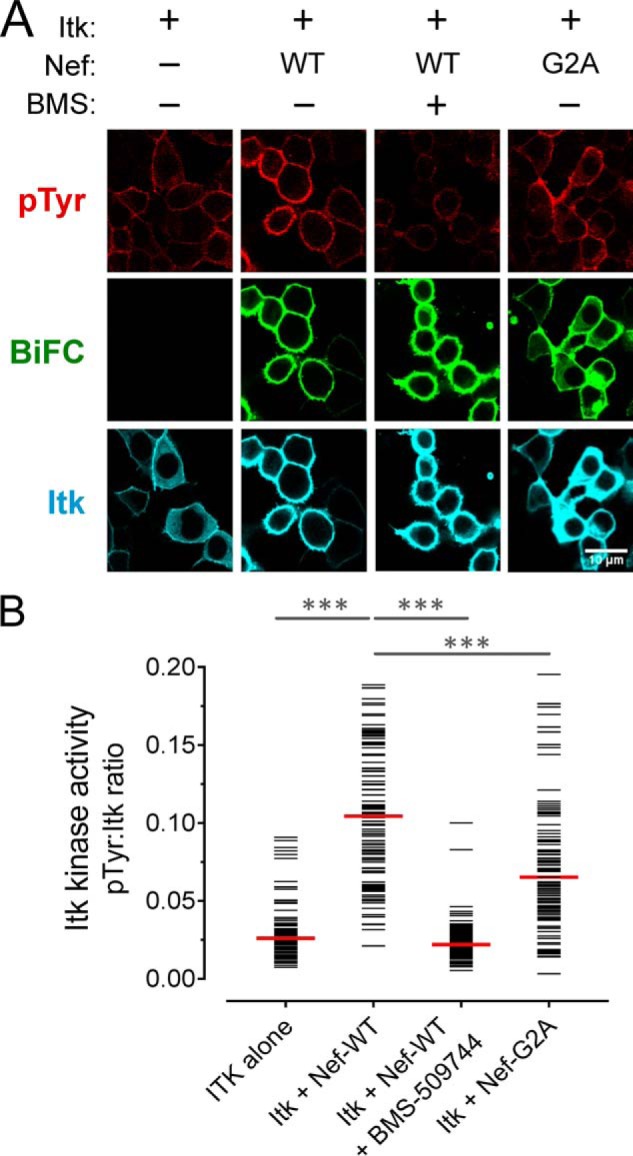
Nef induces constitutive activation of Itk at the cell membrane. A, Itk was expressed either alone or together with WT or myristoylation-defective Nef (G2A) as BiFC pairs in the absence or presence of the Itk inhibitor BMS-509744 (1 μm) in 293T cells. The cells were fixed and stained for confocal microscopy with anti-pTyr antibodies as a measure of kinase activity (red) and anti-V5 antibodies to verify Itk protein expression (blue). Nef interaction with Itk is observed as fluorescence complementation of the YFP variant, Venus (BiFC; green). B, single-cell image analysis. Mean fluorescence intensities for the pTyr and Itk signals were determined for ≥100 cells from each condition using ImageJ. The fluorescence intensity ratio (pTyr:Itk expression) for each cell is shown as a horizontal bar, with the median value indicated by the red bar. Student's t tests were performed on the groups indicated by horizontal lines above the plot; p < 0.0001 in each case (***).
Figure 2.
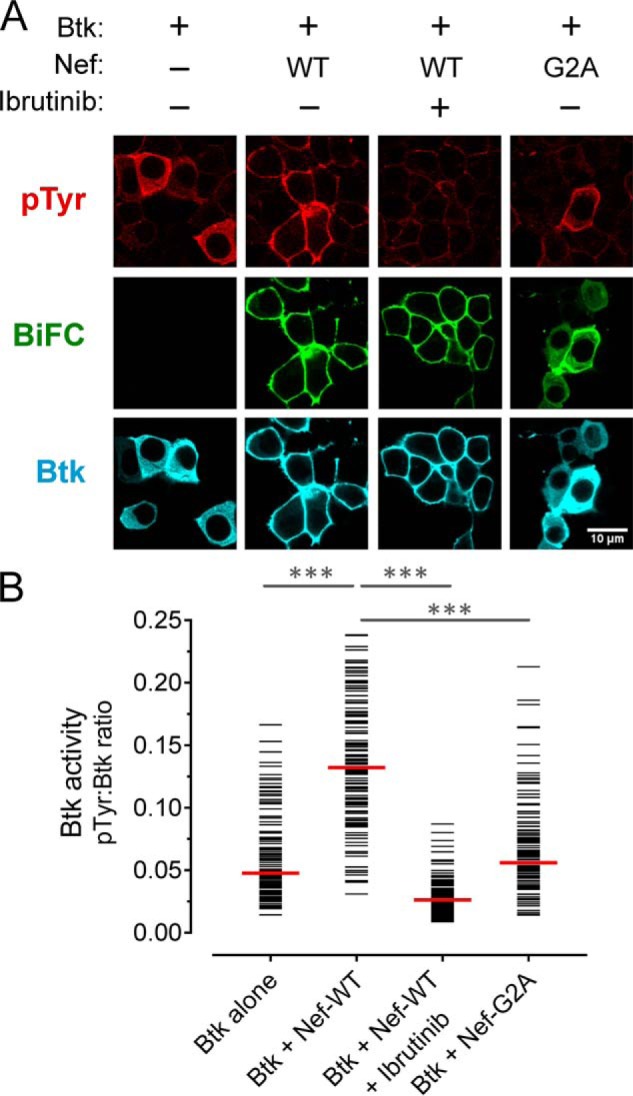
Nef induces constitutive activation of Btk at the cell membrane. A, Btk was expressed either alone or together with WT or myristoylation-defective Nef (G2A) as BiFC pairs in the absence or presence of the Btk inhibitor, ibrutinib (1 μm) in 293T cells. The cells were fixed and stained for confocal microscopy with anti-pTyr antibodies as a measure of kinase activity (red) and anti-V5 antibodies to verify Btk protein expression (blue). Nef interaction with Btk is observed as fluorescence complementation of the YFP variant, Venus (BiFC; green). B, single-cell image analysis was performed as per the legend to Fig. 1. The fluorescence intensity ratio (pTyr:Btk expression) for each cell is presented as a horizontal bar, with each median value indicated by the red bar. Student's t tests were performed on the groups indicated by horizontal lines above the plot; p < 0.0001 in each case (***).
To quantify the results, the anti-pTyr and kinase expression IF signal intensities were measured for a minimum of 100 cells from each condition, and the results are shown as pTyr/kinase–IF signal ratios (Figs. 1B and 2B). This analysis clearly shows that cell populations co-expressing Itk or Btk with Nef had significantly higher ratios compared with those expressing the kinase alone or the inhibitor-treated cells expressing the Nef·kinase complex.
We next investigated whether Nef stimulated Itk and Btk autophosphorylation on their respective activation loop tyrosines (pTyr511 and pTyr551, respectively), an essential step in Tec-family kinase activation. For this experiment, 293T cells were transfected as before and stained with phosphospecific antibodies for the activation loop tyrosines of Itk and Btk in place of the anti-pTyr antibody (Figs. S1 and S2). For both Itk and Btk, co-expression with Nef led to significant increases in activation loop phosphorylation at the cell membrane that was suppressed by the addition of ibrutinib. In addition to HIV-1 Nef, we also included SIV Nef (mac239 isolate) in this experiment. As with HIV-1 Nef, SIV Nef also induced strong membrane-associated autophosphorylation of both Itk and Btk (Figs. S1 and S2). These observations suggest that Tec-family kinase activation is a broadly conserved property of primate lentiviruses.
Active complexes of Itk and Btk with Nef were observed predominantly at the cell membrane, suggesting that membrane association of Nef may be required for kinase activation. Unlike Tec-family kinases, Nef is constitutively associated with the membrane because of myristoylation of a conserved glycine residue at position 2 near the N terminus. To test the role of Nef membrane association in Tec-family kinase activation, we co-expressed a myristoylation-defective Nef mutant (Gly2 to Ala; G2A) with Itk or Btk and monitored interaction as well as kinase expression and activation as before. The Nef-G2A mutant induced significantly lower Itk and Btk activation compared with WT Nef (Figs. 1A and 2A), suggesting that membrane localization is required for kinase activation. The BiFC signal also revealed that Nef-G2A·kinase complexes were present in the cytoplasm in addition to partial localization at the membrane. Although myristoylation is essential for Nef membrane localization and function, a basic patch in the N-terminal region of Nef also contributes to membrane targeting and may be partially responsible for retention at the membrane.
The results presented above show that active complexes of Itk and Btk localize almost exclusively to the cell membrane and that membrane localization may contribute to kinase activation. These findings imply that Nef actively recruits Tec-family kinases to the membrane, potentially short-circuiting the mechanisms normally responsible for regulated kinase activation. Both Itk and Btk have N-terminal pleckstrin homology (PH) domains that bind phosphorylated inositol lipids (30). Under physiological conditions, antigen receptor stimulation leads to phosphatidylinositol 3-kinase (PI3K) activation, triggering phosphatidylinositol 3-phosphate formation in the membrane and subsequent recruitment of Itk and Btk via their PH domains. To assess the impact of Nef on Itk and Btk localization more directly, we used the plot profile tool of the ImageJ image-analysis software package. This tool allowed us to generate a two-dimensional graph of the fluorescence intensities of pixels along a line drawn across the cytoplasm of cells expressing each kinase alone or in the presence of WT Nef or the myristoylation-defective mutant (Nef-G2A). As shown in Fig. 3, cells expressing Itk alone showed localization to both the cytoplasm and the membrane. In the presence of Nef, however, the localization shifted almost exclusively to the membrane; this effect was not observed with the Nef-G2A mutant. Unlike Itk, Btk showed very little membrane localization on its own, whereas co-expression with Nef led to a dramatic shift to the membrane compartment that was also dependent on Nef myristoylation. The ability of Nef to induce translocation of Itk and Btk to the membrane, along with kinase activation, strongly suggests that Nef can short-circuit antigen receptor signaling in HIV-infected cells for the benefit of the virus (see “Discussion”).
Figure 3.
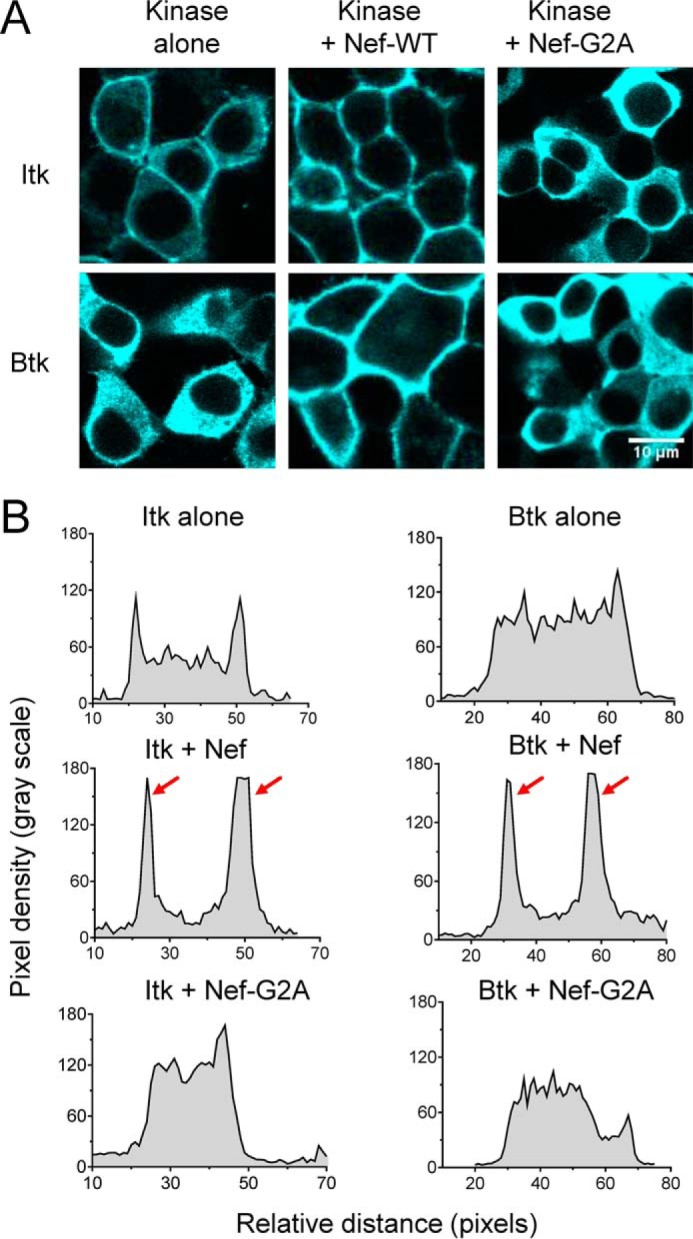
Membrane recruitment of Btk and Itk requires myristoylation of HIV-1 Nef. A, Itk or Btk were expressed either alone or in combination with WT or myristoylation-defective Nef (G2A) in 293T cells. The cells were stained with antibodies to the V5 tag present in each kinase protein, and imaged via confocal microscopy. Representative images are shown. B, using the Plot Profile tool from ImageJ, a line was drawn across the cytoplasm of representative cells from each condition, excluding the nucleus. The change in fluorescence intensity along this line was then plotted as relative pixel density as a function of distance. Very similar results were observed with at least 10 cells from each condition, and a representative example is shown. Strong membrane localization of both Itk and Btk was observed in the presence of WT Nef and is reflected by the prominent peaks indicated by the red arrows.
Nef homodimerization is required for Itk and Btk activation but not membrane recruitment
Previous structural studies have demonstrated that Nef forms homodimers when crystallized in the presence of the regulatory SH3 and SH2 domains of Src-family kinases (20, 21). In these structures, the Nef homodimer interface is formed by the αB helices present in each Nef monomer and involves the side chains of residues Leu112, Tyr115, and Phe121, which come together to form a hydrophobic core (modeled in Fig. 4A). These hydrophobic residues are highly conserved among HIV-1 Nef subtypes, indicating that homodimerization may be an essential feature of HIV-1 Nef that is critical to its functions including activation of Itk and Btk. To test this hypothesis, we generated a panel of mutants in which these conserved hydrophobic residues were replaced with either aspartate (L112D, Y115D, and the corresponding double mutant, LY/DD) or alanine (F121A). WT and mutant Nef proteins were then expressed in recombinant form in Escherichia coli followed by analytical size-exclusion chromatography (Fig. 4B). WT Nef eluted from the size-exclusion column primarily as a dimer, whereas the mutants exist primarily (Y115D) or exclusively (L112D, F121A, and LY/DD) in the monomeric form. We then assessed the impact of the mutations on Nef homodimer formation in cells using the BiFC assay. All four Nef mutants showed a significant reduction in the BiFC signal arising from homodimer formation without affecting Nef protein expression (Fig. 4, C and D). Together with the analytical gel filtration data, these results confirm the importance of these three amino acids in Nef homodimer formation, consistent with the crystal structures.
Figure 4.

Conserved hydrophobic residues in the folded Nef core are required for homodimer formation. A, Nef homodimers present in the X-ray crystal structure of the HIV-1 Nef core in complex with a Src-family kinase SH3 domain (PDB code 1EFN). An overview of the Nef dimer structure is shown on the left, with the αB helices that form the dimer interface highlighted; SH3 domains are not shown for clarity. The αB helices are enlarged on the right. Side chains of Leu112, Tyr115, and Phe121 from each Nef monomer form the hydrophobic core of the interface. B, analytical size-exclusion chromatography of WT Nef and dimerization interface mutants. WT Nef and the L112D, Y115D, F121A, and L112D/Y115D (LY/DD) mutants were expressed in E. coli and purified (see “Experimental procedures”). Each Nef protein was then characterized by analytical size-exclusion chromatography. Elution peaks of standard proteins are indicated by the vertical dotted lines and molecular weight. m, monomer; d, dimer. C, hydrophobic residues in the Nef core are required for homodimer formation in cells. WT Nef and the dimer interface mutants were expressed as BiFC pairs in 293T cells. The cells were stained for Nef expression with an anti-Nef antibody and imaged by confocal microscopy to detect Nef homodimer formation (BiFC, green) and Nef expression as immunofluorescence (Nef-IF, red). D, single-cell image analysis. Mean fluorescence intensities for the BiFC (interaction) and Nef-IF signals were determined with ImageJ for ≥100 cells. The Nef-BiFC:Nef-IF fluorescence intensity ratio for each cell is presented as a horizontal bar, with the median value indicated by the red bar. Student's t tests were performed on the groups indicated by horizontal lines above the plot; p < 0.0001 in each case.
Using these dimerization-defective mutants, we investigated whether Nef dimer formation is required for constitutive activation of Tec-family kinases at the cell membrane. Each of the mutants was co-expressed with Itk or Btk as BiFC pairs in 293T cells as before, and kinase activity was assessed by immunofluorescence imaging with anti-pTyr and anti-kinase antibodies followed by single-cell image analysis. All four dimerization-defective Nef mutants failed to activate Itk (Fig. 5) and Btk (Fig. 6), with pTyr:kinase expression ratios equivalent to those observed with the kinases alone. Based on the BiFC signal, all four Nef mutants remained associated with each kinase protein at the cell membrane. These observations suggest that the Nef mutants most likely form nonproductive 1:1 heterodimers with the kinases, whereas the WT Nef protein induces a 2:2 dimer complex with the kinase and promotes autophosphorylation in trans.
Figure 5.
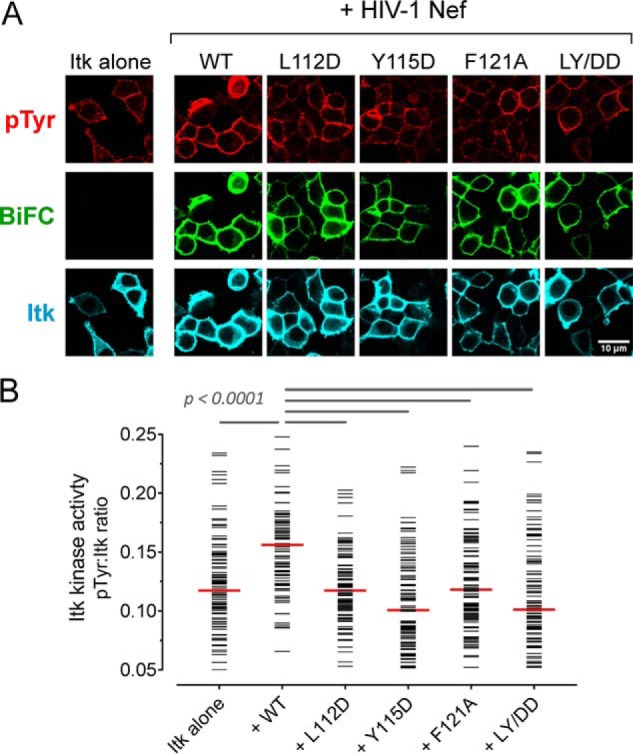
Dimerization-defective Nef mutants interact with Itk but fail to induce kinase activation. A, Itk was expressed either alone or together with WT Nef or dimerization-defective mutants as BiFC pairs in 293T cells. The cells were fixed and stained for confocal microscopy with anti-pTyr antibodies as a measure of kinase activity (red) and anti-V5 antibodies to verify Itk protein expression (blue). Nef interaction with Itk is observed as Venus fluorescence (BiFC; green). B, single-cell image analysis was performed as per the legend to Fig. 1. The fluorescence intensity ratio (pTyr:Itk expression) for each cell is shown as a horizontal bar, with the median value indicated by the red bar. Student's t tests were performed on the groups indicated by horizontal lines above the plot; p < 0.0001 in each case.
Figure 6.
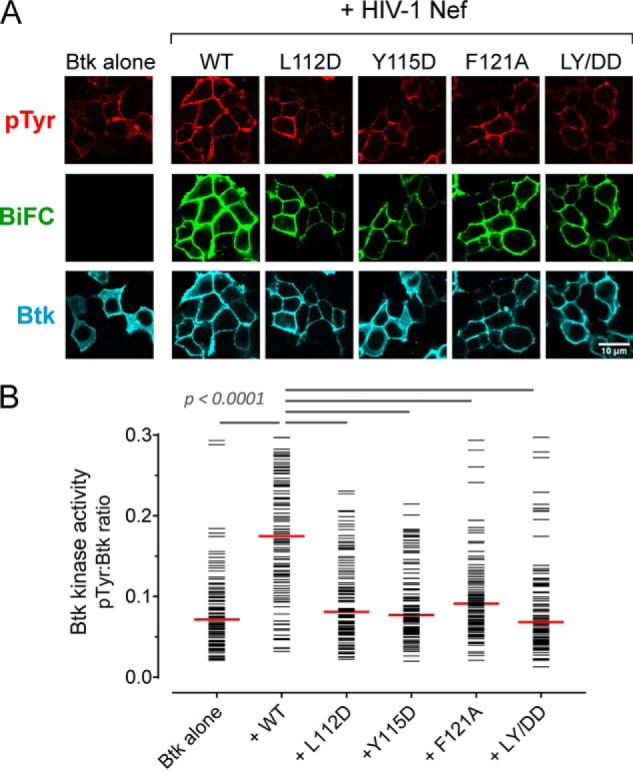
Dimerization-defective Nef mutants interact with Btk but fail to induce kinase activation. A, Btk was expressed either alone or together with WT Nef or Nef dimerization-defective mutants as BiFC pairs in 293T cells. The cells were fixed and stained for confocal microscopy with anti-pTyr antibodies as a measure of kinase activity (red) and anti-V5 antibodies to verify Btk protein expression (blue). Nef interaction with Btk is observed as Venus fluorescence (BiFC; green). B, single-cell image analysis was performed as per the legend to Fig. 2. The fluorescence intensity ratio (pTyr:Btk expression) for each cell is shown as a horizontal bar, with the median value indicated by the red bar. Student's t tests were performed on the groups indicated by horizontal lines above the plot; p < 0.0001 in each case.
Very similar results were obtained when cells were stained with phosphospecific antibodies against the Itk and Btk activation loop phosphotyrosines in place of the general anti-pTyr antibody (Figs. S3 and S4). Not only did the Nef mutants fail to activate the kinases, the Nef-LY/DD and F121A mutants suppressed the autophosphorylation of both Itk and Btk well below the levels observed when the kinases were expressed in the absence of Nef. This observation, coupled with the sustained interaction of the Nef mutants with each kinase at the membrane by BiFC, suggests that dimerization-defective Nef proteins may suppress spontaneous autophosphorylation of Itk and Btk via a dominant-negative mechanism.
Small molecule Nef inhibitors block Nef-dependent Itk and Btk activation
Our group previously reported the discovery of small molecule Nef inhibitors using a high-throughput screening assay based on Nef-dependent activation of the Src-family kinase, Hck (17). The original hit compound from this study, known as B9, and a non-azo B9 analog (compound 2 from Emert-Sedlak et al. (31); see Fig. 7A for structures) bind directly to recombinant HIV-1 Nef in vitro with KD values in the high nanomolar range and inhibit Nef-mediated enhancement of viral infectivity and replication. Both compounds have been shown to restore cell-surface MHC-I to HIV-1–infected CD4 T cells in a Nef-dependent manner, triggering an autologous CTL response in vitro (32). Docking models of both inhibitors with X-ray crystal structures of Nef have suggested that these compounds may recognize a pocket formed by the helical Nef dimer interface (17, 31) (Fig. 7A). To test whether these inhibitors affect Nef-mediated Tec-family kinase activation, we transfected 293T cells with BiFC pairs of Nef and Itk or Btk as before and then treated the cells with each compound at a final concentration of 1 μm or with the DMSO carrier solvent as a negative control. The cells were then imaged for kinase activity as antiphosphotyrosine immunofluorescence, interaction of Nef with each kinase (BiFC signal), and kinase expression by immunofluourescence. Representative confocal micrographs (Fig. 7B) and single-cell image analysis (Fig. 7C) show that both inhibitors significantly inhibited Nef-dependent activation of Itk and Btk without influencing Nef interaction with each kinase. Control experiments showed that the compounds did not affect 293T cell viability at this concentration, did not quench the immunofluorescent signals, and had no effect on basal kinase activity when added to cultures expressing the kinases in the absence of Nef (data not shown). These results suggest that Nef inhibitors in this class interfere with Tec-family kinase activation by affecting Nef homodimer formation in a manner similar to the dimerization-defective mutants. Inhibition of Nef-dependent activation of Itk may explain, in part, the effect of these compounds on HIV-1 replication in T-cell lines as reported previously (17).
Figure 7.
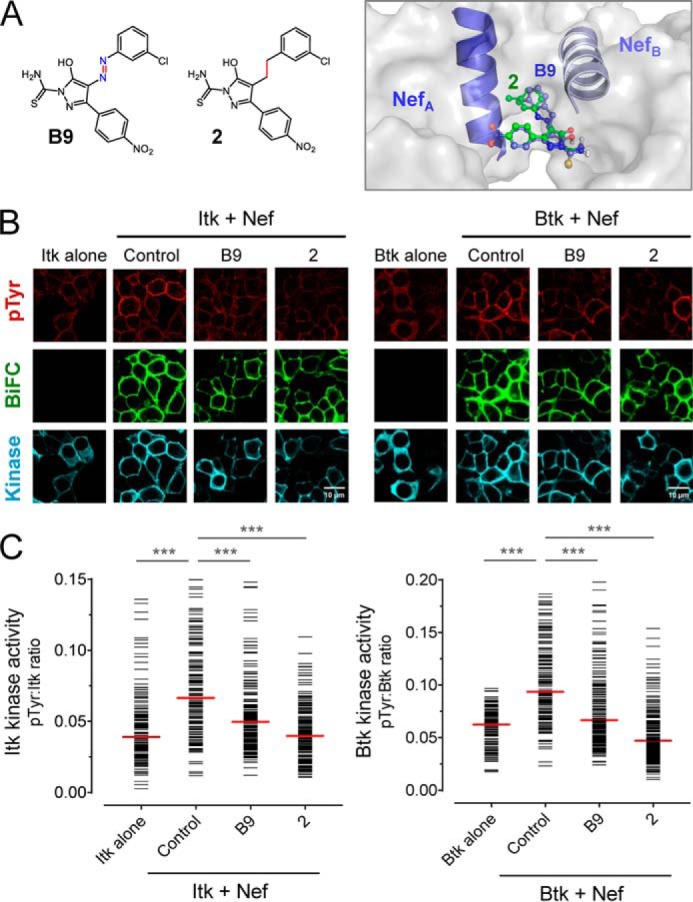
Small molecule Nef inhibitors block Nef-mediated Itk and Btk activation. A, structures of small molecule Nef inhibitors, B9 and compound 2, are shown on the left. Computational docking predicts interaction of both inhibitors with the Nef dimerization interface (model at right; Nef αB helices that form this interface are also shown). B and C, Itk and Btk were expressed in 293T cells either alone or together with WT Nef as BiFC pairs in the absence (Control) or presence of B9 or compound 2 (final concentration of 1.0 μm). Following inhibitor treatment, the cells were fixed and stained for confocal microscopy with anti-pTyr antibodies as a measure of kinase activity (red) and anti-V5 antibodies to verify kinase protein expression (blue). Nef interaction with each kinase was visualized as fluorescence complementation of the YFP variant, Venus (BiFC; green). B, representative images from each experiment. C, single-cell image analysis. Mean fluorescence intensities for the pTyr and Itk or Btk signals were determined for ≥100 cells from each condition using ImageJ. The fluorescence intensity ratio (pTyr:Itk expression) for each cell is shown as a horizontal bar, with the median value indicated by the red bar. Student's t tests were performed on the groups indicated by horizontal lines above the plot; p < 0.0001 in each case (***).
Endogenous Itk activation requires Nef homodimers in HIV-infected T-cell lines and PBMCs
Using transfected 293T cells as a model system, we established that HIV-1 Nef recruits both Itk and Btk to the cell membrane and induces constitutive kinase activity through a dimerization-dependent mechanism. To validate this mechanism in the context of HIV-1 infection, we introduced the same panel of dimerization-defective Nef mutations into an HIV-1 NL4-3 proviral backbone, which carries the closely related SF2 Nef sequence. Our previous work has shown that HIV-1 produced from this provirus is as equally infectious as WT NL4-3, thereby allowing a direct comparison to the NefSF2 isolate used in the 293T cell experiments (33).
The human T-cell line SupT1 was infected with WT HIV-1, the Nef dimerization-defective mutants, as well as a virus that fails to express Nef (ΔNef). Four days later, the cells were primed with T-cell co-receptor antibodies (anti-CD3/CD28), fixed, and immunostained for HIV-1 p24 Gag as well as a phosphospecific antibody for the Itk activation loop phosphotyrosine (pTyr511). Flow cytometry showed that close to 40% of the SupT1 cells infected with WT HIV-1 stained positive for HIV-1 p24, whereas only 25% of cells infected with the ΔNef virus were positive for p24 (Fig. 8A). This difference is likely to reflect the reduced infectivity of the Nef-defective virions, which may result from enhanced incorporation of SERINC proteins from the producer cells (13, 14). All four SupT1 cell populations infected with the Nef dimerization-defective viruses showed even lower proportions of HIV-1 p24+ cells compared with the ΔNef virus, with less than 10% of the cells positive in each case. Similar results were observed following analysis of HIV-1 release into the culture supernatant by p24 Gag AlphaLISA assay (Fig. 8B). In this case, WT HIV-1 showed 4-fold higher replication compared with the ΔNef virus and a 20-fold to nearly 40-fold enhancement compared with the Nef dimerization-defective mutants. We then looked for Nef-dependent differences in endogenous Itk activity across the HIV-infected SupT1 cell populations. Cultures infected with WT HIV-1 showed significantly more cells positive for Itk activation loop phosphorylation (pTyr511) compared with those infected with the ΔNef virus or each of the dimerization-defective mutants (Fig. 8C).
Figure 8.
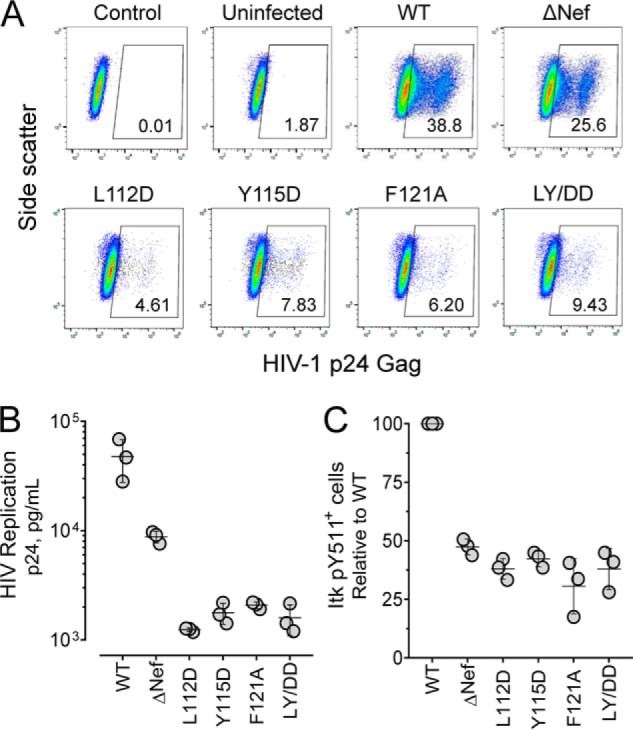
HIV-1 expressing dimerization-defective Nef mutants show attenuated replication and reduced endogenous Itk activation in SupT1 cells. SupT1 cells were infected with HIV-1 expressing WT Nef, dimerization-defective mutants of Nef or virus that does not express Nef (ΔNef; viral input in each case 5000 pg/ml p24). Four days later, infected cells were primed with anti-CD3/anti-CD28 antibodies, fixed, and stained with antibodies to HIV-1 p24 Gag and active Itk (anti-pTyr511 antibody). A, representative flow cytometry plots for unstained, uninfected cells (Control), uninfected stained cells (Uninfected), and cells infected with WT, ΔNef, and dimerization-defective Nef mutant viruses. The number in the gating box indicates the percentage of p24+ infected cells present in each culture. B, viral replication was quantified as p24 Gag release into the culture supernatant. The results from three independent determinations are shown with the horizontal bar representing the mean values ± S.E. Student's t tests show that p24 values for ΔNef and each Nef mutant virus are significantly different from WT (p < 0.05 in each case). In addition, comparison of the p24 values for ΔNef with each of the Nef mutants is also significant (p < 0.0005 in each case). C, percentage of Itk pTyr511+ cells present in each infected (p24+) cell population. The results of three independent experiments are plotted as percentages of pTyr511+ cells relative to control cells infected with WT HIV-1. Mean values are shown as the horizontal bar ± S.E. The values for ΔNef and each Nef mutant virus are significantly different from WT (p < 0.0005 in each case).
In a final series of experiments, we extended these observations using donor PBMCs as host cells. PBMCs were first activated with PHA and IL-2 and then infected with WT, ΔNef, or Nef dimerization-defective viruses for 4 days. Two days prior to harvesting, PBMCs were primed with anti-CD3 and anti-CD28 antibodies as before and then stained for p24 Gag and autophosphorylated Itk (anti-pTyr511) and analyzed by flow cytometry (Fig. 9A). The PBMC population infected with WT HIV-1 was ∼7% positive for p24, and this proportion was reduced by more than half in cells infected with the ΔNef virus. The proportion of p24+ cells was diminished even further in PBMCs infected with the dimerization-defective mutants, with the exception of the Y115D virus. Viral replication in the PBMC culture, assessed as p24 Gag release into the supernatant, was diminished nearly 8-fold in cells infected with the ΔNef virus. Replication was reduced even further in cells infected with the Nef dimerization-defective viruses, with the greatest reduction observed with the double mutant, LY/DD (26-fold lower; Fig. 9B). Cultures infected with WT HIV-1 showed significantly more Itk pTyr511+ cells compared with those infected with the ΔNef or Nef dimerization-defective viruses (Fig. 9C). Taken together, these data support a role for Nef homodimer formation in Itk activation to facilitate viral replication. Although these experiments do not identify the specific point in the viral life cycle affected by the Nef dimerization mutants, prior work with Itk inhibitors demonstrates that Itk kinase activity enhances viral entry, transcription, and egress of newly synthesized virions (25, 26). Nevertheless, dimerization-defective Nef mutants are likely to interfere with other signaling pathways dependent on Nef homodimer formation, including activation of Hck and other protein kinases.
Figure 9.
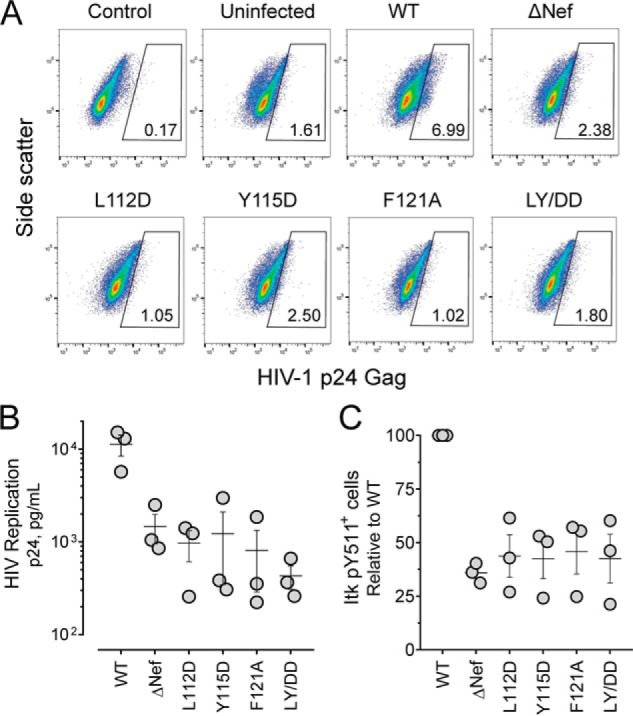
HIV-1–expressing dimerization-defective Nef mutants show attenuated replication and endogenous Itk activation in donor PBMCs. PBMCs were isolated from normal donors and stimulated with PHA and IL-2 for 3 days. The cells were then infected with HIV-1–expressing WT Nef, dimerization-defective mutants of Nef or virus that does not express Nef (ΔNef; viral input in each case 40 pg/ml p24). Infected PBMCs were primed with anti-CD3/anti-CD28 antibodies on day 5, and on day 7 they were fixed and stained for HIV-1 p24 Gag and active Itk (anti-pTyr511 antibody) followed by flow cytometry. A, representative flow cytometry plots for unstained, uninfected cells (Control), uninfected stained cells (Uninfected), and cells infected with WT, ΔNef, and dimerization-defective Nef mutant viruses. The number in the gating box indicates the percentage of p24+ infected cells present in each culture. B, viral replication was quantified as p24 Gag release into the culture supernatant. The results from three independent determinations are shown with the horizontal bar representing the mean value ± S.E. Student's t tests show that p24 values for ΔNef and each Nef mutant virus are significantly different from WT (p < 0.05 in each case). This experiment was repeated three times with comparable results; a representative example is shown. C, percentage of Itk pTyr511+ cells present in each infected (p24+) cell population. This experiment was performed three times, and the results are plotted as percentages of pTyr511+ cells relative to control cells infected with WT HIV-1 ± S.E. The values for ΔNef and each Nef mutant virus are significantly different from WT (p < 0.01 in each case).
Discussion
In this study, we demonstrate that HIV-1 Nef interacts with the Tec-family kinases Itk and Btk at the cell membrane, resulting in autophosphorylation and constitutive kinase activity. Mutants of Nef that are defective for homodimer formation fail to induce kinase activation, supporting a mechanism in which the Nef homodimer engages two kinase molecules, thereby juxtaposing their kinase domains for autophosphorylation via a trans mechanism. These Nef mutants retain the ability to interact with Itk and Btk at the membrane, demonstrating that membrane localization and Nef interaction alone are not sufficient to induce kinase activation. Dimerization-defective Nef mutants reduced HIV-1 replication below the level observed in the absence of Nef expression in a T-cell line. This observation is consistent with a dominant-negative suppressor effect, in which dimerization-defective Nef mutants bind to Itk (and possibly other kinases) and trap it in a nonfunctional, monomeric complex. Although our BiFC results support a mechanism in which Nef recruits Itk and Btk to the membrane for sustained activation, the possibility also exists that they may interact initially in the cytoplasm and traffic to the membrane as a complex. Future studies employing live-cell imaging will be required to test this possibility. A model of the Nef-induced mechanism of Itk activation is shown in Fig. 10A.
Figure 10.
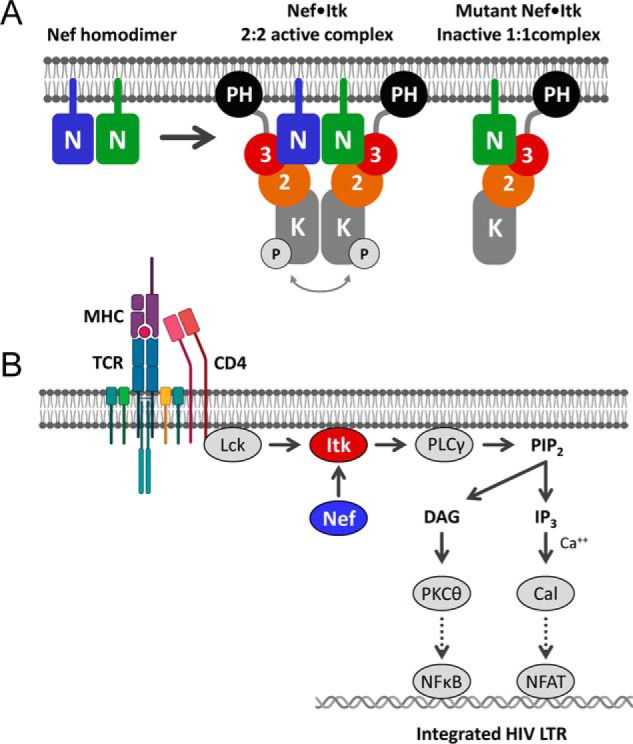
Models of Nef-mediated Itk activation and consequences for HIV-1 transcription. A, Nef forms homodimers at the cytoplasmic face of the plasma membrane, which stabilize Itk homodimers as a 2:2 complex. Regulatory domain displacement and kinase domain juxtaposition contribute to sustained kinase activation via autophosphorylation in trans. Dimerization-defective Nef mutants retain interaction with Itk at the membrane but form inactive 1:1 complexes. N, Nef; 3, Src homology 3 domain; 2, Src homology 2 domain; K, kinase domain; P, activation loop phosphorylation. B, proposed consequences of constitutive Itk activation by Nef for HIV-1 transcription in CD4 T cells. The T-cell receptor (TCR) complex is normally activated by antigen-loaded MHC molecules. The TCR activates the T cell–specific Src-family kinase Lck, which is associated with the cytosolic tail of the co-receptor, CD4. Active Lck directly phosphorylates Itk on its activation loop, which in turn phosphorylates and activates phospholipase Cγ (PLCγ). Active PLCγ generates the second messenger diacylglycerol (DAG) and inositol triphosphate (IP3) via hydrolysis of membrane phosphatidylinositol 4,5-bisphosphate (PIP2), ultimately leading to activation of protein kinase Cθ (PKCθ) and the calcium-dependent protein serine/threonine phosphatase, calcineurin (Cal), respectively. Protein kinase Cθ promotes activation of NF-κB via the CARMA1/BCL10/MALT1 complex (not shown), whereas calcineurin dephosphorylates NFAT to drive nuclear localization. Both NF-κB and NFAT participate in transcription of the integrated HIV-1 provirus. The data presented here support direct activation of Itk by Nef at the membrane downstream of the TCR. Additional details of TCR signaling are omitted for clarity. The cartoon was adapted from Gaud et al. (46). LTR, long terminal repeat.
Direct and sustained activation of Itk signaling by HIV-1 Nef may short-circuit normal T-cell receptor signaling for the benefit of the virus. TCR signaling is physiologically activated upon interaction of the TCR α and β subunits with cognate peptide-bound major histocompatibility complexes on antigen-presenting cells (27). This interaction exposes immunoreceptor tyrosine-based activation motifs in the TCR ζ-chain, which is phosphorylated by the T cell Src-family kinase, Lck. Immunoreceptor tyrosine-based activation motif tyrosine phosphorylation induces recruitment of the ZAP-70 tyrosine kinase through its SH2 domains, which become fully active upon Lck-mediated phosphorylation. ZAP-70 then phosphorylates the linker for activation of T cells, which recruits additional effector proteins related to Erk and calcium signaling. Together with the CD28 co-receptor, the TCR also recruits and activates PI3K, resulting in PIP3 production in the membrane and subsequent recruitment of Itk through its PH domain. Proximity to the membrane results in Lck-mediated phosphorylation of the Itk activation loop as required for kinase activation. Our data show that HIV-1 Nef bypasses this complex sequence of events, driving direct Itk recruitment to the cell membrane and kinase activation. Itk and Btk signaling are linked to activation of NF-κB, NFAT, and other transcription factors downstream (30, 34). The NF-κB transcription factor is essential for transcription of integrated primate lentiviruses, and Nef contributes to the early stages of HIV-1 transcription through the NF-κB pathway (35). Constitutive activation of Itk and Btk by Nef may therefore trigger NF-κB, as well as other transcription factors linked to viral gene expression in both CD4+ T cells and macrophages, respectively. Indeed, older work has linked Nef to NFAT activation through a calcium-dependent mechanism (36). The proximal substrate for Itk at the plasma membrane is phospholipase-Cγ, which induces intracellular calcium mobilization via IP3. Nef-mediated Itk activation may therefore represent the initial event that triggers this signaling pathway. The point at which Nef may short-circuit the TCR signaling cascade to enhance T cell activation and viral transcription is illustrated in Fig. 10B.
Our results support a mechanism in which Nef recruits Itk and Btk to the cell membrane through direct protein–protein interaction. Although previous work has implicated Tec-family kinase SH3 domains in the interaction mechanism, mutation of the Nef PXXPXR motif essential for Src-family kinase recruitment only partially reduced interaction with Itk and Btk (26). In the present study, we observed that in addition to HIV-1 Nef, SIV Nef also strongly interacts with Itk and Btk at the membrane and induces robust kinase activation. Notably, SIV Nef does not bind to the SH3 domain or affect the activity of Hck (37), suggesting that the molecular mechanisms of Nef interaction with Tec versus Src family members may be distinct.
Nef is constitutively associated with the cell membrane by virtue of N-terminal myristoylation, and mutants lacking this post-translational modification are defective for Itk and Btk membrane recruitment as well as activation. However, previous studies have demonstrated that Nef also interacts with the regulatory 85-kDa subunit of PI3K (p85), resulting in enhanced PI3K activity and PIP3 formation in the membrane (38). In this way, Nef-mediated PI3K activation may also contribute to the relocalization of Tec-family kinases from the cytoplasm to the membrane through their PH domains. Structural work has established that the PH domain of Btk interacts with the kinase domain in an autoinhibitory fashion that is relieved by phosphoinositide binding (39). Interaction with Nef may relieve this negative regulatory influence by displacing the PH domain, in a manner analogous to regulatory SH3 domain displacement associated with Nef-dependent activation of the Src-family kinase, Hck (16, 40). Furthermore, membrane PIP3 may also drive Btk dimer formation as part of a unique activation mechanism not shared by Itk (41). In this case, Nef may exploit and enhance this natural mechanism of Btk activation by stabilizing preformed dimers at the membrane.
Part of the evidence supporting a role for Nef homodimer formation in the Tec-family kinase activation mechanism involves Nef mutants that are attenuated for homodimer formation in the BiFC assay. The three amino acids that were mutated, Leu112, Phe121, and Tyr115, all contribute to the helical dimerization interface present in two distinct X-ray crystal structures of Nef in complex with an SH3 domain or the dual SH3-SH2 domain from Hck (PDB files 1EFN and 4U5W, respectively). Note that these Nef residues do not make direct contact with the kinases, and BiFC data show that Nef·kinase complex formation is maintained with these mutants, despite their inability activate the kinase. Prior use of these same mutants in other experimental systems may suggest a previously unrecognized role for Nef homodimerization and Tec-family kinase activation. For example, the Nef L112A and F121A mutations also impair interaction with the endocytic regulatory protein Dynamin-2 (42), as well as down-regulation of CD4 (33). Wu et al. (43) reported SERINC protein-independent restriction of HIV-1 replication in the T-lymphoblast cell line MOLT-3 that is counteracted by Nef, supporting the existence of a novel restriction factor in these cells. This study also showed that HIV-1 expressing Nef mutants L112A and F121A replicated very poorly in MOLT-3 cells, raising the possibility that part of the replication defect may be due to failure to activate the Itk pathway as shown here.
In summary, our results show that Nef recruits Itk and Btk to the cell membrane, resulting in their constitutive activation through a unique mechanism that requires the Nef homodimer. In CD4+ T cells, the major host-cell type for HIV-1 replication, this effect of Nef may enhance normal mechanisms of Itk activation through the TCR, bypassing the requirements for Lck, ZAP-70, and PI3K for membrane recruitment, leading to enhanced viral transcription from the HIV-1 LTR. Small molecules that bind directly to Nef also suppress Nef-mediated Itk activation, providing a plausible explanation for the potent inhibition of HIV-1 replication by these compounds in T-cell lines (17, 31) and donor PBMCs (44). Future work will address the structure of the Nef·Itk complex to provide additional insight regarding the unique mechanism by which Nef hijacks this kinase pathway for the benefit of HIV-1.
Experimental procedures
Expression vectors
Full-length, sequence-verified human Tec-family kinase cDNA clones were obtained from the Dana-Farber/Harvard Cancer Center PlasmID DNA Resource Core (Btk, HsCD00346954; Itk, HsCD00021352). The coding regions for full-length Itk and Btk were amplified by PCR and fused in-frame with a V5 epitope tag followed by the C-terminal coding fragment of the Venus protein (Venus residues Ala154 to Lys238) at their C termini as described (26). Complementary HIV-1 Nef (SF2) and SIV Nef (mac239) expression constructs fused the N-terminal coding fragment of the Venus protein (residues Val2 to Asp173) to the Nef C termini as described (26). Nef dimerization interface mutants (L112D, Y115D, F121A, and LY/DD), and the myristoylation-defective mutant (G2A) were created using the QuikChange II XL site-directed mutagenesis kit (Agilent). All PCR products were subcloned into the mammalian expression vector, pCDNA3.1(−) (Thermo Fisher/Invitrogen).
Cell culture, reagents, and antibodies
Human embryonic kidney 293T cells and the SupT1 T cells were obtained from the American Type Culture Collection. 293T cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS; Gemini Bio-Products). SupT1 cells were cultured in RPMI 1640 medium supplemented with 10% FBS and l-glutamine. Cell culture media and supplements were purchased from Thermo Fisher/Invitrogen. The Itk inhibitor BMS-509744 was purchased from Calbiochem (catalog no. 41-982-05MG), and the Btk inhibitor ibrutinib was purchased from Selleckchem (catalog no. S2680). Discovery, synthesis, and characterization of the hydroxypyrazole Nef inhibitor B9 (17) and a non-azo B9 analog (compound 2 from Emert-Sedlak et al. (31)) are described in detail elsewhere.
Primary antibodies were obtained from Thermo Fisher (V5 tag mouse monoclonal, catalog no. R960-25), Sigma (V5 tag rabbit polyclonal, catalog no. AB3792), Abcam (BTK pY551 Rabbit monoclonal, catalog no. ab40770), Santa Cruz (pTyr antibody pY99, catalog no. sc-7020), and the National Institutes of Health AIDS Reagent Program (HIV-1 Nef monoclonal antibody 6.2, catalog no. 1539). Phosphoflow antibodies were purchased from BD Pharmingen (phycoerythrin-conjugated mouse anti-BTK pTyr551/interleukin-2–inducible T-cell kinase pTyr511, catalog no. 558219) and Beckman Coulter (HIV Gag flow antibody Kc57-FITC, catalog no. 6604665). Secondary antibodies were obtained from Southern Biotech (goat anti-rabbit IgG mouse, catalog no. 4050-07; goat anti-mouse IgG, catalog no. 1031-07; and goat anti-mouse IgG, catalog no. 1031-04), and Thermo Fisher/Molecular Probes (Pacific Blue goat anti-mouse IgG, catalog no. P31582; and Pacific Blue goat anti-rabbit IgG, catalog no. P10994).
BiFC assay and immunofluorescence
Human 293T cells were plated in 35-mm microwell dishes (MatTek, catalog no. P35G-1.5-14-C) and cultured overnight. The cells were then transfected with expression vectors for individual BiFC fusion proteins or as BiFC pairs using X-tremeGENE 9 DNA transfection reagent (Sigma–Aldrich). For inhibitor studies, the cells were treated with Nef inhibitors or the DMSO carrier solvent (0.1% final concentration) 4 h post-transfection. 40 h post-transfection, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 2% BSA in PBS overnight. The cells were immunostained with anti-V5, anti-pTyr, or anti-pY551 (all antibodies diluted 1:1000 in PBS with 2% BSA) for 1 h at room temperature. The cells were washed and stained with secondary antibodies conjugated to Texas Red or Pacific Blue at dilutions of 1:500 and 1:1000, respectively. Immunostained images were acquired using confocal microscopy (Fluoview FV1000, Olympus) with a 60× objective using x-y scan mode. Single-cell image analysis was performed with the Java-based image processing program, ImageJ. Immunofluorescence intensities with each antibody were measured for at least 100 cells, and single-cell data were calculated as the fluorescence ratio of either kinase activity (pTyr or pTyr551/511) to kinase protein (Btk or Itk) or BiFC (interaction) to protein signals.
HIV-1 replication assays
The HIV-1 proviral constructs used in this study are based on a NL4-3 backbone in which the NL4-3 Nef ORF is replaced with that of Nef from the closely related HIV-1 B-clade isolate, SF2. This was done to match the Nef allele used in experiments with transfected 293T cells with those using HIV-1. HIV-1 stocks were produced in 293T cells transfected with a pUC18-based proviral clone and the XtremeGENE 9 transfection reagent. Viral supernatants were harvested 72 h post-transfection and amplified in the T-cell line MT2 as described elsewhere (18). Viral titers were quantified by HIV-1 p24 AlphaLISA assay (PerkinElmer, catalog no. AL291F) according to the manufacturer's protocol. For HIV-1 replication assays, SupT1 cells were infected with WT or mutant viruses (5000 pg p24/ml) and incubated for 4 days. Normal donor PBMCs were isolated from whole blood using Ficoll-Paque PLUS (GE Healthcare) and activated with 5 μg/ml PHA (Sigma, catalog no. L1668) and 200 U/ml IL-2 (Thermo Fisher, catalog no. CB-40043B) for 3 days. PBMCs were infected with WT or mutant viruses (40 pg/ml p24) for 4 days. Viral supernatants were collected, and viral replication was measured by AlphaLISA assay for HIV-1 p24.
T-cell stimulation and flow cytometry
SupT1 cells and donor PBMCs were prepared and infected with WT and mutant forms of HIV-1 as described above. SupT1 cells were harvested on day 4 and stimulated with 1 μg/ml anti-CD3 (clone OKT3, Thermo Fisher, catalog no. 16-0037-81) and 1 μg/ml anti-CD28 (Clone CD28.2, BD Pharmingen, catalog no. 556620) at 37 °C for 5 min followed by cross-linking with 1 μg/ml goat anti-mouse Fc IgG (Thermo Fisher, catalog no. Pl31168) at 37 °C for 5 min. Donor PBMCs were stimulated with 1 μg/ml anti-CD3 and 1 μg/ml anti-CD28 for 48 h 2 days after viral infection. The cells were harvested and cross-linked with 1 μg/ml goat anti-mouse Fc IgG at 37 °C for 5 min. After stimulation, both SupT1 cells and donor PBMCs were fixed and permeabilized for 20 min (BD Cytofix/Cytoperm, catalog no. 554714), followed by staining with p24 Gag and Itk pTyr511 antibodies diluted in PBS containing 2% FBS on ice for 1 h. Flow cytometry analysis was performed using a BD AccuriC6 Flow cytometer, and the data were processed using the FlowJo software package.
Expression and purification of recombinant Nef proteins and size-exclusion chromatography
Recombinant pET-21b(+) expression vectors containing the coding regions of HIV-1 Nef (SF2 allele) WT plus the L112D, Y115D, LY/DD, and F121A mutant forms were used to transform the E. coli strain Rosetta2(DE3)pLysS (EMD Millipore). Single colonies were used to inoculate starter cultures of LB medium (5.0 ml) and grown overnight at 37 °C. Starter cultures were then used to inoculate 2 liters of LB medium and grown to an A600 of 0.6. The cultures were cooled at room temperature for 1 h followed by addition of isopropyl β-d-thiogalactopyranoside to a final concentration of 1 mm. Nef protein expression was then induced at 17 °C for 16 h. Recombinant Nef protein was purified by ion exchange and gel filtration as described elsewhere (20) with gel filtration conducted at pH 7.4 instead of pH 8.3. The WT Nef protein eluted as two peaks from the preparative gel filtration column, corresponding to a mixture of dimeric and monomeric states that were isolated separately. The mutant Nef proteins were isolated as single monomeric peaks. Purified Nef proteins were concentrated and stored at −80 °C. Analytical size-exclusion chromatography of each purified Nef protein was performed as described elsewhere (45) in 10 mm Hepes, pH 7.4, containing 150 mm NaCl with Nef proteins at a final concentration of 12.5 μm.
Statistical analysis
Single cell analysis was performed on at least 100 cells/condition using ImageJ and used to calculate ratios of anti-pTyr or anti-pTyr511/551 to kinase protein fluorescence intensity. Ratios for individual cells are presented in all figures as horizontal bars with the horizontal red lines representing the median values. For each experiment, a representative result is shown from at least three independent biological replicates (n = 3). HIV-1 replication data and flow cytometry results are presented as mean values ± S.E. from three biological replicates (n = 3), each measured in triplicate. Where applicable, significant differences between groups were evaluated using unpaired Student's t test (GraphPad Prism v. 7.04).
Author contributions
W. F. L., M. A., S. T. S., and T. E. S. conceptualization; W. F. L. and T. E. S. data curation; W. F. L., M. A., S. T. S., and T. E. S. formal analysis; W. F. L., S. T. S., and T. E. S. validation; W. F. L., M. A., and S. T. S. investigation; W. F. L., M. A., and T. E. S. visualization; W. F. L. and M. A. methodology; W. F. L., M. A., and T. E. S. writing-original draft; W. F. L., S. T. S., and T. E. S. writing-review and editing; S. T. S. and T. E. S. supervision; T. E. S. funding acquisition; T. E. S. project administration.
Supplementary Material
This work was supported by National Institutes of Health Grants AI057083 and AI126617 (to T. E. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
- SIV
- simian immunodeficiency virus
- Itk
- interleukin-2–inducible T-cell kinase
- Btk
- Bruton's tyrosine kinase
- BiFC
- bimolecular fluorescence complementation
- IF
- immunofluorescence
- PH
- pleckstrin homology
- PI3K
- phosphatidylinositol 3-kinase
- PBMC
- peripheral blood mononuclear cell
- FBS
- fetal bovine serum
- PHA
- phytohemagglutinin.
References
- 1. Basmaciogullari S., and Pizzato M. (2014) The activity of Nef on HIV-1 infectivity. Front. Microbiol. 5, 232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pawlak E. N., and Dikeakos J. D. (2015) HIV-1 Nef: a master manipulator of the membrane trafficking machinery mediating immune evasion. Biochim. Biophys. Acta 1850, 733–741 10.1016/j.bbagen.2015.01.003 [DOI] [PubMed] [Google Scholar]
- 3. Kestler H. 3rd, Ringler D. J., Mori K., Panicali D. L., Sehgal P. K., Daniel M. D., and Desrosiers R. C. (1991) Importance of the nef gene for maintenance of high viral loads and for development of AIDS. Cell 65, 651–662 10.1016/0092-8674(91)90097-I [DOI] [PubMed] [Google Scholar]
- 4. Kirchhoff F., Greenough T. C., Brettler D. B., Sullivan J. L., and Desrosiers R. C. (1995) Absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N. Engl. J. Med. 332, 228–232 10.1056/NEJM199501263320405 [DOI] [PubMed] [Google Scholar]
- 5. Deacon N. J., Tsykin A., Solomon A., Smith K., Ludford-Menting M., Hooker D. J., McPhee D. A., Greenway A. L., Ellett A., Chatfield C., Lawson V. A., Crowe S., Maerz A., Sonza S., Learmont J., et al. (1995) Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 270, 988–991 10.1126/science.270.5238.988 [DOI] [PubMed] [Google Scholar]
- 6. Foster J. L., and Garcia J. V. (2008) HIV-1 Nef: at the crossroads. Retrovirology 5, 84 10.1186/1742-4690-5-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aiken C., Konner J., Landau N. R., Lenburg M. E., and Trono D. (1994) Nef induces CD4 endocytosis: requirement for a critical dileucine motif in the membrane-proximal CD4 cytoplasmic domain. Cell 76, 853–864 10.1016/0092-8674(94)90360-3 [DOI] [PubMed] [Google Scholar]
- 8. Venzke S., Michel N., Allespach I., Fackler O. T., and Keppler O. T. (2006) Expression of Nef downregulates CXCR4, the major coreceptor of human immunodeficiency virus, from the surface of target cells and thereby enhances resistance to superinfection. J. Virol. 80, 11141–11152 10.1128/JVI.01556-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sloan R. D., Donahue D. A., Kuhl B. D., Bar-Magen T., and Wainberg M. A. (2010) Expression of Nef from unintegrated HIV-1 DNA downregulates cell surface CXCR4 and CCR5 on T-lymphocytes. Retrovirology. 7, 44 10.1186/1742-4690-7-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Veillette M., Désormeaux A., Medjahed H., Gharsallah N. E., Coutu M., Baalwa J., Guan Y., Lewis G., Ferrari G., Hahn B. H., Haynes B. F., Robinson J. E., Kaufmann D. E., Bonsignori M., Sodroski J., et al. (2014) Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J. Virol. 88, 2633–2644 10.1128/JVI.03230-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Collins K. L., Chen B. K., Kalams S. A., Walker B. D., and Baltimore D. (1998) HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391, 397–401 10.1038/34929 [DOI] [PubMed] [Google Scholar]
- 12. Schwartz O., Maréchal V., Le Gall S., Lemonnier F., and Heard J. M. (1996) Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 2, 338–342 10.1038/nm0396-338 [DOI] [PubMed] [Google Scholar]
- 13. Rosa A., Chande A., Ziglio S., De Sanctis V., Bertorelli R., Goh S. L., McCauley S. M., Nowosielska A., Antonarakis S. E., Luban J., Santoni F. A., and Pizzato M. (2015) HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526, 212–217 10.1038/nature15399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Usami Y., Wu Y., and Göttlinger H. G. (2015) SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 526, 218–223 10.1038/nature15400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trible R. P., Emert-Sedlak L., and Smithgall T. E. (2006) HIV-1 Nef selectively activates SRC family kinases HCK, LYN, and c-SRC through direct SH3 domain interaction. J. Biol. Chem. 281, 27029–27038 10.1074/jbc.M601128200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Briggs S. D., Sharkey M., Stevenson M., and Smithgall T. E. (1997) SH3-mediated Hck tyrosine kinase activation and fibroblast transformation by the Nef protein of HIV-1. J. Biol. Chem. 272, 17899–17902 10.1074/jbc.272.29.17899 [DOI] [PubMed] [Google Scholar]
- 17. Emert-Sedlak L. A., Narute P., Shu S. T., Poe J. A., Shi H., Yanamala N., Alvarado J. J., Lazo J. S., Yeh J. I., Johnston P. A., and Smithgall T. E. (2013) Effector kinase coupling enables high-throughput screens for direct HIV-1 Nef antagonists with antiretroviral activity. Chem. Biol. 20, 82–91 10.1016/j.chembiol.2012.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Narute P. S., and Smithgall T. E. (2012) Nef alleles from all major HIV-1 clades activate Src-family kinases and enhance HIV-1 replication in an inhibitor-sensitive manner. PLoS One 7, e32561 10.1371/journal.pone.0032561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Emert-Sedlak L., Kodama T., Lerner E. C., Dai W., Foster C., Day B. W., Lazo J. S., and Smithgall T. E. (2009) Chemical library screens targeting an HIV-1 accessory factor/host cell kinase complex identify novel antiretroviral compounds. ACS Chem. Biol. 4, 939–947 10.1021/cb900195c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alvarado J. J., Tarafdar S., Yeh J. I., and Smithgall T. E. (2014) Interaction with the Src homology (SH3-SH2) region of the Src-family kinase Hck structures the HIV-1 Nef dimer for kinase activation and effector recruitment. J. Biol. Chem. 289, 28539–28553 10.1074/jbc.M114.600031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee C.-H., Saksela K., Mirza U. A., Chait B. T., and Kuriyan J. (1996) Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH3 domain. Cell 85, 931–942 10.1016/S0092-8674(00)81276-3 [DOI] [PubMed] [Google Scholar]
- 22. Arold S., Franken P., Strub M. P., Hoh F., Benichou S., Benarous R., and Dumas C. (1997) The crystal structure of HIV-1 Nef protein bound to the Fyn kinase SH3 domain suggests a role for this complex in altered T cell receptor signaling. Structure 5, 1361–1372 10.1016/S0969-2126(97)00286-4 [DOI] [PubMed] [Google Scholar]
- 23. Moroco J. A., Alvarado J. J., Staudt R. P., Shi H., Wales T. E., Smithgall T. E., and Engen J. R. (2018) Remodeling of HIV-1 Nef structure by Src-family kinase binding. J. Mol. Biol. 430, 310–321 10.1016/j.jmb.2017.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Andreotti A. H., Schwartzberg P. L., Joseph R. E., and Berg L. J. (2010) T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb. Perspect. Biol 2, a002287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Readinger J. A., Schiralli G. M., Jiang J. K., Thomas C. J., August A., Henderson A. J., and Schwartzberg P. L. (2008) Selective targeting of ITK blocks multiple steps of HIV replication. Proc. Natl. Acad. Sci. U.S.A. 105, 6684–6689 10.1073/pnas.0709659105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tarafdar S., Poe J. A., and Smithgall T. E. (2014) The accessory factor Nef links HIV-1 to Tec/Btk kinases in an Src homology 3 domain-dependent manner. J. Biol. Chem. 289, 15718–15728 10.1074/jbc.M114.572099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Courtney A. H., Lo W. L., and Weiss A. (2018) TCR signaling: mechanisms of initiation and propagation. Trends Biochem. Sci. 43, 108–123 10.1016/j.tibs.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin T. A., McIntyre K. W., Das J., Liu C., O'Day K. D., Penhallow B., Hung C. Y., Whitney G. S., Shuster D. J., Yang X., Townsend R., Postelnek J., Spergel S. H., Lin J., Moquin R. V., et al. (2004) Selective Itk inhibitors block T-cell activation and murine lung inflammation. Biochemistry 43, 11056–11062 10.1021/bi049428r [DOI] [PubMed] [Google Scholar]
- 29. Roskoski R., Jr. (2016) Ibrutinib inhibition of Bruton protein-tyrosine kinase (BTK) in the treatment of B cell neoplasms. Pharmacol. Res. 113, 395–408 10.1016/j.phrs.2016.09.011 [DOI] [PubMed] [Google Scholar]
- 30. Mohamed A. J., Yu L., Bäckesjö C. M., Vargas L., Faryal R., Aints A., Christensson B., Berglöf A., Vihinen M., Nore B. F., and Smith C. I. (2009) Bruton's tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 228, 58–73 10.1111/j.1600-065X.2008.00741.x [DOI] [PubMed] [Google Scholar]
- 31. Emert-Sedlak L. A., Loughran H. M., Shi H., Kulp J. L. 3rd, Shu ST III, Zhao J., Day B. W., Wrobel J. E., Reitz A. B., and Smithgall T. E. (2016) Synthesis and evaluation of orally active small molecule HIV-1 Nef antagonists. Bioorg. Med. Chem. Lett. 26, 1480–1484 10.1016/j.bmcl.2016.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mujib S., Saiyed A., Fadel S., Bozorgzad A., Aidarus N., Yue F. Y., Benko E., Kovacs C., Emert-Sedlak L., Smithgall T. E., and Ostrowski M. A. (2017) Pharmacologic HIV-1 Nef blockade enhances the recognition and elimination of latently HIV-1 infected CD4 T cells by autologous CD8 T cells. J. Clin. Invest. Insight 2, 93684 10.1172/jci.insight.93684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Poe J. A., and Smithgall T. E. (2009) HIV-1 Nef dimerization is required for Nef-mediated receptor downregulation and viral replication. J. Mol. Biol. 394, 329–342 10.1016/j.jmb.2009.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang T., Lu Y., Polk A., Chowdhury P., Murga-Zamalloa C., Fujiwara H., Suemori K., Beyersdorf N., Hristov A. C., Lim M. S., Bailey N. G., and Wilcox R. A. (2017) T-cell receptor signaling activates an ITK/NF-κB/GATA-3 axis in T-cell lymphomas facilitating resistance to chemotherapy. Clin. Cancer Res. 23, 2506–2515 10.1158/1078-0432.CCR-16-1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heusinger E., and Kirchhoff F. (2017) Primate lentiviruses modulate NF-κB activity by multiple mechanisms to fine-tune viral and cellular gene expression. Front. Microbiol. 8, 198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Manninen A., Renkema G. H., and Saksela K. (2000) Synergistic activation of NFAT by HIV-1 nef and the Ras/MAPK pathway. J. Biol. Chem. 275, 16513–16517 10.1074/jbc.M910032199 [DOI] [PubMed] [Google Scholar]
- 37. Collette Y., Arold S., Picard C., Janvier K., Benichou S., Benarous R., Olive D., and Dumas C. (2000) HIV-2 and SIV nef proteins target different Src family SH3 domains than does HIV-1 Nef because of a triple amino acid substitution. J. Biol. Chem. 275, 4171–4176 10.1074/jbc.275.6.4171 [DOI] [PubMed] [Google Scholar]
- 38. Linnemann T., Zheng Y. H., Mandic R., and Peterlin B. M. (2002) Interaction between Nef and phosphatidylinositol-3-kinase leads to activation of p21-activated kinase and increased production of HIV. Virology 294, 246–255 10.1006/viro.2002.1365 [DOI] [PubMed] [Google Scholar]
- 39. Devkota S., Joseph R. E., Boyken S. E., Fulton D. B., and Andreotti A. H. (2017) An autoinhibitory role for the pleckstrin homology domain of interleukin-2–inducible tyrosine kinase and its interplay with canonical phospholipid recognition. Biochemistry 56, 2938–2949 10.1021/acs.biochem.6b01182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moarefi I., LaFevre-Bernt M., Sicheri F., Huse M., Lee C.-H., Kuriyan J., and Miller W. T. (1997) Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature 385, 650–653 10.1038/385650a0 [DOI] [PubMed] [Google Scholar]
- 41. Chung J. K., Nocka L. M., Decker A., Wang Q., Kadlecek T. A., Weiss A., Kuriyan J., and Groves J. T. (2019) Switch-like activation of Bruton's tyrosine kinase by membrane-mediated dimerization. Proc. Natl. Acad. Sci. U.S.A. 116, 10798–10803 10.1073/pnas.1819309116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pizzato M., Helander A., Popova E., Calistri A., Zamborlini A., Palù G., and Göttlinger H. G. (2007) Dynamin 2 is required for the enhancement of HIV-1 infectivity by Nef. Proc. Natl. Acad. Sci. U.S.A. 104, 6812–6817 10.1073/pnas.0607622104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu Y., Olety B., Weiss E. R., Popova E., Yamanaka H., and Gottlinger H. (2019) Potent enhancement of HIV-1 replication by Nef in the absence of SERINC3 and SERINC5. MBio. 10 10.1128/mbio.01071-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi H., Tice C. M., Emert-Sedlak L., Chen L., Li W. F., Carlsen M., Wrobel J. E., Reitz A. B., and Smithgall T. E. (2020) Tight-binding hydroxypyrazole HIV-1 Nef inhibitors suppress viral replication in donor mononuclear cells and reverse Nef-mediated MHC-I downregulation. ACS Infect. Dis. 6, 302–312 10.1021/acsinfecdis.9b00382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu M., Alvarado J. J., Augelli-Szafran C. E., Ptak R. G., and Smithgall T. E. (2018) A single beta-octyl glucoside molecule induces HIV-1 Nef dimer formation in the absence of partner protein binding. PLoS One 13, e0192512 10.1371/journal.pone.0192512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gaud G., Lesourne R., and Love P. E. (2018) Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 18, 485–497 10.1038/s41577-018-0020-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.